

Abstract
Introduction
Phosphorylated tau (p‐tau) in cerebrospinal fluid (CSF) is an established Alzheimer's disease (AD) biomarker. Novel immunoassays targeting N‐terminal and mid‐region p‐tau181 and p‐tau217 fragments are available, but head‐to‐head comparison in clinical settings is lacking.
Methods
N‐terminal‐directed p‐tau217 (N‐p‐tau217), N‐terminal‐directed p‐tau181 (N‐p‐tau181), and standard mid‐region p‐tau181 (Mid‐p‐tau181) biomarkers in CSF were evaluated in three cohorts (n = 503) to assess diagnostic performance, concordance, and associations with amyloid beta (Aβ).
Results
CSF N‐p‐tau217 and N‐p‐tau181 had better concordance (88.2%) than either with Mid‐p‐tau181 (79.7%–82.7%). N‐p‐tau217 and N‐p‐tau181 were significantly increased in early mild cognitive impairment (MCI)‐AD (A+T–N–) without changes in Mid‐p‐tau181 until AD‐dementia. N‐p‐tau217 and N‐p‐tau181 identified Aβ pathophysiology (area under the curve [AUC] = 94.8%–97.1%) and distinguished MCI‐AD from non‐AD MCI (AUC = 82.6%–90.5%) signficantly better than Mid‐p‐tau181 (AUC = 91.2% and 70.6%, respectively). P‐tau biomarkers equally differentiated AD from non‐AD dementia (AUC = 99.1%–99.8%).
Discussion
N‐p‐tau217 and N‐p‐tau181 could improve diagnostic accuracy in prodromal‐AD and clinical trial recruitment as both identify Aβ pathophysiology and differentiate early MCI‐AD better than Mid‐p‐tau181.
Keywords: Alzheimer's disease, biomarker, cerebrospinal fluid, dementia, memory clinic, phosphorylated tau‐181, phosphorylated tau‐217, prodromal Alzheimer's
Abbreviations
- Aa
amino acids
- AD
Alzheimer's disease
- AUC
Area under the curve
- Aβ
amyloid beta
- CU
cognitively unimpaired
- DLB
Lewy body dementia
- ELISA
enzyme linked immunosorbent assays
- FTD
frontotemporal dementia
- IP‐MS
immunoprecipitation‐mass spectrometry
- MCI
mild cognitive impairment
- MSD
Meso Scale Discovery
- PET
positron emission tomography
- p‐tau181
tau phosphorylation at threonine‐181
- p‐tau217
tau phosphorylation at threonine‐217
- VaD
vascular dementia
1. INTRODUCTION
Alzheimer's disease (AD) has two pathological hallmarks: amyloid beta (Aβ) plaques and tau neurofibrillary tangles. 1 , 2 In living individuals, these pathologies are detectable by positron emission tomography (PET) imaging or cerebrospinal fluid (CSF) analysis using immunoassays that measure Aβ42 (or Aβ42/Aβ40 ratio), total‐tau, and phosphorylated tau‐181 (p‐tau181). 3 These biomarkers are included in diagnostic criteria as biological evidence of AD. 4 However, an abnormal decrease of CSF Aβ42 is also present in approximately 30% of cognitively unimpaired (CU) elderly 5 and up to 50% of dementia with Lewy bodies (DLB) patients. 6 Furthermore, CSF total‐tau is a marker of neuronal injury that is also increased in other neurological disorders. 7 , 8 , 9 Conversely, CSF p‐tau181 is highly specific for AD pathology, 2 , 3 , 10 and is unchanged in pure tauopathies including tau‐related frontotemporal lobar degeneration. 11 Similar results have been reported for these biomarkers in blood. 2 , 12 , 13 , 14 , 15 , 16 , 17 , 18
Aβ pathology is the earliest detectable change in AD, 4 while established p‐tau181 becomes abnormal in late mild cognitive impairment (MCI) and dementia stages. 19 , 20 Nonetheless, animal‐model studies suggest that other p‐tau forms may increase earlier in the disease process concomitant with emerging Aβ pathology. 21 , 22 In human CSF, p‐tau199, p‐tau212/p‐tau214, p‐tau231, and p‐tau231/p‐tau235 had similar diagnostic performances as p‐tau181. 23 , 24 P‐tau217 quantified by immunoprecipitation‐mass spectrometry (IP‐MS) was highly increased in AD CSF compared to minimal levels in controls. 25 , 26 , 27 P‐tau217 correlated better with Aβ pathology than p‐tau181 and improved separation between AD and non‐AD disorders. 26 , 27 In preclinical familial AD, CSF p‐tau217 began to increase almost concurrent with initial Aβ changes. 28 Another study showed that p‐tau217 correlated better with tau‐PET and Aβ‐PET than p‐tau181. 29 Together, p‐tau217 may be a marker of incipient Aβ pathophysiology in AD. However, further study is needed, including head‐to‐head comparison against novel and established p‐tau181 biomarkers. Notably, a novel p‐tau181 biomarker 12 , 13 , 14 , 15 had equal performance as CSF p‐tau217 in identifying Aβ abnormalities in preclinical AD when only subtle Aβ pathological changes were detectable, 30 but comparison of these biomarkers in clinical AD is lacking.
In this study, we performed a head‐to‐head comparison of different p‐tau biomarker performances in MCI‐AD and AD dementia. We evaluated, in three prospective cohorts, the: (1) patterns of changes in p‐tau biomarkers in MCI‐AD and AD dementia, (2) accuracies in differentiating early MCI‐AD from non‐AD MCI, and (3) associations of p‐tau biomarkers with Aβ pathophysiology.
2. METHODS
2.1. Description of p‐tau biomarkers studied
Four p‐tau biomarkers were studied (Figure 1A): (1) a novel p‐tau217 (N‐p‐tau217) measuring tau phosphorylated at T217 and containing the N‐terminal amino acids (aa) 6‐18 epitope, and (2) a novel p‐tau181 biomarker (N‐p‐tau181) targeting tau phosphorylated at T181 and bearing the 6‐18aa epitope. These assays used identical reagents (except capture antibodies) to enable direct comparison. Established p‐tau181 assays were used as a reference and included (3) the INNOTEST enzyme‐linked immunosorbent assay (ELISA) and (4) the fully automated Lumipulse method (both by Fujirebio, Ghent, Belgium) that target mid‐region epitopes (Mid‐p‐tau181).
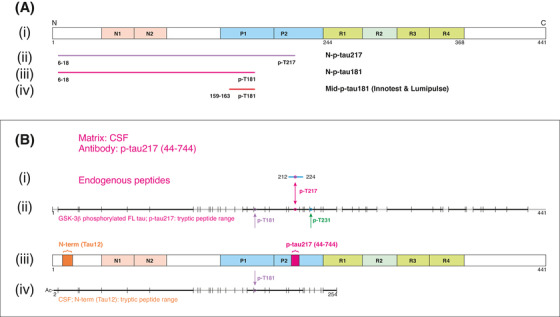
FIGURE 1.
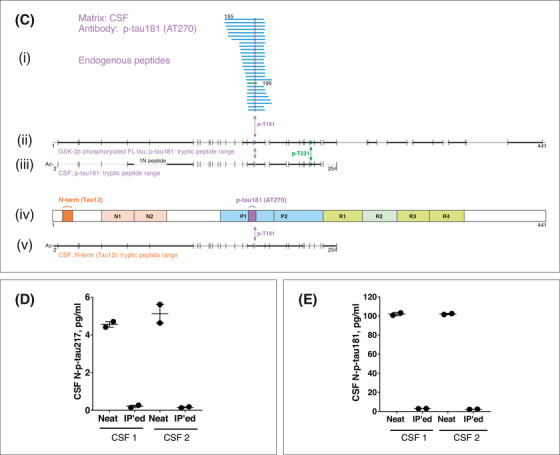
Description and validation of the p‐tau assays studied. A, (i) Schematic illustration of full‐length tau‐441 with the different regions marked. (ii), Antibodies used in the N‐p‐tau217 assay. The capture antibody specifically recognizes tau phosphorylated at threonine‐217 while the detection antibody binds the N‐terminal 6‐QEFEVMEDHAGT‐18 epitope. (iii) For the N‐p‐tau181 assay, the capture antibody specifically recognizes phosphorylation at threonine‐181 and the detection antibody targets the N‐terminal 6‐QEFEVMEDHAGT‐18 epitope. (iv) The Mid‐p‐tau181 assay (commercially available from INNOTEST) uses a capture antibody that is specific to tau phosphorylated at threonine‐181 and a detection antibody directed at the 159‐PPGQK‐163 epitope. The Lumipulse Mid‐p‐tau181 assay uses a similar antibody combination as the INNOTEST assay but the identity and exact epitopes of these antibodies are not published. 40 B, A schematic illustration of full‐length tau‐441 with the different regions and epitopes of the N‐p‐tau217 antibody pair marked (iii). (i) In human CSF immunoprecipitated with the p‐tau217 antibody, an endogenous peptide (amino acid 212‐224) was identified (blue line); this peptide was phosphorylated at threonine‐217 as indicated by the purple circle. In (ii), the range of tryptic peptides identified from glycogen synthase kinase (GSK)‐3β‐phosphorylated full‐length tau‐441 (the assay calibrator; SignalChem #TO8‐50FN), immunoprecipitated using the p‐tau217 antibody. Cleavage positions for trypsin are indicated with vertical lines and the identified peptides are indicated in black while sequence portions not detected are shown in gray. (iv) The range of tryptic peptides identified from human cerebrospinal fluid (CSF) immunoprecipitated using the detection antibody, Tau12. The detected peptide sequence was from the acetylated N‐terminus up to amino acid 254, including peptides phosphorylated at threonine‐217. C, The illustration shows a schematic of full‐length tau‐441 with the antibodies used in the N‐p‐tau181 assay shown in (iv). In human CSF immunoprecipitated with AT270, only endogenous peptides phosphorylated at threonine‐181 were identified (i). These peptides were in the amino acid range 155 to 196 (blue lines with phosphorylation site indicated by purple circles). (ii) The range of tryptic peptides identified from GSK‐3β‐phosphorylated full‐length tau‐441 (the assay calibrator; SignalChem # TO8‐50FN) immunoprecipitated using the AT270 antibody. (iii) The range of tryptic peptides identified from human CSF, both immunoprecipitated using the AT270 antibody. Cleavage positions for trypsin are indicated with vertical lines, the identified peptides are indicated in black while sequence portions not detected are gray. In (v) is shown the range of tryptic peptides identified from human CSF immunoprecipitated using Tau12. Here, the detected range was from the acetylated N‐terminus up to amino acid 254, including peptides phosphorylated at threonine‐181. D, Aliquots from two different CSF samples were analyzed untreated (neat) or immunodepleted with the capture and detection antibodies (IP'ed) used in the N‐p‐tau217 assay. More than 95% of the measurable N‐p‐tau217 levels were lost after immunodepletion. E, Immunodepletion of two different CSF samples with the N‐p‐tau181 assay antibodies led to the removal of more than 94% of the available N‐p‐tau181 signal in each sample
2.2. Study participants
The discovery cohort included biomarker‐positive AD patients and biomarker‐negative controls from the Sahlgrenska University Hospital, Mölndal, Sweden (Table 1).
TABLE 1.
Demographic characteristics and CSF p‐tau concentrations for the discovery and validation cohorts
| Discovery | Validation | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Discovery cohort | Ljubljana cohort | Paris cohort | |||||||||
| Patient group | Aβ– CU n = 17 | AD dementia n = 16 | Aβ– CU n = 25 | Non‐AD MCI n = 72 | MCI‐AD n = 55 | AD dementia n = 114 | Aβ– CU n = 25 | Non‐AD MCI n = 41 | MCI‐AD n = 15 | AD dementia n = 94 | Non‐AD dementia n = 24 |
| Age, years, mean (SD) | 66.7 (9.0) | 80.2 (6.7)* | 67.8 (5.7) | 74.4 (6.9)* | 74.2 (8.4)* | 73.4 (7.8)* | 64.4 (8.7) | 66.7 (10.6) # | 74.1 (6.7)* | 71.8 (8.6)* | 66.4 (7.7) # |
| Sex, F, n (%) | 6 (35) | 5 (31) | 14 (56) | 33 (46) | 33 (60) | 66 (58) | 17 (68) | 24 (58.5) | 10 (66.7) | 56 (59.6) | 9(37.5) |
| MMSE, mean (SD) | – | – | 29 (2) | 25 (3) # | 22 (6)* | 20 (6)* | 27 (5) | 25 (3) # | 22 (6) # | 20 (6)* | 24 (4) # |
| Aβ42, pg/mL, mean (SD) | 712 (119) | 375 (85)* | 877 (277) | 1014 (1120) # | 450 (152)* | 461 (160)* | 1098 (331) | 1089 (420) # | 438.9 (101)* | 513.4 (166)* | 1020 (356) # |
| Aβ42/40, mean (SD) | – | – | 0.090 (0.08) | 0.087 (0.09) # | 0.047 (0.07) *,# | 0.038 (0.07)* | 0.094 (0.006) | 0.090 (0.011) # | 0.046 (0.007)* | 0.041 (0.008)* | 0.091 (0.011) # |
| Total tau, pg/mL, mean (SD) | 214 (68) | 656 (85)* | 225 (63) | 267 (73) | 329 (48) | 927 (430) | 242 (68) | 304.0 (149) # | 339 (64)* | 738.5 (3746)* | 341 (332) # |
| Mid‐p‐tau181, pg/mL, mean (SD) | 33 (10) | 72 (8)* | 39 (11) | 43 (9) # | 50 (7) # | 111 (35)* | 32 (8) | 37.3 (15) # | 52.7 (6.4)* | 117 (54)* | 33 (11) # |
| N‐p‐tau181, pg/mL, mean (SD) | 279.0 (69.9) | 1314.0 (441.1)* | 257.7 (78.5) | 305.3 (99.5) # | 468.0 (148.4) # | 1401 (714.8)* | 210.7 (60.4) | 241.1 (100.7) # | 241.1 (100.7)* | 1254.0 (697.0)* | 230.1 (102.1) # |
| N‐p‐tau217, pg/mL, mean (SD) | 2.4 (1.6) | 22.2 (6.8)* | 3.5 (2.5) | 4.3 (2.7) # | 12.5 (6.3) # | 40.8 (19.5)* | 2.9 (1.5) | 3.5 (2.0) # | 10.9 (3.8)* | 28.0 (14.8)* | 3.2 (2.3) # |
Abbreviations: AD, Alzheimer's disease; Aβ, amyloid beta; CSF, cerebrospinal fluid; CU, cognitively unimpaired; MCI, mild cognitive impairment; MMSE, Mini‐Mental State Examination; p‐tau181, tau phosphorylated at threonine‐181; SD, standard deviation.
Notes: Differences between the groups were tested using analysis of variance followed by Tukey's post hoc test (continuous variables) or contingency chi‐square test (sex). Significant differences compared to CU (*) or AD (#) in the same cohort.
The validation cohorts were clinic‐based prospective memory center cohorts, first from the University Medical Centre, Ljubljana, Slovenia, including AD dementia, MCI‐AD, non‐AD MCI, and Aβ– CU. The second validation cohort, from the Lariboisière Fernand‐Widal University Hospital (Paris, France), additionally included Aβ– non‐AD dementia (frontotemporal dementia [FTD], DLB, and vascular dementia [VaD]). Medial temporal lobe atrophy analysis was available for Paris Cohort (see supporting information). Participants underwent comprehensive clinical examination including neurological, neuropsychological, CSF, and magnetic resonance imaging assessments. CSF biomarker results and respective diagnostic criteria 31 , 32 were used to establish reliable diagnosis of AD in both validation cohorts, and of non‐AD disorders in the Paris cohort. 33 , 34 , 35 MCI‐AD participants had decreased Aβ42/Aβ40 and normal Mid‐p‐tau181 and total‐tau. Aβ– CU, non‐AD MCI, and non‐AD dementia participants had normal CSF biomarker profile. Non‐AD MCI included psychiatric and non‐neurodegenerative disorders.
Throughout the article, MCI‐AD refers to early MCI‐AD (A+T–N–) and non‐AD MCI to MCI without AD‐type pathology (A–T–N–).
2.3. Measurement of CSF core AD biomarkers
In the discovery cohort, Aβ42, Mid‐p‐tau181, and total‐tau were measured using the INNOTEST® β‐AMYLOID(1‐42), PHOSPHO‐TAU(181P), and hTAU Ag ELISAs. In both validation cohorts, Aβ42 and Aβ40 were measured with LUMIPULSE® G1200 (Fujirebio), and Mid‐p‐tau181 and total‐tau with INNOTEST and LUMIPULSE G1200 assays, respectively, for the Ljubljana and Paris cohorts. Measurements were performed by board‐certified scientists following manufacturers’ instructions.
2.4. Development and validation of N‐p‐tau217 and N‐ptau181 biomarkers
A rabbit polyclonal antibody specific for p‐tau217 (#44‐744, Invitrogen) was used as capture, conjugated to paramagnetic beads (#103207, Quanterix). The mouse monoclonal antibody Tau12 (#806502, BioLegend) raised against the N‐terminal epitope 6‐18aa was used for detection. 36 Antibody specificity was independently validated. 37 The assay calibrator was phosphorylated recombinant full‐length tau‐441 (#TO8‐50FN, SignalChem). Calibrators and specimens were diluted with assay diluent (Tau 2.0 buffer; #101556, Quanterix). Analytical validation and assay measurement protocol are described in supporting information.
The N‐p‐tau181 assay, validated for blood, 12 was further validated for CSF (supporting information).
RESEARCH IN CONTEXT
Systematic review: We reviewed PubMed and related sources for p‐tau studies, using the terms “Alzheimer,” “phospho‐tau,” “p‐tau,” and “cerebrospinal fluid.” Established p‐tau (Mid‐p‐tau181) biomarkers were increased in late mild cognitive impairment (MCI) to Alzheimer's disease (AD) dementia. A single study showed that N‐p‐tau217 and N‐p‐tau181 were similarly increased in preclinical AD when Mid‐p‐tau181 remained normal. No head‐to‐head comparison of new versus established p‐tau biomarkers in symptomatic AD was found.
Interpretation: N‐p‐tau217 and N‐p‐tau181 distinguished MCI‐AD (A+T–N–) from non‐AD‐MCI (A–T–N–) and amyloid beta (Aβ)+ from Aβ– cases more accurately than Mid‐p‐tau181. In support, concordance analyses showed that N‐p‐tau217 and N‐p‐tau181 became abnormal in MCI‐AD when Mid‐p‐tau181 was still unchanged.
Future direction: N‐p‐tau217 identified Aβ abnormalities in prodromal AD better than Mid‐p‐tau181 but not N‐p‐tau181. These novel biomarkers (N‐p‐tau217 and N‐p‐tau181) can complement Mid‐p‐tau181 in AD diagnosis and prognosis. Future studies will validate these findings and study the longitudinal trajectories of the new p‐tau biomarkers relative to Aβ changes.
2.5. N‐p‐tau217 and N‐p‐tau181 measurements in clinical cohorts
N‐p‐tau217 and N‐p‐tau181 were measured blinded on Simoa HD‐X using the above‐described in‐house assays at the Department of Psychiatry and Neurochemistry, University of Gothenburg, Mölndal, Sweden. CSF collection and processing followed standard procedures. 38 Randomized samples were thawed and vortexed before diluting up to 16‐fold (N‐p‐tau181) and 4‐fold (N‐p‐tau217) with assay diluent. Signal variations within and between analytical runs were assessed using two internal quality control samples analyzed in duplicate at the beginning and the end of each run. The within‐ and between‐run variations for N‐p‐tau217 were 1.7% to 8.6% and 9.5% to 18.5% respectively. For N‐p‐tau181, the within‐ and between‐run variations were 3.9% to 13.4% and 5.4% to 18.5%.
2.6. Ethical clearance
The discovery, first, and second validation studies were approved by the ethics committees at the University of Gothenburg (#EPN140811); the Ministry of Health, Republic of Slovenia (0120‐442/2017/3); and the Bichat Hospital at the Paris University, respectively.
2.7. Statistical analyses
Statistical analyses were performed with SPSS v26 (IBM, Armonk, New York, USA), Prism v7.03 (GraphPad, San Diego, California, USA), and MedCalc (Ostend, Belgium). Non‐parametric tests were used for non‐normally distributed data. Spearman correlation and the χ2 test were used for continuous and categorical variables, respectively. Group differences were examined using the Mann‐Whitney test (two categories) or the Kruskal‐Wallis test with Dunn's multiple comparison (multiple categories). Non‐AD dementia (FTD, DLB, and VaD) patients were analyzed as one group. Fold changes were calculated by dividing p‐tau concentrations by the mean concentration of the Aβ– CU group. P‐tau diagnostic accuracies were evaluated by area under the curve (AUC) analysis and the results statistically compared head‐to‐head using the DeLong test package in MedCalc. In concordance analyses, overall percentage of (dis)agreement was calculated as the sum of participants classified as “positive” or “negative” over the total number of participants. Concordance was evaluated with Cohen's κ coefficient, with κ = 0.61–0.80 indicating substantial agreement. Two‐sided P < .05 was considered statistically significant.
3. RESULTS
3.1. Analytical performance of the N‐p‐tau217 and N‐p‐tau181 assays
CSF samples diluted linearly comparing samples two‐ or four‐fold diluted to identical samples measured undiluted (Figure S1 in supporting information). Exogenously added phosphorylated recombinant tau was measureable with high recovery (N‐p‐tau217 = 91.9–102.8%; N‐p‐tau181 = 98.6%–111.4%; Figures S2–S3 in supporting information). Assay specificity was demonstrated using IP‐MS, showing that the antibodies specifically recognize the intended epitopes (Figure 1B–C). Each assay was specific to the antibody pair used: immuno‐depletion of the target analyte removed 94% to 98% of the measurable signals (Figure 1D–E). Additionally, the assays demonstrated robust repeatability in the clinical studies (Table S1 in supporting information).
3.2. Cohort characteristics
We studied p‐tau biomarker performance in a discovery cohort (n = 33) and two independent validation cohorts (n = 266 and n = 199; Table 1). The discovery cohort included 16 AD dementia participants with abnormal CSF core biomarkers and 17 Aβ– elderly controls with normal biomarker levels (P < .0001 each). The AD dementia participants were older (P < .0001). The Ljubljana cohort included Aβ– CU (n = 25), non‐AD MCI (n = 72), MCI‐AD (n = 55), and AD dementia (n = 114). The Paris cohort included Aβ– CU (n = 25), non‐AD MCI (n = 41), MCI‐AD (n = 15), AD dementia (n = 94), and non‐AD dementia (FTD [n = 11], DLB [n = 10], VaD [n = 3]). MCI‐AD individuals in both validation cohorts had decreased Aβ42. AD dementia individuals had AD CSF profiles based on defined cut‐offs (Table S2 in supporting information). Aβ– CU, non‐AD MCI, and non‐AD dementia participants had normal core biomarkers. Cognitive impairment assessed by Mini‐Mental State Examination (MMSE) increased with disease severity in both cohorts. In the Paris cohort, AD dementia and MCI‐AD participants were older than Aβ– CU (P ≤ .0023). There were no sex differences between groups (χ2 test; P ≥ .0930).
3.3. Increases in p‐tau biomarkers in the AD pathological process
All p‐tau biomarkers were increased in AD dementia compared to Aβ– CU (P < .0001; Figure 2A–I) and non‐AD MCI (P < .0001; Figure 2D–I). In both validation cohorts, N‐p‐tau217 was significantly elevated in MCI‐AD compared to Aβ– CU (P ≤ .0124, Figure 2G,D). N‐p‐tau181 showed mild albeit significant increases in MCI‐AD in both cohorts (P ≤ .0254, Figure 2E,H). Mid‐p‐tau181 showed minor non‐significant changes in MCI‐AD in either cohort (Figure 2F,I). N‐p‐tau217 was increased in MCI‐AD compared to non‐AD MCI (P ≤ .0217, Figure 2D,G), as was N‐p‐tau181 (P ≤ .0380; Figure 2E,H). Mid‐p‐tau181 did not differ between MCI‐AD and non‐AD MCI in either cohort. In the Paris cohort, all p‐tau biomarkers were increased in AD dementia compared to non‐AD dementia (P < .0001, Figure 2G–I). P‐tau concentrations were each similar in the respective Aβ– groups (Figure 2A–I).
FIGURE 2.
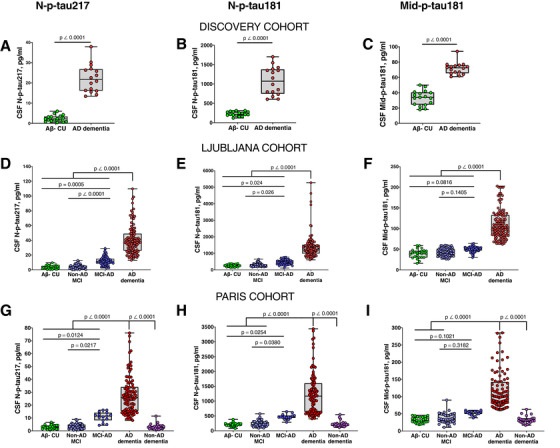
Concentrations of p‐tau biomarkers in the three cohorts. (A), (D), and (G) show N‐p‐tau217 concentrations in the discovery, Ljubljana, and Paris cohorts, respectively. The levels of N‐p‐tau181 in the discovery, Ljubljana, and Paris cohorts are given in (B), (E), and (H), respectively. The plots in (C), (F), and (I) show Mid‐p‐tau181 concentrations in the discovery, Ljubljana, and Paris cohorts, respectively. Participants in each cohort were classified according to clinical diagnosis and amyloid pathology. Group differences were compared using a two‐tailed Mann‐Whitney test (the discovery cohort) or Kruskal‐Wallis test followed by the Dunn's multiple comparison test (Ljubljana and Paris cohorts). Note that p‐tau concentrations were estimated from known concentrations of the assay calibrators. Assays that measure different p‐tau epitopes, those quantified on different analytical platforms as well as assays targeting different tau fragments (N‐terminal versus mid‐region p‐tau species) are therefore likely to give non‐identical values. For these reasons, the p‐tau concentrations measured by the different assays should not be compared by absolute pg/ml levels but rather according to their diagnostic performances and associations with Alzheimer's disease–type pathophysiologies
N‐p‐tau217 had the highest mean fold increases in all cohorts and between groups, followed by N‐p‐tau181 and Mid‐p‐tau181 (Figure S4 in supporting information). For AD dementia, N‐p‐tau217 fold change was 9.2 to 11.5 compared to 3.2 to 6.0 for N‐p‐tau181 and 2.2 to 3.6 for Mid‐p‐tau181 (Table S3 in supporting information). In the validation cohorts, fold changes in MCI‐AD were 3.5 to 3.7 for N‐p‐tau217, 1.8 to 2.2 for N‐p‐tau181, and 1.3 to 1.6 for Mid‐p‐tau181 (Table S3). For AD dementia and MCI‐AD, N‐p‐tau217 fold changes were higher than those for N‐p‐tau181 and Mid‐p‐tau181 (all P < .05, Table S3).
3.4. Association of p‐tau variants with amyloid biomarkers
P‐tau biomarkers were inversely correlated with Aβ42/Aβ40 in the validation cohorts (Table S4 in supporting information) and Aβ42 in the discovery cohort (Figure S5 in supporting information). However, N‐p‐tau217 had the strongest correlation with Aβ42/Aβ40 in the Ljubljana and Paris cohorts (r = –0.813 and r = –0.820, respectively, P < .0001; Table S4). In comparison, the correlation of N‐p‐tau181 with Aβ42/Aβ40 was r = –0.783 to r = –0.819 (P < .0001) while that of Mid‐p‐tau181 was r = –0.736 to r = –0.802 (P < .0001; Table S4). In AD dementia, N‐p‐tau217 correlated with Aβ42/Aβ40 in both validation cohorts (Ljubljana: r = –0.420; Paris: r = –0.375, P < .001), compared to r = –0.321 to r = –0.450 (P ≤ .0019) for N‐p‐tau181 and r = –0.170 to r = –0.372 (P ≤ .0003) for Mid‐p‐tau181. In MCI‐AD, all p‐tau forms showed weak correlations with Aβ42/Aβ40 but only Mid‐p‐tau181 reached significance in the Ljubljana cohort (r = –0.353, P = .0088). There was no correlation between p‐tau and Aβ42/Aβ40 in Aβ– groups.
3.5. Accuracies of p‐tau variants to identify Aβ pathology and differentiate MCI‐AD from non‐AD MCI
In the Ljubljana cohort, N‐p‐tau217 identified individuals with decreased Aβ42/Aβ40 (AUC = 97.1% [95% confidence interval [CI] = 95.4%–98.8%]) equally accurately as N‐p‐tau181 (AUC = 94.8% [95%CI = 92.4%–97.4%], P = .1567) but stronger than Mid‐p‐tau181 (AUC = 91.2% [95%CI = 88.0%–94.4%] P = .0029; Figure 3A). N‐p‐tau217 separated MCI‐AD from Aβ– CU with higher accuracy (AUC = 93.0% [95%CI = 87.7%–98.1%]) than Mid‐p‐tau181 (AUC = 79.8% [95%CI = 67.8%–91.7%], P = .0194) but not N‐p‐tau181 (AUC = 89.1% [95%CI = 82.0%–96.1%], P = .3840; Figure 3B). N‐p‐tau217 more accurately distinguished MCI‐AD from non‐AD MCI (AUC = 90.5% [95%CI = 85.1%–95.8%]) than Mid‐p‐tau181 (AUC = 70.6% [95%CI = 61.6%–79.7%] P = .0004) but not N‐p‐tau181 (AUC = 82.6% [95%CI = 75.3%–90.0%]; P = .1049, Figure 3C).
FIGURE 3.
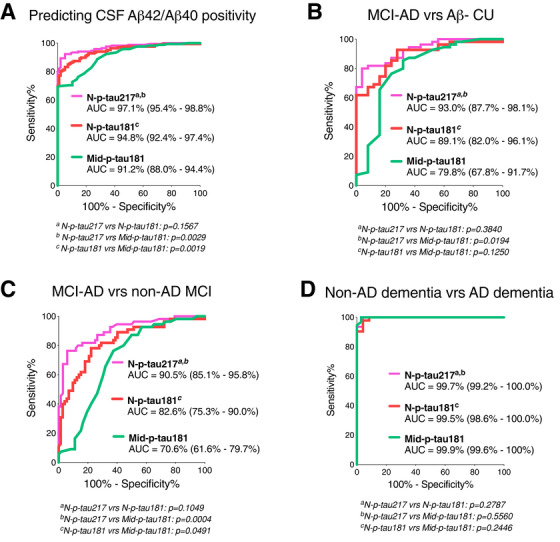
Accuracies of p‐tau biomarkers in identifying increased amyloid pathology, separating mild cognitive impairment‐Alzheimer's disease (MCI‐AD) from non‐AD MCI, and distinguishing AD dementia from amyloid beta (Aβ)– non‐AD. A, Area under the curves (AUC) comparing the predictive capacities of N‐p‐tau217, N‐p‐tau181, and Mid‐p‐tau181 to correctly identify individuals with increased Aβ pathology (assessed by cerebrospinal fluid [CSF] Aβ42/Aβ40 ratio). (B) and (C) depict AUC showing the abilities of the different p‐tau biomarkers to distinguish between individuals with MCI‐AD and Aβ– cognitively unimpaired (CU) groups and the MCI‐AD and non‐AD MCI groups, respectively. D, Diagnostic accuracies of p‐tau variants in separating between AD dementia and non‐AD dementia (including dementia with Lewy bodies, frontotemporal dementia, and vascular dementia patients). AUC values representing diagnostic accuracies for the different p‐tau biomarkers were statistically compared head‐to‐head using the DeLong test package in the MedCalc software. P values < .05 were considered statistically significant
3.6. Accuracies in separating AD dementia from Aβ– CU and non‐AD dementia
In the Paris cohort, N‐p‐tau217 discriminated AD dementia from non‐AD dementia patients (AUC = 99.7% [95%CI = 99.2%–100%]) with similar accuracy as N‐p‐tau181 (AUC = 99.5% [95%CI = 98.6%–100%], P = .2787) and Mid‐ptau181 (AUC = 99.9% [95%CI = 99.6%–100%], P = .5560; Figure 3D). The results were unchanged when non‐AD dementia cases were stratified by dementia types (data not shown).
3.7. Correlation between p‐tau biomarkers and with total‐tau
In all cohorts, N‐p‐tau217 was highly correlated with N‐p‐tau181 (r = 0.913 to r = 0.935, P < .0001; Table 2). These correlations were stronger in Aβ+ than Aβ– cases, with similar observation for MCI (Table 2).
TABLE 2.
Spearman's correlation of N‐p‐tau217 with other p‐tau forms and total‐tau
| N‐p‐tau181 | Mid‐p‐tau181 a | Total‐tau a | |
|---|---|---|---|
| Discovery cohort | |||
| Whole cohort | 0.916 (P < .0001) | 0.847 (P < .0001) | 0.823 (P < .0001) |
| Aβ– CU | 0.512 (P = .0376) | 0.338 (P = .1831) | 0.234 (P = .3629) |
| AD dementia | 0.918 (P < .0001) | 0.5026 (P < .0001) | 0.533 (P = .0397) |
| Ljubljana cohort | |||
| Whole cohort | 0.913 (P < .0001) | 0.851 (P < .0001) | 0.857 (P < .0001) |
| All Aβ– | 0.348 (P = .0006) | 0.144 (P = .1698) | 0.182 (P = .0807) |
| All Aβ+ | 0.894 (P < .0001) | 0.855 (P < .0001) | 0.818 (P < .0001) |
| Aβ– CU | 0.243 (P = .2422) | 0.207 (P = .3209) | 0.327 (P = .1105) |
| Non‐AD MCI | 0.369 (P = .0019) | 0.0629 (P = .6106) | 0.0768 (P = .5334) |
| MCI‐AD | 0.669 (P < .0001) | 0.241 (P = .0757) | 0.186 (P = .1744) |
| AD dementia | 0.749 (P < .0001) | 0.700 (P < .0001) | 0.609 (P < .0001) |
| Paris cohort | |||
| Whole cohort | 0.935 (P < .0001) | 0.930 (P < .0001) | 0.855(P < .0001) |
| All Aβ‐ | 0.564 (P < .0001) | 0.572 (P < .0001) | 0.473 (P < .0001) |
| All Aβ+ | 0.873 (P < .0001) | 0.868 (P < .0001) | 0.858 (P < .0001) |
| Aβ– CU | 0.487 (P = .0136) | 0.578 (P = .0025) | 0.535 (P = .0059) |
| Non‐AD MCI | 0.598 (P < .0001) | 0.529 (P = .0004) | 0.464 (P = .0022) |
| MCI‐AD | 0.814 (P < .0001) | 0.835 (P < .0001) | 0.808 (P < .0001) |
| AD dementia | 0.887 (P < .0001) | 0.875 (P < .0001) | 0.865 (P < .0001) |
| Non‐AD dementia | 0.565 (P = .0040) | 0.545 (P = .0058) | 0.398 (P = .0541) |
Abbreviations: AD, Alzheimer's disease; Aβ, amyloid beta; CU, cognitively unimpaired; MCI, mild cognitive impairment; p‐tau181, tau phosphorylated at threonine‐181.
Mid‐p‐tau181 and total tau were measured using Fujirebio ® Innotest (Ljubljana cohort) or Lumipulse (Paris cohort) assays.
N‐p‐tau217 correlated with Mid‐p‐tau181 in all cohorts (r = 0.847 to r = 0.930, P < .0001; Table 2), with stronger associations in Aβ+ cases (Table 2). Regarding diagnosis, the association was highest in AD cases (r = 0.700 to r = 0.835, P < .0001, Table 2).
N‐p‐tau217 showed strong correlations with total‐tau in all cohorts (discovery r = 0.823 to r = 0.857, P < .0001), as well as in Aβ+ sub‐groups (r = 0.533 to r = 0.857, P < .0001). The correlations were similarly high for MCI‐AD (r = 0.808 in Paris) and AD dementia (r = 0.609‐0.865, P < .0001; Table 2). Similar correlations were recorded for N‐p‐tau181 and Mid‐p‐tau181 versus total‐tau (Table S5 in supporting information).
3.8. Concordance between p‐tau biomarkers
N‐p‐tau217 and N‐p‐tau181 had an overall agreement of 88.2% (negative agreement (–/–): n = 81/262 [30.9%]; positive agreement (+/+): n = 150/262 [57.3%], κ = 0.746; Figure 4A). The overall agreement of N‐p‐tau217 with Mid‐p‐tau181 was relatively lower –79.7% (negative agreement (–/–): n = 91/262 [34.7%]; positive agreement (+/+): n = 118/262 [45.0%], κ = 0.606; Figure 4B). The concordance of N‐p‐tau181 with Mid‐p‐tau181 was 82.7% (negative agreement: n = 103/266 [38.7%]; positive agreement: n = 117/266 [44.0%], κ = 0.662; Figure 4C). A higher proportion of cases were positive for N‐p‐tau217 and negative for Mid‐p‐tau181 (n = 52/262, 19.8%) and N‐p‐tau181 (n = 20/262, 7.6%) compared to those negative for N‐p‐tau217 but positive for N‐p‐tau181 (n = 11/262, 4.2%) and Mid‐p‐tau181 (n = 1/262, 0.4%). According to diagnostic groups, cases that were N‐p‐tau217‐positive but negative for N‐p‐tau181 or Mid‐p‐tau181 were mostly MCI‐AD (N‐p‐tau181 negative: 13 MCI‐AD out of 20 discordant participants; Mid‐p‐tau181 negative: 40 MCI‐AD among 52 discordant individuals).
FIGURE 4.
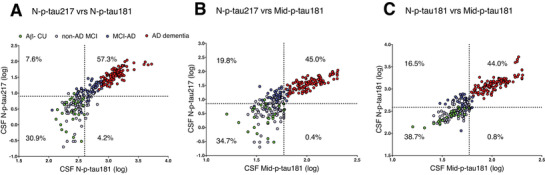
Concordance among the three p‐tau assays. (A) N‐p‐tau217 versus N‐p‐tau181, (B) N‐p‐tau217 versus Mid‐p‐tau181, and (C) N‐p‐tau181 versus Mid‐p‐tau181 in the Ljubljana cohort. On each plot, the percentage of concordant cases are given in the upper right and lower left quadrants while the percent of discordant cases are shown in the upper left and lower right quadrants. Assay cut‐offs were set as the concentrations of the 95th percentage individual in the amyloid beta–negative cognitively unimpaired group.
3.9. Association of p‐tau with cognitive decline and neurodegeneration
N‐p‐tau217 inversely correlated with MMSE in the Ljubljana and Paris cohorts (r = –0.490 and r = –0.419, respectively, P < .0001; Table S6 in supporting information). N‐p‐tau181 and Mid‐p‐tau181 showed similar respective correlations (Ljubljana: r = –0.481 and r = –0.357; Paris: r = –0.428 and r = –0.405, P < .0001). In the Ljubljana cohort, the correlation was significant in Aβ+ cases for N‐p‐tau217 and N‐p‐tau181 (r = –0.222, P = .0215 and r = –0.279, P = .0040, respectively). In the Paris cohort, N‐p‐tau217, N‐p‐tau181, and Mid‐p‐tau181 were each correlated to medial temporal lobe atrophy (r = 0.334, r = 0.335, and r = 0.305, respectively, P < .001, Table S6).
4. DISCUSSION
In this study we compared the diagnostic performance of CSF p‐tau biomarkers in clinical settings. P‐tau217 and p‐tau181, which were partnered with an N‐terminal detection antibody, increased in MCI‐AD while the conventional mid‐region p‐tau181 assays remained in normal ranges. In agreement, N‐p‐tau217 and N‐p‐tau181 identified increased Aβ pathophysiology, separated MCI‐AD from non‐AD MCI, and differentiated MCI‐AD from Aβ– CU significantly better than Mid‐p‐tau181. Similar performances of N‐p‐tau217 and N‐p‐tau181 imply the advantage of N‐p‐tau217 relies on the p‐tau181 biomarker to which it is compared. N‐p‐tau217 and N‐p‐tau181 appear to be earlier markers of AD pathophysiology that could aid in establishing if prodromal cognitive decline is due to AD. All p‐tau variants showed near‐perfect capacities in distinguishing AD from Aβ– non‐ADs, indicating comparable AD specificity.
Tau phosphorylation was first reported as a CSF biomarker for AD in a 1995 study presenting an ELISA based on antibody pairs targeting mid‐region tau, showing p‐tau181 increases in AD. 39 This study informed the development of gold‐standard commercial p‐tau181 immunoassays (eg, INNOTEST, Lumipulse, and Elecsys). 40 , 41 Recently, CSF was found to also contain N‐terminal to mid‐region species with biomarker potential. 24 , 42 , 43 , 44 We recently developed the assay targeting N‐p‐tau181 which is metabolized into both CSF and blood. 12 , 13 , 14 , 15 , 30 Among other p‐tau forms, 23 , 24 , 45 p‐tau217 was suggested to be a potentially superior AD biomarker because it better: (1) correlated with tau and Aβ‐PET than p‐tau181 29 (2) identified Aβ+ individuals from preclinical stage, 27 , 28 and (3) distinguished AD from non‐AD. 27 , 29 However, studies reporting improved p‐tau217 performances compared novel, research‐grade p‐tau immunoassays, 29 IP‐MS assays, 27 , 28 or IP‐MS p‐tau217 versus Mid‐p‐tau181 ELISA. 27 , 28 Further evidence is needed and requires direct comparison to the most clinically characterized Mid‐p‐tau181 biomarkers.
Consequently, we compared a novel N‐p‐tau217 biomarker head‐to‐head against three p‐tau181 assays: one of two commercial Mid‐p‐tau181 assays (INNOTEST and Lumipulse), and an ultrasensitive N‐p‐tau181 Simoa‐based biomarker that shares identical analytical features with N‐p‐tau217 (ie, same detector antibody, assay buffers, and platform). We corroborate previous findings that p‐tau217 highly correlates with p‐tau181 and total‐tau, displays larger fold changes than p‐tau181, and has greater capacity to identify Aβ+ cases. 27 , 29 However, N‐p‐tau217 and N‐p‐tau181 became abnormal earlier than Mid‐p‐tau181, suggesting that both have improved associations with Aβ pathophysiology, especially at the initial stages of the disease process. N‐p‐tau217 and N‐p‐tau181 start increasing almost concurrently with Aβ changes, with Mid‐p‐tau181 becoming abnormal later. 20 Most MCI‐AD cases had increased N‐p‐tau217 (80.0%) and N‐p‐tau181 (61.8%) compared to 9.3% for Mid‐p‐tau181. This may indicate that N‐p‐tau217 becomes abnormal marginally ahead of N‐p‐tau181, although these differences were not statistically significant. Agreeably, we showed in a preclinical AD study that N‐p‐tau217 and N‐p‐tau181 were both better associated with changes in Aβ‐PET and CSF Aβ42/Aβ40 (Elecsys) than Mid‐p‐tau181. 30
All p‐tau biomarkers equally separated AD dementia from non‐AD dementia, showing that their performance differences are limited to the AD spectrum. Altogether, N‐p‐tau217 and N‐p‐tau181 are equally increased in preclinical and MCI‐AD in association with Aβ changes and hence closely track early AD‐related Aβ and tau abnormalities in pre‐dementia stages while Mid‐p‐tau181 monitors established tau pathology in AD‐dementia.
A major challenge in memory clinics is to identify cognitive impairment due to AD in a heterogeneous population presenting with memory complaints. MCI has various outcomes 46 including progression to AD dementia (10%–15% of cases annually 47 ), stability or improvement, or development of other dementia. Brain imaging and neuropsychological assessment remain essential for diagnosis; however, both have limited value to distinguish amyloid‐positivity. 48 Even with CSF testing, Mid‐p‐tau181 is only changed in late prodromal AD and Aβ positivity does not always signal MCI‐AD, 19 , 20 , 49 reinforcing potential clinical utility of the early and AD‐specific N‐p‐tau217 and N‐p‐tau181 biomarkers.
Another crucial contribution is the early identification of AD‐type tau pathology in Aβ+ patients. Currently, inclusion for anti‐amyloid therapeutic trials relies on Aβ+ positivity. 50 Therefore, MCI Aβ+ patients are recruited without being sure of the presence of AD tau pathology and consequently some have low risks of progressing to AD during the trial. Identifying abnormal tau phosphorylation in MCI could be advantageous to “enrich” the trial population by selecting only individuals on the AD continuum, thereby improving the reliability of biomarker‐based outcome measures.
Despite their statistically inseparable clinical performances, N‐p‐tau217 had higher fold changes than N‐p‐tau181, suggesting that pathophysiological changes in the former occur over a wider biological spectrum. However, this point has limited clinical value because both biomarkers become abnormal starting from MCI‐AD, hence clinically validated cut‐off values should identify abnormal concentrations. Moreover, the assessment of fold changes is not feasible in routine clinical settings.
One could argue that the similar performances of N‐p‐tau217 and N‐p‐tau181 might be due to a potential lesser sensitivity of our N‐p‐tau217 assay than previously published assays. Importantly, the assays target different tau fragments. Nonetheless, N‐p‐tau217 showed larger fold changes in AD dementia (9.2–11.5) than the one reported by Janelidze et al. 29 using a Lilly‐developed assay (fold change = 8.6), meaning our new assay appears to have a wider dynamic range. Moreover, while our N‐p‐tau181 had AD fold changes up to 6.0, the Lilly‐developed p‐tau181 assay had a fold change of 3.7, closer to the 2.2 to 4.6 we report for Mid‐p‐tau181. 29 These points support the argument that the significant improvement of p‐tau217 depends on the assay held as a standard for comparison. A head‐to‐head comparison study between the different p‐tau217 assays would help us gain further insights.
The results suggest that pathophysiological changes resulting in the release of novel N‐terminal p‐tau biomarkers into CSF occur early in the AD continuum, ahead of Mid‐p‐tau181. This could be due to differential brain processing and metabolism of distinct p‐tau forms resulting from, for example, distinctions in phosphorylation kinetics, truncation, active secretion, and release. Indeed, p‐tau biomarker changes are dynamic in normal individuals and across the AD pathological process. 26 , 28 , 44 Furthermore, these biomarkers associate better with Aβ pathology because they become progressively abnormal earlier than Mid‐p‐tau181 in the disease process, in agreement with recent reports. 27 , 28 , 29 , 30 In vivo animal‐model studies also support such differences in p‐tau dynamics with respect to Aβ abnormalities. 21 , 22
This study has several strengths including its focus on clinical settings, corroborating findings from three independent cohorts, using unselected, routinely archived clinical samples. Furthermore, we compared the performance of two novel p‐tau biomarkers versus two established Mid‐p‐tau181 assays and investigated the ability of AD biomarkers to identify early MCI‐AD. Limitations include lack of PET data that prevented comparison of p‐tau performance in relation to PET biomarkers. Nonetheless, CSF biomarkers are more widely used in clinical settings and become abnormal earlier than PET biomarkers. Additionally, potential differences in analytical technologies (N‐p‐tau217 and N‐p‐tau181 on Simoa and ELISA/electrochemiluminiscence for Mid‐p‐tau181) contributing to the observed results cannot be discounted. Nonetheless, corroborating results were reported using assays developed with Meso‐Scale Discovery technology. 29 Finally, lack of direct comparison of N‐p‐tau217 with the Lilly‐developed p‐tau217 prevented head‐to‐head characterization of these assays.
In conclusion, we compared p‐tau biomarkers, showing that N‐p‐tau217 and N‐p‐tau181 are both increased in early MCI‐AD, identify individuals with Aβ pathology, and separate early MCI‐AD from non‐AD MCI more accurately than Mid‐p‐tau181. The inseparable accuracies of N‐p‐tau217 and N‐p‐tau181 suggest that they can both support AD diagnosis starting from the prodromal stage. These results suggest that different p‐tau biomarkers change at distinct stages of the AD pathological process, and support the idea of therapeutically targeting specific p‐tau at defined stages. Other important clinical implications of these novel biomarkers include their potential uses for prognosis, progression monitoring, and as outcome measures in therapeutic trials.
CONFLICTS OF INTEREST
Henrik Zetterberg has served at scientific advisory boards for Denali, Roche Diagnostics, Wave, Samumed, Siemens Healthineers, Pinteon Therapeutics, and CogRx; and has given lectures in symposia sponsored by Fujirebio, Alzecure, and Biogen. Kaj Blennow has served as a consultant or on advisory boards for Axon, Biogen, CogRx, Lilly, MagQu, Novartis, and Roche Diagnostics. Henrik Zetterberg and Kaj Blennow are co‐founders of Brain Biomarker Solutions in Gothenburg AB, a GU Ventures‐based platform company at the University of Gothenburg. The other authors declare no competing interest.
Supporting information
Supporting Information
ACKNOWLEDGMENTS
TKK holds a research fellowship from the BrightFocus Foundation (#A2020812F) and is further supported by the Swedish Alzheimer Foundation (Alzheimerfonden; #AF‐930627), the Swedish Brain Foundation (Hjärnfonden; #FO2020‐0240), the Swedish Dementia Foundation (Demensförbundet), Gamla Tjänarinnor Foundation, the Aina (Ann) Wallströms and Mary‐Ann Sjöbloms Foundation, the Agneta Prytz‐Folkes & Gösta Folkes Foundation (#2020‐00124), the Gun and Bertil Stohnes Foundation, and the Anna Lisa and Brother Björnsson's Foundation. AE was supported by the Department of Neurology, University Medical Centre Ljubljana, Ljubljana, Slovenia. AV is supported by Fondation Ophtalmologique Adolphe de Rothschild, Fondation Chatrier, Association des Anciens Internes des Hôpitaux de Paris, the Swedish Dementia Foundation (Demensförbundet), and the Anna Lisa and Brother Björnsson's Foundation. NJA is supported by the Swedish Alzheimer Foundation (Alzheimerfonden; #AF‐931009) and the Swedish Dementia Foundation (Demensförbundet). HZ is a Wallenberg Scholar supported by grants from the Swedish Research Council (#2018‐02532), the European Research Council (#681712), Swedish State Support for Clinical Research (#ALFGBG‐720931), the Alzheimer Drug Discovery Foundation (ADDF), USA (#201809‐2016862), and the UK Dementia Research Institute at UCL. KB is supported by the Swedish Research Council (#2017‐00915), the Alzheimer Drug Discovery Foundation (ADDF), USA (#RDAPB‐201809‐2016615), the Swedish Alzheimer Foundation (#AF‐742881), Hjärnfonden, Sweden (#FO2017‐0243), the Swedish state under the agreement between the Swedish government and the County Councils, the ALF‐agreement (#ALFGBG‐715986), and European Union Joint Program for Neurodegenerative Disorders (JPND2019‐466‐236). The funders had no role in data collection, analysis, or decision to publish.
Karikari TK, Emeršič A, Vrillon A, et al. Head‐to‐head comparison of clinical performance of CSF phospho‐tau T181 and T217 biomarkers for Alzheimer's disease diagnosis. Alzheimer's Dement. 2021;17:755–767. 10.1002/alz.12236
Thomas K. Karikari, Andreja Emeršič, and Agathe Vrillon equally contributed in this study.
REFERENCES
- 1. Spillantini MG, Goedert M. Tau pathology and neurodegeneration. Lancet Neurol. 2013;12:609‐622. [DOI] [PubMed] [Google Scholar]
- 2. Ashton NJ, Hye A, Rajkumar AP, et al. An update on blood‐based biomarkers for non‐Alzheimer neurodegenerative disorders. Nat Rev Neurol;2020:1‐20. [DOI] [PubMed] [Google Scholar]
- 3. Blennow K, Hampel H, Weiner M, Zetterberg H. Cerebrospinal fluid and plasma biomarkers in Alzheimer disease. Nat Rev Neurol. 2010;6:131‐144. [DOI] [PubMed] [Google Scholar]
- 4. Jack CR, Bennett DA, Blennow K, et al. NIA‐AA research framework: toward a biological definition of Alzheimer's disease. Alzheimers Dement. 2018;14:535‐562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Chételat G, La Joie R, Villain N, et al. Amyloid imaging in cognitively normal individuals, at‐risk populations and preclinical Alzheimer's disease. NeuroImage Clin. 2013;2:356‐365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. van Steenoven I, Aarsland D, Weintraub D, et al. Cerebrospinal fluid Alzheimer's disease biomarkers across the spectrum of lewy body diseases: results from a large multicenter cohort. J Alzheimers Dis. 2016;54:287‐295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Skillbäck T, Rosén C, Asztely F, Mattsson N, Blennow K, Zetterberg H. Diagnostic performance of cerebrospinal fluid total tau and phosphorylated tau in creutzfeldt‐jakob disease: results from the Swedish mortality registry. JAMA Neurol. 2014;71:476‐483. [DOI] [PubMed] [Google Scholar]
- 8. Hesse C, Rosengren L, Andreasen N, et al. Transient increase in total tau but not phospho‐tau in human cerebrospinal fluid after acute stroke. Neurosci Lett. 2001;297:187‐190. [DOI] [PubMed] [Google Scholar]
- 9. Franz G, Beer R, Kampfl A, et al. Amyloid beta 1‐42 and tau in cerebrospinal fluid after severe traumatic brain injury. Neurology. 2003;60:1457‐1461. [DOI] [PubMed] [Google Scholar]
- 10. Skillbäck T, Farahmand BY, Rosén C, et al. Cerebrospinal fluid tau and amyloid‐β1‐42 in patients with dementia. Brain. 2015;138:2716‐2731. [DOI] [PubMed] [Google Scholar]
- 11. Rosso SM, van HE, Pijnenburg YAL, et al. Total tau and phosphorylated tau 181 levels in the cerebrospinal fluid of patients with frontotemporal dementia due to P301L and G272V tau mutations. Arch Neurol. 2003;60:1209‐1213. [DOI] [PubMed] [Google Scholar]
- 12. Karikari TK, Pascoal TA, Ashton NJ, et al. Blood phosphorylated tau 181 as a biomarker for Alzheimer's disease: a diagnostic performance and prediction modelling study using data from four prospective cohorts. Lancet Neurol. 2020;19:422‐433. [DOI] [PubMed] [Google Scholar]
- 13. O'Connor A, Karikari TK, Poole T, et al. Plasma phospho‐tau181 in presymptomatic and symptomatic familial Alzheimer's disease: a longitudinal cohort study. Mol Psychiatry; 2020:1‐10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Benussi A, Karikari TK, Ashton N, et al. Diagnostic and prognostic value of serum NfL and p‐Tau181 in frontotemporal lobar degeneration. J Neurol Neurosurg Psychiatry. 2020. [DOI] [PubMed] [Google Scholar]
- 15. Lantero Rodriguez J, Karikari TK, Suárez‐Calvet M, et al. Plasma p‐tau181 accurately predicts Alzheimer's disease pathology at least 8 years prior to post‐mortem and improves the clinical characterisation of cognitive decline. Acta Neuropathol. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Janelidze S, Mattsson N, Palmqvist S, et al. Plasma P‐tau181 in Alzheimer's disease: relationship to other biomarkers, differential diagnosis, neuropathology and longitudinal progression to Alzheimer's dementia. Nat Med. 2020;26:379‐386. [DOI] [PubMed] [Google Scholar]
- 17. Thijssen EH, La Joie R, Wolf A, et al. Diagnostic value of plasma phosphorylated tau181 in Alzheimer's disease and frontotemporal lobar degeneration. Nat Med. 2020;26:387‐397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Nakamura A, Kaneko N, Villemagne VL, et al. High performance plasma amyloid‐β biomarkers for Alzheimer's disease. Nature. 2018;554:249‐254. [DOI] [PubMed] [Google Scholar]
- 19. Hansson O, Zetterberg H, Buchhave P, Londos E, Blennow K, Minthon L. Association between CSF biomarkers and incipient Alzheimer's disease in patients with mild cognitive impairment: a follow‐up study. Lancet Neurol. 2006;5:228‐234. [DOI] [PubMed] [Google Scholar]
- 20. Buchhave P, Minthon L, Zetterberg H, Wallin AK, Blennow K, Hansson O. Cerebrospinal fluid levels of β‐amyloid 1‐42, but not of tau, are fully changed already 5 to 10 years before the onset of Alzheimer dementia. Arch Gen Psychiatry. 2012;69:98‐106. [DOI] [PubMed] [Google Scholar]
- 21. Mattsson‐Carlgren N, Andersson E, Janelidze S, et al. Aβ deposition is associated with increases in soluble and phosphorylated tau that precede a positive Tau PET in Alzheimer's disease. Sci Adv. 2020;6:eaaz2387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Maia LF, Kaeser SA, Reichwald J, et al. Changes in amyloid‐β and tau in the cerebrospinal fluid of transgenic mice overexpressing amyloid precursor protein. Sci Transl Med. 2013;5:194re2‐194re2. [DOI] [PubMed] [Google Scholar]
- 23. Hampel H, Buerger K, Zinkowski R, et al. Measurement of phosphorylated tau epitopes in the differential diagnosisof Alzheimer disease: a comparative cerebrospinal fluid study. Arch Gen Psychiatry. 2004;61:95‐102. [DOI] [PubMed] [Google Scholar]
- 24. Meredith Jr JE, Sankaranarayanan S, Guss V, et al. Characterization of novel CSF tau and ptau biomarkers for Alzheimer's disease. PLOS ONE. 2013;8:e76523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hanger DP, Betts JC, Loviny TLF, Blackstock WP, Anderton BH. New phosphorylation sites identified in hyperphosphorylated tau (paired helical filament‐tau) from Alzheimer's disease brain using nanoelectrospray mass spectrometry. J Neurochem. 1998;71:2465‐2476. [DOI] [PubMed] [Google Scholar]
- 26. Barthélemy NR, Mallipeddi N, Moiseyev P, Sato C, Bateman RJ. Tau phosphorylation rates measured by mass spectrometry differ in the intracellular brain versus extracellular cerebrospinal fluid compartments and are differentially affected by Alzheimer's disease. Front Aging Neurosci. 2019;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Barthélemy NR, Bateman RJ, Hirtz C, et al. Cerebrospinal fluid phospho‐tau T217 outperforms T181 as a biomarker for the differential diagnosis of Alzheimer's disease and PET amyloid‐positive patient identification. Alzheimers Res Ther. 2020;12:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Barthélemy NR, Li Y, Joseph‐Mathurin N, et al. A soluble phosphorylated tau signature links tau, amyloid and the evolution of stages of dominantly inherited Alzheimer's disease. Nat Med. 2020;26:398‐407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Janelidze S, Stomrud E, Smith R, et al. Cerebrospinal fluid p‐tau217 performs better than p‐tau181 as a biomarker of Alzheimer's disease. Nat Commun. 2020;11:1‐12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Suárez‐Calvet M, Karikari TK, Ashton NJ, et al. ALFA study (2020) Novel tau biomarkers phosphorylated at T181, T217 or T231 rise in the initial stages of the preclinical Alzheimer’s continuum when only subtle changes in Aβ pathology are detected. EMBO Mol Med, 10.15252/emmm.202012921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Dubois B, Feldman HH, Jacova C, et al. Advancing research diagnostic criteria for Alzheimer's disease: the IWG‐2 criteria. Lancet Neurol. 2014;13:614‐629. [DOI] [PubMed] [Google Scholar]
- 32. Albert MS, DeKosky ST, Dickson D, et al. The diagnosis of mild cognitive impairment due to Alzheimer's disease: recommendations from the national institute on aging‐Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. 2011;7:270‐279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. McKeith IG, Boeve BF, Dickson DW, et al. Diagnosis and management of dementia with Lewy bodies. Neurology. 2017;89:88‐100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chare L, Hodges JR, Leyton CE, et al. New criteria for frontotemporal dementia syndromes: clinical and pathological diagnostic implications. J Neurol Neurosurg Psychiatry. 2014;85:865‐870. [DOI] [PubMed] [Google Scholar]
- 35. Sachdev P, Kalaria R, O'Brien J, et al. Diagnostic criteria for vascular cognitive disorders: a VASCOG statement. Alzheimer Dis Assoc Disord. 2014;28:206‐218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Horowitz PM, Patterson KR, Guillozet‐Bongaarts AL, et al. Early N‐terminal changes and caspase‐6 cleavage of tau in Alzheimer's disease. J Neurosci. 2004;24:7895‐7902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ercan E, Eid S, Weber C, et al. A validated antibody panel for the characterization of tau post‐translational modifications. Mol Neurodegener. 2017;12:87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Vanderstichele H, Bibl M, Engelborghs S, et al. Standardization of preanalytical aspects of cerebrospinal fluid biomarker testing for Alzheimer's disease diagnosis: a consensus paper from the Alzheimer's biomarkers standardization initiative. Alzheimers Dement. 2012;8:65‐73. [DOI] [PubMed] [Google Scholar]
- 39. Blennow K, Wallin A, Agren H, Spenger C, Siegfried J, Vanmechelen E. Tau protein in cerebrospinal fluid: a biochemical marker for axonal degeneration in Alzheimer disease? Mol Chem Neuropathol. 1995;26:231‐245. [DOI] [PubMed] [Google Scholar]
- 40. Leitão MJ, Silva‐Spínola A, Santana I, et al. Clinical validation of the Lumipulse G cerebrospinal fluid assays for routine diagnosis of Alzheimer's disease. Alzheimers Res Ther. 2019;11:91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lifke V, Kollmorgen G, Manuilova E, et al. Elecsys® Total‐Tau and Phospho‐Tau (181P) CSF assays: analytical performance of the novel, fully automated immunoassays for quantification of tau proteins in human cerebrospinal fluid. Clin Biochem. 2019;72:30‐38. [DOI] [PubMed] [Google Scholar]
- 42. Cicognola C, Brinkmalm G, Wahlgren J, et al. Novel tau fragments in cerebrospinal fluid: relation to tangle pathology and cognitive decline in Alzheimer's disease. Acta Neuropathol. 2018;137:279‐296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Chen Z, Mengel D, Keshavan A, et al. Learnings about the complexity of extracellular tau aid development of a blood‐based screen for Alzheimer's disease. Alzheimers Dement. 2018;15:487‐496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Sato C, Barthélemy NR, Mawuenyega KG, et al. Tau kinetics in neurons and the human central nervous system. Neuron. 2018;97:1284‐1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hu YY, He SS, Wang X, et al. Levels of nonphosphorylated and phosphorylated tau in cerebrospinal fluid of Alzheimer's disease patients : an ultrasensitive bienzyme‐substrate‐recycle enzyme‐linked immunosorbent assay. Am J Pathol. 2002;160:1269‐1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Langa KM, Levine DA. The diagnosis and management of mild cognitive impairment: a clinical review. JAMA. 2014;312:2551‐2561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Mitchell AJ, Shiri‐Feshki M. Rate of progression of mild cognitive impairment to dementia–meta‐analysis of 41 robust inception cohort studies. Acta Psychiatr Scand. 2009;119:252‐265. [DOI] [PubMed] [Google Scholar]
- 48. Alves L, Cardoso S, Silva D, et al. Neuropsychological profile of amyloid‐positive versus amyloid‐negative amnestic mild cognitive impairment. J Neuropsychol;2020:e12218. [DOI] [PubMed] [Google Scholar]
- 49. Cognat E, Liger FM, Troussière A‐C, et al. What is the clinical impact of cerebrospinal fluid biomarkers on final diagnosis and management in patients with mild cognitive impairment in clinical practice? Results from a nation‐wide prospective survey in France. BMJ Open. 2019;9:e026380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Tolar M, Abushakra S, Sabbagh M. The path forward in Alzheimer's disease therapeutics: reevaluating the amyloid cascade hypothesis. Alzheimers Dement. 2019. [DOI] [PubMed] [Google Scholar]
- 51. Karikari TK, Benedet AL, Ashton NJ, et al. (2020) Diagnostic performance and prediction of clinical progression of plasma phospho‐tau181 in the Alzheimer’s Disease Neuroimaging Initiative. Mol Psychiatry, 10.1038/s41380-020-00923-z. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information