

Abstract
Activated T cells drive a range of immune‐mediated inflammatory diseases. LAG‐3 is transiently expressed on recently activated CD4+ and CD8+ T cells. We describe the engineering and first‐in‐human clinical study (NCT02195349) of GSK2831781 (an afucosylated humanized IgG1 monoclonal antibody enhanced with high affinity for Fc receptors and LAG‐3 and antibody‐dependent cellular cytotoxicity capabilities), which depletes LAG‐3 expressing cells. GSK2831781 was tested in a phase I/Ib, double‐blind, placebo‐controlled clinical study, which randomized 40 healthy participants (part A) and 27 patients with psoriasis (part B) to single doses of GSK2831781 (up to 0.15 and 5 mg/kg, respectively) or placebo. Adverse events were generally balanced across groups, with no safety or tolerability concern identified. LAG‐3+ cell depletion in peripheral blood was observed at doses ≥ 0.15 mg/kg and was dose‐dependent. In biopsies of psoriasis plaques, a reduction in mean group LAG‐3+ and CD3+ T‐cell counts was observed following treatment. Downregulation of proinflammatory genes (IL‐17A, IL‐17F, IFNγ, and S100A12) and upregulation of the epithelial barrier integrity gene, CDHR1, was observed with the 5 mg/kg dose of GSK2831781. Psoriasis disease activity improved up to day 43 at all GSK2831781 doses (0.5, 1.5, and 5 mg/kg) compared with placebo. Depletion of LAG‐3‐expressing activated T cells is a novel approach, and this first clinical study shows that GSK2831781 is pharmacologically active and provides encouraging early evidence of clinical effects in psoriasis, which warrants further investigation in T‐cell‐mediated inflammatory diseases.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
☑ LAG‐3 is transiently expressed on activated T cells.
WHAT QUESTION DID THIS STUDY ADDRESS?
☑ Depletion of LAG‐3+ cells is a potential treatment strategy to selectively deplete activated T cells, for T‐cell‐mediated inflammatory diseases. We investigated GSK2831781 (an afucosylated, humanized IgG1 anti‐LAG‐3 mAb enhanced for affinity and antibody‐dependent cell cytotoxicity), in a phase I study to assess its safety/tolerability in healthy individuals and patients with psoriasis, and its potential efficacy in patients with psoriasis.
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
☑ No safety/tolerability concern was identified. Following treatment with GSK2831781, LAG‐3+ and CD3+ T‐cell counts were reduced in peripheral blood and in psoriasis plaque biopsies; additionally, expression of proinflammatory genes was reduced, and expression of genes associated with skin barrier function was increased.
HOW MIGHT THIS CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE?
☑ GSK2831781 was pharmacologically active and demonstrated that depletion of LAG‐3+ T cells is a therapeutic approach warranting further investigation in T‐cell‐mediated inflammatory diseases.
Activated pathogenic T cells play an important role in immune‐mediated inflammatory diseases, as evidenced by favorable clinical outcomes with several T‐cell‐targeted therapies. 1 , 2 , 3 , 4 Plaque psoriasis, a chronic autoimmune skin disease, is driven by excessive IL‐17A, TNF‐α, and IL‐23, as shown by successful therapeutic blockade of these cytokines. 5 Th17 cells play a dominant role in disease and drives keratinocyte hyperplasia, which typifies psoriasis plaque histopathology. 5 Skin resident T cells are implicated in the maintenance and recurrence of psoriatic plaque lesions 6 and depleting such subsets may lead to long‐term remission. Pan‐T‐cell‐targeted therapies carry some drawbacks, such as increased risk of infection, 7 so more selective ways of targeting activated, pathogenic T cells are needed to improve risk–benefit profile.
LAG‐3 or CD223 was originally described as a marker of activation on T cells and natural killer (NK) cells. 8 It was subsequently confirmed as an inhibitory transmembrane receptor that negatively regulates T‐cell activation and is upregulated as an immune checkpoint to balance T‐cell co‐stimulation, thereby limiting inflammation and autoimmunity. 9 , 10 , 11 It is expressed on activated CD4+ (predominantly Th17 and Th1 memory), 12 CD8+ T cells, minor subsets of NK cells, 8 , 13 , 14 a subset of activated regulatory T (Treg) cells, T regulatory type 1 (Tr1) cells, 15 , 16 , 17 , 18 , 19 , 20 and plasmacytoid dendritic cells. 21 , 22 Soluble LAG‐3 (sLAG‐3) is found at low concentrations in human serum 23 , 24 , 25 , 26 , 27 ; however, its biological function is unclear.
LAG‐3 blockade has been investigated as a therapeutic strategy for checkpoint blockade immunotherapy and as a potential cancer treatment. 28 A contrasting strategy of depleting activated LAG‐3+ T cells was explored preclinically using cytotoxic antibodies to delay acute rejection in a rodent heart allotransplant model 29 and to reduce skin inflammation in a tuberculin‐induced delayed type hypersensitivity (DTH) model in nonhuman primates. 30 Because LAG‐3 is rapidly upregulated following T‐cell activation 31 and has low or undetectable levels on resting lymphocytes, it might be possible to leverage LAG‐3 as a marker of pathogenic lymphocytes that mediate chronic inflammation in humans, thereby targeting these cells for depletion.
Here, we report the development of GSK2831781, a high affinity (sub nanomolar), humanized IgG1 monoclonal antibody (mAb) targeting LAG‐3, enhanced for antibody‐dependent cell cytotoxicity (ADCC) compared with the parental antibody, using POTELLIGENT technology (BioWa, Princeton, NJ), which generated an afucosylated mAb. It was hypothesized that GSK2831781 would differentially deplete tissue‐resident effector pathogenic T cells with relatively high LAG‐3 expression (over lymphocytes expressing low or no LAG‐3), impact downstream immunopharmacology, and translate to clinical improvement. The hypothesis was investigated clinically in a first‐time‐in‐human trial (GSK study ID: 200630; NCT02195349). The primary objective was to assess the safety and tolerability of a single i.v. dose of GSK2831781 from 0.0003 to 5 mg/kg. Key secondary objectives included evaluation of GSK2831781 pharmacology, pharmacokinetics (PKs), immunogenicity, and effect on psoriasis disease activity.
METHODS
Development of GSK2831781
Engineering, expression, and characterization of GSK2831781 are described in Supplementary Methods .
Study design
A phase I/Ib, randomized, double‐blind (sponsor unblinded), placebo‐controlled, first‐time‐in‐human two‐part study (GSK study 200630; NCT02195349) was conducted at three centers in the United Kingdom and Germany from July 30, 2014, to March 7, 2018 (Figure 1 ). Part A enrolled healthy participants into six GSK2831781 dose cohorts. Cohorts 1–5 (0.0003–0.15 mg/kg) were screened for the absence of pre‐existing anti‐drug antibodies (ADAs; ADA‐negative). Cohort 6 (0.15 mg/kg) were selected for presence of pre‐existing ADA (ADA‐positive). Part B enrolled patients with psoriasis into three cohorts. Cohort 7 (0.5 mg/kg) was stratified by pre‐existing ADA status; stratification was removed for cohorts 8 and 9 (1.5 and 5 mg/kg) owing to a lower than predicted incidence of pre‐existing ADA and lack of observed impact of pre‐existing ADA on safety and PK to that point.
Figure 1.
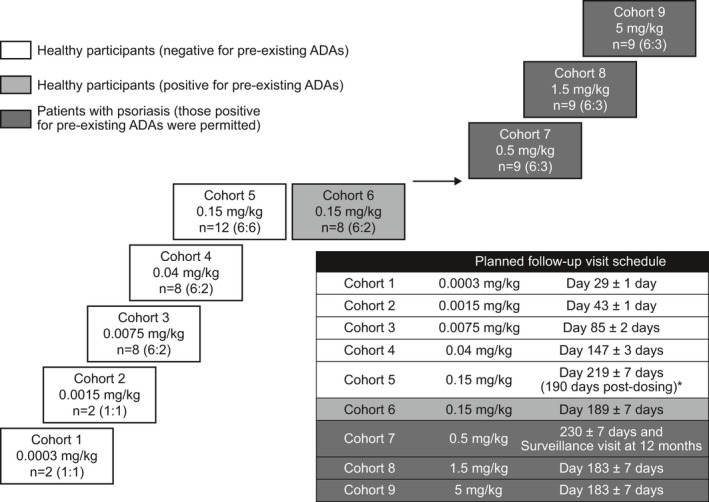
Clinical study design. *Cohort 5 included a pre‐treatment delayed‐type hypersensitivity challenge. Dosing with GSK2831781 occurred on day 29, hence, follow‐up is correspondingly longer relative to study day 1. ADA, anti‐drug antibodies; n, total participants (GSK2831781:placebo).
Participants were block‐randomized to treatment groups with sentinel dosing (one active and one placebo for each cohort). GSK2831781 (or placebo) was administered via a 2‐hour i.v. infusion.
The protocol was approved by national or regional ethics committees. The study was conducted in accordance with the International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use Good Clinical Practice guidelines and applicable country‐specific requirements. Written informed consent was obtained. Anonymized individual participant data and study documents can be requested for further research from www.clinicalstudydatarequest.com.
Study population
Full inclusion and exclusion criteria are provided in Supplementary Methods . Part A included healthy men aged 18–65 years. Part B included women or men aged 18−75 years with chronic plaque psoriasis for ≥6 months, covering ≥3% body surface area at screening and day 1, with 2 accessible psoriatic plaques each with a Psoriatic Lesion Severity Sum (PLSS) of ≥ 5 (≥ 2 for the induration component, ≥ 1 for redness, and ≥ 1 for scaling) 32 : 1 index plaque for monitoring of clinical activity and the other a biopsy plaque. Patients were excluded if at high risk of requiring rescue with topical psoriasis therapies or systemic immunosuppressants, or if the washout conditions of any prior psoriasis treatment were not met.
Safety and tolerability
The primary objective was to assess the safety and tolerability via monitoring of adverse events (AEs), vital signs, physical examinations, routine hematology, clinical chemistry, urinalysis, and electrocardiograms. Serum cytokine levels (IL‐6, IL‐8, TNF‐α, and IFN‐γ) were assessed to 48 hours; granulocyte‐colony stimulating factor was assessed throughout. Peripheral blood T (CD3+, CD4+, and CD8+), B (CD45+, CD3−, and CD19+), and NK (CD45+, CD3−, CD16+, and CD56+) cells were measured by flow cytometry. Further details are provided in Supplementary Methods .
Measurement of antidrug antibodies
Anti‐GSK2831781 antibodies were assessed using a validated electrochemiluminescence bridging immunoassay. Samples identified as potentially ADA‐positive in a screening assay were further analyzed to confirm ADA specificity, and if confirmed positive, titer. Assay sensitivity of 24.45 ng/mL was assessed using a polyclonal positive control antibody, which was specific for GSK2831781. At 500 ng/mL positive control, the assay tolerated up to GSK2831781 13.5 μg/mL and up to sLAG‐3 100 ng/mL. Monomeric sLAG‐3 precludes false‐positive ADA results. 33
Pharmacokinetics
Blood samples for PKs were collected at nominal times relative to the start of GSK2831781 infusion and analyzed using a validated affinity capture, high performance liquid chromatography mass spectrometry/mass spectrometry method. The limits of quantification were 10−5,000 ng/mL. PK parameters were derived using WinNonlin version 8.0 (Pharsight, Sunnyvale, CA).
Monitoring of LAG‐3+ cells in blood
Blood samples for analysis of target engagement, T‐cell depletion, and immune cell phenotyping were analyzed by flow cytometry. Target engagement (receptor occupancy) of GSK2831781 T‐cell surface LAG‐3 in blood and consequent depletion of LAG‐3+ T cells (mechanism of action) were evaluated using a noncompetitive and a competitive anti‐LAG‐3 antibody in the flow cytometric analysis of CD4+, CD45RA− (central memory and effector memory cells).
Measurement of total sLAG‐3 in blood
Total sLAG‐3 (sLAG‐3 free/unbound and bound to GSK2831781) was analyzed in serum using an electrochemiluminescence immunoassay. Total sLAG‐3 concentrations (ng/mL) were obtained using a standard curve; lower and upper limits of quantification were 0.244 and 250 ng/mL, respectively.
Pharmacodynamic effect in skin
In healthy participants, a DTH model was used (ref. 34 and Supplementary Methods ). Psoriasis skin punch biopsies (6 mm) were taken predose at day 1 and at day 29. Serial skin sections were stained for LAG‐3, CD3, and the proliferation marker, Ki67. LAG‐3+ and CD3+ cell counts in dermis and epidermis, and Ki67+ cell counts in epidermis were manually quantified by immunohistochemistry. Epidermal thickness was measured following hematoxylin‐eosin staining.
Transcriptomic analysis was performed to evaluate changes in gene expression. Briefly, total RNA was prepared from whole blood and psoriatic skin lesion biopsies and used for cDNA amplification for quantitative real‐time polymerase chain reaction (qRT‐PCR) analysis against a panel of TaqMan Gene Expression Assays (Thermo Fisher Scientific, Loughborough, UK) comprising housekeeping genes and genes of interest, using the Fluidigm BioMark HD microfluidic PCR platform (Fluidigm, South San Francisco, CA), as detailed in Supplementary Methods .
Psoriasis disease activity
The clinical activity of psoriasis lesions was assessed using the PLSS (of the index plaque) 32 and Psoriasis Area and Severity Index (PASI), 35 as detailed in Supplementary Methods .
Sample size and statistical analysis
As the primary end point was safety and tolerability, the sample size was based on feasibility and no formal sample size calculations were conducted. However, to understand the properties of the design, the upper limit of the exact 95% confidence interval (CI) of the incidence rate was determined. A maximum of 6 participants received each active dose and therefore if a safety event was observed in 0 of 6 of a GSK2831781 group, the upper limit of the exact 95% CI would indicate that a true incidence rate of 46.5% could not be ruled out. Whereas, if 1 of 6 of the same safety event in the GSK2831781 group was to be observed, the upper limit of the exact 95% CI would indicate that a true incidence rate of 62.9% could not be ruled out. Sample size sensitivity analysis for safety is described in Table S1 . As the study was not powered on the pharmacodynamic or clinical efficacy end points, no formal hypothesis testing was performed. Descriptive summaries are reported (see [Link] , [Link] , [Link] , [Link] , [Link] , [Link] , [Link] , [Link] , [Link] for detailed listings), including point estimates and 95% CI to provide the direction and the magnitude of the effect of GSK2831781. Estimations of the difference of change from baseline for each treatment arm to placebo were made for selected parameters from lesional biopsies (including CD3+ and LAG‐3+ cell counts, Ki67, and epidermal thickness) and clinical parameters PLSS and PASI, by fitting a mixed‐effect model for repeated measures with fixed effects of treatment, visit, treatment‐by‐visit interaction, and baseline. Unstructured covariance structure was used. Epigenetic cell count data were generated after the study had reported and analyzed post hoc.
RESULTS
Characterization of GSK2831781
GSK2831781, an afucosylated IgG1 mAb, was humanized from a murine anti‐LAG‐3 chimeric parent mAb and further modified to improve affinity for LAG‐3 and ADCC. GSK2831781 exhibited enhanced affinity for LAG‐3 and human FcγIIIa receptors (data not shown), and 30‐fold enhanced killing potency of a LAG‐3‐expressing T‐cell line compared with anti‐LAG‐3 antibodies with wild‐type fucosylation (murine anti‐LAG‐3 chimeric parent mAb and its humanized variant GSK2831780; data not shown).
Antigen‐stimulated T‐cell proliferation was reduced in a dose‐dependent manner by GSK2831781. Blockade of NK‐mediated cell killing with anti‐CD16 countered this reduction in proliferation, suggesting ADCC was the dominant mechanism of killing (Figure 2 ). GSK2831781 inhibited the interaction between LAG‐3 and its cognate MHCII ligand in a dose‐dependent manner, demonstrating potential for competitive antagonism (data not shown). GSK2831781 did not increase proliferation, so we concluded it did not antagonize the role of LAG‐3 as a checkpoint inhibitory receptor.
Figure 2.
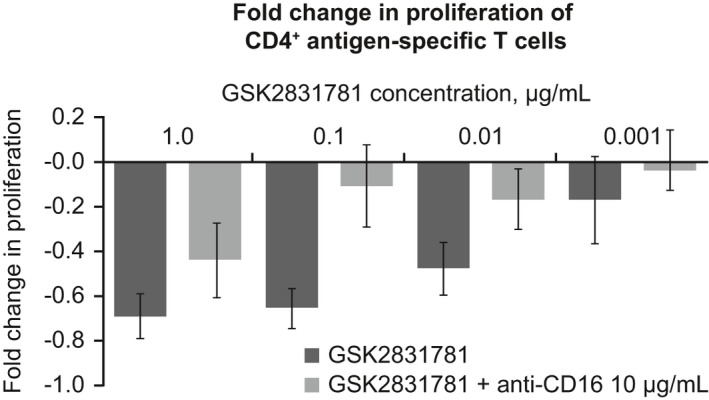
Characterization of GSK2831781, an affinity‐enhanced and ADCC‐enhanced afucosylated IgG1 LAG‐3‐specific mAb. Fold change in the proliferation of CD4+ antigen‐specific T cells in the presence of GSK2831781 alone and with anti‐CD16, relative to untreated antigen stimulation controls. ADCC, antibody‐dependent cellular cytotoxicity; IgG1, immunoglobulin G1; mAb, monoclonal antibody; PBMC, peripheral blood mononuclear cell.
Patient disposition and demographics
Part A randomized 40 healthy participants (of 330 screened) and part B randomized 27 patients with psoriasis (of 197 screened; Figure 2 ). All randomized participants completed the study (Figure S1 ). The high initial screen failure rate for part A was principally due to the requirement for evidence of childhood vaccinations. The initial high screen failure rate in part B was mainly due to the requirement to stratify for pre‐existing ADAs. Baseline patient characteristics are summarized in Table 1 .
Table 1.
Baseline demographics and clinical characteristics of healthy participants (part A) and patients with psoriasis (part B)
| Part A: Healthy participants (N = 40) | Placebo (n = 14) | GSK2831781 | ||||
|---|---|---|---|---|---|---|
| 0.0003 mg/kg (n = 1) | 0.0015 mg/kg (n = 1) | 0.0075 mg/kg (n = 6) | 0.04 mg/kg (n = 6) | 0.15 mg/kg (n = 12) | ||
| Age, years, mean (SD) | 46.4 (12.3) | 21.0 (−) | 53.0 (−) | 46.7 (20.8) | 39.7 (12.5) | 50.7 (10.1) |
| Male, n (%) | 14 (100) | 1 (100) | 1 (100) | 6 (100) | 6 (100) | 12 (100) |
| Weight, kg, mean (SD) | 85.5 (9.6) | 70.7 (−) | 87.0 (−) | 84.2 (10.8) | 82.6 (12.0) | 79.2 (8.9) |
| Part B: Patients with psoriasis (N = 27) | Placebo (n = 9) | GSK2831781 | ||
|---|---|---|---|---|
| 0.5 mg/kg (n = 6) | 1.5 mg/kg (n = 6) | 5 mg/kg (n = 6) | ||
| Age, years, mean (SD) | 45.3 (13.1) | 47.2 (8.2) | 48.5 (13.6) | 51.8 (5.4) |
| Male, n (%) | 5 (56.0) | 6 (100) | 5 (83.0) | 5 (83.0) |
| Weight, kg, mean (SD) | 76.9 (14.6) | 88.7 (15.3) | 83.6 (12.0) | 94.4 (10.4) |
| PLSS, mean (SD) | 7.1 (1.1) | 6.5 (1.4) | 7.7 (1.4) | 6.5 (0.8) |
| PASI, mean (SD) | 10.3 (4.5) | 6.8 (2.4) | 14.1 (6.6) | 9.2 (4.1) |
| Total BSA, mean (SD) | 12.7 (8.9) | 6.7 (5.0) | 18.2 (10.5) | 10.7 (6.9) |
BSA, body surface area; PASI, Psoriasis Area Severity Index; PLSS, Psoriasis Lesion Severity Score.
Safety and tolerability
No safety or tolerability concern was identified following a single i.v. dose of GSK2831781 up to 5 mg/kg. AEs were reported in 17 of 26 (65%) healthy participants receiving GSK2831781 and 8 of 14 (57%) receiving placebo in part A, and 16 of 18 (89%) of patients with psoriasis receiving GSK2831781 and 9 of 9 (100%) receiving placebo in part B (Table 2 ). AEs were balanced across treatment groups, except for back pain in part B, reported by 6 of 18 (33%) patients in the GSK2831781 group and none in the placebo group; each episode was mild or moderate with no temporal relationship to dosing, and no episode was considered related to study treatment by the investigators. There was no imbalance in incidence of infections between groups. There was one serious AE of worsening of osteoarthritis, occurring 4 days after randomization to GSK2831781 at a dose of 5 mg/kg; the patient had a medical history of knee osteoarthritis and subsequently elected to have surgery. This event was not considered related to the study drug. There were no withdrawals or deaths.
Table 2.
Summary of adverse events (MedDRA preferred term) occurring in ≥ 2 participants per cohort in healthy participants (part A) or patients with psoriasis (part B)
| Part A: Healthy participants (N = 40) a | Placebo (n = 14) | GSK2831781 | |||||
|---|---|---|---|---|---|---|---|
|
0.0075 mg/kg (ADA‐ve) (n = 6) |
0.04 mg/kg (ADA‐ve) (n = 6) |
0.15 mg/kg (ADA‐ve) (n = 6) |
0.15 mg/kg (ADA + ve) (n = 6) |
0.15 mg/kg (combined) (n = 12) |
Total (n = 24) | ||
| Any event, n (%) | 8 (57) | 5 (83) | 6 (100) | 3 (50) | 3 (50) | 6 (50) | 17 (65) |
| Headache, n (%) | 2 (14) | 1 (17) | 3 (50) | 0 | 1 (17) | 1 (8) | 5 (19) |
| Back pain, n (%) | 2 (14) | 1 (17) | 0 | 0 | 2 (33) | 2 (17) | 3 (12) |
| Nasopharyngitis, n (%) | 2 (14) | 0 | 2 (33) | 1 (17) | 0 | 1 (8) | 3 (12) |
| Abdominal pain, n (%) | 0 | 3 (50) | 0 | 0 | 0 | 0 | 3 (12) |
| Oropharyngeal pain, n (%) | 0 | 2 (33) | 0 | 0 | 1 (17) | 1 (8) | 3 (12) |
| Part B: Patients with psoriasis (N = 27) | Placebo (n = 9) | GSK2831781 | |||
|---|---|---|---|---|---|
| 0.5 mg/kg (n = 6) | 1.5 mg/kg (n = 6) | 5 mg/kg (n = 6) | Total (n = 18) | ||
| Any event, n (%) | 9 (100) | 4 (67) | 6 (100) | 6 (100) | 16 (89) |
| Headache, n (%) | 4 (44) | 1 (17) | 3 (50) | 2 (33) | 6 (33) |
| Nasopharyngitis, n (%) | 3 (33) | 2 (33) | 2 (33) | 2 (33) | 6 (33) |
| Back pain, n (%) | 0 | 2 (33) | 1 (17) | 3 (50) | 6 (33) |
| Oral herpes, b n (%) | 1 (11) | 2 (33) | 0 | 1 (17) | 3 (17) |
| Diarrhea, n (%) | 1 (11) | 2 (33) | 0 | 0 | 2 (11) |
| Oropharyngeal pain, n (%) | 0 | 0 | 2 (33) | 0 | 2 (11) |
| Post‐procedural inflammation, c n (%) | 0 | 0 | 0 | 2 (33) | 2 (11) |
ADA, anti‐drug antibodies; ADA‐ve, ADA negative; ADA + ve, ADA positive; AE, adverse event; MedDRA, Medical Dictionary for Regulatory Activities.
No AEs were reported for cohorts 1 (0.0003 mg/kg, n = 1) and 2 (0.0015 mg/kg, n = 1).
Three of 18 (17%) patients receiving GSK2831781 reported oral herpes infection, and 3 of 9 (33%) placebo‐treated patients reported AEs of oral herpes, herpes zoster, and ophthalmic herpes zoster.
Related to skin biopsy site.
There was no clinically significant pattern in vital signs, electrocardiograms, or laboratory parameters, nor in serum IL‐6, IL‐8, IFN‐γ, or granulocyte‐colony stimulating factor concentrations. Minor, transient increases in serum TNF‐α were observed at all doses of GSK2831781 from 6 hours post dose, returning toward baseline by 48 hours, with no clear dose−response relationship; all values fell within the normal range (0.0–32.5 pg/mL 36 ).
There was no biologically meaningful change in peripheral blood T‐cell counts (including CD4+ and CD8+ subsets) or B cells. Minor, transient reductions (mean ~ 50% of baseline) in NK cells were seen within 6 hours of dosing; counts were recovering within 72 hours and returned to baseline within 7–14 days.
Immunogenicity
Antidrug antibodies were observed in 8 of 44 (18%) of GSK2831781‐treated participants at any time after dosing; 7 of these were treatment‐induced, and 1 was treatment‐boosted (Table S2 ). These were generally of low titer, with most responses near the sensitivity of the assay (equivalent to a titer of 100). There was no evidence that pre‐existing ADA increased the risk of treatment‐boosted ADA and there was no effect of ADA on the incidence of AEs (Table 2 ) or the PK profile (Table S3 ) in this single‐dose study.
Pharmacokinetics and target engagement
PKs were nonlinear, with the nonlinear process(es) saturated at doses ≥ 0.5 mg/kg (Figure 3a ). Because of this nonlinearity, the terminal phase could not be determined and therefore terminal half‐life and associated parameters (including area under the concentration time‐curve from time zero to infinity (AUC0–∞)) could not be derived, and so AUC from time 0 to the time of the last quantifiable concentration (AUC0– t) is reported instead. Maximum plasma concentration (Cmax), but not AUC0− t, increased proportionally with dose (Table S3 ). Total sLAG‐3 levels (free and GSK2831781‐complexed) increased upon dosing with GSK2831781, plateauing at doses ≥ 1.5 mg/kg (indicating target saturation; Figure 3b ). The terminal decline in sLAG‐3 concentration occurred at the same rate as the terminal decline in plasma GSK2831781 concentration, again suggesting that target‐mediated drug disposition could explain the nonlinearity. Target engagement (membrane LAG‐3 binding to competitive antibody) in peripheral blood occurred in a dose‐dependent manner at doses as low as ≥0.04 mg/kg (competitive antibody data not shown).
Figure 3.
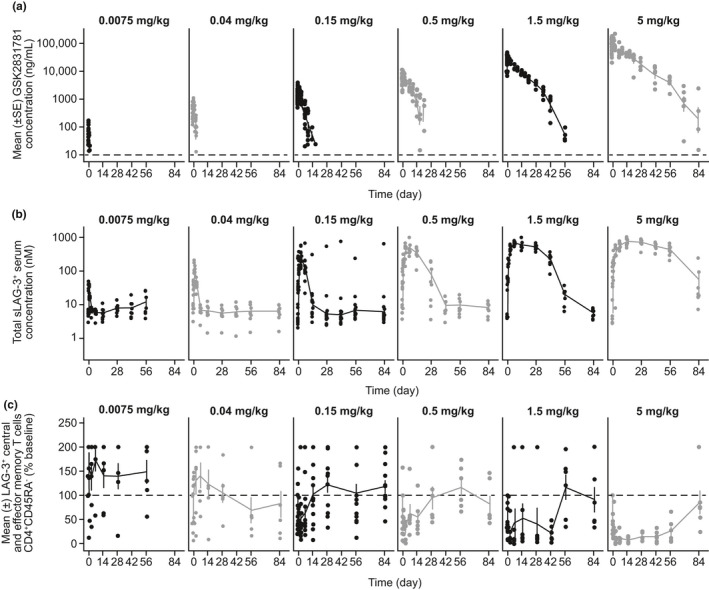
Pharmacokinetics and pharmacodynamics of GSK2831781 in blood. (a) Plasma concentration‐time profile of GSK2831781. (b) Individual serum concentration‐time profiles of total sLAG‐3. (c) Depletion of LAG‐3+ T cells over time in blood following dosing with GSK2831781*. Only data up to 84 days after dose are shown. *Due to high variability in the data, all CD4+CD45RA− data > 2 times the individual baseline is shown at 200%. Dashed line in a represents lower limit of quantification of assay (10 ng/mL), b represents the individual’s baseline shown at 200%. Dashed line in a represents lower limit of quantification of assay (10 ng/mL), in b it represents the individual’s baseline count normalized to 100%.
GSK2831781 depletes circulating LAG‐3+ T cells in a dose‐dependent manner
At baseline, mean numbers of circulating LAG‐3+ central and effector memory T cells were variable, ranging from 282 to 1,799 cells/mL of blood. Depletion of these T cells by GSK2831781 was dose‐dependent from 0.15 mg/kg (Figure 3c ), with near‐complete depletion at doses of 1.5 and 5 mg/kg. This was followed by repletion from days 43−57, coinciding with reduced GSK2831781 plasma concentrations (Figure 3c ).
DTH model
There was no difference in the change from baseline in skin induration between placebo and the 0.15 mg/kg dose of GSK2831781 following tuberculin purified protein derivative challenge/re‐challenge in healthy participants. At this dose, transient partial LAG‐3+ T‐cell depletion was observed in blood, with no LAG‐3+ T‐cell depletion in skin (Figure S2 ); therefore, skin pharmacology data were considered insufficient to evaluate the mechanism of action.
GSK2831781 depletes LAG‐3+ and CD3+ cells in psoriasis plaques
At day 1, psoriasis plaque biopsy LAG‐3+ cell counts were low (compared with CD3+ cell counts) in all groups. There were trends to a dose‐dependent reduction in LAG‐3+ cells in the dermis and epidermis of psoriatic lesions, with complete depletion in 3 of 3 (dermis) and 3 of 5 (epidermis) postdose samples at the 5 mg/kg dose of GSK2831781 (Figure 4 ).
Figure 4.
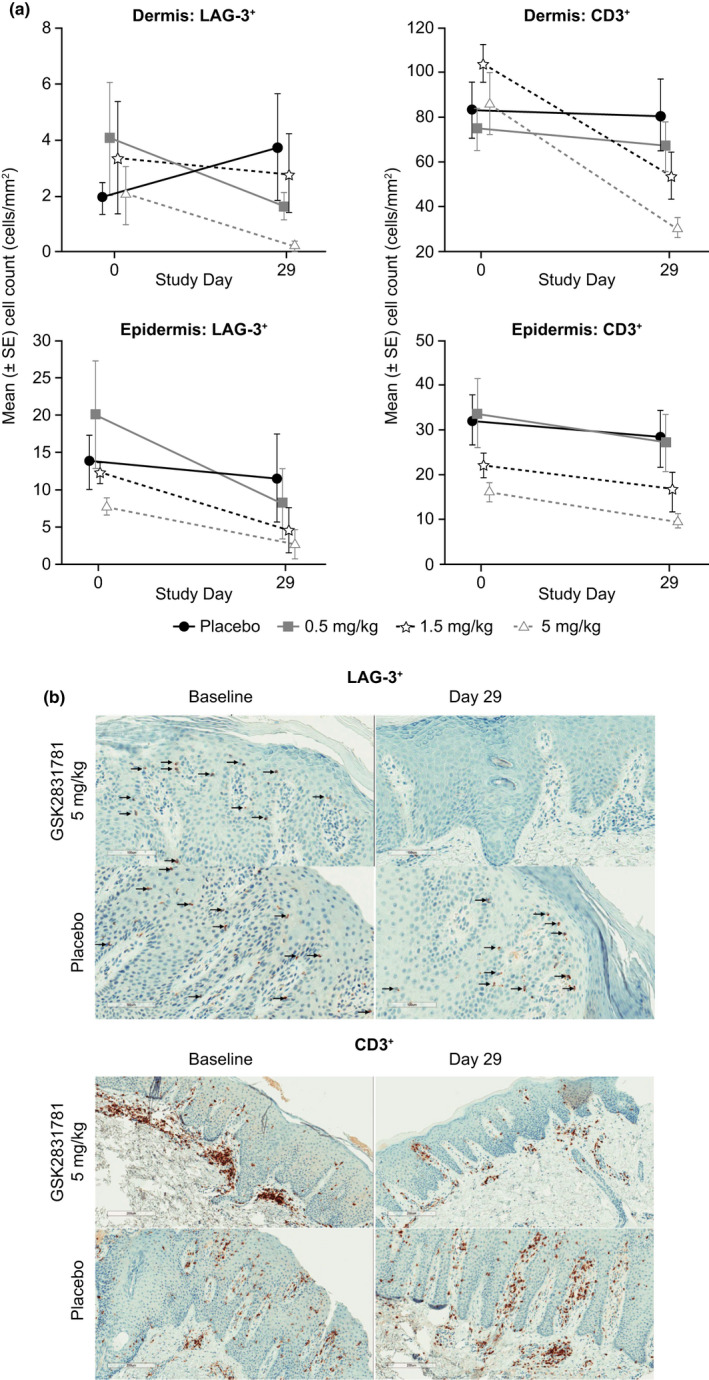
GSK2831781 depletes LAG‐3+ and CD3+ T cells in psoriatic skin lesions. (a) Observed mean (±SE) LAG‐3+ and (±SE) CD3+ cell counts in the dermis and epidermis following administration of GSK2831781 or placebo. (b) Representative IHC photomicrographs of LAG‐3+ (×20 magnification; arrows point to LAG‐3+ cells) and CD3+ (×10 magnification) stained psoriatic skin biopsies. IHC, immunohistochemistry.
A dose‐dependent reduction in CD3+ T cells was observed in the dermis that was significant at the 5 mg/kg dose (estimated mean difference from placebo in change from baseline of CD3+ cells −53.34 cells/mm2 (95% CI −91.83 to −14.85; Figure 4 ).
Pharmacologic markers: Ki67 and epidermal thickness
Keratinocyte proliferation, determined by Ki67 staining of the epidermis, was decreased at day 29 with GSK2831781 vs. placebo (Table S4 ). Epidermal thickness values were highly variable: there was a larger mean reduction with placebo than GSK2831781 (Table S3 ).
Depletion of LAG‐3+ cells is accompanied by changes in gene expression in skin
Analyzing genes associated with the T helper (Th)1/Th17 signaling axis implicated in psoriasis pathogenesis, we found treatment with GSK2831781 at 5 mg/kg was associated with > 1.5‐fold downregulation of proinflammatory genes (IL‐17A, IL‐17F, IFNγ, and S100A12), whereas IL‐12 and IL‐23, which direct Th‐cell differentiation, were unchanged (Figure S3, Table S5 ).
Analyzing genes associated with skin tight junctions, which are dysfunctional in psoriasis, we found upregulation (≥ 1.5‐fold change) of CDHR1 with GSK2831781 (Figure S3 , Table S5 ), which may signify improved skin barrier function. There were no changes in Treg‐associated IL‐10 and FOXP3 (Figure S3 ; Table S5 ).
Epigenetic quantification 37 of skin leukocytes by qRT‐PCR showed that, consistent with immunohistochemistry data (Figure 4 ), there was a dose‐dependent trend toward a reduction in CD3+ T cells and a weak trend toward a reduction in LAG‐3+ cells at 5 mg/kg (Figure 4b ). A weak trend toward reduction in CD4+ T‐cell numbers was also seen. Consistent with qRT‐PCR analysis, IL‐17A+ cells were reduced in the 5 mg/kg group. A weak trend toward reduction in FoxP3+ cells suggested that LAG‐3 may also mark a subpopulation of Tregs (Figure S3 ).
Clinical end points
PLSS and PASI scores showed an improvement to day 43 in all GSK2831781 treatment groups compared with placebo (Figure 5a ). At day 29, there was a reduction from baseline in PLSS and PASI scores in the 5 mg/kg group compared with placebo (estimated difference from placebo at day 29: PLSS − 2.0, 95% CI −3.57 to −0.44; PASI −3.3, 95% CI −6.28 to −0.35; Table S6 ). There was a return toward baseline in both measures after day 57, consistent with drug clearance and repletion of LAG‐3+ cells. The change in psoriasis in one illustrative patient who received a 1.5 mg/kg dose of GSK2831781 is shown (Figure 5b ).
Figure 5.
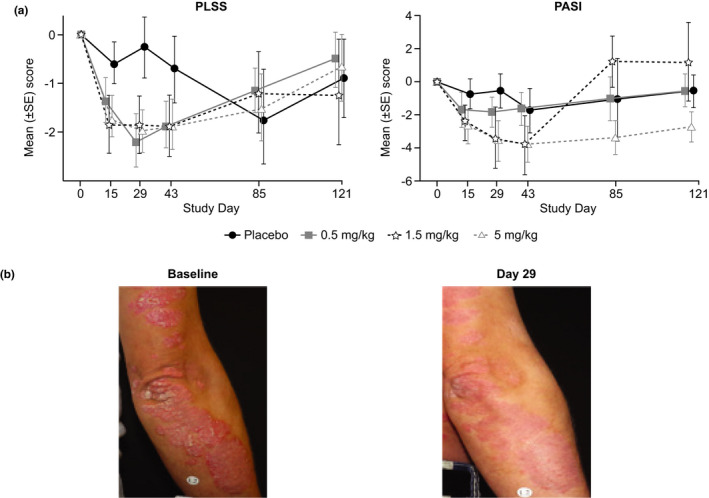
PLSS and PASI scores over time in patients with psoriasis. (a) Observed mean (±SE) change from baseline* in PLSS and PASI following administration of GSK2831781 or placebo to patients with psoriasis. (b) Illustrative photograph of one participant showing a response following a GSK2831781 1.5 mg/kg. *No assessment at day 85 for 0.5 mg/kg treatment group. PASI, Psoriasis Activity Severity Index; PLSS, Psoriasis Lesion Severity Score.
Four participants received disease‐modifying topical therapies after the protocol‐restricted period (defined as day 43). Two were placebo‐treated patients. One patient who received GSK2831781 0.5 mg/kg was treated with hydrocortisone from days 111 to 113; and one patient who received 5 mg/kg of GSK2831781 was treated with clobetasol for pityriasis from days 199 to 214. In both cases, this exposure was long after clearance of GSK2831781. No individual withdrew from the study due to worsening of psoriasis, or the need for prohibited rescue medication.
DISCUSSION
This clinical study is the first to report the pharmacology and clinical improvement in psoriasis disease activity following the targeted depletion of immune cells expressing LAG‐3 (a negative regulator) as a marker of recent cell activation using a novel mAb GSK2831781. The role of LAG‐3+ T cells in autoimmune disease is incompletely understood but relatively higher LAG‐3 expression in tissues (as compared with blood) is a marker of pathogenic autoreactive T cells that are chronically activated. 12 Given the interest in manipulating LAG‐3 in both oncology and autoinflammatory diseases, its function and impact is of considerable scientific interest. In order to deplete LAG‐3+ cells, we generated an ADCC‐enhanced afucosylated antibody using POTELLIGENT technology with high affinity for LAG‐3, GSK2831781.
No safety or tolerability concern was identified up to a maximum single 5 mg/kg i.v. dose of GSK2831781, and notably, no increase in infection was observed. In keeping with the low proportion of LAG‐3+ cells in blood, no consistent or sustained change in blood lymphocyte or T‐cell counts was observed, other than a minor early transient reduction in NK cells.
Dose‐dependent depletion of LAG‐3+ cells was observed in blood and psoriatic skin, indicating that GSK2831781 is capable of depleting LAG‐3+ cells both peripherally and in inflamed tissue. Depletion of LAG‐3+ cells in blood was near‐complete after single doses of 1.5–5 mg/kg, and lasted for several weeks, with repletion to baseline soon after drug clearance.
The PKs of GSK2831781 were nonlinear, with nonlinear process(es) saturated at doses ≥ 0.5 mg/kg. Nonlinear PKs are often observed with mAbs, which is related to target‐mediated drug disposition, in this case, likely through binding of GSK2831781 to its target, LAG‐3, on T cells. Because the number of LAG‐3+ T cells in blood is low (~ 1,000 cells/μL) compared with the concentration of GSK2831781, the extent of nonlinearity we observed was not anticipated and cannot be explained solely by binding to LAG‐3 on T cells (both in blood and in tissue). A tissue cross‐reactivity study using immunohistochemical techniques assessed specific and nonspecific binding of GSK2831781 with human tissues; no potential off‐target binding was identified, and binding in the human tonsil, which was used as positive control for the study, was confirmed. However, GSK2831781 also binds sLAG‐3, which is found at low concentrations in human serum. 23 , 24 , 25 , 26 , 27 GSK2831781/sLAG‐3 complexes may be cleared more slowly than free sLAG‐3, which would also explain the observation that total sLAG‐3 concentrations increase upon administration of GSK2831781 and decrease with declining GSK2831781 concentrations. Increases in sLAG‐3 are unlikely to be from shedding of LAG‐3 from T cells given the low number of LAG‐3+ T cells.
Although overall, LAG‐3+ cells constitute only a small proportion of T cells present in psoriasis lesions, their depletion led to an overproportionate reduction in tissue CD3+ cells, implying a broad impact on the disease process than might be anticipated based on the number of LAG‐3+ cells in tissue at any one time. Although expression of LAG‐3 is usually transient upon antigen‐dependent T‐cell activation, 24 its dynamic expression in inflamed tissues where there may be persistent antigen expression, is not known. Therefore, we cannot determine whether the reduction in dermal T cells was due to either direct depletion (via asynchronous transient expression of LAG‐3 on the broader population of T cells over time), or the downstream effects of depleting a small but auto‐reactive subset of LAG‐3+ T cells, or both.
The amelioration of clinical parameters and of the histological and transcriptomic biomarkers of inflammation (following treatment with GSK2831781) are in contrast with literature that has previously focused on the role of LAG‐3 expression on Tregs and IL‐10+ Tr1 cells. 16 , 18 , 38 , 39 , 40 Two key lines of evidence from the current clinical study suggest that the expression of LAG‐3 on Tregs or Tr1 cells in patients with active inflammation is limited and/or not a dominant mode of tolerance. First, there was no consistent reduction in IL‐10 or FOXP3 expression in skin biopsies post‐treatment and, second, psoriasis activity was improved rather than exacerbated. Together, these data suggest that LAG‐3 may preferentially mark pro‐inflammatory pathogenic T cells in this disease context. Epigenetic analysis revealed that FoxP3+ Tregs showed a minor trend toward reduction, although this was accompanied by a reduction in IL‐17A+ cells in the skin. Thus, GSK2831781 may deplete both Tregs and Th17 cells, modulating anti‐inflammatory and pro‐inflammatory arms, respectively, but ultimately ameliorate disease by abrogating the numerically dominant pathogenic cells. No immune‐related AEs were recorded in this study, although only a single dose of GSK2831781 was administered to a small number of participants. Checkpoint inhibitors, including LAG‐3, have been associated with the functional state of T‐cell exhaustion mainly in chronic viral infection, parasitic disease, and cancer. 41 , 42 , 43 , 44 , 45 The role of exhausted T cells in autoimmunity is unclear; however, we hypothesize that similar to chronic infection, persistent exposure to antigen under autoimmune conditions may induce auto‐reactive T cells to upregulate inhibitory receptors, such as LAG‐3. Thus, by utilizing LAG‐3 as a marker for depletion, our therapeutic strategy may selectively eliminate auto‐reactive pathogenic T cells and thereby block the central node of autoimmunity.
There were some limitations to the study. As this was a first‐time‐in‐human study, cohort sizes at each dose level were necessarily small and clinical evaluations envisaged as exploratory. The number of biopsies was chosen to minimize patient burden, and sampling of uninvolved (nonlesional) skin would have improved our understanding of LAG‐3+ cells, epidermal thickness, and the biomarkers in uninflamed tissue. In addition, there was difficulty with processing biopsy material, which reduced the number of paired samples for the calculation of change from baseline. Last, technical assay difficulties prevented co‐staining of biopsies for LAG‐3+/CD3+ cells, so direct evaluation of the proportion of recently activated (LAG‐3‐expressing) CD3+ cells at baseline and following treatment could not be performed.
In this first clinical experience with GSK2831781, single doses up to 5 mg/kg were well‐tolerated and caused a dose‐related depletion of LAG‐3+ T cells in blood and a reduction of LAG‐3+ and CD3+ cells in psoriatic skin. There were further positive downstream effects on pro‐inflammatory and epithelial integrity transcripts in psoriatic plaques, and GSK2831781 was associated with a reduction in disease activity in patients with mild‐to‐moderate psoriasis. These favorable mechanistic, pharmacological, and clinical outcomes in psoriasis may also be realized in other T‐cell‐mediated inflammatory diseases.
Funding
This study was funded by GlaxoSmithKline (GSK; NCT02195349; GSK study 200630). Medical writing support (in the form of writing assistance, including development of the initial draft based on author direction, assembling tables and figures, collating authors’ comments, grammatical editing, and referencing) was provided by Olga Conn, PhD, and Ann Kerrigan, PhD, of Fishawack Indicia Ltd., UK, and was funded by GSK.
Conflict of Interest
J.E., D.J.B.M., N.S., A.R., L.L., K.L., K.L.N., S.T., S.A.H., L.F., K.E., Y.C., R.A., M.F., T.M.W., S.J.B., N.W., and R.M.T. are employees of, and hold stocks/share options in, GSK. C.B., C.J.D., E.C., J.S., and C.O.S.S. were employees of GSK at the time of the study and hold stocks/share options in GSK. T.S.S. and T.G.H. were employees of GSK at the time of the study. R.F. and M.A. were employees of Parexel International at the time of the study; Parexel International was funded for the conduct of the study by GSK. There are patents and patent applications related to the development of GSK2831781 and its medical uses thereof. M.C. declared no competing interests for this work.
Author Contributions
All authors wrote the manuscript. J.E., C.B., T.G.H., A.R., L.L., K.L., K.L.N., S.A.H., S.J.B., T.S.S., J.S., C.O.S.S., N.W., and R.M.T designed the research. R.F., M.A., M.C., S.T., and R.A. performed the research. J.E., D.J.B.M., N.S., L.L., K.L.N., S.T., S.A.H., L.F., K.E., Y.C., and R.M.T. analyzed the data. C.J.D., E.C., M.F., T.M.W., and S.J.B. contributed new reagents/analytical tools (preclinical antibody development).
Data and Materials Availability
For the clinical portion of the paper, anonymized individual participant data and study documents can be requested for further research from www.clinicalstudydatarequest.com. All reasonable requests for materials used in this research will be fulfilled, providing that written agreement is executed between GSK and the requester (and his/her affiliated institution). Such inquiries or requests should be directed to the corresponding author.
Supporting information
Figure S1
Figure S2
Figure S3
Table S1
Table S2
Table S3
Table S4
Table S5
Table S6
Supinfo
Acknowledgments
GSK2831781 is derived from Immutep Ltd’s parent antibody, a depleting anti‐LAG‐3 antibody technology that was exclusively licensed to GlaxoSmithKline (GSK) in 2010. POTELLIGENT Technology (licensed by BioWa, Inc., Princeton, NJ, USA) was utilized to generate an afucosylated antibody. The authors thank the staff at the study sites and all study participants. Medical writing support (in the form of writing assistance, including development of the initial draft based on author direction, assembling tables and figures, collating authors’ comments, grammatical editing, and referencing) was provided by Olga Conn, PhD, and Ann Kerrigan, PhD, of Fishawack Indicia Ltd., UK, and was funded by GSK. The authors thank all past and present project team members for their contributions, including Tejash Shah for formulation development work; Alan Lewis for designing the humanized antibody variants; Mark Creighton‐Gutteridge for generation of EL4 cell lines expressing LAG‐3; Gladstone Thompson for preparation of reagents used in clinical study; Daniel Gibson for undertaking early production of large scale material post‐candidate selection; Dan Sikkema for supporting biomarker assay development; Cherilyn Caucci for antidrug antibody assay validation; Lisa Saville for anti‐drug antibody testing; Boyenoh Gaye and Mary Birchler for sLAG‐3 data quality check; Chris Parkinson for input on clinical trial regulations and quality management; Jonathan Ayer, Virginia Norris, Dagmar Wilsmann‐Theis, Annelize Koch, Angela Sinn, Pablo Forte Soto, Chiara Zecchin, Sally Egginton, and Kirsty Hicks for design/conduct of the clinical study and/or clinical data acquisition; and Frances Warren and Sally Rumley for operational oversight of the clinical study.
References
- 1. Baker, K.F. & Isaacs, J.D. Novel therapies for immune‐mediated inflammatory diseases: what can we learn from their use in rheumatoid arthritis, spondyloarthritis, systemic lupus erythematosus, psoriasis, Crohn's disease and ulcerative colitis? Ann. Rheum. Dis. 77, 175–187 (2018). [DOI] [PubMed] [Google Scholar]
- 2. Duijvestein, M. et al. Novel therapies and treatment strategies for patients with inflammatory bowel disease. Curr. Treat. Options Gastroenterol. 16, 129–146 (2018). [DOI] [PubMed] [Google Scholar]
- 3. McKay, C. , Kondratuk, K.E. , Miller, J.P. , Stumpf, B. & Boh, E. Biologic therapy in psoriasis: navigating the options. Cutis 102, 13–17 (2018). [PubMed] [Google Scholar]
- 4. Alnaimat, F. , Sidhu, P. & Sarkar, S. T‐cell targeted therapies in autoimmune diseases. Drug Dev. Res. 72, 585–597 (2011). [Google Scholar]
- 5. Lowes, M.A. , Suárez‐Fariñas, M. & Krueger, J.G. Immunology of psoriasis. Annu. Rev. Immunol. 32, 227–255 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gallais Sérézal, I. et al. A skewed pool of resident T cells triggers psoriasis‐associated tissue responses in never‐lesional skin from patients with psoriasis. J. Allerg. Clin. Immunol. 143, 1444–1454 (2019). [DOI] [PubMed] [Google Scholar]
- 7. Salvana, E.M. & Salata, R.A. Infectious complications associated with monoclonal antibodies and related small molecules. Clin. Microbiol. Rev. 22, 274–290 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Triebel, F. et al. LAG‐3, a novel lymphocyte activation gene closely related to CD4. J. Exp. Med. 171, 1393–1405 (1990). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Workman, C.J. & Vignali, D.A. The CD4‐related molecule, LAG‐3 (CD223), regulates the expansion of activated T cells. Eur. J. Immunol. 33, 970–979 (2003). [DOI] [PubMed] [Google Scholar]
- 10. Workman, C.J. & Vignali, D.A. Negative regulation of T cell homeostasis by lymphocyte activation gene‐3 (CD223). J. Immunol. 174, 688–695 (2005). [DOI] [PubMed] [Google Scholar]
- 11. Okazaki, T. et al. PD‐1 and LAG‐3 inhibitory co‐receptors act synergistically to prevent autoimmunity in mice. J. Exp. Med. 208, 395–407 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Slevin, S.M. et al. Lymphocyte activation gene (LAG)‐3 is associated with mucosal inflammation and disease activity in ulcerative colitis. J. Crohns Colitis 14, 1446–1461 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Baixeras, E. et al. Characterization of the lymphocyte activation gene 3‐encoded protein. A new ligand for human leukocyte antigen class II antigens. J. Exp. Med. 176, 327–337 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Huard, B. , Gaulard, P. , Faure, F. , Hercend, T. & Triebel, F. Cellular expression and tissue distribution of the human LAG‐3‐encoded protein, an MHC class II ligand. Immunogenetics 39, 213–217 (1994). [DOI] [PubMed] [Google Scholar]
- 15. Huang, C.T. et al. Role of LAG‐3 in regulatory T cells. Immunity 21, 503–513 (2004). [DOI] [PubMed] [Google Scholar]
- 16. Brockmann, L. et al. Molecular and functional heterogeneity of IL‐10‐producing CD4(+) T cells. Nat. Commun. 9, 5457 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Camisaschi, C. et al. LAG‐3 expression defines a subset of CD4(+)CD25(high)Foxp3(+) regulatory T cells that are expanded at tumor sites. J. Immunol. 184, 6545–6551 (2010). [DOI] [PubMed] [Google Scholar]
- 18. Gagliani, N. et al. Coexpression of CD49b and LAG‐3 identifies human and mouse T regulatory type 1 cells. Nat. Med. 19, 739–746 (2013). [DOI] [PubMed] [Google Scholar]
- 19. Huang, W. , Solouki, S. , Carter, C. , Zheng, S.G. & August, A. Beyond type 1 regulatory T cells: co‐expression of LAG3 and CD49b in IL‐10‐producing T cell lineages. Front. Immunol. 9, 2625 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Okamura, T. et al. CD4+CD25‐LAG3+ regulatory T cells controlled by the transcription factor Egr‐2. Proc. Natl. Acad. Sci. USA 106, 13974–13979 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Camisaschi, C. et al. Alternative activation of human plasmacytoid DCs in vitro and in melanoma lesions: involvement of LAG‐3. J. Invest. Dermatol. 134, 1893–1902 (2014). [DOI] [PubMed] [Google Scholar]
- 22. Workman, C.J. et al. LAG‐3 regulates plasmacytoid dendritic cell homeostasis. J. Immunol. 182, 1885–1891 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Triebel, F. LAG‐3: a regulator of T‐cell and DC responses and its use in therapeutic vaccination. Trends Immunol. 24, 619–622 (2003). [DOI] [PubMed] [Google Scholar]
- 24. Annunziato, F. et al. Expression and release of LAG‐3‐encoded protein by human CD4+ T cells are associated with IFN‐gamma production. FASEB J. 10, 769–776 (1996). [DOI] [PubMed] [Google Scholar]
- 25. Lienhardt, C. et al. Active tuberculosis in Africa is associated with reduced Th1 and increased Th2 activity in vivo. Eur. J. Immunol. 32, 1605–1613 (2002). [DOI] [PubMed] [Google Scholar]
- 26. Scala, E. et al. Lymphocyte activation gene‐3 (LAG‐3) expression and IFN‐gamma production are variably coregulated in different human T lymphocyte subpopulations. J. Immunol. 161, 489–493 (1998). [PubMed] [Google Scholar]
- 27. Li, N. , Workman, C.J. , Martin, S.M. & Vignali, D.A. Biochemical analysis of the regulatory T cell protein lymphocyte activation gene‐3 (LAG‐3; CD223). J. Immunol. 173, 6806–6812 (2004). [DOI] [PubMed] [Google Scholar]
- 28. Wierz, M. et al. Dual PD1/LAG3 immune checkpoint blockade limits tumor development in a murine model of chronic lymphocytic leukemia. Blood 131, 1617–1621 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Haudebourg, T. , Dugast, A.S. , Coulon, F. , Usal, C. , Triebel, F. & Vanhove, B. Depletion of LAG‐3 positive cells in cardiac allograft reveals their role in rejection and tolerance. Transplantation 84, 1500–1506 (2007). [DOI] [PubMed] [Google Scholar]
- 30. Poirier, N. et al. Antibody‐mediated depletion of lymphocyte‐activation gene‐3 (LAG‐3(+) )‐activated T lymphocytes prevents delayed‐type hypersensitivity in non‐human primates. Clin. Exp. Immunol. 164, 265–274 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Workman, C.J. , Rice, D.S. , Dugger, K.J. , Kurschner, C. & Vignali, D.A. Phenotypic analysis of the murine CD4‐related glycoprotein, CD223 (LAG‐3). Eur. J. Immunol. 32, 2255–2263 (2002). [DOI] [PubMed] [Google Scholar]
- 32. Boy, M.G. et al. Double‐blind, placebo‐controlled, dose‐escalation study to evaluate the pharmacologic effect of CP‐690,550 in patients with psoriasis. J. Invest. Dermatol. 129, 2299–2302 (2009). [DOI] [PubMed] [Google Scholar]
- 33. Sierro, S. , Romero, P. & Speiser, D.E. The CD4‐like molecule LAG‐3, biology and therapeutic applications. Expert Opin. Ther. Targets 15, 91–101 (2011). [DOI] [PubMed] [Google Scholar]
- 34. Belson, A. et al. Characterisation of the clinical and activated T cell response to repeat delayed‐type hypersensitivity skin challenges in human subjects, with KLH and PPD, as a potential model to test T cell‐targeted therapies. Inflamm. Res. 65, 389–404 (2016). [DOI] [PubMed] [Google Scholar]
- 35. Fredriksson, T. & Pettersson, U. Severe psoriasis–oral therapy with a new retinoid. Dermatologica 157, 238–244 (1978). [DOI] [PubMed] [Google Scholar]
- 36. Arican, O. , Aral, M. , Sasmaz, S. & Ciragil, P. Serum levels of TNF‐alpha, IFN‐gamma, IL‐6, IL‐8, IL‐12, IL‐17, and IL‐18 in patients with active psoriasis and correlation with disease severity. Mediators Inflamm. 2005, 273–279 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Baron, U. et al. Epigenetic immune cell counting in human blood samples for immunodiagnostics. Sci. Transl. Med. 10, eaan3508 (2018). [DOI] [PubMed] [Google Scholar]
- 38. Brockmann, L. et al. IL‐10 receptor signaling is essential for TR1 cell function in vivo. J. Immunol. 198, 1130–1141 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kim, J. , Lee, J. , Gonzalez, J. , Fuentes‐Duculan, J. , Garcet, S. & Krueger, J.G. Proportion of CD4(+)CD49b(+)LAG‐3(+) type 1 regulatory T cells in the blood of psoriasis patients inversely correlates with Psoriasis Area and Severity Index. J. Invest. Dermatol. 138, 2669–2672 (2018). [DOI] [PubMed] [Google Scholar]
- 40. Bauché, D. et al. LAG3(+) regulatory T cells restrain interleukin‐23‐producing CX3CR1(+) gut‐resident macrophages during group 3 innate lymphoid cell‐driven colitis. Immunity 49, 342–352.e5 (2018). [DOI] [PubMed] [Google Scholar]
- 41. McLane, L.M. , Abdel‐Hakeem, M.S. & Wherry, E.J. CD8 T cell exhaustion during chronic viral infection and cancer. Annu. Rev. Immunol. 37, 457–495 (2019). [DOI] [PubMed] [Google Scholar]
- 42. Andrews, L.P. , Marciscano, A.E. , Drake, C.G. & Vignali, D.A. LAG3 (CD223) as a cancer immunotherapy target. Immunol. Rev. 276, 80–96 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Richter, K. , Agnellini, P. & Oxenius, A. On the role of the inhibitory receptor LAG‐3 in acute and chronic LCMV infection. Int. Immunol. 22, 13–23 (2010). [DOI] [PubMed] [Google Scholar]
- 44. Butler, N.S. et al. Therapeutic blockade of PD‐L1 and LAG‐3 rapidly clears established blood‐stage Plasmodium infection. Nat. Immunol. 13, 188–195 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Long, L. et al. The promising immune checkpoint LAG‐3: from tumor microenvironment to cancer immunotherapy. Genes Cancer 9, 176–189 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1
Figure S2
Figure S3
Table S1
Table S2
Table S3
Table S4
Table S5
Table S6
Supinfo