

SUMMARY
Drugs selectively targeting CB2 hold promise for treating neurodegenerative disorders, inflammation and pain while avoiding psychotropic side effects mediated by CB1. The mechanisms underlying CB2 activation and signaling are poorly understood but critical for drug design. Here, we report the cryo-EM structure of human CB2-Gi signaling complex bound to the agonist WIN 55,212–2. The 3-D structure reveals the binding mode of WIN 55,212–2 and structural determinants for distinguishing CB2 agonists from antagonists, which are supported by a pair of rationally designed agonist and antagonist. Further structural analyses with computational docking results uncover the differences between CB2 and CB1 in receptor activation, ligand recognition and Gi-coupling. These findings are expected to facilitate rational structure-based drug discovery targeting the cannabinoid system.
In Brief
The 3-D structure of the agonist-bound CB2-Gi signaling complex provides insight into the key residues involved with ligand recognition and the distinction of agonists and antagonists critical to facilitate rational drug design targeting the cannabinoid system.
Graphical Abstract
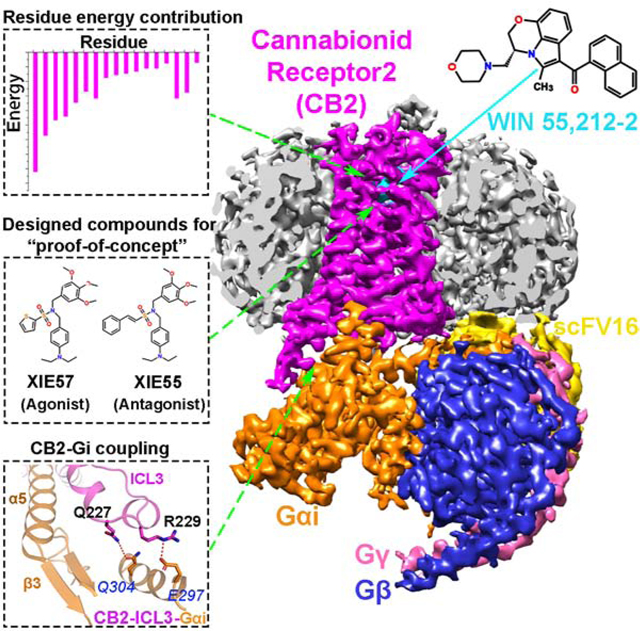
INTRODUCTION
The endocannabinoid system (ECS) includes two major G protein-coupled receptors (GPCRs), cannabinoid receptor subtypes 1 and 2 (CB1 and CB2), which mediate the biophysiological activities of phytocannabinoid Δ9-tetrahydrocannabinol (THC) and several endogenous cannabinoids. ECS is a complex and homeostatic system that is involved in a broad spectrum of physiological and pathological processes such as appetite regulation (Kirkham, 2005), peripheral energy metabolism (Seeley and Woods, 2003), pain and inflammation (Calignano et al., 1998, Walter and Stella, 2004), cardiovascular regulation (Fulmer and Thewke, 2018), musculoskeletal disorders (Idris and Ralston, 2012), and cancer (Sledzinski et al., 2018). Efforts have been made to develop selective CB1 and CB2 ligands that can modulate these biological functions and treat associated diseases. While some CB2-selective agonists have been developed showing certain efficacy in animal models or in vitro assays (Leleu-Chavain et al., 2019, Nettekoven et al., 2016, Scott et al., 2019, Mukhopadhyay et al., 2016, Mugnaini et al., 2019), there are no such drugs on the market for clinical use besides the phytocannabinoids and their analogs (Maroon and Bost, 2018, Yang et al., 2012b).
As a therapeutic target, CB2 has a notable advantage over CB1 in its pattern of expression. CB1 is primarily expressed in the central nervous system (CNS) and is the primary receptor responsible for the psychotropic effects of THC and the deleterious psychiatric side effects of CB1-targeting drugs. The CB1 inverse agonists Rimonabant and Taranabant (MK-0364) were developed as anti-obesity drugs, yet both produce crippling CNS side effects of anxiety, depression and suicidal ideation (Aronne et al., 2010, Proietto et al., 2010, Moreira and Crippa, 2009), and were consequently either withdrawn from the market or dropped in clinical trials. In contrast, CB2 is predominantly expressed in peripheral tissues of the immune system, such as the spleen and thymus, where it modulates immune suppression, apoptosis and cell migration (Yang et al., 2012a, Galiegue et al., 1995). Modest levels of CB2 expression have also been observed in several areas of the brain, such as the ventral tegmental area dopamine neurons and the hippocampal CA3 neurons (Onaivi et al., 2006), which regulate cravings of drug addiction and memory processes, respectively (Chen et al., 2017). Thus, it is believed that targeting CB2 receptor and CB2 structure-based design offer promising avenues of new drug discovery for treatments of a number of disorders while avoiding the severe psychiatric side effects associated with CB1 (Seely et al., 2011).
Activated CB2 mainly couples to the Gi/o family and induces Gi/o-mediated signaling pathways, although Gq/11 coupling has also been reported (Ibsen et al., 2017). CB2 antagonists/inverse agonists have been reported to promote osteoclast apoptosis and to prevent bone loss (Idris et al., 2005). On the other hand, a large body of evidence has indicated a broad therapeutic application of CB2 agonists in neurodegenerative disorders (Aso and Ferrer, 2016, Gómez-Gálvez et al., 2016), drug abuse or addiction (Chen et al., 2017), cardiovascular diseases (Fulmer and Thewke, 2018), neuroinflammation and neuropathic pain (Guimaraes et al., 2018, Turcotte et al., 2016, Guindon and Hohmann, 2008). However, the lack of a well-defined CB2 ligand activation and signaling mechanism has hindered development of synthetic CB2 agonists.
WIN 55,212–2 ((R)-(+)-[2,3-Dihydro-5-methyl-3-(4-morpholinylmethyl)pyrrolo[1,2,3-de]-1,4-benzoxazin-6-yl]-1-naphthalenylmethanone) is a representative agonist of cannabinoid receptors CB1 and CB2 (Ki=62.3nM and 3.3nM, respectively) and it has been used extensively to investigate cannabinoid receptor function. In animal models, WIN 55,212–2 has demonstrated efficacy in treating several disease states, including pain (Mohammadi Vosough et al., 2019), seizure (Citraro et al., 2016), and cancer (Roberto et al., 2019). Unlike most CB1/CB2 agonists, WIN 55,212–2 activates G protein signaling pathways without eliciting CB2 internalization, a property that putatively avoids drug tolerance development. Here, we report a 3.2 Å cryo-EM structure of human CB2 bound to the potent agonist WIN 55,212–2 in complex with Gi. Analyses of the ligand binding pocket, the CB2-Gi signaling complex, and conformational dynamics of activated CB2 are conducted by comparisons and contrasts with previously reported structures of inverse agonist-bound CB2 and inverse agonist-/agonist-bound CB1. We developed and applied an innovative residual energy calculation algorithm to aid in the design of a pair of selective CB2 agonist and inverse agonist in order to validate our putative mechanism of receptor activation involving the structural arrangement of critical residues. Our results provide insightful information to facilitate structure-based design and discovery of high-affinity/high-selectivity CB2 ligands that can be developed as CB2-targeting therapeutic agents.
RESULTS
Cryo-EM Structure of the CB2-WIN 55,212–2-Gi-scFv16 Complex
To obtain a stable complex of CB2 bound to WIN 55,212–2 and Gi, we co-expressed human CB2 with heterotrimeric Gi protein in insect cells (Figure S1). The receptor-Gi complexes were assembled on the membrane by incubation with WIN 55,212–2 and apyrase. In addition, the antibody fragment scFv16 was added to stabilize the nucleotide-free Gi complex by binding to the interface between Gαi and Gβ (Maeda et al., 2018). The CB2-WIN 55,212–2-Gi-scFv16 complex was then purified through sequential steps of affinity chromatography to homogeneity, yielding a relatively thermostable complex suitable for single particle cryo-EM analysis (Figure S1).
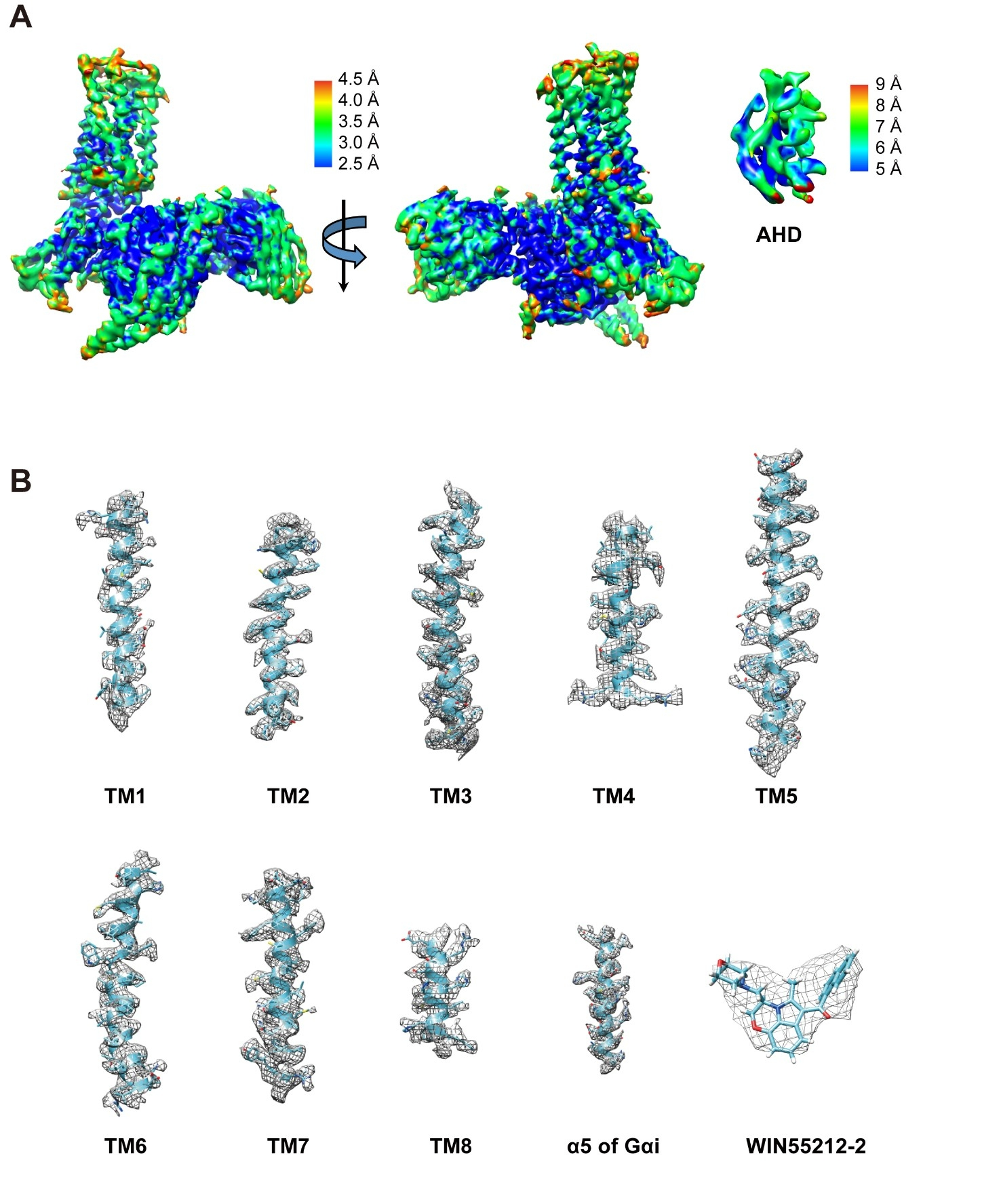
The structure of the CB2-WIN 55,212–2-Gi-scFv16 complex (Figure 1) was determined from 772k particles to a nominal resolution of 3.2 Å (Figure S2), which allowed an unambiguous assignment of the TM domain of CB2, Gi protein, as well as the antibody fragment in the EM density maps (Figure S3). The α-helical domain (AHD) of Gαi, which is poorly resolved in most cryo-EM GPCR-G protein complex structures, is resolved in our CB2-Gi complex structure with a relatively low resolution compared to that of the core structure of this complex. The overall structures were built by rigid body fit of an online homology building model (Kelley et al., 2015). Apart from the AHD of the Gi protein, the majority of the amino acid side chains were well resolved in the final models against the EM density map with excellent geometry (TABLE S1).
Figure 1. Cryo-EM Structure of CB2-Gi Coupling in Complex with WIN 55,212–2.

(A) Cut-through view of cryo-EM density map of CB2-WIN 55,212–2-Gi-scFv16 complex. The unsharpened cryo-EM density map at 0.015 contour level shown as light gray surface indicates a micelle. The colored complex cryo-EM density map is shown at 0.022 contour level.
(B) Cartoon representation of the CB2-Gi in complex with antibody scFv16 is shown with lipids in gray stick representation. Magenta, CB2; cyan, WIN-55,212–2; orange, Gαi; blue, Gβ; pink, Gγ; gold, scFv16.
WIN 55,212–2 Interactions in CB2 Ligand-Binding Pocket
The EM density map shows the binding mode of the CB2 agonist WIN 55,212–2 in the orthosteric binding site in the TM domain of CB2. The orientation of the agonist was determined with both the electron density and the chemical and geometric constraints of the binding pocket of CB2 (Figure 2). To further validate the binding pose of WIN 55,212–2 in the ligand binding pocket of CB2, we conducted molecular docking (Figure S4A) using three different software/algorithms: Schrödinger-Glide, Auto Dock, and SYBYL-Surflex Dock. The pose of WIN 55,212–2 depicted in our complex structure (Figure S4A) was scored as the top one or two from all three algorithms, with the lowest binding energies. We also performed molecular dynamics simulation and the results showed that the pose of WIN 55,212–2 in the orthosteric ligand binding pocket of CB2 is stable (Figure S4B, S4C and S4D). The binding interface between WIN 55,212–2 and CB2 is also supported by previously reported mutagenesis-based and structural studies (Li et al., 2019, Zhang et al., 2011, McAllister et al., 2004, Singh et al., 2002).
Figure 2. WIN 55,212–2 Binding in CB2 Orthosteric Ligand-Binding Site.

(A) The overview of CB2 in complex with WIN 55,212–2. Cyan sphere model: chemical structure of WIN 55,212–2. Magenta cartoon: cryo-EM structure of CB2.
(B) Detailed interactions of WIN 55,212–2 (cyan) within CB2 (magenta). The residues involved in the binding pocket of CB2 are mainly hydrophobic (magenta sticks) and are derived from TM2-TM3, TM5-TM7, and ECL2.
As shown in Figure 2, the binding pocket of WIN 55,212–2, buried in the TM region, is formed by residues from TM2-TM3 and TM5-TM7 and capped by the extracellular loop 2 (ECL2). The naphthalene moiety of WIN 55,212–2 extends between TM2 and TM3 and is predicted to form strong π-π interactions with F912.61 and F942.64 and hydrophobic interactions with F872.57, H952.65, P184ECL2, and F2817.35. These results are consistent with the findings from Li et al. (Li et al., 2019), who reported that F872.57, F912.61, F942.64, and H952.65 in TM2 are important for the recognition of CB2 antagonist AM10257. Furthermore, our cryo-EM data reveal that the core structure of WIN 55,212–2 (2,3-dihydro-[1,4]oxazino[2,3,4-hi]indole) points downward and engages in π-π interactions with F1173.36 and W2586.48. It also interacts with I1103.29, V1133.32, F183ECL2, V2616.51, and M2656.55 via hydrophobic interactions, which have been shown previously to play key roles in the ligand binding of CB2. The morpholine moiety of WIN 55,212–2, which adopts the chair conformation, approaches TM5 and ECL2 to form additional hydrophobic interactions with critical residues that have been reported to function in ligand binding (Feng et al., 2014), including F183ECL2, I186ECL2, and W1945.43.
CB2 Activation Mechanism
WIN 55,212–2 shares a certain similarity with the antagonist AM10257 in binding to the receptor, but noteworthy differences also exist. From the structural alignment shown in Figure 3A, we may infer that AM10257 (antagonist) and WIN 55,212–2 (agonist) share a similar binding pocket and that most of the interactions with the receptor are conserved (Figure 3A). In addition, the volume of the WIN 55,212–2 binding site within active CB2 (~415 Å3) is similar to that of the AM10257 binding site (antagonist, ~447 Å3). One significant difference is that AM10257 (antagonist, orange sticks) inserts deeper (2.8 Å) into the binding pocket compared to WIN 55,212–2 (agonist, cyan sticks), which results in different conformations of the toggle switch residue W2586.48. W2586.48 is a highly conserved residue in class A GPCRs and has been reported to have a crucial role in GPCR activation (Lin and Sakmar, 1996). In comparison to the inactive CB2, our data showed that W2586.48 undergoes a 64° clockwise rotation and F1173.36 endures a 10° counterclockwise rotation in the acti ve CB2 (Figure 3A and 3B). We also note that the distance between W2586.48 and WIN 55,212–2 is about 5.0 Å, greater than that observed for AM10257 (~3.8 Å). Therefore, the steric effects of CB2 ligands on W2586.48 appear to play critical roles in determining ligand efficacy.
Figure 3. CB2 Activation by WIN 55,212–2.

(A) Superposition of the WIN 55,212–2 (cyan)-activated CB2 (magenta) complex with antagonist (AM10257, orange)-bound CB2 receptor (green) (PDB: 5ZTY, resolution: 2.8 Å).
(B) WIN 55,212–2 bound at the CB2 orthosteric pocket makes direct contacts with residues F1173.36 and W2586.48. The subtle rotation of F1173.36 to interact with the 2,3-dihydro-[1,4]oxazino[2,3,4-hi]indole moiety of WIN 55,212–2 allows W2586.48 to undergo a large rotation, with a consequent outward movement of the cytoplasmic end of TM6 that serves to create a cavity for G protein binding. Green cartoon: the inactive CB2 crystal structure (PDB: 5ZTY, resolution: 2.8 Å). Magenta cartoon: the active CB2 cryo-EM structure. There is relatively little rearrangement in residues V1213.40, L2015.50, and L2546.44, different from the corresponding residue Pro5.50 of β2AR and μOR, which is involved in packing interactions with Ile3.40 and Phe6.44 during the activation of these GPCRs.
(C) The energy contribution of key residues involved in the binding pockets of inactive and active CB2. Green bars: calculated energy contributions of key residues based on the inactive CB2 crystal structure (PDB: 5ZTY). Magenta bars: calculated energy contributions of key residues using the active CB2 cryo-EM structure.
To further explore the role of critical residues on CB2 activation, we have calculated the individual residue energy contribution to determine how much certain residues contribute to the binding of WIN 55,212–2. Briefly, the total binding energy of WIN 55,212–2 at CB2 is the sum of intra-ligand free energy (the energy of WIN 55,212–2) and inter-ligand free energy (the energy between WIN 55,212–2 and residues in CB2), with the latter component further divided into energy contributions from each residue. We quantitatively calculated and compared the residual energy contributions between the inactive and active CB2. Although most of the residues contribute equally to the recognition of both antagonist-AM10257 (Figure 3C, green bars) and agonist-WIN 55,212–2 (Figure 3C, magenta bars), three residues, W1945.43, F1173.36, and W2586.48, were found to potentially play important roles in distinguishing agonist from antagonist. This occurs in a manner not requiring direct interaction since they are too far from the WIN 55,212–2 molecule to contribute significantly to its recognition as an agonist (Figure 3C). Several mutagenesis studies from our group and others (Zhang et al., 2011, McAllister et al., 2004, Singh et al., 2002) also provided evidence of the importance of these three residues in terms of WIN 55,212–2 binding and CB2 activation, further validating our residual energy contribution algorithm. This derivation aids in predicting how to design a functional agonist/antagonist pair based on 3-D structure.
To further investigate and validate the structural findings, we rationally designed and synthesized two structurally related CB2-selective compounds and also measured the binding affinity Ki to CB1 and CB2, XIE55 (CB2 Ki=138 nM, CB1/CB2>1000) and XIE57 (CB2 Ki=639 nM, CB1/CB2>1000). They share the same pharmacophore but with different chemical moieties: XIE55 has a phenylethylene group whereas XIE57 has a thiophene ring (Figure 4A and Figure S5). Congruently, cAMP functional assays determined that XIE55 behaves as an inverse agonist and XIE57 acts as a partial agonist of CB2. Molecular docking also showed that these two ligands adopt similar binding poses to interact with CB2 (Figure 4C). The most different feature is that agonist XIE57 does not extend sufficiently deep to constrain the conformation of W2586.48 with a distance of 5.5 Å, while inverse agonist XIE55 approaches closer to W2586.48 to form potentially strong π-π interactions (3.0 Å), which is consistent with our structural analysis of the action of WIN 55,212–2 and AM10257, addressed above. This pair of small molecules (XIE55 and XIE57) serves as a “proof-of-concept” of our method and investigation that can be applied by other researchers in the field for design and discovery of CB2 functional ligands.
Figure 4. Pharmacology and Molecular Docking of the Designed CB2 Agonist and Antagonist.

(A) Chemical structures of the rationally designed antagonist XIE55 and agonist XIE57.
(B) XIE57 acts as a CB2 agonist while XIE55 behaves as a CB2 antagonist, as determined by cAMP assay. The dose response curves for CP55940, a known agonist, and SR144528, a known inverse agonist, are also shown. CP55940 EC50: 45.5 ± 24 nM, Ki: 1–2 nM; SR144528 EC50: 10.5 ± 4.5 nM, Ki: 0.6 nM; XIE57 EC50: 6.9 ± 2.9 μM, Ki: 639 nM; XIE55 EC50: 1.5 ± 0.4 μM, Ki: 138 nM. Data are presented as mean ± SEM of at least 3 experiments performed in duplicate.
(C) The docking of XIE55 and XIE57 in WIN 55,212–2-bound CB2 structure (WIN 55,212–2 is removed).
Differences between CB2 and CB1
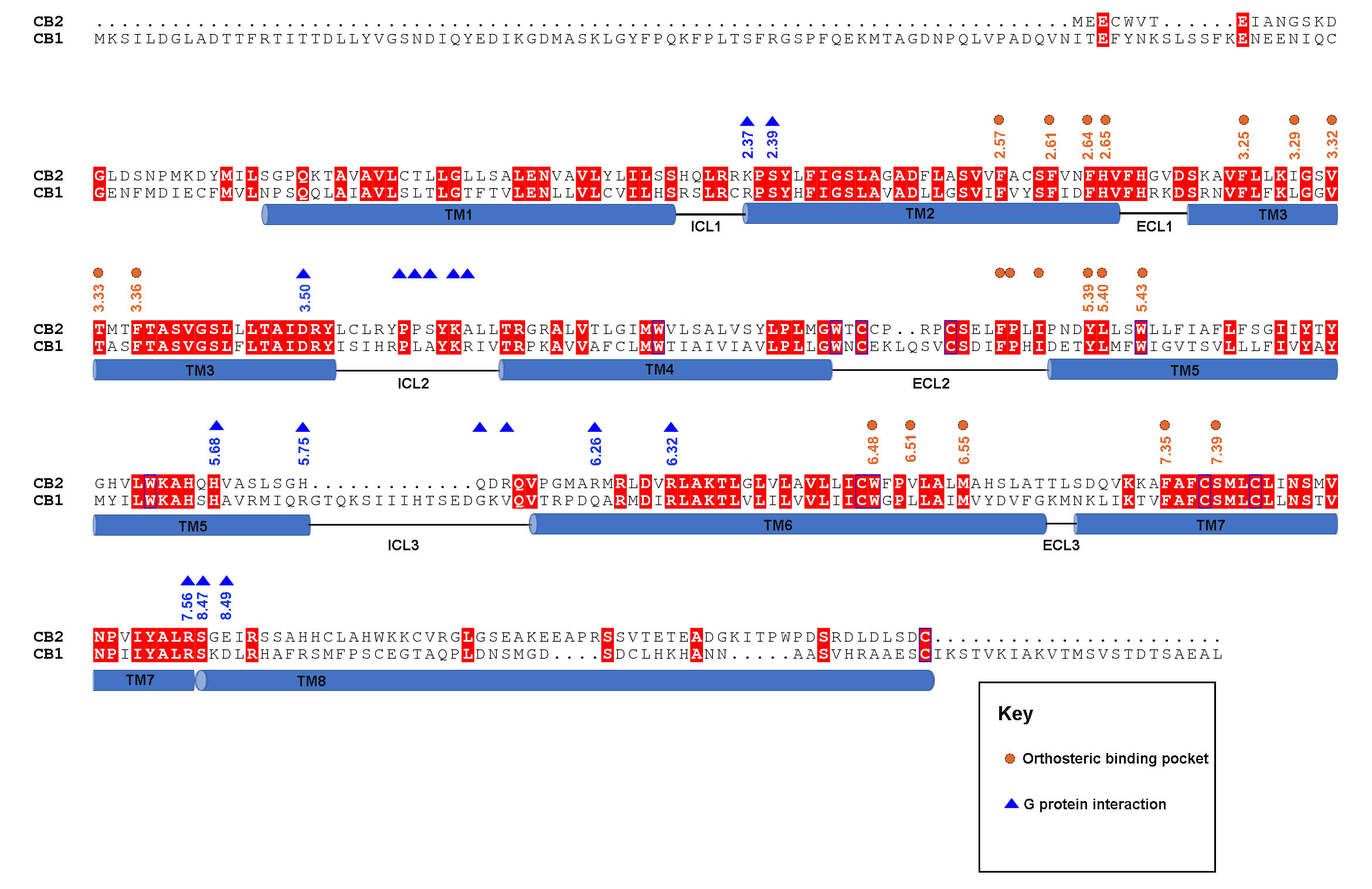
In order to explore the differences in receptor activation between CB2 and CB1, we aligned the agonist-bound CB2-Gi and agonist-bound CB1-Gi complexes. Although the two ligands, WIN 55,212–2 and MDMB-Fubinaca (FUB), overlap very well, the critical “toggle switch” residues W6.48 and F3.36 show differences. Specifically, W3566.48 in CB1 is positioned about 2.3 Å up towards the extracellular surface, and F2003.36 in CB1 is rotated 68° compared to the analogous res idues in CB2 (Figure 5A). As a result, the distance between W3566.48 and F2003.36 in CB1 (4.8 Å) is longer than that in CB2 (3.4 Å). Similarly, the different arrangement of the “toggle switch” causes a 74° counterclockwise rotation of F202 5.51 in TM5 of CB2 in comparison to the corresponding residue L2875.51 in CB1. Other residues involved in the orthosteric binding in CB receptors, especially those from TM2 and ECL2, show some conformational differences as well (Figure 5A). Interestingly, the conformation and position of ECLs remain largely unchanged during receptor activation, which can likely be attributed to the C174-C179 disulfide bond and a rigid motif of “PXP” (P176 and P178). We also observed modest upward shifts of residues on the cytoplasmic ends of TM5 and TM6 of CB2 relative to those of CB1 (Figure 5B), which leads to a shift of Y7.53 in the NPXXY motif away from Y5.58 and L6.41 in CB2 compared to that in CB1. Such structural differences in the ligand-binding pockets are associated with observed differences in the conformation of TM5 and TM6 and the Gi coupling interface between CB1 and CB2, indicating the activation mechanisms of CB1 and CB2 are not the same. All these structural alterations may be also associated with the distinct interactions of G-proteins between CB1 and CB2 (discussed below).
Figure 5. Structure Difference Between Agonist-bound CB2 and Agonist-bound CB1.

(A) A comparison of binding pockets between CB1-Gi and CB2-Gi complexes. Marine cartoon: the CB1 structure (PDB: 6N4B, resolution: 3.0 Å). Magenta cartoon: the CB2 structure. Green sticks: MDMB-Fubinaca. Cyan sticks: WIN 55,212–2.
(B) Residue arrangements in the cytoplasmic end of TM5/TM6/TM7 between CB1-Gi and CB2-Gi structures. Marine cartoon: the CB1 structure (PDB: 6N4B, resolution: 3.0 Å). Purple cartoon: the CB2 structure. Arrows indicate the movements of residues from CB1 to CB2.
To compare the selectivity between CB2 and CB1, we conducted molecular docking studies and calculated the residual energy contribution for GW-405,833 (L-768,242) on active CB2 and inactive CB1 (PDB: 5TGZ) (Hua et al., 2016). GW-405,833, a dual functional compound (Dhopeshwarkar et al., 2017), acts as a highly selective partial agonist on CB2 (Ki= 3.92 ± 1.58 nM) but as an antagonist on CB1 (Ki= 4772 ± 1676 nM). The docking poses of GW-405,833 on these two receptors are quite different, as shown in Figure 6A. Moreover, the binding energy of GW-405,833 on CB2 (−10.83 kcal/mol) is greater than that on CB1 (−6.66 kcal/mol), which is consistent with the reported binding affinities for CB2 and CB1. Sequentially, the residual energy contributions for GW-405,833 binding on both CB2 and CB1 were calculated and compared (Figure 6B). Several residues on inactive CB1, including P102 (N-terminus), M103 (N-terminus), I105 (N-terminus), F1702.57, and W3566.48, are important for GW-405,833 binding. In particular, W3566.48 on CB1 contributes greatly (−0.94 kcal/mol) to the recognition of GW-405,833 as an antagonist. However, the energy contribution of the corresponding residue W2586.48 on CB2 for partial agonist GW-405,833 is −0.15 kcal/mol (Figure 6B), which is similar to that for WIN 55,212–2. Importantly, L182ECL2, F183ECL2, P184ECL2, and I186ECL2 from ECL2 show greater contributions to the binding of GW-405,833 on CB2 than the corresponding residues on CB1 (Figure 6B), supporting findings from previous studies (Raitio et al., 2005).
Figure 6. Comparison of Docking Results of CB2 Selective Partial Agonist GW-405,833 (L-768,242) on Active CB2 and Inactive CB1.

(A) Comparison of docking poses and detailed interactions of CB2 selective partial agonist GW-405,833 on active CB2 (magenta) and inactive CB1 (gray, PDB: 5TGZ). It is reported that GW-405,833 acts as CB2 agonist (Ki=3.92 ± 1.58 nM) and CB1 antagonist (Ki=4772 ± 1676 nM).
(B) Comparison of residual energy contribution on GW-405,833 between active CB2 (magenta bars) and inactive CB1 conformations (gray bars).
CB2-Gi Coupling and G Protein Selectivity
The overall cryo-EM structure of the CB2-Gi complex reveals a similar interaction mode between the receptor and G protein compared to other Gi/Gs-coupled receptors (Figure S6). The C-terminal α5 helix of the Gαi subunit inserts into the cavity at the cytoplasmic site of CB2 to form the major interaction interface with residues from TM2, ICL2, TM3, TM5, and TM6 of the receptor. When aligned with the reported μ-opioid receptor (μOR)-Gi (PDB: 6DDE) and β2-adrenergic receptor (β2AR)-Gs (PDB:3SN6) complexes (Rasmussen et al., 2011), the α5 helix in CB2-Gi is rotated by 18° and 14°, respectively, along the axis of the membrane (Figure S6A and 6B). However, several striking differences of Gi protein coupling between CB2 and CB1 can be observed. For example, the N-terminus of the α5 helix of Gαi in CB2-Gi is closer to TM5 (Figure 7A) and inserts deeper into the receptor binding cavity than those in Rhodopsin-Gi (PDB: 6CMO)(Kang et al., 2018) and CB1-Gi (PDB: 6N4B)(Li et al., 2019) complexes, as pointed out above. As a result, CB2 forms a more extensive hydrogen-bonding network with the α5 helix of Gαi than that found in the structure of CB1-Gi (Figures 7B and 7C). Specifically, compared to the five hydrophilic interactions between CB1 and Gαi (Li et al., 2019), eight pairs of hydrogen bonds can be observed between CB2 and α5 helix (Figure 7B). K672.37 and S692.39 in the intracellular side of TM2 are observed to form hydrogen bonds with D350 and C351 in α5 helix, respectively. K142ICL2 in ICL2 interacts with both N347 and D350 via strong hydrogen bond interactions. R1313.50, a residue of the “ionic lock” in TM3 also forms a hydrogen bond with C351. In addition, H2195.68 and H2265.75 in TM5 approach D341 and D337 in α5 to form two hydrogen bonds. Finally, we also observed a hydrogen bond between R2426.32 and F354. In contrast to CB1, ICL3 in CB2 is well-structured and directly interacts with Gi. As shown in Figure 7D, an additional α-helix is formed by residues from ICL3 and TM6 including R229ICL3, Q230ICL3, V2316.21, and P2326.22. Two polar residues from ICL3, Q227ICL3, and R229ICL3, form additional hydrogen bonds with Q304 and E297 of Gαi, which are absent in the CB1-Gi complex (Figure S7).
Figure 7. Comparison of the Interface between CB2 and Gα as well as CB1 and Gα.

(A) Overlay of CB2 or CB1 and Gi-α5 structures. Marine cartoon: the CB1 structure (PDB: 6N4B, resolution: 3.0 Å). Purple cartoon: the CB2 structure. Orange: Gi-α5/Gi-αN protein of CB2. Green: Gi-α5/Gi-αN protein of CB1.
(B) The hydrogen bond network in the complex of CB2-Gα. Magenta cartoon: the CB2 structure. Orange: Gα protein. Residues from α5 are highlighted in blue font. The residues belonging to CB2 are highlighted in black font.
(C) The hydrogen bond network in the complex of CB1-Gα. Marine cartoon: the CB1 structure (PDB: 6N4B, resolution: 3.0 Å). Green: Gα protein. Residues from α5 are highlighted in blue font. The residues belonging to CB1 are highlighted in black font.
(D) Detailed interactions between ICL3 of CB2 and Gα. Magenta cartoon: the CB2 structure. Orange: Gα protein. The hydrogen bonds are highlighted by red dash lines.
(E) Interactions between ICL2 of CB2 and Gα. Magenta cartoon: the CB2 structure. Orange: Gα protein. The hydrogen bond is highlighted by a red dash line. The hydrophobic interaction is represented by a blue dash line.
(F) Interactions between ICL2 of CB1 and Gα. Marine cartoon: the CB1 structure. Green: Gα protein. The hydrophobic interaction is represented by a blue dash line.
Although CB1 and μOR primarily signal through the Gi/o family, previous studies suggested that they can also couple to Gs (Kumar et al., 2019). However, CB2 does not couple to the Gs family. On the other hand, ICL2 point mutations P139F, P139M, or P139L allows CB2 to couple to Gs in a CRE-driven luciferase assay (Zheng et al., 2013). Moreover, a CB2 P139L mutant could activate ERK through both Gi- and Gs-mediated pathways (Zheng et al., 2013). Interestingly, when L222 of CB1 (corresponding to P139 of CB2) is mutated to proline, Gs coupling is lost but coupling to Gi is retained (Timossi et al., 2002). These studies, together with previous cross-linking studies (Mnpotra et al., 2014), suggest an important role of P139ICL2 in the G protein selectivity and coupling for CB2.
In our cryo-EM structure, P139ICL2 in CB2 interacts with L194 in the β2-β3 loop of Gi but is away from the hydrophobic pocket formed by L194, I343, T340, and F336 in Gi (Figure 7E). It has been suggested that for CB1, the corresponding residue of P139ICL2 in CB2, L222ICL2 (Figure 7F), may engage a similar hydrophobic pocket when coupled to Gs, which is important for Gs recognition (Kumar et al., 2019). However, the P139ICL2 in CB2 may not permit interactions with the hydrophobic pocket in Gs due to the rigid motif of “P138ICL2P139ICL2” motif, leading to the high selectivity of CB2 for Gi over Gs. Consistently, mutation of CB2 P139ICL2 to bulkier residues, such as Phe or Leu, might allow such coupling interactions with Gs to be regained.
DISCUSSION
Delineating the CB2 structural basis for ligand recognition and G protein recruitment will facilitate the rational design and development of drugs with high affinity and selectivity, as well as optimal therapeutic effects. Here, we report the cryo-EM structure of an agonist-bound CB2-Gi signaling complex, and the detailed interactions between the potent agonist WIN 55,212–2 and the receptor, the key residues determining ligand selectivity and efficacy, the differences of activation mechanisms between CB2 and CB1, and the unique molecular features of the CB2-Gi protein interaction.
Our 3-D cryo-EM structure of CB2 reveals that WIN 55,212–2 occupies the same orthosteric binding pocket as antagonist AM10257 in CB2 and stabilizes the CB2-Gi complex in its active conformation through interference with the “toggle switch” residue W2586.48, which is critical for distinguishing agonists from antagonists. ECL2 and residues from TM2, TM3, and TM6 play important roles in CB2 ligand recognition and ligand selectivity. This provides an advanced strategy for the rational design of CB2 selective agonists. The divergent conformation of W6.48 in CB1 versus CB2 is associated with a series of distinct residue rearrangements in the intracellular side of TM5, TM6 and TM7 of CB2, as addressed above, which distinguish the activation/ Gi signaling mechanism of CB2 from that of CB1. While several synthetic CB2-selective agonists, including thiophene-containing compounds have recently been reported (Scott et al., 2019, Leleu-Chavain et al., 2019, Nettekoven et al., 2016, Mugnaini et al., 2019, Mukhopadhyay et al., 2016), few have entered clinical trials and none have yet been approved by FDA. The results presented herein are likely to aid in the development of potent and selective CB2 ligands with clinical therapeutic potential.
Comparison of the CB2-Gi complex with CB1-Gi, Rhodopsin-Gi, μOR-Gi, and β2AR-Gs complexes reveals certain similar overall interaction profiles for receptor-G protein binding. Although CB2 and CB1 share great structural similarity, their interactions with the α5 helix of the G protein remain distinct, reflecting the versatility of Gi coupling. P139ICL2 in CB2 and the homologous residue in other GPCRs are critical for G protein coupling, and the unique motif of “P138ICL2P139ICL2” in ICL2 of CB2 contributes to its Gi coupling specificity. The well-resolved ICL3 structure provides detailed information relating to CB2 interaction with G protein. Our findings, along with the published CB1 structures and CB2 inactive structures, fulfill the complete profile of cannabinoid system receptors and should aid in the rational design of drugs targeting CB2.
STAR★METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Xiang-Qun Xie (xix15@pitt.edu). All unique/stable reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines
Chinese hamster ovary (CHO)-K1 cells (PerkinElmer) were cultured in DMEM/F-12 medium (GIBCO) supplemented with 10% fetal bovine serum at 37°C wi th 5% CO2. Spodoptera frugiperda Sf9 cells (Novagen) were cultured in ESF 921 serum-free medium (Expression Systems) in 27°C. CHO-K1 cells over-expressing CB2 were used in radioligand-binding assays and cAMP functional assays. Sf9 cells were used exclusively for recombinant protein expression.
METHOD DETAILS
Constructs
The full-length gene coding sequence of wild type human CB2 was cloned into pFastBac vector (ThermoFisher) with an N-terminal FLAG tag followed by β2-AR N-terminal tail region (BN, hereafter) as fusion protein and a TEV cleavage site, along with a His8 tag at the C-terminal to facilitate expression and purification. The prolactin precursor sequence was inserted into the N-terminus as a signaling peptide to anchor CB2 to the membrane and improve CB2 expression. A dominant-negative bovine Gαi1 (Gαi1_4M) construct was generated by site-directed mutagenesis to incorporate mutations S47N, G203A, E245A and A326S to decrease the affinity of nucleotide binding of Gαβγ complex (Liu et al., 2016). All three G protein complex components, Gαi1_4M, rat Gβ1 and bovine Gγ2, were cloned into pFastBac vector separately. ScFv16 coding sequence was cloned into pFastBac vector (ThermoFisher) with a GP67 signaling peptide inserted into the N-terminus and a TEV cleavage site-His8 at the C-terminus.
Protein complex expression, formation, and purification
Purification of scFv16 was conducted as previously described (Koehl et al., 2018) with a subtle change. Briefly, secreted scFv16 from baculovirus-infected Sf9 insect cells (Novagen) was purified using Ni-NTA and size exclusion chromatography. After balancing the pH and removing the chelating agents by Ni2+ and Ca2+, the cell culture supernatant was loaded onto Ni-NTA. The nickel resin was first washed with 20mM HEPES pH 7.2, 100mM NaCl, 50mM imidazole for 10 column volumes and then eluted in buffer containing 250mM imidazole. The eluted sample was treated with TEV protease (homemade) followed by dialysis in 20mM HEPES pH 7.2, 100mM NaCl overnight at 4°C and then reloaded onto Ni-NTA resin to remove cleaved octa-histidine tag. The flow-through was collected and applied to a HiLoad Superdex 200, 10/60 column (GE Healthcare). The monomeric peak fractions were concentrated and fast-frozen by liquid nitrogen.
CB2, Gαi1_4M, His8-tagged Gβ1 and Gγ2 were co-expressed in Sf9 insect cells (Novagen) using the Bac-to-Bac baculovirus expression system (Novagen). Cell cultures were grown in ESF 921 serum-free medium (Expression Systems) at 27°C to a density of 4 × 106 cells mL−1and then infected with the four types of baculoviruses expressing CB2, Gαi1_4M, His8-tagged Gβ1 and Gγ2 at the ratio of 1:1:1:1. After infection at 27°C for 48h, the cells were collected by centrifugation at 2000 rpm (ThermoFisher, H12000) for 20 min and kept frozen at −80°C for further usage.
For the purification of CB2-Gi complex, cell pellets from 1.5L culture were thawed at room temperature and suspended by French press in 20 mM HEPES pH 7.2, 50 mM NaCl, 5 mM CaCl2. Complex was formed on cell membrane in the presence of 2 μM WIN 55,212–2 (Tocris) and treated with apyrase (25 mU/ml, NEB), followed by incubation for 1.5 h at room temperature. Cell membranes were collected by ultra-centrifugation at 100,000 x g for 35 min. The membranes were then re-suspended and solubilized in buffer containing 20 mM HEPES, pH 7.2, 100 mM NaCl, 5 mM CaCl2, 10% Glycerol, 0.5% (w/v) dodecyl-β-D-maltoside (DDM, Anatrace), 0.15% (w/v) cholesteryl hemisuccinate TRIS salt (CHS, Anatrace), 0.5% (w/v) 3-[(3-cholamido-propyl)-dimethylammonio]-1-propanesulfonate (CHAPS, Anatrace), 0.1%(w/v) digitonin (Sigma Aldrich), 2 μM WIN 55,212–2 and 25 mU mL−1 apyrase for 2.5 h at 4°C. The supernatant was isolated by centrifugation at 100,000 × g for 45 min and then incubated for 3h at 4°C with pre-equilibrated 5mL FLAG resin. After batch binding, the FLAG resin with immobilized protein complex was manually loaded onto a gravity column (Bio-Rad). The resin was first washed with 10 column volumes of 20 mM HEPES, pH 7.2, 100 mM NaCl, 0.05% DDM (w/v), 0.01% CHS (w/v), 0.05% CHAPS, 0.1% digitonin(w/v), 2μM WIN 55,212–2. Detergent was then exchanged on resin by two washing steps in 20 mM HEPES, pH 7.2, 100 mM NaCl, 2μM WIN 55,212–2 supplemented with different detergents: first 0.02% DDM (w/v), 0.004% CHS (w/v), 0.02% CHAPS, 0.1% digitonin, then 0.1% digitonin for 10 column volumes each. Subsequently, the FLAG resin with bound material was gently resuspended in 20 mM HEPES, pH 7.2, 100 mM NaCl, 0.1% digitonin, 2μM WIN 55,212–2, treated with TEV protease and 2.5 mg scFv16 overnight at 4°C. Re leased protein from FLAG beads was collected next day, concentrated and then loaded onto a Superdex 200, 10/300 GL increase column (GE Healthcare) pre-equilibrated with buffer containing 20 mM HEPES, pH 7.2, 100 mM NaCl, 0.075% digitonin, 2μM WIN 55,212–2. The eluted fractions of monomeric complex were pooled and concentrated for cryo-EM grids preparation. The final yield of purified complex is approximately 1mg per liter of insect cell culture.
Sample preparation and EM data collection
A droplet (3.0 μl) of the purified CB2-WIN 55,212–2-Gi-scFv16 complex at a concentration of about 7.0 mg ml−1 was applied to a glow-discharged holey carbon grid (Quantifoil R1.2/1.3, Au 300 mesh), and subsequently vitrified using a Vitrobot Mark IV (FEI Company). Cryo-EM movie stacks were collected on a Titan Krios microscope operated at 300 kV under EFTEM mode. Nanoprobe with 0.9 μm illumination area was used. Data were recorded on a post-GIF Gatan K2 summit camera at a nominal magnification of 130,000, using super-resolution counting model at physical pixel size of 1.029 Å. BioQuantum energy filter was operated in the zero-energy-loss mode with an energy slit width of 20 eV. The total accumulative electron dose is ~83 e-/ Å2 fractioned over 40 subframes with a total exposure time of 8 seconds. The target defocus range was set to −1.0 ~ −2.5 μm. A total of 8,810 image stacks were collected with two datasets.
Image processing and 3-D reconstructions
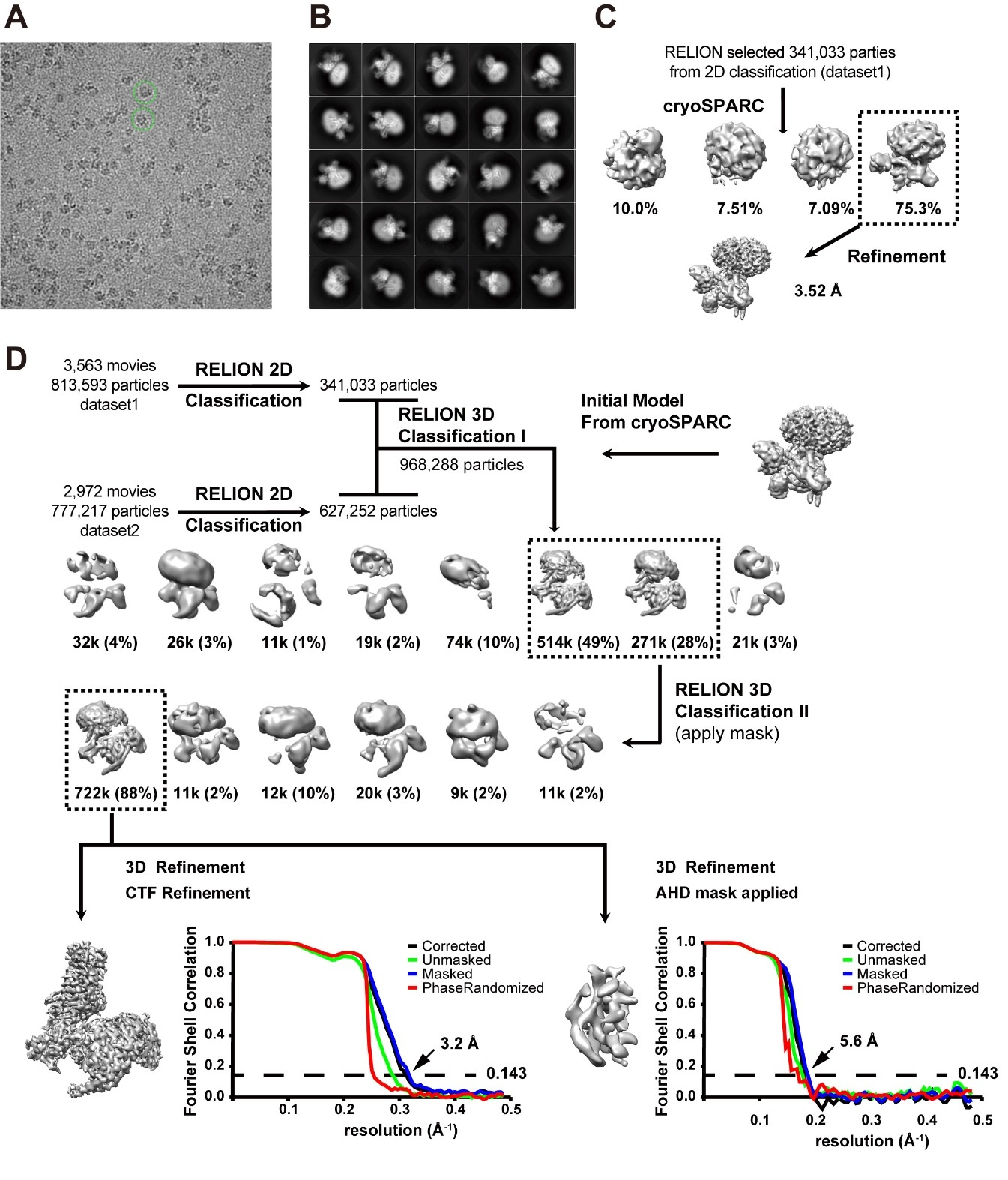
The movie stacks were corrected for drift and beam-induced motion by MotionCor2 (Zheng et al., 2017) with 2× binning, which generated drift-corrected summed images with and without electron-dose weighting. Each micrograph was manually inspected to remove bad pictures that were contaminated by crystalline ice or other forms of visible contamination, and CTF parameters were estimated by CTFFIND4 (Rohou and Grigorieff, 2015) using non-dose-weighted images. After sorting, micrographs with maximum estimated resolution beyond 4.0 Å were discarded, and good motion-corrected summed images with dose-weighting were used for all other image processing in RELION 3.0 (Zivanov et al., 2018) and cryoSPARC (Punjani et al., 2017). Global and local resolution estimates were calculated in RELION using the gold-standard Fourier shell correlation (FSC = 0.143) criterion (Rosenthal and Henderson, 2003). About 3.5K manually picked parties were used to generate the auto picking 2D reference. The two datasets were performed auto picking and particle extraction, separately in RELION3.0. Particles extracted from each sub-dataset were 4 times downscaled and subjected to reference-free 2D classification to remove false picks and obvious junk classes leaving behind approximately 341,033 particles from dataset1 and 627,252 particles from dataset2 (Figure S2). The selected particles were re-centered and re-extracted for further 3-D processing. The initial model was generated in cryoSPARC (Figure S2) with re-extracted particles from dataset1. The totally good 968,288 particles were 3-D classified with 8 classes in RELION3.0 with 60 Å low pass filter of the initial model. Two good classes (~812k particles) were selected for another round of 3-D classification with CB2/Gi/scFv16 mask. A final set of 772k homogeneous CB2/Gi/scFv16 complex particles was selected to perform 3-D refinements in RELION3.0. The final model was refined to an overall resolution of 3.2 Å. The map was sharpened with a B-factor of −98 Å2 (Figure S3). Local resolution estimates were determined by ResMap software (Kucukelbir et al., 2014) (Figure S3). The final set of homogeneous CB2/Gi/scFv16 complex particles were also subjected to AHD focused refinement to generate a better map for AHD domain (Figure S3).
Model building and refinement
The initial Gi protein and scFv16 model were adopted from the cryo-EM structure of μ-opioid receptor-Gi Protein complex (PDB: 6DDE), and initial CB2 model was generated by an online homology model building tool (Kelley et al., 2015). All models were docked into the electron microscopy density map, followed by iterative manual adjustment and rebuilding in COOT (Emsley and Cowtan, 2004), and real space refinement using Phenix programs (Adams et al., 2010). The model statistics were validated using Phenix Comprehensive validation (Adams et al., 2010). Structural figures were prepared in Chimera and PyMOL (https://pymol.org/2/). The final refinement statistics are provided in the Supplemental Table S1. The extent of any model overfitting during refinement was measured by refining the final model against one of the half-maps and by comparing the resulting map versus model FSC curves with the two half-maps and the full model.
Protein thermostability assay
A fluorescence detection assay was conducted using the thiol-specific fluorochrome N-[4-(7-diethylamino-4-methyl-3-coumarinyl)-phenyl]-maleimide (CPM) to determine thermal stability of CB2-Gi-scFv16 complex. The CPM reacts with the native cysteines embedded in the protein. CPM dye (Sigma Aldrich) was dissolved at 4 mg/mL in DMSO as stock, and then diluted 1:40 with CPM dilution buffer containing 20 mM HEPES, pH 7.2, 100 mM NaCl. The protein (30–40 ng) was diluted in CPM dilution buffer supplemented with 0.1% digitonin to a final volume of 150μL. 10μL of the diluted dye was added and mixed together with protein sample gently. After incubation in ice for 10 min to allow fully equilibration of the reaction system, the mixture was transferred into a sub-micro quartz fluorimeter cuvette (Starna Cells, Inc.). The melting curve was recorded by heating the mixture from 20°C to 90 °C with a ramp rate of 2 °C/min in a Cary Eclipse Fluo rescence Spectrophotometer (Agilent Technologies). The excitation wavelength was set at 387 nm and the emission wavelength was 463 nm. Data analysis was performed by Prism 7 (GraphPad), Tm were determined by fitting the curve to the Boltzmann sigmoidal equation.
Synthesis and characterization of XIE55 and XIE57
(E)-N, N-diethyl-4-(((3,4,5- trimethoxybenzyl)imino)methyl)aniline (1)
3,4,5-Trimethoxybenzylamine (1972 mg, 10 mmol) was added slowly to a solution of 4-(diethylamino) benzaldehyde (1770 mg, 10 mmol) and methanol (20 mL). The reaction mixture was stirred and refluxed for 12 hours. The reaction mixture was cooled to room temperature, and the solvent was removed by evaporation in vacuum to give the crude compound 1, which was used in the next step without further purification.
N, N-diethyl-4-(((3,4,5-trimethoxybenzyl)amino)methyl)aniline (2)
The crude compound 1 was dissolved in methanol (20 mL), and NaBH4 (570 mg, 15 mmol) was added. The mixture was stirred continuously for 12 hours at room temperature. The reaction solution was poured into water and extracted with ethyl acetate. The combined organic layers were washed with water and brine and then dried over Na2SO4. The residue was purified by flash chromatography (ethyl acetate/petroleum ether, 1:2) on silica gel to obtain the intermediate 2 used in the following steps.
(E)-N-(4-(diethylamino)benzyl)-2-phenyl-N-(3,4,5-trimethoxybenzyl)ethene-1-sulfonamide (XIE55)
XIE55 was prepared from the intermediate compound 2 (358 mg, 1.0 mmol) and (E)-2-phenylethene-1-sulfonyl chloride (202 mg, 1.0 mmol). The residue was purified by flash chromatography (ethyl acetate/petroleum ether, 1:2) on silica gel to obtain final product XIE55. White solid (278 mg, yield: 53%). m.p 95–97°C; 1H NMR (400 MHz, DMSO-d6) δ7.65 (t, J = 2.4 Hz, 2H), 7.44–7.42 (m, 3H), 7.38 (s, 1H), 7.26 (s, 1H), 7.09 (d, J = 8.80 Hz, 2H), 6.59 (d, J = 8.40 Hz, 2H), 6.47 (s, 2H), 4.22 (s, 2H), 4.19 (s, 2H), 3.65 (s, 6H), 3.59 (s, 3H), 3.30–3.25 (m, 4H), 1.06 (t, J = 7.20 Hz, 6H). 1H NMR (400 MHz, CDCl3) δ7.44 – 7.37 (m, 6H), 6.64 (d, J = 8.80 Hz, 2H), 6.56–6.502 (m, 3H), 4.28 (s, 4H), 3.85 (s, 3H), 3.82 (s, 6H), 1.18 (t, J = 6.80 Hz, 6H); 13C NMR (600 MHz, CDCl3): δ 153.29, 147.54, 140.51, 137.37, 132.88, 131.88, 130.57, 130.47, 129.00, 128.05, 125.70, 121.63, 111.47, 105.55, 60.86, 56.10, 50.02, 49.85, 44.33, 12.54; MS (ESI): m/z 525.33 (M + H) +.
N-(4-(diethylamino)benzyl)-N-(3,4,5-trimethoxybenzyl)thiophene-2-sulfonamide (XIE57)
XIE57 was prepared from the intermediate compound 2 (358 mg, 1.0 mmol) and thiophene-2-sulfonyl chloride (182 mg, 1.0 mmol). The residue was purified by flash chromatography (ethyl acetate/petroleum ether, 1:2) on silica gel to obtain final product XIE57. Yellow solid (270 mg, yield: 54%). m.p 77–80°C; 1H NMR (400 MHz, DMSO-d6) δ 8.03–8.02 (m, 1H), 7.76–7.75 (m, 1H), 7.28–7.25 (m, 1H), 6.96 (d, J = 8.40 Hz, 2H), 6.55 (d, J = 8.80 Hz, 2H), 6.28 (s, 2H), 4.20 (s, 2H), 4.17 (s, 2H), 3.61 (s, 6H), 3.60 (s, 3H), 3.30–3.26 (m, 4H), 1.06 (t, J = 11.20 Hz, 6H); 13C NMR (600 MHz, CDCl3): δ 153.06, 147.44, 141.71, 137.18, 131.72, 131.57, 131.33, 130.27, 127.28, 121.26, 111.36, 105.50(2C), 60.84, 56.00, 50.84, 50.73, 44.30, 12.52; MS (ESI): m/z 505.28 (M + H) +.
CB2 radioligand competition binding assay
Nonradioactive ligands were diluted in binding buffer and supplemented with 10% dimethyl sulfoxide (DMSO) and 0.4% methylcellulose. Each assay plate well contained a total of 200 μL of reaction mixture composed of 5 μg of CB2 membrane protein, labeled [3H] CP-55,940 ligand at a final concentration of 3 nM, and the unlabeled ligand at variable dilutions, as stated above. Plates were incubated at 30°C for 1 h with gentle shaking. The reaction was terminated by rapid filtration through Unifilter GF/B filter plates using a Unifilter Cell Harvester (PerkinElmer). After the plate was allowed to dry overnight, 30 μL of MicroScint-0 cocktail (PerkinElmer) was added to each well and the radioactivity was counted using a PerkinElmer TopCounter. All assays were performed in duplicate and data points were represented as mean ± SEM. Bound radioactivity data was analyzed for Ki values using nonlinear regression analysis via GraphPad Prism 7 software.
cAMP functional assay
Cellular cAMP levels were measured according to the reported method with modifications using LANCE cAMP 384 kits (PerkinElmer) (Zhang et al., 2011, Ouyang et al., 2013). The assay is based on competition between a Europium-labeled cAMP trace complex and total cAMP for binding sites on cAMP-specific antibodies labeled with a fluorescent dye. CB2 receptor wild type (WT)-transfected CHO cells were seeded into a 384-well white ProxiPlates with a density of 2000 cells per well in 5 μL of RPMI-1640 medium containing 1% dialyzed FBS, 25 mM HEPES, 100 μg/mL penicillin, 100 U/ml streptomycin and 200 μg/mL of G-418. After culture overnight, 2.5 μL of cAMP antibody and RO20–1724 (final concentration, 50 μM) in stimulation buffer (DPBS 1x, containing 0.1% BSA) was added to each well, followed by addition of either 2.5 μL compound or forskolin (final 5 μM) for agonist-inhibited adenylate cyclase (AC) activity assay. After incubation at room temperature for 45 min, 10 μL of detection reagent was added into each well. The plate was then incubated for 1 h at room temperature and measured in Synergy H1 hybrid reader (BioTek) with excitation at 340 nm and emission at 665 nm. Each cAMP determination was made via at least three independent experiments, each in triplicate. EC50 values were determined by nonlinear regression, dose-response curves (GraphPad Prism 7).
Quantitative characterization of binding residues on CB2
Ligand binding always involves its interaction with key binding residues, the changes in the features of ligand binding site, particular rearrangements of the protein structure, etc. Thus, several key aspects of ligand binding can be explored quantitatively in a special binding region of a crystal/Cryo-EM structure, including the involved key residues, residual energy contribution, energy term, etc. Starting with the code base of the current stable version 2.2.3 of idock (Li et al., 2012, Chen et al., 2019) that adopts the exact scoring function of AutoDock Vina (Trott and Olson, 2010), we developed an even more efficient variant integrating the ability to calculate also the residual contributions of the binding energy. Our adaptation skips the CPU-intensive precalculation of free energy grid map in the case of crystal/Cryo-EM structures with a ligand bound because the need for testing a massive number of conformational candidates is no longer there. Thereafter, the program populates the precisely calculated per-atom-pair free energy for every residue and produces the results. The improvement could reduce total time consumption by 95%~97% for most input. We will discuss this method in depth in our future publications.
Molecular docking
The docking program Surflex-Dock GeomX (SFXC) in SYBYL-X 2.0 was applied to construct receptor-ligand complexes in which the docking scores were expressed in -log10 (Kd). The main protocols or parameters of docking were addressed in our previous publications (Feng et al., 2014, Feng et al., 2015). Briefly, the docking parameters used were as follows: (a) the “number of starting conformations per ligand” was set to 10, and the “number of max conformations per fragment” was set to 20; (b) the “maximum number of rotatable bonds per molecule” was set to 100; (c) flags were turned on at “pre-dock minimization,” “post-dock minimization,” “molecule fragmentation,” and “soft grid treatment”; (d) “activate spin alignment method with density of search” was set to 9.0; and (e) the “number of spins per alignment” was set to 12.
MD simulations
The MD simulation system consists of one copy of human CB2 transmembrane domain and the WIN 55212–2 ligand, 240 POPC (1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine) lipids, 0.15 M NaCl, and 17082 TIP3P (Jorgensen et al., 1983) water molecules. The lipids, ions and water molecules were added using CHARMM-GUI (www.charm-gui.org). The detailed force field set up and MD protocol can be found elsewhere (Yin et al., 2018). In brief, protein, lipid and the ligand are described by AMBER FF14SB (Maier et al., 2015) LIPID14 (Dickson et al., 2014) and GAFF (Wang et al., 2004) force fields, respectively.
MD simulation was performed using the AMBER18 software package (Case et al., 2018). The MD system was first relaxed by a set of minimizations by removing possible steric clashes. There were three phases for the subsequent NPT (constant particle number, pressure and temperature) MD simulations: the relaxation phase (1 nanosecond for each temperature from 50 to 250 K at a step of 50 K), the equilibrium phase (25 nanoseconds, 298 K), and the sampling phase (200 nanoseconds). 2000 snapshots were evenly selected from the sampling phase for MM-GBSA binding free energy decomposition analysis. Integration of the equations of motion was conducted at a time step of 1 fs for the relaxation phase and 2 fs for the equilibrium and sampling phases.
For each MD snapshot, the molecular mechanical (MM) energy (EMM) and the MM-GB/SA solvation free energy were calculated without further minimization (Wang et al., 2006, Hou et al., 2011a, Hou et al., 2011b). The interaction energies between each residue and the ligand were calculated with the solvent effect being taken into account using a MM-GBSA solvation model developed previously (Hawkins et al., 1996). Key parameters controlling the MM-GBSA analyses were the following: external dielectric constant ~ 80; internal dielectric constant ~ 1; and the surface tension for estimating the nonpolar solvation energy ~ 0.005.
Figure preparation
Structural figures were prepared in UCSF Chimera (https://www.cgl.ucsf.edu/chimera/) and PyMOL (https://pymol.org/2/).
QUANTIFICATION AND STATISTICAL ANALYSIS
All reported resolutions in cryo-EM studies are based upon the 0.143 Fourier Shell Correlation criterion. Error bars in Figure 4B represent standard error of the mean for three independent experiments.
DATA AND CODE AVAILABILITY
The cryo-EM density map has been deposited in the Electron Microscopy Data Bank (EMDB) under accession code EMD-20470 and model coordinates have been deposited in the Protein Data Bank (PDB) under accession number 6PT0.
Supplementary Material
(A) Snake model of the CB2 construct used in this paper. (B) Size exclusion chromatography profile and SDS-PAGE analysis of CB2-Gi-scFv16-WIN 55,212–2 complex; the peak of complex elution is marked with a star and the peak of extra scFv16 is marked with an arrow. Fractions between two dashed lines were pooled and concentrated for cryo-EM experiments. (C) CPM assay of CB2-Gi-scFv16 activated by WIN 55,212–2 to test the half denaturing temperature, Tm value. Tm was determined by fitting the curve to Boltzmann sigmoidal equation.
{kind=link}
(A) Representative micrographs CB2-WIN 55,212–2-Gi-scFv16 complex with particles of 190 Å diameter highlighted in a green circle. (B) Representative images of 2D class-averages. (C) Cryo-electron microscopy data processing workflow by cryoSPARC to generate an unbiased Ab initio 3D map. Boxed 3D classes were selected for further processing. (D) Cryo-electron microscopy data processing workflow by RELION. Boxed 3D classes were selected for further processing. Half-map Fourier Shell Correlation (FSC) plots generated by RELION was shown at the down right corner. An overall resolution was set at 3.2 Å at 0.143 FSC, and the α-helical domain (AHD) focused refinement at 5.8 Å at 0.143 FSC.
{kind=link}
(A) Cryo-EM map colored according to local resolution, which was calculated from half-maps by ResMap software. The density map is shown at 0.022 contour level. (B) Density maps of TM1–8 in CB2/WIN 55,212–2, α5 of Gαi and WIN 55,212–2 ligand, showing at 0.016 contour level.
{kind=link}
(A) To further validate the binding pose of WIN 55,212–2, we first conducted molecular docking using three different software/algorithms, including Schrödinger-Glide (left panels), Auto Dock (middle panels), and SYBYL-Surflex Dock (right panels) to conduct the studies. All the docking results showed that the pose of WIN 55,212–2 presented in our manuscript (pose 1) ranked at the top one or two with lowest energy (−10.28, −13.26, and −11.73 kcal/mol, respectively) compared to the alternative pose 2 (−4.02, −7.19, and −7.04 kcal/mol, respectively). (B) Root-mean-square deviation (RMSD) approximate time plots for all (black), all excluding the residues 215–241 loop (red), helices (green), ligand with (blue) and without (purple) least-square fitting. (C) Hotspot residues identified by MM-GBSA decomposition. A residue is recognized as a hotspot when its interaction with the ligand is stronger than −1.0 kcal/mol. The hotspots residues according to their MM-GBSA interaction energies with the ligand are shown as brownish sticks. The stronger the interaction, the more reddish the residue is colored, while the weaker the interaction the more blueish the residue. (D) Representative MD structure (blue cartoon and brownish sticks), having the smallest RMSD to the average structure of MD snapshots, aligned to the Cryo-EM structure (grey cartoon and greenish sticks).
{kind=link}
Reagents and conditions: (a) Methanol, 70°C reflux. (b) NaBH4, room temperature. (c) TEA, DCM, (E)-2-phenylethene-1- sulfonyl chloride, room temperature. (d) TEA, DCM, Thiophene-2-sulfonyl chloride, room temperature.
{kind=link}
(A)Comparison of the relative orientations of Gi bound to CB2 (magenta), CB1 (PDB: 6N4B, marine), μOR (PDB: 6DDE, yellow), and Rhodopsin (PDB: 6CMO, red) when aligned on the receptor.
(B)Comparison of CB2-Gi complex with β2AR-Gs complex (PDB:3SN6, light pink) when aligned with the receptor.
{kind=link}
Sequence alignments between CB2 and CB1 highlight the orthosteric binding pocket residues and the TM6 kink residues for receptor activation, as well as the Gi-interface residues for Gi coupling. Secondary structure elements are annotated underneath the sequences.
{kind=link}
Table S1. Data collection, model refinement and validation. Related to Figure 1.
Highlights.
3.2 Å Cryo-EM structure of the CB2-Gi complex bound to potent agonist WIN 55,212–2
Algorithm developed for quantitative characterization of binding residues
Structural determinants for distinguishing CB2 agonists from antagonists
CB2-Gi binding features and different activation mechanisms between CB2 and CB1
ACKNOWLEDGMENTS
The project is supported by funding to the Xie Laboratory and Center from the NIH NIDA (P30 DA035778A1) and NIH (R01 DA025612) (to XIE), NIH grant 1R35GM128641 (to C.Z.), and 1R01GM127710 (to H.E.X), the Jay and Betty Van Andel Foundation (to H.E.X.), and XDB08020303 (to H. E. X.). The cryo-EM data were collected at the David Van Andel Advanced Cryo-Electron Microscopy Suite at the Van Andel Research Institute. Y. Z. was supported by the UCAS Joint PhD Training Program.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- ADAMS PD, AFONINE PV, BUNKOCZI G, CHEN VB, DAVIS IW, ECHOLS N, HEADD JJ, HUNG LW, KAPRAL GJ, GROSSE-KUNSTLEVE RW, MCCOY AJ, MORIARTY NW, OEFFNER R, READ RJ, RICHARDSON DC, RICHARDSON JS, TERWILLIGER TC & ZWART PH 2010. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr, 66, 213–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ARONNE LJ, TONSTAD S, MORENO M, GANTZ I, ERONDU N, SURYAWANSHI S, MOLONY C, SIEBERTS S, NAYEE J, MEEHAN AG, SHAPIRO D, HEYMSFIELD SB, KAUFMAN KD & AMATRUDA JM 2010. A clinical trial assessing the safety and efficacy of taranabant, a CB1R inverse agonist, in obese and overweight patients: a high-dose study. Int J Obes (Lond), 34, 919–35. [DOI] [PubMed] [Google Scholar]
- ASO E. & FERRER I. 2016. CB2 cannabinoid receptor as potential target against Alzheimer’s disease. Frontiers in neuroscience, 10, 243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CALIGNANO A, LA RANA G, GIUFFRIDA A. & PIOMELLI D. 1998. Control of pain initiation by endogenous cannabinoids. Nature, 394, 277–81. [DOI] [PubMed] [Google Scholar]
- CASE DA, BEN-SHALOM IY, BROZELL SR, CERUTTI DS, CHEATHAM I, T. E, CRUZEIRO VWD, DARDEN TA, DUKE RE, GHOREISHI D, GILSON MK, GOHLKE H, GOETZ AW, GREENE D, HARRIS R, HOMEYER N, IZADI S, KOVALENKO A, KURTZMAN T, LEE TS, LEGRAND S, LI P, LIN C, LIU J, LUCHKO T, LUO R, MERMELSTEIN DJ, MERZ KM, MIAO Y, MONARD G, NGUYEN C, NGUYEN H, OMELYAN I, ONUFRIEV A, PAN F, QI R, ROE DR, ROITBERG A, SAGUI C, SCHOTT-VERDUGO S, SHEN J, SIMMERLING CL, SMITH J, SALOMON-FERRER R, SWAILS J, WALKER RC, WANG J, WEI H, WOLF RM, WU X, XIAO L, D.M. Y. & KOLLMAN PA 2018. AMBER 2018. University of California, San Francisco. [Google Scholar]
- CHEN D-J, GAO M, GAO F-F, SU Q-X & WU J. 2017. Brain cannabinoid receptor 2: expression, function and modulation. Acta Pharmacologica Sinica, 38, 312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHEN M, JING Y, WANG L, FENG Z. & XIE X-Q 2019. DAKB-GPCRs: An Integrated Computational Platform for Drug Abuse Related GPCRs. Journal of chemical information modeling. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CITRARO R, RUSSO E, LEO A, RUSSO R, AVAGLIANO C, NAVARRA M, CALIGNANO A. & DE SARRO G. 2016. Pharmacokinetic-pharmacodynamic influence of N-palmitoylethanolamine, arachidonyl-2′-chloroethylamide and WIN 55,212–2 on the anticonvulsant activity of antiepileptic drugs against audiogenic seizures in DBA/2 mice. European journal of pharmacology, 791, 523–534. [DOI] [PubMed] [Google Scholar]
- DHOPESHWARKAR A, MURATAEVA N, MAKRIYANNIS A, STRAIKER A. & MACKIE K. 2017. Two Janus cannabinoids that are both CB2 agonists and CB1 antagonists. Journal of Pharmacology Experimental Therapeutics, 360, 300–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DICKSON CJ, MADEJ BD, SKJEVIK AA, BETZ RM, TEIGEN K, GOULD IR & WALKER RC 2014. Lipid14: The Amber Lipid Force Field. J Chem Theory Comput, 10, 865–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- EMSLEY P. & COWTAN K. 2004. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr, 60, 2126–32. [DOI] [PubMed] [Google Scholar]
- FENG Z, ALQARNI MH, YANG P, TONG Q, CHOWDHURY A, WANG L. & XIE X-Q 2014. Modeling, molecular dynamics simulation, and mutation validation for structure of cannabinoid receptor 2 based on known crystal structures of GPCRs. Journal of chemical information modeling, 54, 2483–2499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FENG Z, PEARCE LV, XU X, YANG X, YANG P, BLUMBERG PM & XIE X-Q 2015. Structural insight into tetrameric hTRPV1 from homology modeling, molecular docking, molecular dynamics simulation, virtual screening, and bioassay validations. Journal of chemical information modeling, 55, 572–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FULMER ML & THEWKE DP 2018. The Endocannabinoid System and Heart Disease: The Role of Cannabinoid Receptor Type 2. Cardiovascular Haematological Disorders-Drug Targets, 18, 34–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GALIEGUE S, MARY S, MARCHAND J, DUSSOSSOY D, CARRIERE D, CARAYON P, BOUABOULA M, SHIRE D, LE FUR G. & CASELLAS P. 1995. Expression of central and peripheral cannabinoid receptors in human immune tissues and leukocyte subpopulations. Eur J Biochem, 232, 54–61. [DOI] [PubMed] [Google Scholar]
- GÓMEZ-GÁLVEZ Y, PALOMO-GARO C, FERNÁNDEZ-RUIZ J. & GARCÍA C. 2016. Potential of the cannabinoid CB2 receptor as a pharmacological target against inflammation in Parkinson’s disease. Progress in Neuro-Psychopharmacology Biological Psychiatry, 64, 200–208. [DOI] [PubMed] [Google Scholar]
- GUIMARAES S, GODBOUT JP & SHERIDAN JF 2018. Repeated Social Defeat-Induced Neuroinflammation, Anxiety-like behavior and Resistance to Fear Extinction were Attenuated by the Cannabinoid Receptor Agonist WIN55, 212–2. Neuropsychopharmacology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GUINDON J. & HOHMANN A. 2008. Cannabinoid CB2 receptors: a therapeutic target for the treatment of inflammatory and neuropathic pain. British journal of pharmacology, 153, 319–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HAWKINS GD, CRAMER CJ & TRUHLAR DG 1996. Parametrized models of aqueous free energies of solvation based on pairwise descreening of solute atomic charges from a dielectric medium. Journal of Physical Chemistry, 100, 19824–19839. [Google Scholar]
- HOU T, WANG J, LI Y. & WANG W. 2011a. Assessing the performance of the MM/PBSA and MM/GBSA methods. 1. The accuracy of binding free energy calculations based on molecular dynamics simulations. J Chem Inf Model, 51, 69–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HOU T, WANG J, LI Y. & WANG W. 2011b. Assessing the performance of the molecular mechanics/Poisson Boltzmann surface area and molecular mechanics/generalized Born surface area methods. II. The accuracy of ranking poses generated from docking. J Comput Chem, 32, 866–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HUA T, VEMURI K, PU M, QU L, HAN GW, WU Y, ZHAO S, SHUI W, LI S. & KORDE A. 2016. Crystal structure of the human cannabinoid receptor CB1. Cell, 167, 750–762. e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- IBSEN MS, CONNOR M. & GLASS M. 2017. Cannabinoid CB1 and CB2 Receptor Signaling and Bias. Cannabis Cannabinoid Res, 2, 48–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- IDRIS AI & RALSTON SH 2012. Role of cannabinoids in the regulation of bone remodeling. Front Endocrinol (Lausanne), 3, 136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- IDRIS AI, VAN ‘T HOF RJ, GREIG IR, RIDGE SA, BAKER D, ROSS RA & RALSTON SH 2005. Regulation of bone mass, bone loss and osteoclast activity by cannabinoid receptors. Nat Med, 11, 774–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JORGENSEN WL, CHANDRASEKHAR J, MADURA JD, IMPEY RW & KLEIN ML 1983. Comparison of simple potential functions for simulating liquid water. The Journal of Chemical Physics, 79, 926. [Google Scholar]
- KANG Y, KUYBEDA O, DE WAAL PW, MUKHERJEE S, VAN EPS N, DUTKA P, ZHOU XE, BARTESAGHI A, ERRAMILLI S. & MORIZUMI T. 2018. Cryo-EM structure of human rhodopsin bound to an inhibitory G protein. Nature, 558, 553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KELLEY LA, MEZULIS S, YATES CM, WASS MN & STERNBERG MJ 2015. The Phyre2 web portal for protein modeling, prediction and analysis. Nat Protoc, 10, 845–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KIRKHAM TC 2005. Endocannabinoids in the regulation of appetite and body weight. Behav Pharmacol, 16, 297–313. [DOI] [PubMed] [Google Scholar]
- KUCUKELBIR A, SIGWORTH FJ & TAGARE HD 2014. Quantifying the local resolution of cryo-EM density maps. Nat Methods, 11, 63–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KUMAR KK, SHALEV-BENAMI M, ROBERTSON MJ, HU H, BANISTER SD, HOLLINGSWORTH SA, LATORRACA NR, KATO HE, HILGER D. & MAEDA S. 2019. Structure of a Signaling Cannabinoid Receptor 1-G protein complex. Cell, 176, 448–458. e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LELEU-CHAVAIN N, BAUDELET D, HELOIRE VM, ROCHA DE, RENAULT N, BARCZYK A, DJOUINA M, BODY-MALAPEL M, CARATO P. & MILLET R. 2019. Benzo[d]thiazol-2(3H)-ones as new potent selective CB2 agonists with anti-inflammatory properties. Eur J Med Chem, 165, 347–362. [DOI] [PubMed] [Google Scholar]
- LI H, LEUNG K-S & WONG M-H idock: A multithreaded virtual screening tool for flexible ligand docking. 2012 IEEE Symposium on Computational Intelligence in Bioinformatics and Computational Biology (CIBCB), 2012. IEEE, 77–84. [Google Scholar]
- LI X, HUA T, VEMURI K, HO JH, WU Y, WU L, POPOV P, BENCHAMA O, ZVONOK N, LOCKE K, QU L, HAN GW, IYER MR, CINAR R, COFFEY NJ, WANG J, WU M, KATRITCH V, ZHAO S, KUNOS G, BOHN LM, MAKRIYANNIS A, STEVENS RC & LIU ZJ 2019. Crystal Structure of the Human Cannabinoid Receptor CB2. Cell, 176, 459–467 e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LIN SW & SAKMAR TPJB 1996. Specific tryptophan UV-absorbance changes are probes of the transition of rhodopsin to its active state. 35, 11149–11159. [DOI] [PubMed] [Google Scholar]
- LIU P, JIA M-Z, ZHOU XE, DE WAAL PW, DICKSON BM, LIU B, HOU L, YIN Y-T, KANG Y-Y & SHI Y. 2016. The structural basis of the dominant negative phenotype of the Gα i1 β 1 γ 2 G203A/A326S heterotrimer. Acta Pharmacologica Sinica, 37, 1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MAEDA S, KOEHL A, MATILE H, HU H, HILGER D, SCHERTLER GFX, MANGLIK A, SKINIOTIS G, DAWSON RJP & KOBILKA BK 2018. Development of an antibody fragment that stabilizes GPCR/G-protein complexes. Nat Commun, 9, 3712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MAIER JA, MARTINEZ C, KASAVAJHALA K, WICKSTROM L, HAUSER KE & SIMMERLING C. 2015. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J Chem Theory Comput, 11, 3696–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MAROON J. & BOST J. 2018. Review of the neurological benefits of phytocannabinoids. Surgical neurology international, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MCALLISTER SD, HURST DP, BARNETT-NORRIS J, LYNCH D, REGGIO PH & ABOOD ME 2004. Structural mimicry in class A G protein-coupled receptor rotamer toggle switches: the importance of the F3.36(201)/W6.48(357) interaction in cannabinoid CB1 receptor activation. J Biol Chem, 279, 48024–37. [DOI] [PubMed] [Google Scholar]
- MNPOTRA JS, QIAO Z, CAI J, LYNCH DL, GROSSFIELD A, LEIOATTS N, HURST DP, PITMAN MC, SONG ZH & REGGIO PH 2014. Structural basis of G protein-coupled receptor-Gi protein interaction: formation of the cannabinoid CB2 receptor-Gi protein complex. J Biol Chem, 289, 20259–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MOHAMMADI VOSOUGH E, BARADARAN RAHIMI V, MASOUD SA, MIRKARIMI HR, DEMNEH MK, ABED A, BANAFSHE HR & ASKARI VR 2019. Evaluation of protective effects of non-selective cannabinoid receptor agonist WIN 55,212–2 against the nitroglycerine-induced acute and chronic animal models of migraine: A mechanistic study. [DOI] [PubMed] [Google Scholar]
- MOREIRA FA & CRIPPA JA 2009. The psychiatric side-effects of rimonabant. Braz J Psychiatry, 31, 145–53. [DOI] [PubMed] [Google Scholar]
- MUGNAINI C, RABBITO A, BRIZZI A, PALOMBI N, PETROSINO S, VERDE R, DI MARZO V, LIGRESTI A. & CORELLI F. 2019. Synthesis of novel 2-(1-adamantanylcarboxamido)thiophene derivatives. Selective cannabinoid type 2 (CB2) receptor agonists as potential agents for the treatment of skin inflammatory disease. Eur J Med Chem, 161, 239–251. [DOI] [PubMed] [Google Scholar]
- MUKHOPADHYAY P, BAGGELAAR M, ERDELYI K, CAO Z, CINAR R, FEZZA F, IGNATOWSKA-JANLOWSKA B, WILKERSON J, VAN GILS N, HANSEN T, RUBEN M, SOETHOUDT M, HEITMAN L, KUNOS G, MACCARRONE M, LICHTMAN A, PACHER P. & VAN DER STELT M. 2016. The novel, orally available and peripherally restricted selective cannabinoid CB2 receptor agonist LEI-101 prevents cisplatin-induced nephrotoxicity. Br J Pharmacol, 173, 446–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NETTEKOVEN M, ADAM JM, BENDELS S, BISSANTZ C, FINGERLE J, GRETHER U, GRUNER S, GUBA W, KIMBARA A, OTTAVIANI G, PULLMANN B, ROGERS-EVANS M, ROVER S, ROTHENHAUSLER B, SCHMITT S, SCHULER F, SCHULZ-GASCH T. & ULLMER C. 2016. Novel Triazolopyrimidine-Derived Cannabinoid Receptor 2 Agonists as Potential Treatment for Inflammatory Kidney Diseases. ChemMedChem, 11, 179–89. [DOI] [PubMed] [Google Scholar]
- ONAIVI ES, ISHIGURO H, GONG JP, PATEL S, PERCHUK A, MEOZZI PA, MYERS L, MORA Z, TAGLIAFERRO P, GARDNER E, BRUSCO A, AKINSHOLA BE, LIU QR, HOPE B, IWASAKI S, ARINAMI T, TEASENFITZ L. & UHL GR 2006. Discovery of the presence and functional expression of cannabinoid CB2 receptors in brain. Ann N Y Acad Sci, 1074, 514–36. [DOI] [PubMed] [Google Scholar]
- OUYANG Q, TONG Q, FENG R, MYINT K-Z, PENG YANG& XIE X-Q 2013. Trisubstituted Sulfonamides: a New Chemotype for Development of Potent and Selective CB2 Receptor Inverse Agonists. ACS Medicinal Chemistry Letters, February 22, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PROIETTO J, RISSANEN A, HARP JB, ERONDU N, YU Q, SURYAWANSHI S, JONES ME, JOHNSON-LEVONAS AO, HEYMSFIELD SB, KAUFMAN KD & AMATRUDA JM 2010. A clinical trial assessing the safety and efficacy of the CB1R inverse agonist taranabant in obese and overweight patients: low-dose study. Int J Obes (Lond), 34, 1243–54. [DOI] [PubMed] [Google Scholar]
- PUNJANI A, RUBINSTEIN JL, FLEET DJ & BRUBAKER MA 2017. cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat Methods, 14, 290–296. [DOI] [PubMed] [Google Scholar]
- RAITIO K, SALO O, NEVALAINEN T, POSO A. & JARVINEN T. 2005. Targeting the cannabinoid CB2 receptor: mutations, modeling and development of CB2 selective ligands. Current medicinal chemistry, 12, 1217–1237. [DOI] [PubMed] [Google Scholar]
- RASMUSSEN SG, DEVREE BT, ZOU Y, KRUSE AC, CHUNG KY, KOBILKA TS, THIAN FS, CHAE PS, PARDON E. & CALINSKI D. 2011. Crystal structure of the β 2 adrenergic receptor–Gs protein complex. Nature, 477, 549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ROBERTO D, KLOTZ LH & VENKATESWARAN V. 2019. Cannabinoid WIN 55,212-2 induces cell cycle arrest and apoptosis, and inhibits proliferation, migration, invasion, and tumor growth in prostate cancer in a cannabinoid-receptor 2 dependent manner. The Prostate, 79, 151–159. [DOI] [PubMed] [Google Scholar]
- ROHOU A. & GRIGORIEFF N. 2015. CTFFIND4: Fast and accurate defocus estimation from electron micrographs. J Struct Biol, 192, 216–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ROSENTHAL PB & HENDERSON R. 2003. Optimal determination of particle orientation, absolute hand, and contrast loss in single-particle electron cryomicroscopy. J Mol Biol, 333, 721–45. [DOI] [PubMed] [Google Scholar]
- SCOTT CE, TANG Y, ALT A, BURFORD NT, GERRITZ SW, OGAWA LM, ZHANG L. & KENDALL DA 2019. Identification and biochemical analyses of selective CB2 agonists. Eur J Pharmacol, 854, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SEELEY RJ & WOODS SC 2003. Monitoring of stored and available fuel by the CNS: implications for obesity. Nat Rev Neurosci, 4, 901–9. [DOI] [PubMed] [Google Scholar]
- SEELY KA, PRATHER PL, JAMES LP & MORAN JH 2011. Marijuana-based drugs: innovative therapeutics or designer drugs of abuse? Molecular interventions, 11, 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SINGH R, HURST DP, BARNETT-NORRIS J, LYNCH DL, REGGIO PH & GUARNIERI F. 2002. Activation of the cannabinoid CB1 receptor may involve a W6.48/F3.36 rotamer toggle switch. Journal of Peptide Research, 60, 357–370. [DOI] [PubMed] [Google Scholar]
- SLEDZINSKI P, ZEYLAND J, SLOMSKI R. & NOWAK A. 2018. The current state and future perspectives of cannabinoids in cancer biology. Cancer Med, 7, 765–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TIMOSSI C, MALDONADO D, VIZCAıNO A, LINDAU-SHEPARD B, CONN PM & ULLOA-AGUIRRE A. 2002. Structural determinants in the second intracellular loop of the human follicle-stimulating hormone receptor are involved in Gs protein activation. Molecular cellular endocrinology, 189, 157–168. [DOI] [PubMed] [Google Scholar]
- TROTT O. & OLSON AJ 2010. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. Journal of computational chemistry, 31, 455–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TURCOTTE C, BLANCHET M-R, LAVIOLETTE M. & FLAMAND N. 2016. The CB 2 receptor and its role as a regulator of inflammation. Cellular Molecular Life Sciences, 73, 4449–4470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WALTER L. & STELLA N. 2004. Cannabinoids and neuroinflammation. Br J Pharmacol, 141, 775–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WANG JM, HOU TJ & XU XJ 2006. Recent Advances in Free Energy Calculations with a Combination of Molecular Mechanics and Continuum Models. Current Computer-Aided Drug Design, 2, 287–306. [Google Scholar]
- WANG JM, WOLF RM, CALDWELL JW, KOLLMAN PA & CASE DA 2004. Development and testing of a general amber force field. Journal of Computational Chemistry, 25, 1157–1174. [DOI] [PubMed] [Google Scholar]
- YANG P, MYINT KZ, TONG Q, FENG R, CAO H, ALMEHIZIA AA, ALQARNI MH, WANG L, BARTLOW P, GAO Y, GERTSCH J, TERAMACHI J, KURIHARA N, ROODMAN GD, CHENG T. & XIE XQ 2012a. Lead discovery, chemistry optimization, and biological evaluation studies of novel biamide derivatives as CB2 receptor inverse agonists and osteoclast inhibitors. J Med Chem, 55, 9973–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YANG P, WANG L. & XIE X-Q 2012b. Latest advances in novel cannabinoid CB2 ligands for drug abuse and their therapeutic potential. Future medicinal chemistry, 4, 187–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YIN J, CHAPMAN K, CLARK LD, SHAO Z, BOREK D, XU Q, WANG J. & ROSENBAUM DM 2018. Crystal structure of the human NK1 tachykinin receptor. Proc Natl Acad Sci U S A, 115, 13264–13269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZHANG YX, WANG LR, XIE ZJ, SCHRIBEIT B, LAZO JS & XIE X-Q 2011. Mutagenesis and Computer Modeling Studies of A GPCR Conserved Residue W5.43(194) in Ligand Recognition and Signal Transduction For CB2 Receptor. International Immunopharmacology, 11, 1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZHENG C, CHEN L, CHEN X, HE X, YANG J, SHI Y. & ZHOU N. 2013. The second intracellular loop of the human cannabinoid CB2 receptor governs G protein coupling in coordination with the carboxyl terminal domain. PloS one, 8, e63262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZHENG SQ, PALOVCAK E, ARMACHE JP, VERBA KA, CHENG Y. & AGARD DA 2017. MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat Methods, 14, 331–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZIVANOV J, NAKANE T, FORSBERG BO, KIMANIUS D, HAGEN WJ, LINDAHL E. & SCHERES SH 2018. New tools for automated high-resolution cryo-EM structure determination in RELION-3. Elife, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(A) Snake model of the CB2 construct used in this paper. (B) Size exclusion chromatography profile and SDS-PAGE analysis of CB2-Gi-scFv16-WIN 55,212–2 complex; the peak of complex elution is marked with a star and the peak of extra scFv16 is marked with an arrow. Fractions between two dashed lines were pooled and concentrated for cryo-EM experiments. (C) CPM assay of CB2-Gi-scFv16 activated by WIN 55,212–2 to test the half denaturing temperature, Tm value. Tm was determined by fitting the curve to Boltzmann sigmoidal equation.
(A) Representative micrographs CB2-WIN 55,212–2-Gi-scFv16 complex with particles of 190 Å diameter highlighted in a green circle. (B) Representative images of 2D class-averages. (C) Cryo-electron microscopy data processing workflow by cryoSPARC to generate an unbiased Ab initio 3D map. Boxed 3D classes were selected for further processing. (D) Cryo-electron microscopy data processing workflow by RELION. Boxed 3D classes were selected for further processing. Half-map Fourier Shell Correlation (FSC) plots generated by RELION was shown at the down right corner. An overall resolution was set at 3.2 Å at 0.143 FSC, and the α-helical domain (AHD) focused refinement at 5.8 Å at 0.143 FSC.
(A) Cryo-EM map colored according to local resolution, which was calculated from half-maps by ResMap software. The density map is shown at 0.022 contour level. (B) Density maps of TM1–8 in CB2/WIN 55,212–2, α5 of Gαi and WIN 55,212–2 ligand, showing at 0.016 contour level.
(A) To further validate the binding pose of WIN 55,212–2, we first conducted molecular docking using three different software/algorithms, including Schrödinger-Glide (left panels), Auto Dock (middle panels), and SYBYL-Surflex Dock (right panels) to conduct the studies. All the docking results showed that the pose of WIN 55,212–2 presented in our manuscript (pose 1) ranked at the top one or two with lowest energy (−10.28, −13.26, and −11.73 kcal/mol, respectively) compared to the alternative pose 2 (−4.02, −7.19, and −7.04 kcal/mol, respectively). (B) Root-mean-square deviation (RMSD) approximate time plots for all (black), all excluding the residues 215–241 loop (red), helices (green), ligand with (blue) and without (purple) least-square fitting. (C) Hotspot residues identified by MM-GBSA decomposition. A residue is recognized as a hotspot when its interaction with the ligand is stronger than −1.0 kcal/mol. The hotspots residues according to their MM-GBSA interaction energies with the ligand are shown as brownish sticks. The stronger the interaction, the more reddish the residue is colored, while the weaker the interaction the more blueish the residue. (D) Representative MD structure (blue cartoon and brownish sticks), having the smallest RMSD to the average structure of MD snapshots, aligned to the Cryo-EM structure (grey cartoon and greenish sticks).
Reagents and conditions: (a) Methanol, 70°C reflux. (b) NaBH4, room temperature. (c) TEA, DCM, (E)-2-phenylethene-1- sulfonyl chloride, room temperature. (d) TEA, DCM, Thiophene-2-sulfonyl chloride, room temperature.
(A)Comparison of the relative orientations of Gi bound to CB2 (magenta), CB1 (PDB: 6N4B, marine), μOR (PDB: 6DDE, yellow), and Rhodopsin (PDB: 6CMO, red) when aligned on the receptor.
(B)Comparison of CB2-Gi complex with β2AR-Gs complex (PDB:3SN6, light pink) when aligned with the receptor.
Sequence alignments between CB2 and CB1 highlight the orthosteric binding pocket residues and the TM6 kink residues for receptor activation, as well as the Gi-interface residues for Gi coupling. Secondary structure elements are annotated underneath the sequences.
Table S1. Data collection, model refinement and validation. Related to Figure 1.
Data Availability Statement
The cryo-EM density map has been deposited in the Electron Microscopy Data Bank (EMDB) under accession code EMD-20470 and model coordinates have been deposited in the Protein Data Bank (PDB) under accession number 6PT0.