

Abstract
Dopamine beta hydroxylase (DBH) deficiency is an extremely rare autosomal recessive disorder with severe orthostatic hypotension, that can be treated with L‐threo‐3,4‐dihydroxyphenylserine (L‐DOPS). We aimed to summarize clinical, biochemical, and genetic data of all world‐wide reported patients with DBH‐deficiency, and to present detailed new data on long‐term follow‐up of a relatively large Dutch cohort. We retrospectively describe 10 patients from a Dutch cohort and 15 additional patients from the literature. We identified 25 patients (15 females) from 20 families. Ten patients were diagnosed in the Netherlands. Duration of follow‐up of Dutch patients ranged from 1 to 21 years (median 13 years). All patients had severe orthostatic hypotension. Severely decreased or absent (nor)epinephrine, and increased dopamine plasma concentrations were found in 24/25 patients. Impaired kidney function and anemia were present in all Dutch patients, hypomagnesaemia in 5 out of 10. Clinically, all patients responded very well to L‐DOPS, with marked reduction of orthostatic complaints. However, orthostatic hypotension remained present, and kidney function, anemia, and hypomagnesaemia only partially improved. Plasma norepinephrine increased and became detectable, while epinephrine remained undetectable in most patients. We confirm the core clinical characteristics of DBH‐deficiency and the pathognomonic profile of catecholamines in body fluids. Impaired renal function, anemia, and hypomagnesaemia can be part of the clinical presentation. The subjective response to L‐DOPS treatment is excellent and sustained, although the neurotransmitter profile in plasma does not normalize completely. Furthermore, orthostatic hypotension as well as renal function, anemia, and hypomagnesaemia improve only partially.
Keywords: dopamine beta hydroxylase (DBH) deficiency, epinephrine, hypomagnesaemia, L‐DOPS, neurogenic orthostatic hypotension, neurotransmitter disorders, norepinephrine
SYNOPSIS.
In this overview article, clinical presentation and treatment recommendations of dopamine beta hydroxylase (DBH)‐deficiency are presented, and several new observations regarding female fertility, anemia, kidney function, and serum electrolytes are described.
1. INTRODUCTION
Dopamine beta hydroxylase (DBH) deficiency (MIM#223360) is an autosomal recessive neurometabolic disorder due to pathogenic variants in the DBH gene. A gene transcript is present in (nor)adrenergic neurosecretory vesicles in the central and peripheral nervous system, the retina, and chromaffin cells of the adrenal medulla, where the enzyme DBH (EC 1.14.17.1) converts dopamine to norepinephrine. 1 , 2 DBH‐deficiency leads to undetectable levels of norepinephrine and epinephrine and increased levels of dopamine in the central and autonomous nervous system and peripheral organs 3 , 4 , 5 (Figure 1). Key symptom in DBH‐deficiency is profound orthostatic hypotension, 3 while patients—surprisingly—have no clear central nervous system manifestations, like neurocognitive abnormalities 6 or major sleep disturbances. 7
FIGURE 1.
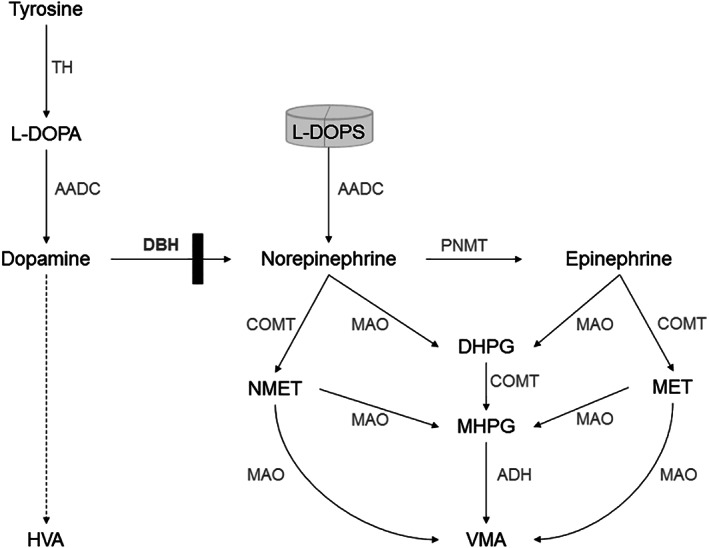
Simplified scheme of catecholamine synthesis and breakdown in DBH‐deficiency. Figure 1 depicts the simplified scheme of the biosynthesis and catabolism of catecholamines (dopamine, norepinephrine, and epinephrine), and shows the metabolic block in DBH‐deficiency (black bar). The artificial compound L‐DOPS can bypass this block because it can be converted to norepinephrine using the enzyme AADC. Breakdown of catecholamines can involve multiple steps (dashed arrows) and differs in different body compartments. HVA is the major stable end‐product of dopamine catabolism in cerebrospinal fluid, blood and urine. MHPG is the major end‐product of (nor)epinephrine catabolism in cerebrospinal fluid. NMET and MET are breakdown products of norepinephrine and epinephrine in the peripheral circulation and urine, VMA is the major common end‐product of (nor)epinephrine catabolism. AADC, aromatic l‐amino acid decarboxylase; ADH, alcohol dehydrogenase; DBH, dopamine beta hydroxylase; DHPG, dihydroxyphenylglycol; COMT: catechol‐O‐methyltransferase; HVA, homovanillic acid; L‐DOPA, L‐3,4 dihydroxyphenylalanine; L‐DOPS: l‐threo‐3,4‐dihydroxyphenylserine; MAO, monoamine oxidase; MET, metanephrine; MHPG, 3‐methoxy‐4‐hydroxyphenylglycol; NMET, normetanephrine; PNMT, phenylethanolamine N‐methyltransferase; VMA, vanillylmandelic acid
DBH‐deficiency is an extremely rare disorder. From its first description in the mid‐1980s, both in the United States of America 4 and the Netherlands, 5 only about 20 additional patients have been described. Despite its rarity, however, DBH‐deficiency offers a unique opportunity to study the role of catecholamines in the human autonomic and central nervous system. DBH‐deficiency leads to a selective and functional noradrenergic failure of a structurally normal sympathetic nervous system. 8 , 9 This is nicely illustrated by normal sweating patterns in patients with DBH‐deficiency, 4 , 10 a function of the sympathetic nervous system mediated by acetylcholine instead of norepinephrine. 11 Of utmost importance, DBH‐deficiency is a treatable disorder, in which the metabolic block can be bypassed by oral supplementation with L‐threo‐3,4‐dihydroxyphenylserine (L‐DOPS/Droxidopa) 12 (Figure 1). This makes DBH‐deficiency a diagnosis not to be missed.
Literature on DBH‐deficiency consists mainly of case reports, and many patients are presented in multiple publications. In order to summarize the available body of evidence, we performed a literature review on DBH‐deficiency with attention to clinical presentation, biochemical, and genetic diagnosis and treatment, and combined this with a retrospective chart review of a relatively large Dutch cohort of patients with DBH‐deficiency. The latter includes also data on long‐term follow‐up during treatment. In this overview, clinical presentation and treatment recommendations are summarized, and several new observations regarding female fertility, anemia, kidney function, and serum electrolytes are described.
2. METHODS
We performed a literature search on DBH‐deficiency using Pubmed indexed for Medline, Cochrane library, clinicaltrials.gov, and WHO clinical trial registry through August 2019 with search terms “dopamine beta hydroxylase deficiency,” “DBH deficiency” and [dopamine beta hydroxylase deficiency] (MESH). Neither language nor date filters were used in the initial search. Reference lists of key papers were manually screened for additional publications.
Patients from the Dutch cohort were diagnosed and are followed by four university clinics in the Netherlands.
Patients were included in this study if they had a confirmed genetic diagnosis of DBH‐deficiency and a plasma catecholamine profile with low/undetectable in levels of norepinephrine and epinephrine and increased levels of dopamine. If only one of the two diagnostic biomarkers was available, patients were included only if clinical description was of a clear purely noradrenergic failure.
We evaluated the published case reports and medical charts of the Dutch patients for clinical symptoms, initial diagnosis, age of onset, age of diagnosis, diagnostic tests, blood pressure values, treatment and treatment response, and long‐term follow‐up. Orthostatic hypotension was defined as a sustained reduction of systolic blood pressure of at least 20 mm Hg or diastolic blood pressure of at least 10 mm Hg within 3 minutes of standing. 13
We classified levels of catecholamines (norepinephrine, epinephrine, and dopamine) and their breakdown products in plasma, urine, and cerebrospinal fluid arbitrarily as low if decreased but >10% of local lower reference value, very low if <10% of local lower reference value, extremely low if <5% of local lower reference value or below detection limit, high if increased but <2 times local upper reference value, very high if >2 times local upper reference value, and extremely high if >5 times local upper reference value.
We recorded all reported routine biochemical measurements including hematological parameters, serum creatinine, and electrolytes. Kidney function was estimated by calculating the estimated glomerular filtration rate (eGFR), using the updated bedside Schwartz formulae (CKiD) for children, 14 and the CKD‐EPI formula for adults. 15 Estimated GFR (mL/min/1.73 m2) was classified as normal if ≥90, mildly decreased if 60 to 89, moderately decreased if 30 to 59, severely decreased if 15 to 29, and kidney failure if <15 mL/min/1.73 m2 (or dialysis) according to the National Kidney Foundation practice guidelines. 16 We used Dutch national reference values for blood hemoglobin and magnesium: hemoglobin: 7.5 to 10 mmol/L for females and 8.5 to 11 mmol/L for males, magnesium 0.7 to 1.0 mmol/L. We calculated fractional excretion of magnesium by the following general formula: (magnesium (urine, mmol/L) × creatinine (plasma, μmol/L))/(0.7 × magnesium (plasma, mmol/L) × creatinine (urine, μmol/L)) × 100, with normal values 2% to 4%. 17
Statistical analysis was performed using IBM SPSS statistics 22. Data were explored by descriptive statistics and scatter plots. To compare means, independent samples t‐test was used.
This study was retrospective, in accordance with the Declaration of Helsinki, and approved by the research ethics committee of the Radboud University Medical Center (2019‐5281). All Dutch patients gave informed consent for retrospective analysis of their medical files, and for anonymized publication of data.
3. RESULTS
The literature search ultimately revealed 39 publications containing information that could be deduced to individual patients (Figure S1). One patient was excluded because no genetic analysis was reported and he had an atypical clinical presentation (fixed heart rate on tilt table test suggesting concomitant parasympathetic failure). 18 In total, we identified 22 unique patients from 18 families reported in the literature. Furthermore, we identified three Dutch patients of whom data had never been published before, including a twin brother of a previously reported patient. Characteristics of all 25 patients (15 females) with DBH‐deficiency from 20 families are shown in Table S1 and summarized below. Data represent a combination of literature review and chart review unless specified otherwise.
3.1. Clinical presentation
3.1.1. Clinical symptoms and signs
All patients with DBH‐deficiency had severe orthostatic hypotension. Orthostatic complaints (mostly described as reduced standing time, lightheadedness, fainting spells, and exercise intolerance) started in early childhood (1‐4 years of age) in the majority of patients (n = 18), at school age (4‐12 years) in four patients, and in two patients during early adulthood of which one patient had onset of complaints after start of olanzapine. One patient who was diagnosed at age 40 years because of a positive family history did not report complaints, but had severe orthostatic hypotension on measurement and improved in overall functioning with L‐DOPS treatment. Orthostatic symptoms became more severe during adolescence in 11 patients.
Blood pressure within 3 minutes of standing fell to a mean of 72 mmHg systolic (range 44‐115, SD 15) and 49 mm Hg diastolic (range 20‐75, SD 13, with undetectably low diastolic blood pressure in three patients). Overall, the untreated drop in systolic and diastolic blood pressure within 3 minutes of standing was profound: mean systolic difference 58 mm Hg (range 32‐115 mm Hg, SD 21), mean diastolic difference 38 mm Hg (range 18‐74, SD 18). Supine blood pressure (office measurements) was <135/90 mm Hg in all patients (mean 109/68 mm Hg SD 13/11).
Eyelid ptosis was present in 18 patients and was absent in two patients who did have a mild miosis. In five patients, ptosis was not described. Ptosis could be unilateral or asymmetric. Other autonomic symptoms and signs were not reported systematically but included intermittent diarrhea and chronic nasal congestion. Nycturia was reported in three patients.
Information on the neonatal period was available for 14 patients and was reported as normal in five. Five patients (including one pair of twins) were born preterm and had low birth weight. Childhood hypoglycemia was reported in four patients.
Dysmorphic features were described non‐systematically and included brachydactyly and/or short hands or feet (n = 7), high palate (n = 6), mild hypertelorisme (n = 1), and micrognathia (n = 1). Additional dysmorphic features including coloboma in patient 14 were thought to be due to a concomitant mosaic 11p13 deletion.
Reproductive function in males was described in 5/10 patients and was abnormal in three (difficulty maintaining erections and/or retrograde ejaculation). Menarche and menstrual cycle were described in 7/15 female patients and were normal in all. Primary infertility before diagnosis was described in two female patients, it persisted during L‐DOPS treatment in one. The other patient had an early abortion during treatment with L‐DOPS. One other patient became pregnant with assisted fertility treatment before diagnosis. Spontaneous pregnancy before diagnosis was described in two patients, they delivered without complications, but lactation did not start.
Cognitive development was described in 18 patients, and found to be normal in 16. One patient had mildly delayed motor and speech milestones with normal mental capacities subsequently. One patient with a complicated neonatal period after premature birth and a severe infection at age 6 months had mild intellectual disability. In five patients extensive neurocognitive testing was performed, with a mean total IQ of 106 (range 98‐119). 6
Three patients were initially diagnosed as having epilepsy with generalized seizures and were treated with anti‐epileptic drugs. Their interictal EEGs were normal and seizures only occurred on assuming the upright position. One additional patient with epilepsy was reported, with focal onset seizures with rainbow visual aura, without information on triggers for the attacks. Two patients first came under medical attention because of anemia, three patients because of decreased kidney function. Median age of diagnosis of DBH‐deficiency was 24 years (range 13‐73, SD 13.5).
3.2. Diagnostic tests
3.2.1. Catecholamine measurements in plasma, urine, and CSF
In plasma, norepinephrine and dopamine measurements were reported in 24/25 patients, epinephrine in 22/25 patients (Table S1). Norepinephrine was decreased in all patients: extremely low in 20 and very low in four. Epinephrine was decreased in 21 patients: extremely low in 18, very low in two, and low in one patient. In one patient (pt 23), plasma epinephrine surprisingly was within the normal range. Dopamine was increased in all patients: extremely high in 12 patients, very high in seven, high in one patient. Norepinephrine and epinephrine remained extremely low when changing to standing position (reported in eight patients). Dopamine further increased on standing position in seven patients, and remained extremely high without further increase in one.
Urine measurements of norepinephrine, epinephrine, and dopamine were reported in six, five, and four patients, respectively, with a profile similar to plasma: very low to extremely low levels of norepinephrine and epinephrine, and very high levels of dopamine. The one patient (pt 23) with normal plasma epinephrine also had normal urinary epinephrine levels. Metanephrine (a metabolite of epinephrine, see Figure 1) in plasma was also normal in this patient and urinary dopamine was only slightly elevated. Catecholamines in cerebrospinal fluid were reported in three patients, and showed undetectable levels of norepinephrine and epinephrine, and increased dopamine (3‐20‐fold increase) in all.
3.2.2. DBH enzyme activity
DBH enzyme activity in plasma was reported in 18 patients and absent or severely decreased in all (Table S1). In three patients, DBH enzyme activity was tested in CSF and also absent there.
3.2.3. Molecular diagnosis
Molecular diagnosis was reported in 19 patients from 16 families; 10 different pathogenic variants were identified. In 12 patients, compound heterozygous pathogenic variants were found. The most frequently reported pathogenic variant (19 patients total, seven homozygous) is the intronic mutation c.339+2T>C (also reported as IVS1+2T>C) leading to aberrant splicing. 19 Table 1 shows all reported pathogenic variants. Distribution of pathogenic variants is schematically represented in Figure 2. Due to small sample size and relatively large number of variants, no phenotype/genotype correlations can be made.
TABLE 1.
Pathogenic DBH variants reported in patients with DBH‐deficiency
| NR | Location | DNA (NM_000787.4) | Protein (NP_000778.3) | Reference |
|---|---|---|---|---|
| 1 | Intron 1 | c.339+2T>C a | None (affects splicing) | Kim et al 19 |
| 2 | Exon 1 | c.301G>A | p.(Val101Met) b | Kim et al 19 |
| 3 | Exon 2 | c.342C>A | p.(Asp114Glu) | Kim et al 19 |
| 4 | Exon 3 | c.617del | p.(Glu206Glyfs*82) | Deinum et al 20 |
| 5 | Exon 4 | c.806G>T | p.(Cys269Phe) | Deinum et al 20 |
| 6 | Exon 6 | c.1033G>A | p.(Asp345Asn) b | Kim et al 19 |
| 7 | Exon 6 | c.1085C>A | p.(Ala362Glu) | Kim et al 19 |
| 8 | Intron 8 | c.1374+2_1374+20del | None (affects splicing) | Deinum et al 20 |
| 9 | Exon 9 | c.1409_1410delinsTG | p.(Thr470Met) | Bartoletti‐Stella et al 21 |
| 10 | Exon 11 | c. 1667A>G | p.(Tyr556Cys) | Deinum et al 20 |
Note: Pathogenic DBH variants reported in patients with DBH‐deficiency. Numbers in column 1 correspond with numbers in column 12 (Molecular diagnosis) of Table S1. RefSeq IDs are NM_000787.4 for DNA and NP_000778.3 for protein.
Also reported as IVS1+2T>C.
This has only been described in trans with c.1033G>A, one of the two may not be pathogenic.
FIGURE 2.
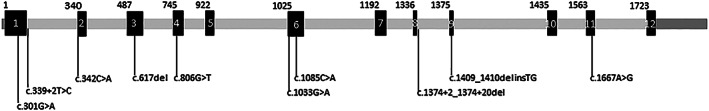
Schematic representation of pathogenic variants in the dopamine beta hydroxylase gene and pathogenic variants in DBH deficiency. Introns are shown in gray, exons in black and numbered 1 to 12, and UTR's in dark‐gray. Intron lengths are not to scale, while exon sizes are. The coding DNA positions of the first bases of each exon are shown above the schematic. In DBH‐deficiency, 2 intronic pathogenic variants and 8 exonic pathogenic variants have been described sofar, and are depicted under the schematic. The HGVS nomenclature is done using reference sequence NM_000787.4. The respective protein variants are given in Table 1
3.2.4. Biochemical parameters: kidney function, hemoglobin, and electrolytes
Kidney function, hemoglobin, and electrolytes have sparsely been reported in the literature and therefore data is mainly derived from chart review of the Dutch patients. Kidney function (eGFR) before treatment was available in all Dutch patients (Table 2) and in two additional patients from the literature. Decreased kidney function before treatment was present in all patients: mildly decreased in seven (of which three patients had a range including normal values >90 mL/min/1.73 m2), moderately decreased in four, and moderately to severely decreased in one patient.
TABLE 2.
Characteristics of the Dutch cohort with DBH‐deficiency and follow‐up of catecholamines, kidney function, hemoglobin, and magnesium before and during treatment
| Patient number | Sex (M/F) | FU before diagnosis (y) | Age at diagnosis (y) | FU after diagnosis (y) | Catechol‐amines (plasma) | Kidney function | Anemia | Magnesium | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Before treatment NE/E/DA | During treatment NE/E/DA | Before treatment | During treatment | Before treatment | During treatment | Serum Mg2+ before treatment | Fractional Mg2+ excretion before treatment (%) | Serum Mg2+ during treatment | Fractional Mg2+ excretion during treatment (%) | |||||
| 3 | F | 1 | 26 | 21 |
↓↓↓ ↓↓↓ ↑↑↑ |
↓↓ − ↑↑ |
↓ |
N− ↓↓ |
+ | + | N | |||
| 4 | F | 15 | 20 | 17 |
↓↓↓ ↓↓↓ ↑↑↑ |
↓↓ ↓↓↓ − |
N− ↓↓ |
N− ↓↓ |
+ | + | N−↓ | |||
| 8 | F | 3 | 34 | 18 |
↓↓↓ ↓↓↓ ↑ |
↓ ↓↓↓ ↑↑ |
N−↓ | N−↓ | + | + | N |
↑ |
N |
N |
|
9 Sib 10,11 |
M | 1 | 45 | 11 |
↓↓̂ ↓̂ ↑↑ |
− − − |
↓↓− ↓↓↓ |
↓↓− ↓↓↓ |
+ | + | ↓↓ | ↓ |
↑↑ a |
|
|
10 Sib 9,11 |
F | 4 | 36 | 11 |
↓↓̂ ↓↓↓ ↑↑ |
− − − |
↓ |
↓− ↓↓ |
+ | + | N | N |
N |
|
|
11 Sib 9,10 |
F | 9 | 40 | 15 |
↓↓↓ ↓↓↓ ↑ |
↓ ↓↓↓ − |
↓↓ |
↓− ↓↓ |
+ | + | ↓↓ |
↑↑ |
↓− ↓↓ |
↑− ↑↑ |
|
19 Sib 25 |
F | 23 | 23 | 15 |
↓↓↓ ↓↓↓ ↑↑↑ |
↓↓ ↓↓ ↑↑ |
↓↓ |
↓− ↓↓ |
+ | + | N−↓ |
↑ |
N−↓ |
↑ |
| 23 | F | 2 | 38 | 6 |
↓↓↓ N ↑↑↑ |
↓↓ N ↑↑ |
↓− ↓↓ |
↓− ↓↓ |
++ | ++ | ↓ | N−↓ | ||
| 24 | M | 0 | 13 | 1 |
urine ↓↓↓ ↓↓↓ ↑↑↑ |
− | ↓ | ↓ | − | − | N | ↑ | N |
↑ |
|
25 Sib 19 |
M | 0 | 35 | 1 |
↓↓↓ ↓↓↓ ↑↑↑ |
− ↓↓↓ ↑↑ |
N−↓ | N−↓ | + | + | N |
↑ |
N |
↑ |
Note: Representation of follow‐up data of the Dutch patients. Catecholamines: NE: norepinephrine, E: epinephrine, DA: dopamine. ↓: low (below reference value), ↓↓: very low (< 10% of local lower reference value), ↓↓↓: extremely low (< 5% of local lower reference value or below detection limit), ↑↑: very high (> two times local upper reference value), ↑↑↑: extremely high (> five times local upper reference limit), N: normal, ^probable overestimation due to technical reasons. 6 Kidney function: N: normal (eGFR>90 mL/min/1.73 m2), ↓: mildly decreased (eGFR 60‐90 mL/min/1.73 m2), ↓↓: moderately decreased (eGFR 30–59 mL/min/1.73 m2), ↓↓↓: severely decreased (eGFR 15–29 mL/min/1.73 m2). Anemia: − absent, + present (mild), ++ present (moderate). Magnesium: ↓: mildly decreased, ↓↓: moderately decreased ↑: increased, N = normal. Fractional magnesium excretion: normal value 2% to 4%. n = normal, ↑ = 4% to 10% ↑↑ > 10%.
Abbreviations: F, female. FU, follow‐up. M, male; y, years.
Fractional magnesium excretion calculation unreliable due to eGFR<40 mL/min/1.73 m2.
Hemoglobin before treatment was recorded in all Dutch patients (Table 2) and in four additional patients from the literature review. Mean hemoglobin level was below lower limit of normal for age and sex in 12/14 patients, while mean corpuscular volume was normal in 10/10 patients. Mild thrombocytopenia (100‐149 × 109/L) was reported in four patients before treatment, leukocytes were normal in all patients except for one who had mild leucopenia.
3.2.5. Electrolytes
Serum magnesium was not reported in the literature but available from chart review of all Dutch patients (Table 2). Two siblings evidently had hypomagnesaemia, while serum magnesium in the third sibling was normal. In three additional patients, serum magnesium was mildly decreased before treatment. Compared to mean serum magnesium level in a large general adult follow‐up cohort (0.86 mmol/L, range 0.34‐1.17, SD 0.06 22 ), mean magnesium levels in patients with DBH‐deficiency were much lower: untreated mean 0.60 mmol/L, range 0.33‐0.87, SD 0.18. Available serum sodium and potassium levels were normal in all patients. Fractional magnesium excretion could be calculated for five patients before treatment and was increased in all (range 5.0‐15.1%; Table 2).
3.2.6. Other additional testing
Cerebral MRI was performed in five patients as part of a research protocol, 6 and described in two additional patients, 21 , 23 and was structurally normal. EMG and nerve conduction studies were reported as normal in four patients and abnormal in one patient with a concomitant genetically confirmed hereditary neuropathy. Extensive physiologic autonomic function tests were performed in several patients and are excellently summarized in a review. 3
3.3. Treatment and follow‐up
3.3.1. L‐DOPS: effect, dose, and tolerability
All patients were treated with L‐DOPS, and remained on the same dose of L‐DOPS over the years. Doses ranged from 400 to 1800 mg/day, given in 2 to 3 doses. For the Dutch cohort, treatment follow‐up ranged from 1 to 21 years (median 13 years; Table 2). Treatment response was favorable and sustained in all patients and mostly subjectively described as “orthostatic symptoms disappeared” or “symptoms improved greatly.” Side effects were not reported apart from one patient with possible increase of nightmares on L‐DOPS. In the Netherlands, L‐DOPS was prescribed for compassionate use and imported from Sumitomo pharmaceuticals, Japan.
3.3.2. Other clinical features during treatment
Mild ptosis persisted under L‐DOPS treatment (reported in 7 patients). Response of diarrhea and nasal obstruction to L‐DOPS treatment was not reported in the literature or medical charts. Quality of life was not objectively recorded by questionnaires, but a great and continuous improvement in overall functioning was reported by all patients, for example, becoming able to work‐full time and to do sports. In one patient, fatigue remained present, especially during menstrual periods.
3.3.3. Blood pressure measurements under L‐DOPS treatment
Office orthostatic blood pressure measurements during treatment with L‐DOPS were not systematically reported. In some patients, it was stated that orthostatic hypotension disappeared. When exact blood pressure measurements were available for analysis, all patients (n = 7) still fulfilled the criteria for orthostatic hypotension during treatment in the great majority of measurements, with a mean systolic (supine minus standing) difference of 38 mm Hg (range 8‐60) and a mean diastolic difference of 21 mm Hg (range 4‐40). Overall, this was a less pronounced drop than before treatment was installed. There was no significant increase in supine office blood pressure during L‐DOPS treatment. However, in the Dutch patient who was diagnosed during adolescence L‐DOPS 100 mg twice daily led to mild hypertension, necessitating to decrease the dose to 75 mg twice daily, with still adequate clinical response. Figure 3 shows representative 24‐hours blood pressure measurements in one patient, before and during L‐DOPS treatment. Before treatment, there is no nightly dip in blood pressure. During treatment, there is no increase in overall blood pressure, and there is a more physiological dip in blood pressure during the night although still less than expected.
FIGURE 3.
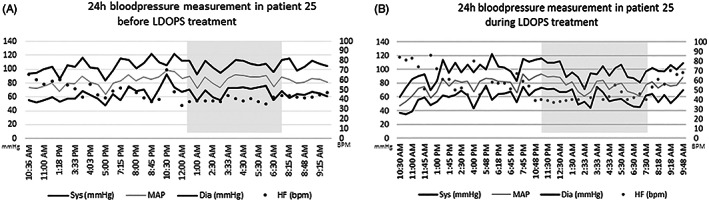
Twenty‐four hours bloodpressure measurements in one patient before and during L‐DOPS treatment. Twenty‐four hours blood pressure measurements in one patient before (left panel) and during (right panel L‐DOPS treatment. The night is shaded in gray. bpm, beats per minute; Dia, diastolic blood pressure; HF, heart frequency; MAP, mean arterial pressure; Sys: systolic blood pressure
3.3.4. Follow‐up of catecholamines during treatment
Norepinephrine and epinephrine levels in plasma before and during treatment were reported in 13 patients. Follow‐up for the Dutch cohort is shown in Table 2. With L‐DOPS, plasma norepinephrine increased in all patients: it became detectable but still below the lower limit of normal in 11, and reached normal levels in two. Epinephrine in plasma remained undetectable in nine and became detectable but still below lower limit of normal in three patients. Patient 23 had normal levels of epinephrine before and during L‐DOPS treatment. Plasma dopamine before and during treatment was reported in 12 patients and decreased with L‐DOPS in all, but in 10 patients remained above upper limit of normal.
3.3.5. Kidney function, hematological parameters, and electrolytes during treatment
For biochemical parameters under L‐DOPS treatment, only data from chart review of the Dutch patients was available (Table 2). With L‐DOPS treatment, there was an improvement of kidney function in only three patients.
In four patients, there was an increase of hemoglobin during treatment with L‐DOPS, with no other associated treatments at the time (eg, erythropoetin, ferrofumarate). There was no change in leucocyte and thrombocyte counts during treatment.
Overall, serum magnesium showed a significant increase under treatment (treated mean 0.67 mmol/L, range 0.37‐0.98, SD 0.15, P = .019). Fractional magnesium excretion could be calculated for seven patients during treatment and was normal in two patients who both had normal serum magnesium levels. In one patient (pt 9), calculation of fractional magnesium excretion was unreliable because of eGFR <40 (24 mL/min/1.73 m2; Table 2).
3.3.6. Mortality
One fatal case is extensively described in the literature (pt 13). This man had a severe phenotype with frequent syncopes from early childhood, and died at age 28 years after maximum 4 years of L‐DOPS treatment. Autopsy showed isolated microfoci of cortical gliosis, cardiac arteriolar smooth muscle hypertrophy, dispersed fibrosis in the cardiac conduction system, and sclerotic renal glomeruli. There was absent DBH immunoreactivity in the central nervous system (ventrolateral medulla) and the authors felt that his early death might have resulted from cardiac dysrhythmia. Furthermore, three deceased patients have been reported in a review (age 20; suspected suicide, and age 57 and 63 years; suspected natural cause, not specified). 3 One Dutch patient died at age 60 years due to urogenital cancer.
4. DISCUSSION
We describe the clinical characteristics of 25 patients with DBH‐deficiency and detailed follow‐up data of the 10 Dutch patients within this group. The study gives a comprehensive overview of clinical presentation, diagnosis, treatment, and long‐term follow‐up of this extremely rare neurogenic orthostatic hypotension syndrome, and has—for the first time—disclosed the high prevalence of decreased kidney function, hypomagnesaemia, and anemia in DBH‐deficiency.
Core clinical characteristics of DBH‐deficiency are confirmed, 3 namely a combination of profound orthostatic hypotension with onset mostly in early childhood, and eyelid ptosis. Patients often present with nonspecific symptoms and long diagnostic delays are common. Nonetheless, the clue to the diagnosis, that is, orthostatic hypotension, can easily be tested, and underscores the value of a complete physical examination in patients with poorly understood complaints.
In patients with a clinical suspicion of DBH‐deficiency, measurement of plasma catecholamines is the first ancillary diagnostic test. The pathognomonic metabolite profile shows undetectable levels of norepinephrine and epinephrine, and increased dopamine. This profile was found in all but one patient, who had thus far unexplained normal levels of epinephrine in plasma (and urine), despite previously reported pathogenic DBH variants, absent DBH enzyme activity, and a classical clinical phenotype.
The differential diagnosis of orthostatic hypotension varies from volume depletion and drug effects to primary autonomic failure. The latter includes a wide variety of neurological disorders, varying from pure autonomic failure, neurodegenerative disease (eg, multiple system atrophy), toxic and paraneoplastic neuropathies, amyloidosis, inflammatory disease (eg, Guillain Barré syndrome) and genetical disorders such as Fabry disease and DBH‐deficiency. 24 Other than in late adulthood, in childhood true orthostatic hypotension is rare and a genetic cause should be considered early in the diagnostic work‐up. 25 , 26 Recently, a novel young onset cause of orthostatic hypotension caused by mutations in CYB561 was identified. 27 Although clinically quite similar to DBH deficiency, this disorder is characterized by normal plasma dopamine in the presence of severely reduced levels of norepinephrine and epinephrine. 28
To diagnose DBH‐deficiency, a clinical presentation with severe young‐onset orthostatic hypotension, undetectable levels of norepinephrine and epinephrine and increased levels of dopamine in plasma, and pathogenic variants in the DBH gene should be present. Although the diagnosis of DBH‐deficiency can be supported by demonstration of absent to severely decreased DBH enzyme activity, it should be noted that plasma DBH enzyme activity is absent in 4 % of the healthy population due to a specific variant (−1021C>T) of the DBH gene, leading to pseudodeficiency with normal levels of plasma norepinephrine. 29 It is not necessary to test catecholamines in urine and cerebrospinal fluid for the diagnosis of DBH‐deficiency.
In the Dutch cohort, it is clearly shown that impaired kidney function is part of the clinical presentation of DBH‐deficiency. Our initial hypothesis was that this is caused by chronic, intermittent renal hypoperfusion due to severe orthostatic hypotension. However, the results of the kidney biopsy in one patient with DBH‐deficiency, which showed only discrete fibrosis, make this a less likely explanation 30 because renal hypoxia would cause clear histological abnormalities. 31 , 32 Renal failure in peripheral autonomic failure is attributed to supine hypertension, 33 but this is not found in patients with DBH‐deficiency. A potential damaging factor to the kidneys, which is quite unique to patients with DBH‐deficiency, is the very high level of dopamine intracellularly in the proximal tubular cells. In high concentrations, dopamine and intermediate dopamine metabolites are thought to be autotoxic, 34 possibly because of increased radical oxygen species production. 35 This might also explain why there is only partial improvement of kidney function after treatment is started, because dopamine levels remain evidently increased in most patients.
Anemia is also clearly part of the clinical presentation of DBH‐deficiency in most Dutch patients. It is mostly mild, and can improve on treatment with L‐DOPS. In general, chronic kidney disease is often associated with anemia due to relative erythropoietin deficiency, especially in moderate to severe kidney failure. 36 However, we also found anemia in patients with DBH‐deficiency who only had mildly decreased kidney function and therefore this is not likely to be the only explanation. Sympathetic innervation of the bone marrow plays an important role in hematopoiesis. Adrenergic signals induce mobilization of hematopoetic stem cells, and continuous trafficking of stem cells between bone marrow and blood compartments likely contributes to the maintenance of normal hematopoiesis. 37 Also in several other autonomic disorders, anemia with or without reduced kidney function has been described. 38 The precise mechanism of anemia in DBH‐deficiency and why it does not resolve completely with treatment in most patients is unknown.
Hypomagnesaemia was present in five Dutch patients with DBH‐deficiency before and during treatment. Fractional magnesium excretion was increased in four Dutch patients. Magnesium handling depends on gastro‐intestinal uptake and renal excretion, with fine regulation of magnesium transport in the distal tubule. 39 The precise pathophysiology, clinical significance and optimal treatment strategy of hypomagnesaemia in patients with DBH‐deficiency needs further investigation.
While plasma norepinephrine showed the expected—although incomplete—increase on L‐DOPS treatment, epinephrine remained below the detection limit in four Dutch patients during follow‐up and showed a slight increase in only one patient. This is puzzling because physiologically, it is expected that patients with DBH‐deficiency who are treated with L‐DOPS can synthesize epinephrine in the adrenal medulla. Lamotte et al showed that in patients with neurogenic orthostatic hypotension treated with L‐DOPS, norepinephrine excretion is much higher than expected and hypothesized that this is due to norepinephrine built up in the cytoplasm of proximal tubular cells exiting to the interstitium of the kidney. 40 Probably, norepinephrine excretion is also increased in patients with DBH‐deficiency under L‐DOPS treatment, preventing further metabolization to epinephrine.
Healthy pregnancy and delivery is possible in patients with DBH‐deficiency before and during L‐DOPS treatment, and with the limited data at hand continuing L‐DOPS during pregnancy appears safe and desirable. However, in the Dutch cohort three female patients had problems conceiving which might indicate that the rate of female fertility problems is increased before and during treatment with L‐DOPS. Oocytes express DBH, can take up dopamine and convert it to norepinephrine. 41 Oocyte derived norepinephrine may be necessary to regulate follicular development and oocyte maturation. 41 , 42 Higher levels of dopamine might lead to more reactive oxygen species in human follicular cells. 43 Hypothetically both mechanisms (absence of norepinephrine and presence of high dopamine concentrations in oocytes) could lead to decreased fertility in DBH‐deficiency. Another issue concerning reproduction is that successful lactation has not been described in patients with DBH‐deficiency, probably due to the increased levels of dopamine that inhibit the secretion of prolactin. 44
DBH‐deficiency is a treatable disorder: L‐DOPS leads to an almost immediate and sustained clinical response. In adults, Federal Drug Administration (FDA) approved dosing schedules can be followed, starting with 100 mg two to three times daily, increasing to a maximum of 1800 mg divided in three doses. Dose should be titrated individually based on clinical response, and last dose should be given more than 4 hours before bedtime to avoid nightly hypertension. 45 In our experience, more than 1200 mg per day is rarely needed. Temporary increase of daily dose during periods with more complaints, for example, illness or hot weather, can be of use (personal observation JD). For children, a starting dose of 2 mg/kg/d divided in 2 to 3 doses seems reasonable, eventually increasing to 5 to 10 mg/kg/d in 2 to 3 doses, with careful monitoring of side effects and blood pressure. L‐DOPS is excellently tolerated in patients with DBH‐deficiency. 45 Although the FDA approved L‐DOPS for neurogenic orthostatic hypotension in adults, in many countries including the Netherlands L‐DOPS is not registered and should be prescribed off‐label or for compassionate use.
Treatment of DBH‐deficiency with L‐DOPS greatly improves quality of life and exercise intolerance. However, catecholamine levels, kidney function, anemia, and hypomagnesaemia do not or only partially respond to treatment. Furthermore, ptosis often does not disappear, and blood pressure does not normalize; many patients still have (less pronounced) orthostatic hypotension on office measurements. The clinical relevance of these observations is unclear. Because of the extreme rarity of this disorder, clinical trials to determine optimum treatment strategy in DBH‐deficiency seem hard to effectuate. We think it is important that routine biochemical parameters including kidney function, hematological parameters, and electrolytes are regularly followed, while structured outcome measures like the orthostatic hypotension questionnaire, 46 could be used to monitor treatment response. Whether 24‐hours blood pressure measurements during L‐DOPS treatment are useful to improve long‐term outcome, for example, by aiming at a more physiological blood pressure course including the nocturnal dip, should be further investigated.
In conclusion, DBH‐deficiency is an extremely rare but fascinating cause of neurogenic orthostatic hypotension. It should be suspected in patients with profound, young‐onset orthostatic hypotension and ptosis. We have shown that decreased kidney function, hypomagnesaemia, and anemia are part of the clinical picture at presentation. DBH‐deficiency is a treatable disorder: L‐DOPS strongly improves the clinical symptoms and quality of life, even though orthostatic hypotension does not disappear. Laboratory parameters, including catecholamine profile in plasma, impaired kidney function, anemia, and hypomagnesaemia only partially respond to L‐DOPS treatment. The questions whether long‐term outcome can be improved by optimizing dosing strategies of L‐DOPS, and which parameters should be used to guide this, remain to be answered.
CONFLICT OF INTEREST
Tessa Wassenberg declares financial activities outside the submitted work: support for conference visits (travel, accommodation, and registration fees) to SSIEM 2018 Athens and SSIEM 2019 Rotterdam from Sanofi Genzyme, and speaker honorarium from PTC. Jaap Deinum, Frans J. van Ittersum, Erik‐Jan Kamsteeg, Maartje Pennings, Marcel M. Verbeek, Ron A. Wevers, Mirjam E. van Albada, Ido P. Kema, Jorie Versmissen, Ton van den Meiracker, Jacques W.M. Lenders, Leo Monnens, Michèl A. Willemsen declare that they have no conflict of interest.
ETHICS STATEMENT
Informed consent statement: All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1975, as revised in 2000. Informed consent was obtained from all Dutch patients for being included in the study.
Animal rights: This article does not contain any studies with animal subjects performed by any of the authors.
Supporting information
Figure S1 Literature search flowchart. Flow‐chart of literature search. Literature search performed on Pubmed, Cochrane database, clinicaltrials.gov, and WHO trial registry. The 39 publications that ultimately where identified containing unique patient information can be found in de reference list of Table S1.
Table S1 Clinical characteristics of 25 patients with DBH‐deficiency
ACKNOWLEDGEMENTS
The authors thank all patients for their contribution. No funding was received for this research.
Wassenberg T, Deinum J, van Ittersum FJ, et al. Clinical presentation and long‐term follow‐up of dopamine beta hydroxylase deficiency. J Inherit Metab Dis. 2021;44:554–565. 10.1002/jimd.12321
Communicating Editor: Georg Hoffmann
DATA AVAILABILITY STATEMENT
Anonymized data will be shared by reasonable request from any qualified investigator for purposes of replicating procedures and results.
REFERENCES
- 1. Nagatsu T. Genes for human catecholamine‐synthesizing enzymes. Neurosci Res. 1991;12(2):315‐345. [DOI] [PubMed] [Google Scholar]
- 2. Levin EY, Levenberg B, Kaufman S. The enzymatic conversion of 3,4‐dihydroxyphenylethylamine to norepinephrine. J Biol Chem. 1960;235:2080‐2086. [PubMed] [Google Scholar]
- 3. Robertson D, Garland EM. Dopamine beta‐hydroxylase deficiency. In: Pagon RA, Adam MP, Bird TD, Dolan CR, Fong CT, Stephens K, eds. GeneReviews. Seattle, WA: University of Washington; 1993. [PubMed] [Google Scholar]
- 4. Robertson D, Goldberg MR, Onrot J, et al. Isolated failure of autonomic noradrenergic neurotransmission. Evidence for impaired beta‐hydroxylation of dopamine. N Engl J Med. 1986;314(23):1494‐1497. [DOI] [PubMed] [Google Scholar]
- 5. Man in't Veld AJ, Boomsma F, Moleman P, Schalekamp MA. Congenital dopamine‐beta‐hydroxylase deficiency. A novel orthostatic syndrome. Lancet. 1987;1(8526):183‐188. [DOI] [PubMed] [Google Scholar]
- 6. Jepma M, Deinum J, Asplund CL, et al. Neurocognitive function in dopamine‐beta‐hydroxylase deficiency. Neuropsychopharmacology. 2011;36(8):1608‐1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Tulen JH, Man in't Veld AJ, Mechelse K, Boomsma F. Sleep patterns in congenital dopamine beta‐hydroxylase deficiency. J Neurol. 1990;237(2):98‐102. [DOI] [PubMed] [Google Scholar]
- 8. Donadio V, Liguori R, Incensi A, et al. Skin biopsy and microneurography disclose selective noradrenergic dysfunction due to dopamine‐beta‐hydroxylase deficiency. Auton Neurosci. 2016;197:56‐59. [DOI] [PubMed] [Google Scholar]
- 9. Rea RF, Biaggioni I, Robertson RM, Haile V, Robertson D. Reflex control of sympathetic nerve activity in dopamine beta‐hydroxylase deficiency. Hypertension. 1990;15(1):107‐112. [DOI] [PubMed] [Google Scholar]
- 10. Magnifico F, Misra VP, Murray NM, Mathias CJ. The sympathetic skin response in peripheral autonomic failure—evaluation in pure failure, pure cholinergic dysautonomia and dopamine‐beta‐hydroxylase deficiency. Clinical Autonomic Research. 1998;8(3):133‐138. [DOI] [PubMed] [Google Scholar]
- 11. Wehrwein EA, Orer HS, Barman SM. Overview of the anatomy, physiology, and pharmacology of the autonomic nervous system. Compr Physiol. 2016;6(3):1239‐1278. [DOI] [PubMed] [Google Scholar]
- 12. Biaggioni I, Robertson D. Endogenous restoration of noradrenaline by precursor therapy in dopamine‐beta‐hydroxylase deficiency. Lancet. 1987;2(8569):1170‐1172. [DOI] [PubMed] [Google Scholar]
- 13. Freeman R, Wieling W, Axelrod FB, et al. Consensus statement on the definition of orthostatic hypotension, neurally mediated syncope and the postural tachycardia syndrome. Auton Neurosci. 2011;161(1–2):46‐48. [DOI] [PubMed] [Google Scholar]
- 14. Mian AN, Schwartz GJ. Measurement and estimation of glomerular filtration rate in children. Adv Chronic Kidney Dis. 2017;24(6):348‐356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Levey AS, Stevens LA, Schmid CH, et al. A new equation to estimate glomerular filtration rate. Ann Intern Med. 2009;150(9):604‐612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Levey AS, Coresh J, Balk E, et al. National Kidney Foundation practice guidelines for chronic kidney disease: evaluation, classification, and stratification. Ann Intern Med. 2003;139(2):137‐147. [DOI] [PubMed] [Google Scholar]
- 17. Elliott C, Newman N, Madan A. Gentamicin effects on urinary electrolyte excretion in healthy subjects. Clin Pharmacol Ther. 2000;67(1):16‐21. [DOI] [PubMed] [Google Scholar]
- 18. Gentric A, Fouilhoux A, Caroff M, Mottier D, Jouquan J. Dopamine B hydroxylase deficiency responsible for severe dysautonomic orthostatic hypotension in an elderly patient. J Am Geriatr Soc. 1993;41(5):550‐551. [DOI] [PubMed] [Google Scholar]
- 19. Kim CH, Zabetian CP, Cubells JF, et al. Mutations in the dopamine beta‐hydroxylase gene are associated with human norepinephrine deficiency. Am J Med Genet. 2002;108(2):140‐147. [PubMed] [Google Scholar]
- 20. Deinum J, Steenbergen‐Spanjers GC, Jansen M, et al. DBH gene variants that cause low plasma dopamine beta hydroxylase with or without a severe orthostatic syndrome. Journal of medical genetics. 2004;41(4):e38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bartoletti‐Stella A, Chiaro G, Calandra‐Buonaura G, et al. A patient with PMP22‐related hereditary neuropathy and DBH‐gene‐related dysautonomia. J Neurol. 2015;262(10):2373‐2381. [DOI] [PubMed] [Google Scholar]
- 22. Kieboom BCT, Ligthart S, Dehghan A, et al. Serum magnesium and the risk of prediabetes: a population‐based cohort study. Diabetologia. 2017;60(5):843‐853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cheshire WP Jr, Dickson DW, Nahm KF, Kaufmann HC, Benarroch EE. Dopamine beta‐hydroxylase deficiency involves the central autonomic network. Acta Neuropathol. 2006;112(2):227‐229. [DOI] [PubMed] [Google Scholar]
- 24. Brown TP. Pure autonomic failure. Pract Neurol. 2017;17(5):341‐348. [DOI] [PubMed] [Google Scholar]
- 25. Axelrod FB. Genetic autonomic disorders. Semin Pediatr Neurol. 2013;20(1):3‐11. [DOI] [PubMed] [Google Scholar]
- 26. Stewart JM, Boris JR, Chelimsky G, et al. Pediatric disorders of orthostatic intolerance. Pediatrics. 2018;141(1):e20171673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. van den Berg MP, Almomani R, Biaggioni I, et al. Mutations in CYB561 causing a novel orthostatic hypotension syndrome. Circ Res. 2018;122(6):846‐854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Shibao CA, Garland EM, Black BK, et al. Congenital absence of norepinephrine due to CYB561 mutations. Neurology. 2020;94(2):e200‐e204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zabetian CP, Anderson GM, Buxbaum SG, et al. A quantitative‐trait analysis of human plasma‐dopamine beta‐hydroxylase activity: evidence for a major functional polymorphism at the DBH locus. Am J Hum Genet. 2001;68(2):515‐522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wassenberg T, Willemsen M, Dijkman H, Deinum J, Monnens L. Congenital eyelid ptosis, decreased glomerular filtration, and orthostatic hypotension: questions. Pediatr Nephrol. 2017;32(7):1169‐1170. 10.1007/s00467-016-3494-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Nangaku M. Hypoxia and tubulointerstitial injury: a final common pathway to end‐stage renal failure. Nephron Exp Nephrol. 2004;98(1):e8‐e12. [DOI] [PubMed] [Google Scholar]
- 32. Fine LG, Norman JT. Chronic hypoxia as a mechanism of progression of chronic kidney diseases: from hypothesis to novel therapeutics. Kidney Int. 2008;74(7):867‐872. [DOI] [PubMed] [Google Scholar]
- 33. Garland EM, Gamboa A, Okamoto L, et al. Renal impairment of pure autonomic failure. Hypertension. 2009;54(5):1057‐1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Goldstein DS, Kopin IJ, Sharabi Y. Catecholamine autotoxicity. Implications for pharmacology and therapeutics of Parkinson disease and related disorders. Pharmacol Ther. 2014;144(3):268‐282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Acquier AB, Mori Sequeiros Garcia M, Gorostizaga AB, Paz C, Mendez CF. Reactive oxygen species mediate dopamine‐induced signaling in renal proximal tubule cells. FEBS Lett. 2013;587(19):3254‐3260. [DOI] [PubMed] [Google Scholar]
- 36. Fishbane S, Spinowitz B. Update on anemia in ESRD and earlier stages of CKD: Core curriculum 2018. Am J Kidney Dis. 2018;71(3):423‐435. [DOI] [PubMed] [Google Scholar]
- 37. Garcia‐Garcia A, Korn C, Garcia‐Fernandez M, et al. Dual cholinergic signals regulate daily migration of hematopoietic stem cells and leukocytes. Blood. 2019;133(3):224‐236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Biaggioni I, Robertson D, Krantz S, Jones M, Haile V. The anemia of primary autonomic failure and its reversal with recombinant erythropoietin. Ann Intern Med. 1994;121(3):181‐186. [DOI] [PubMed] [Google Scholar]
- 39. de Baaij JH, Hoenderop JG, Bindels RJ. Regulation of magnesium balance: lessons learned from human genetic disease. Clin Kidney J. 2012;5(Suppl 1):i15‐i24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lamotte G, Holmes C, Sullivan P, Goldstein DS. Substantial renal conversion of L‐threo‐3,4‐dihydroxyphenylserine (droxidopa) to norepinephrine in patients with neurogenic orthostatic hypotension. Clin Autonom Res. 2019;29(1):113‐117. [DOI] [PubMed] [Google Scholar]
- 41. Mayerhofer A, Smith GD, Danilchik M, et al. Oocytes are a source of catecholamines in the primate ovary: evidence for a cell‐cell regulatory loop. Proc Natl Acad Sci U S A. 1998;95(18):10990‐10995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Greiner M, Paredes A, Rey‐Ares V, Saller S, Mayerhofer A, Lara HE. Catecholamine uptake, storage, and regulated release by ovarian granulosa cells. Endocrinology. 2008;149(10):4988‐4996. [DOI] [PubMed] [Google Scholar]
- 43. Saller S, Kunz L, Berg D, et al. Dopamine in human follicular fluid is associated with cellular uptake and metabolism‐dependent generation of reactive oxygen species in granulosa cells: implications for physiology and pathology. Hum Reprod. 2014;29(3):555‐567. [DOI] [PubMed] [Google Scholar]
- 44. Fitzgerald P, Dinan TG. Prolactin and dopamine: what is the connection? A review article. J Psychopharmacol. 2008;22(2 Suppl):12‐19. [DOI] [PubMed] [Google Scholar]
- 45. Strassheim V, Newton JL, Tan MP, Frith J. Droxidopa for orthostatic hypotension: a systematic review and meta‐analysis. J Hypertens. 2016;34(10):1933‐1941. [DOI] [PubMed] [Google Scholar]
- 46. Kaufmann H, Malamut R, Norcliffe‐Kaufmann L, Rosa K, Freeman R. The orthostatic hypotension questionnaire (OHQ): validation of a novel symptom assessment scale. Clin Autonom Res. 2012;22(2):79‐90. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Literature search flowchart. Flow‐chart of literature search. Literature search performed on Pubmed, Cochrane database, clinicaltrials.gov, and WHO trial registry. The 39 publications that ultimately where identified containing unique patient information can be found in de reference list of Table S1.
Table S1 Clinical characteristics of 25 patients with DBH‐deficiency
Data Availability Statement
Anonymized data will be shared by reasonable request from any qualified investigator for purposes of replicating procedures and results.