

Abstract
Trastuzumab deruxtecan (DS‐8201) is a human epidermal growth factor receptor 2 (HER2)–targeting antibody–drug conjugate with a novel enzyme‐cleavable linker, a topoisomerase I inhibitor payload, and a drug‐to‐antibody ratio of ≈ 8. We have characterized the population pharmacokinetics (PK) of trastuzumab deruxtecan and released drug (topoisomerase I inhibitor) in patients with HER2‐positive breast cancer or other solid tumor malignancies. This analysis includes pooled data from five clinical studies with 639 patients. Trastuzumab deruxtecan doses ranged from 0.8 to 8.0 mg/kg every 3 weeks. Serum concentrations of trastuzumab deruxtecan and released drug were analyzed using a sequential two‐step approach, with the nonlinear mixed‐effects modeling methods. Covariate assessment was based upon stepwise forward‐addition and backward‐elimination process, followed by both univariate and multivariate analysis quantifying their impact on steady‐state exposure of trastuzumab deruxtecan and released drug. A two‐compartment model with linear elimination best described PK profiles of intact trastuzumab deruxtecan, while a one‐compartment model with time‐varying release‐rate constant and linear elimination described released‐drug PK profiles. Statistically significant covariates (country, tumor size, sex, formulation, age, body weight, albumin, total bilirubin, and aspartate aminotransferase) resulted in < 20% change in steady‐state area under the concentration‐time curve of trastuzumab deruxtecan and released drug, except for increased body weight (95th percentile, 86 kg) and decreased albumin (5th percentile, 31 g/L). Analysis of patients stratified by country, race, renal function, and hepatic function found no clinically meaningful differences in steady‐state exposure of intact trastuzumab deruxtecan or released drug. Overall, results suggest that no dose adjustment based on tested covariates or in specific patient populations is warranted.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
☑ Trastuzumab deruxtecan is a novel anti–human epidermal growth factor receptor 2 (HER2) antibody–drug conjugate delivering targeted cytotoxic activity associated with topoisomerase I inhibition. It is approved for patients with previously treated HER2‐positive, unresectable or metastatic breast cancer who received previous therapy.
WHAT QUESTION DID THIS STUDY ADDRESS?
☑ We aimed to characterize population pharmacokinetics (PK), assess the impact of potential covariates on intact trastuzumab deruxtecan and released drug, and estimate individual post hoc PK parameters for use in an exposure–response analysis.
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
☑ This is the first report describing the population PK characteristics of trastuzumab deruxtecan. Statistically significant covariates included in PK models appeared not to have a clinically meaningful impact on the steady‐state exposure of intact trastuzumab deruxtecan or released drug, and no clinically significant differences in exposure were observed when patients were stratified by country, race, renal function, or hepatic function.
HOW MIGHT THIS CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE?
☑ Trastuzumab deruxtecan can be used in all patients at the approved dosage without needing dose adjustment based on tested covariates or in specific patient populations.
Overall, mortality rates of breast cancer have been decreasing as a result of early detection and improved treatment options. 1 However, among the 15–20% of patients whose tumors are positive for human epidermal growth factor receptor 2 (HER2) expression, historically outcomes are poorer, and rates of recurrence are higher. 2 , 3 The current standard of care for first‐line treatment of HER2‐positive unresectable or metastatic breast cancer consists of HER2‐targeted agents trastuzumab and pertuzumab combined with taxanes. After treatment failure, trastuzumab emtansine, an antibody–drug conjugate (ADC), is typically used as second‐line therapy. 4 , 5 , 6
Trastuzumab deruxtecan (DS‐8201) is a rationally designed ADC that addresses limitations of previous‐generation ADCs. It combines a humanized anti‐HER2 antibody backbone with a topoisomerase I inhibitor payload (an exatecan derivative) through the use of an enzymatically cleavable peptide‐based linker. 7 The anti‐HER2 antibody component of trastuzumab deruxtecan is a human monoclonal immunoglobulin G1 produced with reference to the same amino acid sequence as trastuzumab. 8 Importantly, trastuzumab deruxtecan contains a peptide‐based linker designed to be preferentially cleaved by lysosomal enzymes. These enzymes, including cathepsins B and L, are overexpressed in tumor cells, which facilitates cleavage of free drug in tumor cells while limiting drug release in plasma. 7 Unlike trastuzumab emtansine, the released drug (topoisomerase I inhibitor) associated with trastuzumab deruxtecan has high cell‐membrane permeability, potentially allowing for a potent cytotoxic effect on neighboring tumor cells regardless of target expression; this is known as the bystander effect. 7 Additionally, the released drug has a short half‐life designed to minimize broad systemic exposure. 9 In a comparison, the topoisomerase I inhibitor used in trastuzumab deruxtecan was found to more potently inhibit DNA topoisomerase I (concentration of drug producing 50% inhibition: 0.31 µM) than SN‐38, the sacituzumab govitecan payload (concentration of drug producing 50% inhibition: 2.78 µM), an ADC payload with a similar mechanism of action. 8 The use of this linker‐payload system also enabled conjugation of eight molecules of deruxtecan with each antibody, a higher ratio than that of most other ADCs, which typically range from 2 to 4. This facilitates higher delivery of released drug to targeted cells, and preclinical studies have demonstrated this can be achieved without an increase in toxicity. 8
The efficacy and safety of trastuzumab deruxtecan were evaluated in the registrational phase II (A Study of DS‐8201a in Metastatic Breast Cancer Previously Treated With Trastuzumab Emtansine (T‐DM1) (DESTINY‐Breast01)) study of patients with HER2‐positive breast cancer who had received previous treatment with trastuzumab emtansine. Based on the results of this trial, 10 trastuzumab deruxtecan was recently approved by the US Food and Drug Administration (FDA) for the treatment of adult patients with HER2‐positive, unresectable or metastatic breast cancer who have received two or more prior anti‐HER2–based regimens in the metastatic setting.
To evaluate trastuzumab deruxtecan dosing in patient subgroups of interest, we characterized the population pharmacokinetics (PK) of trastuzumab deruxtecan and released drug in patients with HER2‐positive advanced breast cancer or other HER2‐expressing advanced solid tumors.
Methods
Analysis data set and sampling schedules
Trastuzumab deruxtecan was administered every 3 weeks (dose range, 0.8–8.0 mg/kg) with a treatment cycle length of 21 days. This analysis was performed using data from 4 phase I studies (J101, 11 J102, 12 A103, 13 A104 14 ) and a phase II study (DESTINY‐Breast01). 10 Details regarding the study population, dose regimens, and PK sampling scheme are provided in Table S1 . Serum concentrations of intact trastuzumab deruxtecan were determined by a validated electrochemiluminescence assay, with lower limit of quantification of 400 ng/mL, and serum concentrations of released drug were determined by a liquid chromatography–tandem mass spectrometry method, with lower limit of quantification of 0.01 ng/mL. Patients who received at least one trastuzumab deruxtecan dose and had at least one postdose PK sample of intact trastuzumab deruxtecan or released drug were included in the analysis. Collectively, the final analysis data set included a total of 11,434 intact trastuzumab deruxtecan concentrations and 11,480 released drug concentrations from 639 patients.
Population PK analysis
Population PK modeling for intact trastuzumab deruxtecan and released drug was conducted using the first‐order conditional estimation with interaction method in NONMEM (version 7.3) (ICON Development Solutions, Ellicott City, MD). Xpose and Perl‐Speaks‐NONMEM (PsN) (version 4.6.0) (Department of Pharmacy, Uppsala University, Uppsala, Sweden) was used for model diagnostics and facilitation of NONMEM tasks, such as covariate testing. R (version 3.5.1) (R Core Team, Vienna, Austria) was used for data exploration, postprocessing of NONMEM results, and simulations.
A sequential two‐step approach was used in this analysis. First, PK profiles of intact trastuzumab deruxtecan were fitted to the model; next, PK parameter estimates, including the fixed‐ and random‐effects terms of trastuzumab deruxtecan, were fixed, and released‐drug parameters were then estimated.
For the base‐model determination, several structure compartment models with linear or nonlinear clearance were explored. Between‐subject variability (BSV) was modeled using an exponential random effect on all PK parameters. Residual variability included additive and proportional error terms for trastuzumab deruxtecan and a proportional error term only for released drug. Covariates evaluated (listed in Table S2 ) were based on known characteristics of trastuzumab deruxtecan and released drug, as well as prior knowledge about trastuzumab emtansine.
Correlations between covariates were first explored to identify confounded covariates. Correlations between post hoc individual random effects and covariates were then explored using pairwise correlations. Analysis‐of‐variance tests for categorical covariates and linear regressions for continuous covariates were conducted to identify possible covariate relationships significant at P < 0.05. Significance levels of P ≤ 0.01 (a drop in objective function value (OFV) ≥ 6.63 when adding a covariate) and P ≤ 0.001 (an increase in OFV ≥ 10.83 when removing a covariate) were used in the stepwise forward‐addition and backward‐elimination processes, respectively, for the selection of covariates in the final model.
Model selection was based on OFV, goodness‐of‐fit plots, precision in parameter estimates, and scientific plausibility. Inspection of goodness of fit and prediction‐corrected visual predictive checks (pcVPC) were done for major steps of model building, including the base and final models. Evaluation of model robustness and parameter precision was conducted using nonparametric bootstrapping. A total of 200 replicated data sets were generated and then fitted to the model to obtain the parameter estimate and 95% confidence interval (CI). Sensitivity analysis was conducted by reincluding outliers (conditioned weighted residuals > 5 in the base model) and rerunning the final models.
Model application
The impact of identified statistically significant covariates on intact trastuzumab deruxtecan and released‐drug exposures at steady state, i.e., area under the concentration‐time curve (AUC), maximum concentration (Cmax), and minimum concentration (Cmin), was examined in both univariate and multivariate fashion. For the univariate approach, simulations were performed by varying each covariate value one at a time and keeping other covariates as constant (that is, categorical covariates were set at the reference categories and continuous covariate values were set to the median values in the data set). Exposures of intact trastuzumab deruxtecan and released drug at the 5th, 25th, 75th, and 95th percentiles of the continuous covariates, or at each level of the categorical covariates, were then compared with the exposure estimates for a reference patient (with typical covariate values) via a forest plot. A total of 1,000 simulations were conducted for each covariate effect.
For the multivariate approach, individual intact trastuzumab deruxtecan and released‐drug exposures were calculated from the individual post hoc PK parameters, and then summarized and compared among subgroups of interest, such as Asian vs. non‐Asian, Japanese vs. non‐Japanese Asian vs. non‐Japanese other, hepatic function categories, 15 and renal function categories. 16 Specifically, individual post hoc PK parameters for all patients with breast cancer included in the final models for intact trastuzumab deruxtecan and released drug were used to simulate the corresponding individual concentration‐time profiles with the 5.4‐mg/kg every‐3‐weeks dosing regimen.
Results
Population PK analysis data sets
Among 639 patients whose data were included in the analysis data set, 512 (80.1%) had unresectable and/or metastatic breast cancer and 445 (69.6%) had HER2‐positive, unresectable and/or metastatic breast cancer. PK profiles stratified by study and dose level are presented in Figure 1a–f and for intact trastuzumab deruxtecan and released drug, respectively. Intact trastuzumab deruxtecan concentration follows a biphasic pattern, suggesting a two‐compartment model would describe the data. PK profiles of the released drug appear to be driven by intact trastuzumab deruxtecan elimination kinetics. Summaries of baseline categorical and continuous covariates by study are shown in Tables S3 and S4 , respectively. Summaries of baseline categorical and continuous covariates by trastuzumab deruxtecan dose level are shown in Tables S5 and S6 , respectively.
Figure 1.
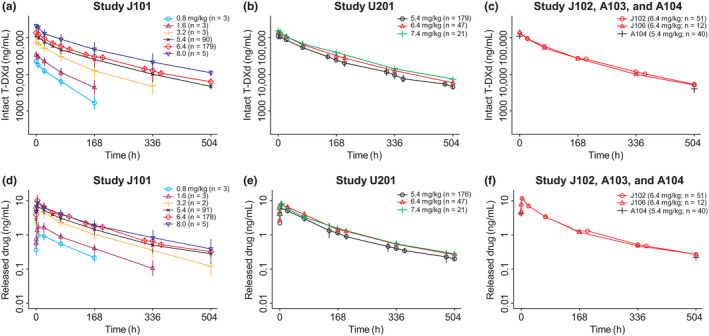
Observed pharmacokinetic profiles by study and dose following first dose of (a–c) intact T‐DXd (trastuzumab deruxtecan) and (d–f) released drug. Note: Points are geometric mean concentrations at nominal time points after the first dose. Vertical lines are ±1 standard error in the logarithmic domain. The plots show the curves over the first 504 hours. T‐DXd, trastuzumab deruxtecan.
Development of intact trastuzumab deruxtecan model
A two‐compartment model with linear elimination was found to adequately describe the PK profiles of intact trastuzumab deruxtecan (Figure 2 ). BSV was evaluated on all PK parameters, including clearance (CLintact), central volume of distribution (V1,intact), distribution clearance (Qintact), and peripheral volume of distribution (V2,intact).
Figure 2.
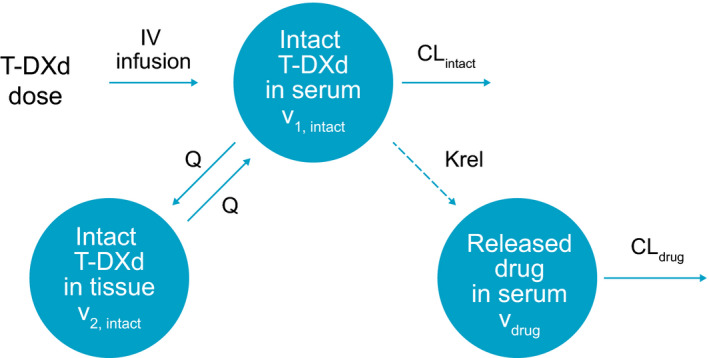
Pharmacokinetic model structure. CLdrug, released drug clearance; CLintact, intact T‐DXd clearance; IV, intravenous; Krel, release‐rate constant; Q, intercompartment distribution clearance for intact T‐DXd; T‐DXd, trastuzumab deruxtecan; V1, intact, intact T‐DXd central volume of distribution; V2, intact, intact T‐DXd peripheral volume of distribution; Vdrug, released drug volume of distribution.
The following covariate relationships were significant at P < 0.05, on the basis of the initial analysis‐of‐variance or linear regressions:
CLintact: baseline tumor size, albumin, lactate dehydrogenase (LDH), HER2 status, tumor type, body weight, age, sex, race, and country
V1,intact: body weight, sex, race, and country
V2,intact: trastuzumab deruxtecan formulation, body weight, race, and country
Among these covariates, race and country were highly confounded (correlation coefficient, −0.81). Race was removed, while country was retained in the subsequently stepwise covariate modeling, because country was more significant on all PK parameters. During the next stepwise forward‐addition and backward‐elimination processes, the effect of age, tumor type, HER2 status, LDH, or trastuzumab deruxtecan formulation was removed, resulting in the final model that included the following parameter‐covariate relationships:
Therefore, increased body weight, increased baseline tumor size, and male sex were associated with increased CLintact. Patients with increased baseline albumin and Japanese patients had decreased CLintact. The V1,intact was greater in males and patients with increased body weight, and the V2,intact was lower in Japanese patients.
No parameter‐covariate relationships were included for Qintact. Parameters for the final intact trastuzumab deruxtecan model are shown in Table 1 together with results of the bootstrap analysis and sensitivity analysis. The relative standard error (RSE) for the PK parameters ranged from < 1% to 27%, indicating good precision of model parameter estimates. The BSV was 25%, 16%, 20%, and 66% for CLintact, V1,intact, Qintact, and V2,intact, respectively, and the shrinkage ranged from 5.5% to 28.6%. Parameter estimates of the final intact trastuzumab deruxtecan model were in good agreement with and fell within the 95% CI of the bootstrap analysis. Parameter estimates were comparable overall between the sensitivity analysis and final trastuzumab deruxtecan model run, except that in the sensitivity analysis, there was a smaller effect size and less precision of the estimated Japan country effect on clearance. The goodness‐of‐fit diagnostics (Figure S1 a) and pcVPC (Figure 3a ) also demonstrated that the final model provided adequate description of the observed data.
Table 1.
Parameter estimates from the final population pharmacokinetics model for intact trastuzumab deruxtecan
| Parameter | Final intact T‐DXd model | Bootstrap analysis a | Sensitivity analysis with inclusion of outliers | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Estimate | Between‐patient variability | Estimate | Estimate | ||||||
| Typical value | RSE b (%) | Magnitude (%CV) | RSE b (%) | Shrinkage c (%) | Typical value | 95% CI | Typical value | RSE b (%) | |
| Clearance (CLintact, L/day) | 0.421 | 1.59 | 25.1 | 3.10 | 5.87 | 0.420 | 0.405–0.434 | 0.388 | 2.35 |
| Central volume of distribution (V1,intact, L) | 2.77 | 0.793 | 15.8 | 2.58 | 5.53 | 2.78 | 2.74–2.81 | 2.80 | 1.07 |
| Distributional clearance (Qintact, L/day) | 0.199 | 1.91 | 30.0 | 5.04 | 28.6 | 0.200 | 0.190–0.212 | 0.197 | 2.20 |
| Peripheral volume of distribution (V2,intact, L) | 5.16 | 5.15 | 65.6 | 5.42 | 23.1 | 5.17 | 4.40–5.97 | 5.52 | 6.25 |
| Albumin on CLintact | –0.533 | 13.0 | — | — | — | –0.509 | –0.686 to −0.315 | –0.524 | 21.8 |
| Country (Japan) on CLintact d | –0.0970 | 22.0 | — | — | — | –0.096 | –0.148 to −0.0468 | –0.0180 | 215 |
| Sex (male) on CLintact | 0.174 | 27.3 | — | — | — | 0.171 | 0.0955–0.267 | 0.194 | 47.2 |
| Tumor size on CLintact | 0.0710 | 21.1 | — | — | — | 0.072 | 0.0310–0.108 | 0.060 | 37.5 |
| Body weight on CLintact | 0.370 | 14.8 | — | — | — | 0.379 | 0.234–0.516 | 0.404 | 17.6 |
| Sex (male) on V1,intact | 0.197 | 11.7 | — | — | — | 0.193 | 0.141–0.254 | 0.208 | 16.3 |
| Body weight on V1,intact | 0.489 | 6.89 | — | — | — | 0.492 | 0.432–0.551 | 0.505 | 9.38 |
| Country (Japan) on V2,intact d | –0.262 | 19.6 | — | — | — | –0.256 | –0.397 to −0.0759 | –0.305 | 19.1 |
| Residual variability e | |||||||||
| Proportional residual error SD | 0.163 | 0.370 | — | — | — | 0.163 | 0.154–0.172 | 0.209 | 0.360 |
| Additive residual error SD (ng/mL) | 1,181 | 2.90 | — | — | — | 1,192 | 1,048–1,331 | 784 | 4.94 |
| Between‐patient variability | |||||||||
| Variance of CLintact | 0.0630 | 6.20 | — | — | — | 0.062 | 0.0539–0.0732 | 0.112 | 5.68 |
| Covariance of CLintact and V1,intact f | 0.0210 | 9.58 | — | — | — | 0.0215 | 0.0181–0.0256 | 0.023 | 8.52 |
| Variance of V1,intact | 0.0250 | 5.16 | — | — | — | 0.0245 | 0.0210–0.0298 | 0.0360 | 3.84 |
| Variance of Qintact | 0.0900 | 10.1 | — | — | — | 0.0897 | 0.0538–0.111 | 0.110 | 10.3 |
| Variance of V2,intact | 0.430 | 10.8 | — | — | — | 0.426 | 0.304–0.529 | 0.575 | 10.2 |
CI, confidence interval; CLintact, intact T‐DXd clearance; CV, coefficient of variation; Qintact, intact T‐DXd distribution clearance; RSE, relative standard error; T‐DXd, trastuzumab deruxtecan; V1,intact, intact T‐DXd central volume of distribution; V2intact, intact T‐DXd peripheral volume of distribution; —, not applicable.
200 bootstrapped data sets were used.
RSE of parameter estimate is calculated as 100 × (SE/typical value); RSE of between‐patient variability magnitude is presented on %CV scale and approximated as 100 × (SE/variance estimate)/2.
Shrinkage (%) is calculated as 100 × (1 − variance of post hoc/estimated variance).
The country covariate was initially tested with three categories (Japan, non‐Japan Asia, and non‐Japan other) but was reduced to two categories (Japan vs. non‐Japan) at the final intact T‐DXd model refinement stage. Because there was no difference in their covariate effect on clearance and volume of distribution, the non‐Japan Asia and non‐Japan other categories were combined into the non‐Japan category.
Overall residual variability shrinkage was estimated to be 7.97%.
The correlation coefficient between CLintact and V1,intact was estimated as 0.539.
Figure 3.
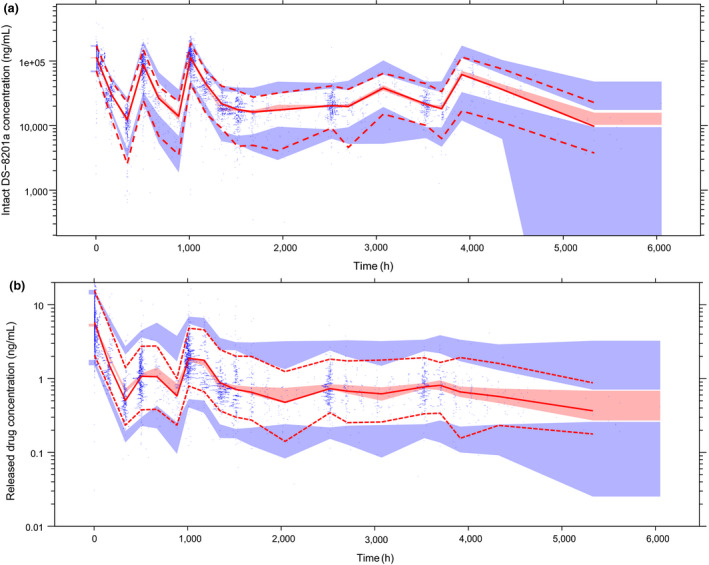
Prediction‐corrected visual predictive check (pcVPC) of observed vs. simulated exposure to (a) intact T‐DXd (trastuzumab deruxtecan) and (b) released drug. Note: Solid line corresponds to prediction‐corrected observed median; dashed lines correspond to the 2.5th and 97.5th percentiles of prediction‐corrected observations; shaded regions correspond to 95% of the simulated (n = 1,000) values of the predicted median (pink) and 2.5th and 97.5th percentiles (blue). Blue dots correspond to prediction‐corrected observed data.
Development of released‐drug model
The structural model for released drug was a one‐compartment model, with first‐order release rate from trastuzumab deruxtecan and first‐order elimination (Figure 2 ). The time‐course of intact trastuzumab deruxtecan concentrations, adjusted for the drug–antibody ratio and molar mass, was the input to the released‐drug model. Released‐drug concentrations were observed to decrease over treatment cycles; therefore, time‐varying release‐rate constant (Krel) was considered. Based on an evaluation of various functions, Krel was modeled according to the following, including a component describing a gradual decline per cycle with a power function, and a second component describing a further constant reduction in Krel after cycle 1:
This combination enabled an adequate description of released‐drug concentrations over the entire time‐course of data. The net effect of both components is a reduction of 39% in Krel at cycle 10 and 45% at cycle 20, compared with cycle 1.
Following the same covariate assessment procedures as described for trastuzumab deruxtecan, the following parameter‐covariate relationships were found to be statistically significant and were included in the final model for released drug:
The model suggests that clearance of released drug (CLdrug) is increased with increasing body weight, and clearance is decreased with increasing bilirubin, aspartate aminotransferase (AST), and coadministration of itraconazole or ritonavir. Volume of distribution of released drug (Vdrug) was fixed at 17 L/m2 according to the literature‐reported value of exatecan mesylate, since this parameter was not uniquely identifiable. 8 Vdrug is higher in older participants and participants receiving frozen liquid drug product 2 and lower in participants receiving frozen liquid drug product 1.
Parameters for the final released‐drug model are shown in Table 2 , together with results of the bootstrap analysis, and sensitivity analysis. Model parameters were estimated with good precision; the RSE for PK parameters ranged from 1.6% to 28%, shrinkage ranged from 16% to 38%, and parameter estimates of the final released‐drug model were in good agreement with and fell within the 95% CIs from bootstrap analysis. BSV percent coefficient of variation was 25.4%, 42.0%, 37.6%, and 22.9% for CLdrug, Vdrug, Krel, and fraction of decrease in Krel, respectively. No significant differences were noted between the sensitivity analysis and final released‐drug model estimates. The goodness‐of‐fit diagnostics (Figure S1 b) and pcVPC (Figure 3b ) support the overall adequate prediction performance by the final model, except that there are slight underpredictions for the high concentrations.
Table 2.
Parameter estimates from the final population pharmacokinetics model for released drug
| Parameter | Final released‐drug model | Bootstrap analysis a | Sensitivity analysis with inclusion of outliers | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Estimate | Between‐patient variability | Estimate | Estimate | ||||||
| Typical value | RSE b (%) | Magnitude (%CV) | RSE b (%) | Shrinkage c (%) | Typical value | 95% CI | Typical value | RSE b (%) | |
| Released‐drug clearance (CLdrug, L/h) | 19.2 | 4.41 | 25.4 | 6.31 | 37.8 | 19.1 | 17.8–20.4 | 19.1 | 4.50 |
| Released‐drug volume of distribution (Vdrug, L) |
Fixed 17•BSA |
— | 42.0 | 4.64 | 19.9 |
Fixed 17•BSA |
— |
Fixed 17•BSA |
— |
| Release rate (Krel, 1/h) | 0.0159 | 4.42 | 37.6 | 4.56 | 16.0 | 0.0159 | 0.0146–0.0172 | 0.0158 | 4.37 |
| Fraction of Krel when cycle > 1 | 0.830 | 1.62 | 22.9 | 4.01 | 17.5 | 0.829 | 0.803–0.859 | 0.828 | 1.85 |
| Cycle effect on Krel | –0.137 | 5.23 | — | — | — | –0.137 | –0.160 to −0.114 | –0.139 | 5.40 |
| Ritonavir on CLdrug | –0.113 | 13.9 | — | — | — | –0.112 | –0.236 to −0.0177 | –0.113 | 15.2 |
| Itraconazole on CLdrug | –0.103 | 19.0 | — | — | — | –0.104 | –0.159 to −0.0535 | –0.103 | 20.9 |
| AST on CLdrug | –0.219 | 14.5 | — | — | — | –0.221 | –0.290 to −0.150 | –0.206 | 15.2 |
| Total bilirubin on CLdrug | –0.207 | 18.4 | — | — | — | –0.209 | –0.294 to −0.128 | –0.191 | 19.7 |
| Body weight on CLdrug | 0.440 | 18.9 | — | — | — | 0.448 | 0.299–0.576 | 0.480 | 17.6 |
| Age on Vdrug | 0.562 | 16.8 | — | — | — | 0.558 | 0.377–0.750 | 0.644 | 15.4 |
| FL‐DP2 formulation on Vdrug | 0.255 | 28.0 | — | — | — | 0.258 | 0.115–0.392 | 0.278 | 27.1 |
| FL‐DP1 formulation on Vdrug | –0.212 | 20.4 | — | — | — | –0.215 | –0.289 to −0.124 | –0.209 | 21.1 |
| Residual variability d | |||||||||
| Proportional residual error SD | 0.279 | 0.483 | — | — | — | 0.279 | 0.272–0.288 | 0.298 | 0.380 |
| Between‐patient variability | |||||||||
| Variance of CLdrug | 0.0647 | 12.6 | — | — | — | 0.0623 | 0.0340–0.102 | 0.0705 | 12.1 |
| Variance of Vdrug | 0.176 | 9.28 | — | — | — | 0.175 | 0.141–0.211 | 0.178 | 9.73 |
| Variance of Krel | 0.142 | 9.13 | — | — | — | 0.142 | 0.116–0.175 | 0.140 | 9.57 |
| Variance of fraction of Krel when cycle> 1 | 0.0524 | 8.01 | — | — | — | 0.0525 | 0.0438–0.0635 | 0.0692 | 6.84 |
AST, aspartate aminotransferase; BSA, body surface area; CI, confidence interval; CLdrug, released drug clearance; CV, coefficient of variation; FL‐DP1, frozen liquid drug product 1; FL‐DP2, frozen liquid drug product 2; Krel, release‐rate constant; RSE, relative standard error; Vdrug, released drug volume of distribution; —, not applicable.
200 bootstrapped data sets were used.
RSEs of parameter estimate are calculated as 100 × (SE/typical value); RSEs of between‐patient variability magnitude are presented on %CV scale and approximated as 100 × (SE/variance estimate)/2.
Shrinkage (%) is calculated as 100 × (1 − variance of post hoc/estimated variance).
Overall residual variability shrinkage was estimated to be 8.74%.
Impact of covariates on intact trastuzumab deruxtecan exposure
Figure 4 shows the results of a forest plot of relative changes in exposure (Cmax, Cmin, and AUC at steady state) when covariates were varied one at a time. Ten dosing cycles were simulated to obtain represented steady‐state profiles, since Krel difference became very small after cycle 10 (< 1% difference when compared with cycle 9). All covariates had effects less than 20% (i.e., exposure ratio was within the 0.8–1.25 range) except for patients with ≥ 86 kg (95th percentile) body weight who had an approximately 28% increase in AUC relative to a patient with the median body weight (57.8 kg), and patients with 31 g/L (5th percentile) albumin had a 23% decrease in Cmin relative to a patient with the median albumin (40 g/L).
Figure 4.
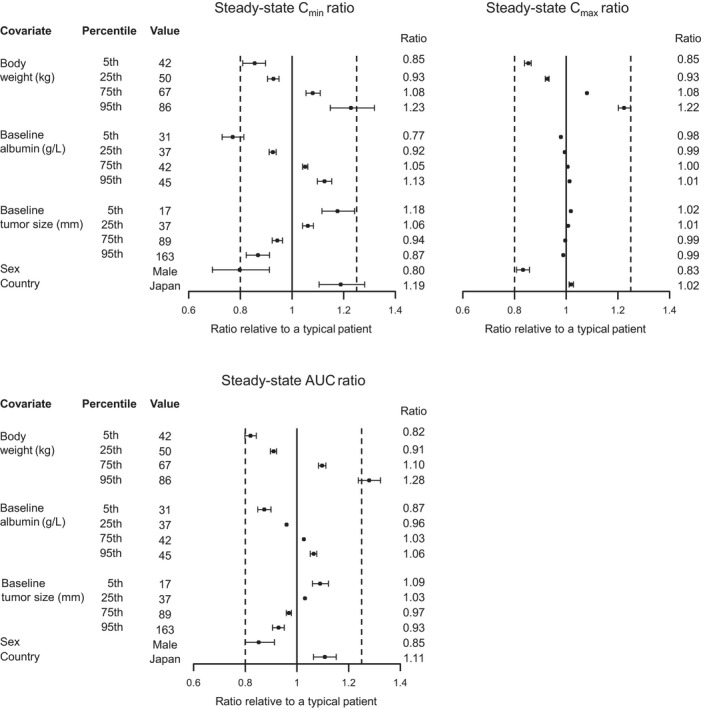
Forest plot of covariate effects on intact T‐DXd (trastuzumab deruxtecan) exposure. Note: First and second dashed vertical lines correspond to ratios of 0.8 and 1.25, respectively. The solid vertical line corresponds to a ratio of 1 and represents the typical patient. Points and whiskers represent the median and 90% confidence interval, respectively. A typical patient is defined as a female from a non‐Japan country with body weight 57.8 kg, albumin 40 g/L, and baseline tumor size 57 mm. AUC, area under the concentration‐time curve; Cmax, maximum concentration; Cmin, minimum concentration.
Trastuzumab deruxtecan is FDA approved for patients with HER2‐positive metastatic breast cancer. Comparisons of steady‐state exposure of intact trastuzumab deruxtecan with the 5.4‐mg/kg every‐3‐weeks dosing regimen and stratified by country and race are presented in Figure S2 . Exposure appears similar between patients regardless of country or race.
Impact of covariates on released‐drug exposure
In a similar fashion, a forest plot of relative changes in released‐drug exposure (Cmax, Cmin, and AUC) when covariates were varied one at a time is presented in Figure 5 . All covariates except extreme values of baseline body weight had effects less than 20%. Specifically, patients with 86 kg (95th percentile) body weight had an approximately 30% increase in AUC and 26% increase in Cmax relative to a patient with the median body weight (57.8 kg).
Figure 5.
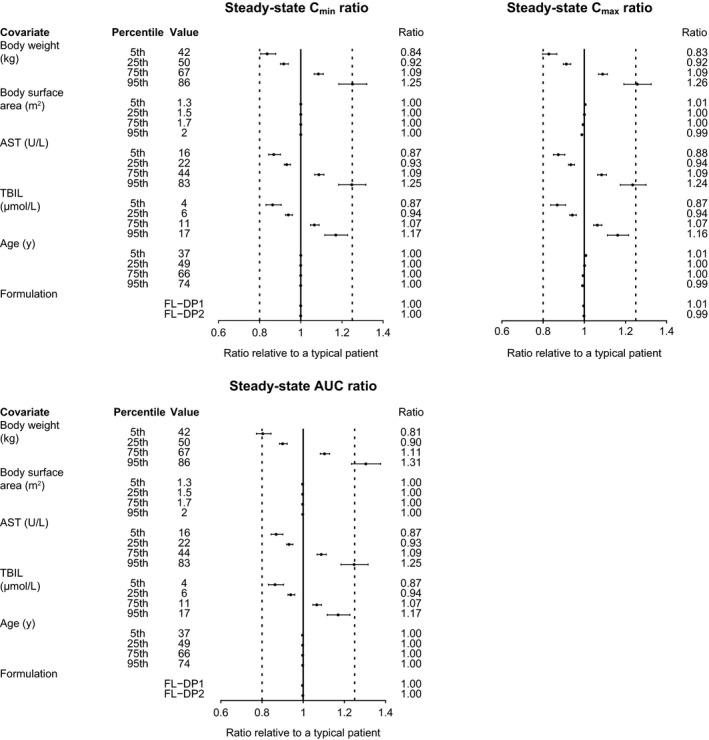
Forest plot of covariate effects on released‐drug exposure. Note: First and second dashed vertical lines correspond to ratios of 0.8 and 1.25, respectively. The solid vertical line corresponds to a ratio of 1 and represents the typical patient. Points and whiskers represent the estimate and 90% CI, respectively. A typical patient is defined as a 57‐year‐old female with body weight 57.8 kg, TBIL 8 μM, and AST 30 U/L and who was administered the Lyo‐DP formulation of T‐DXd. Covariate effects of ritonavir or itraconazole coadministration are not presented because they were assessed in the dedicated drug–drug interaction study (Study A104; N = 40). Ten cycles were simulated to represent steady‐state profiles. AST, aspartate aminotransferase; AUC, area under the concentration‐time curve; CI, confidence interval; Cmax, maximum concentration; Cmin, minimum concentration; FL‐DP1, frozen liquid drug product 1; FL‐DP2, frozen liquid drug product 2; Lyo‐DP, lyophilized powder drug product; TBIL, total bilirubin; T‐DXd, trastuzumab deruxtecan.
Steady‐state exposure of released drug with the 5.4‐mg/kg every‐3‐weeks dosing regimen was similar when patients were stratified by country, race, hepatic function, and renal function (Figures S3 and S4 ).
Discussion
Trastuzumab deruxtecan is a novel ADC recently approved by the FDA for patients with HER2‐positive, unresectable or metastatic breast cancer who have received ≥ 2 prior anti‐HER2–based regimens in the metastatic setting and by Japan’s Ministry of Health, Labor, and Welfare for patients with HER2‐positive, unresectable or recurrent breast cancer after prior chemotherapy. 17 , 18 This is the first report to characterize the population PK profiles of trastuzumab deruxtecan and released drug, based on data from 639 patients from five clinical trials in HER2‐positive, unresectable and/or metastatic breast cancer and other solid tumors. The released drug showed formation limited kinetics and its PK profile was driven by trastuzumab deruxtecan. However, the released drug is not expected to alter the PK of trastuzumab deruxtecan. Considering the large, complex data set in this analysis, a sequential two‐step modeling approach was taken because it is more efficient, can significantly reduce computing time, avoids common model convergence issues, and helps improve model stability. In addition, a comparison of this sequential two‐step modeling and a simultaneous two‐analyte modeling suggested very similar model parameter estimates for both trastuzumab deruxtecan and released drug (Daiichi Sankyo, data on file).
We have demonstrated that the PK of intact trastuzumab deruxtecan is adequately described by a two‐compartment model with linear elimination. Observed exposure (Cmax and AUC) of intact trastuzumab deruxtecan was shown to increase proportionally over a dose range of 3.2 to 8.0 mg/kg, while a trend of nonlinear elimination was apparent at lower doses of 0.8 and 1.6 mg/kg, suggesting potential target‐mediated drug disposition at these lower doses. Such characteristics are common for monoclonal antibodies 19 , 20 and have also been reported for trastuzumab. 21 During the initial base‐model exploration, linear, nonlinear, parallel linear, and nonlinear elimination were evaluated. Because limited data from the lower‐dose groups (three patients each in the 0.8‐mg/kg and 1.6‐mg/kg groups) were included in the current analysis, incorporation of a nonlinear component of elimination did not result in a marked improvement in the model fitting. Lower doses are also deemed to be suboptimal for efficacy and would not likely be included in the current dosing regimen. Furthermore, the nonlinear component accounted for < 10% of the total elimination over the observed concentration range. Therefore, a model with linear clearance was used for trastuzumab deruxtecan. This was considered appropriate for the current analysis data set and purpose of the current modeling work based on the reasons mentioned above as well as model qualification.
Body weight, albumin, tumor size, sex, and country were identified as statistically significant covariates for intact trastuzumab deruxtecan. Trastuzumab deruxtecan clearance increases with increasing body weight; this is expected and consistent with current knowledge about monoclonal antibodies. 22 However, because trastuzumab deruxtecan is administered by weight‐based dosing (i.e., drug amount administered increases with increasing weight), the apparent net effect of increased body weight is slightly increased exposure. The 5th to 95th percentile body weight range of 42–86 kg corresponded to an 18% decrease in steady‐state AUC at body weight 42 kg and a 28% increase in steady‐state AUC at body weight 86 kg relative to a typical patient. Albumin is also an expected covariate for clearance of monoclonal antibodies, 22 possibly due to elevated protein turnover associated with cancer‐related cachexia, for which low albumin is an indicator. 22 , 23 The baseline albumin effect on trastuzumab deruxtecan exposure is less than 20%, except for the extreme low value of albumin, where patients with 31 g/L (5th percentile) albumin had a 23% decrease in Cmin relative to a patient with the median albumin (40 g/L). Such an effect is not considered clinically meaningful. Although baseline tumor size, being Japanese, and being male were identified as statistically significant covariates for trastuzumab deruxtecan, their effects were all contained within the 0.8 to 1.25 ratio range and therefore were not clinically relevant. Comparison of post hoc individual exposures also confirmed similar trastuzumab deruxtecan exposure between patients regardless of country or race. No clear or direct explanations were seen for the identified effects of male sex or Japan on the volume of distribution of trastuzumab deruxtecan; such results could be data driven.
Exploratory data analysis suggested that the intact trastuzumab deruxtecan exposure tended to be slightly lower (by ~ 10%) in patients with gastric cancer than in patients with breast cancer; however, tumor type was not shown as a statistically significant covariate for trastuzumab deruxtecan. There were differences between the breast and gastric cancer patient populations in average body weight and sex ratio, which may account for the difference in exposure. In addition, the majority (80%) of patients included in this data set were patients with breast cancer; therefore, further assessment may be needed with additional data from patients with other tumor types. Studies evaluating the safety and efficacy of trastuzumab deruxtecan in HER2‐expressing advanced gastric or gastroesophageal junction adenocarcinoma, 24 , 25 HER2‐positive or HER2‐mutated, unresectable and/or metastatic non‐small cell lung cancer, 26 , 27 and HER2‐expressing advanced colorectal cancer are also ongoing 28 , 29 and may provide additional insight into the PK of trastuzumab deruxtecan in these other tumor types.
The released‐drug PK profile was driven by intact trastuzumab deruxtecan, and the kinetics of released drug manifested in the early upswing to peak concentrations in the first 6 hours after dose (Figure 1 ). Structural models with or without a time‐varying release rate were tested during the base‐model development stage. The model with a constant release rate showed largely biased predictions and a much higher OFV than the selected released‐drug base model; hence, a time‐varying component was incorporated. Based on an evaluation of various functions, the release rate was modeled to decrease over time using two components: one describes a constant reduction in release rate after cycle 1 and the other describes a gradual decline per cycle. This combination of two terms enabled an adequate description of released‐drug concentrations over the entire time‐course of data, and was consistent with the results of an approximately twofold difference in Cmin in intact trastuzumab deruxtecan between cycle 1 and steady state and a 1.4‐fold difference in Cmin in released‐drug levels between cycle 1 and steady state, determined from the individual simulated profiles with the 5.4‐mg/kg every‐3‐weeks dosing regimen (Figure S3 d–f). The decline of the release‐rate constant slows at later cycles: the release rate decreases by 39% after 10 cycles and 45% after 20 cycles, and it decreases to around half of the release rate after the 40th cycle.
The decrease in release rate over time is potentially driven by multiple mechanisms. Since trastuzumab deruxtecan contains a linker that is cleaved selectively in tumor cells, 8 changes in tumor size (e.g., tumor shrinkage) over time can impact drug release and clearance. In the current analysis, a semimechanistic time‐varying Krel model was initially considered; for example, decrease of Krel was modeled as a function of tumor shrinkage. This approach did not result in successful model convergence, presumably due to limited longitudinal tumor‐size data (tumor sum‐of‐lesion diameters was assessed every 6 weeks / 2 cycles in the first 24 weeks, then every 12 weeks afterward). Subsequently, empirical models were explored and found to provide an adequate description of the observed concentration‐time profiles of released drug. Similar empirical approaches have been used for other ADCs—including brentuximab vedotin, with which released drug was assumed to form through ADC proteolytic degradation and deconjugation and to decrease exponentially over time. 30 Further evaluation may be warranted when more data from other ongoing clinical studies are available.
The released drug showed formation rate‐limited kinetics. Body weight, age, formulation, AST, and total bilirubin (TBIL) were identified as statistically significant covariates; however, as in the case with intact trastuzumab deruxtecan, they do not appear to have a clinically meaningful impact on the steady‐state exposure of released drug and do not present a compelling indication for any dose adjustment. Similar to the findings for trastuzumab deruxtecan, increased body weight was correlated with increased clearance of released drug, but the net effect of increased body weight is slightly increased exposure of released drug because of the weight‐based dosing scheme. Age and formulation were identified as statistically significant covariates on the volume of distribution of released drug but had no effect on released‐drug AUC and Cmin and a limited effect on Cmax. This is because the released‐drug time course is primarily driven by trastuzumab deruxtecan elimination kinetics, and the volume of distribution affects only the initial rate of rise. Increased TBIL and AST were associated with decreased clearance and corresponded with a 25% and 16–17% increase in released‐drug exposure, respectively, at the 95th percentile values of AST and TBIL. The impact of these changes is not expected to be clinically meaningful because the post hoc individual AUC ratio between patients with mild hepatic impairment (according to National Cancer Institute (NCI) organ dysfunction working group criteria) and those with normal hepatic function was 1.07. No effect of creatinine clearance was identified on any of the PK parameters of released drug. These findings are consistent with current knowledge that released drug undergoes minimal renal excretion and is mainly eliminated through the hepatobiliary pathway. Overall, the exposure ranges observed in patients with mild renal impairment and those with normal renal function were similar, but there was a slightly lower (~ 10%) released‐drug exposure in mild renal impairment, which may be explained by the lower body weight of patients with mild renal impairment vs. those with normal renal function (~ 10 kg less). These results support that no dose adjustment is required in patients with mild or moderate renal or hepatic impairment. Presently no data are available for patients with severe renal or hepatic impairment. Data for patients with moderate hepatic impairment are limited; due to potentially increased exposure, close monitoring for increased toxicities related to the released drug is recommended.
Concomitant use of itraconazole and ritonavir was associated with an 11.5% and 12.7% increase in released‐drug AUC, respectively. These results agree with the observed data from the phase I study evaluating these specific drug–drug interactions. 14 Itraconazole is a strong CYP3A inhibitor, and ritonavir is a dual organic‐anion‐transporting polypeptide 1B (OATP1B)/CYP3A inhibitor. The released drug is a substrate of CYP3A, and its hepatic update is mediated by OATP1B. The results from this analysis, as well as observed data, suggest that inhibition of CYP3A and OATP1B has small effects on the exposure of released drug, and therefore strong inhibitors of CYP3A and inhibitors of OATP1B can be used without trastuzumab deruxtecan dose adjustment.
Individual post hoc steady‐state exposures in Japanese, non‐Japanese Asian, and non‐Asian patients were similar, with geometric mean ratios ranging from 0.965 to 1.08 comparing Japanese vs. non‐Asian patients and from 0.901 to 0.918 comparing non‐Japanese Asian vs. non‐Asian patients. Therefore, no dose adjustments were recommended on the basis of race.
Overall, the PK properties of trastuzumab deruxtecan are more similar to those of trastuzumab emtansine and trastuzumab than to those of small‐molecule therapies, with relatively slow clearance and a small volume of distribution mostly confined to plasma. Trastuzumab deruxtecan, similar to trastuzumab emtansine, is cleared at a faster rate than trastuzumab (clearance, 0.421 L/day with trastuzumab deruxtecan vs. 0.676 L/day with trastuzumab emtansine vs. 0.225 L/day with trastuzumab), which could be attributed to additional mechanisms of clearance for ADCs vs. unconjugated monoclonal antibodies. The central volume of distribution of trastuzumab deruxtecan (2.77 L) is similar to that of trastuzumab emtansine (3.13 L) and unconjugated trastuzumab (2.95 L). 31 , 32
In conclusion, the PK of intact trastuzumab deruxtecan was adequately described by a two‐compartment model with linear elimination from central compartment, and the PK of released drug was characterized by a one‐compartment model with time‐varying release rate and linear elimination. Although some statistically significant covariates were identified in the population PK analysis (country, tumor size, sex, formulation, age, body weight, albumin, TBIL, and AST), none had a clinically meaningful impact on steady‐state exposure of intact trastuzumab deruxtecan or released drug. Likewise, stratification of patients by country, race, renal function, and hepatic function did not reveal any clinically meaningful differences in the steady‐state exposure of intact trastuzumab deruxtecan or released drug. Based on these findings, there is no recommendation for dose adjustment based on tested covariates or specific populations.
Funding
Sponsored by Daiichi Sankyo, Inc and AstraZeneca Pharmaceuticals.
Conflicts of Interest
O.Y., T.G., M.A.T., and F.L. report employment with and equity ownership in Daiichi Sankyo, Inc; S.E. and K.Y. report employment with Daiichi Sankyo, Co, Ltd; Y.X. and R.W. report receiving consulting fees from Daiichi Sankyo, Inc; R.W. reports consulting fees outside of the submitted work from undisclosed sponsors.
Author Contributions
O.Y., S.E., K.Y., T.G., R.W., and F.L. wrote the manuscript. O.Y., K.Y., T.G., M.A.T., and R.W. designed the research. O.Y. and R.W. performed the research. O.Y., Y.X., S.E., K.Y., M.A.T., and R.W. analyzed the data.
Supporting information
Supplementary Material
Acknowledgments
The authors thank Alana Reed, PhD, of ArticulateScience LLC, for providing medical editorial support funded by AstraZeneca Pharmaceuticals, LP, Gaithersburg, MD, USA, in accordance with Good Publication Practice (GPP3) guidelines (http://www.ismpp.org/gpp3).
References
- 1. Aggarwal, N. & Sloane, B.F. Cathepsin B: multiple roles in cancer. Proteomics Clin. Appl. 8, 427–437 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Howlader, N. et al. US incidence of breast cancer subtypes defined by joint hormone receptor and HER2 status. J. Natl. Cancer Inst. 106, dju055 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Slamon, D.J. , Clark, G.M. , Wong, S.G. , Levin, W.J. , Ullrich, A. & McGuire, W.L. Human breast cancer: correlation of relapse and survival with amplification of the HER‐2/neu oncogene. Science 235, 177–182 (1987). [DOI] [PubMed] [Google Scholar]
- 4. NCCN Clinical Practice Guidelines in Oncology. Breast Cancer. V4.2020 <https://www.nccn.org/professionals/physician_gls/pdf/breast.pdf> (2020). Accessed June 4, 2020.
- 5. Aihara, T. et al. The Japanese Breast Cancer Society Clinical Practice Guideline for systemic treatment of breast cancer, 2015 edition. Breast Cancer 23, 329–342 (2016). [DOI] [PubMed] [Google Scholar]
- 6. Cardoso, F. et al. 4th ESO‐ESMO International Consensus Guidelines for Advanced Breast Cancer (ABC 4). Ann. Oncol. 29, 1634–1657 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Doi, T. et al. Safety, pharmacokinetics, and antitumour activity of trastuzumab deruxtecan (DS‐8201), a HER2‐targeting antibody‐drug conjugate, in patients with advanced breast and gastric or gastro‐oesophageal tumours: a phase 1 dose‐escalation study. Lancet Oncol. 18, 1512–1522 (2017). [DOI] [PubMed] [Google Scholar]
- 8. Ogitani, Y. et al. DS‐8201a, A Novel HER2‐targeting ADC with a novel DNA topoisomerase I inhibitor, demonstrates a promising antitumor efficacy with differentiation from T‐DM1. Clin. Cancer Res. 22, 5097–5108 (2016). [DOI] [PubMed] [Google Scholar]
- 9. Nakada, T. , Sugihara, K. , Jikoh, T. , Abe, Y. & Agatsuma, T. The latest research and development into the antibody‐drug conjugate, [fam‐] trastuzumab deruxtecan (DS‐8201a), for HER2 cancer therapy. Chem. Pharm. Bull. 67, 173–185 (2019). [DOI] [PubMed] [Google Scholar]
- 10. Modi, S. et al. Trastuzumab deruxtecan in previously treated HER2‐positive breast cancer. N. Engl. J. Med. 382, 610–621 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tamura, K. et al. Trastuzumab deruxtecan (DS‐8201a) in patients with advanced HER2‐positive breast cancer previously treated with trastuzumab emtansine: a dose‐expansion, phase 1 study. Lancet Oncol. 20, 816–826 (2019). [DOI] [PubMed] [Google Scholar]
- 12. Yamashita, T. et al. A phase 1, multicenter, open‐label study to assess the effect of [fam‐] trastuzumab deruxtecan (T‐DXd; DS‐8201a) on QTc and pharmacokinetics in subjects with HER2‐expressing metastatic and/or unresectable breast cancer [abstract P1–18‐12]. Cancer Res. 80 (2020). [Google Scholar]
- 13. Chang, D.‐Y. et al. Safety and pharmacokinetic results from a phase 1, multicenter, open‐label study of [fam‐] trastuzumab deruxtecan (T‐DXd; DS‐8201a) in subjects with advanced HER2‐positive breast cancer [abstract C041]. Mol. Cancer Ther. 18 (2019). [Google Scholar]
- 14. Bang, Y.‐J. et al. Pharmacokinetics (PK), safety, and efficacy of [fam‐] trastuzumab deruxtecan with OATP1B/CYP3A inhibitors in subjects with HER2‐expressing advanced solid tumours [abstract 330P]. Ann. Oncol. 30, v116–v117 (2019). [Google Scholar]
- 15. Mansfield, A.S. et al. The effect of hepatic impairment on outcomes in phase I clinical trials in cancer subjects. Clin. Cancer Res. 22, 5472–5479 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. US Food and Drug Administration . Guidance for industry: pharmacokinetics in patients with impaired renal function—study design, data analysis, and impact on dosing and labeling <https://www.fda.gov/media/78573/download> (2010). Accessed June 4, 2020.
- 17. ecancer . FDA approves new treatment option for patients with HER2‐positive breast cancer who have progressed on available therapies <https://ecancer.org/en/news/17123‐fda‐approves‐new‐treatment‐option‐for‐patients‐with‐her2‐positive‐breast‐cancer‐who‐have‐progressed‐on‐available‐therapies> (2019). Accessed June 4, 2020.
- 18. BioSpace . ENHERTU approved in Japan for treatment of patients with HER2 positive unresectable or metastatic breast cancer <https://www.biospace.com/article/releases/enhertu‐approved‐in‐japan‐for‐treatment‐of‐patients‐with‐her2‐positive‐unresectable‐or‐metastatic‐breast‐cancer‐/> (2020). Accessed June 4, 2020.
- 19. Dirks, N.L. & Meibohm, B. Population pharmacokinetics of therapeutic monoclonal antibodies. Clin. Pharmacokinet. 49, 633–659 (2010). [DOI] [PubMed] [Google Scholar]
- 20. Mould, D.R. & Sweeney, K.R.D. The pharmacokinetics and pharmacodynamics of monoclonal antibodies—mechanistic modeling applied to drug development. Curr. Opin. Drug Discov. Devel. 10, 84–96 (2007). [PubMed] [Google Scholar]
- 21. Cosson, V.F. , Ng, V.W. , Lehle, M. & Lum, B.L. Population pharmacokinetics and exposure‐response analyses of trastuzumab in patients with advanced gastric or gastroesophageal junction cancer. Cancer Chemother. Pharmacol. 73, 737–747 (2014). [DOI] [PubMed] [Google Scholar]
- 22. Bajaj, G. , Suryawanshi, S. , Roy, A. & Gupta, M. Evaluation of covariate effects on pharmacokinetics of monoclonal antibodies in oncology. Br. J. Clin. Pharmacol. 85, 2045–2058 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ryman, J.T. & Meibohm, B. Pharmacokinetics of monoclonal antibodies. CPT Pharmacometrics Syst. Pharmacol. 6, 576–588 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Iwata, H. et al. Trastuzumab deruxtecan (DS‐8201a) in subjects with HER2‐expressing solid tumors: Long‐term results of a large phase 1 study with multiple expansion cohorts [abstract 2501]. J. Clin. Onco. 36, 2501 (2018). [Google Scholar]
- 25. Yamaguchi, K. et al. A randomized phase 2, multicenter, open‐label study of trastuzumab deruxtecan (DS‐8201a) in subjects with HER2‐expressing gastric cancer [abstract TPS4133]. J. Clin. Oncol. 36 (2018). [Google Scholar]
- 26. Jänne, P. et al. A phase 2 study of DS‐8201a in HER2‐overexpressing or ‐mutated advanced non‐small‐cell lung cancer [abstract S476]. J. Thorac. Oncol. 13 (2018). [Google Scholar]
- 27. Tsurutani, J. et al. Updated results of phase 1 study of DS‐8201a in HER2‐expressing or ‐mutated advanced non‐small‐cell lung cancer [abstract S234]. J. Thorac. Oncol. 13 (2018). [Google Scholar]
- 28. Yoshino, T. et al. Updated results of phase I study of trastuzumab deruxtecan (DS‐8201a) in HER2‐expressing advanced colorectal cancer [abstract 563P]. Annal. Oncol. 29 (2018). [Google Scholar]
- 29. Yoshino, T. , Siena, S. , Dalal, R. , Okuda, Y. , Yamamoto, E. & Grothey, A. A multicenter, multicohort, phase 2 study of trastuzumab deruxtecan (DS‐8201a) in subjects with HER2‐expressing metastatic colorectal cancer—trial in progress [abstract 295P]. Ann. Oncol. 29 (2018). [Google Scholar]
- 30. Li, H. , Han, T.H. , Hunder, N.N. , Jang, G. & Zhao, B. Population pharmacokinetics of brentuximab vedotin in patients with CD30‐expressing hematologic malignancies. J. Clin. Pharmacol. 57, 1148–1158 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lu, D. et al. Population pharmacokinetics of trastuzumab emtansine (T‐DM1), a HER2‐targeted antibody‐drug conjugate, in patients with HER2‐positive metastatic breast cancer: clinical implications of the effect of covariates. Cancer Chemother. Pharmacol. 74, 399–410 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bruno, R. , Washington, C.B. , Lu, J.‐F. , Lieberman, G. , Banken, L. & Klein, P. Population pharmacokinetics of trastuzumab in patients with HER2+ metastatic breast cancer. Cancer Chemother. Pharmacol. 56, 361–369 (2005). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material