

Abstract
The treatment of multiple sclerosis (MS), the most common chronic inflammatory, demyelinating and neurodegenerative disease of the central nervous system (CNS), continues to transform. In recent years, a number of novel and increasingly effective disease‐modulatory therapies (DMTs) have been approved, including oral fumarates and selective sphingosine 1‐phosphate modulators, as well as cell‐depleting therapies such as cladribine, anti‐CD20 and anti‐CD52 monoclonals. Amongst DMTs in clinical development, inhibitors of Bruton's tyrosine kinase represent an entirely new emerging drug class in MS, with three different drugs entering phase III trials. However, important remaining fields of improvement comprise tracking of long‐term benefit‐risk with existing DMTs and exploration of novel treatment targets relating to brain inherent disease processes underlying the progressive neurodegenerative aspect of MS, which accumulating evidence suggests start already early in the disease process. The aim here is to review current therapeutic options in relation to an improved understanding of the immunopathogenesis of MS, also highlighting examples where controlled trials have not generated the desired results. An additional aim is to review emerging therapies undergoing clinical development, including agents that interfere with disease processes believed to be important for neurodegeneration or aiming to enhance reparative responses. Notably, early trials now have shown initial evidence of enhanced remyelination both with small molecule compounds and biologicals. Finally, accumulating evidence from clinical trials and post‐marketing real‐world patient populations, which underscore the importance of early high effective therapy whilst maintaining acceptable tolerability, is discussed.
Keywords: multiple sclerosis, immunomodulatory therapy, biologics, remyelination, benefit‐risk, randomized controlled trial

Introduction
Multiple sclerosis (MS), a chronic inflammatory, demyelinating and neurodegenerative disease of the central nervous system (CNS), affects approximately 2.5 million people worldwide, with a significant societal economic impact and high burden for patients and their relatives [1, 2, 3]. MS reduces life expectancy with 7–14 years, with infections and suicide being important contributors, but with trends for a closing gap to the general population [2, 4]. The typical disease onset is in the third and fourth decades of life, with women being affected two to three times more often than men. MS is also more frequent amongst people of European descent, especially in those residing at higher latitudes, where the prevalence can reach above 200 per 100 000 [5]. MS disease risk is affected by complex gene–environment interactions, where allelic variants in the human leukocyte antigen (HLA)‐complex, history of infection with Epstein–Barr virus (EBV)/mononucleosis, smoking and low sun exposure/vitamin D levels have the greatest impact (for references, see [6]). The complexity of these interactions likely also explains a high degree of heterogeneity in disease characteristics across individuals, although factors regulating disease severity are still largely unknown [7]. Three major MS disease phenotypes are traditionally recognized as follows: relapsing–remitting MS (RRMS), secondary progressive MS (SPMS) and primary progressive MS (PPMS) [1, 2]. Most patients present with RRMS, characterized by at least partly reversible episodes of neurological deficits, usually lasting days to months. After a first such event, criteria for a clinically isolated syndrome (CIS) is fulfilled, whilst two episodes are required for a diagnosis of clinically definite RRMS, even if imaging or cerebrospinal fluid findings sometimes can substitute for a second clinical event [8]. After some years or many decades with RRMS, most patients will experience a chronic progression of disabilities with or without overlaid relapses, SPMS. In 5–10%, with a higher relative proportion of men and onset later in life, disease is progressive from onset, PPMS (Fig. 1).
Fig. 1.

In most patients, MS starts as a relapsing–remitting disease (RRMS), which is termed Clinically Isolated Syndrome (CIS) after a first bout of clinical symptoms. This is often preceded by a phase of subclinical disease activity that can be detected with magnetic resonance imaging (MRI; yellow stars denote MRI signs of active inflammation). In later stages of RRMS, patients accumulate persistent disabilities, where recent evidence suggests that a progressive component may start already soon after diagnosis (light grey). This underlying progressive disease component becomes more pronounced at later stages, when the disease converts to secondary progressive MS (SPMS). The fact that inflammatory disease activity, as reflected by frequency of bouts or MRI activity, diminishes over time suggests a shift from adaptive to innate or local disease mechanisms, which may explain the relative loss of efficacy of disease‐modulatory treatments (DMT).
Drugs that improve long‐term outcomes of MS are termed disease‐modulatory therapies (DMTs). Until recently, they were restricted to the RRMS subtype, whilst the progressive phenotype was considered unresponsive to most forms of immunomodulation. A key pathological finding in MS is the chronic accumulation of demyelinating lesions in the CNS, which can be visualized by magnetic resonance imaging (MRI; Fig. 2). Such focal lesions represent a proxy of important clinical outcomes, such as relapse rates and disability accumulation, and therefore represent the most important para‐clinical monitoring tool [9]. In context of treatment, they also denote an objective disease feature intimately connected with inflammatory disease activity and therefore often are used as primary and important secondary outcomes in phase II and III trials, respectively [10]. In addition, more recent revisions of progressive MS phenotype definitions stratify a disease course into inflammatory ‘active’ or ‘not active’ based on occurrence of clinical relapses or evidence of neuroradiological disease activity, that is contrast‐enhancing lesions or newly formed lesions compared to a previous scan, in order to identify patients likely to benefit from DMT [11, 12]. Major efforts have been invested in identifying additional monitoring tools, where most progress have been made with a soluble protein marker of ongoing neuro‐axonal degeneration termed neurofilament light [13, 14]. Access to accurate disease monitoring tools is of importance not only in drug development, but also in clinical practice, since development of disability is a late and largely irreversible phenomenon. Consequently, the importance of protecting the CNS from further damage is increasingly appreciated as key to improve long‐term outcomes. Equally important, data on the comparative safety of different types of DMTs are far from complete. Thus, whilst frequency of more common side effects usually are detected already during clinical development, more rare events or those that affect special patient groups must be recorded in large‐scale post‐marketing studies. Collectively, whilst we today have an increasing number of DMT options, the knowledge base for assessing long‐term benefit‐risk across different DMTs and patient groups is still restricted. In addition, the increasing complexity of DMT landscape represents a challenge when discussing therapeutic options with individual patients [15].
Fig. 2.
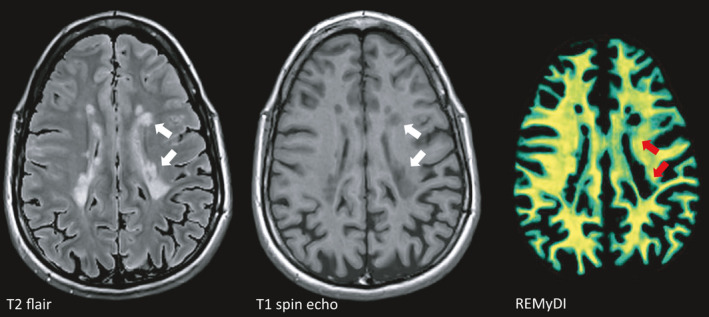
Magnetic resonance imaging (MRI) is the most important non‐clinical tool to monitor disease progression. On T2 flair‐weighted images, accumulation of hyperintense (bright) lesions around the ventricles is one of the hallmark signs of MS. These lesions appear hypointense (dark) on T1‐weighted images as a sign of more pronounced tissue destruction. Rapid Estimation of Myelin for Diagnostic Imaging (REMyDI) represents a novel technique to visualize myelin integrity using standard MRI equipment [63]. In this image, complete or near complete loss of myelin is seen in T1 hypointense lesions, but more widespread affection of myelin is evident also in white matter areas outside of focal lesions (green areas as contrasted by yellow areas with intact myelin). Images courtesy of Tobias Granberg, KI, Sweden.
Pathogenic mechanisms as therapeutic targets
Disease mechanism in earlier disease phases
Traditionally, MS has been regarded as a predominately T‐cell‐mediated disease, largely based on data from experimental autoimmune encephalomyelitis (EAE), a widely used animal model of MS [16]. Thus, EAE studies have shown that effector CD4+ and CD8+ T cells secreting interferon‐γ (IFN‐γ), interleukin‐17 (IL‐17) and granulocyte–macrophage colony‐stimulating factor (GM‐CSF) are important in EAE, and such T‐cell populations are also increased in MS; see, for example, [17, 18, 19]. However, limitations with the EAE model include the necessity of using active immunization protocols, the inability to simultaneously study important environmental triggers such as EBV and the difficulty to mimic progressive disease that in humans evolves over decades. Therefore, EAE has been questioned as being of relevance for identification of therapeutic targets, but nevertheless must be seen as relevant for dissection of disease mechanisms and exploring the mode of action of therapies [16, 20, 21].
The emerging understanding of the immunopathology of MS reveals that T‐cell effector responses are shaped by complex bidirectional interactions with other immune cells, especially B and myeloid linage cells [22, 23]. Selective targeting of certain T‐cell populations has been tested in clinical trials, but not taken further in clinical development; see, for example, [24]. In contrast, less specific targeting of T‐ and/or B‐cell populations using monoclonal antibodies or lymphocyte‐specific cytostatics is the basis of several currently used DMTs. A notable observation in this context is that B cells seem to play a non‐redundant role in this immune communication loop [25]. The relevance of this notion is also underscored by the striking efficacy of B‐cell‐deleting therapies [26, 27, 28].
In order to gain pathogenic potential, disease‐driving cells must be recruited from lymphoid organs to blood, circulate to brain vessels and migrate across the blood–brain barrier (BBB), which under physiological conditions impedes macromolecules and cells from accessing the brain tissue [29]. However, to engage with brain vessel endothelia activated immune cells upregulate a molecular machinery of chemokine receptors and adhesion molecules [30, 31]. The fact that therapeutic targeting of immune cell trafficking at different levels has proven fruitful in MS further supports the notion that certain immune cell populations are responsible for clinical relapses and the formation of focal inflammatory brain lesions, plaques, which represent the hallmark sign of MS (Fig. 2). Even if inflammation traditionally has been thought to target mainly the brain white matter, increasingly sensitive MRI techniques and histopathological studies also reveal extensive brain cortical engagement [32, 33, 34]. Plaques are typically centred around post‐capillary venules and involve the breakdown of BBB integrity, with dense infiltration of lymphocytes, mainly CD8+ T cells, activated microglia and macrophages containing myelin debris and reactive, scar‐forming astrocytes [35, 36, 37]. Plaques can be classified into active, chronic active and chronic inactive, based on their histopathological appearance, suggesting that the acute inflammatory phase is followed by demyelination, oligodendrocyte death, reactive gliosis and degeneration of axons [35].
The exact antigenic targets in MS are still not known. EAE studies have shown that immunization with myelin proteins such as myelin basic protein (MBP), proteolipid protein and myelin oligodendrocyte glycoprotein can elicit MS‐like disease [16]. MS patients have been shown to display functionally distinct clones of T cells recognizing myelin epitopes, even if such cells can be detected also in controls [38]. Major efforts have been invested in trying to translate immune tolerance studies into effective MS treatments, but so far with limited success. However, an experimental compound composed of random oligomers of amino acids enriched in myelin originally developed to induce EAE in animals contrary to expectations was shown to reduce disease activity in MS [39]. This compound, glatiramer acetate, is believed to shift T‐cell responses from Th1 and Th2 and to induce more neuroprotective facets of inflammation [40].
Disease mechanism in later disease phases
Aside of cellular immune interactions in the periphery that cause relapses and formation of focal brain lesions, chronic inflammation within the CNS tissue is thought to contribute to the progressive facet of MS [41], (Fig. 3). The notion of ‘trapped inflammation’, which at least partly is de‐coupled from adaptive immune responses in the periphery, has gained increasing interest [37]. This may also explain the poor effect in progressive stages of MS of currently available DMTs, which typically exert a systemic anti‐inflammatory action [42, 43]. Supporting evidence includes histopathological observations of demyelinating cortical injury, with evidence of a gradually increasing neuronal and myelin loss, as well as activated microglial in tissue closer to the ventricular system, perhaps indicative of presence in CSF of toxic compounds [44, 45]. Such cortical injuries have also been spatially associated with areas of meningeal inflammation and follicular structures composed of B‐cell infiltrates [46]. Another feature of ‘trapped inflammation’ is the slowly expanding lesions or ‘rim lesions’, that is slowly expanding chronic lesions with an active border zone characterized by activated microglia and increased densities of transected axons [47].
Fig. 3.

Periodic entry of encephalitogenic T and B cells into the brain tissue is believed to cause acute focal lesions, which typically are centred around veins in the white matter. However, with more advanced disease other types of pathology become more prominent. These include slowly expanding chronic lesions that feature an active border zone, diffuse damage to myelin and axonal connections in the so called normal appearing white matter and accumulation of follicular structures in the meninges with signs of subpial demyelination. Whilst acute lesions explain occurrence of bouts, these other pathological mechanisms are thought to contribute to progressive worsening in disability. Current MS therapies reduce the risk of acute lesions and relapses, whilst their effect on other disease processes is more uncertain.
So called ‘shadow plaques’ are chronic lesions where a variable degree of remyelination has taken place [48, 49]. Evidence of such reparative mechanism even in the adult represents a foundation for therapies aiming to promote remyelination. The capacity for remyelination is highest in younger persons and becomes reduced with ageing and conversion into a progressive disease stage, where advanced molecular studies have provided insights into characteristics of the oligodendrocyte pool in patients with MS and controls, respectively [50, 51]. In order to address remyelination as a therapeutic target, encouraging progress has been made in imaging techniques that allow brain myelin fractions to be determined; see, for example, [52] (Fig. 2).
An important requisite for remyelination is the preservation of axonal integrity. In fact, amongst different neuropathological MS features, neuro‐axonal loss is of particular importance, since it likely represents the closest pathological proxy of accumulation of irreversible clinical disability. An enigmatic pathological finding in MS is the occurrence of widespread involvement of the so called normal appearing white matter, that is the much larger volume of tissue outside of focal lesions [53, 54]. This features transected axons and reduced axonal densities, diffuse loss of myelinating cells, sparse immune cell infiltrates and widespread activation microglia and astrocytes. It can be detected already in early stages of RRMS, but becomes more prominent in later disease stages, thus representing another mechanism that may explain the transition from RRMS to SPMS. Interestingly, the extent of such damage is not closely correlated with numbers, volumes or appearance of focal lesions, suggesting it to be regulated differently [54]. Underlying mechanisms may include energy depletion due to mitochondrial dysfunction, increased oxidative and excitotoxic stress, accumulation of iron, loss of trophic support from oligodendrocytes, complement activation and negative effects mediated by chronically activated microglia and astrocytes [41]. It may be speculated if this represents a correlate of a feature recently described in RRMS, that is progression independent of relapse activity [55]. Thus, disability accumulation in the setting of a clinical trial with a B‐cell‐depleting agent mostly occurred independent of relapse (and new focal lesion) activity, unlike earlier assumptions that such worsening occurs in a step‐wise fashion associated with relapses [1].
Collectively, features that have been associated with transition to progressive disease include exhaustion of remyelinating potential, diffuse engagement of the normal appearing grey and white matter, compartmentalized inflammation within the CNS that includes clones of CD8+ T and B/plasma cells, and more traditional neurodegenerative disease processes (Fig. 3). Importantly, however, these processes likely commence well before the clinical presentation of a progressive disease course and development of more sensitive diagnostic techniques will be needed to explore if these disease processes can be targeted with novel therapies [12, 14].
Treatment of MS – current and emerging drug classes
In this section, an overview of approved DMTs and drug candidates that have been tested in clinical trials for MS is presented and stratified into drug/target classes (Fig. 4). The basis for the selection was a search in ClinicalTrials.gov with the search string ‘multiple sclerosis’, which identified 2153 studies as of 6 July 2020, with the addition of relevant additional studies not listed in this database through reference searches. Studies not reported as peer‐reviewed scientific reports are referred to their ClinicalTrials.gov identifier.
Fig. 4.
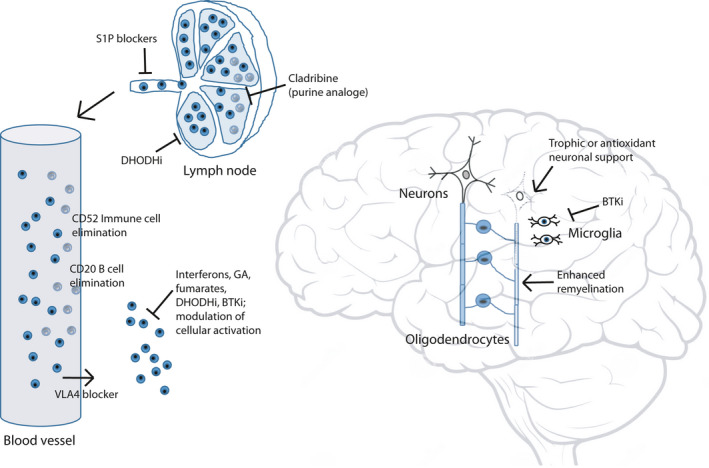
Encephalitogenic T and B cells can be targeted in lymphoid organs or the circulation. Elimination of certain lymphocyte populations is the therapeutic action of cladribine, and anti‐CD52 (both T and B cells) and anti‐CD20 (mainly B cells) monoclonals, whilst DHODHi limit the proliferation of activated lymphocytes. S1P and VLA‐4 blockers on the other hand interfere with the recruitment of lymphocytes to the circulation and brain tissue, respectively. A larger therapeutic group instead targets the activation of immune cells by, for example, affecting expression of MHC molecules and cytokines or interfering with intracellular activation pathways. MS therapies under clinical development include drugs that aim to provide enhanced remyelination or neuroprotection, or dampen the activity of microglia. Abbreviations; BTKi, Bruton's tyrosine kinase inhibitor; DHODHi, dihydroorotate dehydrogenase inhibitor; GA, glatiramer acetate; S1P, sphingosine 1‐phosphate; VLA‐4, very late antigen‐4.
Modulators of inflammatory mediators
Interferons
Based on the assumption that MS had a viral origin, several studies tested the effects of interferons (IFNs) in MS in the late 1970s and 1980s [56]. Most of these studies applied different preparations of type II (α/β) IFNs, whilst one study reported increased relapse frequency in patients treated with IFN‐γ [57]. Further development led to the first breakthrough in the treatment of RRMS with the approval of (IFN‐β‐1b, Betaseron/Betaferon, now also generic Extavia) in 1993, soon followed by two IFN‐β‐1a preparations (Avonex and Rebif). Controlled trials have shown a reduction in annualized relapse rate (ARR) in the range of 30–40% compared with placebo and a relatively innocuous safety profile, even if tolerability is negatively affected by influenza‐like side effects and the need for self‐administered injections [58, 59, 60]. More recently, a pegylated version of IFN‐β‐1a with twice‐monthly subcutaneous injections was approved (Plegridy), based on a phase III study showing a 36% reduction in ARR compared with placebo [61].
Cytokine and chemokine modulators
In light of increasing data on the inflammatory environment in MS, a number of different attempts at modulating cytokine and chemokine responses have been made, several of which not progressing to interpretable or published results, for example, PF‐06342674 (NCT02045732), an interleukin‐7 receptor inhibitor, VAY736 (NCT02038049), a B‐cell activating factor receptor inhibitor, and a study with low‐dose interleukin‐2 (IL‐2; NCT02424396). This is also the case for modulation of C‐C chemokine receptor type 2 (CCR2) and chemokine (C‐C motif) ligand 2 signalling, which has been attributed with a pathogenic role for recruitment of monocytes and T cells in experimental models of MS using different approaches [62]. Thus, a phase II trial with plozalizumab, an antagonist of CCR2 with evidence of ameliorated synovial inflammation in rheumatoid arthritis [63], has been conducted, but not reported (NCT01199640).
Blocking of the IL‐2 receptor with monthly subcutaneous 150 mg daclizumab (Zinbryta), a drug previously approved for prevention of transplant rejections, was shown to reduce ARR with 45% compared with weekly IFN‐β1a in a phase III study [64]. The safety profile showed higher incidences of infections, cutaneous events and elevated liver enzymes compared to IFN‐β1a, but daclizumab was nevertheless approved in 2016 for RRMS both in the United States and EU. However, in 2018 the marketing authorization was withdrawn due to reports of serious and sometimes fatal immune reactions affecting different organs, also including events of severe CNS inflammation occurring as a rebound after stopping treatment [65].
Expression by encephalitogenic T cells of both GM‐CSF and IL‐17 has been implicated in MS; see, for example, [19, 66]. A phase 1b safety trial with a blocker of GM‐CSF (otilimab) has been conducted in RRMS [67]. A larger phase II trial with (secukinumab), a blocker of IL‐17A, did not meet its primary end point, but displayed a moderate degree of reduction of MRI lesion activity [68]. In contrast, ustekinumab, a monoclonal binding the p40 subunit of the IL‐12/IL‐23 receptor and approved for psoriasis and inflammatory bowel disease, did not show efficacy on MRI‐based parameters [69]. Similar results were obtained with briakinumab, a competing IL‐12/IL‐23 blocker [70]. Furthermore, tabalumab, a blocker of BAFF, did not show evidence of beneficial therapeutic effects in a phase II trial, as referred in Baker et al. [71].
Hence, in contrast to autoimmune diseases such as inflammatory bowel disease, psoriasis and rheumatoid arthritis, inhibition of individual cytokines in MS has not proven to be a fruitful strategy, daclizumab being an exception, but also having an intolerable safety profile. On the contrary, there is some evidence that such modulation may increase inflammatory disease activity. Thus, an early phase II trial with lenercept, a blocker of TNFα, reported increased relapse activity [72]. This is also supported by real‐world data reporting an increased risk of neuroinflammatory events in patients treated with TNFα blockers for other indications; see, for example, [73]. Interestingly, studies in MS with atacicept, a recombinant fusion protein targeting B‐cell‐activating B‐lymphocyte stimulator (BLyS) and A proliferation‐inducing ligand (APRIL), also found evidence of a disease‐activating effect [74, 75]. Collectively, these observations support the notion of differences in disease mechanisms between MS and other autoimmune diseases that translate into clinically significant diversity in therapeutic responses.
Immune cell migration inhibitors
Non‐selective sphingosine 1‐phosphate modulators
Two important drug classes in MS are based on interference with recruitment of encephalitogenic cells to the CNS. Fingolimod (Gilenya) is a small molecule drug that was developed from myriocin, a fungus‐derived compound, and found to interrupt sphingosine 1‐phosphate (S1P) signalling, in turn inhibiting egress of immune cells from lymph nodes [76]. Fingolimod first was found to have a modest effect on transplant rejection, but subsequently completed a successful trial programme in RRMS. In two phase III studies, fingolimod was compared with placebo, showing a 48% and 60% reduced ARR, respectively, and in a third study reduced ARR with 39% compared to weekly IFN‐β‐1a [77, 78, 79]. A more recent phase III trial in paediatric MS showed a reduction in ARR of 82% with 0.5 mg fingolimod (0.25 mg with body weight ≤ 40 kg) compared with weekly IFN‐β‐1a [80]. In contrast, the active arm was not superior to placebo in a phase III trial in PPMS [42]. In the EU, Gilenya is authorized for use in adults and children aged ≥ 10 years with highly active RRMS and in the United States for relapsing forms of MS (RMS; includes CIS, RRMS and SPMS with relapses) in the same patient groups (Table 1). The safety profile of fingolimod includes effects on heart conduction, elevated liver enzymes and increased risks of macular oedema and certain infections, such as herpes viruses. Rare opportunistic infections, for example, cryptococcal meningitis and progressive multifocal leukoencephalopathy (PML) caused by JC virus, have been observed, and potentially, there is an increased risk of skin cancers.
Table 1.
Currently approved MS disease‐modulatory therapies
| DMT class | Substance name | Brand name | Administration route/frequency | Type | Year of approval EU/US | Indication | ARR in phase III trials | References | Major safety issues highlighted in label | |
|---|---|---|---|---|---|---|---|---|---|---|
| EU | US | |||||||||
| Interferons | Interferon β‐1a | Rebif | s.c., 3 × week (22 or 44 μg) | Recombinant protein | 1998/1996 | RRMS, CIS (only 44 μg) | RMS | −32% vs PBO | [60] | Depression, hepatic injury |
| Avonex | i.m. 1 × week | Recombinant protein | 1997/1996 | RRMS, CIS | RMS, CIS | −32% vs PBO | [59] | |||
| Pegylated interferon β‐1a | Plegridy | s.c. 2 × month | Recombinant protein | 2014/2014 | RRMS | RMS | −36% vs PBO | [61] | Depression, hepatic injury | |
| Interferon β‐1b | Betaferon/Betaseron | s.c., every other day | Recombinant protein | 1995/1993 | RRMS, CIS | RMS, CIS | −34% vs PBO | [58] | Depression, hepatic injury | |
| Extavia (generic) | RMS, CIS | N/A | ||||||||
| Glatiramer acetate | Glatiramer acetate | Copaxone | s.c., 1 × day 20 mg or 3 × week 40 mg | Amino acid oligomers | 2004/1996 | RRMS a | RRMS, CIS | −30% vs PBO | [138] | Injection‐site lipoatrophy |
| Glatiramer Mylan | s.c., 1 × day 20 mg or 3 × week 40 mg | RMS | n.s vs Copaxone | [140] | ||||||
| Glatopa | s.c., 1 × day 20 mg or 3 × week 40 mg | RMS | ||||||||
| Oral immunomodulators | Dimethyl fumarate | Tecfidera | oral, 2 × day | Fumarate/Nrf2 agonist | 2014/2013 | RRMS | RMS | −45/−53% vs PBO | [127, 128] | Flushing, gastrointestinal disturbances, lymphopenia, (PML) |
| Diroximel fumarate | Vumerity | oral, 2 × day | Fumarate/Nrf2 agonist | /2019 | RMS, CIS | N/A | [131] | Lymphopenia, (PML) | ||
| Monomethyl fumarate | Bafiertam | oral, 2 × day | Fumarate/Nrf2 agonist | /2020 | RMS, CIS | N/A | [133] | Lymphopenia, (PML) | ||
| Teriflunomide | Aubagio | oral, 1 × day | DHODH inhibitor | 2013/2012 | RRMS | RMS | −31/−36% vs PBO b | [121, 122] | Hepatic injury, alopecia, nausea, teratogenicity (polyneuropathy) | |
| Cell migration modulators | Fingolimod | Gilenya | oral, 1 × day | S1P inhibitor | 2011/2010 | POMS, RRMS, highly active or second line | POMS, RMS |
−48/−60% vs PBO −39% vs interferon |
[77, 78, 79]] | Reduced heart rate, infections, hepatic injury, macular oedema, teratogenicity, (PML) |
| Ozanimod | Zeposia | oral, 1 × day | S1P inhibitor | 2020/2020 | RRMS | RMS, CIS | −39/−49% vs interferon | [82, 83] | Reduced heart rate, infections, hepatic injury, macular oedema, teratogenicity, (PML) | |
| Siponimod | Mayzent | oral, 1 × day | S1P inhibitor | 2020/2019 | Active SPMS | RMS | −21% vs PBO (SPMS) c | [84] | Reduced heart rate, infections, hepatic injury, macular oedema, teratogenicity, (PML) | |
| Natalizumab | Tysabri | i.v., 1 × month | Monoclonal anti‐VLA4 | 2006/2004 | RRMS, highly active or second line | RMS, second line | −69% vs PBO | [87] | Infections, PML | |
| Cell depleting | Alemtuzumab | Lemtrada | i.v., induction | Monoclonal anti‐CD52 | 2013/2014 | RRMS, highly active or second line | RMS, third line | −50/−54% vs interferon | [94, 95] | Autoimmune reactions, serious infusion reactions, cerebrovascular disease |
| Cladribine | Mavenclad | oral, induction | Purine analog | 2017/2019 | RRMS, highly active | RMS, second line | −58% vs PBO | [100] | Infections, cancer, teratogenicity | |
| Mitoxantrone | Novantrone | i.v., induction | Cytotoxic chemotherapy | 1998/1998 | RMS, highly active and worsening | Relapsing SPMS, worsening RRMS | N/A | [104, 105] | Cardiotoxicity, cancer, teratogenicity | |
| Ocrelizumab | Ocrevus | i.v., 2x year | Monoclonal anti‐CD20 | 2018/2018/2020 | RRMS, PPMS | RMS, PPMS |
−45% vs interferon −24% vs PBO (PPMS) c |
[26, 27] | Infections, cancer | |
| Ofatumumab | Kesimpta | s.c., 1 × month | Monoclonal anti‐CD20 | RMS, CIS | −50/−60% vs teriflunomide | [28] | Infections | |||
Abbreviations: ARR, annualized relapse rate; CIS, clinically isolated syndrome (one episode of demyelinating event with high risk of converting to multiple sclerosis); DHODH, dihydroorotate dehydrogenase; DMT, disease‐modulatory therapy; i.m., intramuscular injection; i.v., intravenous infusion; n.s, non‐significant; Nrf2, nuclear factor erythroid 2‐related factor 2; PBO, placebo; PML, progressive multifocal leukoencephalopathy; POMS, paediatric onset MS; PPMS, primary progressive multiple sclerosis; RMS, relapsing forms of multiple sclerosis; RRMS, relapsing–remitting multiple sclerosis; s.c., subcutaneous injection; S1P, sphingosine‐1‐phosphate; SPMS, secondary progressive multiple sclerosis; VLA4, very late antigen 4.
Nationally authorized medicinal product within the EU.
14 mg dose.
Reduction in hazard ratio for 3 months confirmed disability progression.
Selective sphingosine 1‐phosphate modulators
The S1P family of receptors comprises five subtypes, where fingolimod is a non‐selective modulator of S1P receptors 1, 3, 4 and 5, reviewed in [81]. S1P receptor 1 is expressed on lymphocytes, and second‐generation S1P modulators are more selective for this subtype. Oral once‐daily 0.92 mg ozanimod (Zeposia), a S1P receptor 1 blocker with a weaker effect on the type 3 receptor subtype, was recently approved for RRMS in the EU and for RMS in the United States, based on two phase III trials showing a 39% and 49% reduced ARR, respectively, compared with weekly IFN‐β‐1a [82, 83]. In contrast to fingolimod, no clinically significant bradycardia or second‐ or third‐degree atrioventricular blocks were reported in the trial programme. Siponimod (Mayzent), a selective modulator of S1P receptors 1 and 5, was first tested in a phase II trial in RRMS, followed by a placebo‐controlled phase III trial in SPMS showing a 21 % reduced risk of 3 months confirmed disability progression [84]. In the United States, Mayzent has the same label as Zeposia, whilst it is only approved for active forms of SPMS in the EU (Table 1). Similar to Gilenya, Mayzent was associated with higher frequencies of elevated liver enzymes, bradycardia and bradyarrhythmia at treatment initiation, macular oedema, hypertension and varicella zoster reactivation. Ponesimod, a selective S1P receptor 1 modulator, recently completed a phase III trial showing a 30% reduced ARR compared to teriflunomide [85]. Two further S1P modulators, ceralifimod (NCT01081782) and amiselimod, have been tested in phase II trials, but not progressed into phase III testing [86]. Apart from a more selective targeting of S1P receptor subtypes, a further advantage with second‐generation S1P modulators is a significantly shorter half‐life [81]. On the other hand, S1P receptor 1‐independent effects of fingolimod have been attributed to its effect on CNS cells such as astrocytes in experimental models [81].
Adhesion molecule modulators
A major breakthrough in the treatment of RRMS came in the mid‐2000s with the approval of natalizumab (Tysabri), a monoclonal binding the α4‐integrin of the very late antigen‐4 (VLA‐4) protein, in turn effectively abrogating the transmigration of immune cells over the BBB. In a phase III trial, a monthly infusion of 300 mg natalizumab reduced ARR with 69% compared with placebo [87]. In contrast, natalizumab was not superior to placebo for reducing disability worsening in SPMS [43]. In both the EU and United States, Tysabri is indicated for highly active RRMS or as a second‐line DMT (Table 1). A major safety concern with natalizumab is the increased risk of PML. This risk, however, can be stratified based on presence of antibodies to JC virus in blood, which are present in a majority in most patient populations, in turn restricting the usefulness of natalizumab [88, 89]. Additional integrin modulators include vatelizumab, a monoclonal blocker of α2‐integrin, as well as firategrast and zaurategrast, two oral α4β‐integrin modulators. Whilst vatelizumab did not meet its primary imaging outcome in a RRMS trial (NCT02222948), it was shown to increase the frequency of regulatory T cells [90]. A placebo‐controlled phase II trial of firategrast at different doses demonstrated a reduction of new contrast‐enhancing MRI lesions with higher doses [91]. In contrast, development of zaurategrast was terminated after unsatisfactory interim results of phase II trial (NCT00484536), even if modulation of immune cell populations in blood was shown in a biomarker study [92].
Cell‐depleting/induction therapies
Anti‐CD52 monoclonals
Both subsets of T and B cells have been implicated in the pathogenesis of MS, providing a rational for therapeutic targeting. The CD52 antigen, targeted by the monoclonal alemtuzumab (Lemtrada), is present on a variety of immune cells, including memory T and B cells [93]. In the Care‐MS I and II trials, an induction cycle of five repeated infusions of 12 mg alemtuzumab at baseline followed by three infusions after one year was compared with continuous subcutaneous IFN‐β‐1a over two years, showing a 54% and 50% reduction in ARR, respectively [94, 95]. A minority of patients required an additional treatment cycle due to continued disease activity, and long‐term follow‐up of the Care‐MS cohorts has shown long‐term durability of treatment effects, including low brain atrophy rates [96, 97]. Unfortunately, important safety concerns have arisen, which include a high rate of non‐MS autoimmune conditions, composed mostly thyroid disorders, as well as acute infusion‐related cardiovascular events [98, 99]. Given the need for careful patient selection in order to maintain an acceptable benefit‐risk, the label for alemtuzumab has recently been revised (Table 1). A modified anti‐CD52 monoclonal, GZ402668, has undergone early testing in MS with the objective to reduce infusion‐related reactions (NCT02977533).
Cladribine
An alternative induction type of DMT is cladribine (Mavenclad), a purine antimetabolite with selectivity for lymphocytes lacking a rescue pathway for purine synthesis. A phase III trial in RRMS showed that a body weight‐adjusted dose cycle of oral 3.5 mg tablets given at baseline and after one year reduced ARR with 58% compared with placebo [100]. In a second trial, cladribine significantly reduced the risk of conversion to definite MS in patients with CIS compared with placebo [101]. Adverse events include lymphopenia and herpes reactivation, but there was also an imbalance in the number of detected malignancies, however, not exceeding the expected rate during long‐term follow‐up [102]. In the EU, Mavenclad is indicated for patients with highly active RRMS, whilst the US label recommends it to be used in patients with an inadequate response to a prior DMT (Table 1).
Mitoxantrone
The first induction therapy to be approved in MS was mitoxantrone (Novantrone), a DNA‐intercalating agent used in cancer treatment, which was approved for relapsing SPMS and disability worsening in RRMS based on two smaller trials [103, 104]. Due to cardiotoxicity and increased rates of haematological malignancies, mitoxantrone hardly has any place in the treatment algorithm of MS today [105, 106].
CD19/20 monoclonals
Ocrelizumab (Ocrevus), an anti‐CD20 monoclonal depleting B cells, became the first DMT to be approved for PPMS, in addition to RRMS (Table 1). In the two phase III Opera trials for RRMS, 600 mg ocrelizumab every 6 months reduced ARR with 45% compared to subcutaneous IFN‐β‐1a, whilst, in the Oratorio trial in PPMS, the proportion of patients with 3 months confirmed disability progression was reduced with 24% compared with placebo [26, 27]. Rituximab, an older, related monoclonal approved for rheumatic and haematological conditions recognizing the same antigen on the CD20 protein, had previously been tested in a placebo‐controlled phase II trial in RRMS and a phase II/III trial in PPMS [107, 108]. The latter study indicated a similar degree of reduction in progression rate, however, not reaching the level of statistical significance. Results from the Asclepios I and II phase III trials with a third anti‐CD20 monoclonal, ofatumumab, were recently reported, demonstrating that 20 mg of ofatumumab administered weekly by self‐administered subcutaneous injections reduced ARR with 50% and 60%, respectively, compared with teriflunomide [28]. Apart from a different mode of administration, ofatumumab also recognizes another epitope on the CD20 protein compared to ocrelizumab and rituximab. Ublituximab is a novel anti‐CD20 monoclonal that is glycoengineered for enhanced B‐cell targeting through antibody‐dependent cellular cytotoxicity, which may allow lowering of doses and shortening infusion times. In a phase II trial, a dose‐escalating protocol of ublituximab resulted in profound B‐cell depletion and abrogation of MRI activity [109]. Concerns with anti‐CD20 therapies mainly regard infusion reactions, hypogammaglobulinemia and increased risks of infections; see, for example, [110]. Apart from B cells anti‐CD19, antibodies also target plasmablasts and certain plasma cell populations. Inebilizumab, an anti‐CD19 monoclonal antibody, has undergone early testing in RRMS, but is now being developed for neuromyelitis optica and myasthenia gravis, two autoantibody‐mediated inflammatory neurological diseases [111, 112].
Autologous haematopoietic stem cell transplantation
The use of autologous haematopoietic stem cell transplantation (AHSCT) in MS has emerged as a treatment option almost entirely based on empiric evidence from observational studies [113, 114]. In most current AHSCT protocols, haematopoietic stem cells are first collected, after which a conditioning regime is used to deplete bone marrow cells. After wash out of cytostatic drugs, autologous stem cells are given back, usually together with anti‐thymocyte globulins to kill any remaining T cells. It is not until recently a first randomized trial comparing AHSCT with standard care was completed in 110 patients, showing a hazard ratio of 0.07 for disability progression in the AHSCT arm [115]. A major concern has been the risks of adverse events, in particular treatment‐related mortality, which has been estimated at 2.1% in a meta‐analysis [113]. In contrast, no treatment‐related mortality was detected in a recent nationwide registry‐linkage study of a contemporary cohort, however, finding a long‐lasting effect on infection risk [116]. In current guidelines, AHSCT is considered an experimental treatment option for younger patients with high inflammatory activity, usually in context of an insufficient response to regular high effective DMTs, and a limited accumulated level of disability [117].
Targeting of EBV‐infected cells
An advanced cell therapy for elimination of EBV‐infected cells is ATA188, which comprise HLA‐matched donor T cells that have been sensitized against EBV antigens and given to recipients. Preliminary safety and efficacy data from a small trial in progressive MS were recently presented [118], in turn corroborating safety data from a previous small trial [119].
Immunomodulators with intracellular mechanisms of action
Dihydroorotate dehydrogenase inhibitors
Dihydroorotate dehydrogenase inhibitors interfere in the pyrimidine biosynthesis pathway, reducing the proliferation of T lymphocytes, but also exert more complex immunomodulatory effects, including modulation of cytokine production [120]. Teriflunomide (Aubagio) is the active metabolite of leflunomide, a drug used in rheumatoid and psoriatic arthritis, where a 14 mg tablet once daily is approved for RRMS and a 7 or 14 mg tablet for RMS in the EU and United States, respectively. Approval was based on two phase III trials comparing 7 and 14 mg teriflunomide with placebo, showing a 31% (both doses) and a 22/36% reduction in ARR, respectively [121, 122]. In a small additional phase III trial, RRMS patients were randomized to 7 or 14 mg teriflunomide, or subcutaneous IFN‐β‐1a, finding a similar ARR between the 14 mg group and the comparator, but a higher ARR (+86%) in the 7 mg arm [123]. The time to treatment failure, which was the primary outcome, did not differ between treatment arms. Teriflunomide has a complex pharmcokinetic profile that includes enterohepatic re‐uptake, resulting in clinically significant plasma concentrations remaining many months after stopping drug intake, unless applying rapid elimination by administration of cholestyramine or activated charcoal. Due to suspected teratogenicity, it should be used with care in fertile women. A second‐generation dihydroorotate dehydrogenase inhibitor, vidofludimus, has been tested in pre‐clinical MS models and a phase I study, and recently reported phase II study outcomes [124, 125].
Fumarates
Fumarates derive from fumaric acid, which occur naturally in certain mushrooms, lichen and moss and are named after the earth smoke plant (Fumaria officinalis). Different preparations of fumarates have been used to treat psoriasis since long, whilst not until much later an effect on MS disease activity was discovered [126]. A special oral preparation of dimethyl fumarate (DMF; Tecfidera) has been approved for RRMS and RMS in the EU and United States, respectively (Table 1). In two placebo‐controlled phase III trials, 240 mg of DMF taken twice daily reduced ARR with 45 and 53%, respectively [127, 128]. The mechanism of action of DMF includes effects on nuclear factor erythroid 2‐related factor 2, a transcription factor involved in cellular redox reactions, and an increase in oxidative responses in myeloid cells, in turn downregulating activated lymphocytes [66, 129]. Flushing, abdominal pain, diarrhoea and nausea are common and have a negative effect on tolerability. In addition, rare cases of PML cases have been recorded, often, but not always associated with concomitant lymphopenia [130]. A second‐generation fumarate, diroximel fumarate (Vumerity), was recently approved in the United States, based on comparative studies with DMF demonstrating less gastrointestinal side effects, whilst still yielding similar concentrations of the active metabolite monomethyl fumarate (MMF) [131, 132]. Similarly, a delayed‐release preparation of MMF (Bafiertam) has been launched in the United States based on bioavailability and gastrointestinal tolerability studies in healthy subjects [133].
Bruton's tyrosine kinase inhibitors
Inhibitors of Bruton's tyrosine kinase (BTK) represent an emerging drug class in MS. BTK is involved in intracellular signalling downstream of cell surface receptors in both lymphocytes (including the B cell and Fc‐γ receptors) and cells of the innate immune system, which may allow to modulate immune reactions both in the periphery and in the brain. So far, evobrutinib has completed a phase II study in RRMS, demonstrating a superior effect on imaging, but not clinical outcomes compared with placebo and with phase III testing ongoing [134]. Tolebrutinib recently completed a dose‐finding study and will enter phase III testing soon (NCT03889639). Fenebrutinib has been tested in other autoimmune conditions and will now directly enter phase III trials in RRMS and PPMS [135]. Finally, BIIB091 has completed a phase I testing [136].
Modulation of costimulatory activation
The effect of modulating costimulatory activation of T cells has been tested in a placebo‐controlled phase II trial in RRMS with the CTLA4‐Ig fusion protein abatacept. The study was terminated prematurely due to slow recruitment, but did not suggest a beneficial effect on imaging outcomes [137].
Tolerization therapies
Glatiramer acetate
Tolerization therapies have a long history in MS clinical research, even if the exact antigens still are unknown. Based on the unexpected finding that a random mixture of four amino acids enriched in myelin ameliorated experimental MS‐like disease in animals, glatiramer acetate (GA; Copaxone) became the second drug class to be approved for MS after interferons. The approval of GA was based on a small placebo‐controlled phase III trial in RRMS, later supported by a larger study in CIS and a trial comparing two different GA formulations against placebo [138, 139, 140]. Further, large‐scale head‐to‐head studies comparing interferons with GA have shown a similar effect on clinical outcomes [141, 142]. Initially given as a 20 mg daily subcutaneous injection, a three times weekly administration of 40 mg GA was found to reduce the frequency of injection‐site reactions whilst retaining a similar effect on ARR (−34% compared to placebo) [143, 144]. The exact mechanism of action of GA is not known, but is thought to involve a shift towards a more neuroprotective immune response [40]. Apart from tolerability issues relating to frequency of injections, the safety profile is innocuous.
Other tolerization therapies
More recent attempts at inducing immune tolerance for myelin antigens include ATX‐MS‐1467, which is a mixture of four immunodominant peptide fragments from MBP. In two smaller dose‐escalation trials, preliminary evidence of a beneficial effect on imaging outcomes was observed without major adverse events [145]. In contrast, trials with tiplimotide, an altered peptide ligand derived from MBP, had to be halted due to disease exacerbation likely linked to encephalitogenic potential of the administered peptide [146]. A third example is a relatively large placebo‐controlled phase II trial with a DNA vaccine encoding MBP, which only showed a trend towards a beneficial effect in one of the dose groups [147]. Recently, preliminary safety results regarding an innovative approach of linking myelin peptides to red blood cells have been reported [148].
Miscellaneous
Laquinimod is a quinoline derivative with similarities to linomide, for which late stage trials for RRMS were interrupted due to an emerging safety signal [149]. Laquinimod is believed to modulate innate immune responses including microglia and was shown to reduce ARR modestly, but with a relatively stronger effect on risk of disability progression in a first phase III trial in RRMS, however, not replicated by a subsequent trial [150, 151]. Similarly, minocycline, an antibiotic with additional inhibitory effects on microglial activation, displayed a lowered risk of conversion from CIS to definite MS compared with placebo at 6 but not 24 months and was not superior to placebo when added to subcutaneous IFN‐β‐1a [152, 153]. Rolipram, originally developed as an anti‐depressant, is an inhibitor of phosphodiesterase‐4 inhibitor, which has been shown to suppress Th1 autoimmunity in EAE. However, a clinical trial was prematurely terminated due to poor tolerability and a suspected increase in disease activity [154]. Polyphenon E, a green tea extract containing high levels of antioxidant epigallocatechin‐gallate, did not show evidence of a beneficial effect, whilst also being hepatotoxic [155]. Based on accumulating evidence of an important role of viruses, in particular EBV, in the pathogenesis of MS, raltegravir, an anti‐retroviral drug, was tested in a small phase II trial, however, not providing evidence of an effect [156]. In a larger phase II trial temelimab, a monoclonal binding the human endogenous retrovirus‐W protein, which has been associated with nerve and myelin damage in MS brain tissue, did not achieve its primary outcome, however, with some evidence of tissue protective effects on MRI‐based measures [157, 158]. In an ongoing phase II study, temelimab is now tested as add‐on to rituximab in relapsing forms of MS (NCT04480307).
Regenerative and neuroprotective strategies
Oral small molecules
An expanding number of substances have been tested or are currently undergoing testing for improving remyelination or providing neuroprotection. Clemastine, a first‐generation anti‐histaminic with anti‐muscarinic properties being identified as a remyelination promoting substance in a high‐throughput screen, was shown to improve nerve conduction in MS‐related chronic optic neuropathy in a phase II trial [159, 160]. Additional studies are ongoing, including a multiarm study comprising also pioglitazone, dantrolene and hydroxychloroquine (NCT03109288, NCT02521311). Similarly, a small but positive effect on remyelination was shown with a brain penetrant histamine H3 receptor blocker, GSK239512, in a phase II study [161]. In contrast, a multiarm phase II trial in SPMS testing three different drugs with potential neuroprotective effects, amiloride, fluoxetine and riluzole, did not result in superior outcomes compared with placebo [162]. Ongoing studies include two trials examining the potential of nanocrystalline gold to treat remyelination failure and impaired neuronal redox state (NCT03993171, NCT03536559) [163].
Biologicals
Monoclonal antibodies blocking proteins that inhibit axonal growth and myelination include elezanumab recognizing repulsive guidance molecule A, opicinumab, directed at leucine‐rich repeat and immunoglobulin domain‐containing Nogo receptor‐interacting protein 1 (LINGO‐1), recombinant human immunoglobulin M22 for which the target antigen is not known, and VX15/2503, an anti‐semaphorin 4D antibody [164, 165, 166, 167, 168]. Except for opicinumab, trial data are still limited and results from the two opicinumab trials have been mixed. This is also the case for ozanezumab, which bind neurite outgrowth inhibitor (NOGO‐A), where trials in MS have been terminated and a phase II trial in amyotrophic lateral sclerosis was negative [169]. Similarly, erythropoietin, a growth factor with neuroprotective effects in experimental models, was not effective in a phase II progressive MS study [170].
Emerging treatment concepts
Since the launch of interferons more than 20 years ago, there has been an intense debate of the optimal use of MS DMTs. In part, this relates to the high costs that most DMTs command, but more recently also relating to the benefit‐risk with long‐term use. One of the early discussion points that still is pertinent is how to evaluate the clinical and cost effectiveness combined with the difficulty to extrapolate results from trials with relatively short duration; see, for example, [171]. It is clear that increasing disability affecting both physical and mental capacities exerts a heavy toll on affected individuals, but also leads to high societal costs [3]. Disability evolves over many years, and the typical two‐year duration of a phase III trial gives limited sensitivity to detect differences across treatment arms. More recent data from long‐term extension studies of phase III trials, however, demonstrate that differences in risk of disability progression between the more effective DMT and the comparator remain, suggesting that timing of start of effective therapy is key to minimize long‐term consequences of MS; see, for example, [172]. This notion is also supported by observations outside of controlled trials, where disability accumulation and conversion to progressive disease in real‐world cohorts of patients can be reduced with early start of highly effective DMTs, for example alemtuzumab, natalizumab and anti‐CD20 monoclonals [173, 174]. Another important aspect is that disability accumulation today mainly occurs independent of relapses, also implying that absence of relapse activity not necessarily can be interpreted as a sufficient treatment response [55]. This contrasts with current treatment guidelines and reimbursement policies, which normally recommend so called first‐line DMTs, such as interferons, GA, teriflunomide or DMF, unless there are signs of highly active disease [175, 176]. It is important to consider also the safety and tolerability profile of DMTs. So far, there are more limited data on comparative safety outcomes across different therapies, but it is evident that anti‐CD20 DMTs benefit from experiences with long‐term use in rheumatic disorders, as well as more recent studies in MS [110, 177]. Notably, rituximab, an anti‐CD20 DMT, has by far the lowest rate of treatment interruptions when compared with multiple currently available alternatives [178, 179]. Whilst rituximab is not formally approved for MS, it is increasingly used off‐label for this indication in some countries based on data from early controlled trials and larger observational studies; for references, see [180]. This also points towards another aspect of MS DMTs, namely that increasing competition not has resulted in lowered drug costs. On the contrary, US Medicaid MS DMT costs almost tripled from 2011 to 2017 as a result of a shift to more expensive drugs and lack of price reductions for DMTs going off patent [181]. Even in high‐income countries, such as the United States, patients may express greater concerns for out‐of‐pocket drug costs than drug efficacy or side effects [182]. Collectively, rituximab therefore combines a high degree of efficacy with a known safety profile, but also has a drastically lower cost than currently available alternatives.
It is still not clear to what degree DMTs primarily acting on the adaptive arm of the immune system also may affect brain inherent disease processes, which become more important in later disease stages. Ocrelizumab, an anti‐CD20 monocolnal, was the first DMT to be approved for PPMS, even if the relative effect compared with placebo was relatively modest [27]. In light of this, there are now attempts to address if higher doses may give a better therapeutic response [183], even if the lack of effect of earlier trials with intrathecal administration of anti‐CD20 drugs tempers expectations [184, 185, 186]. Another approach is to target simultaneously both the adaptive and innate immune systems, which is fuelling the hopes around brain penetrant inhibitors of BTK. So far, however, there is hardly any clinical evidence that modulation of microglia or other innate immune responses affects disease progression in MS. On the other hand, there are preliminary evidence for efficacy of certain remyelination‐enhancing agents [160, 161], even if the clinical relevance still remains uncertain. However, an increasing knowledge of the molecular regulation of oligodendrocyte differentiation and the myelination process likely will increase chances of developing reparative strategies [187]. Taken together, it may then be speculated if the future treatment of MS will combine a highly effective DMT acting on the adaptive immune system, such as anti‐CD20 or agents specifically targeting disease‐driving immune cells, with drugs that target disease processes in the brain tissue or enhance reparative responses.
Conflicts of interest
The author is or has been clinical investigator in trials with cladribine, daclizumab, dimethylfumarate, evobrutinib, fingolimod, firategrast, natalizumab, ocrelizumab, ofatumumab, opicinumab, ponesimod, siponimod, temelimab, teriflunomide and tolebrutinib; has served as steering committee member in trials with cladribine; received research support from Sanofi Genzyme, Merck KgaA, Novartis and UCB; and received personal fees for serving as chairman of DMC in studies with Parexel.
Piehl F (Karolinska Institutet; Stockholm Health Services, Stockholm, Sweden). Current and emerging disease‐modulatory therapies and treatment targets for multiple sclerosis (Review). J Intern Med. 2021;289:771–791. 10.1111/joim.13215
References
- 1. Compston A, Coles A. Multiple sclerosis. Lancet 2008; 372: 1502–17. [DOI] [PubMed] [Google Scholar]
- 2. Filippi M, Bar‐Or A, Piehl F et al. Multiple sclerosis . Nat Rev Dis Primers 2018; 4: 43. [DOI] [PubMed] [Google Scholar]
- 3. Kobelt G, Thompson A, Berg J et al. European Multiple Sclerosis P. New insights into the burden and costs of multiple sclerosis in Europe. Mult Scler 2017; 23: 1123–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Burkill S, Montgomery S, Hajiebrahimi M, Hillert J, Olsson T, Bahmanyar S. Mortality trends for multiple sclerosis patients in Sweden from 1968 to 2012. Neurology 2017; 89: 555–62. [DOI] [PubMed] [Google Scholar]
- 5. Koch‐Henriksen N, Sorensen PS. The changing demographic pattern of multiple sclerosis epidemiology. Lancet Neurol 2010; 9: 520–32. [DOI] [PubMed] [Google Scholar]
- 6. Olsson T, Barcellos LF, Alfredsson L. Interactions between genetic, lifestyle and environmental risk factors for multiple sclerosis. Nat Rev Neurol 2017; 13: 25–36. [DOI] [PubMed] [Google Scholar]
- 7. Jokubaitis VG, Butzkueven H. A genetic basis for multiple sclerosis severity: Red herring or real? Mol Cell Probes 2016; 30: 357–65. [DOI] [PubMed] [Google Scholar]
- 8. Thompson AJ, Banwell BL, Barkhof F et al. Diagnosis of multiple sclerosis: 2017 revisions of the McDonald criteria. Lancet Neurol 2018; 17: 162–73. [DOI] [PubMed] [Google Scholar]
- 9. Sastre‐Garriga J, Pareto D, Battaglini M et al. MAGNIMS consensus recommendations on the use of brain and spinal cord atrophy measures in clinical practice. Nat Rev Neurol 2020; 16: 171–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sormani MP, Bruzzi P. MRI lesions as a surrogate for relapses in multiple sclerosis: a meta‐analysis of randomised trials. Lancet Neurol 2013; 12: 669–76. [DOI] [PubMed] [Google Scholar]
- 11. Lublin FD, Reingold SC, Cohen JA et al. Defining the clinical course of multiple sclerosis: the 2013 revisions. Neurology 2014; 83: 278–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Pardini M, Cutter G, Sormani MP. Multiple sclerosis: clinical trial design 2019. Curr Opin Neurol 2019; 32: 358–64. [DOI] [PubMed] [Google Scholar]
- 13. Khalil M, Teunissen CE, Otto M et al. Neurofilaments as biomarkers in neurological disorders. Nat Rev Neurol 2018; 14: 577–89. [DOI] [PubMed] [Google Scholar]
- 14. Sormani MP, Haering DA, Kropshofer H et al. Blood neurofilament light as a potential endpoint in Phase 2 studies in MS. Ann Clin Transl Neurol 2019; 6: 1081–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Berger JR, Markowitz C. Deciding on the best multiple sclerosis therapy: tough choices. JAMA Neurol 2018; 75: 1461–2. [DOI] [PubMed] [Google Scholar]
- 16. Baxter AG. The origin and application of experimental autoimmune encephalomyelitis. Nat Rev Immunol 2007; 7: 904–12. [DOI] [PubMed] [Google Scholar]
- 17. Kebir H, Kreymborg K, Ifergan I et al. Human TH17 lymphocytes promote blood‐brain barrier disruption and central nervous system inflammation. Nat Med 2007; 13: 1173–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. van Langelaar J, van der Vuurst de Vries RM, Janssen M et al. T helper 17.1 cells associate with multiple sclerosis disease activity: perspectives for early intervention. Brain 2018; 141: 1334–49. [DOI] [PubMed] [Google Scholar]
- 19. Galli E, Hartmann FJ, Schreiner B et al. GM‐CSF and CXCR4 define a T helper cell signature in multiple sclerosis. Nat Med 2019; 25: 1290–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sriram S, Steiner I. Experimental allergic encephalomyelitis: a misleading model of multiple sclerosis. Ann Neurol 2005; 58: 939–45. [DOI] [PubMed] [Google Scholar]
- 21. Steinman L, Zamvil SS. How to successfully apply animal studies in experimental allergic encephalomyelitis to research on multiple sclerosis. Ann Neurol 2006; 60: 12–21. [DOI] [PubMed] [Google Scholar]
- 22. Codarri L, Greter M, Becher B. Communication between pathogenic T cells and myeloid cells in neuroinflammatory disease. Trends Immunol 2013; 34: 114–9. [DOI] [PubMed] [Google Scholar]
- 23. Li R, Patterson KR, Bar‐Or A. Reassessing B cell contributions in multiple sclerosis. Nat Immunol 2018; 19: 696–707. [DOI] [PubMed] [Google Scholar]
- 24. Olsson T, Edenius C, Ferm M et al. Depletion of Vbeta5.2/5.3 T cells with a humanized antibody in patients with multiple sclerosis. Eur J Neurol 2002; 9: 153–64. [DOI] [PubMed] [Google Scholar]
- 25. Jelcic I, Al Nimer F, Wang J et al. Memory B cells activate brain‐homing, autoreactive CD4(+) T cells in multiple sclerosis. Cell 2018; 175: 85–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hauser SL, Bar‐Or A, Comi G et al. Ocrelizumab versus Interferon Beta‐1a in Relapsing Multiple Sclerosis. N Engl J Med 2017; 376: 221–34. [DOI] [PubMed] [Google Scholar]
- 27. Montalban X, Hauser SL, Kappos L et al. Ocrelizumab versus placebo in primary progressive multiple sclerosis. N Engl J Med 2017; 376: 209–20. [DOI] [PubMed] [Google Scholar]
- 28. Hauser SL, Bar‐Or A, Cohen JA et al. Ofatumumab versus teriflunomide in multiple sclerosis. N Engl J Med 2020; 383: 546–57. [DOI] [PubMed] [Google Scholar]
- 29. Marchetti L, Engelhardt B. Immune cell trafficking across the blood‐brain barrier in the absence and presence of neuroinflammation. Vasc Biol 2020; 2: H1–H18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Larochelle C, Lecuyer MA, Alvarez JI et al. Melanoma cell adhesion molecule‐positive CD8 T lymphocytes mediate central nervous system inflammation. Ann Neurol 2015; 78: 39–53. [DOI] [PubMed] [Google Scholar]
- 31. Pare A, Mailhot B, Levesque SA et al. IL‐1beta enables CNS access to CCR2(hi) monocytes and the generation of pathogenic cells through GM‐CSF released by CNS endothelial cells. Proc Natl Acad Sci USA 2018; 115: E1194–E203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Magliozzi R, Howell OW, Reeves C et al. A Gradient of neuronal loss and meningeal inflammation in multiple sclerosis. Ann Neurol 2010; 68: 477–93. [DOI] [PubMed] [Google Scholar]
- 33. Harrison DM, Roy S, Oh J et al. Association of Cortical Lesion Burden on 7‐T Magnetic Resonance Imaging With Cognition and Disability in Multiple Sclerosis. JAMA Neurol 2015; 72: 1004–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Granberg T, Fan Q, Treaba CA et al. In vivo characterization of cortical and white matter neuroaxonal pathology in early multiple sclerosis. Brain 2017; 140: 2912–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Frischer JM, Weigand SD, Guo Y et al. Clinical and pathological insights into the dynamic nature of the white matter multiple sclerosis plaque. Ann Neurol 2015; 78: 710–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. van Nierop GP, van Luijn MM, Michels SS et al. Phenotypic and functional characterization of T cells in white matter lesions of multiple sclerosis patients. Acta Neuropathol 2017; 134: 383–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Machado‐Santos J, Saji E, Troscher AR et al. The compartmentalized inflammatory response in the multiple sclerosis brain is composed of tissue‐resident CD8+ T lymphocytes and B cells. Brain 2018; 141: 2066–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Cao Y, Goods BA, Raddassi K et al. Functional inflammatory profiles distinguish myelin‐reactive T cells from patients with multiple sclerosis. Sci Transl Med 2015; 7: 287ra74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bornstein MB, Miller A, Slagle S et al. A pilot trial of Cop 1 in exacerbating‐remitting multiple sclerosis. N Engl J Med 1987; 317: 408–14. [DOI] [PubMed] [Google Scholar]
- 40. Aharoni R. The mechanism of action of glatiramer acetate in multiple sclerosis and beyond. Autoimmun Rev 2013; 12: 543–53. [DOI] [PubMed] [Google Scholar]
- 41. Mahad DH, Trapp BD, Lassmann H. Pathological mechanisms in progressive multiple sclerosis. Lancet Neurol 2015; 14: 183–93. [DOI] [PubMed] [Google Scholar]
- 42. Lublin F, Miller DH, Freedman MS et al. Oral fingolimod in primary progressive multiple sclerosis (INFORMS): a phase 3, randomised, double‐blind, placebo‐controlled trial. Lancet 2016; 387: 1075–84. [DOI] [PubMed] [Google Scholar]
- 43. Kapoor R, Ho PR, Campbell N et al. Effect of natalizumab on disease progression in secondary progressive multiple sclerosis (ASCEND): a phase 3, randomised, double‐blind, placebo‐controlled trial with an open‐label extension. Lancet Neurol 2018; 17: 405–15. [DOI] [PubMed] [Google Scholar]
- 44. Haider L, Zrzavy T, Hametner S et al. The topograpy of demyelination and neurodegeneration in the multiple sclerosis brain. Brain 2016; 139: 807–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lassmann H. Pathogenic mechanisms associated with different clinical courses of multiple sclerosis. Front Immunol 2018; 9: 3116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Howell OW, Reeves CA, Nicholas R et al. Meningeal inflammation is widespread and linked to cortical pathology in multiple sclerosis. Brain 2011; 134: 2755–71. [DOI] [PubMed] [Google Scholar]
- 47. Dal‐Bianco A, Grabner G, Kronnerwetter C et al. Slow expansion of multiple sclerosis iron rim lesions: pathology and 7 T magnetic resonance imaging. Acta Neuropathol 2017; 133: 25–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Patrikios P, Stadelmann C, Kutzelnigg A et al. Remyelination is extensive in a subset of multiple sclerosis patients. Brain 2006; 129: 3165–72. [DOI] [PubMed] [Google Scholar]
- 49. Albert M, Antel J, Bruck W, Stadelmann C. Extensive cortical remyelination in patients with chronic multiple sclerosis. Brain Pathol 2007; 17: 129–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Jakel S, Agirre E, Mendanha Falcao A et al. Altered human oligodendrocyte heterogeneity in multiple sclerosis. Nature 2019; 566: 543–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Yeung MSY, Djelloul M, Steiner E et al. Dynamics of oligodendrocyte generation in multiple sclerosis. Nature 2019; 566: 538–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ouellette R, Mangeat G, Polyak I et al. Validation of rapid magnetic resonance myelin imaging in multiple sclerosis. Ann Neurol 2020; 87: 710–24. [DOI] [PubMed] [Google Scholar]
- 53. Trapp BD, Peterson J, Ransohoff RM, Rudick R, Mork S, Bo L. Axonal transection in the lesions of multiple sclerosis. N Engl J Med 1998; 338: 278–85. [DOI] [PubMed] [Google Scholar]
- 54. Kutzelnigg A, Lucchinetti CF, Stadelmann C et al. Cortical demyelination and diffuse white matter injury in multiple sclerosis. Brain 2005; 128: 2705–12. [DOI] [PubMed] [Google Scholar]
- 55. Kappos L, Wolinsky JS, Giovannoni G et al. Contribution of relapse‐independent progression vs relapse‐associated worsening to overall confirmed disability accumulation in typical relapsing multiple sclerosis in a pooled analysis of 2 randomized clinical trials. JAMA neurology 2020; 77: 1132–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Jacobs L, Johnson KP. A brief history of the use of interferons as treatment of multiple sclerosis. Arch Neurol 1994; 51: 1245–52. [DOI] [PubMed] [Google Scholar]
- 57. Panitch HS, Hirsch RL, Schindler J, Johnson KP. Treatment of multiple sclerosis with gamma interferon: exacerbations associated with activation of the immune system. Neurology 1987; 37: 1097–102. [DOI] [PubMed] [Google Scholar]
- 58. Interferon beta‐1b is effective in relapsing‐remitting multiple sclerosis. I. Clinical results of a multicenter, randomized, double‐blind, placebo‐controlled trial. The IFNB Multiple Sclerosis Study Group. Neurology 1993; 43: 655–61. [DOI] [PubMed] [Google Scholar]
- 59. Jacobs LD, Cookfair DL, Rudick RA et al. A phase III trial of intramuscular recombinant interferon beta as treatment for exacerbating‐remitting multiple sclerosis: design and conduct of study and baseline characteristics of patients. Multiple Sclerosis Collaborative Research Group (MSCRG). Mult Scler 1995; 1: 118–35. [DOI] [PubMed] [Google Scholar]
- 60. Randomised double‐blind placebo‐controlled study of interferon beta‐1a in relapsing/remitting multiple sclerosis. PRISMS (Prevention of Relapses and Disability by Interferon beta‐1a Subcutaneously in Multiple Sclerosis) Study Group. Lancet 1998; 352: 1498–504. [PubMed] [Google Scholar]
- 61. Calabresi PA, Kieseier BC, Arnold DL et al. Pegylated interferon beta‐1a for relapsing‐remitting multiple sclerosis (ADVANCE): a randomised, phase 3, double‐blind study. Lancet Neurol 2014; 13: 657–65. [DOI] [PubMed] [Google Scholar]
- 62. Mahad D, Callahan MK, Williams KA et al. Modulating CCR2 and CCL2 at the blood‐brain barrier: relevance for multiple sclerosis pathogenesis. Brain 2006; 129: 212–23. [DOI] [PubMed] [Google Scholar]
- 63. Vergunst CE, Gerlag DM, Lopatinskaya L et al. Modulation of CCR2 in rheumatoid arthritis: a double‐blind, randomized, placebo‐controlled clinical trial. Arthritis Rheum 2008; 58: 1931–9. [DOI] [PubMed] [Google Scholar]
- 64. Kappos L, Wiendl H, Selmaj K et al. Daclizumab HYP versus Interferon Beta‐1a in Relapsing Multiple Sclerosis. N Engl J Med 2015; 373: 1418–28. [DOI] [PubMed] [Google Scholar]
- 65. Stork L, Bruck W, von Gottberg P et al. Severe meningo‐/encephalitis after daclizumab therapy for multiple sclerosis. Mult Scler 2019; 25: 1618–32. [DOI] [PubMed] [Google Scholar]
- 66. Luckel C, Picard F, Raifer H et al. IL‐17(+) CD8(+) T cell suppression by dimethyl fumarate associates with clinical response in multiple sclerosis. Nat Commun 2019; 10: 5722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Constantinescu CS, Asher A, Fryze W et al. Randomized phase 1b trial of MOR103, a human antibody to GM‐CSF, in multiple sclerosis. Neurol Neuroimmunol Neuroinflamm 2015; 2: e117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Havrdova E, Belova A, Goloborodko A et al. Activity of secukinumab, an anti‐IL‐17A antibody, on brain lesions in RRMS: results from a randomized, proof‐of‐concept study. J Neurol 2016; 263: 1287–95. [DOI] [PubMed] [Google Scholar]
- 69. Segal BM, Constantinescu CS, Raychaudhuri A et al. Repeated subcutaneous injections of IL12/23 p40 neutralising antibody, ustekinumab, in patients with relapsing‐remitting multiple sclerosis: a phase II, double‐blind, placebo‐controlled, randomised, dose‐ranging study. Lancet Neurol 2008; 7: 796–804. [DOI] [PubMed] [Google Scholar]
- 70. Vollmer TL, Wynn DR, Alam MS, Valdes J. A phase 2, 24‐week, randomized, placebo‐controlled, double‐blind study examining the efficacy and safety of an anti‐interleukin‐12 and ‐23 monoclonal antibody in patients with relapsing‐remitting or secondary progressive multiple sclerosis. Mult Scler 2011; 17: 181–91. [DOI] [PubMed] [Google Scholar]
- 71. Baker D, Pryce G, James LK, Schmierer K, Giovannoni G. Failed B cell survival factor trials support the importance of memory B cells in multiple sclerosis. Eur J Neurol 2020; 27: 221–8. [DOI] [PubMed] [Google Scholar]
- 72. Neurology TNF neutralization in MS: results of a randomized, placebo‐controlled multicenter study . The Lenercept Multiple Sclerosis Study Group and The University of British Columbia MS/MRI Analysis Group. Neurology 1999; 53: 457–65. [PubMed] [Google Scholar]
- 73. Kopp TI, Delcoigne B, Arkema EV et al. Risk of neuroinflammatory events in arthritis patients treated with tumour necrosis factor alpha inhibitors: a collaborative population‐based cohort study from Denmark and Sweden. Ann Rheum Dis 2020; 79: 566–72. [DOI] [PubMed] [Google Scholar]
- 74. Kappos L, Hartung HP, Freedman MS et al. Atacicept in multiple sclerosis (ATAMS): a randomised, placebo‐controlled, double‐blind, phase 2 trial. Lancet Neurol 2014; 13: 353–63. [DOI] [PubMed] [Google Scholar]
- 75. Sergott RC, Bennett JL, Rieckmann P et al. ATON: results from a Phase II randomized trial of the B‐cell‐targeting agent atacicept in patients with optic neuritis. J Neurol Sci 2015; 351: 174–8. [DOI] [PubMed] [Google Scholar]
- 76. Brinkmann V, Davis MD, Heise CE et al. The immune modulator FTY720 targets sphingosine 1‐phosphate receptors. J Biol Chem 2002; 277: 21453–7. [DOI] [PubMed] [Google Scholar]
- 77. Kappos L, Radue EW, O'Connor P et al. A placebo‐controlled trial of oral fingolimod in relapsing multiple sclerosis. N Engl J Med 2010; 362: 387–401. [DOI] [PubMed] [Google Scholar]
- 78. Calabresi PA, Radue EW, Goodin D et al. Safety and efficacy of fingolimod in patients with relapsing‐remitting multiple sclerosis (FREEDOMS II): a double‐blind, randomised, placebo‐controlled, phase 3 trial. Lancet Neurol 2014; 13: 545–56. [DOI] [PubMed] [Google Scholar]
- 79. Cohen JA, Barkhof F, Comi G et al. Oral fingolimod or intramuscular interferon for relapsing multiple sclerosis. N Engl J Med 2010; 362: 402–15. [DOI] [PubMed] [Google Scholar]
- 80. Chitnis T, Arnold DL, Banwell B et al. Trial of fingolimod versus interferon Beta‐1a in pediatric multiple sclerosis. N Engl J Med 2018; 379: 1017–27. [DOI] [PubMed] [Google Scholar]
- 81. Cohan S, Lucassen E, Smoot K, Brink J, Chen C. Sphingosine‐1‐phosphate: its pharmacological regulation and the treatment of multiple sclerosis. A Review Article. Biomedicines 2020; 8: 227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Cohen JA, Comi G, Selmaj KW et al. Safety and efficacy of ozanimod versus interferon beta‐1a in relapsing multiple sclerosis (RADIANCE): a multicentre, randomised, 24‐month, phase 3 trial. Lancet Neurol 2019; 18: 1021–33. [DOI] [PubMed] [Google Scholar]
- 83. Comi G, Kappos L, Selmaj KW et al. Safety and efficacy of ozanimod versus interferon beta‐1a in relapsing multiple sclerosis (SUNBEAM): a multicentre, randomised, minimum 12‐month, phase 3 trial. Lancet Neurol 2019; 18: 1009–20. [DOI] [PubMed] [Google Scholar]
- 84. Kappos L, Bar‐Or A, Cree BAC et al. Siponimod versus placebo in secondary progressive multiple sclerosis (EXPAND): a double‐blind, randomised, phase 3 study. Lancet 2018; 391: 1263–73. [DOI] [PubMed] [Google Scholar]
- 85. Kappos L. Efficacy and safety of ponesimod compared to teriflunomide in patients with relapsing multiple sclerosis: results of the randomized, active‐controlled, double‐blind, parallel‐group phase 3 OPTIMUM study. ECTRIMS Online Library 2019; 93. [Google Scholar]
- 86. Kappos L, Arnold DL, Bar‐Or A et al. Safety and efficacy of amiselimod in relapsing multiple sclerosis (MOMENTUM): a randomised, double‐blind, placebo‐controlled phase 2 trial. Lancet Neurol 2016; 15: 1148–59. [DOI] [PubMed] [Google Scholar]
- 87. Polman CH, O'Connor PW, Havrdova E et al. A randomized, placebo‐controlled trial of natalizumab for relapsing multiple sclerosis. N Engl J Med 2006; 354: 899–910. [DOI] [PubMed] [Google Scholar]
- 88. Plavina T, Subramanyam M, Bloomgren G et al. Anti‐JC virus antibody levels in serum or plasma further define risk of natalizumab‐associated progressive multifocal leukoencephalopathy. Ann Neurol 2014; 76: 802–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Olsson T, Achiron A, Alfredsson L et al. Anti‐JC virus antibody prevalence in a multinational multiple sclerosis cohort. Mult Scler 2013; 19: 1533–8. [DOI] [PubMed] [Google Scholar]
- 90. Breuer J, Schneider‐Hohendorf T, Ostkamp P et al. VLA‐2 blockade in vivo by vatelizumab induces CD4+FoxP3+ regulatory T cells. Int Immunol 2019; 31: 407–12. [DOI] [PubMed] [Google Scholar]
- 91. Miller DH, Weber T, Grove R et al. Firategrast for relapsing remitting multiple sclerosis: a phase 2, randomised, double‐blind, placebo‐controlled trial. Lancet Neurol 2012; 11: 131–9. [DOI] [PubMed] [Google Scholar]
- 92. Wolf C, Sidhu J, Otoul C et al. Pharmacodynamic consequences of administration of VLA‐4 antagonist CDP323 to multiple sclerosis subjects: a randomized, double‐blind phase 1/2 study. PLoS One 2013; 8: e58438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Katsavos S, Coles A. Alemtuzumab as treatment for multiple sclerosis. Cold Spring Harb Perspect Med 2018; 8: a032029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Cohen JA, Coles AJ, Arnold DL et al. Alemtuzumab versus interferon beta 1a as first‐line treatment for patients with relapsing‐remitting multiple sclerosis: a randomised controlled phase 3 trial. Lancet 2012; 380: 1819–28. [DOI] [PubMed] [Google Scholar]
- 95. Coles AJ, Twyman CL, Arnold DL et al. Alemtuzumab for patients with relapsing multiple sclerosis after disease‐modifying therapy: a randomised controlled phase 3 trial. Lancet 2012; 380: 1829–39. [DOI] [PubMed] [Google Scholar]
- 96. Arnold DL, Fisher E, Brinar VV et al. Superior MRI outcomes with alemtuzumab compared with subcutaneous interferon beta‐1a in MS. Neurology 2016; 87: 1464–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Havrdova E, Arnold DL, Cohen JA et al. Alemtuzumab CARE‐MS I 5‐year follow‐up: Durable efficacy in the absence of continuous MS therapy. Neurology 2017; 89: 1107–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Holmoy T, Fevang B, Olsen DB, Spigset O, Bo L. Adverse events with fatal outcome associated with alemtuzumab treatment in multiple sclerosis. BMC Res Notes 2019; 12: 497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Scappaticcio L, Castellana M, Virili C et al. Alemtuzumab‐induced thyroid events in multiple sclerosis: a systematic review and meta‐analysis. J Endocrinol Invest 2020; 43: 219–29. [DOI] [PubMed] [Google Scholar]
- 100. Giovannoni G, Comi G, Cook S et al. A placebo‐controlled trial of oral cladribine for relapsing multiple sclerosis. N Engl J Med 2010; 362: 416–26. [DOI] [PubMed] [Google Scholar]
- 101. Leist TP, Comi G, Cree BA et al. Effect of oral cladribine on time to conversion to clinically definite multiple sclerosis in patients with a first demyelinating event (ORACLE MS): a phase 3 randomised trial. Lancet Neurol 2014; 13: 257–67. [DOI] [PubMed] [Google Scholar]
- 102. Cook S, Vermersch P, Comi G et al. Safety and tolerability of cladribine tablets in multiple sclerosis: the CLARITY (CLAdRIbine Tablets treating multiple sclerosis orallY) study. Mult Scler 2011; 17: 578–93. [DOI] [PubMed] [Google Scholar]
- 103. Hartung HP, Gonsette R, Konig N et al. Mitoxantrone in progressive multiple sclerosis: a placebo‐controlled, double‐blind, randomised, multicentre trial. Lancet 2002; 360: 2018–25. [DOI] [PubMed] [Google Scholar]
- 104. Edan G, Miller D, Clanet M et al. Therapeutic effect of mitoxantrone combined with methylprednisolone in multiple sclerosis: a randomised multicentre study of active disease using MRI and clinical criteria. J Neurol Neurosurg Psychiatry 1997; 62: 112–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Fleischer V, Salmen A, Kollar S et al. Cardiotoxicity of mitoxantrone treatment in a german cohort of 639 multiple sclerosis patients. J Clin Neurol 2014; 10: 289–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Ellis R, Brown S, Boggild M. Therapy‐related acute leukaemia with mitoxantrone: four years on, what is the risk and can it be limited? Mult Scler 2015; 21: 642–5. [DOI] [PubMed] [Google Scholar]
- 107. Hauser SL, Waubant E, Arnold DL et al. B‐cell depletion with rituximab in relapsing‐remitting multiple sclerosis. N Engl J Med 2008; 358: 676–88. [DOI] [PubMed] [Google Scholar]
- 108. Hawker K, O'Connor P, Freedman MS et al. Rituximab in patients with primary progressive multiple sclerosis: results of a randomized double‐blind placebo‐controlled multicenter trial. Ann Neurol 2009; 66: 460–71. [DOI] [PubMed] [Google Scholar]
- 109. Fox E, Lovett‐Racke AE, Gormley M et al. A phase 2 multicenter study of ublituximab, a novel glycoengineered anti‐CD20 monoclonal antibody, in patients with relapsing forms of multiple sclerosis. Mult Scler 2020; 1352458520918375. Online ahead of print. PMID: 32351164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Luna G, Alping P, Burman J et al. Infection risks among patients with multiple sclerosis treated with fingolimod, natalizumab, rituximab, and injectable therapies. JAMA Neurol 2020; 77: 184–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Agius MA, Klodowska‐Duda G, Maciejowski M et al. Safety and tolerability of inebilizumab (MEDI‐551), an anti‐CD19 monoclonal antibody, in patients with relapsing forms of multiple sclerosis: Results from a phase 1 randomised, placebo‐controlled, escalating intravenous and subcutaneous dose study. Mult Scler 2019; 25: 235–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Cree BAC, Bennett JL, Kim HJ et al. Inebilizumab for the treatment of neuromyelitis optica spectrum disorder (N‐MOmentum): a double‐blind, randomised placebo‐controlled phase 2/3 trial. Lancet 2019; 394: 1352–63. [DOI] [PubMed] [Google Scholar]
- 113. Sormani MP, Muraro PA, Schiavetti I et al. Autologous hematopoietic stem cell transplantation in multiple sclerosis: A meta‐analysis. Neurology 2017; 88: 2115–22. [DOI] [PubMed] [Google Scholar]
- 114. Muraro PA, Martin R, Mancardi GL, Nicholas R, Sormani MP, Saccardi R. Autologous haematopoietic stem cell transplantation for treatment of multiple sclerosis. Nat Rev Neurol 2017; 13: 391–405. [DOI] [PubMed] [Google Scholar]
- 115. Burt RK, Loh Y, Cohen B et al. Autologous non‐myeloablative haemopoietic stem cell transplantation in relapsing‐remitting multiple sclerosis: a phase I/II study. Lancet Neurol 2009; 8: 244–53. [DOI] [PubMed] [Google Scholar]
- 116. Alping P. Safety outcomes after treatment with alemtuzumab or autologous hematopoietic stem cell transplantation in multiple sclerosis patients. Actrims Online Library 2020; P0491. [Google Scholar]
- 117. Arrambide G, Iacobaeus E, Amato MP et al. Aggressive multiple sclerosis (2): Treatment. Mult Scler 2020; 1352458520924595. Online ahead of print. PMID: 32530385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Bar‐Or A. Phase I study of ATA188, an off‐the‐shelf, allogeneic Epstein‐Barr virus‐targeted T‐cell immunotherapy for progressive forms of multiple sclerosis. Actrims Online Library 2020; P0226. [Google Scholar]
- 119. Pender MP, Csurhes PA, Smith C et al. Epstein‐Barr virus‐specific T cell therapy for progressive multiple sclerosis. JCI Insight 2020; 5: e144624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Klotz L, Eschborn M, Lindner M et al. Teriflunomide treatment for multiple sclerosis modulates T cell mitochondrial respiration with affinity‐dependent effects. Sci Transl Med 2019; 11: eaao5563. [DOI] [PubMed] [Google Scholar]
- 121. O'Connor P, Wolinsky JS, Confavreux C et al. Randomized trial of oral teriflunomide for relapsing multiple sclerosis. N Engl J Med 2011; 365: 1293–303. [DOI] [PubMed] [Google Scholar]
- 122. Confavreux C, O'Connor P, Comi G et al. Oral teriflunomide for patients with relapsing multiple sclerosis (TOWER): a randomised, double‐blind, placebo‐controlled, phase 3 trial. Lancet Neurol 2014; 13: 247–56. [DOI] [PubMed] [Google Scholar]
- 123. Vermersch P, Czlonkowska A, Grimaldi LM et al. Teriflunomide versus subcutaneous interferon beta‐1a in patients with relapsing multiple sclerosis: a randomised, controlled phase 3 trial. Mult Scler 2014; 20: 705–16. [DOI] [PubMed] [Google Scholar]
- 124. Muehler A, Peelen E, Kohlhof H, Groppel M, Vitt D. Vidofludimus calcium, a next generation DHODH inhibitor for the Treatment of relapsing‐remitting multiple sclerosis. Mult Scler Relat Disord 2020; 43: 102129. [DOI] [PubMed] [Google Scholar]
- 125. Fox RJ. Top‐line results of EMPhASIS, a Phase 2 clinical trial of vidofludimus calcium (IMU‐838) in relapsing‐remitting multiple sclerosis. Actrims Online Library 2020; P0241. [Google Scholar]
- 126. Linker RA, Lee DH, Stangel M, Gold R. Fumarates for the treatment of multiple sclerosis: potential mechanisms of action and clinical studies. Expert Rev Neurother 2008; 8: 1683–90. [DOI] [PubMed] [Google Scholar]
- 127. Fox RJ, Miller DH, Phillips JT et al. Placebo‐controlled phase 3 study of oral BG‐12 or glatiramer in multiple sclerosis. N Engl J Med 2012; 367: 1087–97. [DOI] [PubMed] [Google Scholar]
- 128. Gold R, Kappos L, Arnold DL et al. Placebo‐controlled phase 3 study of oral BG‐12 for relapsing multiple sclerosis. N Engl J Med 2012; 367: 1098–107. [DOI] [PubMed] [Google Scholar]
- 129. Carlstrom KE, Zhu K, Ewing E et al. Gsta4 controls apoptosis of differentiating adult oligodendrocytes during homeostasis and remyelination via the mitochondria‐associated Fas‐Casp8‐Bid‐axis. Nat Commun 2020; 11: 4071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Jordan AL, Yang J, Fisher CJ, Racke MK, Mao‐Draayer Y. Progressive multifocal leukoencephalopathy in dimethyl fumarate‐treated multiple sclerosis patients. Mult Scler 2020; 1352458520949158. Online ahead of print. PMID: 32808554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Naismith RT, Wundes A, Ziemssen T et al. Diroximel fumarate demonstrates an improved gastrointestinal tolerability profile compared with dimethyl fumarate in patients with relapsing‐remitting multiple sclerosis: results from the randomized, double‐blind, phase III EVOLVE‐MS‐2 study. CNS Drugs 2020; 34: 185–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Naismith RT, Wolinsky JS, Wundes A et al. Diroximel fumarate (DRF) in patients with relapsing‐remitting multiple sclerosis: Interim safety and efficacy results from the phase 3 EVOLVE‐MS‐1 study. Mult Scler 2020; 26: 1729–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Wynn D, Lategan TW, Sprague TN, Rousseau FS, Fox EJ. Monomethyl fumarate has better gastrointestinal tolerability profile compared with dimethyl fumarate. Multiple sclerosis and related disorders 2020; 45: 102335. [DOI] [PubMed] [Google Scholar]
- 134. Montalban X, Arnold DL, Weber MS et al. Placebo‐controlled trial of an oral BTK inhibitor in multiple sclerosis. N Engl J Med 2019; 380: 2406–17. [DOI] [PubMed] [Google Scholar]
- 135. Hauser S. Examination of fenebrutinib, a highly selective BTKi, on disease progression of multiple sclerosis. Actrims Online Library 2020; P0211. [Google Scholar]
- 136. Scaramozza M. A Phase 1 study of BIIB091, a Bruton’s tyrosine kinase (BTK) inhibitor, in healthy adult participants: preliminary results. Actrims Online Library 2020; P0186. [Google Scholar]
- 137. Khoury SJ, Rochon J, Ding L et al. ACCLAIM: A randomized trial of abatacept (CTLA4‐Ig) for relapsing‐remitting multiple sclerosis. Mult Scler 2017; 23: 686–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Johnson KP, Brooks BR, Cohen JA et al. Copolymer 1 reduces relapse rate and improves disability in relapsing‐remitting multiple sclerosis: results of a phase III multicenter, double‐blind placebo‐controlled trial. The Copolymer 1 Multiple Sclerosis Study Group. Neurology 1995; 45: 1268–76. [DOI] [PubMed] [Google Scholar]
- 139. Comi G, Martinelli V, Rodegher M et al. Effect of glatiramer acetate on conversion to clinically definite multiple sclerosis in patients with clinically isolated syndrome (PreCISe study): a randomised, double‐blind, placebo‐controlled trial. Lancet 2009; 374: 1503–11. [DOI] [PubMed] [Google Scholar]
- 140. Cohen J, Belova A, Selmaj K et al. Equivalence of generic glatiramer acetate in multiple sclerosis: a randomized clinical trial. JAMA Neurol 2015; 72: 1433–41. [DOI] [PubMed] [Google Scholar]
- 141. O'Connor P, Filippi M, Arnason B et al. 250 microg or 500 microg interferon beta‐1b versus 20 mg glatiramer acetate in relapsing‐remitting multiple sclerosis: a prospective, randomised, multicentre study. Lancet Neurol 2009; 8: 889–97. [DOI] [PubMed] [Google Scholar]
- 142. Mikol DD, Barkhof F, Chang P et al. Comparison of subcutaneous interferon beta‐1a with glatiramer acetate in patients with relapsing multiple sclerosis (the REbif vs Glatiramer Acetate in Relapsing MS Disease [REGARD] study): a multicentre, randomised, parallel, open‐label trial. Lancet Neurol 2008; 7: 903–14. [DOI] [PubMed] [Google Scholar]
- 143. Wolinsky JS, Borresen TE, Dietrich DW et al. GLACIER: An open‐label, randomized, multicenter study to assess the safety and tolerability of glatiramer acetate 40 mg three‐times weekly versus 20 mg daily in patients with relapsing‐remitting multiple sclerosis. Mult Scler Relat Disord 2015; 4: 370–6. [DOI] [PubMed] [Google Scholar]
- 144. Khan O, Rieckmann P, Boyko A, Selmaj K, Zivadinov R, Group GS. Three times weekly glatiramer acetate in relapsing‐remitting multiple sclerosis. Ann Neurol 2013; 73: 705–13. [DOI] [PubMed] [Google Scholar]
- 145. Chataway J, Martin K, Barrell K et al. Effects of ATX‐MS‐1467 immunotherapy over 16 weeks in relapsing multiple sclerosis. Neurology 2018; 90: e955–e62. [DOI] [PubMed] [Google Scholar]
- 146. Bielekova B, Goodwin B, Richert N et al. Encephalitogenic potential of the myelin basic protein peptide (amino acids 83–99) in multiple sclerosis: results of a phase II clinical trial with an altered peptide ligand. Nat Med 2000; 6: 1167–75. [DOI] [PubMed] [Google Scholar]
- 147. Garren H, Robinson WH, Krasulova E et al. Phase 2 trial of a DNA vaccine encoding myelin basic protein for multiple sclerosis. Ann Neurol 2008; 63: 611–20. [DOI] [PubMed] [Google Scholar]
- 148. Martin R. Antigenspecific MS treatment revisited – recent development and prospects. Ectrims Online Library 2019; 281901. [Google Scholar]
- 149. Tan IL, Lycklama a Nijeholt GJ, Polman CH, Ader HJ, Barkhof F. Linomide in the treatment of multiple sclerosis: MRI results from prematurely terminated phase‐III trials. Mult Scler 2000; 6: 99–104. [DOI] [PubMed] [Google Scholar]
- 150. Comi G, Jeffery D, Kappos L et al. Placebo‐controlled trial of oral laquinimod for multiple sclerosis. N Engl J Med 2012; 366: 1000–9. [DOI] [PubMed] [Google Scholar]
- 151. Vollmer TL, Sorensen PS, Selmaj K et al. A randomized placebo‐controlled phase III trial of oral laquinimod for multiple sclerosis. J Neurol 2014; 261: 773–83. [DOI] [PubMed] [Google Scholar]
- 152. Metz LM, Li DKB, Traboulsee AL et al. Trial of minocycline in a clinically isolated syndrome of multiple sclerosis. N Engl J Med 2017; 376: 2122–33. [DOI] [PubMed] [Google Scholar]
- 153. Sorensen PS, Sellebjerg F, Lycke J et al. Minocycline added to subcutaneous interferon beta‐1a in multiple sclerosis: randomized RECYCLINE study. Eur J Neurol 2016; 23: 861–70. [DOI] [PubMed] [Google Scholar]
- 154. Bielekova B, Richert N, Howard T et al. Treatment with the phosphodiesterase type‐4 inhibitor rolipram fails to inhibit blood–brain barrier disruption in multiple sclerosis. Mult Scler 2009; 15: 1206–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Lovera J, Ramos A, Devier D et al. Polyphenon E, non‐futile at neuroprotection in multiple sclerosis but unpredictably hepatotoxic: Phase I single group and phase II randomized placebo‐controlled studies. J Neurol Sci 2015; 358: 46–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Gold J, Marta M, Meier UC et al. A phase II baseline versus treatment study to determine the efficacy of raltegravir (Isentress) in preventing progression of relapsing remitting multiple sclerosis as determined by gadolinium‐enhanced MRI: The INSPIRE study. Multi Scler Relat Disord 2018; 24: 123–8. [DOI] [PubMed] [Google Scholar]
- 157. Kremer D, Gruchot J, Weyers V et al. pHERV‐W envelope protein fuels microglial cell‐dependent damage of myelinated axons in multiple sclerosis. Proc Natl Acad Sci USA 2019; 116: 15216–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Hartung HP. Neuroprotective effects of temelimab in relapsing‐remitting MS patients extend to 96 weeks. Ectrims Online Library 2019; P1379. [Google Scholar]
- 159. Mei F, Fancy SPJ, Shen YA et al. Micropillar arrays as a high‐throughput screening platform for therapeutics in multiple sclerosis. Nat Med 2014; 20: 954–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Green AJ, Gelfand JM, Cree BA et al. Clemastine fumarate as a remyelinating therapy for multiple sclerosis (ReBUILD): a randomised, controlled, double‐blind, crossover trial. Lancet 2017; 390: 2481–9. [DOI] [PubMed] [Google Scholar]
- 161. Schwartzbach CJ, Grove RA, Brown R, Tompson D, Then Bergh F, Arnold DL. Lesion remyelinating activity of GSK239512 versus placebo in patients with relapsing‐remitting multiple sclerosis: a randomised, single‐blind, phase II study. J Neurol 2017; 264: 304–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Chataway J, De Angelis F, Connick P et al. Efficacy of three neuroprotective drugs in secondary progressive multiple sclerosis (MS‐SMART): a phase 2b, multiarm, double‐blind, randomised placebo‐controlled trial. Lancet Neurol 2020; 19: 214–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Robinson AP, Zhang JZ, Titus HE et al. Nanocatalytic activity of clean‐surfaced, faceted nanocrystalline gold enhances remyelination in animal models of multiple sclerosis. Sci Rep 2020; 10: 1936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Cree B. Elezanumab in patients with different disease courses of multiple sclerosis: study design and baseline analysis from two phase 2 studies. Actrims Online Library 2020; P0210. [Google Scholar]
- 165. Cadavid D, Mellion M, Hupperts R et al. Safety and efficacy of opicinumab in patients with relapsing multiple sclerosis (SYNERGY): a randomised, placebo‐controlled, phase 2 trial. Lancet Neurol 2019; 18: 845–56. [DOI] [PubMed] [Google Scholar]
- 166. Petrillo J, Balcer L, Galetta S, Chai Y, Xu L, Cadavid D. Initial impairment and recovery of vision‐related functioning in participants with acute optic neuritis from the RENEW trial of opicinumab. J Neuroophthalmol 2019; 39: 153–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Eisen A, Greenberg BM, Bowen JD, Arnold DL, Caggiano AO. A double‐blind, placebo‐controlled, single ascending‐dose study of remyelinating antibody rHIgM22 in people with multiple sclerosis. Mult Scler J Exp Transl Clin 2017; 3: 2055217317743097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. LaGanke C, Samkoff L, Edwards K et al. Safety/tolerability of the anti‐semaphorin 4D Antibody VX15/2503 in a randomized phase 1 trial. Neurol Neuroimmunol Neuroinflamm 2017; 4: e367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Meininger V, Genge A, van den Berg LH et al. Safety and efficacy of ozanezumab in patients with amyotrophic lateral sclerosis: a randomised, double‐blind, placebo‐controlled, phase 2 trial. Lancet Neurol 2017; 16: 208–16. [DOI] [PubMed] [Google Scholar]
- 170. Schreiber K, Magyari M, Sellebjerg F et al. High‐dose erythropoietin in patients with progressive multiple sclerosis: A randomized, placebo‐controlled, phase 2 trial. Mult Scler 2017; 23: 675–85. [DOI] [PubMed] [Google Scholar]
- 171. Clegg A, Bryant J. Immunomodulatory drugs for multiple sclerosis: a systematic review of clinical and cost effectiveness. Expert Opin Pharmacother 2001; 2: 623–39. [DOI] [PubMed] [Google Scholar]
- 172. Giovannoni G. Long‐term reduction of relapse rate and 48‐week confirmed disability progression after 6.5 years of ocrelizumab treatment in patients with RMS. iu Online Library 2020; P0216. [Google Scholar]
- 173. Harding K, Williams O, Willis M et al. Clinical outcomes of escalation vs early intensive disease‐modifying therapy in patients with multiple sclerosis. JAMA neurology 2019; 76: 536–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. He A, Merkel B, Brown JWL et al. Timing of high‐efficacy therapy for multiple sclerosis: a retrospective observational cohort study. Lancet Neurol 2020; 19: 307–16. [DOI] [PubMed] [Google Scholar]
- 175. Montalban X, Gold R, Thompson AJ et al. ECTRIMS/EAN Guideline on the pharmacological treatment of people with multiple sclerosis. Mult Scler 2018; 24: 96–120. [DOI] [PubMed] [Google Scholar]
- 176. Rae‐Grant A, Day GS, Marrie RA et al. Practice guideline recommendations summary: Disease‐modifying therapies for adults with multiple sclerosis: Report of the Guideline Development, Dissemination, and Implementation Subcommittee of the American Academy of Neurology. Neurology 2018; 90: 777–88. [DOI] [PubMed] [Google Scholar]
- 177. Alping P, Askling J, Burman J et al. Cancer risk for fingolimod, natalizumab, and rituximab in multiple sclerosis patients. Ann Neurol 2020; 87: 688–99. [DOI] [PubMed] [Google Scholar]
- 178. Granqvist M, Boremalm M, Poorghobad A et al. Comparative effectiveness of rituximab and other initial treatment choices for multiple sclerosis. JAMA neurology 2018; 75: 320–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Virtanen S. Comparative effectiveness of different DMT strategies in new onset RRMS; First results of the CombatMS trial. Actrims Online Library 2020; P0198. [Google Scholar]
- 180. Ineichen BV, Moridi T, Granberg T, Piehl F. Rituximab treatment for multiple sclerosis. Mult Scler 2020; 26: 137–52. [DOI] [PubMed] [Google Scholar]
- 181. Hartung DM, Johnston KA, Geddes J, Bourdette DN. Effect of generic glatiramer acetate on spending and use of drugs for multiple sclerosis. Neurology 2020; 94: e1407–e14. [DOI] [PubMed] [Google Scholar]
- 182. Callaghan BC, Reynolds E, Banerjee M et al. Out‐of‐pocket costs are on the rise for commonly prescribed neurologic medications. Neurology 2019; 92: e2604–e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Hauser S. Rationale and design of two Phase IIIb studies of ocrelizumab at higher than the approved dose in patients with RMS and PPMS. Actrims Online Library 2020; P0230. [Google Scholar]
- 184. Komori M, Lin YC, Cortese I et al. Insufficient disease inhibition by intrathecal rituximab in progressive multiple sclerosis. Ann Clin Transl Neurol 2016; 3: 166–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185. Bergman J, Burman J, Gilthorpe JD et al. Intrathecal treatment trial of rituximab in progressive MS: An open‐label phase 1b study. Neurology 2018; 91: e1893–e901. [DOI] [PubMed] [Google Scholar]
- 186. Bhargava P, Wicken C, Smith MD et al. Trial of intrathecal rituximab in progressive multiple sclerosis patients with evidence of leptomeningeal contrast enhancement. Mult Scler Relat Disord 2019; 30: 136–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187. Lubetzki C, Zalc B, Williams A, Stadelmann C, Stankoff B. Remyelination in multiple sclerosis: from basic science to clinical translation. Lancet Neurol 2020; 19: 678–88. [DOI] [PubMed] [Google Scholar]