

Abstract
Ravulizumab every 8 weeks showed non‐inferiority to eculizumab every 2 weeks in a 26‐week, phase 3, randomized controlled trial in adults with paroxysmal nocturnal hemoglobinuria (PNH) who were clinically stable on eculizumab (NCT03056040). We report results from the first 26 weeks of the extension period in which patients continued ravulizumab (n = 96) or switched from eculizumab to ravulizumab (n = 95). At week 52, mean (SD) lactate dehydrogenase levels increased 8.8% (29%) with ravulizumab‐ravulizumab and 5.8% (27%) with eculizumab‐ravulizumab from primary evaluation period baseline. During the extension period, four patients (ravulizumab‐ravulizumab, n = 3; eculizumab‐ravulizumab, n = 1) experienced breakthrough hemolysis, but none associated with serum free C5 ≥ 0.5 μg/mL. Mean Functional Assessment of Chronic Illness Therapy (FACIT)‐Fatigue scores remained stable through week 52. During the extension period, proportions of patients avoiding transfusion remained stable (ravulizumab‐ravulizumab, 86.5%; eculizumab‐ravulizumab, 83.2%); 81.2% and 81.1%, respectively, had stabilized hemoglobin. All patients maintained serum free C5 levels < 0.5 μg/mL. Adverse events were generally similar between groups, and rates were lower in the extension period. Adults with PNH on stable eculizumab therapy who received ravulizumab over 52 weeks experienced durable efficacy, with consistent efficacy in patients who received eculizumab during the primary evaluation period and then switched to ravulizumab. Ravulizumab was well tolerated.
Keywords: breakthrough hemolysis, complement inhibitor, hemoglobin, paroxysmal nocturnal hemoglobinuria, quality of life, transfusion
Novelty Statements.
Ravulizumab every 8 weeks showed non‐inferiority to eculizumab every 2 weeks in a 26‐week, phase 3, randomized controlled trial in adults with paroxysmal nocturnal hemoglobinuria (PNH) who were clinically stable on eculizumab; this work reports results from the first 26 weeks of the extension period in which the durability of ravulizumab efficacy and safety (1 year of treatment) and the efficacy and safety of ravulizumab after switching from eculizumab were assessed.
Ravulizumab was well tolerated with durable efficacy through 52 weeks in adults with PNH who were clinically stable on therapy with eculizumab.
These results suggest that patients with PNH who are clinically stable on eculizumab can switch safely and effectively to weight‐based dosing of ravulizumab.
1. INTRODUCTION
Paroxysmal nocturnal hemoglobinuria (PNH) is a rare genetic hematologic disorder caused by uncontrolled activation of the terminal complement pathway. 1 In patients with PNH, mutations in the phosphatidylinositol glycan class A (PIGA) gene lead to deficiency in glycosylphosphatidylinositol (GPI) anchor proteins and the subsequent reduction in GPI‐linked proteins (eg, CD55, CD59) on the cell surface, making red blood cells more vulnerable to complement‐mediated intravascular hemolysis and activating platelets, monocytes, and granulocytes. 1 , 2 Severe intravascular hemolysis results in high concentrations of free hemoglobin in plasma; depletion of nitric oxide; and clinical sequelae, including thrombosis, dysphagia, abdominal pain, erectile dysfunction, renal impairment, pulmonary hypertension, and hypertension. 2 In untreated patients, clinical symptoms are associated with reduced quality of life (QoL) and increased incidence of hospitalization, 3 as well as increased risk of thrombosis and death. 4
Eculizumab, a humanized monoclonal antibody that blocks terminal C5 activation, 5 was the first therapy for PNH approved by the US Food and Drug Administration and European Medicines Agency in 2007 and has been established as a standard of care for PNH management. Intravenous infusion of eculizumab every 2 weeks (q2w) is associated with a rapid reduction in lactate dehydrogenase (LDH) levels (a marker for hemolysis) and decreased transfusion requirements, as well as significantly improved QoL. 6 , 7 , 8 Patients receiving long‐term treatment with eculizumab also show significantly longer survival versus untreated patients, with survival rates similar to those in an age‐ and sex‐matched normal population. 9
Despite treatment with approved dosages of eculizumab, approximately 11%–27% of patients may experience breakthrough hemolysis, 10 , 11 , 12 which typically occurs when serum levels of eculizumab are reduced, just before administration of the next dose, or in patients with complement hyperactivation due to infection or trauma. 13 The risk for breakthrough hemolysis, together with the q2w dosing schedule, may impose a substantial burden on patients and have negative effects on prognosis and QoL.
Ravulizumab is a long‐acting C5 inhibitor developed through targeted engineering to provide immediate, complete, and sustained C5 inhibition with 8‐week dosing intervals 13 and is approved in the United States, European Union, Japan, Canada, Brazil, and Australia for the treatment of adults with PNH. In the largest phase 3 randomized study of eculizumab‐experienced patients with PNH, intravenous infusion of ravulizumab every 8 weeks (q8w) was non‐inferior to eculizumab q2w after 26 weeks for all primary and key secondary endpoints, including percentage change in LDH levels, proportion of patients with breakthrough hemolysis, mean change in Functional Assessment of Chronic Illness Therapy (FACIT)‐Fatigue scores, proportion of patients avoiding transfusion, and proportion of patients with stabilized hemoglobin. 14 As expected, no significant treatment differences were observed in this non‐inferiority study; however, five patients receiving eculizumab had breakthrough hemolysis events compared with no patient who received ravulizumab. 14 After 26 weeks of treatment, all patients in the phase 3 study had the option to receive weight‐based dosing of ravulizumab in an open‐label extension.
The aim of this study was to assess the durability of ravulizumab efficacy and safety for up to 52 weeks and to assess the efficacy and safety of ravulizumab after switching from eculizumab in complement inhibitor–experienced adults with PNH.
2. METHODS
2.1. Study Design
This was an open‐label extension of a phase 3, randomized, active‐controlled trial conducted at 49 sites in 11 countries (ClinicalTrials.gov Identifier: NCT03056040). Detailed methods have been previously described. 14 The protocol was approved by the institutional review board or independent ethics committee at each participating site. The trial was conducted in accordance with the principles of Good Clinical Practice according to the International Council for Harmonisation Harmonized Tripartite Guideline.
The study included a 4‐week screening period and a 26‐week primary evaluation period, followed by an extension period. During the primary evaluation period, patients were stratified by history of transfusion and were randomly assigned (1:1) to switch to weight‐based dosing of ravulizumab q8w or to continue receiving eculizumab 900 mg q2w. Patients in the ravulizumab arm received a weight‐based loading dose on day 1 (2400 mg for patients weighing ≥ 40 to < 60 kg, 2700 mg for ≥ 60 to < 100 kg, and 3000 mg for ≥ 100 kg), followed by weight‐based maintenance doses on day 15 and q8w thereafter (3000 mg for patients weighing ≥ 40 to < 60 kg, 3300 mg for ≥ 60 to < 100 kg, and 3600 mg for ≥ 100 kg). 14 During the extension period, which is reported in this manuscript, patients who received ravulizumab in the primary evaluation period continued on ravulizumab q8w, and all patients who received eculizumab were switched to ravulizumab q8w (loading dose on day 183, followed by maintenance doses on day 197 and q8w thereafter).
2.2. Patients
Male and female patients with PNH confirmed by flow cytometry who were ≥ 18 years of age were eligible for inclusion if they were clinically stable on treatment with eculizumab for ≥ 6 months, had LDH levels ≤ 1.5 × the upper limit of normal (ULN) at screening (measured at trough eculizumab level), and had been vaccinated against meningococcal infections within 3 years before, or at the time of, study drug initiation. Patients were excluded if they were < 40 kg at screening; had LDH levels > 2 × ULN or major adverse vascular event (MAVE) in the 6 months before day 1; had platelet count < 30 × 109/L or neutrophil count < 500/μL at screening; or had a history of bone marrow transplantation, Neisseria meningitidis infection, unexplained or recurrent infection, or human immunodeficiency virus infection. All patients provided written informed consent.
2.3. Outcomes
The primary efficacy endpoint was the percentage change in LDH levels from the primary evaluation period baseline at study visit. Secondary endpoints included the proportion of patients with breakthrough hemolysis; change from baseline in QoL as measured by the FACIT‐Fatigue scale (range, 0–52; higher scores indicate less fatigue) and European Organisation for Research and Treatment of Cancer, Quality of Life Questionnaire–Core 30 (EORTC QLQ‐C30) global health status, physical functioning, and fatigue subscales (range, 0–100; higher scores for functional scales indicate better QoL; lower scores for symptom scales indicate lower symptom levels); proportion of patients avoiding transfusion; and proportion of patients with stabilized hemoglobin levels. Breakthrough hemolysis was defined as at least one new or worsening symptom or sign of intravascular hemolysis in the presence of LDH levels ≥ 2 × ULN after prior reduction of LDH to < 1.5 × ULN on treatment. QoL scales were administered at baseline and at weeks 1, 4, 10, 18, 26, and 52. Patients were considered to have achieved transfusion avoidance if they remained transfusion free and did not require a transfusion per protocol‐specified guidelines. Stabilized hemoglobin was defined as avoidance of a ≥ 2 g/dL decrease in hemoglobin levels from baseline in the absence of transfusion. Additional endpoints included levels of serum free C5 and safety, including adverse events (AEs), serious AEs (SAEs), MAVEs, and immunogenicity, up to the 52‐week data cutoff. Laboratory samples were collected predose on the same visit day.
2.4. Statistical Analysis
For the purposes of data analysis, study visits were defined using windows in order to align adjacent visits for patients in the two treatment arms. Any measurement within the window was assigned to that visit (eg, week 36 analysis visit included day 239 visit for the ravulizumab‐ravulizumab arm and day 253 visit for the eculizumab‐ravulizumab arm). Descriptive statistics were used to summarize outcomes as of the data cutoff (7 September 2018). Efficacy was analyzed in the extension set (ie, all patients who entered the extension period), and safety was analyzed in the safety set (ie, all patients who received at least one dose of study drug). For breakthrough hemolysis, transfusion avoidance, and stabilized hemoglobin, patients who withdrew because of lack of efficacy were considered non‐responders.
3. RESULTS
3.1. Patients
A total of 208 patients were screened for eligibility, and 195 were randomly assigned and received treatment with ravulizumab (n = 97) or eculizumab (n = 98). Of the 191 patients who completed the 26‐week primary evaluation period, all 96 patients in the ravulizumab arm and 95 patients in the eculizumab arm entered the extension period and received ravulizumab (Figure S1). As of the data cutoff (7 September 2018), two patients discontinued treatment (ravulizumab‐ravulizumab, n = 1 [patient decision]; eculizumab‐ravulizumab, n = 1 [physician decision]). Patient demographics and baseline clinical characteristics have been previously reported 14 and were generally similar between treatment arms.
3.2. Primary Endpoint
Patients in both treatment arms showed durable responses for percentage change in LDH levels, similar to that observed during the primary evaluation period. At week 52, patients in the ravulizumab‐ravulizumab arm had a mean (SD) 8.8% (29%) increase in LDH levels from primary evaluation baseline, whereas patients in the eculizumab‐ravulizumab arm had a 5.8% (27%) increase in LDH levels from primary evaluation baseline (Figure 1A ). Mean LDH levels were maintained at approximately 1 × ULN during the extension period (Figure 1B ).
FIGURE 1.
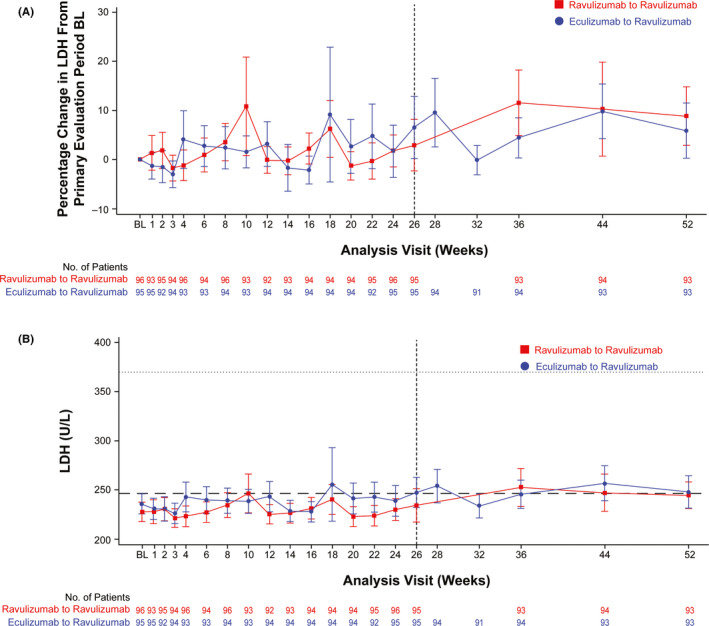
(A) Mean (95% CI) percentage change from primary evaluation period BL in LDH levels. Dashed vertical line indicates the end of the primary evaluation period. (B) Mean (95% CI) LDH levels over time. Dashed horizontal line indicates 1 × ULN, and dotted horizontal line indicates 1.5 × ULN. BL = baseline; LDH = lactate dehydrogenase; ULN = upper limit of normal
3.3. Secondary Endpoints
During the extension period, three patients in the ravulizumab‐ravulizumab group and one patient in the eculizumab‐ravulizumab group experienced breakthrough hemolysis (Table 1 ). None of the events of breakthrough hemolysis in patients receiving ravulizumab were associated with serum free C5 ≥ 0.5 μg/mL, and 2/3 and 1/1 were associated with infection in the ravulizumab‐ravulizumab and eculizumab‐ravulizumab arms, respectively. The two BTH events between weeks 27 to 52 in the ravulizumab‐ravulizumab arm were associated with the following infections: upper respiratory tract infection (one patient) and nasopharyngitis (one patient). The third BTH event in this treatment arm was unrelated to identified concomitant infection to explain the cause of BTH. In the eculizumab‐ravulizumab arm, one BTH event occurred between weeks 27 and 52, which was associated with upper respiratory tract infection. Of the patients who did not have breakthrough hemolysis during the primary evaluation period, 97% in the ravulizumab‐ravulizumab arm and 100% in the eculizumab‐ravulizumab arm had no breakthrough hemolysis during the extension period. One patient who received eculizumab and experienced breakthrough hemolysis with infections and no free C5 elevation during the primary evaluation period experienced one event of breakthrough hemolysis during the extension period while receiving ravulizumab. Mean (SD) FACIT‐Fatigue scores achieved at the end of the primary evaluation period (week 26; ravulizumab‐ravulizumab, 44.1 [8.49]; eculizumab‐ravulizumab, 41.5 [10.19]) were similar to those observed at the end of open‐label treatment (week 52; ravulizumab‐ravulizumab, 43.3 [8.62]; eculizumab‐ravulizumab, 40.7 [10.67]; Figure S2). At week 52, mean (SD) EORTC QLQ‐C30 subscale scores were similar between treatment groups (ravulizumab‐ravulizumab vs eculizumab‐ravulizumab: global health status, 74.4 [17.12] vs 71.2 [20.87]; physical functioning, 89.3 [18.58] vs 86.7 [17.37]; fatigue, 22.6 [23.73] vs 27.0 [25.82]; Figure S3). During the extension period, 86.5% and 83.2% of patients in the ravulizumab‐ravulizumab and eculizumab‐ravulizumab arms, respectively, avoided transfusion (Table 1 ); 92.9% and 93.8%, respectively, of those who avoided transfusion during the primary evaluation period maintained the response during the extension period. A total of 81.2% and 81.1% of patients in the ravulizumab‐ravulizumab and eculizumab‐ravulizumab arms, respectively, achieved stabilized hemoglobin (Table 1 ); 90.3% and 91.7%, respectively, of those who achieved stabilized hemoglobin during the primary evaluation period maintained the response during the extension period.
TABLE 1.
Summary of Efficacy Endpoints
| Ravulizumab‐Ravulizumab | Eculizumab‐Ravulizumab | |||
|---|---|---|---|---|
|
Primary Evaluation Period (n = 97) a |
Extension Period (n = 96) |
Primary Evaluation Period (n = 98) a |
Extension Period (n = 95) |
|
| Breakthrough hemolysis, patients, n (%) | 0 | 3 (3.1) | 5 (5.1) | 1 (1.1) b |
| Breakthrough hemolysis, events, n | 0 | 3 | 7 | 1 |
| Serum free C5 ≥ 0.5 μg/mL | 0 | 0 | 4 c | 0 |
| Infection (with no Serum free C5 elevation) | 0 | 2 d | 2 e | 1 b |
| Unknown | 0 | 1 | 1 | 0 |
| Transfusion avoidance, n (%) | 85 (87.6) | 83 (86.5) | 81 (82.7) | 79 (83.2) |
| Hemoglobin stabilization, n (%) | 74 (76.3) | 78 (81.2) | 74 (75.5) | 77 (81.1) |
Data from the primary evaluation period (weeks 0–26) are based on the full analysis set.
The 1 patient who experienced breakthrough hemolysis during the extension period had previously experienced breakthrough hemolysis during the primary evaluation period. Between baseline and week 26, this patient experienced influenza‐like symptoms; between weeks 27 and 52, the patient experienced an upper respiratory tract infection.
One patient in the eculizumab‐ravulizumab arm between BL and week 26 with inadequate C5 inhibition also had concomitant infection.
The 2 BTH events were associated with the following infections reported from week 27 to week 52 in the ravulizumab‐ravulizumab arm: nasopharyngitis (1 patient) and upper respiratory tract infection (1 patient).
The following symptoms and infections were reported from BL to week 26 in the eculizumab‐ravulizumab arm: flu‐like symptoms (1 patient) and acute pyelonephritis (1 patient).
3.4. Serum free C5 Levels
Patients in both treatment groups had Serum free C5 levels < 0.5 µg/mL through the extension period. In the ravulizumab‐ravulizumab arm, patients continued to maintain Serum free C5 levels < 0.5 µg/mL (Figure 2A ). In the eculizumab‐ravulizumab arm, patients showed improved Serum free C5 control, as none had suboptimal terminal complement inhibition (Serum free C5 levels ≥ 0.5 µg/mL) after switching to ravulizumab (Figure 2B ).
FIGURE 2.
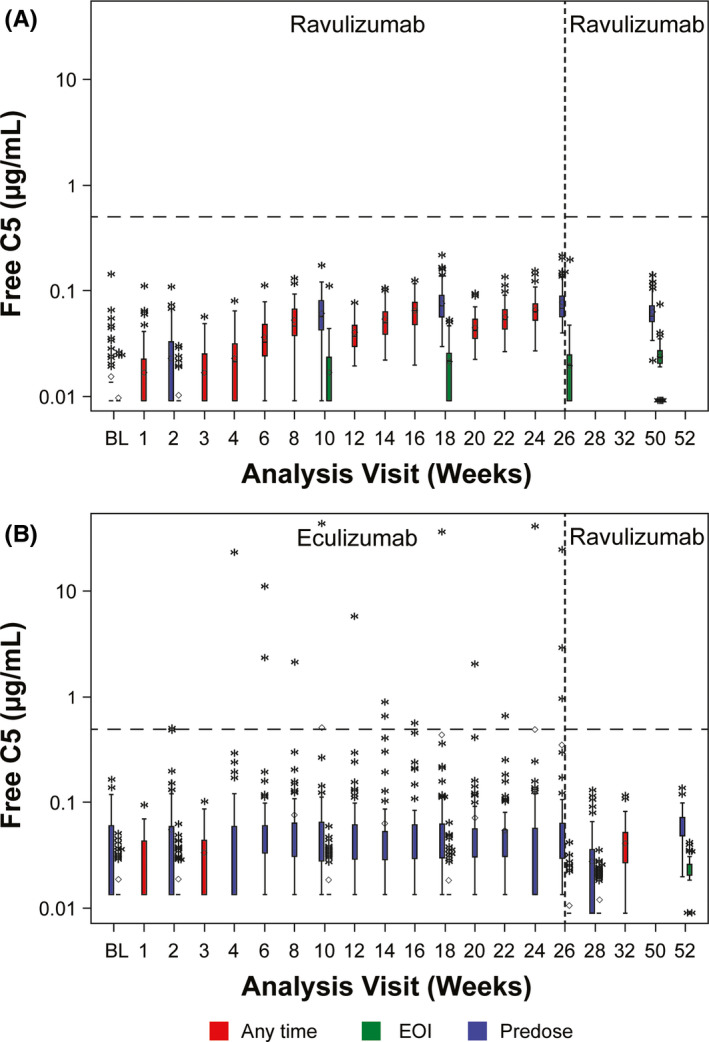
Serum free C5 levels through 52 weeks in the (A) ravulizumab‐ravulizumab arm and (B) eculizumab‐ravulizumab arm. Horizontal line in the middle of each box indicates the median, and a diamond indicates the mean. Top and bottom borders of the box represent the 75th and 25th percentiles, respectively, and whiskers represent the 1.5 interquartile range of the lower and upper quartiles. Asterisks represent values outside the interquartile range. Dashed horizontal lines indicate Serum free C5 concentration of 0.5 μg/mL. Dashed vertical line indicates the end of the primary evaluation period. BL = baseline; EOI = end of infusion
3.5. Safety
The incidence of treatment‐emergent AEs (TEAEs) was lower in the extension period compared with the primary evaluation period (Table 2 ). The most common TEAEs in the ravulizumab‐ravulizumab arm during the extension period were fatigue (n = 13 [13.5%]), upper respiratory tract infection (n = 9 [9.4%]), and headache, nasopharyngitis, diarrhea, and pyrexia (n = 6 [6.3%] each). In the eculizumab‐ravulizumab arm, fatigue (n = 13 [13.7%]), headache (n = 10 [10.5%]), upper respiratory tract infection (n = 8 [8.4%]), and nasopharyngitis (n = 7 [7.4%]) were the most common TEAEs (Table 2 and Table S1). One patient in each treatment arm experienced a TEAE considered as a MAVE during the extension period: thrombophlebitis in a patient in the ravulizumab‐ravulizumab arm (deemed unrelated to catheter; patient recovered fully) and cerebral infarction in a patient in the eculizumab‐ravulizumab arm (event resolved with treatment). There was one incident of thrombosis in the eculizumab‐ravulizumab arm, which was deemed unrelated to study drug. No meningococcal infections were reported through the 52‐week cutoff date.
TABLE 2.
Summary of AEs
| AE, n (%) | Ravulizumab‐Ravulizumab | Eculizumab‐Ravulizumab | ||
|---|---|---|---|---|
|
Primary Evaluation Period (n = 97) |
Extension Period (n = 96) |
Primary Evaluation Period (n = 98) |
Extension Period (n = 95) |
|
| Any TEAE | 89 (91.8) | 76 (79.2) | 86 (87.8) | 71 (74.7) |
| TEAE considered as a MAVE | 0 | 1 (1.0) | 0 | 1 (1.1) |
| TEAE leading to study drug discontinuation | 0 | 0 | 0 | 0 |
| Any SAE | 4 (4.1) | 8 (8.3) | 8 (8.2) | 5 (5.3) |
| SAE leading to study drug discontinuation | 0 | 0 | 0 | 0 |
| Death | 0 | 0 | 0 | 0 |
| TEAEs occurring in ≥ 5% of patients in either treatment group during the extension period | ||||
| Fatigue | 7 (7.2) | 13 (13.5) | 7 (7.1) | 13 (13.7) |
| URTI | 18 (18.6) | 9 (9.4) | 11 (11.2) | 8 (8.4) |
| Nasopharyngitis | 21 (21.6) | 6 (6.3) | 20 (20.4) | 7 (7.4) |
| Headache | 27 (27.8) | 6 (6.3) | 19 (19.4) | 10 (10.5) |
| Diarrhea | 9 (9.3) | 6 (6.3) | 7 (7.1) | 5 (5.3) |
| Pyrexia | 9 (9.3) | 6 (6.3) | 5 (5.1) | 6 (6.3) |
| Dizziness | 3 (3.1) | 2 (2.1) | 7 (7.1) | 6 (6.3) |
| Back pain | 4 (4.1) | 1 (1.0) | 4 (4.1) | 6 (6.3) |
| Pain in extremity | 5 (5.2) | 4 (4.2) | 3 (3.1) | 5 (5.3) |
| Anemia | 6 (6.2) | 1 (1.0) | 3 (3.1) | 5 (5.3) |
| SAEs occurring in at least 1 patient during the extension period | ||||
| Pyrexia | 0 | 2 (2.1) a | 3 (3.1) | 0 |
| Influenza | 1 (1.0) | 1 (1.0) | 0 | 1 (1.1) |
| Pharyngitis | 0 | 1 (1.0) | 0 | 0 |
| Pneumonia | 0 | 1 (1.0) | 0 | 0 |
| Colitis | 1 (1.0) | 1 (1.0) | 0 | 0 |
| Tibia fracture | 0 | 1 (1.0) | 0 | 0 |
| Foot deformity | 0 | 1 (1.0) | 0 | 0 |
| Hemolysis | 0 | 0 | 2 (2.0) | 1 (1.1) |
| Anemia | 0 | 0 | 0 | 1 (1.1) |
| Liver disorder | 0 | 0 | 0 | 1 (1.1) |
| Gastroenteritis | 0 | 0 | 0 | 1 (1.1) |
AE, adverse event; MAVE, major adverse vascular event; SAE, serious AE; TEAE, treatment‐emergent AE; URTI = upper respiratory tract infection.
One event was considered possibly related to treatment.
The incidence of individual SAEs generally remained stable between the primary evaluation and extension periods (Table 2 ). During the extension period, the only SAEs to occur in more than one patient overall were pyrexia (ravulizumab‐ravulizumab, n = 2; eculizumab‐ravulizumab, n = 0) and influenza (ravulizumab‐ravulizumab, n = 1; eculizumab‐ravulizumab, n = 1). No patients died during the study. All but one SAE (possibly related pyrexia starting on day 195 and resolved by day 199 following treatment) in the ravulizumab‐ravulizumab arm were considered unrelated or unlikely to be related to study treatment. No SAE led to study drug interruption or discontinuation. No new treatment‐emergent antidrug antibody–positive response was reported through the 52‐week cutoff date.
4. DISCUSSION
In adults with PNH who are clinically stable on treatment with eculizumab, switching to ravulizumab showed durable efficacy through 52 weeks of treatment and continued to be well tolerated. Efficacy outcomes achieved at the end of the primary evaluation period—including percentage change in LDH levels, rates of breakthrough hemolysis, FACIT‐Fatigue and EORTC QLQ‐C30 scores, transfusion avoidance, and hemoglobin stabilization—were maintained through the extension period. Importantly, patients who had Serum free C5 levels ≥ 0.5 μg/mL while receiving eculizumab showed complete terminal complement inhibition (Serum free C5 levels < 0.5 μg/mL) after switching to ravulizumab. Ravulizumab continued to be well tolerated through 52 weeks, with no new safety concerns.
Treatment with eculizumab is associated with decreased intravascular hemolysis, as determined by rapid and sustained reductions in LDH levels. 6 , 8 , 9 In the current study, patients were required to be clinically stable on eculizumab for ≥ 6 months and have LDH levels ≤ 1.5 × ULN at screening; accordingly, mean LDH levels at baseline were within the normal range (120–246 U/L). 14 During the primary evaluation period, ravulizumab showed non‐inferiority to eculizumab with respect to LDH normalization, and LDH levels generally remained within the normal range (1 × ULN) for both treatment arms during the extension period. These results align with those of the phase 3 study of previously untreated patients with PNH in which patients randomly assigned to ravulizumab or eculizumab showed rapid decreases from baseline in LDH levels that were maintained within the normal range through the 26‐week treatment period. 15
Despite treatment with eculizumab, a subset of patients still experience breakthrough hemolysis, which may require dosing intervals to be shortened to < 14 days or individual dosages to be increased. 10 , 11 , 12 Low levels of hemolysis while receiving eculizumab treatment are believed to be associated with suboptimal Serum free C5 inhibition and residual activation of the terminal pathway, with densely packed C3b molecules competing with eculizumab for C5 priming. 16 , 17 In this trial, rates of breakthrough hemolysis were low overall (<5% during the extension period) and were generally similar between the primary evaluation and extension periods. Furthermore, of the five patients treated with eculizumab who experienced breakthrough hemolysis in the primary evaluation period, four did not experience breakthrough hemolysis during the extension period. Breakthrough hemolysis in eculizumab‐treated patients frequently occurs in the setting of bacterial or viral infections, 13 , 17 , 18 and, during the extension period of this study, two of the three events of breakthrough hemolysis in the ravulizumab‐ravulizumab arm and the one event in the eculizumab‐ravulizumab arm were associated with infection. A recent article by Brodsky et al 19 analyzing breakthrough hemolysis events in the primary evaluation period of the phase 3 randomized studies of eculizumab versus ravulizumab in adults with PNH showed that none of the breakthrough hemolysis events experienced by patients treated with ravulizumab were related to suboptimal free C5 inhibition, whereas 11 of 22 breakthrough hemolysis events in patients treated with eculizumab were related to suboptimal free C5 inhibition. Importantly, in the current extension study, no patient treated with ravulizumab experienced breakthrough hemolysis associated with suboptimal free C5 inhibition. Mean Serum free C5 levels were maintained at < 0.5 μg/mL during the extension period, inclusive of patients who experienced suboptimal Serum free C5 inhibition while receiving eculizumab during the primary evaluation period.
The number of patients avoiding transfusion was as expected for patients who entered the study clinically stable on eculizumab. At baseline, 13% of patients randomly assigned to ravulizumab and 12% of patients randomly assigned to eculizumab had received at least one transfusion within 1 year preceding the first dose of study drug. 14 The proportion of patients avoiding transfusions in the extension period remained stable. During the primary evaluation period, 87.6% and 82.7% of patients in the ravulizumab‐ravulizumab and eculizumab‐ravulizumab arms, respectively, avoided transfusion. In the extension period, 86.5% (ravulizumab‐ravulizumab arm) and 83.2% (eculizumab‐ravulizumab arm) avoided transfusion.
Patients with PNH report severe fatigue, 20 and treatment with eculizumab is associated with significant improvements in the FACIT‐Fatigue scale and EORTC QLQ‐C30 subscale scores. 6 , 8 Because patients were required to be clinically stable on eculizumab before study entry, FACIT‐Fatigue scores were relatively high at baseline (ravulizumab‐ravulizumab, 43; eculizumab‐ravulizumab, 41; range, 0–52; high scores indicate less fatigue). FACIT‐Fatigue scores reported at the end of the primary evaluation period were maintained through the extension period, suggesting that switching to ravulizumab did not affect levels of patient‐reported fatigue. Similarly, EORTC QLQ‐C30 scores also confirmed relatively high QoL at the start of the primary evaluation period, with similar scores reported at the end of the primary evaluation and extension periods.
Ravulizumab was well tolerated during the extension period, and comparable percentages of patients experienced TEAEs and SAEs between treatment arms. The safety profile of ravulizumab during the extension period was consistent with previous reports of eculizumab 8 , 10 and of ravulizumab 15 in previously untreated patients with PNH. The number of patients who discontinued the study during the extension period was low (one patient per treatment arm), and neither patient discontinued because of an AE. With the exception of fatigue, TEAE rates in the extension period were lower than those from the primary evaluation period.
This was the largest phase 3 study of patients with PNH who were previously receiving stable therapy with eculizumab. Analysis of the safety and efficacy of switching from eculizumab to ravulizumab is clinically meaningful, as eculizumab is the current standard of care for patients with PNH, and in this study, patients had been receiving treatment with eculizumab for a mean of 5.8 years. 14 Additional strengths include the range of clinically relevant and patient‐reported outcomes assessed. There were no pregnancies before data cutoff; therefore, no conclusions can be made regarding maternal or fetal safety when switching from eculizumab to ravulizumab. Further, efficacy and safety of ravulizumab beyond 52 weeks are currently unknown.
In conclusion, ravulizumab q8w was well tolerated, with durable efficacy in adults with PNH who were clinically stable on therapy with eculizumab. Maintenance of LDH levels, FACIT‐Fatigue scores, transfusion avoidance, and stabilized hemoglobin, together with relatively low levels of breakthrough hemolysis, were seen through 52 weeks of ravulizumab treatment. Importantly, all patients treated with ravulizumab achieved complete Serum free C5 inhibition. Results suggest that patients clinically stable on eculizumab can switch safely to weight‐based dosing of ravulizumab.
DISCLOSURES
Austin G. Kulasekararaj: Honoraria from Alexion Pharmaceuticals, Inc., Amgen, Celgene, Novartis, and Ra Pharma; Board of Directors or advisory board member for Alexion Pharmaceuticals, Inc., Amgen, Celgene, Novartis, and Ra Pharma; and consulting fees from Achillion, Akari Therapeutics, Alexion Pharmaceuticals, Inc., Celgene, and Novartis. Anita Hill: At the time of study: honoraria and/or consulting fees from Akari Therapeutics, Alexion Pharmaceuticals, Inc., Apellis, Bioverativ, Novartis, Ra Pharma, Regeneron, and Roche; current employee of Alexion Pharmaceuticals, Inc. Saskia Langemeijer: No conflicts of interest. Richard Wells: Honoraria and research funding from Alexion Pharmaceuticals, Inc., Celgene, and Novartis and consulting fees from Alexion Pharmaceuticals, Inc. F. Ataúlfo González Fernández: Honoraria, consulting/speakers bureau fees, and research funding from Alexion Pharmaceuticals, Inc. Anna Gaya: Honoraria and consulting fees from Alexion Pharmaceuticals, Inc., Novartis, and Merck Sharpe & Dohme and Board of Directors or advisory board member for Alexion Pharmaceuticals, Inc. Emilio Ojeda Gutierrez: Honoraria and consulting fees from Alexion Pharmaceuticals, Inc. Caroline I. Piatek: Honoraria from and Board of Directors or advisory board member for Alexion Pharmaceuticals, Inc., Dova, and Rigel; research funding from Alexion Pharmaceuticals, Inc., and Incyte; and speakers bureau for Dova. Lindsay Mitchell: Honoraria from Alexion Pharmaceuticals, Inc., and Novartis. Kensuke Usuki: Honoraria and research funding from Alexion Pharmaceuticals, Inc., Kyowa‐Kirin, and Novartis and research funding from Apellis, Chugai Pharmaceutical, and Roche. Alberto Bosi: Travel expenses from Alexion Pharmaceuticals, Inc. Robert Brodsky: Research funding from Achillion and Board of Directors or advisory board member for and grant funding from Alexion Pharmaceuticals, Inc. Masayo Ogawa: Employee and stockholder of Alexion Pharmaceuticals, Inc. Ji Yu: Employee and stockholder of Alexion Pharmaceuticals, Inc. Stephan Ortiz: Employee and stockholder of Alexion Pharmaceuticals, Inc. Alexander Röth: Honoraria from Alexion Pharmaceuticals, Inc., Bioverativ, Novartis, and Roche; consulting fees from Alexion Pharmaceuticals, Inc., Apellis, Bioverativ, Novartis, Roche, and Sanofi; Board of Directors or advisory board member for Alexion Pharmaceuticals, Inc., Apellis, Novartis, Roche, and Sanofi; and research funding from Roche. Jong Wook Lee: Honoraria, consulting fees, and research support (to Seoul St. Mary's Hospital) from Alexion Pharmaceuticals, Inc.; Board of Directors or advisory board member for Alexion Pharmaceuticals, Inc.; and research funding from Achillion. Régis Peffault de Latour: Consultancies, honoraria, and research funding from Alexion Pharmaceuticals, Inc., Novartis, and Pfizer and research funding from Amgen.
Supporting information
Supplementary Material
ACKNOWLEDGMENTS
Portions of this work were presented at the 61st American Society of Hematology Annual Meeting, Orlando, FL, December 7–10, 2019. This study was supported by funding from Alexion Pharmaceuticals, Inc. The authors thank Arshad Mujeebuddin, MD, of Alexion Pharmaceuticals, Inc., for his support with defining the safety strategy for the study and for reviewing the manuscript. Medical writing and editorial support for development of this manuscript was provided by Krystina Neuman, PhD, at ICON plc (North Wales, PA, USA), and funded by Alexion Pharmaceuticals, Inc. Editorial support/critical review was provided by Gabriela Marcheva, PharmD, at Alexion Pharmaceuticals, Inc.
Kulasekararaj AG, Hill A, Langemeijer S, et al. One‐year outcomes from a phase 3 randomized trial of ravulizumab in adults with paroxysmal nocturnal hemoglobinuria who received prior eculizumab. Eur J Haematol.2021;106:389–397. 10.1111/ejh.13564
DATA AVAILABILITY STATEMENT
Alexion will consider requests for disclosure of clinical study participant‐level data provided that participant privacy is assured through methods such as data deidentification, pseudonymization, or anonymization (as required by applicable law), and if such disclosure was included in the relevant study informed consent form or similar documentation. Qualified academic investigators may request participant‐level clinical data and supporting documents (statistical analysis plan and protocol) pertaining to Alexion‐sponsored studies. Further details regarding data availability and instructions for requesting information are available in the Alexion Clinical Trials Disclosure and Transparency Policy at https://alexionclinicaltrials.com/Disclosure‐and‐Transparency‐Policy; the Data Request Form is available at https://alexion.com/contact‐alexion/medical‐information.
REFERENCES
- 1. Brodsky RA. Paroxysmal nocturnal hemoglobinuria. Blood. 2014;124:2804‐2811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Devalet B, Mullier F, Chatelain B, Dogne JM, Chatelain C. Pathophysiology, diagnosis, and treatment of paroxysmal nocturnal hemoglobinuria: a review. Eur J Haematol. 2015;95:190‐198. [DOI] [PubMed] [Google Scholar]
- 3. Schrezenmeier H, Muus P, Socie G, et al. Baseline characteristics and disease burden in patients in the International Paroxysmal Nocturnal Hemoglobinuria Registry. Haematologica. 2014;99:922‐929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Jang JH, Kim JS, Yoon SS, et al. Predictive factors of mortality in population of patients with paroxysmal nocturnal hemoglobinuria (PNH): results from a Korean PNH registry. J Korean Med Sci. 2016;31:214‐221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Rother RP, Rollins SA, Mojcik CF, Brodsky RA, Bell L. Discovery and development of the complement inhibitor eculizumab for the treatment of paroxysmal nocturnal hemoglobinuria. Nat Biotechnol. 2007;25:1256‐1264. [DOI] [PubMed] [Google Scholar]
- 6. Brodsky RA, Young NS, Antonioli E, et al. Multicenter phase 3 study of the complement inhibitor eculizumab for the treatment of patients with paroxysmal nocturnal hemoglobinuria. Blood. 2008;111:1840‐1847. [DOI] [PubMed] [Google Scholar]
- 7. Hillmen P, Hall C, Marsh JC, et al. Effect of eculizumab on hemolysis and transfusion requirements in patients with paroxysmal nocturnal hemoglobinuria. N Engl J Med. 2004;350:552‐559. [DOI] [PubMed] [Google Scholar]
- 8. Hillmen P, Young NS, Schubert J, et al. The complement inhibitor eculizumab in paroxysmal nocturnal hemoglobinuria. N Engl J Med. 2006;355:1233‐1243. [DOI] [PubMed] [Google Scholar]
- 9. Kelly RJ, Hill A, Arnold LM, et al. Long‐term treatment with eculizumab in paroxysmal nocturnal hemoglobinuria: sustained efficacy and improved survival. Blood. 2011;117:6786‐6792. [DOI] [PubMed] [Google Scholar]
- 10. Hillmen P, Muus P, Röth A, et al. Long‐term safety and efficacy of sustained eculizumab treatment in patients with paroxysmal nocturnal haemoglobinuria. Br J Haematol. 2013;162:62‐73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Nakayama H, Usuki K, Echizen H, Ogawa R, Orii T. Eculizumab dosing intervals longer than 17 days may be associated with greater risk of breakthrough hemolysis in patients with paroxysmal nocturnal hemoglobinuria. Biol Pharm Bull. 2016;39:285‐288. [DOI] [PubMed] [Google Scholar]
- 12. Peffault de Latour R, Fremeaux‐Bacchi V, Porcher R, et al. Assessing complement blockade in patients with paroxysmal nocturnal hemoglobinuria receiving eculizumab. Blood. 2015;125:775‐783. [DOI] [PubMed] [Google Scholar]
- 13. Sheridan D, Yu ZX, Zhang Y, et al. Design and preclinical characterization of ALXN1210: a novel anti‐C5 antibody with extended duration of action. PLoS One. 2018;13:e0195909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kulasekararaj AG, Hill A, Rottinghaus ST, et al. Ravulizumab (ALXN1210) vs eculizumab in C5‐inhibitor‐experienced adult patients with PNH: the 302 study. Blood. 2019;133:540‐549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lee JW, Sicre de Fontbrune F, Wong Lee Lee L, et al. Ravulizumab (ALXN1210) vs eculizumab in adult patients with PNH naive to complement inhibitors: the 301 study. Blood. 2019;133:530‐539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Harder MJ, Kuhn N, Schrezenmeier H, et al. Incomplete inhibition by eculizumab: mechanistic evidence for residual C5 activity during strong complement activation. Blood. 2017;129:970‐980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Harder MJ, Hochsmann B, Dopler A, et al. Different levels of incomplete terminal pathway inhibition by eculizumab and the clinical response of PNH patients. Front Immunol. 2019;10:1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Brodsky RA. Eculizumab: another breakthrough. Blood. 2017;129:922‐923. [DOI] [PubMed] [Google Scholar]
- 19. Brodsky RA, Peffault de Latour R, Rottinghaus ST, et al. Characterization of breakthrough hemolysis events observed in the phase 3 randomized studies of ravulizumab versus eculizumab in adults with paroxysmal nocturnal hemoglobinuria. Haematologica. 2020. 10.3324/haematol.2019.236877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Escalante CP, Chisolm S, Song J, et al. Fatigue, symptom burden, and health‐related quality of life in patients with myelodysplastic syndrome, aplastic anemia, and paroxysmal nocturnal hemoglobinuria. Cancer Med. 2019;8:543‐553. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material
Data Availability Statement
Alexion will consider requests for disclosure of clinical study participant‐level data provided that participant privacy is assured through methods such as data deidentification, pseudonymization, or anonymization (as required by applicable law), and if such disclosure was included in the relevant study informed consent form or similar documentation. Qualified academic investigators may request participant‐level clinical data and supporting documents (statistical analysis plan and protocol) pertaining to Alexion‐sponsored studies. Further details regarding data availability and instructions for requesting information are available in the Alexion Clinical Trials Disclosure and Transparency Policy at https://alexionclinicaltrials.com/Disclosure‐and‐Transparency‐Policy; the Data Request Form is available at https://alexion.com/contact‐alexion/medical‐information.