

Abstract
Chronic kidney disease (CKD) differentially affects the pharmacokinetics (PK) of nonrenally cleared drugs via certain pathways (e.g., cytochrome P450 (CYP)2D6); however, the effect on CYP2C8‐mediated clearance is not well understood because of overlapping substrate specificity with hepatic organic anion‐transporting polypeptides (OATPs). This study used physiologically based pharmacokinetic (PBPK) modeling to delineate potential changes in CYP2C8 or OATP1B activity in patients with CKD. Drugs analyzed are predominantly substrates of CYP2C8 (rosiglitazone and pioglitazone), OATP1B (pitavastatin), or both (repaglinide). Following initial model verification, pharmacokinetics (PK) of these drugs were simulated in patients with severe CKD considering changes in glomerular filtration rate (GFR), plasma protein binding, and activity of either CYP2C8 and/or OATP1B in a stepwise manner. The PBPK analysis suggests that OATP1B activity could be decreased up to 60% in severe CKD, whereas changes to CYP2C8 are negligible. This improved understanding of CKD effect on clearance pathways could be important to inform the optimal use of nonrenally eliminated drugs in patients with CKD.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
☑ Our recent analysis of reported clinical renal impairment studies indicates that CKD differentially affects the PK of certain nonrenally cleared drugs (e.g., CYP2D6 substrates). However, the effect on CYP2C8 could not be confirmed due to competing disposition mediated by hepatic OATP transporters for drugs investigated.
WHAT QUESTION DID THIS STUDY ADDRESS?
☑ Can effects of CKD on CYP2C8 and OATP1B be quantified using PBPK modeling of drugs whose hepatic clearances are differentially mediated by these two processes?
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
☑ Our study suggests that in patients with severe CKD, OATP1B activity may be decreased up to 60%, whereas CYP2C8 activity is minimally affected.
HOW MIGHT THIS CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE?
☑ Quantitative understanding of the effect of CKD on various clearance pathways is important to inform the need to conduct clinical PK studies for dose selection of nonrenally eliminated drugs in patients with CKD.
Chronic kidney disease (CKD), characterized by a progressive loss of kidney function over time, is a global public health issue.1 Impaired kidney function observed in patients with CKD may alter systemic drug exposure. Despite efforts to develop and use dosage adjustment recommendations for renally eliminated drugs, impaired kidney function is associated with an increasing risk of adverse drug events.2 Therefore, it is critical to understand the mechanism of altered drug disposition in patients with CKD in order to guide optimization of therapeutic regimens in these highly vulnerable patients.
Impaired kidney function not only alters the rate of drug elimination from the kidneys, but it may also influence drug disposition by modifying the function of enzymes and transporters in extrarenal organs, such as the liver.3, 4, 5 Regulatory agencies recommend conducting clinical renal impairment studies for nonrenally eliminated drugs in patients with the “worst‐case scenario” kidney function category to determine the need for possible dosing adjustment in patients with CKD.6, 7 However, there are no generalizable well‐defined guidances pertaining to dedicated CKD study designs. This may be due to inconsistent pharmacokinetic (PK) alterations observed in patients with CKD for nonrenally cleared drugs, which is likely also why dose adjustment of nonrenally cleared drugs is still not a common practice for patients with CKD.
The main challenge associated with assessing the PK of nonrenally eliminated drugs is a poor understanding of the complex overlapping substrate specificity of most drugs, and the mechanisms by which and to what extent the activities of related metabolic enzymes and transporters may be changed in patients with impaired kidney function. Uremic toxins usually accumulate in plasma of patients with kidney diseases. These circulating toxins and associated pathophysiological changes have been reported to alter hepatic clearance of pharmaceuticals by modifying activities of metabolizing enzymes, such as cytochrome P450 (CYP) and UDP‐glucuronosyltransferase (UGT) enzymes, and the function of active uptake/efflux membrane transporters.4, 8 It is also reported that drug absorption and plasma protein binding are altered in patients with CKD.5, 8, 9, 10 The uremic toxin‐mediated impact on enzymes/transporters is supported by experimental endstage renal disease (ESRD) animal models.3 Although not all transporter‐related observations in animals can be translated to humans,11 limited direct evidence in humans also support this hypothesis.5, 8, 9, 10
Our recent studies have shown that CKD differentially affects the PK of certain nonrenally cleared drugs.12, 13 Briefly, CYP2D6‐mediated and hepatic organic anion‐transporting polypeptides (OATP)1B‐mediated drug clearance generally decreases as kidney function declines (up to 79%), whereas CKD has minimal or variable effect on CYP1A2‐, CYP2C9‐, CYP2C19‐, or CYP3A4‐mediated clearance.12, 13 For CYP2C8‐mediated clearance, a similar decreasing trend was observed as kidney function declines, but the interpretation of such trends is challenging due to the overlap observed between some CYP2C8 and OATP1B substrates.13 Quantifying the individual contribution of hepatic transporters and metabolic enzymes is critical to improve our understanding of the effect of CKD on the PK of drugs cleared by these nonrenal pathways and, ultimately, to guide dosing adjustments in patients with CKD.
Physiologically based pharmacokinetic (PBPK) modeling and simulations are useful for predicting both systemic and tissue concentration‐time profiles.14, 15, 16, 17 Organ dysfunctions, including impaired kidney function, can be accounted for by modifying physiological (system) parameters of the PBPK models.5, 18, 19, 20, 21 The effect of impaired kidney function on the PK of nonrenally cleared drugs may then be predicted using PBPK models by taking into account known alterations in the function of related enzymes and transporters in patients with CKD.5, 18 Thus, PBPK modeling and simulations provide a practical tool to better understand the underlying mechanism by which system parameters may be affected and to what degree the change may be observed in patients with impaired kidney function.
In the current study, we used PBPK modeling and simulations to evaluate potential changes in the activity of hepatic CYP2C8 and OATP1B (OATP1B1 and OATP1B3) in patients with impaired kidney function. Four drugs that are predominantly metabolized by CYP2C8 and/or transported by OATPs (largely by OATP1B1 and OATP1B3) and with clinical data in patients with CKD, namely rosiglitazone (CYP2C8 substrate), pioglitazone (CYP2C8 substrate), pitavastatin (OATP1B substrate), and repaglinide (CYP2C8/OATP1B dual substrate), were selected for the analysis.
Results
PBPK models in healthy populations
The model‐simulated plasma concentration‐time profiles of four substrate drugs, rosiglitazone, pioglitazone, pitavastatin, and repaglinide, were compared with the corresponding clinically observed data in healthy populations (Figure 1 , Supplementary Reading List S1 ). The exposure changes caused by CYP2C8 and OATP1B inhibitors, gemfibrozil, and its major metabolite, gemfibrozil 1‐O‐β‐glucuronide, on four victim (substrate) drugs, and the OATPs inhibitor, cyclosporine, on pitavastatin and repaglinide were simulated. The effects of SLCO1B1 polymorphism on systemic exposure of OATP1B substrates pitavastatin and repaglinide were also simulated (Table 1 ). The corresponding predicted area under the concentration‐time curve ratio (AUCR)s (with/without inhibitor or poor transporters (PTs)/extensive transporters (ETs) are summarized in Table 1 . R values (i.e., the ratio of simulated AUCRs and observed AUCRs) under these several conditions are within 0.62–1.20, indicating that the substrate models well recovered the clinical data.
Figure 1.
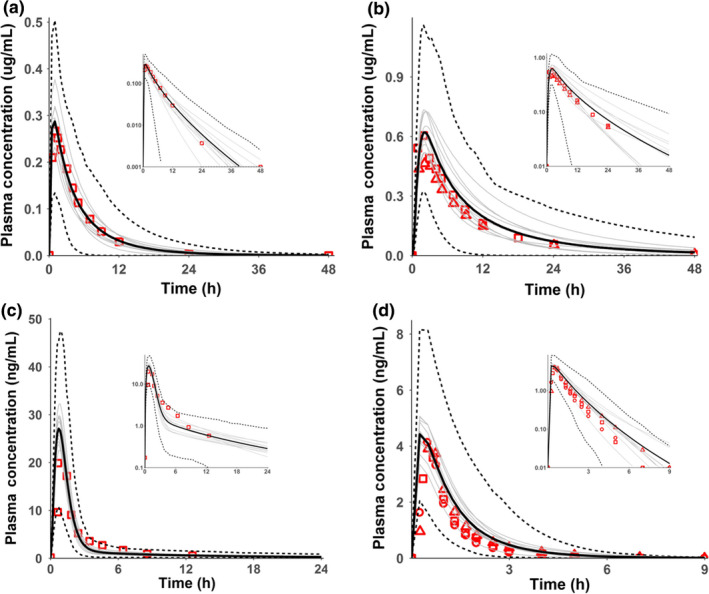
The simulated drug plasma concentration‐time profile in healthy controls (or populations) of (a) single oral dose of 4 mg rosiglitazone, (b) single oral dose of 15 mg pioglitazone, (c) single oral dose of 2 mg pitavastatin, and (d) single oral dose of 0.25 mg repaglinide. The solid lines are the simulated mean values, dotted lines are the 5th and 95th percentiles and grey lines are for different trials. Data points are observed values from different studies. Semi‐log scales are shown as insets.
Table 1.
The effect of SLCO1B1 polymorphism or inhibitors on AUC changes of substrate drugs
| Drug | SLCO1B1 polymorphism or drug inhibitor | Simulated AUCR | Observed AUCR | R value |
|---|---|---|---|---|
| Rosiglitazone | Gemfibrozil | 2.41 | 2.36 | 1.02 |
| Pioglitazone | Gemfibrozil | 3.84 | 3.2 | 1.20 |
| Pitavastatin | SLCO1B1 polymorphism (c. 521 CC vs. c. 521 TT) | 1.90a | 3.08a | 0.62a |
| Gemfibrozil | 1.58 | 1.45 | 1.09 | |
| Cyclosporineb | 3.29 | 4.55 | 0.72 | |
| Repaglinide | SLCO1B1 polymorphism (c. 521 CC vs. c. 521 TT) | 1.88 | 1.83 | 0.97 |
| Gemfibrozilc | 3.22 | 5.0 | 0.64 | |
| Cyclosporineb | 2.79 | 2.4 | 1.16 |
References in the Table and superscripts a–c are listed in Table S2 .
AUC, area under the concentration‐time curve; AUCR, ratio of AUC; R value, ratio of the simulated AUCR and observed AUCR.
aThe simulated ratio of SLCO1B1 c. 521 CC and SLCO1B1 c. 521 TT was compared to the observed ratio of “SLCO1B1 *15/*15 and ABCG2 421C/C 421C/A” (mixed with BCRP polymorphism) and “SLCO1B1 *1b/*1b and ABCG2 421C/C” because no purely genotyped “SLCO1B1 *15/*15 and ABCG2 421C/C” data were reported. bThe estimated cyclosporine K i values of OATP1B1/1B3 (0.014/0.007 μΜ) were used with updated unbound fraction = 0.1 (Product Labeling: Drugs@FDA). cThe physiologically based pharmacokinetic models with gemfibrozil‐glucuronide K Ioatp1b of 7.9 μM underestimated the complex repaglinide‐gemfibrozil drug‐drug interactions, as also observed in literature. The simulated AUCRs were 3.79 and 5.15 if K Ioatp1b of 7.9 μM was reduced to the values of 4 μM and 1.48 μM, respectively.
Prediction of the CKD effect on the PK of nonrenally eliminated drugs
The exposure change in patients with severe CKD was simulated using the Simcyp V16 default severe renal impairment populations “Sim‐RenalGFR_less30,” in which changes in relevant physiological parameters (e.g., hematocrit, albumin, and gastric emptying) were implemented compared to the healthy volunteer (HV) population (see Methods for details). The AUCRs based on simulated area under the concentration‐time curves (AUCs) in CKD vs. healthy populations are summarized in Table 2 . Overall, the exposure changes in patients with severe CKD were overpredicted for CYP2C8 substrates, rosiglitazone and pioglitazone, whereas underprediction was apparent for OATP1B substrates, pitavastatin and repaglinide, for both unbound and total AUCR when using the V16 default “Sim‐RenalGFR_less30” population (Table 2 ).
Table 2.
Effect of CKD on pharmacokinetics of model substrate drugs
| Enzyme or transporter | Substrate drug | CKD populations | f u a (%) | AUCR (total) | R value | AUCR (unbound) | R value | |||
|---|---|---|---|---|---|---|---|---|---|---|
| HV | CKD | Simulated | Observeda | Simulated | Observeda | |||||
| CYP2C8 | Rosiglitazone | Simcyp (CYP2C8 47%) | 0.16 | 0.22 | 1.44 | 0.81 | 1.78 | 2.47 | 1.11 | 2.23 |
| Modified (CYP2C8 100%)b | 0.16 | 0.22 | 0.93 | 0.81 | 1.14 | 1.58 | 1.11 | 1.42 | ||
| Pioglitazone | Simcyp (CYP2C8 47%) | 3 | 3.5 c | 1.58 | 0.78 | 2.03 | 2.40 | 0.92 c | 2.61 c | |
| Modified (CYP2C8 100%)b | 3 | 3.5 c | 0.90 | 0.78 | 1.15 | 1.36 | 0.92 c | 1.48 c | ||
| OATP | Pitavastatin | Simcyp (CYP2C8 47%, OATP100%) | 0.6 | 0.6 | 0.85 | 1.36 | 0.63 | 1.05 | 1.36 | 0.77 |
| Simcyp (CYP2C8 47%, OATP 60%)d | 0.6 | 0.6 | 1.28 | 1.36 | 0.94 | 1.59 | 1.36 | 1.17 | ||
| Modified (CYP2C8 100%, OATP100%) | 0.6 | 0.6 | 0.84 | 1.36 | 0.62 | 1.04 | 1.36 | 0.77 | ||
| Modified (CYP2C8 100%, OATP60%)d | 0.6 | 0.6 | 1.28 | 1.36 | 0.94 | 1.59 | 1.36 | 1.17 | ||
| CYP2C8/OATP | Repaglinide | Simcyp (CYP2C8 47%, OATP100%) | 3.6 | 3.6 | 1.37 | 2.72 | 0.51 | 1.72 | 2.72 | 0.63 |
| Simcyp (CYP2C8 47%, OATP 50%)d | 3.6 | 3.6 | 2.55 | 2.72 | 0.94 | 3.18 | 2.72 | 1.17 | ||
| Modified (CYP2C8 100%, OATP100%) | 3.6 | 3.6 | 1.08 | 2.72 | 0.40 | 1.35 | 2.72 | 0.50 | ||
| Modified (CYP2C8 100%, OATP60%) | 3.6 | 3.6 | 1.72 | 2.72 | 0.63 | 2.14 | 2.72 | 0.79 | ||
| Modified (CYP2C8 100%, OATP45%) | 3.6 | 3.6 | 2.20 | 2.72 | 0.81 | 2.75 | 2.72 | 1.01 | ||
| Modified (CYP2C8 100%, OATP40%)d | 3.6 | 3.6 | 2.43 | 2.72 | 0.89 | 3.03 | 2.72 | 1.11 | ||
Simcyp default and modified “Sim‐RenalGFR_less30” severe renal impairment populations were used in the simulations with dosing regimen, GFR, average age, and gender matched to the corresponding clinical CKD studies. Simcyp: V16 default “Sim‐RenalGFR_less30” population where the CYP2C8 abundance was reduced to 47% of the HVs. Modified: the CYP2C8 abundance was set back to 100% of HVs in the V16 default “Sim‐RenalGFR_less30” population. The OATP1B abundance was adjusted in the simulations when needed as indicated in parenthesis.
AUCR, area under the concentration‐time curves ratio; CKD, chronic kidney disease; CYP, cytochrome P450; f u, unbound fraction; HV, healthy volunteer; OATP, organic anion‐transporting polypeptide.
aObserved f u and AUCR values were from the dedicated clinical CKD studies: rosiglitazone,51 pitavastatin28 and repaglinide.35 bCYP2C8 abundance as in HV. cPioglitazone f u for patients with CKD were not reported in the clinical study,52 thus predicted f u was used in the simulations. Prediction of f u was based on changes in albumin content reported in severe CKD, assuming that albumin was the main plasma protein involved in binding of this drug, as detailed in ref.13 The “observed” unbound AUCR of 0.92 was calculated based on the estimated fu and the corresponding R values were estimated, shown as italic numbers. dOATP1B function was optimized to reproduce the observed AUCR for pitavastatin and repaglinide.
The AUCRs of rosiglitazone and pioglitazone (CYP2C8 substrates) were approximately twofold overpredicted when using the V16 default “Sim‐RenalGFR_less30” populations (Table 2 ). In contrast, the simulated AUCRs for rosiglitazone and pioglitazone were improved and more comparable to the observed values when the modified “Sim‐RenalGFR_less30” population was used in the simulations (Table 2 ). For the latter, CYP2C8 enzyme abundance was assumed to be similar to the HV population (see Methods).
In the initial analysis, AUCRs were underpredicted for OATP1B substrate pitavastatin when using the V16 default “Sim‐RenalGFR_less30” populations (which assumes no decrease in OATP1B function compared to healthy populations). Further analysis implied that OATP1B abundance needs to be reduced by 40% to better recover the observed exposure changes (when using the modified “Sim‐RenalGFR_less30”; see Methods; Table 2 ).
The simulated AUCRs for repaglinide were ~50% of the observed values when using the V16 default “Sim‐RenalGFR_less30” population, in which CYP2C8 enzyme abundance was reduced to 47% of the value in the healthy population (expression data listed in Table S1 ). The AUCRs were well recovered when OATP1B abundance was decreased to about 50% of the abundance in healthy subjects in the “Sim‐RenalGFR_less30” population (Table 2 ). With the modified “Sim‐RenalGFR_less30” population (in which the CYP2C8 enzyme abundance was set to the same value as in the healthy population; see Methods), a decrease in OATP1B abundance to around 40% of the abundance in the HV population was needed to match the observed AUCRs (Table 2 ).
The repaglinide plasma unbound fraction (f u) is associated with uncertainty and varies from 0.74%22 to 3.6%13 for HVs. Therefore, a sensitivity analysis of the impact of f u on the predicted repaglinide AUCR was performed. Figure 2 shows that simulated AUCRs were not sensitive to changes in plasma f u within values between 0.37% and 7.2%. Previous studies showed that repaglinide AUC is sensitive to the hepatic passive diffusion clearance (CLpd) used in the model, in particular when a “top‐down” approach is applied and an optimization of the active uptake clearance is performed using clinical data.18, 23 Therefore, a sensitivity analysis of CLpd (values ranged between 1 and 40 μL/minute/106 cells) was performed to assess the impact of this parameter on the estimated percentage reduction of OATP1B abundance in patients with severe CKD necessary to recover the AUCR between severe CKD and healthy populations. The simulated AUCRs were not significantly affected over the range of passive diffusion clearance values reported in the literature for repaglinide (Figure S1 ).
Figure 2.
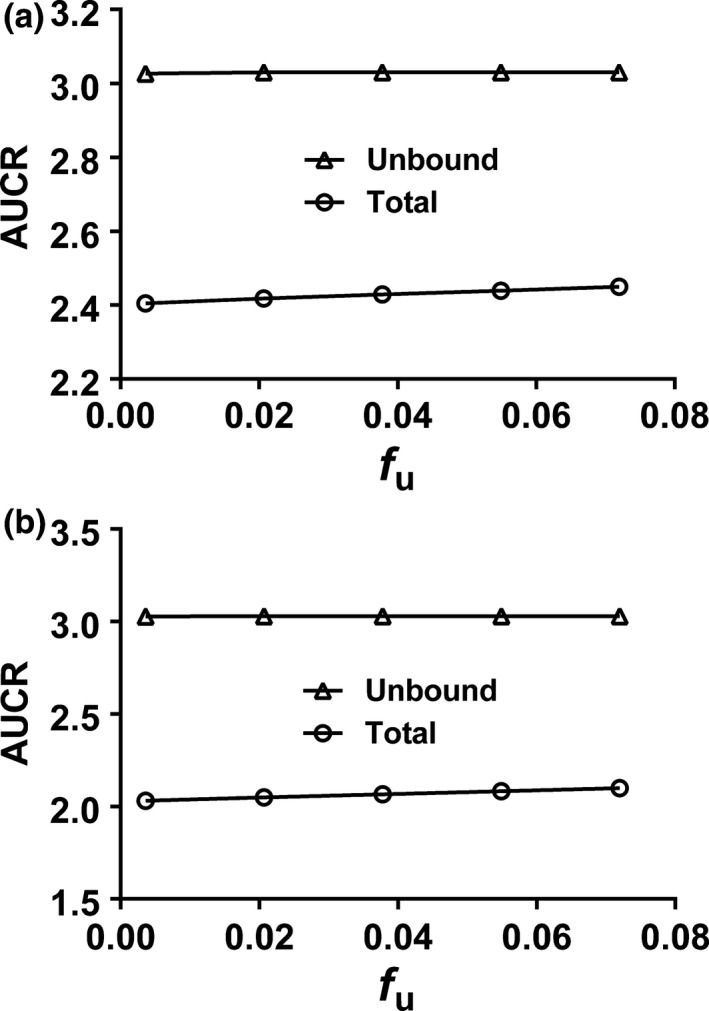
Sensitivity analysis on unbound fraction (f u) for cytochrome P450 CYP2C8 and organic anion‐transporting polypeptide (OATP)1B dual substrate repaglinide with OATP1B abundance decreased to 40% of the healthy populations assuming the same f u values as observed in clinical study35: (a) The CKD populations with the same f u as the healthy populations as observed in the corresponding clinical studies.35 (b) The severe CKD populations with f u estimated based on the f u of HVs using the information on the changed albumin levels.5, 13
Discussion
Recent studies have shown that CKD differentially affects the PK of certain nonrenally cleared drugs.12, 13 Understanding the potential effect of CKD on the activity of CYP2C8 and OATP1B transporters is challenging due to overlapping substrate specificities. To this end, PBPK modeling and simulations were used to evaluate the effect of CKD on the exposure of several drugs cleared nonrenally either by CYP2C8 and/or OATP1B. System‐dependent parameters, such as CYP2C8 and OATP1B abundance in patients with severe CKD, were examined to recover the observed exposure data in an effort to assess possible physiological changes and, thus, to understand the underlying mechanism of exposure changes of nonrenally eliminated drugs in patients with impaired kidney function.
Drug‐drug interaction (DDI) and pharmacogenomic (PGx) studies indicated that both rosiglitazone and pioglitazone are CYP2C8 substrates.24, 25, 26 The PBPK simulations with reduced CYP2C8 abundance (to 47%) overestimated the AUCRs using the V16 default “Sim‐RenalGFR_less30” population. Interestingly, the observed exposure change was recovered well by using a modified severe renal impairment population with hepatic CYP2C8 abundance similar to the healthy populations (i.e., no reduction in CYP2C8 function), suggesting no significant decrease of CYP2C8 function in severe renal impairment patients. Our previous meta‐analysis approach showed a decreasing trend of drug clearance as CKD severity increases for CYP2C8 substrate drugs; however, the data were inconclusive due to the overlapping substrate specificity with OATP1B.13 The present PBPK analysis suggests a negligible change in CYP2C8 function/activity in patients with severe CKD.
Pitavastatin is actively transported into the liver by the hepatic uptake transporter OATP1B, and is suggested to be a clinical probe substrate to investigate OATP1B activity.27, 28, 29, 30 Therefore, change in pitavastatin PK can be primarily linked to altered OATP1B activity. The PBPK simulations in this study suggested that OATP1B activity in patients with severe CKD was decreased to about 60% of HV values.28 This decreased OATP1B function needed to reproduce pitavastatin AUCRs for patients with severe CKD is consistent with our previous meta‐analysis and trends of decreased OATP1B transport activity with increased severity of CKD.13
The CYP2C8 and OATP1B dual substrate repaglinide involves transporter‐enzyme interplay in its hepatic clearance.31, 32, 33, 34 A significant decrease in repaglinide clearance was observed in patients with severe CKD.35 The PBPK simulations with the V16 default “Sim‐RenalGFR_less30” population (with a 47% of CYP2C8 abundance of HVs) suggested that about 50% decrease in OATP1B activity was needed to match the observed AUCRs for repaglinide. In contrast, a decrease to about 40% of OATP1B activity was needed to recover the observed AUCRs when the modified severe CKD population was used. In the latter case, the CYP2C8 abundance was set to the value in healthy populations based on the analysis of CYP2C8 substrates rosiglitazone and pioglitazone (different to the Simcyp default population “Sim‐RenalGFR_less30” with a 47% of CYP2C8 abundance reported in HVs).
Overall, PBPK modeling and simulations of these four drugs in severe CKD populations suggested a minimal decrease in CYP2C8 activity, and about a 40–60% decrease in OATP1B activity. This “middle‐out” simulation analysis provides an insight with respect to the contribution of changes in CYP2C8 vs. OATP1B to the decreased clearance of repaglinide observed in patients with severe CKD. We observed a slight difference in terms of the level of decrease in OATP1B function for pitavastatin and repaglinide, which could be due to clinical study variability. One factor to consider was the plasma protein binding because some uncertainties existed in measured f u of repaglinide (the measured f u value reported in patients with CKD was the same as in the HV population). Changes in plasma protein binding with impaired renal function may have an effect on the predicted exposure changes in patients with CKD and estimated extent of decrease in transporter function. In general, the f u for most drugs we investigated was increased as kidney function declined and this trend seemed to be more profound for drugs with a relatively low f u.13 A sensitivity analysis on f u of repaglinide did not show a significant increase of AUCR to account for the difference between repaglinide and pitavastatin in the estimated decrease in the OATP1B abundance required to recover the observed exposure changes in patients with CKD (Figure 2 ).
It is worth mentioning that a small amount of pitavastatin is renally excreted as unchanged drug,13, 28 which was accounted for in the pitavastatin PBPK model in which the CKD population with decreased glomerular filtration rate (GFR) was matched to the clinical study design. In contrast, CYP2C8/OATP1B dual substrate repaglinide did not have the effect of GFR change in the PBPK model, as repaglinide is extensively cleared through the liver35 (see Methods), and only 0.1% of the parent compound is eliminated renally.13 Although OATP1B‐mediated hepatic uptake is thought to be the rate‐determining step in the clearance of pitavastatin; other transporters/enzymes may also influence the estimated effect on OATP1B activity. Pitavastatin is a substrate of the efflux transporters ABCB1 (multidrug resistance protein 1 (MDR1) or P‐glycoprotein), ABCG2 (BCRP), ABCC3 (MRP3), and ABCC2 (MRP2) and the hepatic uptake transporters SLCO2B1 (OATP2B1) and SLC10A1 (NTCP).36, 37, 38 Uremic toxins in patients with CKD may change the activities of these efflux and uptake transporters in the disposition of pitavastatin, which was not considered in the current modeling approach due to lack of reliable transporter expression data from tissues of patients with renal disease. However, pitavastatin uptake via OATP1B1 is the rate‐determining step for its clearance and, as such, its systemic PK is more dependent on this process. Therefore, the effect on OATP1B is expected to be the major factor contributing to the changes observed in pitavastatin PK in CKD. Further understanding of changes in these system parameters in CKD will refine the prediction of hepatic clearance and implications on dosage regimen in these patients.
Last, metabolism of pitavastatin and repaglinide is handled by different enzymes, which may also contribute to the differences in the degree of exposure changes among them. Repaglinide was shown to be predominantly metabolized by phase I enzymes CYP2C8 and CYP3A4,31, 32, 33, 34 with a minor contribution of UDP‐glucuronosyltransferase (UGT)1A1/1A3 glucuronidation determined from an in vitro study.39 Pitavastatin is minimally metabolized, and its metabolism occurs via glucuronidation by hepatic UGT1A3 and UGT2B7.8, 28, 40 Our previous studies showed that the CKD effect may be pathway dependent and the effect of CKD on CYP3A4 is variable.12, 13 At this stage, we have not incorporated any potential effect of CKD on different UGT enzymes in our simulations due to the lack of knowledge of any potential changes in UGT activity in patients with CKD. However, CKD effect on UGT is likely based on in vitro studies that reported inhibitory effect of uremic toxins on UGT activity.8 Further investigation of the effect of CKD on the UGT abundance or function may be helpful to refine the PBPK predictions.
In this study, kidney function, average age, and gender ratio were matched to the corresponding clinical CKD study designs for the simulations of CKD effects on the exposure changes in patients with impaired kidney function, which may be helpful to refine physiological components of PBPK models for confident predictions of drug PK in patients with CKD. This is important because differences in demographics may result in different PK profile predictions, even though the same extrinsic and intrinsic factors are used in the modeling and simulations. Matching to clinical demographics in simulations allows proper comparisons between the simulated and observed PK in renal impairment studies, which is one of the advantages of utilizing PBPK modeling and simulations because the drug parameters, system parameters, and trial designs are separated in the PBPK modeling.14
The aim of this study was to understand consequences of possible physiological changes in CYP2C8 and OATP1B transporter activity related to impaired kidney function by simulating the observed exposure change in patients with CKD using PBPK modeling. Unfortunately, reliable quantitative data on the decrease in OATP1B expression or uptake linked to the degree of the renal impairment are not available and, therefore, have not yet been incorporated into the Simcyp Simulator. Therefore, potential physiological changes in patients with CKD were incorporated in the models by adjusting system‐dependent parameters (i.e., CYP2C8 or OATP1B expression/function) in the severe renal impairment population relative to the HV population using clinical data for respective drug probes. The predicted trends of exposure changes for the CYP2C8 substrate drugs rosiglitazone and pioglitazone, the OATP1B substrate drug pitavastatin, and the dual CY2C8/OATP1B substrate repaglinide were consistent with our previous meta‐analysis of a large drug subset.13 The analysis showed quantitative discrepancy in the extent of decreased OATP1B abundance needed to recover the observed AUCRs between pitavastatin and repaglinide. This discrepancy may be associated with the modeling approach that focused primarily on the activity of these major proteins and ignored other potential minor pathways that may gain more relevance in patients with CKD. Better understanding of these fundamental system‐related parameters will further improve the quantitative PBPK prediction. In addition, parameter estimation in patients with CKD was limited due to lack of rich clinical data in this population, which may introduce some bias in cases of complex interplay of active transport and metabolism contributing to hepatic elimination.
In summary, PBPK modeling and simulations provide a useful approach to investigate the activities of enzymes and transporters involved in drug clearance in renally impaired patients. Negligible changes in CYP2C8 enzyme function were required to match the observed AUC changes in severe CKD subjects for CYP2C8 substrates rosiglitazone and pioglitazone. In contrast, decreases in OATP1B activity of up to 60% were needed to recover the change in AUC observed in severe CKD subjects. These findings on the effect of CKD on OATP1B activity may be useful in optimizing dose regimen selection for nonrenally cleared drugs in patients with impaired kidney function.
Methods
PBPK modeling and simulations were performed using a population‐based PBPK platform, the Simcyp Simulator (V16R1; Certara UK, Sheffield, UK). Unless otherwise noted, each simulation was performed in 100 subjects (10 trials with 10 subjects for each trial) using the simulator's default virtual population libraries. The PBPK models in populations of HVs were first adopted and verified when needed, and then applied to patients with severe CKD to simulate the CKD effect on drug exposure changes. The severe CKD group was investigated in this study rather than the ESRD group. Consistent with the healthcare standard,41 most ESRD groups were either on regular dialysis during the clinical study period or off dialysis but were on regular dialysis before the CKD study period.12, 13 Because uremic toxins are continuously removed during dialysis and a certain portion of the drug may also be cleared during dialysis, the ESRD groups may not represent the “worst case” scenario compared to nondialyzed CKD groups.7, 41 The overall workflow of PBPK modeling and simulations is presented in Figure 3 .
Figure 3.
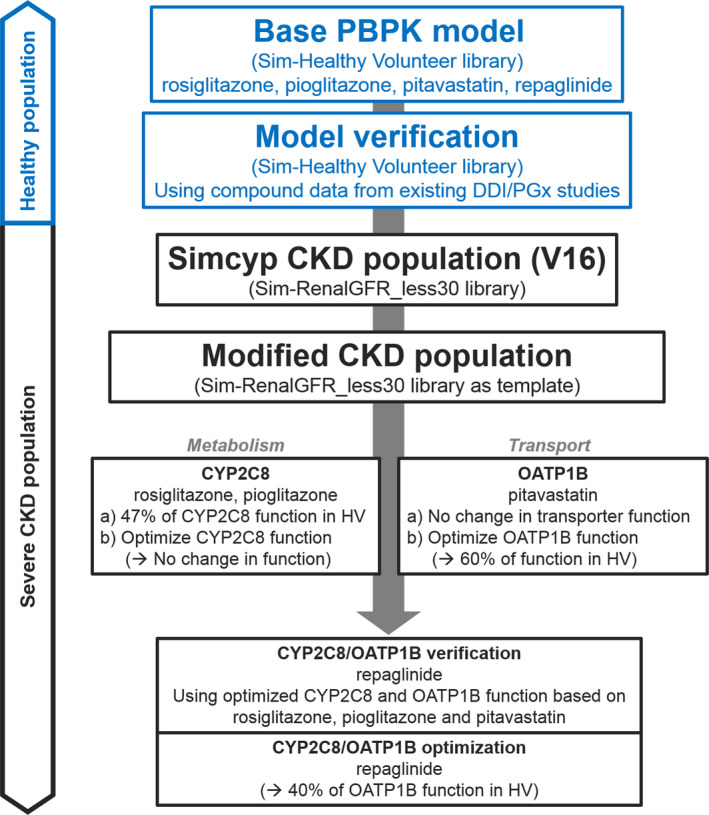
Workflow of physiologically based pharmacokinetic (PBPK) modeling and simulations. The adopted base PBPK models were verified using clinical drug‐drug interaction (DDI) and pharmacogenetic (PGx) studies in a healthy volunteer (HV) population. The verified models were applied to Simcyp Simulator V16 default and modified severe chronic kidney disease (CKD) populations for: (i) cytochrome P450 (CYP)2C8 substrates rosiglitazone and pioglitazone; (ii) organic anion‐transporting polypeptide (OATP)1B substrate pitavastatin; and (iii) CYP2C8/OATP1B dual substrate repaglinide. The CYP2C8 and OATP1B activities were optimized to recover clinical observations.
PBPK modeling and model verification in healthy populations
The base PBPK models for the four substrate drugs (rosiglitazone, pioglitazone, pitavastatin, and repaglinide) were adopted from previous reports.42, 43, 44 Published models used earlier versions of the Simcyp Simulator platform and, therefore, additional verification was carried out. Final model parameters are summarized in Table 3 . Briefly, minimal PBPK distribution models were used for both CYP2C8 substrates rosiglitazone and pioglitazone assuming rapid‐equilibrium between blood and the liver compartments.43, 44 For the OATP1B substrate pitavastatin and the OATP1B/CYP2C8 dual substrate repaglinide, full PBPK distribution models45 were used due to the involvement of the transporter‐mediated hepatic uptake,42, 43, 44, 45 which assumed permeability‐limited distribution into the liver and rapid equilibrium between blood and other tissues. For pitavastatin, a scaling factor of 15 was applied for hepatic active uptake to describe the observed PK profiles from a PGx study46 (Figure S2 ). In the case of repaglinide, a universal K p scalar of 2.42 was applied to all tissue compartments to allow a good agreement with the observed volume of distribution at steady state. This K p scalar is different from the previous report44 due to the changes in tissue composition parameters in the liver (relative volume of wet tissue percentage for neutral lipids), muscle (tissue‐to‐plasma albumin ratio), and adipose (tissue‐to‐plasma albumin ratio) between Simcyp V13R1 and V16R1.
Table 3.
Summary of drug parameters for PBPK models
| Parameters | Rosiglitazonea | Pioglitazoneb | Pitavastatinc | Repaglinided |
|---|---|---|---|---|
| Physicochemical properties | ||||
| Molecular weight (g/mol) | 357.4 | 356.4 | 421 | 452.6 |
| Compound type | Ampholyte | Monoprotic base | Monoprotic acid | Ampholyte |
| Log P | 2.6 | 3.5 | 2.91 | 4.87 |
| pKa | 6.25 and 6.32 | 5.53 | 5.31 | 4.19 and 5.78 |
| Major binding protein | Albumin | Albumin | Albumin | Albumin |
| f u | 0.002 | 0.015 | 0.005 | 0.015 |
| Blood/plasma ratio | 0.57 | 1 | 0.55 | 0.62 |
| Absorption | ||||
| Absorption type | First order | ADAM | ADAM | First order |
| Fraction absorbed | 1 (k a = 3.6/hour) | 0.98 | 0.99 | 0.997 (k a = 2.8/hour) |
| Peff, man (10−4 cm/seconds) | 1.291 | 3.754 | 4.688 | 6.490 |
| Absorption scalar | 1 | 1 | 1 | 1.873 |
| Distribution | ||||
| Distribution model | Minimal PBPK | Minimal PBPK | Full PBPK (Rodgers & Rowland methode) | Full PBPK (Rodgers & Rowland methode) |
| K p scalar | 1 | 2.42f | ||
| Vss (L/kg) | 0.12 | 0.253 | 1.88 | 0.256 |
| Elimination | ||||
| CLint, CYP2C8 (μL/minute/mg protein) | 191 (HLM) | 27.5 (HLM) | 12.98 (rCYP) (μL/minute/pmol) | 93 (HLM) |
| CLint, CYP2C9 (μL/minute/mg protein) | 102 (HLM) | 1.5 (HLM) | 7.93 (rCYP) (μL/minute/pmol) | |
| CLint, CYP2C19 (μL/minute/mg protein) | 6.1 (HLM) | |||
| CLint, CYP3A4 (μL/minute/mg protein) | 38 (HLM) | |||
| CLint, others (μL/minute/mg protein) | 1,453 | |||
| Renal clearance (L/hour) | 0.32 | 0 | 0.129 | 0 |
| Hepatobiliary transport | ||||
| Liver unbound fraction (intra‐/extracellular) | 0.460/0.0096 | 0.143/0.028 | ||
| Passive diffusion (mL/minute/106 cells) | 0.011 | 0.024 | ||
| CLint, active (mL/minute/106 cells) | 0.0584 (OATB1B1), 0.0051 (OATP1B3)g | 0.037 (OATP1B1) | ||
| Scaling factor (OATP active uptake) | 15h | 16.9 | ||
Models were adopted from references.
ADAM, advanced dissolution, absorption, metabolism; CLint, intrinsic clearance; CYP, cytochrome P450; f u, unbound fraction; HLM, human liver microsome; OATP, organic anion‐transporting polypeptide; PBPK, physiologically based pharmacokinetic; rCYP, recombinant cytochrome P450; Vss, volume of distribution at steady state.
aRosiglitazone model adopted from ref. 44, which was originally adopted from Simcyp V13 compound file. bPioglitazone model was adopted from ref. 44. cPitavastatin model was adopted from ref. 42. OATP1B active uptake scaling factor of 15 was applied to recover the reported plasma profiles from pharmacogenomic studies (see Methods). dRepaglinide model was adopted from refs 43, 44. eRodgers and Rowland method (Simcyp Method 2) was used for full PBPK distribution models for both pitavastatin and repaglinide.45 fRevised K p scalar of 2.42 due to the changes in muscle and adipose tissue composition parameters in Simcyp V16. gThe active intrinsic clearance was from reference.53 hOATP1B active update scaling factor of 15 was applied to recover the reported plasma profiles from pharmacogenomic studies (see Methods).
The base models were further verified using observed data from DDI and PGx studies. The PBPK models of the drug interaction perpetrators gemfibrozil (with its metabolite gemfibrozil 1‐O‐β‐glucuronide) and cyclosporine were adopted from the literature.42, 44, 47, 48
The PK profiles of the OATP1B substrates pitavastatin and repaglinide were simulated in subjects with different genotypes of SLCO1B1 using a previously published approach.42 Briefly, it was assumed that the virtual healthy population with genotype SLCO1B1 c.521 TT was comprised of subjects who were ETs (wild type), whereas the population with genotype SLCO1B1 c.521 CC was comprised of PTs (with transporter activity assumed to be 37% of that in ETs (Simcyp V16R1)).
The AUCR (with/without inhibitors in the case of DDI or between PT and ET for PGx simulations of healthy populations) was calculated. Similarly, AUCRRI/HV (patients with severe renal impairment vs. HVs) was calculated for CKD simulations. The performance of PBPK model prediction was evaluated by the ratio of simulated AUCR (AUCRs) and observed AUCR (AUCRo), an R value (R = AUCRs/AUCRo). Although there is no consensus regarding the acceptance criteria, an R value within twofold range (between 0.5 and 2.0) has been frequently used to assess a PBPK model performance.49, 50
PBPK modeling in severe CKD populations
The dosing regimen, average age, gender, and GFR values were matched to the corresponding clinical trial study designs,28, 35, 51, 52 as summarized in Table 4 for both healthy and CKD populations. The reported measured f u values in the corresponding CKD studies28, 35, 51 were used, with the exception of pioglitazone in which f u = 0.035 was predicted (details in ref. 13) due to unavailability of such data in patients with CKD52 (Table 2 ). The details of related clinical studies have been summarized in a previous study13 and the references within. For each simulation, adjustment of the average serum creatinine concentration of related virtual populations was made to obtain a targeted GFR (creatinine clearance) for a specific study. For repaglinide, the Simcyp Simulator default GFR for the healthy population and the average GFR for severe CKD were used because information on GFR was not available from the clinical CKD study35 (Table 4 ). The simulations were first performed with two CYP2C8 substrates (rosiglitazone and pioglitazone) and the OATP1B substrate pitavastatin to investigate the effects of severe CKD on the individual pathways (Figure 3 ). Subsequent simulations were conducted for the OATP1B/CYP2C8 dual substrate, repaglinide (Figure 3 ).
Table 4.
Summary of CKD trial designs of the observed and those used in the simulations
| Drug | Subjects | Dosing regimen | M/F | Age (years) | Kidney function | ||
|---|---|---|---|---|---|---|---|
| Obs | Obs | Obs | Sim | Obs (CLcr, mL/minute) | Sim (GFR, mL/minute/1.73 m2) | ||
| Rosiglitazone | HV | p.o. 8 mg | 8/4 | 51 ± 16 | 50 ± 5 | 93 ± 9 | 93 ± 22 |
| CKD | 7/5 | 49 ± 14 | 49 ± 6 | 20 ± 5 | 20 ± 4 | ||
| Pioglitazone | HV | p.o. 45 mg | 3/3 | 35.7 ± 9.4 | 36 ± 5 | 100 ± 13 | 100 ± 21 |
| CKD | 7/5 | 48.7 ± 16.8 | 48 ± 6 | 15 ± 8 | 16 ± 3 | ||
| Pitavastatin | HV | p.o. 4 mg | 5/3 | 52.0 ± 2.6 | 53 ± 3 | 98 ± 8 | 98 ± 20 |
| CKD | 5/3 | 54.1 ± 15.4 | 54 ± 8 | 23 ± 5 | 23 ± 4 | ||
| Repaglinide | HV | p.o. 2 mg | 5/1 | 31.5 ± 8.4 | 32 ± 6 | NA | 104 ± 20 |
| CKD | 5/1 | 53.0 ± 9.7 | 53 ± 8 | NA | 23 ± 4a | ||
Dosing regimen, gender, average age, and kidney function were matched to the reported clinical trials. CKD population: Simcyp default or modified “Sim‐RenalGFR_less30” populations. Observed CKD trial designs (Obs) were from refs.28, 35, 51, 52
CKD, chronic kidney disease; CLcr, creatinine clearance; GFR, glomerular filtration rate; HV, healthy volunteers; NA, not applicable; Obs, observed; Sim, simulated.
The observed AUC increase was up to 2.7‐fold as kidney function decreased in patients with CKD for the drugs investigated in this study. For the purpose of our analysis, an R value of the range 0.8 and 1.2 (model predicted vs. observed) was considered to be satisfactory for predicting the effect of CKD on drug exposure by the PBPK modeling.
“Bottom‐up” modeling and simulations in severe CKD populations
Once the substrate drug models were verified in the healthy populations, the AUCR in the CKD population was predicted initially using the simulator default severe renal impairment population with GFR of < 30 mL/minute/1.73 m2 (labeled as “Sim‐RenalGFR_less30”) and was compared to the exposure in Simcyp HV populations (“Sim‐Healthy Volunteers”). Several changes in physiological parameters were already implemented in the V16 default (“Sim‐RenalGFR_less30”) CKD population. For example, enzyme abundances of CYP2C8, CYP2C9, CYP2C19, and CYP3A4 were reduced to 47%, 47%, 43%, and 64%, respectively, of the corresponding healthy subjects (Simcyp Simulator, V16R1), in addition to changes of other system parameters, such as human serum albumin, hematocrit, and gastric emptying time (Table S1 ). It is worth mentioning that, due to the lack of published data, OATP1B transporter abundance implemented in the V16 default “Sim‐RenalGFR_less30” population is the same as in the HV population (Simcyp Simulator, V16R1; Table S1 ).
“Middle‐out” modeling and simulations with modified CKD populations
In addition to the V16 default severe CKD population, several modified severe CKD populations were created using the default “Sim‐RenalGFR_less30” as the template. The first change was to set the hepatic CYP2C8 enzyme abundance to the value of virtual healthy population, named as “Modified,” because Simcyp default severe population “Sim‐RenalGFR_less30” (CYP2C8 enzyme abundance was reduced to 47% of the value in healthy populations; see Table S1 ) overpredicted the AUCR for CYP2C8 substrates (see Results). For rosiglitazone and pioglitazone, the simulations were performed assuming no changes in CYP2C8 abundance as a result of severe CKD. Analogous to this approach, the initial simulations for pitavastatin were performed assuming no changes in OATP1B abundance relative to healthy, followed by simulations in which OATP1B abundance was decreased to recover the observed change in the exposure and clearance in this patient group. Subsequently, for the CYP2C8 and OATP1B dual substrate repaglinide, the initial simulations were performed assuming no changes in the OATP1B abundance, followed by simulations with OATP1B abundance decreased to the level observed in the pitavastatin studies, and finally OATP1B function was optimized to recover the observed exposure change in repaglinide CKD study, as illustrated in the workflow shown in Figure 3 .
It is worth noting that other CYP enzymes, such as CYP2C9, CYP2C19, and CYP3A4, may be involved in the drug metabolism as minor pathways for the drugs investigated in this study. To simplify the analysis, the effect of CKD on these pathways was counted for using Simcyp Simulator default CKD settings (Table S1 ).
Funding
This research was supported by the US Food and Drug Administration (FDA) Medical Countermeasures Initiative. Drs. Ming‐Liang Tan, Yunn‐Fang Ho, Thomas D. Nolin, and Aleksandra Galetin were supported in part by an appointment to the Research Participation Program at the Center for Drug Evaluation and Research, administered by the Oak Ridge Institute for Science and Education through an interagency agreement between the US Department of Energy and the FDA. Dr. Nolin is supported in part by the National Institutes of Health, National Institute of General Medical Sciences (R01‐GM107122).
Conflict of Interest
As an Associate Editor for Clinical Pharmacology & Therapeutics, S.‐M.H. was not involved in the review or decision process for this article. S.N. is an employee of the Simcyp Division of Certara UK. Simcyp's research is funded by a consortium of pharmaceutical companies. The other authors declared no conflicts of interest.
Author Contributions
M.‐L.T., P.Z., L.Z., Y.‐F.H., M.V.S.V., S.N., T.D.N., A.G., and S.‐M.H. wrote the manuscript. M.‐L.T., P.Z., L.Z., A.G., and S.‐M.H. designed the research. M.‐L.T. performed the research. M.‐L.T., P.Z., L.Z., T.D.N., A.G., and S.‐M.H. performed data analysis.
Disclaimer
The contents of this article reflect the views of the authors and should not be construed to represent the FDA's views or policies. No official support or endorsement by the FDA is intended or should be inferred. The mention of commercial products, their sources, or their use in connection with material reported herein is not to be construed as either an actual or implied endorsement of such products by the FDA.
Supporting information
Supplementary Reading List S1
Table S1. Selected physiological and biochemical parameter changes for patients with severe CKD in Simcyp V16.
Table S2. References for the observed AUC changes.
Figure S1. Sensitivity analysis of passive diffusion clearance (CLpd), on area under the concentration‐time curves ratio (AUCR; total) between patients with severe chronic kidney disease (CKD; cytochrome P450 (CYP)2C8 100% and organic anion‐transporting polypeptides (OATP)1B 40%) and healthy volunteer (HV) populations for repaglinide.
Figure S2. Hepatic uptake was scaled to fit c. 521 TT data for pitavastatin.
Acknowledgments
The authors thank Drs. Peng Duan, Vicky Hsu, Khondoker Alam, Bilal AbuAsal, Rajnikanth Madabushi, Xinning Yang, and Issam Zineh for their helpful discussions and invaluable comments.
[The copyright line for this article was changed on 25 March 2021 after original online publication].
References
- 1. Weiner, D.E. Public health consequences of chronic kidney disease. Clin. Pharmacol. Ther. 86, 566–569 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Chen, Y.C. et al. Risk factors associated with adverse drug events among older adults in emergency department. Eur. J. Intern. Med. 25, 49–55 (2014). [DOI] [PubMed] [Google Scholar]
- 3. Nolin, T.D. , Naud, J. , Leblond, F.A. & Pichette, V. Emerging evidence of the impact of kidney disease on drug metabolism and transport. Clin. Pharmacol. Ther. 83, 898–903 (2008). [DOI] [PubMed] [Google Scholar]
- 4. Yeung, C.K. , Shen, D.D. , Thummel, K.E. & Himmelfarb, J. Effects of chronic kidney disease and uremia on hepatic drug metabolism and transport. Kidney Int. 85, 522–528 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Rowland Yeo, K. , Aarabi, M. , Jamei, M. & Rostami‐Hodjegan, A. Modeling and predicting drug pharmacokinetics in patients with renal impairment. Expert Rev. Clin. Pharmacol. 4, 261–274 (2011). [DOI] [PubMed] [Google Scholar]
- 6. European Medicines Agency . Draft guideline on the evaluation of the pharmacokinetics of medicinal products in patients with decreased renal function. <http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2014/02/WC500162133.pdf> (2014). Accessed 26 March 2017.
- 7. U.S. Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research (CDER) . Draft Guidance/Guidance for Industry. Pharmacokinetics in Patients with Impaired Renal Function — Study Design, Data Analysis, and Impact on Dosing and Labeling. <http://www.fda.gov/downloads/Drugs/Guidances/UCM204959.pdf> (2010). Accessed 26 March 2017.
- 8. Barnes, K.J. , Rowland, A. , Polasek, T.M. & Miners, J.O. Inhibition of human drug‐metabolising cytochrome P450 and UDP‐glucuronosyltransferase enzyme activities in vitro by uremic toxins. Eur. J. Clin. Pharmacol. 70, 1097–1106 (2014). [DOI] [PubMed] [Google Scholar]
- 9. Nolin, T.D. , Appiah, K. , Kendrick, S.A. , Le, P. , McMonagle, E. & Himmelfarb, J. Hemodialysis acutely improves hepatic CYP3A4 metabolic activity. J. Am. Soc. Nephrol. 17, 2363–2367 (2006). [DOI] [PubMed] [Google Scholar]
- 10. Nolin, T.D. et al. ESRD impairs nonrenal clearance of fexofenadine but not midazolam. J. Am. Soc. Nephrol. 20, 2269–2276 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Grime, K. & Paine, S.W. Species differences in biliary clearance and possible relevance of hepatic uptake and efflux transporters involvement. Drug Metab. Dispos. 41, 372–378 (2013). [DOI] [PubMed] [Google Scholar]
- 12. Yoshida, K. et al. Systematic and quantitative assessment of the effect of chronic kidney disease on CYP2D6 and CYP3A4/5. Clin. Pharmacol. Ther. 100, 75–87 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Tan, M.‐L. et al. Effect of chronic kidney disease on nonrenal elimination pathways: a systematic assessment of CYP1A2, CYP2C8, CYP2C9, CYP2C19 and OATP. Clin. Pharmacol. Ther. 103, 854–867 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Rostami‐Hodjegan, A. Physiologically based pharmacokinetics joined with in vitro‐in vivo extrapolation of ADME: a marriage under the arch of systems pharmacology. Clin. Pharmacol. Ther. 92, 50–61 (2012). [DOI] [PubMed] [Google Scholar]
- 15. Zhao, P. et al. Applications of physiologically based pharmacokinetic (PBPK) modeling and simulation during regulatory review. Clin. Pharmacol. Ther. 89, 259–267 (2011). [DOI] [PubMed] [Google Scholar]
- 16. Zhao, P. , Rowland, M. & Huang, S.M. Best practice in the use of physiologically based pharmacokinetic modeling and simulation to address clinical pharmacology regulatory questions. Clin. Pharmacol. Ther. 92, 17–20 (2012). [DOI] [PubMed] [Google Scholar]
- 17. Galetin, A. , Zhao, P. & Huang, S.M. Physiologically based pharmacokinetic modeling of drug transporters to facilitate individualized dose prediction. J. Pharm. Sci. 106, 2204–2208 (2017). [DOI] [PubMed] [Google Scholar]
- 18. Zhao, P. et al. Evaluation of exposure change of nonrenally eliminated drugs in patients with chronic kidney disease using physiologically based pharmacokinetic modeling and simulation. J. Clin. Pharmacol. 52, 91S–108S (2012). [DOI] [PubMed] [Google Scholar]
- 19. Hsu, V. et al. Towards quantitation of the effects of renal impairment and probenecid inhibition on kidney uptake and efflux transporters, using physiologically based pharmacokinetic modelling and simulations. Clin. Pharmacokinet. 53, 283–293 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hsueh, C.H. , Hsu, V. , Zhao, P. , Zhang, L. , Giacomini, K.M. & Huang, S.M. PBPK modeling of the effect of reduced kidney function on the pharmacokinetics of drugs excreted renally by organic anion transporters. Clin. Pharmacol. Ther. 103, 485–492 (2018). [DOI] [PubMed] [Google Scholar]
- 21. Scotcher, D. , Jones, C.R. , Galetin, A. & Rostami‐Hodjegan, A. Delineating the role of various factors in renal disposition of digoxin through application of physiologically based kidney model to renal impairment populations. J. Pharmacol. Exp. Ther. 360, 484–495 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Jones, H.M. et al. Mechanistic pharmacokinetic modeling for the prediction of transporter‐mediated disposition in humans from sandwich culture human hepatocyte data. Drug Metab. Dispos. 40, 1007–1017 (2012). [DOI] [PubMed] [Google Scholar]
- 23. Gertz, M. , Tsamandouras, N. , Säll, C. , Houston, J.B. & Galetin, A. Reduced physiologically‐based pharmacokinetic model of repaglinide: impact of OATP1B1 and CYP2C8 genotype and source of in vitro data on the prediction of drug‐drug interaction risk. Pharm. Res. 31, 2367–2382 (2014). [DOI] [PubMed] [Google Scholar]
- 24. Cox, P.J. et al. Absorption, disposition, and metabolism of rosiglitazone, a potent thiazolidinedione insulin sensitizer, in humans. Drug Metab. Dispos. 28, 772–780 (2000). [PubMed] [Google Scholar]
- 25. Niemi, M. , Backman, J.T. , Neuvonen, M. & Neuvonen, P.J. Effects of gemfibrozil, itraconazole, and their combination on the pharmacokinetics and pharmacodynamics of repaglinide: potentially hazardous interaction between gemfibrozil and repaglinide. Diabetologia 46, 347–351 (2003). [DOI] [PubMed] [Google Scholar]
- 26. Aquilante, C.L. et al. Impact of the CYP2C8 *3 polymorphism on the drug‐drug interaction between gemfibrozil and pioglitazone. Br. J. Clin. Pharmacol. 75, 217–226 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Prueksaritanont, T. et al. Pitavastatin is a more sensitive and selective organic anion‐transporting polypeptide 1B clinical probe than rosuvastatin. Br. J. Clin. Pharmacol. 78, 587–598 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Morgan, R.E. , Campbell, S.E. , Yu, C.Y. , Sponseller, C.A. & Muster, H.A. Comparison of the safety, tolerability, and pharmacokinetic profile of a single oral dose of pitavastatin 4 mg in adult subjects with severe renal impairment not on hemodialysis versus healthy adult subjects. J. Cardiovasc. Pharmacol. 60, 42–48 (2012). [DOI] [PubMed] [Google Scholar]
- 29. Zhou, Q. , Chen, Q.X. , Ruan, Z.R. , Yuan, H. , Xu, H.M. & Zeng, S. CYP2C9*3(1075A > C), ABCB1 and SLCO1B1 genetic polymorphisms and gender are determinants of inter‐subject variability in pitavastatin pharmacokinetics. Pharmazie 68, 187–194 (2013). [PubMed] [Google Scholar]
- 30. Hirano, M. , Maeda, K. , Shitara, Y. & Sugiyama, Y. Contribution of OATP2 (OATP1B1) and OATP8 (OATP1B3) to the hepatic uptake of pitavastatin in humans. J. Pharmacol. Exp. Ther. 311, 139–146 (2004). [DOI] [PubMed] [Google Scholar]
- 31. Hatorp, V. , Oliver, S. & Su, C.A. Bioavailability of repaglinide, a novel antidiabetic agent, administered orally in tablet or solution form or intravenously in healthy male volunteers. Int. J. Clin. Pharmacol. Ther. 36, 636–641 (1998). [PubMed] [Google Scholar]
- 32. Honkalammi, J. , Niemi, M. , Neuvonen, P.J. & Backman, J.T. Dose‐dependent interaction between gemfibrozil and repaglinide in humans: strong inhibition of CYP2C8 with subtherapeutic gemfibrozil doses. Drug Metab. Dispos. 39, 1977–1986 (2011). [DOI] [PubMed] [Google Scholar]
- 33. Kim, S.‐J. et al. Clarification of the mechanism of clopidogrel‐mediated drug‐drug interaction in a clinical cassette small‐dose study and its prediction based on in vitro information. Drug Metab. Dispos. 44, 1622–1632 (2016). [DOI] [PubMed] [Google Scholar]
- 34. Kalliokoski, A. , Backman, J.T. , Kurkinen, K.J. , Neuvonen, P.J. & Niemi, M. Effects of gemfibrozil and atorvastatin on the pharmacokinetics of repaglinide in relation to SLCO1B1 polymorphism. Clin. Pharmacol. Ther. 84, 488–496 (2008). [DOI] [PubMed] [Google Scholar]
- 35. Marbury, T. et al. Pharmacokinetics of repaglinide in subjects with renal impairment. Clin. Pharmacol. Ther. 67, 7–15 (2000). [DOI] [PubMed] [Google Scholar]
- 36. Fujita, K. et al. Direct inhibition and down‐regulation by uremic plasma components of hepatic uptake transporter for SN‐38, an active metabolite of irinotecan, in humans. Pharm. Res. 31, 204–215 (2006). [DOI] [PubMed] [Google Scholar]
- 37. Vildhede, A. et al. Mechanistic modeling of pitavastatin disposition in sandwich‐cultured human hepatocytes: a proteomics‐informed bottom‐up approach. Drug Metab. Dispos. 44, 505–516 (2016). [DOI] [PubMed] [Google Scholar]
- 38. Hirano, M. , Maeda, K. , Matsushima, S. , Nozaki, Y. , Kusuhara, H. & Sugiyama, Y. Involvement of BCRP (ABCG2) in the biliary excretion of pitavastatin. Mol. Pharmacol. 68, 800–807 (2005). [DOI] [PubMed] [Google Scholar]
- 39. Sall, C. , Houston, J.B. & Galetin, A. A comprehensive assessment of repaglinide metabolic pathways: impact of choice of in vitro system and relative enzyme contribution to in vitro clearance. Drug Metab. Dispos. 40, 1279–1289 (2012). [DOI] [PubMed] [Google Scholar]
- 40. Mutsaers, H.A. et al. Uremic toxins inhibit renal metabolic capacity through interference with glucuronidation and mitochondrial respiration. Biochim. Biophys. Acta 1832, 142–150 (2013). [DOI] [PubMed] [Google Scholar]
- 41. U.S. Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research (CDER) . Transcript from the March 17, 2010 Meeting of the Pharmaceutical Science and Clinical Pharmacology Advisory Committee. <http://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/AdvisoryCommitteeforPharmaceuticalScienceandClinicalPharmacology/UCM210803.pdf> (2010). Accessed 2 April 2015.
- 42. Duan, P. , Zhao, P. & Zhang, L. Physiologically based pharmacokinetic (PBPK) modeling of pitavastatin and atorvastatin to predict drug‐drug interactions (DDIs). Eur. J. Drug Metab. Pharmacokinet. 42, 689 (2017). [DOI] [PubMed] [Google Scholar]
- 43. Varma, M.V. , Lai, Y. , Kimoto, E. , Goosen, T.C. , El‐Kattan, A.F. & Kumar, V. Mechanistic modeling to predict the transporter‐ and enzyme‐mediated drug‐drug interactions of repaglinide. Pharm. Res. 30, 1188–1199 (2013). [DOI] [PubMed] [Google Scholar]
- 44. Varma, M.V.S. , Lin, J. , Bi, Y.‐A. , Kimoto, E. & Rodrigues, A.D. Quantitative rationalization of gemfibrozil drug interactions: consideration of transporters‐enzyme interplay and the role of circulating metabolite gemfibrozil 1‐O‐b‐glucuronide. Drug Metab. Dispos. 43, 1108–1118 (2015). [DOI] [PubMed] [Google Scholar]
- 45. Rodgers, T. & Rowland, M. Physiologically based pharmacokinetic modelling 2: predicting the tissue distribution of acids, very weak bases, neutrals and zwitterions. J. Pharm. Sci. 95, 1238–1257 (2006). [DOI] [PubMed] [Google Scholar]
- 46. Ieiri, I. et al. SLCO1B1 (OATP1B1, an uptake transporter) and ABCG2 (BCRP, an efflux transporter) variant alleles and pharmacokinetics of pitavastatin in healthy volunteers. Clin. Pharmacol. Ther. 82, 541–547 (2007). [DOI] [PubMed] [Google Scholar]
- 47. Gertz, M. et al. Cyclosporine inhibition of hepatic and intestinal CYP3A4, uptake and efflux transporters: application of PBPK modeling in the assessment of drug‐drug interaction potential. Pharm. Res. 30, 761–780 (2013). [DOI] [PubMed] [Google Scholar]
- 48. Varma, M.V. , Lai, Y. , Feng, B. , Litchfield, J. , Goosen, T.C. & Bergman, A. Physiologically based modeling of pravastatin transporter‐mediated hepatobiliary disposition and drug‐drug interactions. Pharm. Res. 29, 2860–2873 (2012). [DOI] [PubMed] [Google Scholar]
- 49. Vieira, M.D. et al. PBPK model describes the effects of comedication and genetic polymorphism on systemic exposure of drugs that undergo multiple clearance pathways. Clin. Pharmacol. Ther. 95, 550–557 (2014). [DOI] [PubMed] [Google Scholar]
- 50. Sager, J.E. , Yu, J. , Ragueneau‐Majlessi, I. & Isoherranen, N. Physiologically based pharmacokinetic (PBPK) modeling and simulation approaches: a systematic review of published models, applications, and model verification. Drug Metab. Dispos. 43, 1823–1837 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Chapelsky, M.C. , Thompson‐Culkin, K. , Miller, A.K. , Sack, M. , Blum, R. & Freed, M.I. Pharmacokinetics of rosiglitazone in patients with varying degrees of renal insufficiency. The Journal of Clinical Pharmacology 43, 252–259 (2003). [DOI] [PubMed] [Google Scholar]
- 52. Budde, K. , Neumayer, H.H. , Fritsche, L. , Sulowicz, W. , Stompôr, T. & Eckland, D. The pharmacokinetics of pioglitazone in patients with impaired renal function. Br. J. Clin. Pharmacol. 55, 368–374 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Hirano, M. , Maeda, K. , Shitara, Y. & Sugiyama, Y. Drug‐drug interaction between pitavastatin and various drugs via OATP1B1. Drug Metab. Dispos. 34, 1229–1236 (2006). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Reading List S1
Table S1. Selected physiological and biochemical parameter changes for patients with severe CKD in Simcyp V16.
Table S2. References for the observed AUC changes.
Figure S1. Sensitivity analysis of passive diffusion clearance (CLpd), on area under the concentration‐time curves ratio (AUCR; total) between patients with severe chronic kidney disease (CKD; cytochrome P450 (CYP)2C8 100% and organic anion‐transporting polypeptides (OATP)1B 40%) and healthy volunteer (HV) populations for repaglinide.
Figure S2. Hepatic uptake was scaled to fit c. 521 TT data for pitavastatin.