

Abstract
Aims
To characterize relationships between apolipoprotein A‐I (apoA‐I) exposure and cholesterol efflux capacity (CEC) and covariate effects following CSL112 (apoA‐I [human]) administration in an integrated population including acute myocardial infarction (AMI) patients.
Methods
A pharmacometric analysis utilized data from seven clinical trials, including patients with AMI, subjects with renal impairment and healthy subjects. A population pharmacokinetic (PK) analysis was performed to relate CSL112 doses to changes in apoA‐I plasma concentrations. Covariate analysis was conducted to identify sources of variability in apoA‐I exposure. Exposure‐response modeling was conducted to describe the relationship between apoA‐I exposure and total or ATP binding cassette transporter A1‐(ABCA1)‐dependent CEC and to identify clinical predictors of CEC.
Results
A two‐compartment model described apoA‐I PK. ApoA‐I clearance was slightly lower in subjects with AMI, whereas baseline apoA‐I was marginally higher in female and Japanese subjects. Covariate effects on apoA‐I exposure were in the order of 10% and thus not clinically relevant. The relationships between apoA‐I exposure and CECs were described by nonlinear models. Simulations showed CEC elevation resulting from apoA‐I exposure increment was comparable in AMI and non‐AMI subjects; no covariate had clinically meaningful effects on CEC. Simulations also demonstrated that CEC in patients with AMI post 6 g CSL112 dosing was substantially elevated compared to placebo and lower dose levels.
Conclusions
The model‐based exposure‐response analysis demonstrated, irrespective of body weight, sex and race, that fixed 6 g CSL112 dosing causes a desired CEC elevation, which may benefit AMI patients by potentially reducing early recurrent cardiovascular event risk.
Keywords: atherosclerosis, clinical trials, modelling and simulation, pharmacokinetics‐pharmacodynamics, pharmacometrics
What is already known about this subject
CSL112 (apolipoprotein A‐I [apoA‐I; human]) is in development to reduce the risk of early recurrent cardiovascular events post acute myocardial infarction (AMI) by rapidly removing plaque cholesterol.
CSL112 substantially elevates apoA‐I and cholesterol efflux capacity (CEC) post AMI; further analysis is needed to confirm the suitability of the dose for all patients.
What this study adds
Sex, body weight, AMI status and race had no clinically relevant effect on CSL112 dose/apoA‐I exposure and apoA‐I exposure/CEC relationships. The nonlinear exposure/CEC relationship supports the hypothesis that the fixed 6 g dose of CSL112 is an appropriate dose that may reduce the risk of early recurrent cardiovascular events.
1. INTRODUCTION
Apolipoprotein A‐I (apoA‐I) is the primary functional component of high‐density lipoprotein (HDL). Its key atheroprotective property is its ability to rapidly remove cholesterol from atherosclerotic plaque via cholesterol efflux. Cholesterol is then transported to the liver for excretion. 1 This process, known as reverse cholesterol transport, is the body's primary mechanism for clearing excess cholesterol that would otherwise accumulate in plaque. 2 One of the major mediators of cholesterol efflux from plaque cells is the ATP binding cassette transporter A1 (ABCA1), and lipid‐poor apoA‐I is the most efficient acceptor of cholesterol via ABCA1. 3
Following an acute myocardial infarction (AMI), cholesterol efflux capacity (CEC) is impaired. 4 , 5 , 6 Having a low CEC has been associated with an increased incidence of cardiovascular events, independently of traditional cardiovascular risk factors, 7 , 8 , 9 and with increased mortality following ST‐elevation myocardial infarction. 10 CSL112 (apoA‐I [human]), which is a novel intravenous formulation of human apoA‐I 11 that upregulates cholesterol CEC, 12 , 13 , 14 is in development for reducing the risk of early recurrent cardiovascular events following an AMI. Upregulating CEC post AMI is hypothesized to rapidly remove cholesterol from plaque, thus reducing the risk of a secondary cardiovascular event by stabilizing plaque. 15
Clinical safety, efficacy, pharmacokinetics (PK) and pharmacodynamics (PD) of CSL112 post single or multiple doses have been investigated in dose ranges of 5‐135 mg kg−1 and 1.7‐6.8 g in seven phase 1 and 2 clinical trials. 12 , 13 , 14 , 16 , 17 , 18 , 19 , 20 It was observed in these clinical trials that intravenous administration of CSL112 resulted in a rapid and approximately dose‐proportional enhancement in plasma apoA‐I exposure followed by a gradual decline back to baseline, with a mean half‐life of 46.4 and 53.9 h observed for 2 g and 6 g infusions of CSL112 in post‐AMI patients (manuscript in preparation). In addition, these trials have demonstrated that CSL112‐enhanced plasma apoA‐I concentration is associated with a rapid and substantial elevation in CEC, predominantly ABCA1‐dependent CEC. In addition, these studies have characterized the PK and PD of CSL112 in both healthy volunteers and subjects with cardiovascular disease (stable and post AMI), and have shown in noncompartmental analysis (NCA) that the PK and PD are largely unaffected by moderate renal impairment (RI). 18 , 19 , 20 The largest clinical trial with CSL112 conducted to date was a phase 2b, multicenter, randomized, placebo‐controlled dose‐ranging study (AEGIS‐I), which investigated the safety and tolerability of multiple doses (four weekly doses) of CSL112 (2 g or 6 g). AEGIS‐I supported the use of the 6 g dose in patients with a recent AMI, with administration of 6 g CSL112 resulting in 2.46‐ and 4.30‐fold elevations in total and ABCA1‐dependent CEC, respectively, 13 immediately after infusion.
As the first quantitative PKPD assessment in the integrated population including patients with AMI in CSL112 development, the objectives of this pharmacometric analysis were to (a) characterize the population PK and exposure‐response (exposure‐CEC) relationships of apoA‐I following CSL112 administration utilizing integrated data from seven phase 1 and 2 studies, and (b) evaluate the potential influence of covariates of interest on these relationships.
2. METHODS
This pharmacometric analysis comprised two parts: (a) population PK analysis to relate CSL112 doses to changes in apoA‐I plasma concentrations in the integrated population including healthy subjects and patients with AMI; and (b) exposure‐response analysis to characterize the relationship between apoA‐I exposure and associated changes in ABCA1‐dependent and total CEC. All seven phase 1 and 2 trials included in this analysis were conducted in accordance with the International Conference on Harmonization Good Clinical Practice guidelines, the Declaration of Helsinki (version of 1996) and local legal/regulatory requirements. Details of individual study methodology, including sampling and assay/reagent information, for those studies have been previously described. 12 , 13 , 14 , 16 , 19 , 20 In those studies, apoA‐I exposure, total CEC and ABCA1‐dependent CEC were measured at multiple time points up to 144 h following the start of a 2‐h infusion, using assays as previously described. 21 Briefly, an immunonephelometric assay was used to assess apoA‐I concentration by measuring turbidity caused by apolipoprotein‐antibody complexes. CEC was assessed by incubating serum for 4 hours with J774 macrophages preloaded with radiolabelled cholesterol with and without induction of ABCA1 levels by cyclic AMP. Assessment of CEC without cyclic AMP induction provides a value for ABCA1‐independent CEC, which is then subtracted from total CEC to calculate ABCA1‐dependent CEC. Liquid scintillation counting was used to quantify the efflux of radioactive cholesterol from the cells. The quantity of radioactive cholesterol incorporated into cellular lipids was calculated by means of isopropanol extraction of control wells not exposed to patient serum. The CECs were reported with a unit of %efflux/4 h. Brief descriptions of the sampling schedules for all studies included are provided in Supporting Information Table S1.
2.1. Clinical study data
PK and PD data, which were sourced from healthy subject with normal or impaired renal function, patients with cardiovascular disease (stable and post AMI), in all seven phase 1 and 2 clinical trials of CSL112, were pooled and used for this analysis (Supporting Information Table S1). As the phase 1/2a studies were intensively sampled and the AEGIS‐I study (AMI patients only) was sparsely sampled, the data from phase 1/2a studies were used to inform model development in this analysis. The demographic and clinical characteristic data from those trials were also included in this analysis for comprehensive evaluation of their effects on CSL112 PKPD. Subjects were included in the population PK analysis if they had at least one post‐dose measurement of apoA‐I. Exposure‐response relationships were assessed using data from four studies: study CSL112_1002, CSL112_1001, AEGIS‐I and CSL112_2001. Subjects were included in the exposure‐response analysis if they had at least one ABCA1‐dependent or total CEC measurement. For details see Supporting Information Methods.
2.2. Development of the population PK model
A nonlinear mixed effects modelling approach using NONMEM (version 7.3) 22 was used to construct the population PK model. All data preparation, summary statistics, graphics, exploratory analyses and post‐processing of NONMEM outputs were performed in R (version 3.2.2). 23 The previously published two‐compartment model 21 was used as the starting point for model building, as supported by graphical evaluations of apoA‐I PK profiles. 12 , 16 PK measurements collected post dose in treated subjects were the total of endogenous and exogenous (from CSL112 administration) apoA‐I plasma concentrations. In subjects assigned to placebo, PK measurements reflected only endogenous apoA‐I plasma concentrations. Because the time course of PK profiles in those seven studies was short (≤35 days), and endogenous apoA‐I secretion rate is stable and not closely associated with body weight, sex, HDL‐cholesterol (HDL‐C) and exercise, 24 , 25 , 26 endogenous apoA‐I concentrations were assumed to be stable over the study period and modelled using a baseline parameter. Differing from the previous model, 21 interoccasion variability (IOV), varying from week to week during the treatment period, was applied to apoA‐I baseline in the current model to reflect natural fluctuation in endogenous apoA‐I level over time. Interindividual variability (IIV), IOV on model parameters and the residual error model were examined in the PK model. The significances of covariates were evaluated using stepwise procedures with forward addition/backward deletion. The magnitude of individual covariate effects was simulated and graphically evaluated in a forest plot by comparing to predefined clinical relevance criteria (bioequivalence range 0.8‐1.25). The assessed IIV (percentage coefficient of variation, %CV) of apoA‐I level and CECs at baseline or post CSL112 administration within dose groups were typically in the range of 10‐30% across the completed clinical trials. 13 , 14 , 21 In fact, CSL112 was well tolerated in a wide dose range (single doses of up to 135 mg kg–1, 14 and multiple doses of up to 6.8 g 21 ) and an acceptable safety profile was observed across the tested dose range. 27 Therefore, if interindividual difference in apoA‐I exposure was within the range of −20% to +25%, or the magnitude of covariate effect was within the range of 80‐125%, the covariate was not considered clinically relevant. A nonparametric bootstrap and prediction‐corrected visual predictive check (VPC) were used to evaluate the stability, robustness and appropriateness of the final PK model. For details of the methodology see Supporting Information Methods.
2.3. Development of the exposure‐response model
Observed plasma apoA‐I concentrations vs CEC relationships were first explored graphically by subpopulation, with total and ABCA1‐dependent efflux evaluated separately. ABCA1‐dependent efflux represents a subfraction of total efflux. Cholesterol efflux following CSL112 administration is predominantly ABCA1‐dependent, 13 , 14 , 21 which due to the upregulation of ABCA1 on atherosclerotic plaque may result in preferential removal of plaque cholesterol. The PKPD model for ABCA1‐dependent and total CEC were developed sequentially using NONMEM. A sigmoid E max model described the relationship between predicted individual apoA‐I exposure (IPRED in the PK model output) and ABCA1‐dependent CEC. For total CEC, a model combining sigmoid E max and linear functions was used to characterize the relationship in which sigmoid function parameters (maximum effect [E max], concentration at half maximum effect [EC50], Gamma) were fixed to the individual estimates from the ABCA1‐dependent CEC model. The functional forms of the exposure‐response relationships in the models are:
| (1) |
| (2) |
where Conc. represents PK model‐estimated individual apoA‐I concentration (IPRED), E max represents maximum effect, EC50 represents the concentration of apoA‐I at half‐maximal response, Gamma is the Hill coefficient, and slope and intercept are the linear function parameters to describe the part of ABCA1‐independent CEC in total CEC. The values of E max, EC50 and Gamma in the total CEC model were fixed to the individual estimates obtained from the ABCA1‐dependent CEC model.
A covariate analysis was conducted to evaluate predictors of CEC. As the covariate analysis was mainly intended to detect potential clinically relevant effects (outside of the range of 0.8‐1.25) to identify opportunities for dose adjustments, covariate effects on CEC elevation were then evaluated using simulations in response to different increment in apoA‐I exposure, which is equal to 50‐125% of a typical subject's baseline. For details of the methodology see Supporting Information Methods.
2.4. Simulations of apoA‐I exposure and CECs within 48 hours post first dose
It was observed that apoA‐I and CEC increase immediately and then gradually decline after CSL112 administration. ApoA‐I returns to baseline levels within 48‐96 hours, depending on the dose and study population. 12 , 13 , 14 , 16 , 19 , 20 , 28 As such, elevations in apoA‐I level and ABCA1‐dependent CEC were sustained for at least 48 hours in post‐AMI patients following a 6 g dose. 28 In addition, elevations in apoA‐I and CEC were similar after the first and fourth CSL112 doses in post‐AMI patients. 28 Suppression of CECs post AMI, followed by a gradual return back to baseline level, has been reported. 4 , 6 Since the risk of major adverse cardiac events (MACE) remains elevated soon after a coronary event, it is critical for those AMI patients to receive apoA‐I treatment as early as possible post AMI. Therefore, exposure and CECs post first dose were analysed due to an interest in assessing the rapid increase immediately post AMI, when CEC has been shown to be suppressed and the MACE risk is highest. Baseline‐corrected apoA‐I AUC0‐48 represents the overall elevation in apoA‐I level over 48 hours. AUCE0‐48/48 h represents the time‐averaged elevation in CEC over 48 hours, which can more reasonably reflect overall elevation in CEC response over 48 hours, in comparison with maximum CEC elevation. In the current analysis, individual empirical Bayesian estimates from the final PKPD models were utilized to obtain baseline‐corrected individual apoA‐I AUC0‐48 and time‐averaged total and ABCA1‐dependent CEC (AUEC0‐48/48 h) after the first dose. These relationships were then examined by sex, race and AMI status. The relationship of baseline‐corrected exposures (AUC0‐48) and time‐averaged CEC (AUEC0‐48/48 h) were also examined by linear or nonlinear models. Consequently, the best model was used to simulate baseline‐corrected apoA‐I AUC0‐48 and time‐averaged AUEC0‐48 values after the first dose in 1000 virtual subjects. The simulated median and 90% prediction interval were overlaid on the individual exposure‐response data and the results were shown graphically to evaluate dose selection in patients with AMI. For details of methodology see Supporting Information Materials.
2.5. Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY, and are permanently archived in the Concise Guide to PHARMACOLOGY 2019/20. 29
3. RESULTS
3.1. Study population and analysis datasets
Study sampling information and subject characteristics are presented in Table 1. The majority of subjects were male (76%), with a median age of 57 years and mean body weight of 83 kg. The majority (94%) of subjects were Caucasian; 2% were Japanese, all of whom were enrolled in the CSL112_1002 study. The population PK data set included a total of 10 108 observed apoA‐I plasma concentrations from 1536 subjects. Of these, 488 subjects received placebo, accounting for 2971 apoA‐I plasma concentrations. The maximum dose levels in the PK dataset were 6 g in AMI subject (AEGIS‐I study), 6.8 g in patients with stable atherosclerotic disease (study 12‐70) and 135 mg kg–1 in healthy subjects (study 09‐63), respectively. For the PKPD (exposure‐response) dataset, there were 6522 total CEC and 6409 ABCA1‐dependent CEC measurements from 1395 subjects. The dose range of the PKPD dataset was up to 6 g.
TABLE 1.
Study details and subject characteristics
| Study | |||||||
|---|---|---|---|---|---|---|---|
| CSL112_1001 | CSL112_1002 | CSLCT‐HDL‐09‐63 | CSLCT‐HDL‐10‐68 | CSLCT‐HDL‐12‐70 | CSL112_2001 | AEGIS‐I | |
| Population | Healthy/RI | Healthy | Healthy | Healthy | Atherosclerosis | AMI and RI | AMI |
| Subjects, n | 32 | 52 | 57 | 36 | 44 | 80 | 1235 |
| ApoA‐I measurements, n | 416 | 724 | 1015 | 1161 | 657 | 360 | 5775 |
| Total CEC measurements, n | 223 | 466 | 0 | 0 | 0 | 293 | 5540 |
| ABCA1‐dependent CEC measurements, n | 218 | 431 | 0 | 0 | 0 | 293 | 5467 |
| Dosing schedule | SD | SD | SD | QW x 4 or BIW x 4 (3.4 g); QW X 4 (6.8 g) | SD | QW × 4 | QW × 4 |
| Dose | Placebo, CSL112 2 and 6 g | Placebo, CSL112 2, 4 and 6 g | Placebo, CSL112 5, 15, 40, 70, 105 and 135 mg kg−1 | Placebo, CSL112 3.4 and 6.8 g | Placebo, CSL112 1.7, 3.4 and 6.8 g | Placebo, CSL112 6 g | Placebo, CSL112 2 and 6 g |
| Male sex, n (%) | 22 (69) | 23 (44) | 36 (63) | 23 (64) | 32 (73) | 54 (68) | 981 (79) |
| Race, n (%) | |||||||
| Caucasian | 32 (100) | 18 (35) | 54 (95) | 35 (97) | 35 (80) | 78 (98) | 1,197 (97) |
| Japanese | 0 (0) | 34 (65) | (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| Other | 0 (0) | 0 (0) | 3 (5) | 1 (3) | 9 (20) | 2 (2) | 38 (3) |
| Age, years | 62 (33, 81) | 27 (20, 40) | 23 (18, 52) | 22 (19, 45) | 60 (40, 77) | 73 (36, 89) | 58 (30, 89) |
| Weight, kg | 83 (51, 107) | 62 (49, 73) | 81 (51, 150) | 72 (56, 106) | 86 (53, 145) | 85 (54, 170) | 84 (50, 183) |
| Baseline CrCL, mL min−1 | 78 (31, 161) | 116 (84, 170) | 139 (84, 344) | 128 (80, 165) | 86 (43, 249) | 54 (24, 143) | 103 (18, 290) |
| Baseline apoA‐I, mg dL−1 | 139 (103, 185) | 141 (98, 189) | 119 (82, 168) | 121 (83, 194) | 127 (77, 179) | 110 (74, 183) | 124 (45, 345) |
| Baseline ALT | 18 (7, 41) | 16.5 (5, 29) | 22 (11, 45) | 13 (6, 35) | 20 (7, 61) | 29 (7, 167) | 19 (6, 86) |
| Baseline AST | 21 (15, 30) | 17 (11, 27) | 20 (13, 48) | 18 (8, 27) | 20 (9, 68) | 28 (10, 266) | 25 (10, 132) |
Values are median (minimum, maximum) unless otherwise stated. Abbreviations: ABCA1, ATP‐binding cassette transporter‐1; ALT, alanine aminotransferase; AMI, acute myocardial infarction; apoA‐I, apolipoprotein A‐I; AST, aspartate aminotransferase; BIW × 4, twice‐weekly dose for eight doses; CEC, cholesterol efflux capacity; CrCL, creatinine clearance; QW × 4, weekly dose for four doses; RI, renal impairment; SD, single dose.
3.2. Population PK model
The final PK parameter estimates for fixed‐effect, the random‐effect parameters and the error model parameters together with the results of the nonparametric bootstrap are presented in Table 2. IIV was estimated on all model parameters except Vp and Q as they could not be estimated precisely (large relative standard error, %RSE). The %RSE of the estimated parameters and IIVs were less than 20%, indicating all parameters were precisely estimated. In addition, IOV on baseline apoA‐I (BL), where each week of the first 4 weeks and the time after the fourth week post BL measurement corresponded to the five occasions, respectively, was implemented in the final model to reduce OFV (∆OFV ~ −970) and %RSE. Residual variability was best described by a proportional error model. There were no significant correlations among PK parameters. The condition number was 28.2, which indicated that the model was not overparameterized and that there was no evidence of collinearity. Shrinkage in CL and Vc was approximately 40%, reflecting few observations per subject in the large phase 2 study (AEGIS‐I). Nevertheless, goodness‐of‐fit plots showed that the model adequately described the data over the tested dose range (Supporting Information Figure S1).
TABLE 2.
Summary of pharmacokinetic (PK) parameter estimates obtained from the final apoA‐I population PK model
| Model | Nonparametric bootstrap (300 replicates) | |||
|---|---|---|---|---|
| Parameter | estimate (%RSE) | Median | 95% CI (lower limit) | 95% CI (upper limit) |
| Fixed effect | ||||
| CL (L h−1) | 0.11 (5.2) | 0.11 | 0.10 | 0.13 |
| Vc (L) | 4.48 (2.2) | 4.86 | 4.65 | 5.09 |
| Q (L h−1) | 0.25 (7.9) | 0.25 | 0.22 | 0.29 |
| Vp (L) | 2.47 (6.6) | 2.53 | 2.22 | 2.90 |
| BL (mg L−1) | 1219 (0.01) | 1216 | 1196 | 1231 |
| AMI on CL | 0.92 (10.8) | 0.87 | 0.72 | 1.08 |
| WT on CL | 1.28 (12.9) | 1.25 | 0.73 | 1.65 |
| WT on Vc | 0.53 (12.2) | 0.54 | 0.41 | 0.65 |
| Total dose≥ 6 g on Vc | 0.82 (2.5) | 0.81 | 0.77 | 0.86 |
| WT on BL | −0.12 (19.6) | −0.12 | −0.17 | −0.08 |
| Female on BL | 1.08 (1.2) | 1.08 | 1.06 | 1.10 |
| Japanese on BL | 1.12 (2.3) | 1.13 | 1.08 | 1.18 |
| Study 1001 on BL | 1.16 (2.3) | 1.17 | 1.12 | 1.22 |
| Random effect | ||||
| IIV on CL (%) | 60.9 (19.8) | 64.0 | 48.1 | 79.1 |
| IIV on Vc (%) | 28.2 (18.9) | 27.9 | 22.1 | 34.3 |
| IIV on BL (%) | 14.6 (14.6) | 14.6 | 13.7 | 16.0 |
| IOV on BL (%) | 11.3 (4.7) | 10.8 | 8.7 | 14.0 |
| Residual error (%) | ||||
| Proportional: AEGIS‐I study | 7.9 (4.5) | 7.9 | 7.3 | 8.5 |
| Proportional: other studies | 5.5 (3.9) | 5.5 | 5.2 | 6.0 |
| η‐shrinkage (%) | ||||
| CL | 45.8 | |||
| Vc | 36.2 | |||
| BL | 18.4 | |||
A prediction‐corrected VPC of the final PK model was performed separately by placebo, single‐dose and multiple‐dose groups (Figure 1) and also by healthy subjects and patients (Supporting Information Figure S2). It is shown that the model adequately captured the central tendency as well as the variability in apoA‐I plasma concentrations over time. In addition, the model captured important features of the data, including an immediate, substantial elevation in apoA‐I plasma concentration following CSL112 administration relative to concentrations in placebo subjects and a slight cumulative increase in apoA‐I following repeated administration (Figure 1).
FIGURE 1.
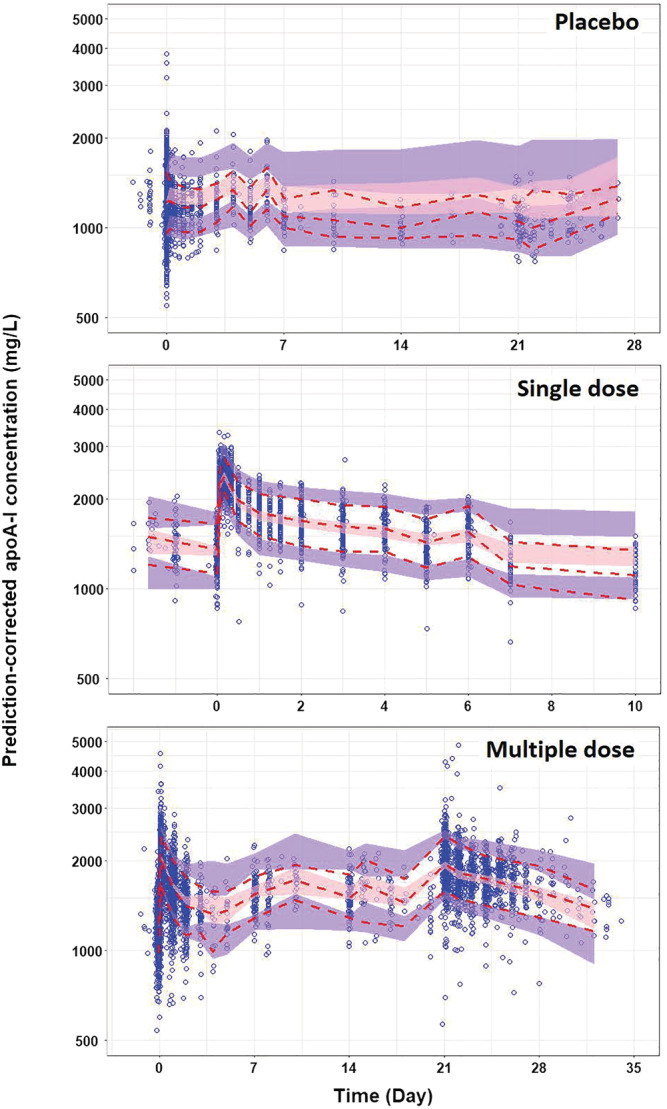
Prediction‐corrected visual predictive check (VPC) obtained through n = 1000 simulations from the final population PK model by regimen: placebo, single doses and multiple doses (QW × 4) of CSL112, and by all subjects, healthy subjects and patients. The blue circles represent the prediction‐corrected observed apoA‐I concentrations. The solid and dashed red lines represent the observed median and 10th‐90th percentile range, respectively. The pink and purple bands represent the 95% confidence interval (CI) of the median prediction, and the 10th and 90th prediction interval, respectively. ApoA‐I, apolipoprotein A‐I; QW × 4, weekly dose for 4 weeks
3.3. CSL112 administration leads to consistent apoA‐I exposure irrespective of weight, race (Japanese or non‐Japanese), sex or AMI status
The covariate analyses revealed several statistically significant relationships with apoA‐I exposure applying power functions in covariate models. Body weight and AMI status were significant covariates on CL, with coefficients of 1.28 and 0.92, respectively. Body weight and dose ≥6 g were significant covariates on Vc, with coefficients of 0.53 and 0.82, respectively. In addition, significant covariates on BL included body weight, Japanese race, sex and study 1001, with coefficients of −0.12, 1.12, 1.08 and 1.16, respectively. As expected, Vc and CL increased with body weight; in line with the latter finding, BL decreased with body weight. There was a relationship between dose and Vc such that total doses of CSL112 over 6 g were associated with lower Vc. CL was 8% lower in subjects who had experienced an AMI compared with healthy subjects and patients with stable atherosclerosis, and there was no overall influence of Japanese race or RI found on apoA‐I PK. However, BL was estimated to be 12% higher in Japanese subjects than non‐Japanese subjects and 8% higher in females than males. In addition, BL was about 16% higher in the CSL112_1001 study population (healthy subjects with normal renal function or moderate RI) compared to other studies, apparently due to unknown interstudy variation.
Although these covariate effects on apoA‐I PK were identified as statistically significant effects, none substantially reduced IIV in apoA‐I PK and all covariates had minimal impact on apoA‐I exposure at steady state (Supporting Information Figure S3). Therefore, they were not deemed to be clinically impactful. This was further demonstrated by examining model‐predicted individual AUC0‐48 after the fourth weekly dose of CSL112 or placebo by race (in healthy subjects) and sex (in subjects with AMI) (Figure 2). There was no significant difference (P > .05) in estimates of AUC0‐48 between healthy Japanese and non‐Japanese subjects (Figure 2A), confirming no clinically relevant impact of race on apoA‐I exposure after multiple doses. In AMI patients (Figure 2B), the median AUC0‐48 in females was 11.1%, 13.0% and 16.5% higher than males in the placebo, 2 g and 6 g groups, respectively (t‐test, P < .01). This is likely due to the higher observed baseline apoA‐I and lower body weight in females in the AEGIS‐I trial. Nevertheless, there was less than a 20% difference in AUC0‐48 between males and females, which suggests that the sex of AMI patients does not have a clinically meaningful effect on apoA‐I exposure after multiple administrations of CSL112.
FIGURE 2.

Box plots of model‐estimated individual apoA‐I exposures (AUC0‐48) following the fourth infusion by dose and race in healthy subjects (A) and by dose and sex in post‐AMI subjects (B), and the median value of AUC0‐48 from each group of post‐AMI subjects (B). Circles denote outlying data. AMI, acute myocardial infarction; AUC0‐48, area under the curve from 0 to 48 hours; Jp, Japanese; M, male; F, female
3.4. Exposure‐response models
Scatter plots of observed total/ABCA1‐dependent CEC against observed apoA‐I concentration by race, AMI and sex are shown in Figure 3. The plots suggested a positive correlation between apoA‐I exposure and CEC that was generally consistent irrespective of covariate. However, ABCA1‐dependent CEC vs apoA‐I concentrations in study CSL112_1002, which included predominantly Japanese subjects, were generally clustered at the lower end of the range (Figure 3B).
FIGURE 3.

Scatter plots of observed apoA‐I concentrations vs observed total and ABCA1‐dependent CEC by race (Japanese and non‐Japanese) (A, B), AMI status (C, D), sex (E, F) and weight (G, H). ApoA‐I, apolipoprotein A‐I; ABCA1, ATP‐binding cassette transporter‐1; AMI, acute myocardial infarction; CEC, cholesterol efflux capacity
Parameter estimates and results of the nonparametric bootstrap for the final exposure‐response models are shown in Table 3. A sigmoidal function, with parameters, E max, EC50 and Gamma, described the relationship between apoA‐I concentrations and ABCA1‐dependent CEC. A combined sigmoid E max and linear function, with intercept and slope parameters, described the relationship between apoA‐I concentrations and total CEC and IIV, was included on all parameters except for intercept due to large (>50%) RSE. Significant covariates on EC50 were AMI, body weight and sex. In addition, AMI was identified as significant covariate on Gamma and slope. Although initial graphical parameter‐covariate evaluations (not shown) revealed effects of Japanese race on PD parameters, once AMI and sex were included in the PD models, Japanese race was no longer significant. Goodness‐of‐fit plots (Supporting Information Figure S4) and prediction‐corrected VPC (Figure 4) suggested that the models reasonably described the data for both the total and ABCA1‐dependent CEC analyses.
TABLE 3.
Summary of exposure‐response parameter estimates for the final total and ABCA1‐dependent CEC
| Parameter | Model | Nonparametric bootstrap (300 replicates) | ||
|---|---|---|---|---|
| estimate (%RSE) | Median | 95% CI (lower limit) | 95% CI (upper limit) | |
| Fixed effect | ||||
| E max (%efflux/4 h) | 11.25 (6.3) | 11.24 | 10.60 | 11.76 |
| EC50 (mg L−1) | 2023 (1.1) | 2024 | 1926 | 2131 |
| Gamma | 6.2 (7.9) | 6.12 | 4.81 | 7.70 |
| Slope (%efflux/4 h/(mg L−1)) | 0.0049 (2.6) | 0.0049 | 0.0047 | 0.0051 |
| Intercept (%efflux/4 h) | 0.52 (18.8) | 0.50 | 0.39 | 0.64 |
| Box‐cox parameter for IIV of E max | −1.61 (27.5) | −1.62 | −4.23 | −0.20 |
| Box‐cox parameter for IIV of Gamma | −2.02 (17.8) | −1.97 | −3.39 | −0.76 |
| AMI on EC50 | 0.83 (10.8) | 0.82 | 0.78 | 0.87 |
| WT on EC50 | −0.18 (12.9) | −0.18 | −0.24 | −0.12 |
| Sex on EC50 | 1.09 (12.2) | 1.09 | 1.05 | 1.13 |
| AMI on Gamma | 0.67 (2.5) | 0.68 | 0.55 | 0.85 |
| AMI on slope | 0.90 (2.3) | 0.89 | 0.86 | 0.92 |
| Random effect | ||||
| IIV on E max (%) | 22.3 (19.8) | 22.0 | 14.2 | 26.1 |
| IIV on EC50 (%) | 15.4 (18.9) | 15.5 | 13.6 | 17.7 |
| IIV on Gamma (%) | 34.6 (14.6) | 34.6 | 26.9 | 44.7 |
| IIV on slope (%) | 14.0 (4.7) | 14.0 | 12.9 | 14.9 |
| Residual error | ||||
| Additive (%efflux/4 h): ABCA1‐dependent efflux | 0.63 (3.2) | 0.62 | 0.52 | 0.75 |
| Proportional (%): ABCA1‐dependent efflux | 33.1 (1.5) | 33.0 | 29.9 | 35.7 |
| Proportional (%): total efflux | 16.7 (0.01) | 16.7 | 16.2 | 17.1 |
| η‐shrinkage (%) | ||||
| E max | 46.7 | |||
| EC50 | 28.5 | |||
| Gamma | 40.3 | |||
| Slope | 26.8 | |||
FIGURE 4.
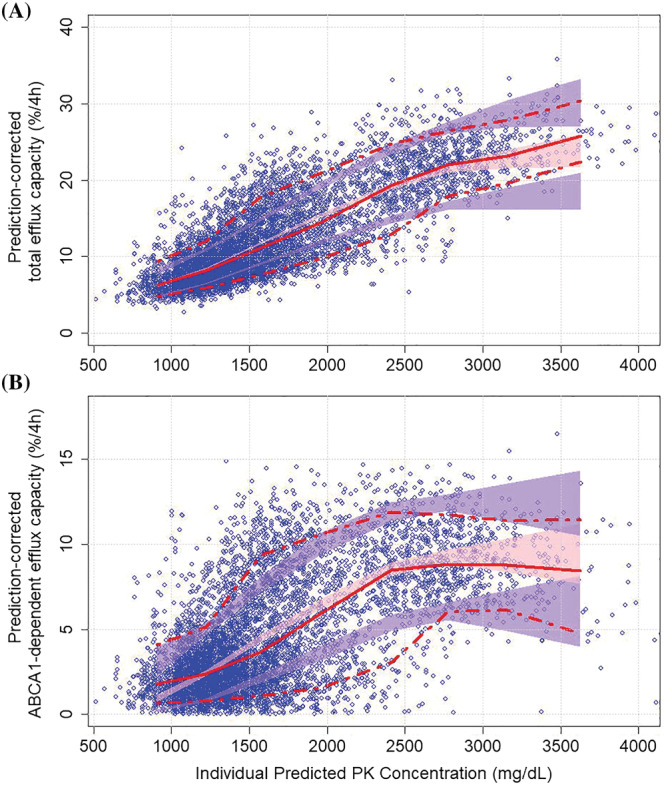
Prediction‐corrected visual predictive check (VPC) of the exposure‐response models for total CEC (A) and ABCA1‐dependent CEC (B). The blue circles represent the prediction‐corrected observed CECs. The solid and dashed red lines represent the observed median and 10th‐90th percentile range, respectively. The pink and purple bands represent the 95% confidence intervals of the 90th, median and 10th percentiles of the simulated data. ABCA1, ATP‐binding cassette transporter 1; CEC, cholesterol efflux capacity
3.5. Limited clinical relevance of covariate effects on CEC elevation
In the covariate analysis of exposure‐response models, no tested clinical factor was identified as a statistically significant covariate on E max in the ABCA1‐dependent CEC model, indicating the maximum response or elevation in ABCA1‐dependent CEC is consistent across subpopulations after CSL112 administration. Lower hill exponent (Gamma) in AMI subjects hints at a steeper exposure‐response relationship in non‐AMI subjects than AMI subjects, suggesting the maximum elevation in ABAC1‐dependent CEC could be reached at relatively moderate apoA‐I exposure in non‐AMI subjects. Interestingly, post‐AMI, high body weight or male subjects have lower potency parameter, EC50, suggesting relatively higher elevation or clinical response in ABCA1‐dependent CEC even with suboptimal elevation in apoA‐I exposure in those subpopulations. The linear function parameter, slope, in the total CEC model was slightly lower in subjects post AMI, suggesting the ABCA1‐independent CEC was suppressed post AMI. The clinical relevance of those statistically significant covariates was appraised by simulations based on the exposure‐response models. As PK simulations showed that apoA‐I exposure was elevated 50‐125% from baseline within 48 hours post 6 g CSL112 dose at steady state, effects of covariates on CEC were evaluated using graphical inspection at different apoA‐I plasma concentrations at 50‐125% increment from the PK model‐estimated typical baseline of 1219 mg L−1. The forest plots showing the magnitude of covariate effects on total and ABCA1‐dependent CEC are presented in Figure 5. Each of the predicted covariate effects fell within 80‐125% of the median prediction for a typical subject, indicating that none of those covariates were clinically relevant for total or ABCA1‐dependent CEC within 48 hours post CSL112 administration at steady state.
FIGURE 5.
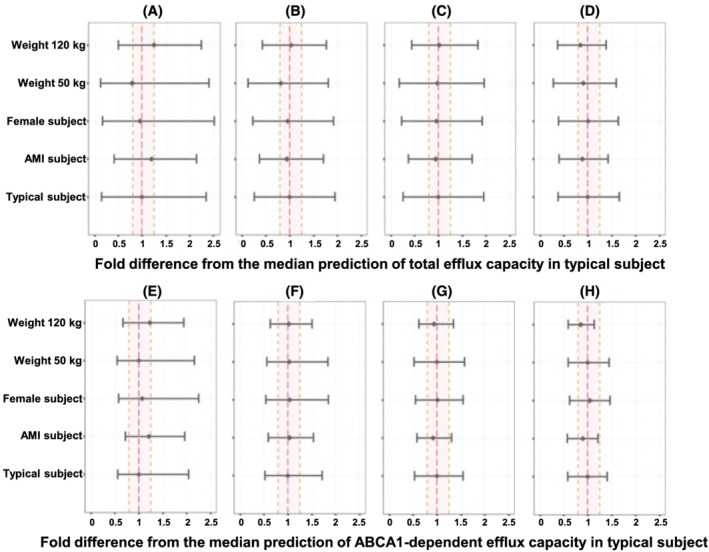
Forest plots of the model‐predicted covariate (AMI, sex and body weight) effects on total (A)‐(D) and ABCA1‐dependent CEC (E)‐(H) in response to different increment in apoA‐I concentration. Typical subjects were defined as non‐Japanese, healthy males with apoA‐I baseline concentration of 121.9 mg dL−1. Baseline‐corrected CECs in response to increment in apoA‐I concentration, which is equal to 50% (A, E), 75% (B, F), 100% (C, G) and 125% (D, H) of a typical subject's baseline, respectively, were simulated and summarized. The covariate effects were displayed as fold change from median predicted CEC in typical subject. For each covariate value, the horizontal line represents the 90% prediction interval for the respective ratio. The vertical dashed lines indicate the predefined clinical relevance criteria (0.80‐1.25, bioequivalence range). ApoA‐I, apolipoprotein A‐I; AMI, acute myocardial infarction; ABCA1, ATP‐binding cassette transporter‐1; CEC, cholesterol efflux capacity
ApoA‐I AUC0‐48 and time‐averaged total and ABCA1‐dependent CEC (AUEC0‐48/48 h) after the first CSL112 dose were estimated based on the individual post hoc parameter estimates from the exposure‐response models. These exposure‐response relationships over 48 hours post dose, stratified by body weight, race, AMI status and sex (not shown), are consistent with the positive correlations observed for the continuous CEC data (Figure 3) in all subpopulations. While Japanese and non‐AMI subjects generally showed smaller elevations in time‐averaged ABCA1‐dependent CEC compared to non‐Japanese and AMI subjects, respectively, the distribution of time‐averaged total CEC was similar between those subpopulations.
A nonlinear mode (power function) best described the relationship between model‐estimated baseline‐corrected individual apoA‐I exposure (AUC0‐48) and time‐averaged CEC (AUEC0‐48/48 h) for both total and ABCA1‐dependent CEC. As shown in Figure 6, baseline‐corrected individual apoA‐I AUC0‐48 and time‐averaged total and ABCA1‐dependent CEC in studies AEGIS‐I and CSL112_1002 were overlaid with the model‐predicted median and 90% prediction interval for 1000 virtual subjects. Individual predictions from AEGIS‐I and CSL112_1002 studies largely fell within the 90% prediction interval and were evenly distributed around the median curve of power model predictions, indicating that the power model well described the nonlinear relationship between apoA‐I exposure and time‐average CECs in the dose range up to 6 g. In comparison to placebo and 2 g dose, exposures associated with the 6 g dose resulted in CEC responses consistently above the median predicted CEC in the placebo group. There is a steep elevation in CEC in response to the low range of baseline‐corrected AUC0‐48; the dose of 6 g CSL112 is associated with a close to doubling of time‐averaged ABCA1‐dependent CEC (AUEC0‐48/48 h) over 48 hours. Although the curve of median prediction in Figure 6 does not display complete saturation, any further increasing apoA‐I exposure results in diminishing returns in cholesterol efflux effect. As the majority (~88%) of model‐estimated individual apoA‐I exposure and time‐averaged CEC in this exposure‐response analysis were from AMI subjects in studies AEGIS‐I, these exposure‐response simulations confirmed the CSL112 6 g dose provided a substantial CEC response in patients with AMI.
FIGURE 6.
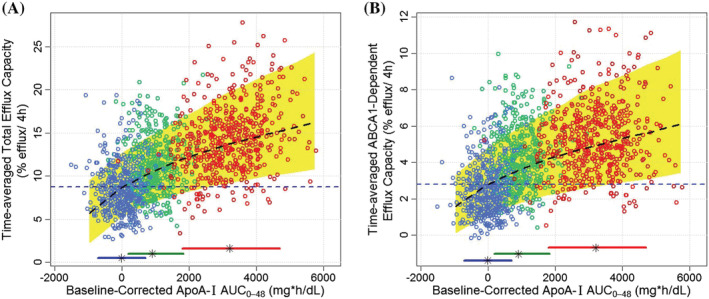
Model‐predicted individual time‐averaged total (A) and ABCA1‐dependent (B) CEC (AUC0‐48/48 h) vs baseline‐corrected apoA‐I exposure (AUC0‐48). The black lines and yellow shaded area represent median and 90% prediction interval, respectively, of power‐model predictions for 1000 virtual subjects. Blue, green and red circles represent individual model estimations for subjects assigned to placebo, 2 g and 6 g, respectively, in the AEGIS‐I and CSL112_1002 studies. Blue horizontal dash lines indicate the efflux capacity threshold, which is the median of predicted CEC in the placebo group. Horizontal bars and black star represent 90% CI and median of individual apoA‐I exposures for placebo and the CSL112 2 g and 6 g doses for subjects in both the AEGIS‐I and CSL112_1002 studies. ApoA‐I, apolipoprotein A‐I; ABCA1, ATP‐binding cassette transporter‐1; AUC0‐48, area under the curve from 0‐48 hours; CEC, cholesterol efflux capacity
4. DISCUSSION
Weekly dosing of CSL112 for 4 weeks was selected as the dosing regimen in CSL112's late phase development. The 1‐week interval between infusions was chosen to maximize continuous exposure to the drug substance, apoA‐I, above baseline levels and to minimize net accumulation of either apoA‐I or its phospholipid/sucrose excipients. The choice of four infusions is based on data demonstrating that the risk of recurrent AMI is highest during the first month post‐index event and that the risk declines after that period, hence the rationale for dosing CSL112 over the course of 1 month. Safety and tolerability assessments of CSL112 that analysed all seven completed clinical studies have shown that CSL112 was well‐tolerated and had an acceptable safety profile across all populations studied, including in the phase 2b study of patients with AMI, which is representative of the target population for the product. 27 , 30 No dose‐response relationship has been observed between the CSL112 dose and the frequency and intensity of treatment‐emergent adverse events (TEAEs) in clinical studies. 27 , 30 The current analysis is the first integrated analysis to quantitatively investigate the relationship between apoA‐I exposure and CEC response in an integrated population including patients with AMI, and to assess whether subpopulations differ in their apoA‐I and CEC response to CSL112.
A two‐compartment model described the PK data adequately, and standard diagnostic plots supported the validity of the population PK model and exposure‐response models. In line with previous reports, 14 , 18 , 21 there was a strong positive correlation between apoA‐I exposure and elevation in CEC, and the present study supports previous observations of a dose‐dependent relationship between these parameters. 18 , 21 Moreover, in the present analysis this relationship was found to be generally consistent across the range of subpopulations (including sex, race, RI and AMI status) tested. Using pooled data from seven phase 1 and 2 studies, the population PK analysis demonstrated that CSL112 consistently elevated apoA‐I levels well above those associated with placebo administration. In addition, based on data pertaining to repeated administration (four weekly doses) gathered predominantly from the AEGIS‐I study, a slight cumulative effect of repeated dosing on apoA‐I levels was confirmed by model predictions.
The population PK analysis revealed several statistically significant covariate effects, but none substantially reduced IIV in apoA‐I PK and all covariates had minimal impact on apoA‐I exposure. Consequently, none of the covariates evaluated were deemed to be clinically meaningful. Covariate analysis indicated that AMI status only had an effect on apoA‐I CL with 8% reduction in individuals with AMI, suggesting that the kinetics of endogenous and CSL112‐sourced apoA‐I was not significantly impacted by AMI status. The population PK analysis found Vc at doses ≥6 g to be slightly lower than that at low‐dose levels, and to be approximately equal to adult plasma volume. This might be the result of a slowing or saturation of the remodelling of CSL112, 11 a process which leads to lower molecular weight forms with higher ability to diffuse out of the bloodstream. Also of interest was that female and Japanese subjects generally had modestly higher baseline (endogenous) concentrations of apoA‐I, which is in line with what is known regarding endogenous HDL and apoA‐I levels in these subpopulations. 31 , 32 Sex differences in lipoprotein levels are well‐established and are thought to be driven by hormonal differences. 32 Data collected between 2007 and 2010 by the National Health and Nutrition Survey in the USA by SRL Inc. in Japan showed endogenous apoA‐I mean (standard deviation) concentrations were 134.9 (29.0) mg/dL in men compared with 151.0 (29.3) mg/dL in women. 31 These data generally align with the results of the present study as endogenous apoA‐I levels were found to be 8% lower in male than in female subjects. In terms of Japanese individuals, hyperalphalipoproteinaemia, which is thought to be largely driven by mutations in the cholesteryl ester transfer protein (CETP) gene, has been shown to be more prevalent in Japan 31 , 33 and this is thought to be a key reason for the higher levels of endogenous HDL observed in this population. 31 Nevertheless, despite statistically significant effects of sex and Japanese race in the population PK analysis, both covariate effects were predicted to have minimal impact on apoA‐I exposure. Furthermore, it should be noted that there was little impact on apoA‐I exposure at the extremes of body weight, supporting the validity of fixed dosing of CSL112 over weight‐based dosing. Four pathways of cellular cholesterol efflux have been identified: passive processes involving simple diffusion and facilitated diffusion (the latter by scavenger receptor class B, type I [SR‐BI]‐mediated pathway), and two active processes via ABCA1 and ABCG1. 34 The two passive efflux pathways and ABCG1‐mediated efflux were measured as ABCA1‐independent efflux in the CEC assay without cAMP induction, as ABCA1 expression in murine J774 macrophages is very low in basal conditions. 35 The total CEC was measured with ABCA1 induction by c‐AMP incubation, and the ABCA1‐dependent CEC was derived from the difference between total and ABAC1‐independent CEC. As ABCA1‐dependent efflux is an active process involving apoA‐I or small HDLs binding to ABCA1 transporter, a nonlinear and saturable relationship between preβ‐1 HDL concentration and ABCA1‐mediated efflux was reported in murine cAMP‐induced J774 macrophages, whereas SR‐BI and ABCG1 efflux show a linear relationship. 36 , 37 In agreement with those findings, our exposure‐response analysis identified that an E max model (shown in equation 1) best describes the nonlinear and saturable relationship between apoA‐I exposure and ABCA1‐dependent CEC in cAMP‐treated J774 macrophages. A linear model was used to describe ABCA1‐independent CEC as part of total CEC (shown in equation 2), measured as the sum of ABCA1‐dependent and independent CECs.
The exposure‐response analysis demonstrated that, in comparison to placebo and 2 g dose, exposures associated with the 6 g dose resulted in CEC responses consistently above the median predicted CEC in the placebo group, confirming robust clinical response at CSL112 dose level of 6 g. The covariate analysis demonstrated that post AMI, high body weight, male gender were predictors of greater elevation in ABCA1‐dependent CEC, especially at suboptimally elevated apoA‐I exposure (eg, 50% increment from apoA‐I baseline). It was found that ABCA1‐dependent CEC in subjects with high body weight are more sensitive (lower EC50) to serum apoA‐I elevation, although the maximum effect (E max) is independent of body weight. This phenomenon is probably associated with higher CEC response to the low apoA‐I baseline in subjects with higher body weight. Although the full explanation for differences in apoA‐I and HDL‐C levels in men vs women is not clear at this time, this covariate analysis still provided a suggestion that CSL112 might provide more beneficial effects on cholesterol homeostasis in male subjects.
In terms of the covariate effect in the total CEC model, less elevation in ABCA1‐independent CEC was found in AMI subjects than those subjects without AMI. Other clinical factors (Japanese race, female gender and low body weight) were not predicted to have any statistically significant effects on ABCA1‐independent CEC following CSL112 administration, despite the higher baseline apoA‐I concentrations observed in these subpopulations. However, the magnitude of covariate effects was within the threshold range of 80‐125% of the median prediction for a typical subject and therefore none of the statistically significant predictors of CEC were found to be clinically relevant at substantially elevated apoA‐I exposures. A limitation of the PKPD analysis was the lack of assessment of concomitant medications as covariates due to lack of data. Previous studies reported that CEC, and particularly ABCA1‐dependent CEC, is suppressed and recovers in the initial days and weeks following an AMI. 4 , 6 , 38 Published studies include a post hoc analysis of the dal‐ACUTE randomized controlled trial of the CETP inhibitor dalcetrapib; in the 150 patients who received placebo in dal‐ACUTE, ABCA1‐dependent CEC was observed to increase by approximately 15% from baseline to week 20 following an AMI. Although the current analysis demonstrated the magnitude of covariate effects on total and ABCA1‐dependent CEC are not clinically relevant (Figure 5), it was found that the occurrence of AMI is associated with slightly higher (~23% higher) elevations in ABCA1‐dependent CEC with 50% increase in apoA‐I exposure after CSL112 administration than those without an AMI (Figure 5A,E). This finding suggests a possible mechanism of recovery or even rebound in CEC following CEC suppression post AMI, although no CEC data prior to AMI occurrence were available in the present analysis to confirm CEC suppression. This is an important finding in the context of the mechanism of action of CSL112 as it is proposed that substantial increases in CEC in these patients will help to increase atherosclerotic plaque stability and prevent recurrent CV events in the days and weeks following an AMI.
Published animal and human studies have demonstrated that 2‐4‐fold elevation of apoA‐I or HDL‐C can produce reductions of plaque cholesterol by as much as 50% in intervals as short as 1 week. 11 , 39 , 40 , 41 , 42 The phase 2b study (AEGIS‐I study) investigated up to 6 g dosing (equal to ~70 mg kg‐1 in an 85 kg individual) in AMI patients, which is close to the saturating dose of ~120 mg kg‐1 for plaque removal in mice. 42 In addition, a 6 g dose allows for suitable safety margins for the excipients of phosphatidylcholine and sucrose. In the AEGIS‐I study, 2.06‐fold, 4.30‐fold and 2.45‐fold elevation in apoA‐I, ABCA1‐dependent CEC and total CEC were observed, respectively, immediately after infusion of CSL112 6 g, suggesting that CSL112 may increase not only the amount of circulating apoA‐I, but also the activity for ABCA1‐dependent efflux on a per‐apoA‐I basis. 13 The current exposure‐response analysis characterized the nonlinear relationship between model‐estimated baseline‐corrected aopA‐I AUC0‐48 and time‐averaged CEC (AUEC0‐48/48 h) by a power model, and model simulations showed a 6 g dose results in greater CEC elevation over the first 48 h post the first dose, compared to placebo and lower dose levels. There is the possibility of diminishing returns in terms of increasing the dose beyond 6 g, with respect to CEC elevation benefit vs safety risks. Therefore, a 6 g dose, which produces an immediate and profound enhancement of CEC, is hypothesized as an appropriate dose to potentially reduce the high rate of early recurrent events following a heart attack. The clinical efficacy of CSL112 6 g dose in AMI patients will thus be investigated in the phase 3 cardiovascular outcomes trial (AEGIS‐II, https://clinicaltrials.gov/ct2/show/NCT03473223). Exposure‐response relationships will be further examined using PK, PD and clinical efficacy data from the larger population following completion of the trial.
In conclusion, pharmacometric results indicated that (a) administration of 6 g CSL112, which was the highest dose tested in patients with AMI, markedly elevates CEC compared to placebo and lower dose levels, based on the modelled quantitative exposure‐response relationship; and (b) effects of tested covariates on apoA‐I exposure or CEC are not expected to be clinically relevant, suggesting no dose adjustment is needed. This analysis provided a quantitative rationale for investigating a 6 g fixed dose of CSL112 in the ongoing phase 3 cardiovascular outcomes trial (AEGIS‐II). These findings are consistent with the results of previous analyses and support the proposed dose strategy for future clinical development of CSL112 in patients with AMI.
ACKNOWLEDGEMENTS
Certara were involved in the design of the analysis presented here and were funded by CSL Behring to conduct the analysis. Medical writing assistance was provided by Steven Foster of Meridian HealthComms Ltd, Plumley, UK, in accordance with Good Publication Practice (GPP3) guidelines, funded by CSL Behring.
COMPETING INTERESTS
B.Z., D.D., P.T., B.K., S.W., A.G. and M.T. are employees of CSL Behring. H.K. and M.P. are employees of Certara.
CONTRIBUTORS
B.Z., M.T., H.K. and M.P. conceived the study, and B.Z. and H.K. conducted the analyses presented here. D.D. and P.T. led clinical trials of CSL112. S.W. and A.G conceived and lead execution of the pharmacodynamics assays. All authors contributed to the writing of the manuscript, approved the manuscript for submission and agree to be accountable for all aspects of the work.
Supporting information
TABLE S1 Summary of studies included in the population PK analysis
TABLE S2 Covariate‐parameter relationships evaluated in the population PK and PKPD analysis
FIGURE S1 Goodness of fit of the final population PK model: observed data vs. population predicted (A) and individual predicted (B) plasma apoA‐I concentrations; CWRES vs. time (C) and population predicted plasma apoA‐I concentration (D). The grey line represents a smoothing function and the dashed black line represents unity. apoA‐I, apolipoprotein A‐I; CWRES, conditional weighted results; PK, pharmacokinetic
FIGURE S2 Prediction‐corrected visual predictive check (VPC) obtained through n=1000 simulations from the final population PK model by regimen: placebo, single doses of CSL112 and multiple doses (QWx4) of CSL112, and by all subjects, healthy subjects and patients. The blue circles represent the prediction‐corrected observed apoA‐I concentrations. The solid and dashed red lines represent the observed median and 10th–90th percentile range, respectively. The pink and purple bands represent the 95% confidence interval (CI) of the median prediction, the 10 and 90th prediction interval, respectively. As shown in Supplementary Table 1, healthy subjects were from study 09‐63, 10‐68, 1001, 1002; patients were from study 12‐70, 2002, AEGIS‐I. QWx4, weekly dose for 4 weeks. ApoA‐I, apolipoprotein A‐I; PK, pharmacokinetic.
FIGURE S3 Forest plots of predicted covariate effects from the final population PK model on apoA‐I baseline (A) and exposures (area under the curve at steady state, AUCtau.ss [B] and concentration at end of infusion, C max.ss [C]) following CSL112 administration. Typical subjects were defined as 84 kg, non‐Japanese, healthy males. All ratios displayed use the respective exposure metric for typical subjects as the reference. For each covariate value, the horizontal line represents the 90% prediction interval for the respective ratio. The vertical dashed lines and pink bands indicate traditional thresholds of bioequivalence (0.80 to 1.25). AMI, acute myocardial infarction; apoA‐I, apolipoprotein A‐I; AUCtau.ss, area under the curve at steady state; C max, maximum concentration; PK, pharmacokinetic
FIGURE S4 Goodness of fit of the final exposure‐response model for total CEC (A)‐(D) and ABCA1‐dependent CEC (E)‐(H): observed CEC vs. population predicted (A, E) and individual predicted (B, F) plasma apoA‐I concentrations; CWRES vs. time (C, G) and population predicted plasma apoA‐I concentration (D, H). The dashed red line represents unity. ABCA1, ATP‐binding cassette transporter‐1; apoA‐I, apolipoprotein A‐I; CEC, cholesterol efflux capacity; CWRES, conditional weighted results
FIGURE S5 Workflow of population PK and PKPD model development ABCA1, ATP‐binding cassette transporter A1; AUC0‐48, area under the curve from 0 to 48 hours; AUEC0‐48, area under the effect curve from 0 to 48 hours; CEC, cholesterol efflux capacity; PD, pharmacodymanic; PK, pharmacokinetic
Zheng B, Duffy D, Tricoci P, et al. Pharmacometric analyses to characterize the effect of CSL112 on apolipoprotein A‐I and cholesterol efflux capacity in acute myocardial infarction patients. Br J Clin Pharmacol. 2021;87:2558–2571. 10.1111/bcp.14666
Trial registration numbers: ClinicalTrials.gov: NCT01129661, NCT01281774, NCT02427035, NCT01499420, NCT02742103, NCT02108262; Australian New Zealand Clinical Trials Registry: ACTRN12618000805279.
The Principal Investigators for the studies were Dr Sepehr Shakib (CSLCT‐HDL‐09‐63), Dr Joanne Marjason (CSLCT‐HDL‐10‐68), Professor Timothy Mant (CSL112_1001), Dr James Kuo (CSL112_1002), Dr Pierluigi Tricoci (CSLCT‐HDL‐10‐70) and Dr C. Michael Gibson (CSL112_2001 and AEGIS‐I). The authors confirm that the Principal Investigators had direct responsibility for the conduct of their respective trials.
[Correction added on 23 January 2021, after first online publication: The copyright line was changed.]
DATA AVAILABILITY STATEMENT
CSL Behring will only consider requests to share Individual Patient Data (IPD) that are received from systematic review groups or bona fide researchers. CSL Behring will not process or act on IPD requests until 12 months after article publication on a public website. An IPD request will not be considered by CSL Behring unless the proposed research question seeks to answer a significant and unknown medical science or patient care question. Applicable country‐specific privacy and other laws and regulations will be considered and may prevent sharing of IPD. Requests for use of the IPD will be reviewed by an internal CSL Behring review committee. If the request is approved, and the researcher agrees to the applicable terms and conditions in a data‐sharing agreement, IPD that has been appropriately anonymized will be made available. Supporting documents including study protocol and the Statistical Analysis Plan will also be provided. For information on the process and requirements for submitting a voluntary data sharing request for IPD, please contact CSL Behring at clinicaltrials@cslbehring.com.
REFERENCES
- 1. Tall AR. An overview of reverse cholesterol transport. Eur Heart J. 1998;19(Suppl A):A31‐A35. [PubMed] [Google Scholar]
- 2. Tabas I. Consequences of cellular cholesterol accumulation: basic concepts and physiological implications. J Clin Invest. 2002;110(7):905‐911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Du XM, Kim MJ, Hou L, et al. HDL particle size is a critical determinant of ABCA1‐mediated macrophage cellular cholesterol export. Circ Res. 2015;116(7):1133‐1142. [DOI] [PubMed] [Google Scholar]
- 4. Ray KK, Ditmarsch M, Kallend D, et al. The effect of cholesteryl ester transfer protein inhibition on lipids, lipoproteins, and markers of HDL function after an acute coronary syndrome: the dal‐ACUTE randomized trial. Eur Heart J. 2014;35(27):1792‐1800. [DOI] [PubMed] [Google Scholar]
- 5. Soares AAS, Tavoni TM, de Faria EC, Remalay AT, Maranhao RC, Sposito AC. HDL acceptor capacities for cholesterol efflux from macrophages and lipid transfer are both acutely reduced after myocardial infarction. Clin Chim Acta. 2018;478:51‐56. [DOI] [PubMed] [Google Scholar]
- 6. Annema W, Willemsen HM, de Boer JF, et al. HDL function is impaired in acute myocardial infarction independent of plasma HDL cholesterol levels. J Clin Lipidol. 2016;10(6):1318‐1328. [DOI] [PubMed] [Google Scholar]
- 7. Rohatgi A, Khera A, Berry JD, et al. HDL cholesterol efflux capacity and incident cardiovascular events. N Engl J Med. 2014;371(25):2383‐2393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Khera AV, Cuchel M, de la Llera‐Moya M, et al. Cholesterol efflux capacity, high‐density lipoprotein function, and atherosclerosis. N Engl J Med. 2011;364(2):127‐135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Liu C, Zhang Y, Ding D, et al. Cholesterol efflux capacity is an independent predictor of all‐cause and cardiovascular mortality in patients with coronary artery disease: a prospective cohort study. Atherosclerosis. 2016;249:116‐124. [DOI] [PubMed] [Google Scholar]
- 10. Guerin M, Silvain J, Gall J, et al. Association of serum cholesterol efflux capacity with mortality in patients with ST‐segment elevation myocardial infarction. J Am Coll Cardiol. 2018;72:3259‐3269. [DOI] [PubMed] [Google Scholar]
- 11. Diditchenko S, Gille A, Pragst I, et al. Novel formulation of a reconstituted high‐density lipoprotein (CSL112) dramatically enhances ABCA1‐dependent cholesterol efflux. Arterioscler Thromb Vasc Biol. 2013;33:2202‐2211. [DOI] [PubMed] [Google Scholar]
- 12. Tricoci P, D'Andrea DM, Gurbel PA, et al. Infusion of reconstituted high‐density lipoprotein, CSL112, in patients with atherosclerosis: safety and pharmacokinetic results from a phase 2a randomized clinical trial. J Am Heart Assoc. 2015;4:e002171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gibson MC, Korjian S, Tricoci P, et al. Safety and tolerability of CSL112, a reconstituted, infusible, plasma‐derived apolipoprotein A‐I, after acute myocardial infarction: the AEGIS‐I trial (apoA‐I event reducing in ischemic syndromes I). Circulation. 2016;134:1918‐1930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gille A, Easton R, D'Andrea D, Wright SD, Shear CL. CSL112 enhances biomarkers of reverse cholesterol transport after single and multiple infusions in healthy subjects. Arterioscler Thromb Vasc Biol. 2014;34:2106‐2114. [DOI] [PubMed] [Google Scholar]
- 15. Silvain J, Kerneis M, Guerin M, Montalescot G. Modulation of cholesterol efflux capacity in patients with myocardial infarction. Curr Opin Cardiol. 2019;34(6):714‐720. [DOI] [PubMed] [Google Scholar]
- 16. Easton R, Gille A, D'Andrea D, Davis R, Wright SD, Shear C. A multiple ascending dose study of CSL112, an infused formulation of ApoA‐I. J Clin Pharmacol. 2014;54:301‐310. [DOI] [PubMed] [Google Scholar]
- 17. Australian New Zealand Clinical Trials Registry (ANZCTR) . A phase 1, randomized, double‐blind, placebo‐controlled study to evaluate the pharmacokinetics, safety, and tolerability of CSL112 in healthy Japanese and Caucasian subjects. Available at: https://www.anzctr.org.au/Trial/Registration/TrialReview.aspx?id=374880&isReview=true. Date last updated: November 22 2018. Date last accessed: 08 May 2019.
- 18. Gille A, Duffy D, Tortorici MA, Wright SD, Deckelbaum LI, D'Andrea DM. Moderate renal impairment does not impact the ability of CSL112 (apolipoprotein A‐I [human]) to enhance cholesterol efflux capacity. J Clin Pharmacol. 2019;59:427‐436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tortorici MA, Duffy D, Evans R, et al. Pharmacokinetics and safety of CSL112 (apolipoprotein A‐I [human]) in adults with moderate renal impairment and normal renal function. Clin Pharmacol Drug Dev. 2018;8(5):628‐636. 10.1002/cpdd.618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gibson CM, Kerneis M, Yee MK, et al. The CSL112‐2001 trial: safety and tolerability of multiple doses of CSL112 (apolipoprotein A‐I [human]), an intravenous formulation of plasma‐derived apolipoprotein A‐I, among subjects with moderate renal impairment after acute myocardial infarction. Am Heart J. 2019;208:81‐90. [DOI] [PubMed] [Google Scholar]
- 21. Gille A, D'Andrea D, Tortorici MA, Hartel G, Wright SD. CSL112 (Apolipoprotein A‐I [human]) enhances cholesterol efflux similarly in healthy individuals and stable atherosclerotic disease patients. Arterioscler Thromb Vasc Biol. 2018;38:953‐963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Beal SL, Sheiner LB, Boeckmann AJ, Bauer RJ. NONMEM users guides. Ellicott City, Maryland, USA: Icon Development Solutions; 1989. ‐2011. [Google Scholar]
- 23. Lindbom L, Pihlgren P, Jonsson EN. PsN‐toolkit‐‐a collection of computer intensive statistical methods for non‐linear mixed effect modeling using NONMEM. Comput Methods Programs Biomed. 2005;79(3):241‐257. [DOI] [PubMed] [Google Scholar]
- 24. Mendivil CO, Furtado J, Morton AM, Wang L, Sacks FM. Novel pathways of apolipoprotein A‐I metabolism in high‐density lipoprotein of different sizes in humans. Arterioscler Thromb Vasc Biol. 2016;36:156‐165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Velez‐Carrasco W, Lichtenstein AH, Li Z, et al. Apolipoprotein A‐I and A‐II kinetic parameters as assessed by endogenous labeling with [(2)H(3)]leucine in middle‐aged and elderly men and women. Arterioscler Thromb Vasc Biol. 2000;20:801‐806. [DOI] [PubMed] [Google Scholar]
- 26. Whyte MB, Shojaee‐Moradie F, Sharaf SE, et al. HDL‐apoA‐I kinetics in response to 16 wk of exercise training in men with nonalcoholic fatty liver disease. Am J Physiol Endocrinol Metab. 2020;318(6):E839‐E847. [DOI] [PubMed] [Google Scholar]
- 27. Capodanno D, Mehran R, Gibson CM, Angiolillo DJ. CSL112, a reconstituted, infusible, plasma‐derived apolipoprotein A‐I: safety and tolerability profiles and implications for management in patients with myocardial infarction. Expert Opin Investig Drugs. 2018;27(12):997‐1005. [DOI] [PubMed] [Google Scholar]
- 28. Tortorici MA, Gille A, Liss C, et al. Direct enhancement of cholesterol efflux in AMI patients: a PKPD substudy of AEGIS‐I. Eur Heart J. 2017;38(Suppl 1):1106. [Google Scholar]
- 29. Alexander SPH, Kelly E, Mathie A, et al. THE CONCISE GUIDE TO PHARMACOLOGY 2019/20: transporters. Br J Pharmacol. 2019;176:S397‐S493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gibson CM, Kastelein JJ, Phillips A, Aylward PE, Yee MK, Tendera M, Nicholls SJ, Pocock S, Goodman SG, Alexander JH, Lincoff AM, Bode C, Duffy D, Heise M, Berman G, Mears SJ, Tricoci P, Deckelbaum LI, Steg PG, Ridker P, Mehran R. Rationale and design of ApoA‐I Event Reducing in Ischemic Syndromes II (AEGIS‐II): A phase 3, multicenter, double‐blind, randomized, placebo‐controlled, parallel‐group study to investigate the efficacy and safety of CSL112 in subjects after acute myocardial infarction In preparation 2020. [DOI] [PubMed]
- 31. Yokoyama S. Unique features of high‐density lipoproteins in the Japanese: in population and in genetic factors. Nutrients. 2015;7:2359‐2381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zitnanova I, Siarnik P, Fullop M, et al. Gender differences in LDL‐ and HDL‐cholesterol subfractions in patients after the acute ischemic stroke and their association with oxidative stress markers. J Clin Biochem Nutr. 2018;63(2):144‐148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Nagano M, Yamashita S, Hirano K, et al. Molecular mechanisms of cholesteryl ester transfer protein deficiency in Japanese. J Atheroscler Thromb. 2004;11(3):110‐121. [DOI] [PubMed] [Google Scholar]
- 34. Yancey PG, Bortnick AE, Kellner‐Weibel G, de la Llera‐Moya M, Phillips MC, Rothblat GH. Importance of different pathways of cellular cholesterol efflux. Arterioscler Thromb Vasc Biol. 2003;23:712‐719. [DOI] [PubMed] [Google Scholar]
- 35. Oram JF, Lawn RM, Garvin MR, Wade DP. ABCA1 is the cAMP‐inducible apolipoprotein receptor that mediates cholesterol secretion from macrophages. J Biol Chem. 2000;275(44):34508‐34511. [DOI] [PubMed] [Google Scholar]
- 36. Phillips MC. Molecular mechanisms of cellular cholesterol efflux. J Biol Chem. 2014;289(35):24020‐24029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. de la Llera‐Moya M, Drazul‐Schrader D, Asztalos BF, Cuchel M, Rader DJ, Rothblat GH. The ability to promote efflux via ABCA1 determines the capacity of serum specimens with similar high‐density lipoprotein cholesterol to remove cholesterol from macrophages. Arterioscler Thromb Vasc Biol. 2010;30:796‐801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Soares AAS, Tavoni TM, de Faria EC, et al. HDL acceptor capacities for cholesterol efflux from macrophages and lipid transfer are both acutely reduced after myocardial infarction. Clin Chim Acta. 2018;478:51‐56. [DOI] [PubMed] [Google Scholar]
- 39. Potteaux S, Gautier EL, Hutchison SB, et al. Suppressed monocyte recruitment drives macrophage removal from atherosclerotic plaques of Apoe−/− mice during disease regression. J Clin Invest. 2011;121(5):2025‐2036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Feig JE, Rong JX, Shamir R, et al. HDL promotes rapid atherosclerosis regression in mice and alters inflammatory properties of plaque monocyte‐derived cells. Proc Natl Acad Sci USA. 2011;108(17):7166‐7171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Cimmino G, Ibanez B, Vilahur G, et al. Up‐regulation of reverse cholesterol transport key players and rescue from global inflammation by ApoA‐I (Milano). J Cell Mol Med. 2009;13(9B):3226‐3235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Murphy AJ, Funt S, Gorman D, Tall AR, Wang N. Pegylation of high‐density lipoprotein decreases plasma clearance and enhances antiatherogenic activity. Circ Res. 2013;113(1):e1‐e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
TABLE S1 Summary of studies included in the population PK analysis
TABLE S2 Covariate‐parameter relationships evaluated in the population PK and PKPD analysis
FIGURE S1 Goodness of fit of the final population PK model: observed data vs. population predicted (A) and individual predicted (B) plasma apoA‐I concentrations; CWRES vs. time (C) and population predicted plasma apoA‐I concentration (D). The grey line represents a smoothing function and the dashed black line represents unity. apoA‐I, apolipoprotein A‐I; CWRES, conditional weighted results; PK, pharmacokinetic
FIGURE S2 Prediction‐corrected visual predictive check (VPC) obtained through n=1000 simulations from the final population PK model by regimen: placebo, single doses of CSL112 and multiple doses (QWx4) of CSL112, and by all subjects, healthy subjects and patients. The blue circles represent the prediction‐corrected observed apoA‐I concentrations. The solid and dashed red lines represent the observed median and 10th–90th percentile range, respectively. The pink and purple bands represent the 95% confidence interval (CI) of the median prediction, the 10 and 90th prediction interval, respectively. As shown in Supplementary Table 1, healthy subjects were from study 09‐63, 10‐68, 1001, 1002; patients were from study 12‐70, 2002, AEGIS‐I. QWx4, weekly dose for 4 weeks. ApoA‐I, apolipoprotein A‐I; PK, pharmacokinetic.
FIGURE S3 Forest plots of predicted covariate effects from the final population PK model on apoA‐I baseline (A) and exposures (area under the curve at steady state, AUCtau.ss [B] and concentration at end of infusion, C max.ss [C]) following CSL112 administration. Typical subjects were defined as 84 kg, non‐Japanese, healthy males. All ratios displayed use the respective exposure metric for typical subjects as the reference. For each covariate value, the horizontal line represents the 90% prediction interval for the respective ratio. The vertical dashed lines and pink bands indicate traditional thresholds of bioequivalence (0.80 to 1.25). AMI, acute myocardial infarction; apoA‐I, apolipoprotein A‐I; AUCtau.ss, area under the curve at steady state; C max, maximum concentration; PK, pharmacokinetic
FIGURE S4 Goodness of fit of the final exposure‐response model for total CEC (A)‐(D) and ABCA1‐dependent CEC (E)‐(H): observed CEC vs. population predicted (A, E) and individual predicted (B, F) plasma apoA‐I concentrations; CWRES vs. time (C, G) and population predicted plasma apoA‐I concentration (D, H). The dashed red line represents unity. ABCA1, ATP‐binding cassette transporter‐1; apoA‐I, apolipoprotein A‐I; CEC, cholesterol efflux capacity; CWRES, conditional weighted results
FIGURE S5 Workflow of population PK and PKPD model development ABCA1, ATP‐binding cassette transporter A1; AUC0‐48, area under the curve from 0 to 48 hours; AUEC0‐48, area under the effect curve from 0 to 48 hours; CEC, cholesterol efflux capacity; PD, pharmacodymanic; PK, pharmacokinetic
Data Availability Statement
CSL Behring will only consider requests to share Individual Patient Data (IPD) that are received from systematic review groups or bona fide researchers. CSL Behring will not process or act on IPD requests until 12 months after article publication on a public website. An IPD request will not be considered by CSL Behring unless the proposed research question seeks to answer a significant and unknown medical science or patient care question. Applicable country‐specific privacy and other laws and regulations will be considered and may prevent sharing of IPD. Requests for use of the IPD will be reviewed by an internal CSL Behring review committee. If the request is approved, and the researcher agrees to the applicable terms and conditions in a data‐sharing agreement, IPD that has been appropriately anonymized will be made available. Supporting documents including study protocol and the Statistical Analysis Plan will also be provided. For information on the process and requirements for submitting a voluntary data sharing request for IPD, please contact CSL Behring at clinicaltrials@cslbehring.com.