

Abstract
Several mutations conferring protection against Alzheimer's disease (AD) have been described, none as profound as the A673T mutation, where carriers are four times less likely to get AD compared to noncarriers. This mutation results in reduced amyloid beta (Aβ) protein production in vitro and lower lifetime Aβ concentration in carriers. Better understanding of the protective mechanisms of the mutation may provide important insights into AD pathophysiology and identify productive therapeutic intervention strategies for disease modification. Aβ(1‐42) protein forms oligomers that bind saturably to a single receptor site on neuronal synapses, initiating the downstream toxicities observed in AD. Decreased formation, toxicity, or stability of soluble Aβ oligomers, or reduction of synaptic binding of these oligomers, may combine with overall lower Aβ concentration to underlie A673T’s disease protecting mechanism. To investigate these possibilities, we compared the formation rate of soluble oligomers made from Icelandic A673T mutant and wild type (wt) Aβ(1‐42) synthetic protein, the amount and intensity of oligomer bound to mature primary rat hippocampal/cortical neuronal synapses, and the potency of bound oligomers to impact trafficking rate in neurons in vitro using a physiologically relevant oligomer preparation method. At equal protein concentrations, mutant protein forms approximately 50% or fewer oligomers of high molecular weight (>50 kDa) compared to wt protein. Mutant oligomers are twice as potent at altering the cellular vesicle trafficking rate as wt at equivalent concentrations, however, mutant oligomers have a >4‐fold lower binding affinity to synaptic receptors (K d = 1,950 vs. 442 nM). The net effect of these differences is a lower overall toxicity at a given concentration. This study demonstrates for the first time that mutant A673T Aβ oligomers prepared with this method have fundamentally different assembly characteristics and biological impact from wt protein and indicates that its disease protecting mechanism may result primarily from the mutant protein's much lower binding affinity to synaptic receptors. This suggests that therapeutics that effectively reduce oligomer binding to synapses in the brain may be beneficial in AD.

Keywords: Alzheimer's disease, Amyloid beta, mutant, neurons, oligomers, therapeutics
Carriers of the A673T mutation are four times less likely to get Alzheimer's disease than noncarriers. We showed that mutant amyloid beta (Aβ) oligomers bind less, and with lower affinity as well as intensity, to synaptic puncta than wt Aβ oligomers. This study demonstrates for the first time that mutant A673T Aβ oligomers have fundamentally different assembly characteristics and biological impact from wt peptide, and indicates that its disease protecting mechanism may result primarily from the mutant peptide's much lower binding affinity to synaptic receptors.

Abbreviations
- AD
Alzheimer's disease
- ANOVA
Analysis of variance
- APP
amyloid precursor protein
- Aβ
amyloid beta
- BACE1
β‐site APP cleaving enzyme 1
- CCD
charge‐coupled device
- CSF
cerebrospinal fluid
- DAPI
4′,6‐diamidino‐2‐phenylindole
- DMSO
dimethyl sulfoxide
- EC50
half maximal effective concentration
- ELISA
enzyme‐linked immunosorbent assay
- HFIP
1,1,1,3,3,3 hexafluoroisopropanol
- MAP2
microtubule associated protein 2
- MTT
3‐(4, 5‐dimethylthiazolyl‐2)‐2, 5‐diphenyltetrazolium bromide
- TFA
trifluoroacetic acid
- wt
wild type
1. INTRODUCTION
Accumulation of amyloid β (Aβ) protein in the brain is one of the hallmark pathologies of Alzheimer's disease (AD) (Selkoe & Hardy, 2016). Aβ is formed from the cleavage of amyloid precursor protein (APP) by proteolytic enzyme secretases. Cleavage by β‐site APP cleaving enzyme 1 (BACE1) generates both a soluble APP fragment and a membrane‐bound fragment. The latter can be subsequently heterogeneously cleaved by γ‐secretase to form Aβ(1‐40) and Aβ(1‐42) proteins (Esler & Wolfe, 2001; Jonsson et al., 2012). With age, a slowdown in the clearance rate (in sporadic AD) or overproduction (in autosomal dominant AD) of Aβ protein results in a higher concentration of Aβ within the brain. At these higher concentrations Aβ self‐associates into insoluble β‐sheet‐rich fibrils, or globular, water‐soluble, metastable intermediates known as oligomers (Urbanc et al., 2004). Most studies concur that on a molar monomer basis, oligomers are more toxic than fibrils (Cline et al., 2018; Goure et al., 2014; Koffie et al., 2011; Murphy & LeVine III, 2010). Moreover there is general agreement in the literature that Aβ oligomers are responsible for initiating the cascade of events that leads to neurotoxicity and neurodegeneration in the disease (Cline et al., 2018; Hayden & Teplow, 2013; Selkoe & Hardy, 2016). In vitro and in vivo data indicate that oligomers bind to synapses and reduce synapse number, ultimately, causing inhibition of synaptic plasticity, long‐term potentiation, and memory formation (Selkoe & Hardy, 2016; Shankar et al., 2008). However, the neurotoxic mechanism of oligomers in AD is still being elucidated.
To date, more than 200 human mutations that cause early‐onset familial AD have been identified. Notably, almost all of these mutations (in APP or presenilin, a key component of the γ‐secretase complex) result in a single phenotype, increased Aβ. The early‐onset mutations either increase the total amount of Aβ or the relative proportion of the longer Aβ(1‐42) form of the protein, the major species deposited in amyloid plaques in AD (Esler & Wolfe, 2001; Glabe, 2008; Hardy, 2017; Jonsson et al., 2012; Lesné et al., 2006; Selkoe, 2001; Stenh et al., 2002). The resultant accumulation of Aβ is identical to what is observed in the sporadic, late‐onset form of the disease, it just occurs earlier in life.
While many causative mutations are known, fewer protective mutations have been identified. Recently published genome‐wide association studies have revealed AD‐associated protective variants located in or near the following genes: APP, APOE, PLCG2, MS4A, MAPT‐KANSL1, RAB10, ABCA1, CCL11, SORL1, NOCT, SCL24A4‐RIN3, CASS4, EPHA1, SPPL2A, NFIC, and PPP4R3A (Andrews et al., 2019; Leigh et al., 2017). However, most confer a very small degree of protection against AD and very few have strong functional evidence of protection (Andrews et al., 2019). Interestingly, the only protective mutation that both significantly lowers AD incidence and also exhibits strong functional evidence of protection, A673T, affects the Aβ sequence, like all of the causative mutations. A673T, an APP coding variant, is also the only known protective mutation that results in lower plasma levels of Aβ(1‐42) [although levels in cerebrospinal fluid (CSF) or brain have not been measured] (Martiskainen et al., 2017).
In an Icelandic population, carriers of A673T were found to have a 4‐fold lower risk of AD as well as a lower risk of non‐AD age‐related cognitive decline (Jonsson et al., 2012). A673T, also known as A2T, results in a substitution of alanine to threonine at position 673 in the N‐terminal Aβ sequence. A673T occurs at the second amino acid residue of the Aβ(1‐42) protein sequence (Figure 1a) (Das et al., 2015; Jonsson et al., 2012; Maloney et al., 2014). This substitution reduces BACE1 cleavage of the APP β site, resulting in 40% lower Aβ(1‐42) accumulation in vitro (Jonsson et al., 2012; Kero et al., 2013; Maloney et al., 2014). The A673T mutation may also promote BACE1‐mediated cleavage of APP at the β’ site (between Thr681 and Gln682), generating Aβ(11‐42), the neurotoxicity of which is not yet fully understood (Jonson et al., 2015; Kimura et al., 2016; Liu et al., 2006).
FIGURE 1.
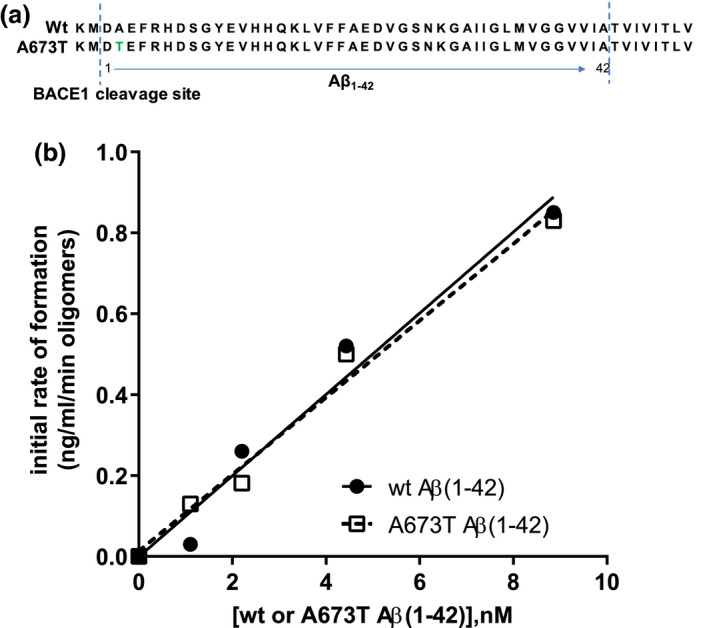
Wt and A673T mutant Aβ(1‐42) oligomers exhibit similar initial rates of assembly. Sequences of wt and mutant Aβ proteins illustrate the A to T substitution at position 2 relative to the β‐secretase cleavage site (a). Assembly of a range of concentrations of wt and A673T mutant Aβ(1‐42) monomeric proteins into oligomers was monitored over time (b). Oligomer content was determined by single‐site binding ELISA after oligomerization was halted by addition of Tween‐20. The slope of the assembly rate of oligomers over the initial 5 min (0, 1, 2, 5 min) was calculated via linear regression. No detectable difference between wt and A673T mutant proteins in initial rate of oligomer formation was observed (linear regression for slope difference, p = .67, F = .195). Dashed and solid lines represent linear regression fits to the data points. Results were obtained from N = 2 independent ELISA experiments (technical replicates)
Recent measurements revealing that A673T mutation carriers have 28% lower levels of Aβ(1‐42) levels in plasma support the model that a reduction in Aβ(1‐42) monomer concentration provides protection against AD (Bussy et al., 2019; Martiskainen et al., 2017). Attempts to recapitulate this lower monomer concentration in AD patients via inhibition of β‐ or γ‐secretase‐mediated production of Aβ or monomer‐selective antibody therapy have not met with clinical success, and the secretase inhibition approach has been largely discontinued due to worsening cognitive function (Panza et al., 2019). The A673T mutation may also offer protection against AD by reducing oligomer and fibril concentrations in the brain, although this has not been directly measured in carriers of this mutation to date. Monoclonal antibodies that lower fibril concentration in the brain are currently being tested in the clinic (Panza et al., 2019).
In addition to lowering Aβ protein concentration (and therefore also lowering the concentration of the most toxic form of the protein, oligomers), the mutation may confer protection from AD in other important ways: reductions in the biophysical assembly rate, synaptic binding, toxicity, or stability of mutant oligomers compared to wt may be additive to the overall lower Aβ protein concentration, and greatly amplify the mutation's disease protective effects. Determining these properties of mutant protein oligomer is important for understanding disease biology and guiding effective therapy design.
When comparing the A673T protective mutation versus wild type (wt) Aβ oligomers, the chosen method of oligomer preparation is vital to the biological relevance of the study. To prepare well‐characterized synthetic Aβ oligomers, synthetic full length Aβ protein must first be rigorously treated to remove the fibril and oligomer structures that assemble even in the solid state, before allowing re‐assembly to occur under well‐controlled conditions. Currently, two main preparation methods are widely used in publications that study the biochemistry of Aβ oligomer assembly: (1) the strong alkaline method (Ryan et al., 2013; Teplow, 2006) and (2) the anhydrous dimethyl sulfoxide (DMSO) method (Klein, 2002). The latter has two advantages: (1) it is more compatible with the pH conditions used in cell culture and (2) it produces oligomer size distributions more similar to those found in humans (Harper et al., 1999; Izzo, Xu, et al., 2014; Jan et al., 2010; LeVine, 2004; Sebollela et al., 2014). Oligomers produced using the DMSO method were shown to better replicate the biological effects of AD brain‐derived oligomers in vitro and in vivo (Izzo, Staniszewski, et al., 2014; Upadhaya et al., 2012; Yang et al., 2017).
The majority of published papers studying A673T mutant oligomer formation that provide a sufficiently detailed description of their oligomer preparation methods have used the strong alkaline method (Colombo et al., 2017; Murray et al., 2016; Poduslo & Howell, 2015; Zhang et al., 2018; Zheng et al., 2015). Collectively, these studies concluded that A673T Aβ oligomers (either Aβ(1‐40) or Aβ(1‐42) or a mixture of both) differ morphologically from their wt counterparts. This result was corroborated in a study that used the DMSO preparation method to facilitate the formation of oligomers under more physiologic conditions (Benilova et al., 2014). In addition, this study used atomic force microscopy to visualize the effect of the A673T mutation on equimolar mixes of A673T Aβ(1‐40) or A673T Aβ(1‐42) oligomers with their wt equivalents. They concluded that A673T oligomers, no matter the species, formed smaller aggregates overall when compared to their wt counterparts (Benilova et al., 2014). However, none of these studies measured the oligomerization rate or functional effects on neurons in vitro of full length pure Aβ(1‐42) A673T mutant oligomers prepared with the DMSO method.
In this study, we measured the effect of the A673T mutation on the oligomerization of the Aβ(1‐42) protein using the DMSO oligomer preparation method to yield synthetic oligomers that most closely resemble those derived from the AD brain. To examine the differences in the rate of oligomer formation of synthetic wt and A673T mutant Aβ oligomers in vitro, we measured oligomerization using a single‐site binding enzyme‐linked immunosorbent assay (ELISA) (LeVine, 2004). To evaluate the interaction between receptors and Aβ proteins, we quantified the amount and intensity of synaptic punctate binding of both oligomer preparations to DIV21 primary hippocampal/cortical neuron and glia cultures in vitro. We also explored the functional effects of the mutation on neurons via a vesicle trafficking assay. To the best of our knowledge, this study is the first of its kind to study the A673T Aβ(1‐42) oligomer assembly rate and functional effects in neurons using the more physiologically relevant method, defining a mechanism by which the A673T mutation protects against AD.
2. METHODS
2.1. Study design
The study was not pre‐registered on any open source registration platforms. All procedures were approved by the Institutional Animal Care and Use Committee at Cognition Therapeutics and were in compliance with the Office of Laboratory Animals Welfare (OLAW Assurance A4611‐01) and the Guide for the Care and Use of Laboratory Animals, Eighth Edition.
2.2. Neuronal cultures
Sprague‐Dawley rats (Taconic Biosciences; RRID: RGD_1566440) were separately housed for 4 days to acclimate, with water and food provided ad libitum. At 18 days of pregnancy they were euthanized by CO2 asphyxiation followed by cervical dislocation, and embryos were removed. Hippocampal/cortical cultures were prepared as previously described (Izzo, Staniszewski, et al., 2014; Izzo, Xu, et al., 2014). Briefly, dissociated embryonic day 18 (E18) hippocampal and cortical cells were plated on 384‐well poly‐D Lysine‐coated plates (Phenix Research, Candler, North Carolina, USA; catalog number MPG‐781946; year of purchase 2014) at a density of 4.6 × 104 cells per cm2 in Neurobasal Media (Life Technologies; catalog number 12348‐017; year of purchase 2014) supplemented with B27 (Life Technologies; catalog number 17504‐044; year of purchase 2014), Glutamax (Life Technologies; catalog number 35050‐061; year of purchase 2014), and antibiotics (penicillin 50 units/ml and streptomycin 50 μg/ml, Life Technologies; catalog number 15070‐063; year of purchase 2014). Cultures were maintained at 37°C in 5% CO2 for 3 weeks with weekly media changes prior to experimentation. Cultures contained 30% ± 8% MAP2 positive neurons (Figure 5). The remainder of cells were astrocytes, microglia, and other glial populations (Figure S1). The number of replicate neuronal culture experiments (cell culture preparations) is specified for each datapoint.
FIGURE 5.
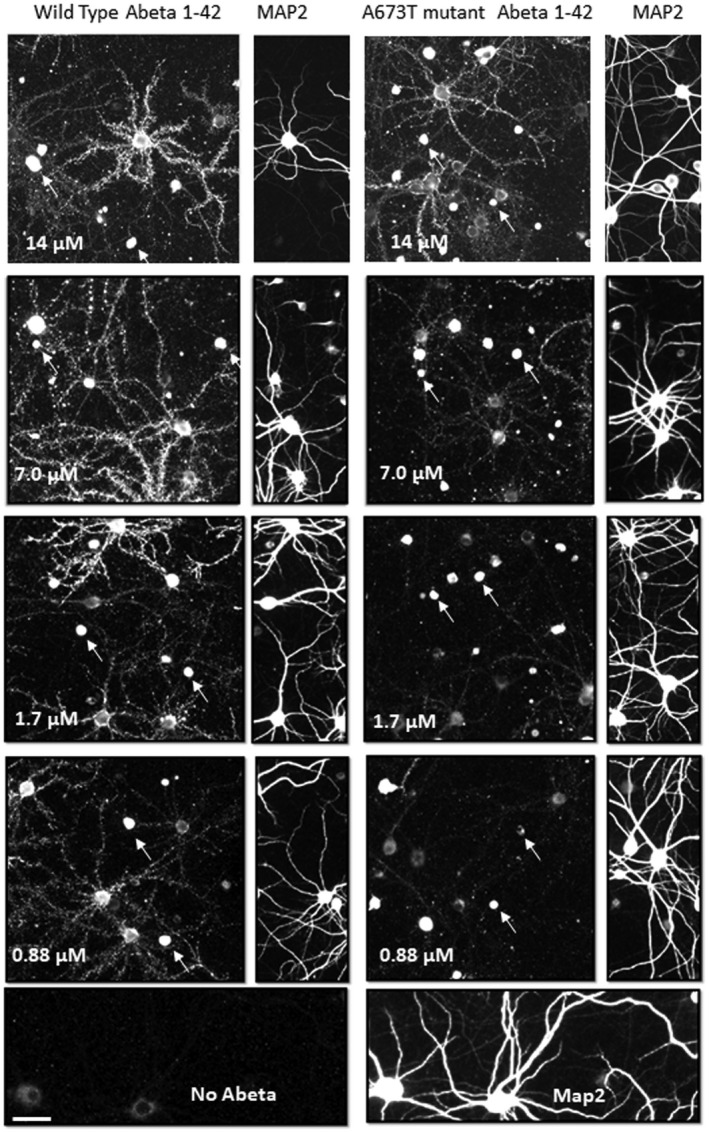
A673T Aβ(1‐42) oligomers bind with lower affinity to synaptic puncta than wt Aβ(1‐42) oligomers. Synthetic wt Aβ(1‐42) oligomers (a) and A673T Aβ(1‐42) oligomers (b) were added to rat neuronal cultures for 60 min at 37°C at total Aβ concentrations ranging from 0.88 to 14 μM. Oligomer binding to synaptic puncta was visualized via quantitative immunofluorescence. A subset of neurons (immunopositive for MAP2) exhibit punctate postsynaptic oligomer binding along their neurites. A673T oligomers labeled significantly fewer puncta than wt Aβ(1‐42) oligomers with the number of puncta for both proteins plateauing at approximately 7 μM (mean Aβ puncta count per neuron for wt = 5.0 × 105 ± 4.4 × 105, A673T = 2.5 × 105 ± 2.0 × 105, p < .05, paired t‐test). For each fluorescent image of bound Aβ, the MAP2 labeling on the right‐hand edge of each image demonstrates a similar density of neurons in each field. Arrows identify examples of Aβ binding to glia (non‐MAP2‐labeled cells). Scale bar = 20 microns. Results were obtained from N = 4 independent neuronal culture experiments
2.3. Aβ(1‐42) protein synthesis
Synthetic human wt and mutant Aβ(1‐42) proteins (custom synthesized at University of Pittsburgh Peptide and Peptoid Synthesis Core Facility; samples provided upon reasonable request) were treated according to previously published methods to remove any structural assemblies that may have formed during the synthesis, isolation, and storage procedures (Klein, 2002; Lambert et al., 2001). All oligomer concentrations are reported as monomer equivalents. An Aβ monomer film was prepared by evaporating the 1,1,1,3,3,3,hexafluoro‐2‐propanol (HFIP) (Sigma‐Aldrich; catalog number 105228; year of purchase 2014) at 20°C from a solution of 253 μg Aβ(1‐42) in HFIP at 20°C for 20 min using N2 gas. The film was then dissolved in 10.12 μL anhydrous DMSO (Sigma‐Aldrich; catalog number D2650; year of purchase 2014) to make a 5 mM working solution. This solution was diluted to 100 μM with cold Basal Media Eagle media (Life Technologies; catalog number 21010; year of purchase 2014), followed by incubation at 4°C for 24 hr to form oligomers. The resulting oligomer preparations were centrifuged at 16 000 g to pellet any insoluble fibrils and the supernatant was diluted in Neurobasal media prior to addition to cultures. All studies using synthetic oligomers were performed with this preparation unless otherwise specified. All lots of Aβ(1‐42) were put through a strict quality control process before being used for experiments, as previously published (Izzo, Staniszewski, et al., 2014).
2.4. Aβ(1‐42) oligomer assembly studies
Upon oligomerization from the monomeric state, Aβ(1‐42) protein can be found in monomeric, oligomeric, and fibril formations. The cleavage of APP generates Aβ protein of various lengths that assemble into structural forms ranging from low molecular weight monomers, to various sizes of oligomers, to high molecular weight fibrils. It is hypothesized that a disruption in the balance of production and clearance of the toxic Aβ oligomer isoform is the underlying cause of AD (Bao et al., 2012; Das et al., 2015; Glabe, 2008; Viola & Klein, 2015; Walsh & Selkoe, 2007; Walsh & Teplow, 2012). Characterization of synthetic wt Aβ(1‐42) and A673T Aβ(1‐42) has shown that, although conformational changes of the intrinsically disordered Aβ vary between the two preparations (Das et al., 2015), there is little effect of the A673T mutation on the detection of the protein when compared with wt Aβ by direct ELISA (Jonsson et al., 2012; Maloney et al., 2014). Monomer was allowed to assemble as described above and briefly below and oligomer content was determined by single‐site binding ELISA (LeVine, 2004) using 4G8 antibody to Aβ(1‐42) (Covance, Gaithersburg, Maryland, USA; catalog number SIG‐39330; RRID:AB_662804; year of purchase 2009). Briefly, wt and mutant Aβ(1‐42) were rigorously disaggregated in HFIP/trifluoroacetic acid (TFA) (Sigma‐Aldrich; catalog number T6508; year of purchase 2014) (LeVine, 2006) and dissolved at 50× the desired final concentration of protein in DMSO. Assembly reactions were initiated upon addition of protein to 1X phosphate buffered saline (20 mM sodium phosphate, 0.145 M NaCl, pH 7.5) and terminated at the desired time by the addition of Tween‐20 (0.1% v/v) (Fisher Bioreagents, Pittsburgh, Pennsylvania, USA; catalog number BP337‐500; year of purchase 2014). A673T and wt Aβ(1‐42) oligomer concentrations were compared via two‐way analysis of variance (ANOVA) and linear regression analysis.
2.5. Western blot analysis
Aβ oligomer preparations were run on a 4%–15% Tris‐HCl nondenaturing gel (Bio‐Rad Laboratories; catalog number 3450029; year of purchase 2014) in Tris‐Glycine buffer (Bio‐Rad Laboratories; catalog number 1610734; year of purchase 2014) at 125V for 110 min and transferred to 0.2 μM nitrocellulose (Bio‐Rad Laboratories; catalog number 1620112; year of purchase 2014) in Tris‐Glycine buffer at 30V for 120 min. The membrane was boiled in PBS for 5 min and blocked with 5% Blotto (Santa Cruz Biotechnology; catalog SC‐2324; year of purchase 2014) overnight at 4°C. The membrane was probed for 1 hr at 20°C with 6E10 monoclonal antibody (Covance; catalog number SIG‐39320; RRID:AB_662798; year of purchase 2014) diluted 1:100 in PBS + 1% Blotto then washed three times for 30 min with PBS + 0.05% Tween. Rabbit anti‐mouse‐HRP secondary antibody (MilliporeSigma, catalog number AP308P, year of purchase 2014) was applied at 1:100 for 1 hr at 20°C. The membrane was washed three times for 30 min with PBS + 0.05% Tween and developed with SuperSignal substrate (Pierce; catalog number 46640; year of purchase 2014) for 5 min and imaged with AlphaView SA (Alpha Innotech) using a CCD camera. Note that under these native, nondenaturing conditions, intrinsically disordered proteins such as Aβ oligomers and monomers run differently than well‐folded proteins (molecular weight protein size standards) and appear at different molecular sizes than expected (Tseng et al., 1999), therefore, monomers were assumed to run at ~25 kDa, although this was not confirmed by an independent control method. Images in Figure 4a show side‐by‐side lanes of wt and A673T mutant proteins prior to addition to cultures on the same gel; images in Figure 4b show a different gel with side‐by‐side lanes of wt and A673T mutant proteins after addition to cultures. Original images were acquired and optimized for publication quality identically.
FIGURE 4.
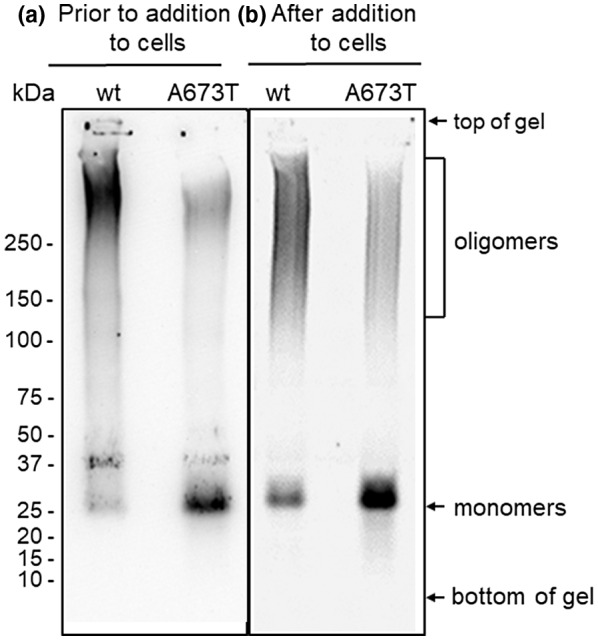
Native western blots of mutant and wt Aβ oligomer preparations before (a) and after (b) addition to hippocampal/cortical neuronal cultures. Western blot using 6E10 to detect wt and A673T Aβ(1‐42) shows mutant protein contains higher concentrations of monomer and lower concentrations of oligomers than wt
2.6. Aβ(1‐42) oligomer binding assay
Neuronal cultures were treated with increasing concentrations of oligomeric protein (wt or mutant) for 60 min at 37°C and automated immunofluorescent imaging was then used to assess the binding of oligomerized wt and mutant Aβ(1‐42) to neurons and glia (MAP2 negative cells) as described above and previously (Izzo, Staniszewski, et al., 2014). Briefly, cells were fixed in 3.75% formaldehyde (Polysciences; catalog number 4,018; year of purchase 2014) and blocked with 5% normal goat serum (Tissue Culture Biologicals; catalog number 701D; year of purchase 2014) and 0.5% Triton X‐100 (Sigma‐Aldrich; catalog number T8787; year of purchase 2014) before being incubated with primary antibodies for Aβ (1 μg/ml 6E10 or 4G8, Covance; catalog numbers SIG‐39320 and SIG‐39330, respectively; RRIDs:AB_662798 and AB_662804, respectively; year of purchase 2014) and MAP2 (0.2 μg/ml; Abcam, Cambridge, Massachusetts, USA; catalog number AB5392; RRID:AB_2138153; year of purchase 2014) and then fluorescently labeled secondary antibodies (2 μg/ml, goat anti‐chicken Alexa Fluor 546 and goat anti‐mouse Alexa Fluor 647, Life Technologies; catalog numbers A11040 and A21235, respectively; RRIDs:AB_2534097 and AB_2535804, respectively; year of purchase 2014). A Cellomics VTi automated microscope with a 20X, 0.75 numerical aperture objective was used to capture images, which were analyzed using the ThermoFisher/Cellomics Neuronal Profiling bioapplication that measured punctate labeling of Aβ along MAP2‐labeled neurites, as well as neuron count, nuclear area, and neurite length. A separate image analysis algorithm was used to measure Aβ binding to non‐MAP2 labelled cells as described previously (Izzo, Staniszewski, et al., 2014); DAPI‐labeled nuclei are used by the image processing algorithm as the basis for identifying cells versus non‐cellular objects such as aggregates. The image processing algorithm then measures Aβ immunofluorescence within the same region as the nuclei, so all Aβ measurements reported are associated with cells. The term ‘MAP2‐negative cells’ therefore refers only to nucleated cell bodies and not to non‐cellular aggregates. All quantification was performed on original images acquired identically as described above. Identical methods were used to optimize all images for publication.
2.7. Trafficking assay
Typically, the 3‐(4, 5‐dimethylthiazolyl‐2)‐2, 5‐diphenyltetrazolium bromide (MTT) assay is used as a measure of toxicity in cultured cells. Yellow tetrazolium salts undergo endocytosis and are then reduced to purple formazan. The amount of formazan reflects the number of metabolically active cells and thus diminished formazan is a measure of cell death. However, the MTT assay can also be used to measure the trafficking rate of endosomal/lysosomal vesicles in response to the addition of synthetic Aβ oligomer (Hong et al., 2007; Izzo, Staniszewski, et al., 2014; Kreutzmann et al., 2010; Liu & Schubert, 1997). One hour after the administration of tetrazolium salt, vehicle‐treated cells are full of formazan‐containing vesicles while synthetic Aβ oligomer‐treated cells have released the contents of their vesicles through exocytosis, leading to observable needle‐shaped crystals at the membrane surface (formed when the water‐insoluble formazan encounters the aqueous extracellular environment). Since Aβ oligomers accelerate the process of exocytosis, reduced intracellular vesicular formazan is observed along with synaptic loss (Izzo, Staniszewski, et al., 2014). The assay was used to measure the potency of A673T mutant and wt Aβ oligomers in altering vesicular trafficking rate and executed as described previously (Izzo, Staniszewski, et al., 2014). Briefly, after being treated with Aβ oligomer preparations, neurons were incubated for 24 hr at 37°C (the time point that achieves the maximal effect (Izzo, Staniszewski, et al., 2014)). They were then incubated with tetrazolium salts (3‐(4,5‐dimethylthiazol‐2yl)‐2,5‐diphenyl tetrazolium bromide, (VWR, Monroeville, Pennsylvania, USA; catalog number 80108‐190; year of purchase 2014); final concentration of 0.75 mM) for 60–90 min. After extraction with 1.6% Tween‐20, absorbance spectrometry was used to quantify the vesicular formazan remaining in cells.
2.8. Blinding, randomization, and statistical analysis
No blinding or randomization of experimental groups was conducted. For replicate experiments utilizing image analysis, at least 100 neurons were sampled using unbiased automated algorithms from four replicate wells for each experimental condition (400–500 neurons per experimental condition). The number of replicates from separate cell culture preparations is reported for each experiment. The number of replicates were determined a priori to attain statistical power of 80% and a p‐value of less than 0.05 (determined using G*Power software, Heinrich Heine University, Düsseldorf, Germany) (Faul et al., 2007). Statistical significance was determined for non‐linear curve fitting by an extra sum of squares F‐test using Prism (GraphPad Software). A D'Agostino‐Pearson normality test was used prior to all statistical analyses. Outliers were not removed.
3. RESULTS
3.1. Characterization of Aβ oligomer preparations via single‐site binding ELISA and western blotting
Quantification of oligomers is essential to understanding the physiological differences between protein variations (Figure 1a). The single‐site binding ELISA used here, in which the epitopes of the capture antibody (monoclonal antibody 4G8) and the detection antibody (biotinylated 4G8) are identical, allows the specific determination of the total quantity of oligomer existent in each Aβ(1‐42) preparation (Esparza et al., 2013; LeVine, 2004; Yang et al., 2013). This assay has been shown to be sensitive to oligomer preparations in concentrations ranging from 0.2 to 1,000 ng/ml (LeVine, 2004). Using this ELISA configuration, there was no detectable difference between the wt and A673T mutant proteins in initial rate of oligomer formation (linear regression for slope difference, p = .67, F = .195; Figure 1b). However, at specified concentrations, monomeric A673T Aβ(1‐42) formed 50% or fewer oligomers than wt Aβ(1‐42) when allowed to assemble for 30 min (two‐way ANOVA p < .0001; Figure 2). Consistent with this finding, mixing of the two protein monomers prior to assembly resulted in oligomer formation proportional to the molar ratio of wt and A673T Aβ(1‐42) concentrations (p < .05 for linearity for all proportions; Figure 3).
FIGURE 2.
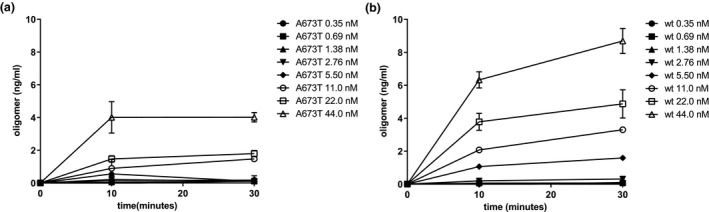
Monomeric A673T Aβ(1‐42) forms fewer oligomers than wt Aβ(1‐42) at specified concentrations and times. Monomeric wt (a) and A673T mutant (b) Aβ(1‐42) proteins were assembled at a series of concentrations in vitro for 10 or 30 min before assembly was stopped with Tween‐20. Oligomer content was determined by single‐site binding ELISA. At specified concentrations, monomeric A673T Aβ(1‐42) formed 50% or fewer oligomers than wt Aβ(1‐42) when allowed to assemble for 30 min (two‐way ANOVA p < .0001). Plotted values are mean ± SD. Results were obtained from N = 3 independent ELISA experiments (technical replicates)
FIGURE 3.
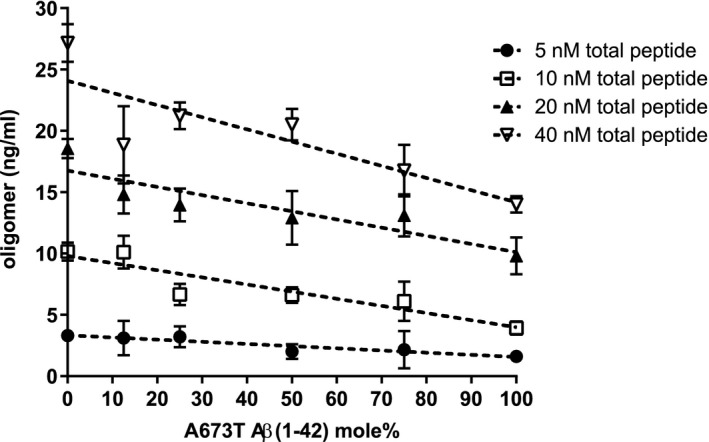
Mixing monomeric A673T mutant Aβ(1‐42) with wt Aβ(1‐42) decreases oligomer formation proportionally. Increasing proportions of monomeric A673T mutant Aβ(1‐42) were mixed with the wt protein in HFIP, disaggregated with TFA, dried, and dissolved in DMSO. Oligomer assembly was initiated to produce total protein concentrations of 5, 10, 20, and 40 nM and incubated for 30 min at 20°C before stopping with Tween‐20. Oligomer content was determined via single‐site ELISA resulting in oligomer formation proportional to the molar ratio of wt and A673T Aβ(1‐42) concentrations (p < .05 for linearity for all proportions). Data points represent mean ± SD. Dashed lines are linear regression fits to the data points. Results were obtained from N = 4 independent ELISA experiments (technical replicates)
Samples of wt and A673T mutant protein (1 μg of total Aβ) were analyzed on non‐denaturing western blots both before being added to cell culture media (Figure 4a) and after (Figure 4b) being incubated in cell culture media for 24 hr. Analysis of Aβ preparations from both conditions confirmed ELISA results shown in Figure 2, and demonstrates that in equimolar samples of protein, mutant protein contains less oligomer and more monomer than wt. Note that under these native, nondenaturing conditions, intrinsically disordered proteins such as Aβ oligomers and monomers run differently than well‐folded proteins (such as molecular weight protein size standards) and appear at different molecular sizes than expected (Tseng et al., 1999).
3.2. Comparison of mutant and wt Aβ(1‐42) oligomer binding to neurons and glia
We previously reported robust characterization of the binding of wt Aβ(1‐42) oligomer made with the same method to synapses on neurons (including colocalization of Aβ oligomers with the synaptic marker synaptophysin (Izzo, Staniszewski, et al., 2014; Figure S1) and demonstrated it to be consistent with a saturable, single‐site binding model (Izzo, Staniszewski, et al., 2014; Izzo, Xu, et al., 2014; Laurén et al., 2009). To examine whether differences in binding to neuronal cultures exist between A673T and wt Aβ(1‐42) protein, neuronal cultures were treated with increasing concentrations of oligomeric protein (wt or mutant, Figure 5), and Aβ binding to synaptic puncta was measured using quantitative immunofluorescence. Similar to previous observations (Izzo, Staniszewski, et al., 2014; Izzo, Xu, et al., 2014), wt Aβ(1‐42) oligomers exhibited saturable single‐site binding (K d = 442 ±70 nM, Figure 6a and b and Table 1), as did A673T oligomers, however, mutant oligomers bound with significantly lower affinity (K d = 1,950 ± 502 nM, one‐site specific binding least squares fit p < .001, Figure 6a and b and Table 1) and exhibited lower intensity binding than wt oligomers (Figure 6c and d). The number of binding puncta per neuron showed that wt oligomers bound to more synaptic puncta at each concentration compared to the A673T oligomers (Figure 6e and f). Taken together, these results suggest that mutant protein oligomers have a lower affinity for synaptic‐binding sites when compared to wt.
FIGURE 6.
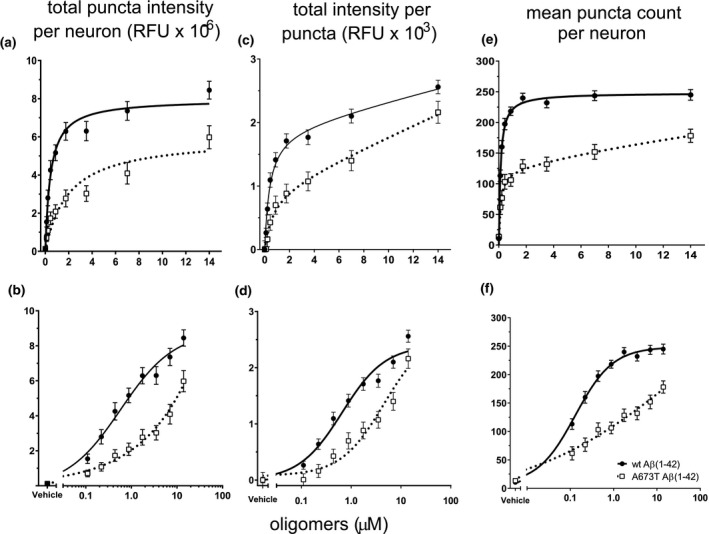
A673T Aβ(1‐42) oligomers bind less, and with lower affinity as well as intensity, to synaptic puncta than wt Aβ(1‐42) oligomers. Synthetic wt Aβ(1‐42) protein and A673T Aβ(1‐42) oligomers were added to rat neuronal cultures for 60 min at 37°C at total Aβ concentrations ranging from 0.88 to 14 μM. Oligomer binding to synaptic puncta was visualized via quantitative immunofluorescence. (a) At equivalent protein concentrations, wt Aβ(1‐42) produced a 3‐fold higher total binding intensity than A673T. (b) Data from (a) displayed on a logarithmic scale to emphasize the difference in binding affinity (wt Aβ(1‐42) = 442 ± 70 nM and A673T mutant = 1,950 ± 732 nM, one‐site specific binding least squares fit model, p < .001). (c) Mean intensity per puncta shows brighter intensity at each concentration for wt oligomer when compared to A673T mutant oligomer on a linear scale. (d) Data from (c) displayed on a logarithmic scale (one‐site specific binding least squares fit model, p < .001). (e) Number of binding puncta per neuron graphed on linear and (f) logarithmic scales show that wt Aβ bound to more puncta at each concentration compared to A673T mutant (one‐site specific binding least squares fit model, p < .001). Wt Aβ(1‐42) oligomers and A673T mutant oligomers are represented by circles with solid lines and open squares with dotted lines, respectively. All data represent mean ± SEM from N = 16 independent neuronal culture experiments. Y‐axis labels are shown at the top of each column of data
TABLE 1.
Binding affinity of wt Aβ(1‐42) and A673T mutant Aβ oligomers to neuronal synaptic puncta
| K d (nM) | B max a | |
|---|---|---|
| wt Aβ(1‐42) oligomers | Site 1:442 ± 70 | 7.98 × 105 ± 0.29 × 105 |
| A673T mutant Aβ(1‐42) oligomers | Site 1:1,955 ± 502 | 5.98 × 105 ± 0.50 × 105 |
intensity in arbitrary fluorescent units.
Additional image analysis parameters from these oligomer binding assays were used to assess any potential toxicity of the oligomer treatments: neuronal counts, neuronal nuclear area, and neuronal neurite length (Figure 7). Small but significant decreases in nuclear area and neurite length were observed only at the highest (non‐physiological) concentration of oligomeric proteins tested (14 µM) with no statistical difference seen between mutant and wt Aβ.
FIGURE 7.
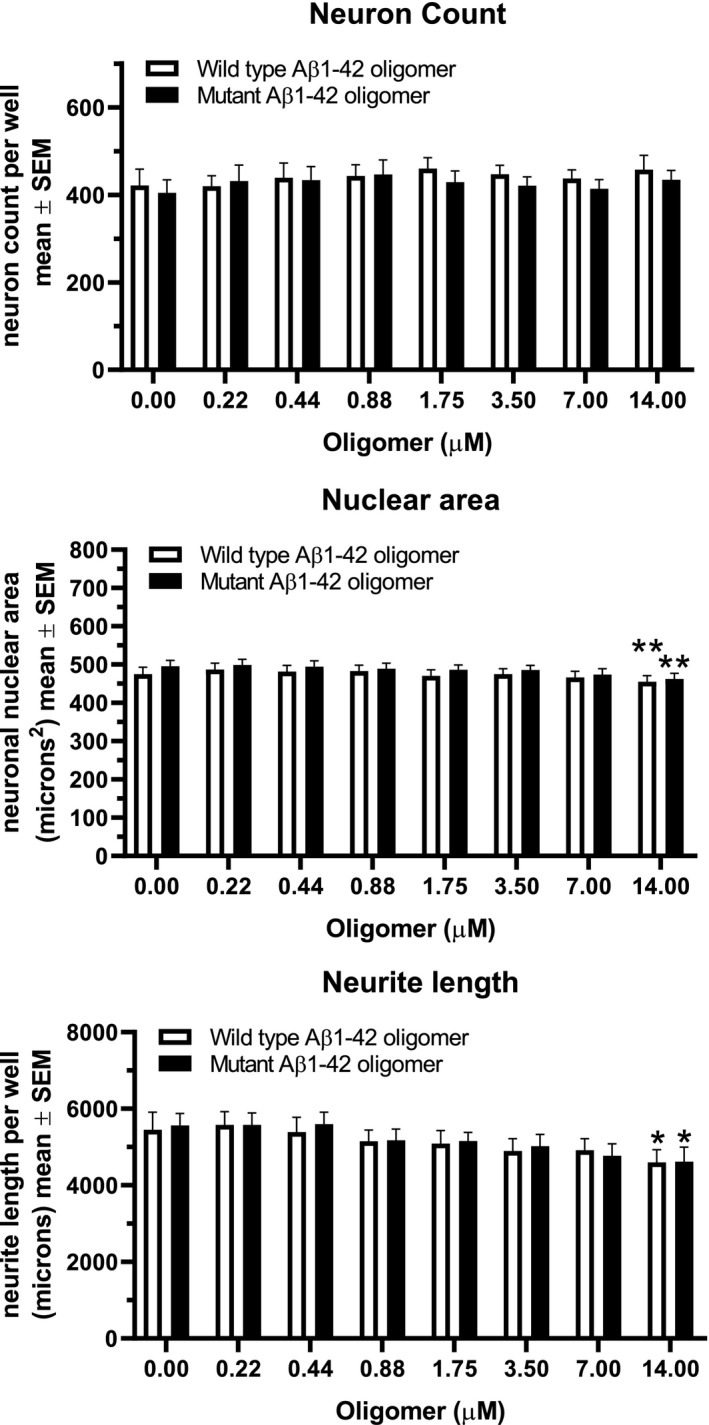
Toxicity measures in neuronal cultures. Neuron count per well, neuronal nuclear area and total neurite length per neuron were measured in Aβ oligomer binding experiments. Small effects on nuclear area and total neurite length were seen only at the highest concentration of oligomers used (14.0 µM, non‐physiological) with no difference seen between oligomers made from wt and mutant proteins (two‐way ANOVA with Tukey's post‐test correction for multiple comparisons). All data represent mean ± SEM from N = 13 independent neuronal culture experiments
We have previously characterized the detectable binding of wt Aβ(1‐42) oligomer made with the same method to subpopulations of glial cell bodies in neuronal cultures (Figure S1 and Figure 6 in Izzo, Staniszewski, et al., 2014). Binding intensity of ascending concentrations of Aβ to non‐MAP2 labeled cells (arrows in Figure 5) was measured. Similar to previous results, about 83% of the non‐neuronal cells had asymmetric or condensed nuclei and exhibited higher Aβ oligomer binding intensity than the remainder of the non‐neuronal cells which had round, symmetrical nuclei (Figure 8). In the non‐neuronal cells with asymmetric or condensed nuclei, no significant difference was seen between the affinity or intensity of wt Aβ and the A673T mutant oligomers (Figure 8a). Similar to previous studies, the binding of Aβ to these non‐neuronal cells fit a 2‐site binding model with a high affinity site (K d = 410 nM for wt, 382 nM for A673T mutant protein oligomers) and a second, unsaturable, low affinity site. Binding to non‐neuronal cells with round, symmetric nuclei was of low intensity and also showed no difference between wt Aβ and the A673T mutant (Figure 8b).
FIGURE 8.
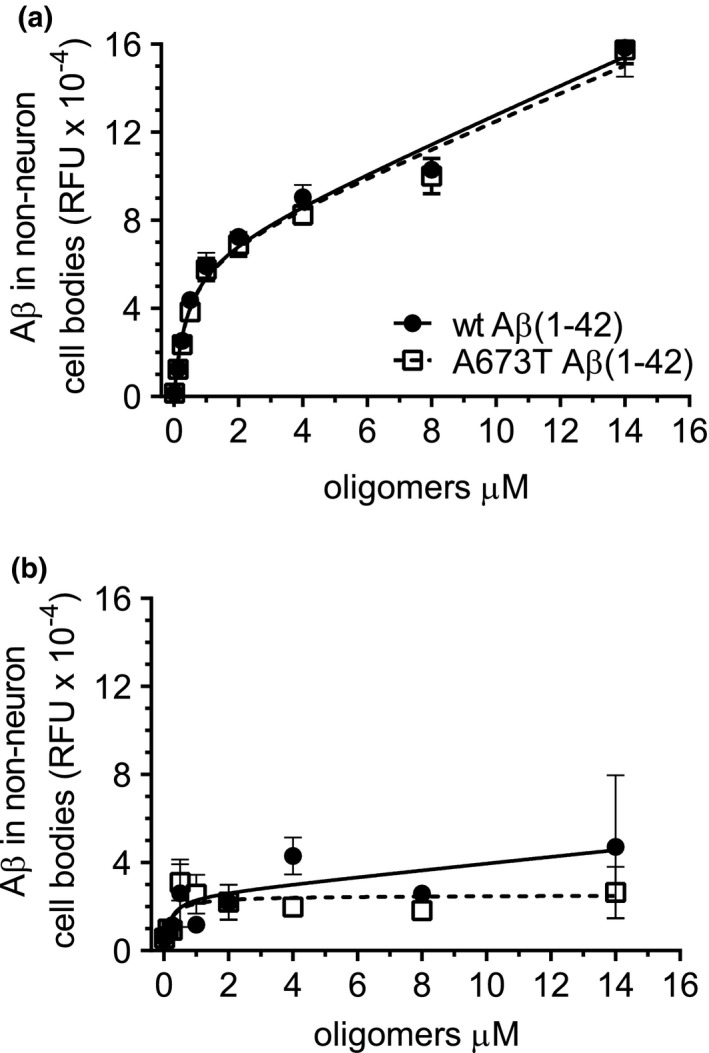
Similar binding affinities of wt Aβ(1‐42) and A673T Aβ(1‐42) mutant oligomers to glial cell bodies. Glial cells were characterized by a lack of MAP2 labeling. Nonlinear regression analysis revealed no significant difference in the binding of wt Aβ(1‐42) and A673T mutant Aβ(1‐42) oligomers to glia (MAP2 negative cells) with condensed or asymmetric nuclei (a) or to glia with round, symmetric nuclei (b). All data represent mean ± SEM from N = 3 independent neuronal culture experiments
3.3. Measurement of mutant and wt Aβ(1‐42) oligomer potency in primary neurons
The potency of mutant and wt synthetic Aβ oligomers to negatively impact trafficking rate is shown in Figure 9. The durations of treatment required to achieve maximal effects on membrane trafficking were similar; however, the mutant protein oligomers’ half maximal effective concentration (EC50 = 2.22 μM) was approximately two times smaller than the wt (EC50 = 4.73 μM, p < .001). These results indicate that the mutant protein oligomers are more potent than wt protein oligomers at inhibiting vesicle trafficking.
FIGURE 9.
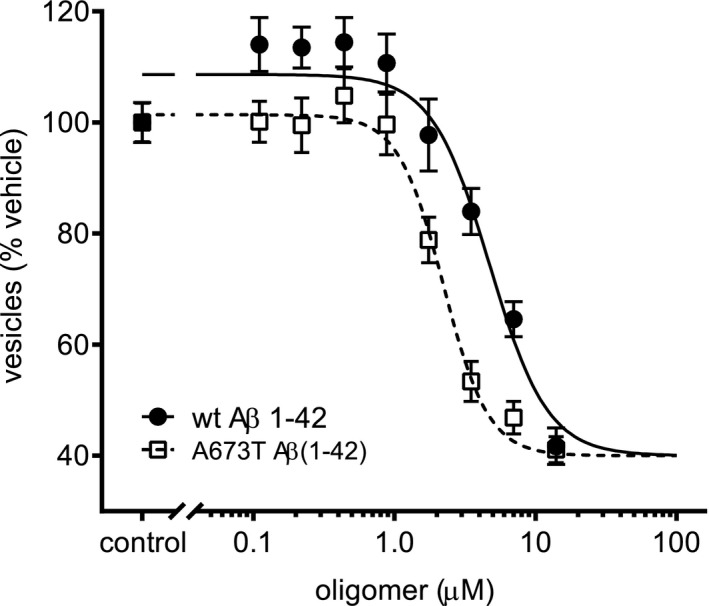
A673T mutant protein is more potent at inducing vesicle trafficking deficits than wt. Wt and mutant Aβ(1‐42) protein oligomer potencies were compared via vesicle trafficking assay. Mature primary neurons, 21 days in vitro, were treated for 24 hr with a particular protein oligomer at varying concentrations as indicated in the plot. The EC50 for A673T mutant and wt protein oligomers were 2.22 and 4.73 μM, respectively (variable slope sigmoidal dose–response curve fit was used to fit the data and an extra sum of squares F‐test was performed to show difference in potency, p < .001). These results indicate that the A673T mutant protein oligomers are more potent than wt protein oligomers at inhibiting vesicle trafficking. All data represent mean ± SEM from N = 16 independent neuronal culture experiments
4. DISCUSSION
The A673T mutation confers a 4‐fold lower risk of AD as well as a lower risk of non‐AD age‐related cognitive decline (Jonsson et al., 2012). Carriers have 28% lower levels of Aβ(1‐42) monomer levels in plasma as a result of lower β‐secretase cleavage efficiency (Bussy et al., 2019; Martiskainen et al., 2017), and presumably lower concentrations of the more toxic oligomer conformation as well. However, it is also possible that other characteristics of mutant protein may lower oligomer toxicity in a manner that is additive to, or even outweighs, overall lower monomer concentration contributions to protection from AD. Understanding the precise molecular basis by which the A673T protective mutation impacts Aβ structure, assembly and biology is essential to both understanding AD and designing the most effective therapeutic strategies, since drug candidates with the same mechanism as protective variants would be expected to have a higher success rate in the clinic (Harper et al., 2015).
This study is the first to examine the A673T mutation effects on both Aβ(1‐42) oligomer assembly rate and neuronal function using a physiologically relevant method of oligomer preparation. Using single‐site binding ELISA and size exclusion chromatography, we demonstrated that A673T mutant Aβ(1‐42) protein forms approximately 50% fewer high molecular weight (>50 kDa) oligomers compared to wt Aβ(1‐42) protein (Figure 2). This is supported by the finding that mixing wt and mutant oligomers together reduces the formation of oligomers in direct proportion to the concentration of mutant protein present. We also observed that A673T Aβ(1‐42) oligomer assembly plateaus at a lower level than wt Aβ(1‐42) oligomer assembly, with a decrease in the propensity to oligomerize that is dependent on the mole fraction of wt and A673T Aβ(1‐42) as measured by ELISA (Figures 2 and 3) and as visualized on western blots (Figure 4). This reduced propensity for A673T Aβ protein to aggregate into toxic oligomeric species could be due to a destabilizing feature of the mutation (Sharma et al., 2018). Our finding that there was no detectable difference between the wt and A673T mutant proteins in initial rate of oligomer formation, yet monomeric A673T Aβ(1‐42) formed 50% or fewer oligomers than wt Aβ(1‐42) after 30 min of assembly suggests that mutant oligomers may be less stable once formed than wt (e.g., oligomer dissociation rate is increased), although we did not examine this directly. Future studies that explicitly measure oligomer stability in physiological conditions are needed to clarify this possibility.
Several other reports have studied the effect of this mutated amino acid residue on the rate of aggregate assembly in vitro (Benilova et al., 2014; Colombo et al., 2017; Lin et al., 2017; Maloney et al., 2014; Murray et al., 2016; Poduslo & Howell, 2015; Somavarapu et al., 2017). Many of these reports show that A673T Aβ(1‐42) proteins aggregate more slowly than wt proteins via thioflavin T fluorescence assays that detect β‐sheet structure characteristic of fibrils (Benilova et al., 2014; Maloney et al., 2014; Murray et al., 2016). Zheng and colleagues demonstrated that A673T Aβ(1‐42) oligomers formed with the alkaline method have different size ranges than wt (Zheng et al., 2015). These findings support our conclusions that A673T Aβ(1‐42) oligomers assemble at a different rate than wt Aβ(1‐42) oligomers.
Aβ oligomers are believed to behave pharmacologically when binding to synapses as they exhibit saturable binding to their target and displacement by small molecule antagonists (Izzo, Staniszewski, et al., 2014; Izzo, Xu, et al., 2014; Maloney et al., 2014). We previously reported that synthetic Aβ(1‐42) oligomers made with the same method bind to synapses in primary hippocampal and cortical neuronal cultures (Izzo, Staniszewski, et al., 2014; Figure S1). In the current study, we found that wt Aβ(1‐42) oligomers bound with greater than 4‐fold higher affinity (K d = 442 nM ± 70nM) to synaptic neuronal puncta than did A673T Aβ(1‐42) oligomers (K d = 1,950 ± 502nM, Figure 5, Figure 6a and b, Table 1). In addition, A673T oligomers bound with lower intensity and to fewer puncta than did wt oligomers (Figure 6c and d). A673T Aβ(1‐42) also displayed a lower total binding intensity than wt Aβ at saturating concentrations and bound to fewer synaptic puncta, indicating the difference in binding is not simply a function of the lower oligomer concentration in the mutant oligomer preparation (Figure 6).
We previously reported that synthetic Aβ(1‐42) oligomers made with the same method bind to cell bodies of a subset of glia (MAP2 negative cells) (Izzo, Staniszewski, et al., 2014; Izzo, Xu, et al., 2014). In the present study, no difference in binding affinities of wt and A673T Aβ(1‐42) oligomers to glial cell bodies was observed (Figures 5 and 8), suggesting that, unlike neuronal synapses, binding to glia may occur via a different mechanism than in neuronal synapses.
Our results indicate that the mutant protein is twice as potent at inhibiting vesicle trafficking than wt (Figure 9), an effect that would be expected to negatively alter neuronal function. Corroborating our findings, Colombo et al. (2017) showed that A673T Aβ(1‐42) aggregates are more toxic than wt Aβ(1‐42) aggregates in a cell viability assay in neuroblastoma cells. Another study reported that the A673T variant results in the production of fewer Aβ(1‐42) oligomers, but that the oligomers produced are equally toxic to wt Aβ(1‐42) (Maloney et al., 2014).
It is important to note that neither wt nor A673T mutant protein produce rapid cell death at physiological concentrations, but instead produce subtler deficits in vesicle trafficking that reflect their impact on synaptic function (Izzo, Staniszewski, et al., 2014). In the present study, we observed a slight reduction in nuclear area and total neurite length (with no differences between the wt and mutant proteins) at non‐physiological (14 µM) concentrations, however this is approximately 100 times higher than the EC50 observed with either wt or mutant Aβ in the trafficking assay.
Other possible differences in the biophysical characteristics or biological impact of wt and A673T Aβ may exist but were not studied. It is possible that the mutated amino acid sequence could produce oligomers with a different conformation, resulting in altered interactions with downstream signaling proteins or even interactions with different downstream proteins which are not detected by the trafficking assay used in these studies. In fact, many studies show that A673T Aβ mutant aggregates are structurally distinct from wt aggregates (Benilova et al., 2014; Colombo et al., 2017; Lin et al., 2017; Murray et al., 2016; Poduslo & Howell, 2015; Zheng et al., 2015). This different shape could lead to binding to different receptors, which could account for lower synaptic affinity binding and/or increased toxicity of mutant protein oligomers. We cannot rule out either possibility.
Internalization of bound oligomers by cells is one potential pathway of lowering oligomer concentration, and is one of four major Aβ monomer clearance pathways (Mohamed & Posse de Chaves, 2011; Pomilio et al., 2016; Yuede et al., 2016). A previous study examining internalization of bound oligomers on primary microglia noted a higher degree of internalization with wt Aβ than A673T Aβ (Maloney et al., 2014). We did not examine internalization in the present study but did observe an equivalent binding of wt and A673T Aβ to glial cell bodies, suggesting that oligomer receptors on the cell body and synapses may be different. As mentioned above, mutant oligomers may have a higher dissociation rate compared to wt, which could affect their concentration. Additionally, increased protease cleavage in the brain is another possible source of lower concentration.
Based on the structural equilibrium between Aβ monomer, oligomer, and fibril, and supported by the A2T mutation's protective effect of reduced monomer production across a lifetime, several therapeutic approaches to lowering Aβ levels have been tested in clinical trials (reviewed recently in Aisen et al., 2020; Long & Holtzman, 2019; Walsh & Selkoe, 2020). Therapeutics that interact primarily with monomer, such as D‐enantiomeric peptide D3 derivatives that bind monomeric Aβ(1‐42), can lower Aβ(1‐42) oligomer and fibril formation and reduce Aβ(1‐42)‐induced cytotoxicity in preclinical model systems, but have challenges achieving stoichiometric concentrations in patients with the high concentrations of monomer that turn over rapidly (Klein et al., 2017). Tramiprosate, a compound that blocks the addition of monomer to fibrils with limited evidence of activity against oligomers under physiological conditions, did not show efficacy in two AD phase 3 trials (Aisen et al., 2011). Lowering Aβ monomer production with β‐ or γ‐secretase inhibitors led to cognitive worsening and this approach has been largely discontinued (Panza et al., 2019). Monoclonal antibodies with high affinity for monomer, such as solanezumab, have not been clinically efficacious (Doody et al., 2014; Honig et al., 2018). These therapeutics generally do not impact fibrillar Aβ load in the brain, leaving a large fibril concentration which can dissociate to form oligomers. N‐terminal monoclonal antibodies such as BAN2401 and aducanumab effectively clear fibril load from patients’ brains as assessed with amyloid PET imaging but also produce edema, and their impact on cognitive decline has yet to be clinically demonstrated (Logovinsky et al., 2016; Sevigny et al., 2016). Small molecule therapeutics that directly target oligomer formation have also been discovered. Hydroxyquinoline compounds block Aβ(1‐42) oligomer assembly directly (LeVine et al., 2009) but have not reached the clinic. Quinoline‐related PBT2, which interacts with metal ions and impacts the oligomeric configurations of Aβ (Ryan et al., 2015), failed to change cognitive function in a phase 2 trial (Villemagne et al., 2017).
Safely lowering Aβ concentrations in Alzheimer's patients without negatively impacting cognitive functioning as occurred following inhibition of β‐ or γ‐secretase enzymes may prove to be difficult, however the present results on the quantitative impact of the mutation on oligomer formation, binding, and functional impact suggest that alternative therapeutic approaches may be even more effective. The decreased APP β‐site cleavage resulting from the A673T mutation results in a 28% reduction in Aβ monomer concentration (Jonsson et al., 2012), which combines with the 50% lower oligomer formation due to the A673T substitution (Figure 2), potentially resulting in a >60% lower total oligomer concentration in A673T carriers than in wt individuals. However, the >4‐fold lower binding affinity of mutant oligomers could lower the amount of oligomer bound to neuronal synaptic receptors by more than 90%, to 10% of the original starting concentration. This is offset by an increase in potency of mutant oligomers at inhibiting membrane trafficking in neurons (making the 10% that do bind twice as effective). While many factors impacting these estimates in humans are unknown, the predominance of lowered binding affinity in quantitatively contributing to the 4‐fold protection of AD conferred by the mutation on carriers suggests that therapeutic approaches that lower binding affinity of oligomers may be productive.
Binding affinity can be lowered by drugs that antagonize or allosterically modulate oligomer receptors. While other structural states of Aβ bind nonsaturably to a variety of proteins, direct binding evidence with well‐characterized physiologically relevant Aβ oligomer preparations demonstrates that they bind saturably to a single receptor site at synapses composed of cellular prion protein, LilRB2, and Nogo (Kim et al., 2013; Park & Strittmatter, 2007; Smith & Strittmatter, 2017). Once bound to these receptor sites, oligomers cause failure of long‐term potentiation, induction of long‐term depression, inhibition of memory formation, and reduction of synapse number and cognitive performance (Selkoe & Hardy, 2016; Shankar et al., 2008). Therapeutics targeting the oligomer receptor constituent proteins LilRB2, prion, and Nogo have not reached the clinic, and it remains unclear whether they can be successfully inhibited or modulated with small molecule therapeutics. We have previously demonstrated that sigma‐2 receptors regulate this oligomer receptor complex, and sigma‐2 allosteric antagonists can destabilize the oligomer receptor binding site, increasing the off‐rate of oligomers (Izzo, Staniszewski, et al., 2014; Izzo, Xu, et al., 2014). Treatment of neurons with drug candidate CT1812 lowers Aβ oligomer binding affinity to neuronal synapses, restoring cognitive function in transgenic AD mice (Izzo et al., 2020). CT1812 is currently in multiple phase II clinical trials in Alzheimer's patients (NCT03522129, NCT03493282, NCT03507790).
This study is the first to examine the effect of the neuroprotective A673T mutation on the biophysical assembly kinetics and biological function in neurons of the most toxic structural form of Aβ protein, using a physiologically relevant oligomer preparation method. Aβ oligomers containing the A673T mutation form approximately 50% less oligomers than wt Aβ but are twice as potent at inhibiting vesicle trafficking rate in mature primary neurons and glia in vitro. However, mutant A673T Aβ oligomers have a >4‐fold lower binding affinity to synaptic receptors than wt. These observations indicate that the protective effect of the A673T mutation derives primarily from mutation‐containing oligomer's much lower binding affinity to synaptic receptors. This suggests that therapeutics that significantly reduce oligomer binding to synapses in the brain may replicate the protective effects of the A673T mutation and be effective treatments for Alzheimer's disease.
Supporting information
Fig S1
ACKNOWLEDGEMENTS
Harry LeVine III and Hank Safferstein are advisors to Cognition Therapeutics Inc. All other authors are full‐time employees of Cognition Therapeutics Inc. The authors report no other conflicts of interest and thank Allison Marin, Ph.D., for providing assistance with the preparation of this manuscript.
All experiments were conducted in compliance with the ARRIVE guidelines.
Limegrover CS, LeVine H III, Izzo NJ, et al. Alzheimer’s protection effect of A673T mutation may be driven by lower Aβ oligomer binding affinity. J Neurochem.2021;157:1316–1330. 10.1111/jnc.15212
FUNDING INFORMATION
HL received support from the National Institute of Neurological Disorders and Stroke (NS080576). All others received support from the Alzheimer's Drug Discovery Foundation (20100501), the National Institute of Neurodegenerative Disease and Stroke (NS083175), the National Institute of Aging (AG037337, AG047059, AG052252, AG052249, AG055247, AG055206, AG06212), and from Cognition Therapeutics, Inc.
REFERENCES
- Aisen, P. S. , Cummings, J. , Doody, R. , Kramer, L. , Salloway, S. , Selkoe, D. J. , Sims, J. , Sperling, R. A. , & Vellas, B. (2020). The future of anti‐amyloid trials. The Journal of Prevention of Alzheimer's Disease, 7, 146–151. [DOI] [PubMed] [Google Scholar]
- Aisen, P. S. , Gauthier, S. , Ferris, S. H. , Saumier, D. , Haine, D. , Garceau, D. , Duong, A. , Suhy, J. , Oh, J. , Lau, W. C. , & Sampalis, J. (2011). Tramiprosate in mild‐to‐moderate Alzheimer’s disease – a randomized, double‐blind, placebo‐controlled, multi‐centre study (the Alphase Study). Archives of Medical Science, 1, 102–111. 10.5114/aoms.2011.20612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews, S. J. , Fulton‐Howard, B. , & Goate, A. (2019). Protective variants in Alzheimer’s disease. Current Genetic Medicine Reports, 7, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao, F. , Wicklund, L. , Lacor, P. N. , Klein, W. L. , Nordberg, A. , & Marutle, A. (2012). Different β‐amyloid oligomer assemblies in Alzheimer brains correlate with age of disease onset and impaired cholinergic activity. Neurobiology of Aging, 33, 825.e1–13. 10.1016/j.neurobiolaging.2011.05.003 [DOI] [PubMed] [Google Scholar]
- Benilova, I. , Gallardo, R. , Ungureanu, A. A. , Castillo, C. V. , Snellinx, A. , Ramakers, M. , Bartic, C. , Rousseau, F. , Schymkowitz, J. , & De, S. B. (2014). The Alzheimer disease protective mutation Ala2Thr modulates kinetic and thermodynamic properties of abeta aggregation. Journal of Biological Chemistry, 289, 30977–30989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bussy, A. , Snider, B. J. , Coble, D. , Xiong, C. , Fagan, A. M. , Cruchaga, C. , Benzinger, T. L. S. , Gordon, B. A. , Hassenstab, J. , Bateman, R. J. , & Morris, J. C. (2019). Effect of apolipoprotein E4 on clinical, neuroimaging, and biomarker measures in noncarrier participants in the Dominantly Inherited Alzheimer Network. Neurobiology of Aging, 75, 42–50. 10.1016/j.neurobiolaging.2018.10.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cline, E. N. , Bicca, M. A. , Viola, K. L. , & Klein, W. L. (2018). The amyloid‐β oligomer hypothesis: Beginning of the third decade. Journal of Alzheimer's Disease, 64, S567–S610. 10.3233/JAD-179941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colombo, L. , Gamba, A. , Cantu, L. , Salmona, M. , Tagliavini, F. , Rondelli, V. , Favero, E. , & Del, B. P. (2017). Pathogenic Abeta A2V versus protective Abeta A2T mutation: Early stage aggregation and membrane interaction. Biophysical Chemistry, 229, 11–18. [DOI] [PubMed] [Google Scholar]
- Das, P. , Murray, B. , & Belfort, G. (2015). Alzheimer’s protective A2T mutation changes the conformational landscape of the Aβ₁₋₄₂ monomer differently than does the A2V mutation. Biophysical Journal, 108, 738–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doody, R. S. , Thomas, R. G. , Farlow, M. , Iwatsubo, T. , Vellas, B. , Joffe, S. , Kieburtz, K. , Raman, R. , Sun, X. , Aisen, P. S. , & Siemers, E. (2014). Phase 3 trials of solanezumab for mild‐to‐moderate Alzheimer’s disease. New England Journal of Medicine, 370, 311–321. [DOI] [PubMed] [Google Scholar]
- Esler, W. P. , & Wolfe, M. S. (2001). A portrait of Alzheimer secretases–new features and familiar faces. Science, 293, 1449–1454. [DOI] [PubMed] [Google Scholar]
- Esparza, T. J. , Zhao, H. , Cirrito, J. R. , Cairns, N. J. , Bateman, R. J. , Holtzman, D. M. , & Brody, D. L. (2013). Amyloid‐β oligomerization in Alzheimer dementia versus high‐pathology controls. Annals of Neurology, 73, 104–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faul, F. , Erdfelder, E. , Lang, A.‐G. , & Buchner, A. (2007). G*Power 3: A flexible statistical power analysis program for the social, behavioral, and biomedical sciences. Behavior Research Methods, 39, 175–191. [DOI] [PubMed] [Google Scholar]
- Glabe, C. G. (2008). Structural classification of toxic amyloid oligomers. Journal of Biological Chemistry, 283, 29639–29643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goure, W. F. , Krafft, G. A. , Jerecic, J. , & Hefti, F. (2014). Targeting the proper amyloid‐beta neuronal toxins: A path forward for Alzheimer’s disease immunotherapeutics. Alzheimer’s Res. Ther., 6, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy, J. (2017). The discovery of alzheimer causing mutations in the APP gene and the formulation of the “Amyloid Cascade Hypothesis”. FEBS Journal, 38, 42–49. [DOI] [PubMed] [Google Scholar]
- Harper, A. R. , Nayee, S. , & Topol, E. J. (2015). Protective alleles and modifier variants in human health and disease. Nature Reviews Genetics, 16, 689–701. [DOI] [PubMed] [Google Scholar]
- Harper, J. D. , Wong, S. S. , Lieber, C. M. , & Lansbury, P. T. (1999). Assembly of Aβ amyloid protofibrils: An in vitro model for a possible early event in Alzheimer’s disease. Biochemistry, 38, 8972–8980. [DOI] [PubMed] [Google Scholar]
- Hayden, E. , & Teplow, D. (2013). Amyloid β‐protein oligomers and Alzheimer’s disease. Alzheimer's Research & Therapy, 5, 60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong, H.‐S. , Maezawa, I. , Yao, N. , Xu, B. , Diaz‐Avalos, R. , Rana, S. , Hua, D. H. , Cheng, R. H. , Lam, K. S. , & Jin, L.‐W. (2007). Combining the rapid MTT formazan exocytosis assay and the MC65 protection assay led to the discovery of carbazole analogs as small molecule inhibitors of Abeta oligomer‐induced cytotoxicity. Brain Research, 1130, 223–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honig, L. S. , Vellas, B. , Woodward, M. , Boada, M. , Bullock, R. , Borrie, M. , Hager, K. , Andreasen, N. , Scarpini, E. , Liu‐Seifert, H. , Case, M. , Dean, R. A. , Hake, A. , Sundell, K. , Poole Hoffmann, V. , Carlson, C. , Khanna, R. , Mintun, M. , DeMattos, R. , … Siemers, E. (2018). Trial of solanezumab for mild dementia due to Alzheimer’s disease. New England Journal of Medicine, 378(4), 321–330. 10.1056/nejmoa1705971 [DOI] [PubMed] [Google Scholar]
- Izzo, N. J. , Staniszewski, A. , To, L. , Fa, M. , Teich, A. F. , Saeed, F. , Wostein, H. , Walko, T. , Vaswani, A. , Wardius, M. , Syed, Z. , Ravenscroft, J. , Mozzoni, K. , Silky, C. , Rehak, C. , Yurko, R. , Finn, P. , Look, G. , Rishton, G. , … Catalano, S. M. (2014). Alzheimer's therapeutics targeting amyloid beta 1–42 oligomers I: Abeta 42 oligomer binding to specific neuronal receptors is displaced by drug candidates that improve cognitive deficits. PLoS ONE, 9(11), e111898. 10.1371/journal.pone.0111898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izzo, N. J. , Xu, J. , Zeng, C. , Kirk, M. J. , Mozzoni, K. , Silky, C. , Rehak, C. , Yurko, R. , Look, G. , Rishton, G. , Safferstein, H. , Cruchaga, C. , Goate, A. , Cahill, M. A. , Arancio, O. , Mach, R. H. , Craven, R. , Head, E. , LeVine, H. , … Catalano, S. M. (2014). Alzheimer's therapeutics targeting amyloid beta 1–42 oligomers II: sigma‐2/PGRMC1 receptors mediate abeta 42 oligomer binding and synaptotoxicity. PLoS ONE, 9(11), e111899. 10.1371/journal.pone.0111899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izzo, N. J. , Yuede, C. M. , Limegrover, C. M. , LaBarbera, K. M. , Rehak, C. , Yurko, R. , Waybright, L. , Look, G. , Rishton, G. , Safferstein, H. , Hamby, M. E. , Williams, C. , Sadlek, K. , Edwards, H. M. , Davis, C. , Grundman, M. , Schneider, L. S. , DeKosky, S. T. , Chelsky, D. , … Catalano, S. M. (2020). Preclinical and clinical biomarker studies of CT1812: A novel approach to Alzheimer’s disease modification. Science Translational Medicine: Submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jan, A. , Hartley, D. M. , & Lashuel, H. A. (2010). Preparation and characterization of toxic Abeta aggregates for structural and functional studies in Alzheimer’s disease research. Nature Protocols, 5, 1186–1209. [DOI] [PubMed] [Google Scholar]
- Jonson, M. , Pokrzywa, M. , Starkenberg, A. , Hammarstrom, P. , & Thor, S. (2015) Systematic Aβ analysis in drosophila reveals high toxicity for the 1–42, 3–42 and 11–42 peptides, and emphasizes N‐ and C‐terminal residues. PLoSOne 10(7), e0133272. 10.1371/journal.pone.0133272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonsson, T. , Atwal, J. K. , Steinberg, S. , Snaedal, J. , Jonsson, P. V. , Bjornsson, S. , Stefansson, H. , Sulem, P. , Gudbjartsson, D. , Maloney, J. , Hoyte, K. , Gustafson, A. , Liu, Y. , Lu, Y. , Bhangale, T. , Graham, R. R. , Huttenlocher, J. , Bjornsdottir, G. , Andreassen, O. A. , … Stefansson, K. (2012). A mutation in APP protects against Alzheimer’s disease and age‐related cognitive decline. Nature, 488(7409), 96–99. 10.1038/nature11283 [DOI] [PubMed] [Google Scholar]
- Kero, M. , Paetau, A. , Polvikoski, T. , Tanskanen, M. , Sulkava, R. , Jansson, L. , Myllykangas, L. , & Tienari, P. J. (2013). Amyloid precursor protein (APP) A673T mutation in the elderly Finnish population. Neurobiology of Aging, 34, 1518.e1–3. [DOI] [PubMed] [Google Scholar]
- Kim, T. , Vidal, G. S. , Djurisic, M. , William, C. M. , Birnbaum, M. E. , Garcia, K. C. , Hyman, B. T. , & Shatz, C. J. (2013). Human LilrB2 is a β‐amyloid receptor and its murine homolog PirB regulates synaptic plasticity in an Alzheimer’s model. Science, 341, 1399–1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura A., Hata S., & Suzuki T. (2016) Alternative selection of β‐site APP‐cleaving enzyme 1 (BACE1) cleavage sites in amyloid β‐protein precursor (APP) harboring protective and pathogenic mutations within the Aβ sequence. Journal of Biological Chemistry 291(46), 24041–24053. 10.1074/jbc.M116.744722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein, A. N. , Ziehm, T. , van Groen, T. , Kadish, I. , Elfgen, A. , Tusche, M. , Thomaier, M. , Reiss, K. , Brener, O. , Gremer, L. , Kutzsche, J. , & Willbold, D. (2017). Optimization of d‐peptides for Aβ monomer binding specificity enhances their potential to eliminate toxic Aβ oligomers. ACS Chemical Neuroscience, 8(9), 1889–1900. 10.1021/acschemneuro.7b00045 [DOI] [PubMed] [Google Scholar]
- Klein, W. L. (2002). Abeta toxicity in Alzheimer’s disease: Globular oligomers (ADDLs) as new vaccine and drug targets. Neurochemistry International, 41, 345–352. [DOI] [PubMed] [Google Scholar]
- Koffie, R. M. , Hyman, B. T. , & Spires‐Jones, T. L. (2011). Alzheimer’s disease: Synapses gone cold. Molecular Neurodegeneration, 6, 63. 10.1186/1750-1326-6-63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreutzmann, P. , Wolf, G. , & Kupsch, K. (2010). Minocycline recovers MTT‐formazan exocytosis impaired by amyloid beta peptide. Cellular and Molecular Neurobiology, 30, 979–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert, M. P. , Viola, K. L. , Chromy, B. A. , Chang, L. , Morgan, T. E. , Yu, J. , Venton, D. L. , Krafft, G. A. , Finch, C. E. , & Klein, W. L. (2001). Vaccination with soluble Abeta oligomers generates toxicity‐neutralizing antibodies. Journal of Neurochemistry, 79, 595–605. [DOI] [PubMed] [Google Scholar]
- Laurén, J. , Gimbel, D. A. , Nygaard, H. B. , Gilbert, J. W. , & Strittmatter, S. M. (2009). Cellular prion protein mediates impairment of synaptic plasticity by amyloid‐β oligomers. Nature, 457, 1128–1132. 10.1038/nature07761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leigh, C. , Valerio, N. , Raiyan, R. K. , Summer, S. H. , & Michael, D. G. (2017). A variant in PPP4R3A protects against alzheimer‐related metabolic decline. Annals of Neurology, 118, 6072–6078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesné, S. , Koh, M. T. , Kotilinek, L. , Kayed, R. , Glabe, C. G. , Yang, A. , Gallagher, M. , & Ashe, K. H. (2006). A specific amyloid‐beta protein assembly in the brain impairs memory. Nature, 440, 352–357. [DOI] [PubMed] [Google Scholar]
- LeVine, H. (2004). Alzheimer’s beta‐peptide oligomer formation at physiologic concentrations. Analytical Biochemistry, 335, 81–90. [DOI] [PubMed] [Google Scholar]
- LeVine, H. (2006). Biotin‐avidin interaction‐based screening assay for Alzheimer’s beta‐peptide oligomer inhibitors. Analytical Biochemistry, 356, 265–272. [DOI] [PubMed] [Google Scholar]
- LeVine, H. , Ding, Q. , Walker, J. A. , Voss, R. S. , & Augelli‐Szafran, C. E. (2009). Clioquinol and other hydroxyquinoline derivatives inhibit Abeta(1–42) oligomer assembly. Neuroscience Letters, 465, 99–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, T. W. , Chang, C. F. , Chang, Y. J. , Liao, Y. H. , Yu, H. M. , & Chen, Y. R. (2017). Alzheimer’s amyloid‐β A2T variant and its Nterminal peptides inhibit amyloid‐β fibrillization and rescue the induced cytotoxicity. PLoS One, 12, 1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, K. , Solano, I. , Mann, D. , Lemere, C. , Mercken, M. , Trojanowski, J. Q. , & Lee, V. M. Y. (2006). Characterization of Abeta11‐40/42 peptide deposition in Alzheimer’s disease and young Down’s syndrome brains: Implication of N‐terminally truncated Abeta species in the pathogenesis of Alzheimer’s disease. Acta Neuropathologica, 112, 163–174. [DOI] [PubMed] [Google Scholar]
- Liu, Y. , & Schubert, D. (1997). Cytotoxic amyloid peptides inhibit cellular 3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyltetrazolium bromide (MTT) reduction by enhancing MTT formazan exocytosis. Journal of Neurochemistry, 69, 2285–2293. [DOI] [PubMed] [Google Scholar]
- Logovinsky, V. , Satlin, A. , Lai, R. , Swanson, C. , Kaplow, J. , Osswald, G. , Basun, H. , & Lannfelt, L. (2016). Safety and tolerability of BAN2401 ‐ A clinical study in Alzheimer’s disease with a protofibril selective Aβ antibody. Alzheimer's Research and Therapy, 8, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long, J. M. , & Holtzman, D. M. (2019). Alzheimer disease: An update on pathobiology and treatment strategies. Cell, 179, 312–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maloney, J. A. , Bainbridge, T. , Gustafson, A. , Zhang, S. , Kyauk, R. , Steiner, P. , van der Brug, M. , Liu, Y. , Ernst, J. A. , Watts, R. J. , & Atwal, J. K. (2014). Molecular mechanisms of Alzheimer disease protection by the A673T allele of amyloid precursor protein. Journal of Biological Chemistry, 289(45), 30990–31000. 10.1074/jbc.m114.589069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martiskainen, H. , Herukka, S.‐K.‐K. , Stančáková, A. , Paananen, J. , Soininen, H. , Kuusisto, J. , Laakso, M. , & Hiltunen, M. (2017). Decreased plasma β‐amyloid in the Alzheimer’s disease APP A673T variant carriers. Annals of Neurology, 82, 128–132. [DOI] [PubMed] [Google Scholar]
- Mohamed, A. , & Posse de Chaves, E. (2011). Aβ internalization by neurons and glia. Journal of Alzheimer's Disease, 2011, 127984–128001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy, M. , & LeVine, H. III (2010). Alzheimer’s disease and the β‐amyloid peptide. J. Alzheimer’s Dis., 19, 311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray, B. , Sorci, M. , Rosenthal, J. , Lippens, J. , Isaacson, D. , Das, P. , Fabris, D. , Li, S. , & Belfort, G. (2016). A2T and A2V Aβ peptides exhibit different aggregation kinetics, primary nucleation, morphology, structure and LTP inhibition. Proteins: Structure, Function, and Bioinformatics, 84, 488–500. [DOI] [PubMed] [Google Scholar]
- Panza, F. , Lozupone, M. , Logroscino, G. , & Imbimbo, B. P. (2019). A critical appraisal of amyloid‐β‐ targeting therapies for Alzheimer disease. Nature Reviews. Neurology, 15, 73–88. [DOI] [PubMed] [Google Scholar]
- Park, J. , & Strittmatter, S. (2007). Nogo receptor interacts with brain APP and Aβ to reduce pathologic changes in Alzheimers transgenic mice. Current Alzheimer Research, 4, 568–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poduslo, J. F. , & Howell, K. G. (2015). Unique molecular signatures of Alzheimer’s disease amyloid β peptide mutations and deletion during aggregate/oligomer/fibril formation. Journal of Neuroscience Research, 93, 410–423. [DOI] [PubMed] [Google Scholar]
- Pomilio, C. , Pavia, P. , Gorojod, R. M. , Vinuesa, A. , Alaimo, A. , Galvan, V. , Kotler, M. L. , Beauquis, J. , & Saravia, F. (2016). Glial alterations from early to late stages in a model of Alzheimer’s disease: Evidence of autophagy involvement in Aβ internalization HHS public access. Hippocampus, 26, 194–210. 10.1002/hipo.22503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan, T. M. , Caine, J. , Mertens, H. D. T. , Kirby, N. , Nigro, J. , Breheney, K. , Waddington, L. J. , Streltsov, V. A. , Curtain, C. , Masters, C. L. , & Roberts, B. R. (2013). Ammonium hydroxide treatment of Aβ produces an aggregate free solution suitable for biophysical and cell culture characterization. PeerJ, 1, e73. 10.7717/peerj.73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan, T. M. , Roberts, B. R. , McColl, G. , Hare, D. J. , Doble, P. A. , Li, Q.‐X. , Lind, M. , Roberts, A. M. , Mertens, H. D. T. , Kirby, N. , Pham, C. L. L. , Hinds, M. G. , Adlard, P. A. , Barnham, K. J. , Curtain, C. C. , & Masters, C. L. (2015). Stabilization of nontoxic A ‐Oligomers: Insights into the mechanism of action of hydroxyquinolines in Alzheimer's disease. Journal of Neuroscience, 35(7), 2871–2884. 10.1523/jneurosci.2912-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sebollela, A. , Mustata, G.‐M. , Luo, K. , Velasco, P. T. , Viola, K. L. , Cline, E. N. , Shekhawat, G. S. , Wilcox, K. C. , Dravid, V. P. , & Klein, W. L. (2014). Elucidating molecular mass and shape of a neurotoxic Aβ oligomer. ACS Chemical Neuroscience, 5(12), 1238–1245. 10.1021/cn500156r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selkoe, D. J. (2001). Alzheimer’s disease: Genes, proteins, and therapy. Physiological Reviews, 81, 741–766. 10.1152/physrev.2001.81.2.741 [DOI] [PubMed] [Google Scholar]
- Selkoe, D. J. , & Hardy, J. (2016). The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Molecular Medicine, 8, 595–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sevigny, J. , Chiao, P. , Bussière, T. , Weinreb, P. H. , Williams, L. , Maier, M. , Dunstan, R. , Salloway, S. , Chen, T. , Ling, Y. , O’Gorman, J. , Qian, F. , Arastu, M. , Li, M. , Chollate, S. , Brennan, M. S. , Quintero‐Monzon, O. , Scannevin, R. H. , Arnold, H. M. , … Sandrock, A. (2016). The antibody aducanumab reduces Aβ plaques in Alzheimer’s disease. Nature, 537, 50–56. 10.1038/nature19323 [DOI] [PubMed] [Google Scholar]
- Shankar, G. M. , Li, S. , Mehta, T. H. , Garcia‐Munoz, A. , Shepardson, N. E. , Smith, I. , Brett, F. M. , Farrell, M. A. , Rowan, M. J. , Lemere, C. A. , Regan, C. M. , Walsh, D. M. , Sabatini, B. L. , & Selkoe, D. J. (2008). Amyloid‐β protein dimers isolated directly from Alzheimer's brains impair synaptic plasticity and memory. Nature Medicine, 14(8), 837–842. 10.1038/nm1782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma, B. , Ranganathan, S. , & Belfort, G. (2018). Weaker N‐terminal interactions for the protective over the causative Aβ peptide dimer mutants. ACS Chemical Neuroscience, 9, 1247–1253. [DOI] [PubMed] [Google Scholar]
- Smith, L. M. , & Strittmatter, S. M. (2017). Binding sites for amyloid‐β oligomers and synaptic toxicity. Cold Spring Harbor Perspectives in Medicine, 7, a024075. 10.1101/cshperspect.a024075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somavarapu, A. K. , Shen, F. , Teilum, K. , Zhang, J. , Mossin, S. , Thulstrup, P. W. , Bjerrum, M. J. , Tiwari, M. K. , Szunyogh, D. , Søtofte, P. M. , Kepp, K. P. , & Hemmingsen, L. (2017). The pathogenic A2V mutant exhibits distinct aggregation kinetics, metal site structure, and metal exchange of the Cu2+ ‐Aβ complex. Chemistry ‐ A European Journal, 23(55), 13591–13595. 10.1002/chem.201703440 [DOI] [PubMed] [Google Scholar]
- Stenh, C. , Nilsberth, C. , Hammarbäck, J. , Engvall, B. , Näslund, J. , & Lannfelt, L. (2002). The Arctic mutation interferes with processing of the amyloid precursor protein. NeuroReport, 13, 1857–1860. 10.1097/00001756-200210280-00005 [DOI] [PubMed] [Google Scholar]
- Teplow, D. B. (2006). Preparation of amyloid β‐protein for structural and functional studies. Methods in Enzymology, 413, 20–33. [DOI] [PubMed] [Google Scholar]
- Tseng, B. P. , Esler, W. P. , Clish, C. B. , Stimson, E. R. , Ghilardi, J. R. , Vinters, H. V. , Mantyh, P. W. , Lee, J. P. , & Maggio, J. E. (1999). Deposition of monomeric, not oligomeric, Abeta mediates growth of Alzheimer’s disease amyloid plaques in human brain preparations. Biochemistry, 38, 10424–10431. [DOI] [PubMed] [Google Scholar]
- Upadhaya, A. R. , Lungrin, I. , Yamaguchi, H. , Fändrich, M. , & Thal, D. R. (2012). High‐molecular weight Aβ oligomers and protofibrils are the predominant Aβ species in the native soluble protein fraction of the AD brain. Journal of Cellular and Molecular Medicine, 16, 287–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urbanc, B. , Cruz, L. , Yun, S. , Buldyrev, S. V. , Bitan, G. , Teplow, D. B. , & Stanley, H. E. (2004). In silico study of amyloid ‐protein folding and oligomerization. Proceedings of the National Academy of Sciences, 101, 17345–17350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villemagne, V. L. , Rowe, C. C. , Barnham, K. J. , Cherny, R. , Woodward, M. , Bozinosvski, S. , Salvado, O. , Bourgeat, P. , Perez, K. , Fowler, C. , Rembach, A. , Maruff, P. , Ritchie, C. , Tanzi, R. , & Masters, C. L. (2017). A randomized, exploratory molecular imaging study targeting amyloid β with a novel 8‐OH quinoline in Alzheimer's disease: The PBT2‐204 IMAGINE study. Alzheimer's & Dementia: Translational Research & Clinical Interventions, 3(4), 622–635. 10.1016/j.trci.2017.10.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viola, K. L. , & Klein, W. L. (2015). Amyloid beta oligomers in Alzheimer’s disease pathogenesis, treatment, and diagnosis. Acta Neuropathologica, 129, 183–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh, D. M. , & Selkoe, D. J. (2007). Abeta oligomers ‐ A decade of discovery. Journal of Neurochemistry, 101, 1172–1184. 10.1111/j.1471-4159.2006.04426.x [DOI] [PubMed] [Google Scholar]
- Walsh, D. M. , & Selkoe, D. J. (2020). Amyloid β‐protein and beyond: The path forward in Alzheimer’s disease. Current Opinion in Neurobiology, 61, 116–124. 10.1016/j.conb.2020.02.003 [DOI] [PubMed] [Google Scholar]
- Walsh, D. M. , & Teplow, D. B. (2012). Alzheimer’s disease and the amyloid β‐protein. Progress in Molecular Biology and Translational Science, 107, 101–124. [DOI] [PubMed] [Google Scholar]
- Yang, T. , Hong, S. , O’Malley, T. , Sperling, R. A. , Walsh, D. M. , & Selkoe, D. J. (2013). New ELISAs with high specificity for soluble oligomers of amyloid β‐protein detect natural Aβ oligomers in human brain but not CSF. Alzheimer's & Dementia, 9, 99–112. 10.1016/j.jalz.2012.11.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, T. , Li, S. , Xu, H. , Walsh, D. M. , & Selkoe, D. J. (2017). Large soluble oligomers of amyloid β‐protein from Alzheimer brain are far less neuroactive than the smaller oligomers to which they dissociate. Journal of Neuroscience, 37, 152–163. 10.1523/JNEUROSCI.1698-16.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuede, C. M. , Lee, H. , Restivo, J. L. , Davis, T. A. , Hettinger, J. C. , Wallace, C. E. , Young, K. L. , Hayne, M. R. , Bu, G. , Li, C.‐Z. , & Cirrito, J. R. (2016). Rapid in vivo measurement of β‐amyloid reveals biphasic clearance kinetics in an Alzheimer’s mouse model. Journal of Experimental Medicine, 213(5), 677–685. 10.1084/jem.20151428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, L. , Trushin, S. , Christensen, T. A. , Tripathi, U. , Hong, C. , Geroux, R. E. , Howell, K. G. , Poduslo, J. F. , & Trushina, E. (2018). Differential effect of amyloid beta peptides on mitochondrial axonal trafficking depends on their state of aggregation and binding to the plasma membrane HHS Public Access. Neurobiology of Diseases, 114, 1–16. 10.1016/j.nbd.2018.02.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng, X. , Liu, D. , Roychaudhuri, R. , Teplow, D. B. , & Bowers, M. T. (2015). Amyloid β‐protein assembly: Differential effects of the protective A2T mutation and recessive A2V Familial Alzheimer’s disease mutation. ACS Chemical Neuroscience, 6, 1732–1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1