

Abstract
Sepsis represents one of the major medical challenges of the 21st century. Despite substantial improvements in the knowledge on pathophysiological mechanisms, this has so far not translated into novel adjuvant treatment strategies for sepsis. In sepsis, both vascular tone and vascular integrity are compromised, and contribute to the development of shock, which is strongly related to the development of organ dysfunction and mortality. In this review, we focus on dipeptidyl peptidase 3 (DPP3) and adrenomedullin (ADM), two molecules that act on the vasculature and are involved in the pathophysiology of sepsis and septic shock. DPP3 is an ubiquitous cytosolic enzyme involved in the degradation of several important signalling molecules essential for regulation of vascular tone, including angiotensin II. ADM is a key hormone involved in the regulation of vascular tone and endothelial barrier function. Previous studies have shown that circulating concentrations of both DPP3 and ADM are independently associated with the development of organ failure and adverse outcome in sepsis. We now discuss new evidence illustrating that these molecules indeed represent two distinct pathways involved in the development of septic shock. Recently, both ADM‐enhancing therapies aimed at improving endothelial barrier function and vascular tone and DPP3‐blocking therapies aimed at restoring systemic angiotensin responses have been shown to improve outcome in various preclinical sepsis models. Given the current lack of effective adjuvant therapies in sepsis, additional research on the therapeutic application of these peptides in humans is highly warranted.
Keywords: cardiovascular regulation, endothelial function, sepsis, vascular disease

Introduction
Despite advances in medical care, sepsis remains a major health problem of the 21st century, with a high mortality and an ever‐increasing incidence [1]. Sepsis is now viewed as an inflammatory disorder, in which a dysregulated host response to infection results in life‐threatening organ dysfunction [2]. In septic shock, the most severe form of sepsis, profound underlying circulatory, cellular and metabolic abnormalities, is associated with an even greater risk of mortality [2, 3]. Septic shock is characterized by increased lactate levels, as well as a necessity for vasopressor therapy to maintain adequate blood pressure and organ perfusion, despite adequate fluid resuscitation [3, 4].
Sepsis consists of a complex, multifaceted pathogenesis, in which the sum of many harmful and protective pathways results in the observed clinical condition [5]. During sepsis, a host response is mounted after pathogen‐associated molecular patterns (PAMPs) are recognized by highly conserved pattern recognition receptors (PRRs) present on immune cells [4, 6]. Activation of these receptors leads to the activation of multiple inflammatory pathways including leucocyte and complement activation, the release of pro‐inflammatory cytokines, reactive oxygen species and damage‐associated molecular patterns (DAMPs) [6, 7]. All these factors ultimately contribute to the development of organ failure, which is the key determinant of sepsis mortality [4].
During sepsis, vascular tone and integrity are compromised, with both factors contributing to the development of shock. Endothelial dysfunction is one of the major hallmarks of sepsis [8]. A profound inflammatory response causes disturbed endothelial cell signalling and endothelial cell death [9, 10]. Subsequent loss of endothelial barrier integrity results in extravasation of fluids and molecules, causing oedema and the loss of intravascular volume. Ultimately, this leads to a decrease in blood pressure, which further contributes to organ failure [4, 8, 9].
Although knowledge on the molecular mechanisms causing sepsis has substantially improved, treatment strategies have remained virtually unchanged for decades, implying that the gap between fundamental knowledge and clinical application has only widened. Treatment consists of adequate and timely antimicrobial therapy, source control, supportive therapies including fluid resuscitation and vasopressor therapy to maintain vascular tone and organ support interventions such as mechanical ventilation and renal replacement therapy [9]. The majority of conducted sepsis trials investigating possible adjuvant treatments focused on attenuating pro‐inflammatory responses [11]. Unfortunately, none of these therapies improved clinical outcome, with some even resulting in increased mortality [12, 13, 14]. This lack of therapeutic benefit observed in clinical sepsis trials can be partially explained by considerable patient heterogeneity, resulting from inter‐individual differences in comorbidity, comedication, source of infection, causative pathogens and timing of onset of the inflammatory response [4, 15]. This heterogeneity impedes evaluation of pathophysiological mechanisms and hampers accurate assessment of pharmacological interventions [16].
Taken together, it is clear that there is still an unmet need for novel treatment strategies for sepsis. Targeted interventions aimed at improving endothelial barrier function and vascular tone may prove highly relevant in this regard [8]. In this review, we focus on dipeptidyl peptidase 3 (DPP3), an ubiquitous cytosolic enzyme involved in the degradation of several important signalling molecules essential for regulation of vascular tone, including angiotensin II [17, 18], and adrenomedullin (ADM), a key hormone involved in the regulation of vascular tone and endothelial barrier function [19]. We describe the general vascular properties of DPP3 and ADM and provide an overview of the current understanding of the different roles of these molecules in sepsis and septic shock. Furthermore, we discuss the potential of DPP3‐ and ADM‐targeted treatments for sepsis patients, as well as the implications of a completed biomarker‐guided trial incorporating ADM measurements on future sepsis trial designs.
Dipeptidyl peptidase 3
DPP3 was the third enzyme in the dipeptidyl peptidase group to be identified when it was first isolated from bovine pituitary tissue more than half a century ago [20]. DPP3 is a zinc‐dependent metallopeptidase capable of hydrolysing a broad spectrum of oligopeptides between three and ten amino acids in length [18]. DPP3 has been implicated in blood pressure regulation [21], inflammation [22] and pain regulation [23, 24] through its capability to hydrolyse and thus inactivate bioactive peptides such as angiotensins, enkephalins and endorphins [18].
DPP3 is ubiquitously expressed in a range of tissues including erythrocytes, leucocytes, lung, heart, kidney, intestines, skeletal muscle, skin, brain, liver and spleen [22, 25, 26, 27, 28, 29, 30, 31, 32]. Whilst DPP3 is classified as a primary cytosolic enzyme [26, 33], membrane‐bound forms of DPP3 have been described in neutrophils, brain tissue and different visceral organs [22, 34]. More recently, specific immunoassays for the detection of DPP3 concentration and enzyme activity in plasma have been developed, which demonstrated the constitutive presence of DPP3 in the circulation (coined cDPP3 for circulating DPP3) [35].
DPP3 exercises its enzymatic function by cleaving a dipeptide fragment from the N‐terminus of its substrates [36]. Its catalytic zinc‐binding domain closely resembles that of other notable but structurally unrelated metallopeptidases such as neprilysin and thermolysin [36]. The catalytic domain of DPP3 is highly preserved between species, underlining its function as an enzyme of biological significance [18]. Of its known substrates, tripeptides are only poorly hydrolysed [37], whilst peptides containing more than ten amino acids cannot be cleaved by DPP3 [20, 34].
cDPP3 has a half‐life of approximately 70 min [35]. The mechanisms through which DPP3 is cleared from the circulation are unknown. Nevertheless, studies on the clearance kinetics of other enzymes suggest that primary endocytosis in the liver followed by further processing in lysosomes [38] could be responsible. Similarly, it is currently unclear to what extent kidney and/or liver dysfunction influence cDPP3 clearance kinetics.
DPP3 as a depressant of the cardiovascular system
The renin–angiotensin–aldosterone system (RAAS) plays a vital role in the regulation of cardiovascular system homeostasis [39]. The primary effector molecule of this system, angiotensin II, affects the function of virtually all organs, and both beneficial and pathological effects have been reported [39, 40, 41]. Acute changes in angiotensin II mainly serve to raise blood pressure through increases in sympathetic tone, endogenous catecholamine and vasopressin release, as well as direct stimulation of vascular smooth muscle cells (VSMCs) [39, 41]. Angiotensin II is also essential to maintain glomerular filtration, especially during periods of reduced renal perfusion [42]. Following chronic stimulation with angiotensin II, the opposite is observed, as this is associated with adverse vascular and cardiac remodelling through induction of hypertrophy as well as fibrosis of VSMCs and cardiomyocytes [39, 40, 41].
Until recently, the interplay between DPP3 and RAAS was only scarcely studied [18, 43]. Multiple studies had already pointed out the putative rapid angiotensin‐scavenging properties of DPP3 based on in vitro experiments [18]. Angiotensin II, angiotensin III, angiotensin IV, angiotensin 1‐5 and angiotensin 1‐7 were all found to be effectively hydrolysed by DPP3 [17, 18, 44], with angiotensin IV (six amino acids in length) being hydrolysed ten times faster than angiotensin II (eight amino acids in length) [21]. However, as reliable assays to measure cDPP3 were not available, these findings could not be confirmed in vivo. Following the recent development of cDPP3 luminometric immunoassays, which also demonstrated the constitutive cDPP3 presence in healthy humans, interest in the field was revitalized [35].
Whereas cDPP3 levels are low in healthy volunteers [35], high cDPP3 concentrations are found in sepsis, septic shock, cardiogenic shock and burn victims exhibiting vasodilatory shock syndrome [35, 45, 46, 47]. In these patient cohorts, admission cDPP3 levels were associated with higher organ dysfunction scores, the development of myocardial dysfunction, refractory shock, acute kidney injury and increased short‐term mortality [35, 45, 46, 47]. Interestingly, a decrease in cDPP3 following treatment was associated with less subsequent organ support requirements and lower mortality in all of these conditions [45].
Based on these clinical associations combined with the known short half‐life and primary cytosolic localization of DPP3 [26, 35], it was hypothesized that high levels of cDPP3, despite adequate supportive treatment, represent a state of ongoing cell death (necrosis) [45] and release of cytosolic DPP3 into the circulation. During shock, upregulation of angiotensin II is a physiologic and potentially life‐saving response aimed at maintaining adequate tissue perfusion [41, 48]. Since the uncontrolled release of DPP3 into the circulation is able to effectively cleave angiotensin II, DPP3 might represent a novel factor contributing to the deterioration of vascular tone in different shock conditions [18]. An overview of the effects of cDPP3 is presented in Fig. 1 and Table 1.
Fig. 1.
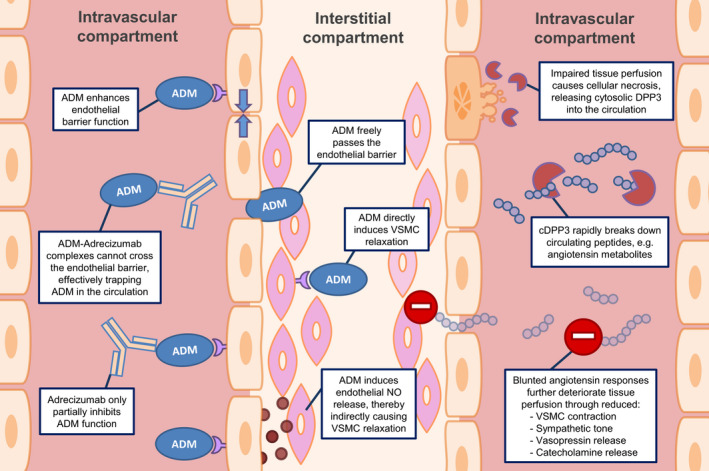
Overview on the effects of adrenomedullin (left part) and circulating dipeptidyl peptidase 3 (right part) on vascular function and the mode of action of the non‐neutralizing adrenomedullin antibody Adrecizumab. ADM = adrenomedullin, VSMC = vascular smooth muscle cell, NO = nitric oxide, cDPP3 = circulating dipeptidyl peptidase 3.
Table 1.
Overview on the biological, pathophysiological, prognostic and therapeutic properties of ADM and DPP3 in sepsis
| ADM | DPP3 | |
|---|---|---|
| General characteristics | ||
| Biological effects | ||
| Metabolism |
|
|
| Prognostic properties in sepsis |
High circulating levels are associated with: |
High circulating levels are associated with: |
| Therapeutic properties in animal models |
ADM administration:
ADM non‐neutralizing antibodies:
ADM‐neutralizing antibodies:
|
DPP3 administration:
DPP3‐neutralizing antibodies:
|
| Underlying mechanisms |
|
|
ADM, adrenomedullin; DPP3, dipeptidyl peptidase 3; cDPP3, circulating dipeptidyl peptidase 3; VSMC, vascular smooth muscle cell.
DPP3 administration and DPP3 antibodies
In a murine model of hypertension induced by continuous infusion of angiotensin II by an implanted micro‐osmotic pump, intravenous administration of DPP3 rapidly normalized blood pressure to a similar extent as the angiotensin receptor blocker candesartan [21]. Prolonged DPP3 infusion also ameliorated the development of cardiac hypertrophy and fibrosis in these hypertensive mice, and reduced urinary albumin excretion and markers of kidney injury [21]. Of note, DPP3 infusion reduced circulating angiotensin II levels to even lower levels than baseline, suggesting that endogenously produced angiotensin II was also effectively cleaved. In DPP3 (−/−) knockout mice, significant upregulation of the classical RAAS was observed, reflected by higher circulating levels of angiotensin II, angiotensin III, angiotensin IV and angiotensin 1‐5, all known substrates of DPP3 [17].
In healthy mice, DPP3 administration provoked rapid deterioration of left ventricular function, as well as increased renal resistance indexes. Following cessation of DPP3 administration, both left ventricular function and cDPP3 levels returned to pre‐infusion levels within approximately 120 min [46]. Additionally, high cDPP3 levels were observed in a murine isoproterenol heart failure model, which were associated with reduced shortening fraction, high resistive renal index and pulmonary congestion [46].
Although much more limited, there are also data on antagonizing DPP3. Interestingly, in the above‐mentioned heart failure model, administration of a neutralizing DPP3 antibody normalized left ventricular function, an effect which was sustained after 24 h and 14 days [46]. Furthermore, administration of the same DPP3 antibody in murine sepsis attenuated sepsis‐induced cardiac dysfunction and improved overall survival [49]. Following the recent findings in these animal models, DPP3 inhibitors are currently being developed for clinical use in septic and cardiogenic shock patients [46]. An overview of the effects of cDPP3‐modulating therapies is presented in Table 1.
Adrenomedullin
ADM is a freely circulating 52‐amino acid peptide, first isolated from human pheochromocytoma tissue more than two decades ago [50]. The formation of biologically active ADM is preceded by a multistep cleavage process. First, a 21‐residue N‐terminal signalling peptide is cleaved of the 185‐amino acid‐long preprohormone (prepro‐ADM), resulting in a 164‐amino acid peptide called pro‐ADM. Pro‐ADM is subsequently cleaved into different fragments, including pro‐ADM N‐terminal 20 peptide (PAMP) [51, 52, 53], midregional pro‐ADM (MR‐pro‐ADM) [54], adrenotensin [55] and a glycine‐extended 53‐amino acid peptide. This last peptide is converted to biologically active ADM through subsequent enzymatic amidation [56]. Although initial studies primarily identified vasodilatory properties of ADM, a myriad of biological functions have since been discovered. Genetic evidence points to the protection of the endothelial barrier as the key function of ADM in vivo [57]. ADM exerts these effects through binding to the ADM1 and ADM2 receptors, heterodimeric complexes consisting of the calcitonin receptor‐like receptor (CRLR) and specific receptor activity‐modifying proteins (RAMP)2 and RAMP3, respectively [58].
ADM is ubiquitously expressed in almost all human tissues [25], with highest ADM concentrations found in the adrenal medullae, cardiac atria and lungs [59, 60]. ADM is produced by multiple cell types including endothelial cells, VSMCs, macrophages, monocytes and renal parenchymal cells [61, 62, 63, 64, 65, 66]. Similar to the ubiquitous expression of the ADM peptide, ADM receptors are also present in multiple tissues including blood vessels, heart, lungs, skeletal muscles and nerve tissues [67, 68, 69, 70].
Studies in rat endothelial cell lines showed that ADM is not stored, but constantly produced. Moreover, it was shown that endothelial cells secrete ADM at a higher rate than VSMCs [71]. ADM has a short half‐life of approximately 22 min [72]. Degradation occurs through cleavage of its N‐terminus by different circulating and membrane‐bound proteases, of which neprilysin is the most important [73, 74, 75]. ADM is also degraded through internalization and degradation of activated ADM receptor complexes, with the lungs being involved as major site of clearance [76, 77, 78].
Adrenomedullin as a regulator of vascular tone
The first discovered effect of ADM was vasodilation, causing reduced peripheral resistance and hypotension in animal studies [50, 79]. Following these initial findings, ex vivo studies demonstrated the direct vasodilatory effects of ADM in isolated blood vessels and isolated organs [80, 81, 82]. Concurrently, in vivo studies in both animals and humans showed that intravenous infusion of ADM decreased blood pressure and induced a compensatory increase in heart rate, as well as enhanced endogenous noradrenaline and renin concentrations, and increased cardiac output [79, 83, 84, 85, 86, 87, 88].
ADM mediates its vasodilatory effects through binding with its target receptors on endothelial cells and VSMCs [80]. Both endothelium‐dependent and endothelium‐independent signalling pathways are implied in ADM’s vasodilatory effects on VSMCs [89]. The latter are caused by direct binding of ADM to its receptors on VSMCs, which leads to increased cyclic adenosine monophosphate (cAMP) and smooth muscle cell relaxation [90, 91]. Several endothelium‐dependent pathways through which ADM causes vasodilatation are all mediated by increased endothelial nitric oxide synthase (eNOS) activity, leading to local nitric oxide (NO) release and consequent vasorelaxation [92, 93].
ADM has also been implicated in central regulation of blood pressure, although studies have yielded contradictive findings. The presence of endogenous ADM in the hypothalamus has been demonstrated [94], and microinjections of ADM into the hypothalamic paraventricular nucleus elicited an immediate and short‐lived decrease in blood pressure in animal studies [95, 96]. In contrast, infusion of ADM directly into intracerebral fluid and microinjections of ADM in the rostral ventrolateral medullae were both found to increase blood pressure in different animal studies [97, 98].
Adrenomedullin as a regulator of endothelial barrier function
The single‐cell layer of vascular endothelium in blood vessels constitutes the barrier between the intravascular and interstitial spaces. The endothelium is essential in regulating the diffusion of molecules and other substrates through paracellular and transcellular transport mechanisms [99, 100]. Because of its location, the endothelium also fulfils unique regulatory functions on local vessel tone, local and systemic inflammatory signalling, and haemostasis and angiogenesis [8, 100, 101].
Following injury, inflammation causes barrier compromise at the endothelial cell‐to‐cell junction level, subsequently allowing for the efflux of inflammatory signal molecules (e.g. cytokines and prostaglandins) and leucocyte infiltration into tissues [8]. These processes are physiological responses paramount to fight off infection locally, as they are required to combat pathogens residing in the tissues. Nevertheless, during sepsis, excessive systemic damage to the endothelial barrier induced by the inflammatory response causes large amounts of intravascular fluids to leak into tissues, leading to oedema formation, which substantially contributes to the development of shock [4, 102].
ADM is essential for endothelial barrier development and maintenance [19]. Knockout mice lacking crucial parts of ADM‐ADM receptor signalling pathways develop lethal hydrops fetalis, indicating inadequate development of the endothelial barrier [103, 104, 105]. In conditional murine knockout models, in which either ADM synthesis by endothelial cells or endothelial ADM receptors were defective, increased vascular permeability and oedema formation were observed [106, 107], further illustrating the relevance of this pathway.
In vitro studies have demonstrated that ADM stabilizes the endothelial barrier through regulation of the actin–myosin cytoskeleton [108]. In cultured human umbilical vein endothelial cells and porcine pulmonary artery endothelial cell monolayers, pretreatment with ADM reduced endothelial hyperpermeability elicited by hydrogen peroxide (H2O2), thrombin and haemolysin A, by attenuating myosin light‐chain phosphorylation, stress fibre formation and subsequent gap formation through a cAMP‐dependent mechanism [109]. ADM also diminished H2O2‐induced oedema formation in isolated perfused rabbit lungs, accompanied by increased cAMP levels in the lung perfusate [109].
Adrenomedullin as a treatment target relevant for sepsis
Circulating ADM levels were found to have prognostic value for clinical outcome in a range of pathophysiological conditions. Whilst various studies have described high ADM levels in patients with congestive heart failure, acute heart failure, cardiogenic shock and sepsis, the highest concentrations are found in patients with septic shock [110, 111, 112, 113, 114]. In septic shock patients, ADM levels correlate with disease severity, mortality and different types of organ dysfunction, including vasopressor/inotrope dependency and need for renal replacement therapy [110, 111, 112, 115, 116]. Moreover, a reduction in ADM following the first day of treatment in the ICU was associated with improvements in organ dysfunction scores and lower 28‐day mortality [116].
Whilst these associations might suggest that ADM plays a detrimental role in sepsis and that neutralizing ADM may be beneficial, no causal relationships should be deducted because of the observational nature of these studies. In the light of the beneficial effects of ADM on endothelial barrier function, the increase in ADM likely represents a failing compensatory response, aimed to protect against inflammation‐induced organ damage in sepsis [117]. Over the last decades, several studies have investigated the effects of ADM administration or other ADM‐targeted therapies in preclinical models of sepsis. Different approaches were used, including ADM administration, modulation of ADM function, and partial or complete neutralization using anti‐ADM antibodies. In this respect, it is important to note that ADM‐related therapies have previously been described as ‘a double‐edged sword’ in sepsis [118]. As mentioned before, apart from stabilization of the endothelial barrier, ADM also has the potential to cause vasodilatation and hypotension, which may contribute to worse outcome of septic shock patients [108, 109, 119, 120, 121]. As such, the beneficial effects of ADM on vascular permeability on the one hand and possible detrimental effects of vasodilatation on the other hand suggest that tight regulation of ADM is required [118]. An overview of the effects of ADM is presented in Fig. 1 and Table 1.
Adrenomedullin administration
Data on ADM administration during inflammatory conditions are limited to animal models. In various endotoxaemia models, ADM administration resulted in improved haemodynamics and reduced vascular leakage, end‐organ damage and mortality [119, 120, 122, 123, 124]. ADM administration also attenuated kidney injury in two different renal injury models [125, 126]. In lung injury models, ADM administration reduced endothelial hyperpermeability, histopathological features and levels of pro‐inflammatory cytokines [121, 127, 128]. Lastly, in an in vivo murine model of shock induced by injection of S. aureus alpha toxins, ADM administration reduced albumin and plasma fluid extravasation, and improved survival [108]. Of note, all the aforementioned studies on ADM were performed using either endotoxaemia or organ injury models, which do not necessarily recapitulate infection. There are also several drawbacks to direct administration of ADM in sepsis and septic shock, limiting its clinical applicability. Because of the aforementioned short half‐life of ADM of only 22 min [72], ADM therapy would have to be applied as prolonged continuous intravenous infusion. Increasing ADM’s half‐life through PEGylation may circumvent this issue [129], but no data on the effects of PEGylated ADM in animal models of sepsis are currently available. Even more important, as ADM possesses potent vasodilatory effects, ADM administration might well induce hypotension in a patient category already at great risk of hypotension‐induced end‐organ failure [118]. An overview of the effects of ADM‐modulating therapies is presented in Table 1.
Adrenomedullin‐binding antibodies
ADM interacts with its receptor through its C‐terminal moiety [130], whilst the N‐terminal part of ADM is thought to be of only minor importance for its agonist function. In preclinical animal studies, several high‐affinity mouse monoclonal anti‐ADM antibodies have been developed, targeting different epitopes of ADM [131]. Interestingly, complete inhibition of ADM signalling by an antibody targeting the C‐terminus of ADM did not improve survival in murine caecal ligation and puncture (CLP) models of sepsis [131], whilst an antibody targeted at the N‐terminus of ADM (HAM1101), which only results in marginal loss of ADM signalling, resulted in a substantial reduction in mortality in the same sepsis model [131]. Subsequent experiments in CLP‐induced sepsis models also demonstrated that this antibody decreased iNOS, but not eNOS expression, reduced catecholamine infusion rates, attenuated kidney dysfunction and improved survival [132, 133]. This partially inhibitory ADM antibody was subsequently humanized (HAM8101) for use in follow‐up human studies and was named Adrecizumab [117].
The proposed mechanism of action through which Adrecizumab exerts its beneficial effects is of special interest; an overview is presented in Fig. 1. In both preclinical models of sepsis and sepsis patients, Adrecizumab causes a potent, dose‐dependent increase in circulating bioactive ADM [133, 134]. This increase in circulating bioactive ADM levels is not caused by increased synthesis, as concentrations of MR‐pro‐ADM (an inactive peptide fragment derived from the same prohormone as ADM) remained unchanged [134]. It is assumed that modulation of the ADM equilibrium between the blood and interstitial compartments accounts for the increase in ADM following administration of Adrecizumab. ADM is a small peptide molecule that can freely cross the endothelial barrier, whilst the large molecular weight of Adrecizumab precludes its free diffusion [117, 134] and remains in the circulation. Subsequently, binding of circulating ADM to the antibody may drain ADM from the interstitial space, by effectively trapping it in the blood compartment. Moreover, antibody binding also increases the half‐life of ADM, likely by limiting its hydrolysis [117, 134, 135]. Despite the partial inhibition of ADM signalling function caused by antibody binding, this is overruled by the much larger increase in circulating bioactive ADM concentrations resulting in an overall increase in ADM activity in the blood compartment. Being confined to the circulation, ADM exerts its beneficial effect on endothelial cell barrier function, whilst the detrimental vasodilatory effects on VSMCs in the interstitial space are reduced [117]. A more detailed description of the proposed mechanism of action of Adrecizumab is provided elsewhere [135].
The Adrenomedullin and Outcome in Sepsis and Septic Shock (AdrenOSS) trials as an example of biomarker‐driven sepsis trial design
Thus far, the typical phase 2 and phase 3 clinical sepsis trial design consisted of enrolling sepsis patients fitting broad inclusion criteria (e.g. current sepsis definitions) not taking into account whether the biological pathways influenced by the specific treatment are activated or inhibited in a specific patient [14, 136]. The use of these broad criteria results in marked population heterogeneity with a large noise‐to‐signal ratio, leading to smaller chances to detect treatment effects even when sample sizes are increased [137]. Consequently, large amounts of resources have gone into studies with limited chances to detect any clinically relevant treatment effects [137, 138, 139].
Population enrichment strategies consist of the preselection of a study population based on patient characteristics specifically associated with the biological pathway modulated by the investigational treatment [136]. Candidate characteristics for population enrichment include the use of biomarkers, imaging or clinical characteristics that correlate with certain disease phenotypes [137, 140]. Because preselecting a population based on biological responses related to the intervention will increase the chance of a trial to detect a treatment effect, it may allow for smaller sample sizes than would be required in unselected populations [137]. This tailoring of trial design to include only a subgroup of patients most likely to benefit from the treatment instead of a ‘one‐drug‐fits‐all’ model is known as precision medicine [137, 141]. Whilst the call for precision medicine in sepsis trial design has been around for more than a decade following the failure of many phase 2 and phase 3 clinical trials, examples of studies actually incorporating these design features have been extremely sparse [138, 139, 141]. A graphic overview of the concept of population enrichment strategies is provided in Fig. 2.
Fig. 2.
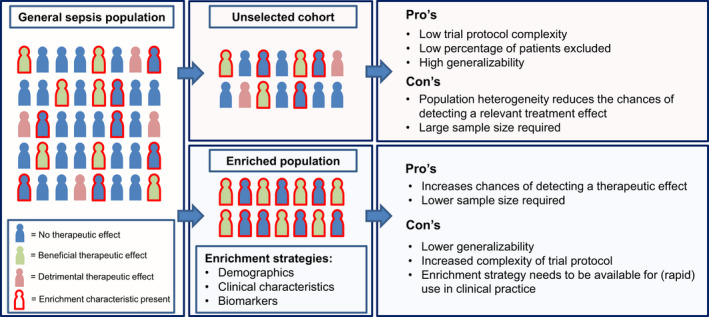
Concept of population enrichment in sepsis trial design. The enrichment characteristic is related to the mode of action of the treatment under study. This can constitute demographic features, clinical characteristics, elevated/depressed biomarkers or a combination of these.
Following phase 1 studies demonstrating a favourable safety and tolerability profile of Adrecizumab [134], design of a follow‐up phase‐2 ‘proof‐of‐concept’ trial in septic shock was initiated. This ‘AdrenOSS‐2’ study represents one of the first examples of a sepsis trial incorporating the use of a biomarker as an enrichment strategy. For AdrenOSS‐2, patients more likely to experience adverse outcome caused by endothelial dysfunction were selected for therapy with Adrecizumab using a biomarker‐driven approach incorporating bedside measurements of bioactive ADM (bio‐ADM, SphingoTec GmbH). This biomarker approach was chosen based on the concept of high ADM levels as a physiological response to maintain endothelial barrier integrity that falls short during sepsis [117, 135].
In order to decide on a specific cut‐off level of bio‐ADM, which would serve as an inclusion criterium, the relationship between initial levels of bio‐ADM and short‐term outcome in sepsis and septic shock patients was first examined in a prospective multicenter cohort study called ‘AdrenOSS‐1’. In this study, serial determinations of bio‐ADM defined a cut‐off value of >70 pg mL−1 as the best predictor of subsequent organ dysfunction and 28‐day mortality [110, 115, 116]. This cut‐off value was selected as an additional inclusion criterium for the AdrenOSS‐2 trial. Moreover, the measurements of other biomarkers (including cDPP3) were also performed in the trial, to examine whether additional population enrichment using these biomarkers would be able to improve future sepsis trial design.
Interestingly, whilst preliminary results of AdrenOSS‐2 presented at the 40th International Symposium on Intensive Care & Emergency Medicine demonstrated beneficial treatment effects of Adrecizumab, including an early reduction in organ dysfunction scores, these treatment effects became more pronounced when patients who also exhibited high pretreatment levels of cDPP3 were excluded [142]. As alluded to before, the uncontrolled release of cytosolic DPP3 into the circulation caused by cellular necrosis during shock leads to decreased vascular tone through inhibition of compensatory angiotensin II responses. This represents a biological pathway, which is not targeted by Adrecizumab. Therefore, when patients with high cDPP3 levels were excluded from the analysis, more pronounced beneficial treatment effects for Adrecizumab were found [142]. These findings are also supported by results from the AdrenOSS‐1 study indicating that bio‐ADM and cDPP3 are independent predictors of mortality in sepsis (unpublished data). In this observational study, both biomarkers combined improved the c‐index for 28‐day mortality to 0.742, whereas it was 0.688 for bio‐ADM and 0.692 for cDPP3 alone (p‐value for added value < 0.0001) (unpublished data). In patients with bio‐ADM < 70 pg mL−1, 16% had elevated cDPP3 (>40 ng mL−1, upper normal range). These patients had a substantially worse outcome (HR: 3.9, 95% confidence interval [CI]: 1.9–8.1, 28‐day survival rate: 71%) compared to patients with both low cDPP3 and low bioactive ADM (28‐day survival rate: 92%). Patients with elevated bio‐ADM but normal cDPP3 (constituting 68% of patients with elevated bio‐ADM) also had a worse outcome than patients with low values in both biomarkers (HR: 2.8, 95% CI: 1.6–4.8, 28‐day survival rate: 78%). Importantly, patients with the highest fatality rate were those who displayed elevated levels of both biomarkers (HR: 7.4, 95% CI: 4.3–12.8; 28‐day survival rate: 54%; reference group: patients who had low levels of both biomarkers) (unpublished data). The Kaplan–Meier curves for these respective groups are displayed in Fig. 3.
Fig. 3.
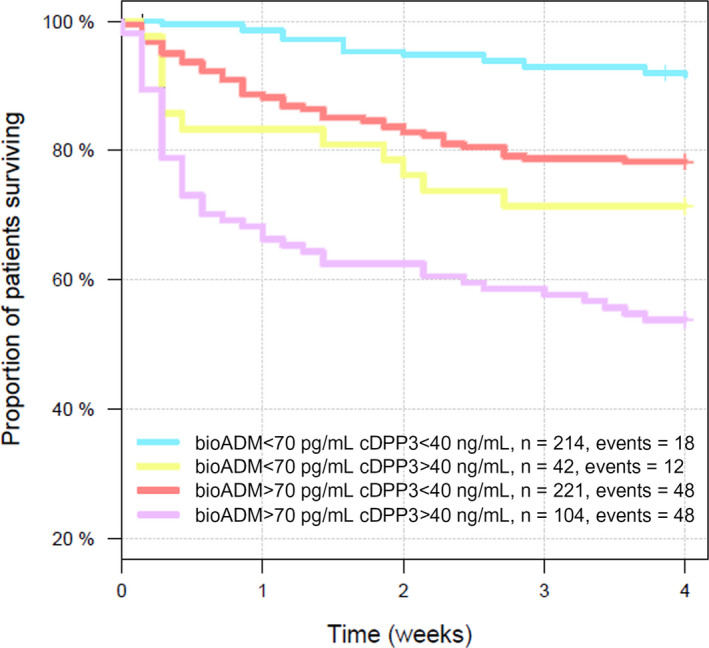
Kaplan–Meier analysis of 28‐day all‐cause mortality in septic shock patients included in the observational AdrenOSS‐1 study. Across the study, 38% of patients displayed elevated bio‐ADM levels, 7% elevated cDPP3 levels, 18% elevated levels of both biomarkers, and 37% elevated low levels of both biomarkers. Bio‐ADM = bioactive adrenomedullin. cDPP3 = circulating dipeptidyl peptidase 3.
These results provide further evidence that ADM and DPP3 represent two distinct pathways involved in the development of organ dysfunction in sepsis and that enrichment strategies combining these biomarkers may improve the therapeutic benefit of therapies targeting ADM and DPP3‐specific pathways.
Conclusion
DPP3 is a ubiquitous, primarily cytosolic enzyme involved in the degradation of several important signal molecules relevant for the regulation of vascular tone, including angiotensin II. ADM is a key hormone involved in the regulation of vascular tone and endothelial barrier function. Increased release of these molecules during sepsis relates to vascular tone and capillary leakage, both independently associated with the development of organ failure and adverse outcome in sepsis. Therefore, these molecules likely represent two unique and distinct pathways of clinical significance involved in the development of septic shock. Both ADM‐enhancing therapies aimed at improving endothelial barrier function and DPP3‐blocking therapies aimed at restoring systemic angiotensin responses have been shown to improve outcome in various preclinical sepsis models. Given the availability of rapid bedside biomarker assays for both DPP3 and ADM, they represent promising opportunities for the conduct of biomarker‐guided sepsis trials. Given the current lack of any adjuvant therapy in sepsis, additional research on the therapeutic application of these peptides in humans is highly warranted.
Conflict of Interest statement
PP received travel and consultancy reimbursement from Adrenomed and 4TEEN4, the companies that produced the ADM and DPP3 bio‐assays and antibodies described. The other authors declare no financial conflicts of interest.
Author contribution
Dirk van Lier: Conceptualization (supporting); Writing‐original draft (lead). Matthijs Kox: Conceptualization (supporting); Writing‐review & editing (supporting). Peter Pickkers: Conceptualization (lead); Writing‐review & editing (lead).
van Lier D, Kox M, Pickkers P (Radboud University Medical Center, Nijmegen, The Netherlands). Promotion of vascular integrity in sepsis through modulation of bioactive adrenomedullin and dipeptidyl peptidase 3 (Review). J Intern Med 2021;289:792–806. 10.1111/joim.13220
References
- 1. Gaieski DF, Edwards JM, Kallan MJ, Carr BG. Benchmarking the incidence and mortality of severe sepsis in the United States. Crit Care Med 2021; 41: 1167–74. [DOI] [PubMed] [Google Scholar]
- 2. Singer M, Deutschman CS, Seymour CW et al. The third international consensus definitions for sepsis and septic shock (Sepsis‐3). JAMA 2016; 315: 801–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Shankar‐Hari M, Phillips GS, Levy ML et al. Developing a new definition and assessing new clinical criteria for septic shock: for the third international consensus definitions for sepsis and septic shock (Sepsis‐3). JAMA 2016; 315: 775–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Angus DC, van der Poll T. Severe sepsis and septic shock. New England J Med 2013; 369: 2063. [DOI] [PubMed] [Google Scholar]
- 5. Abraham E, Singer M. Mechanisms of sepsis‐induced organ dysfunction. Crit Care Med 2007; 35: 2408–16. [DOI] [PubMed] [Google Scholar]
- 6. Frazier WJ. Immunity, inflammation and sepsis: new insights and persistent questions. Crit Care 2011; 15: 124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Matzinger P. The danger model: a renewed sense of self. Science 2002; 296: 301–5. [DOI] [PubMed] [Google Scholar]
- 8. Ince C, Mayeux PR, Nguyen T et al. The endothelium in sepsis. Shock 2016; 45: 259–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gotts JE, Matthay MA. Sepsis: pathophysiology and clinical management. BMJ 2016; 353: i1585. [DOI] [PubMed] [Google Scholar]
- 10. Goldenberg NM, Steinberg BE, Slutsky AS, Lee WL. Broken barriers: a new take on sepsis pathogenesis. Sci Transl Med 2011;3:88ps25. [DOI] [PubMed] [Google Scholar]
- 11. Leentjens J, Kox M, van der Hoeven JG, Netea MG, Pickkers P. Immunotherapy for the adjunctive treatment of sepsis: from immunosuppression to immunostimulation. Time for a paradigm change? Am J Respir Crit Care Med 2013; 187: 1287–93. [DOI] [PubMed] [Google Scholar]
- 12. Warren HS. Strategies for the treatment of sepsis. New Engl J Med 1997; 336: 952–3. [DOI] [PubMed] [Google Scholar]
- 13. Stone R. Search for sepsis drugs goes on despite past failures. Science 1994; 264: 365–7. [DOI] [PubMed] [Google Scholar]
- 14. Fink MP, Warren HS. Strategies to improve drug development for sepsis. Nat Rev Drug Discov 2014; 13: 741–58. [DOI] [PubMed] [Google Scholar]
- 15. Hotchkiss RS, Karl IE. The pathophysiology and treatment of sepsis. New Engl J Med 2003; 348: 138–50. [DOI] [PubMed] [Google Scholar]
- 16. van Lier D, Geven C, Leijte GP, Pickkers P. Experimental human endotoxemia as a model of systemic inflammation. Biochimie 2019; 159: 99–106. [DOI] [PubMed] [Google Scholar]
- 17. Jha S, Taschler U, Domenig O et al. Dipeptidyl peptidase 3 modulates the renin‐angiotensin system in mice. J Biol Chem 2020; 295: 13711–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Prajapati SC, Chauhan SS. Dipeptidyl peptidase III: a multifaceted oligopeptide N‐end cutter. FEBS J 2011; 278: 3256–76. [DOI] [PubMed] [Google Scholar]
- 19. Geven C, Kox M, Pickkers P. Adrenomedullin and adrenomedullin‐targeted therapy as treatment strategies relevant for sepsis. Front Immunol 2018; 9: 292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ellis S, Nuenke JM. Dipeptidyl arylamidase III of the pituitary. Purification and characterization. J Biol Chem 1967; 242: 4623–9. [PubMed] [Google Scholar]
- 21. Pang X, Shimizu A, Kurita S et al. Novel therapeutic role for dipeptidyl peptidase III in the treatment of hypertension. Hypertension 2016; 68: 630–41. [DOI] [PubMed] [Google Scholar]
- 22. Hashimoto J, Yamamoto Y, Kurosawa H, Nishimura K, Hazato T. Identification of dipeptidyl peptidase III in human neutrophils. Biochem Biophys Res Commun 2000; 273: 393–7. [DOI] [PubMed] [Google Scholar]
- 23. Sato H, Kimura K, Yamamoto Y, Hazato T. Activity of DPP III in human cerebrospinal fluid derived from patients with pain. Masui 2003; 52: 257–63. [PubMed] [Google Scholar]
- 24. Barsun M, Jajcanin N, Vukelic B, Spoljaric J, Abramic M. Human dipeptidyl peptidase III acts as a post‐proline‐cleaving enzyme on endomorphins. Biol Chem 2007; 388: 343–8. [DOI] [PubMed] [Google Scholar]
- 25. Uhlen M, Fagerberg L, Hallstrom BM et al. Proteomics. Tissue‐based map of the human proteome. Science 2015; 347: 1260419. [DOI] [PubMed] [Google Scholar]
- 26. Abramic M, Zubanovic M, Vitale L. Dipeptidyl peptidase III from human erythrocytes. Biol Chem Hoppe Seyler 1988; 369: 29–38. [DOI] [PubMed] [Google Scholar]
- 27. Abramic M, Schleuder D, Dolovcak L et al. Human and rat dipeptidyl peptidase III: biochemical and mass spectrometric arguments for similarities and differences. Biol Chem 2000; 381: 1233–43. [DOI] [PubMed] [Google Scholar]
- 28. Grdisa M, Vitale L. Types and localization of aminopeptidases in different human blood cells. Int J Biochem 1991; 23: 339–45. [DOI] [PubMed] [Google Scholar]
- 29. Kar NC, Pearson CM. Dipeptidyl peptidases in human muscle disease. Clin Chim Acta 1978; 82: 185–92. [DOI] [PubMed] [Google Scholar]
- 30. Hopsu‐Havu VK, Jansen CT. Peptidases in the skin. II. Demonstration and partial separation of several specific dipeptide naphthylamidases in the rat and human skin. Arch Klin. Exp Dermatol. 1969; 235(1): 53–62. [PubMed] [Google Scholar]
- 31. Alba F, Arenas JC, Lopez MA. Properties of rat brain dipeptidyl aminopeptidases in the presence of detergents. Peptides 1995; 16: 325–9. [DOI] [PubMed] [Google Scholar]
- 32. Lynn KR. The isolation and some properties of dipeptidyl peptidases II and III from porcine spleen. Int J Biochem 1991; 23: 47–50. [DOI] [PubMed] [Google Scholar]
- 33. Christoforou A, Mulvey CM, Breckels LM et al. A draft map of the mouse pluripotent stem cell spatial proteome. Nat Commun 2016; 7: 8992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lee CM, Snyder SH. Dipeptidyl‐aminopeptidase III of rat brain. Selective affinity for enkephalin and angiotensin. J Biol Chem 1982; 257: 12043–50. [PubMed] [Google Scholar]
- 35. Rehfeld L, Funk E, Jha S, Macheroux P, Melander O, Bergmann A. Novel methods for the quantification of Dipeptidyl Peptidase 3 (DPP3) concentration and activity in human blood samples. J Appl Lab Med 2019; 3: 943–53. [DOI] [PubMed] [Google Scholar]
- 36. Baral PK, Jajcanin‐Jozic N, Deller S, Macheroux P, Abramic M, Gruber K. The first structure of dipeptidyl‐peptidase III provides insight into the catalytic mechanism and mode of substrate binding. J Biol Chem 2008; 283: 22316–24. [DOI] [PubMed] [Google Scholar]
- 37. Bezerra GA, Dobrovetsky E, Viertlmayr R et al. Entropy‐driven binding of opioid peptides induces a large domain motion in human dipeptidyl peptidase III. Proc Natl Acad Sci U S A 2012; 109: 6525–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Smit MJ, Beekhuis H, Duursma AM, Bouma JM, Gruber M. Catabolism of circulating enzymes: plasma clearance, endocytosis, and breakdown of lactate dehydrogenase‐1 in rabbits. Clin Chem 1988; 34: 2475–80. [PubMed] [Google Scholar]
- 39. Mehta PK, Griendling KK. Angiotensin II cell signaling: physiological and pathological effects in the cardiovascular system. Am J Physiol Cell Physiol 2007; 292: C82–97. [DOI] [PubMed] [Google Scholar]
- 40. Benigni A, Cassis P, Remuzzi G. Angiotensin II revisited: new roles in inflammation, immunology and aging. EMBO Mol Med 2010; 2: 247–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Bitker L, Burrell LM. Classic and nonclassic renin‐angiotensin systems in the critically Ill. Crit Care Clin 2019; 35: 213–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hall JE, Coleman TG, Guyton AC, Kastner PR, Granger JP. Control of glomerular filtration rate by circulating angiotensin II. Am J Physiol 1981; 241: R190–7. [DOI] [PubMed] [Google Scholar]
- 43. Menale C, Robinson LJ, Palagano E et al. Absence of dipeptidyl peptidase 3 increases oxidative stress and causes bone loss. J Bone Miner Res 2019; 34: 2133–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Cruz‐Diaz N, Wilson BA, Pirro NT, Brosnihan KB, Marshall AC, Chappell MC. Identification of dipeptidyl peptidase 3 as the Angiotensin‐(1–7) degrading peptidase in human HK‐2 renal epithelial cells. Peptides 2016; 83: 29–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Takagi K, Blet A, Levy B et al. Circulating dipeptidyl peptidase 3 and alteration in haemodynamics in cardiogenic shock: results from the OptimaCC trial. Eur J Heart Fail 2019; 22: 279–286. [DOI] [PubMed] [Google Scholar]
- 46. Deniau B, Rehfeld L, Santos K et al. Circulating dipeptidyl peptidase 3 is a myocardial depressant factor: dipeptidyl peptidase 3 inhibition rapidly and sustainably improves haemodynamics. Eur J Heart Fail 2019; 22: 290–299. [DOI] [PubMed] [Google Scholar]
- 47. Depret F, Amzallag J, Pollina A et al. Circulating dipeptidyl peptidase‐3 at admission is associated with circulatory failure, acute kidney injury and death in severely ill burn patients. Crit Care 2020; 24: 168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Antonucci E, Gleeson PJ, Annoni F et al. Angiotensin II in refractory septic shock. Shock 2017; 47: 560–6. [DOI] [PubMed] [Google Scholar]
- 49. Deniau B, Blet A, Santos K et al. Inhibition of circulating dipeptidyl‐peptidase 3 restores cardiac function in a sepsis‐induced model in rats: A proof of concept study. PLoS One 2020; 15: e0238039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kitamura K, Kangawa K, Kawamoto M et al. Adrenomedullin: a novel hypotensive peptide isolated from human pheochromocytoma. Biochem Biophys Res Commun 1993; 192: 553–60. [DOI] [PubMed] [Google Scholar]
- 51. Washimine H, Kitamura K, Ichiki Y et al. Immunoreactive proadrenomedullin N‐terminal 20 peptide in human tissue, plasma and urine. Biochem Biophys Res Commun 1994; 202: 1081–7. [DOI] [PubMed] [Google Scholar]
- 52. Kitamura K, Kangawa K, Ishiyama Y et al. Identification and hypotensive activity of proadrenomedullin N‐terminal 20 peptide (PAMP). FEBS Lett 1994; 351: 35–7. [DOI] [PubMed] [Google Scholar]
- 53. Nagatomo Y, Kitamura K, Kangawa K, Fujimoto Y, Eto T. Proadrenomedullin N‐terminal 20 peptide is rapidly cleaved by neutral endopeptidase. Biochem Biophys Res Commun 1996; 223: 539–43. [DOI] [PubMed] [Google Scholar]
- 54. Struck J, Tao C, Morgenthaler NG, Bergmann A. Identification of an Adrenomedullin precursor fragment in plasma of sepsis patients. Peptides 2004; 25: 1369–72. [DOI] [PubMed] [Google Scholar]
- 55. Gumusel B, Chang JK, Hyman A, Lippton H. Adrenotensin: an ADM gene product with the opposite effects of ADM. Life Sci 1995; 57: PL87–PL90. [DOI] [PubMed] [Google Scholar]
- 56. Kitamura K, Kato J, Kawamoto M et al. The intermediate form of glycine‐extended adrenomedullin is the major circulating molecular form in human plasma. Biochem Biophys Res Commun 1998; 244: 551–5. [DOI] [PubMed] [Google Scholar]
- 57. Karpinich NO, Hoopes SL, Kechele DO, Lenhart PM, Caron KM. Adrenomedullin function in vascular endothelial cells: insights from genetic mouse models. Curr Hypertens Rev 2011; 7: 228–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Kato J, Kitamura K. Bench‐to‐bedside pharmacology of adrenomedullin. Eur J Pharmacol 2015; 764: 140–8. [DOI] [PubMed] [Google Scholar]
- 59. Ichiki Y, Kitamura K, Kangawa K, Kawamoto M, Matsuo H, Eto T. Distribution and characterization of immunoreactive adrenomedullin in human tissue and plasma. FEBS Lett 1994; 338: 6–10. [DOI] [PubMed] [Google Scholar]
- 60. Sakata J, Shimokubo T, Kitamura K et al. Distribution and characterization of immunoreactive rat adrenomedullin in tissue and plasma. FEBS Lett 1994; 352: 105–8. [DOI] [PubMed] [Google Scholar]
- 61. Kubo A, Minamino N, Isumi Y, Kangawa K, Dohi K, Matsuo H. Adrenomedullin production is correlated with differentiation in human leukemia cell lines and peripheral blood monocytes. FEBS Lett 1998; 426: 233–7. [DOI] [PubMed] [Google Scholar]
- 62. Kubo A, Minamino N, Isumi Y et al. Production of adrenomedullin in macrophage cell line and peritoneal macrophage. J Biol Chem 1998; 273: 16730–8. [DOI] [PubMed] [Google Scholar]
- 63. Minamino N, Shoji H, Sugo S, Kangawa K, Matsuo H. Adrenocortical steroids, thyroid hormones and retinoic acid augment the production of adrenomedullin in vascular smooth muscle cells. Biochem Biophys Res Commun 1995; 211: 686–93. [DOI] [PubMed] [Google Scholar]
- 64. Nagata D, Hirata Y, Suzuki E et al. Hypoxia‐induced adrenomedullin production in the kidney. Kidney Int 1999; 55: 1259–67. [DOI] [PubMed] [Google Scholar]
- 65. Nakayama M, Takahashi K, Murakami O et al. Production and secretion of adrenomedullin in cultured human alveolar macrophages. Peptides 1999; 20: 1123–5. [DOI] [PubMed] [Google Scholar]
- 66. Sugo S, Minamino N, Kangawa K et al. Endothelial cells actively synthesize and secrete adrenomedullin. Biochem Biophys Res Commun 1994; 201: 1160–6. [DOI] [PubMed] [Google Scholar]
- 67. Kobayashi H, Minami S, Yamamoto R et al. Adrenomedullin receptors in rat cerebral microvessels. Brain Res Mol Brain Res 2000; 81: 1–6. [DOI] [PubMed] [Google Scholar]
- 68. Kobayashi H, Shiraishi S, Minami S et al. Adrenomedullin receptors in rat choroid plexus. Neurosci Lett 2001; 297: 167–70. [DOI] [PubMed] [Google Scholar]
- 69. Coppock HA, Owji AA, Bloom SR, Smith DM. A rat skeletal muscle cell line (L6) expresses specific adrenomedullin binding sites but activates adenylate cyclase via calcitonin gene‐related peptide receptors. Biochem J 1996; 318(Pt 1): 241–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Owji AA, Smith DM, Coppock HA et al. An abundant and specific binding site for the novel vasodilator adrenomedullin in the rat. Endocrinology 1995; 136: 2127–34. [DOI] [PubMed] [Google Scholar]
- 71. Isumi Y, Shoji H, Sugo S et al. Regulation of adrenomedullin production in rat endothelial cells. Endocrinology 1998; 139: 838–46. [DOI] [PubMed] [Google Scholar]
- 72. Meeran K, O'Shea D, Upton PD et al. Circulating adrenomedullin does not regulate systemic blood pressure but increases plasma prolactin after intravenous infusion in humans: a pharmacokinetic study. J Clin Endocrinol Metab 1997; 82: 95–100. [DOI] [PubMed] [Google Scholar]
- 73. Lewis LK, Smith MW, Brennan SO, Yandle TG, Richards AM, Nicholls MG. Degradation of human adrenomedullin(1–52) by plasma membrane enzymes and identification of metabolites. Peptides 1997; 18: 733–9. [DOI] [PubMed] [Google Scholar]
- 74. Martinez A, Oh HR, Unsworth EJ et al. Matrix metalloproteinase‐2 cleavage of adrenomedullin produces a vasoconstrictor out of a vasodilator. Biochem J 2004; 383(Pt. 3): 413–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Lisy O, Jougasaki M, Schirger JA, Chen HH, Barclay PT, Burnett JC Jr. Neutral endopeptidase inhibition potentiates the natriuretic actions of adrenomedullin. Am J Physiol 1998; 275: F410–4. [DOI] [PubMed] [Google Scholar]
- 76. Schonauer R, Kaiser A, Holze C et al. Fluorescently labeled adrenomedullin allows real‐time monitoring of adrenomedullin receptor trafficking in living cells. J Pept Sci 2015; 21: 905–12. [DOI] [PubMed] [Google Scholar]
- 77. Nishikimi T, Matsuoka H, Shimada K, Matsuo H, Kangawa K. Production and clearance sites of two molecular forms of adrenomedullin in human plasma. Am J Hypertens 2000; 13: 1032–4. [DOI] [PubMed] [Google Scholar]
- 78. Dupuis J, Caron A, Ruel N. Biodistribution, plasma kinetics and quantification of single‐pass pulmonary clearance of adrenomedullin. Clin Sci (Lond) 2005; 109: 97–102. [DOI] [PubMed] [Google Scholar]
- 79. Ishiyama Y, Kitamura K, Ichiki Y et al. Hemodynamic effects of a novel hypotensive peptide, human adrenomedullin, in rats. Eur J Pharmacol 1993; 241: 271–3. [DOI] [PubMed] [Google Scholar]
- 80. Passaglia P, Gonzaga NA, Tirapelli DP, Tirapelli LF, Tirapelli CR. Pharmacological characterisation of the mechanisms underlying the relaxant effect of adrenomedullin in the rat carotid artery. J Pharm Pharmacol 2014; 66: 1734–46. [DOI] [PubMed] [Google Scholar]
- 81. Nakamura K, Toda H, Terasako K et al. Vasodilative effect of adrenomedullin in isolated arteries of the dog. Jpn J Pharmacol 1995; 67: 259–62. [DOI] [PubMed] [Google Scholar]
- 82. Hirata Y, Hayakawa H, Suzuki Y et al. Mechanisms of adrenomedullin‐induced vasodilation in the rat kidney. Hypertension 1995; 25(4 Pt 2): 790–5. [DOI] [PubMed] [Google Scholar]
- 83. Lainchbury JG, Troughton RW, Lewis LK, Yandle TG, Richards AM, Nicholls MG. Hemodynamic, hormonal, and renal effects of short‐term adrenomedullin infusion in healthy volunteers. J Clin Endocrinol Metab 2000; 85: 1016–20. [DOI] [PubMed] [Google Scholar]
- 84. Nagaya N, Nishikimi T, Uematsu M et al. Haemodynamic and hormonal effects of adrenomedullin in patients with pulmonary hypertension. Heart 2000; 84: 653–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Nakamura M, Yoshida H, Makita S, Arakawa N, Niinuma H, Hiramori K. Potent and long‐lasting vasodilatory effects of adrenomedullin in humans. Comparisons between normal subjects and patients with chronic heart failure. Circulation 1997; 95: 1214–21. [DOI] [PubMed] [Google Scholar]
- 86. Parkes DG, May CN. Direct cardiac and vascular actions of adrenomedullin in conscious sheep. Br J Pharmacol 1997; 120: 1179–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Rademaker MT, Charles CJ, Cooper GJ et al. Combined endopeptidase inhibition and adrenomedullin in sheep with experimental heart failure. Hypertension 2002; 39: 93–8. [DOI] [PubMed] [Google Scholar]
- 88. Kita T, Suzuki Y, Kitamura K. Hemodynamic and hormonal effects of exogenous adrenomedullin administration in humans and relationship to insulin resistance. Hypertens Res 2010; 33: 314–9. [DOI] [PubMed] [Google Scholar]
- 89. Dettmann ES, Vysniauskiene I, Wu R, Flammer J, Haefliger IO. Adrenomedullin‐induced endothelium‐dependent relaxation in porcine ciliary arteries. Invest Ophthalmol Vis Sci 2003; 44: 3961–6. [DOI] [PubMed] [Google Scholar]
- 90. Ross GR, Yallampalli U, Gangula PR et al. Adrenomedullin relaxes rat uterine artery: mechanisms and influence of pregnancy and estradiol. Endocrinology 2010; 151: 4485–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Landry DW, Oliver JA. The pathogenesis of vasodilatory shock. New Engl J Med 2001; 345: 588–95. [DOI] [PubMed] [Google Scholar]
- 92. Shimekake Y, Nagata K, Ohta S et al. Adrenomedullin stimulates two signal transduction pathways, cAMP accumulation and Ca2+ mobilization, in bovine aortic endothelial cells. J Biol Chem 1995; 270: 4412–7. [DOI] [PubMed] [Google Scholar]
- 93. Nishimatsu H, Suzuki E, Nagata D et al. Adrenomedullin induces endothelium‐dependent vasorelaxation via the phosphatidylinositol 3‐kinase/Akt‐dependent pathway in rat aorta. Circ Res 2001; 89: 63–70. [DOI] [PubMed] [Google Scholar]
- 94. Satoh F, Takahashi K, Murakami O et al. Immunocytochemical localization of adrenomedullin‐like immunoreactivity in the human hypothalamus and the adrenal gland. Neurosci Lett 1996; 203: 207–10. [DOI] [PubMed] [Google Scholar]
- 95. Smith PM, Ferguson AV. Adrenomedullin acts in the rat paraventricular nucleus to decrease blood pressure. J Neuroendocrinol 2001; 13: 467–71. [DOI] [PubMed] [Google Scholar]
- 96. Xu Y, Krukoff TL. Decrease in arterial pressure induced by adrenomedullin in the hypothalamic paraventricular nucleus is mediated by nitric oxide and GABA. Regul Pept 2004; 119: 21–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Xu Y, Krukoff TL. Adrenomedullin in the rostral ventrolateral medulla inhibits baroreflex control of heart rate: a role for protein kinase A. Br J Pharmacol 2006; 148: 70–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Saita M, Shimokawa A, Kunitake T et al. Central actions of adrenomedullin on cardiovascular parameters and sympathetic outflow in conscious rats. Am J Physiol 1998; 274: R979–84. [DOI] [PubMed] [Google Scholar]
- 99. Chistiakov DA, Orekhov AN, Bobryshev YV. Endothelial barrier and its abnormalities in cardiovascular disease. Front Physiol 2015; 6: 365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Aird WC. Phenotypic heterogeneity of the endothelium: II. Representative vascular beds. Circ Res 2007; 100: 174–90. [DOI] [PubMed] [Google Scholar]
- 101. Jacob M, Chappell D, Becker BF. Regulation of blood flow and volume exchange across the microcirculation. Crit Care 2016; 20: 319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Deutschman CS, Tracey KJ. Sepsis: current dogma and new perspectives. Immunity 2014; 40: 463–75. [DOI] [PubMed] [Google Scholar]
- 103. Caron KM, Smithies O. Extreme hydrops fetalis and cardiovascular abnormalities in mice lacking a functional Adrenomedullin gene. Proc Natl Acad Sci U S A 2001; 98: 615–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Dackor RT, Fritz‐Six K, Dunworth WP, Gibbons CL, Smithies O, Caron KM. Hydrops fetalis, cardiovascular defects, and embryonic lethality in mice lacking the calcitonin receptor‐like receptor gene. Mol Cell Biol 2006; 26: 2511–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Czyzyk TA, Ning Y, Hsu MS et al. Deletion of peptide amidation enzymatic activity leads to edema and embryonic lethality in the mouse. Dev Biol 2005; 287: 301–13. [DOI] [PubMed] [Google Scholar]
- 106. Ochoa‐Callejero L, Pozo‐Rodrigalvarez A, Martinez‐Murillo R, Martinez A. Lack of adrenomedullin in mouse endothelial cells results in defective angiogenesis, enhanced vascular permeability, less metastasis, and more brain damage. Sci Rep 2016; 6: 33495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Tanaka M, Koyama T, Sakurai T et al. The endothelial adrenomedullin‐RAMP2 system regulates vascular integrity and suppresses tumour metastasis. Cardiovasc Res 2016; 111: 398–409. [DOI] [PubMed] [Google Scholar]
- 108. Brell B, Temmesfeld‐Wollbruck B, Altzschner I et al. Adrenomedullin reduces Staphylococcus aureus alpha‐toxin‐induced rat ileum microcirculatory damage. Crit Care Med 2005; 33: 819–26. [DOI] [PubMed] [Google Scholar]
- 109. Hippenstiel S, Witzenrath M, Schmeck B et al. Adrenomedullin reduces endothelial hyperpermeability. Circ Res 2002; 91: 618–25. [DOI] [PubMed] [Google Scholar]
- 110. Marino R, Struck J, Maisel AS, Magrini L, Bergmann A, Di Somma S. Plasma adrenomedullin is associated with short‐term mortality and vasopressor requirement in patients admitted with sepsis. Crit Care 2014; 18: R34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Chen YX, Li CS. Prognostic value of adrenomedullin in septic patients in the ED. Am J Emerg Med 2013; 31: 1017–21. [DOI] [PubMed] [Google Scholar]
- 112. Guignant C, Voirin N, Venet F et al. Assessment of pro‐vasopressin and pro‐adrenomedullin as predictors of 28‐day mortality in septic shock patients. Intensive Care Med 2009; 35: 1859–67. [DOI] [PubMed] [Google Scholar]
- 113. Ueda S, Nishio K, Minamino N et al. Increased plasma levels of adrenomedullin in patients with systemic inflammatory response syndrome. Am J Respir Crit Care Med 1999; 160: 132–6. [DOI] [PubMed] [Google Scholar]
- 114. Tolppanen H, Rivas‐Lasarte M, Lassus J et al. Adrenomedullin: a marker of impaired hemodynamics, organ dysfunction, and poor prognosis in cardiogenic shock. Ann Intensive Care 2017; 7: 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Caironi P, Latini R, Struck J et al. Circulating biologically Active Adrenomedullin (bio‐ADM) predicts hemodynamic support requirement and mortality during sepsis. Chest 2017; 152: 312–20. [DOI] [PubMed] [Google Scholar]
- 116. Mebazaa A, Geven C, Hollinger A et al. Circulating adrenomedullin estimates survival and reversibility of organ failure in sepsis: the prospective observational multinational Adrenomedullin and Outcome in Sepsis and Septic Shock‐1 (AdrenOSS‐1) study. Crit Care 2018; 22: 354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Geven C, Pickkers P. The mechanism of action of the adrenomedullin‐binding antibody adrecizumab. Crit Care 2018; 22: 159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Kox M, Pickkers P. Adrenomedullin: its double‐edged sword during sepsis slices yet again. Intensive Care Med Exp 2014; 2: 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Temmesfeld‐Wollbruck B, Brell B, David I et al. Adrenomedullin reduces vascular hyperpermeability and improves survival in rat septic shock. Intensive Care Med 2007; 33: 703–10. [DOI] [PubMed] [Google Scholar]
- 120. Temmesfeld‐Wollbruck B, Brell B, zu Dohna C et al. Adrenomedullin reduces intestinal epithelial permeability in vivo and in vitro. Am J Physiol Gastrointest Liver Physiol 2009; 297: G43–51. [DOI] [PubMed] [Google Scholar]
- 121. Muller HC, Witzenrath M, Tschernig T et al. Adrenomedullin attenuates ventilator‐induced lung injury in mice. Thorax 2010; 65: 1077–84. [DOI] [PubMed] [Google Scholar]
- 122. Ertmer C, Morelli A, Rehberg S et al. Exogenous adrenomedullin prevents and reverses hypodynamic circulation and pulmonary hypertension in ovine endotoxaemia. Br J Anaesth 2007; 99: 830–6. [DOI] [PubMed] [Google Scholar]
- 123. Itoh T, Obata H, Murakami S et al. Adrenomedullin ameliorates lipopolysaccharide‐induced acute lung injury in rats. Am J Physiol Lung Cell Mol Physiol 2007; 293: L446–52. [DOI] [PubMed] [Google Scholar]
- 124. Westphal M, Stubbe H, Bone HG et al. Hemodynamic effects of exogenous adrenomedullin in healthy and endotoxemic sheep. Biochem Biophys Res Commun 2002; 296: 134–8. [DOI] [PubMed] [Google Scholar]
- 125. Inal S, Koc E, Ulusal‐Okyay G et al. Protective effect of adrenomedullin on contrast induced nephropathy in rats. Nefrologia 2014; 34: 724–31. [DOI] [PubMed] [Google Scholar]
- 126. Oyar EO, Kiris I, Gulmen S et al. The protective effect of adrenomedullin on renal injury, in a model of abdominal aorta cross‐clamping. Thorac Cardiovasc Surg 2012; 60: 5–10. [DOI] [PubMed] [Google Scholar]
- 127. Muller‐Redetzky HC, Will D, Hellwig K et al. Mechanical ventilation drives pneumococcal pneumonia into lung injury and sepsis in mice: protection by adrenomedullin. Crit Care 2014; 18: R73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Talero E, Di Paola R, Mazzon E, Esposito E, Motilva V, Cuzzocrea S. Anti‐inflammatory effects of adrenomedullin on acute lung injury induced by Carrageenan in mice. Mediators Inflamm. 2012; 2012: 717851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Kubo K, Tokashiki M, Kuwasako K et al. Biological properties of adrenomedullin conjugated with polyethylene glycol. Peptides 2014; 57: 118–21. [DOI] [PubMed] [Google Scholar]
- 130. Watkins HA, Au M, Bobby R et al. Identification of key residues involved in adrenomedullin binding to the AM1 receptor. Br J Pharmacol 2013; 169: 143–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Struck J, Hein F, Karasch S, Bergmann A. Epitope specificity of anti‐Adrenomedullin antibodies determines efficacy of mortality reduction in a cecal ligation and puncture mouse model. Intensive Care Med Exp 2013; 1: 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Wagner K, Wachter U, Vogt JA et al. Adrenomedullin binding improves catecholamine responsiveness and kidney function in resuscitated murine septic shock. Intensive Care Med Exp 2013; 1: 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Geven C, Peters E, Schroedter M et al. Effects of the Humanized Anti‐Adrenomedullin Antibody Adrecizumab (HAM8101) on vascular barrier function and survival in rodent models of systemic inflammation and sepsis. Shock 2018; 50: 648–54. [DOI] [PubMed] [Google Scholar]
- 134. Geven C, van Lier D, Blet A et al. Safety, tolerability and pharmacokinetics/pharmacodynamics of the adrenomedullin antibody adrecizumab in a first‐in‐human study and during experimental human endotoxaemia in healthy subjects. Br J Clin Pharmacol 2018; 84: 2129–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Geven C, Bergmann A, Kox M, Pickkers P. Vascular effects of adrenomedullin and the anti‐adrenomedullin antibody adrecizumab in sepsis. Shock 2018; 50: 132–40. [DOI] [PubMed] [Google Scholar]
- 136. Pickkers P, van der Poll T. What's new in immunostimulating strategies in the ICU. Intensive Care Med. 2019; 45(1): 110–2. [DOI] [PubMed] [Google Scholar]
- 137. Temple R. Enrichment of clinical study populations. Clin Pharmacol Ther 2010; 88: 774–8. [DOI] [PubMed] [Google Scholar]
- 138. Vincent JL. Individual gene expression and personalised medicine in sepsis. Lancet Respir Med 2016; 4: 242–3. [DOI] [PubMed] [Google Scholar]
- 139. Grimaldi D, Vincent JL. Clinical trial research in focus: rethinking trials in sepsis. Lancet Respir Med 2017; 5: 610–1. [DOI] [PubMed] [Google Scholar]
- 140. Seymour CW, Kennedy JN, Wang S et al. Derivation, validation, and potential treatment implications of novel clinical phenotypes for sepsis. JAMA 2019; 321: 2003–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Pickkers P, Kox M. Towards precision medicine for sepsis patients. Crit Care 2017; 21: 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Laterre PF, editor Adrecizumab in patients with high adrenomedullin levels: Results from the Adrenoss‐2 (Phase II) Clinical Trial. e‐ISICEM international symposium on Intensive Care & Emergency Medicine; 2020. 16–09‐2020; online. [Google Scholar]