

Abstract
Aims
Carbamazepine can cause hypersensitivity reactions in ~10% of patients. An immunogenic effect can be produced by the electrophilic 10,11‐epoxide metabolite but not by carbamazepine. Hypothetically, certain single nucleotide polymorphisms might increase the formation of immunogenic metabolites, leading ultimately to hypersensitivity reactions. This study explores the role of clinical and genetic factors in the pharmacokinetics (PK) of carbamazepine and 3 metabolites known to be chemically reactive or formed through reactive intermediates.
Methods
A combination of rich and sparse PK samples were collected from healthy volunteers and epilepsy patients. All subjects were genotyped for 20 single nucleotide polymorphisms in 11 genes known to be involved in the metabolism or transport of carbamazepine and carbamazepine 10,11‐epoxide. Nonlinear mixed effects modelling was used to build a population‐PK model.
Results
In total, 248 observations were collected from 80 subjects. A 1‐compartment PK model with first‐order absorption and elimination best described the parent carbamazepine data, with a total clearance of 1.96 L/h, central distribution volume of 164 L and absorption rate constant of 0.45 h−1. Total daily dose and coadministration of phenytoin were significant covariates for total clearance of carbamazepine. EPHX1‐416G/G genotype was a significant covariate for the clearance of carbamazepine 10,11‐epoxide.
Conclusion
Our data indicate that carbamazepine clearance was affected by total dose and phenytoin coadministration, but not by genetic factors, while carbamazepine 10,11‐epoxide clearance was affected by a variant in the microsomal epoxide hydrolase gene. A much larger sample size would be required to fully evaluate the role of genetic variation in carbamazepine pharmacokinetics, and thereby predisposition to carbamazepine hypersensitivity.
Keywords: carbamazepine, population pharmacokinetics, single nucleotide polymorphisms
What is already known about the subject
Metabolism of carbamazepine leads to the generation of reactive metabolites, such as the 10,11‐epoxide and arene oxides, which have been implicated in the pathogenesis of carbamazepine hypersensitivity reactions such as toxic epidermal necrolysis.
Genetic variants in enzymes of carbamazepine metabolism and drug transporter proteins have been shown to alter plasma drug and metabolite levels and dosage requirements in patients.
Pharmacokinetic modelling can be used to investigate the role of clinical and genetic factors in the metabolism of carbamazepine and its metabolites.
What this study adds
A population pharmacokinetic model with first‐order absorption was developed for carbamazepine and its metabolites carbamazepine 10,11‐epoxide, 2‐hydroxycarbamazepine and 3‐hydroxycarbamazepine.
Genetic variation in microsomal epoxide hydrolase (EPHX1; rs2234922) was associated with reduced clearance of carbamazepine 10,11 epoxide in homozygous GG genotypes compared with GA and AA genotypes.
Genetic variation in drug transporters (ABCB1, ABCC2), cytochrome P450 isoforms (CYP2B6, CYP2C8, CYP2C19, CYP3A4) and other enzymes of drug metabolism (uridine diphosphate glucuronosyltransferase, myeloperoxidase, P450 oxidoreductase) had no effect on clearance of carbamazepine and its metabolites.
1. INTRODUCTION
Carbamazepine (CBZ) is used for the treatment of epilepsy, bipolar disorder and trigeminal neuralgia. 1 Therapy with CBZ is complicated because of the drug's complex pharmacokinetic (PK) profile. 2 , 3 , 4 CBZ is almost completely metabolised in the liver and the major oxidation route is conversion to carbamazepine 10,11‐epoxide (CBZE; Figure 1), which is pharmacologically active 2 and electrophilic. 5 Other metabolites include 2‐hydroxycarbamazepine (2OH‐CBZ), 3‐hydroxycarbamazepine (3OH‐CBZ), 4 2,3‐dihydroxycarbamazepine, and the o‐quinone of the catechol. 2 Multiple cytochrome P450 (CYP) isoforms are involved in the formation of these metabolites. 4 , 6 Other enzymes involved in CBZ metabolism include uridine diphosphate glucuronosyltransferase (UGT2B7), 7 microsomal epoxide hydrolase and myeloperoxidase. 8
FIGURE 1.
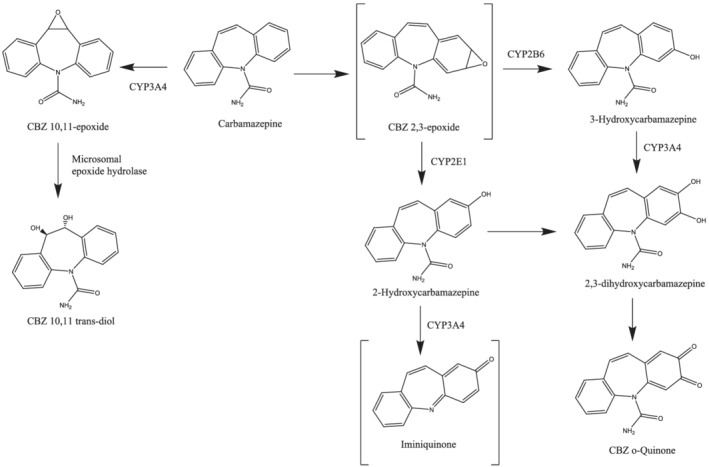
Proposed pathways for the oxidative bioactivation of carbamazepine in humans. The P450 isoforms shown are those reported to be the most active catalysts for the biotransformations. The depiction of the 2,3‐arene oxide as the sole product of aromatic epoxidation is purely representational; the number of arene oxides formed is unknown. The metabolic hydrolysis of carbamazepine 10,11‐epoxide is catalysed by microsomal epoxide hydrolase
Clinically, CBZ has a narrow therapeutic index. It induces multiple CYP isoforms and transporters, 9 and also induces its own metabolism. There is large interindividual variability in plasma levels of CBZ with poor correlation to dose. Patient factors that influence the PK of CBZ include sex, age and total body weight. 10 , 11 , 12 Concomitant medications such as valproic acid, phenytoin, felbamate and phenobarbital have also been associated with variation in metabolism of CBZ. 13 , 14 , 15
CBZ is generally well tolerated, but up to 10% of patients experience a hypersensitivity reaction. 1 Carriage of HLA‐B*15:02 has been associated with increased risk of CBZ‐induced Stevens–Johnson syndrome (SJS) and toxic epidermal necrolysis (TEN) in patients from South‐East Asia while carriage of HLA‐A*31:01 has been associated with increased susceptibility to all phenotypes of CBZ hypersensitivity in European, Japanese and Korean populations. 16 Phenytoin is an aromatic anticonvulsant that is also a known cause of SJS/TEN. Similar to CBZ, carriage of HLA‐B*15:02 is associated with increased susceptibility to hypersenstivity. 17 , 18 More recently, a genome‐wide association study reported a significant association between phenytoin‐induced serious cutaneous adverse reactions and carriage of CYP2C9*3. 19 Delayed clearance of plasma phenytoin in patients with severe cutaneous reactions was identified as providing a mechanistic link to the manifestation of hypersensitivity. 20
Conventionally, it is thought that chemically reactive drug metabolites, such as arene oxides and epoxides, can be cytotoxic or form neo‐antigens with cellular proteins, which activate the immune system, resulting in hypersensitivity reactions. 21 Recently, however, carbamazepine 10,11‐epoxide has also been shown to have a specific immunogenic effect, namely alteration of selective peptide presentation by HLA‐B*15:02, through binding to the protein, a property not demonstrated by CBZ. 22
Theoretically, genetic variants in CBZ metabolism could not only alter the routes of metabolism between different individuals, but they could also increase the formation of chemically reactive and otherwise immunogenic metabolites. 23 Table 1 outlines the studies that have investigated effects of genetic polymorphisms on the metabolism of CBZ. No previous study has comprehensively characterised the effects of genetic variation on metabolism of carbamazepine and its metabolites through PK modelling.
TABLE 1.
Studies that have investigated single nucleotide polymorphisms associated carbamazepine metabolism
| Study reference | Population | No. of subjects | SNP ID | Gene | Nucleotide change/allele | Effect of polymorphism |
|---|---|---|---|---|---|---|
| Ma et al. 2015 24 | Chinese | 166 | rs2242480 | CYP3A4 | *1G | NS |
| rs776746 | CYP3A5 | *3 | NS | |||
| rs1051740 | EPHX1 | c.337 T > C | Higher CBZ maintenance dose, lower CBZ natural logarithmic concentration–dose ratio and higher CBZE concentration | |||
| rs7439366 | UGT2B7 | c.802 T > C | Higher CBZ maintenance dose if also a carrier of ABCC2 c.1249G > A | |||
| rs3740066 | ABCC2 | c.3972C > T |
Higher CBZ maintenance dose and higher CBZE concentration Higher CBZ maintenance dose if also a carrier of UGT2B7 c.802 T > C |
|||
| rs2273697 | c.1249G > A | |||||
| Wang et al. 2015 25 | Chinese | 88 | rs4646440 | CYP3A4 | c.1023 + 608C > T | NS |
| rs2242480 | *1G | NS | ||||
| rs15524 | CYP3A5 | *1D |
Higher dose adjusted CBZ and CBZE concentration in subjects on concomitant PHT/PB Higher dose adjusted CBZ and CBZE concentration in subjects on concomitant PHT/PB |
|||
| *3 | ||||||
| rs776746 | ||||||
| rs1045642 | ABCB1 | c.3435C > T |
NS CBZ/CBZE ratio in subjects on concomitant PHT/PB CBZ/CBZE ratio in subjects on concomitant PHT/PB NS |
|||
| rs2032582 | c.2667G > T | |||||
| rs10234411 | 2,481 + 882A > T | |||||
| rs1128503 | ||||||
| c.1236 T > C | ||||||
| Caruso et al. 2014 26 | Italian | 50 | rs1051740 | EPHX1 | c.337 T > C |
NS NS |
| rs2234922 | c.416A > G | |||||
| rs35599367 | CYP3A4 | *22 | NS | |||
| Zhu et al. 2014 27 | Chinese | 210 | rs35599367 | CYP3A4 | *22 |
NS Lower adjusted levels of CBZ and CBZE |
| rs2242480 | *1G | |||||
| rs776746 | CYP3A5 | *3 | Higher dose adjusted levels of CBZ | |||
| rs1057868 | POR | *28 | NS | |||
| rs2234922 | EPHX1 | c.416A > G |
Lower dose adjusted levels of DiOH‐CBZ and DiOH‐CBZ:CBZE ratio Higher DiOH‐CBZ:CBZE ratio NS |
|||
| rs3738046 | c.128G > C | |||||
| rs1051740 | c.337 T > C | |||||
| Panomvana et al. 2013 28 | Thai | 70 | rs776746 | CYP3A5 | *3 | Trend towards reduced clearance (not significant) |
| Puranik et al. 2013 29 |
African‐American Caucasian |
88 | rs2740574 | CYP3A4 | *1B | Reduced clearance |
| rs776746 | CYP3A5 | *3 | Longer half‐life for CBZ (African Americans) | |||
| rs1051740 | EPHX1 | c.337 T > C |
Lower DiOH‐CBZ:CBZE ratio (African‐Americans) NS |
|||
| rs2234922 | c.416A > G | |||||
| rs7439366 | UGT2B7 | c.802 T > C |
NS NS NS Increased clearance (African‐American) NS |
|||
| rs11302069 | c.1090 + 155delA | |||||
| c.735A > G | ||||||
| rs28365062 | ||||||
| c.372A > G | ||||||
| c.1059C > G | ||||||
| rs28365063 | ||||||
| rs4292394 | ||||||
| rs1045642 | ABCB1 | c.3435C > T |
NS Higher clearance (African‐Americans) NS NS Higher DiOH‐CBZ:CBZE ratio (African‐American) Higher DiOH‐CBZ:CBZE ratio (African‐American) |
|||
| rs1128503 | c.1236 T > C | |||||
| rs2032582 | c.2677G > T | |||||
| rs4148734 | c.702 + 1789C > T | |||||
| rs4148739 | c.2482‐236A > G | |||||
| rs4148740 | ||||||
| c.2686‐1911 T > C | ||||||
| rs2273697 | ABCC2 | c.1249G > A |
Higher clearance (Caucasians) Higher CBZE:CBZ ratio (Caucasian male) Reduced clearance and reduced CBZE:CBZ ratio (African‐American) NS |
|||
| rs3740066 | c.3972C > A | |||||
| rs4148386 | 208‐3523G > A | |||||
| rs8187710 | c.4544G > A | |||||
| rs1523127 | NR1I2 | c.‐131C > A |
NS NS Increased CBZE:CBZ ratio and reduced clearance (African‐American) Reduced clearance Increased clearance (African‐American) Increased CBZE:CBZ ratio |
|||
| rs1523130 | c.‐1,663 T > C | |||||
| rs2461817 | c.‐22‐1425A > C | |||||
| rs3814055 | c.‐1135C > T | |||||
| rs4688040 | c.‐22‐1985G > T | |||||
| rs7643645 | c.‐22‐579A > G | |||||
| Yun et al. 2013 30 | Chinese | 83 | rs1051740 | EPHX1 | c.337 T > C |
NS Higher adjusted plasma CBZ levels |
| c.416A > G | ||||||
| rs2234922 | ||||||
| rs2242480 | CYP3A4 | *1G | NS | |||
| Hung et al. 2012 31 | Taiwanese | 234 | rs1051740 | EPHX1 | c.337 T > C |
Higher maintenance dosage NS |
| rs2234922 | c.416A > G | |||||
| rs7668258 | UGT2B7 | c.‐161 T > C |
NS NS NS NS |
|||
| rs7438135 | c.‐900G > A | |||||
| rs28365062 | c.735A > G | |||||
| rs7439366 | c.802 T > C | |||||
| rs1128503 | ABCB1 | c.1236 T > C |
NS NS NS |
|||
| rs2032582 | c.2677G > T | |||||
| rs1045642 | c.3435C > T | |||||
| rs717620 | ABCC2 | c.‐24C > T |
NS NS |
|||
| c.3972C > T | ||||||
| rs2273697 | ||||||
| Sterjev et al. 2012 32 | North Macedonian | 162 | rs1045642 | ABCB1 | c.3435C > T | Higher CBZ plasma levels |
| Meng et al. 2011 33 | Chinese | 84 | rs1045642 | ABCB1 | c.3435C > T |
Lower plasma CBZ levels NS NS |
| rs1128503 | c.1236 T > C | |||||
| rs2032582 | c.2677G > T | |||||
| Park et al. 2009 34 | Korean | 35 | rs776746 | CYP3A5 | *3 | Reduced clearance |
CBZ: carbamazepine, CBZE: carbamazepine 10,11‐epoxide, DiOH‐CBZ: 10,11‐dihydro‐10,11‐trans‐dihydroxy‐carbamazepine, NS: not significant, PB: phenobarbital, PHT: phenytoin
The aim of this study was to develop a population PK model for CBZ and 3 metabolites, namely CBZE, 2OH‐CBZ and 3OH‐CBZ, in healthy adults and adult patients with epilepsy, to generate population clearance estimates for CBZ and the metabolites. Those metabolites were chosen because they are chemically reactive (CBZE) or their generation is via reactive arene oxide intermediates (2OH‐CBZ and 3OH‐CBZ). 5 The model was used to investigate the influence of physiological factors, concomitant medications, and genetic variation in enzymes and transporters associated with the metabolism and disposition of CBZ.
2. METHODS
2.1. Ethical approval
Study participants were recruited from 2 different studies: PICME I (Pharmacokinetic Investigation into the formation of Carbamazepine Metabolites and carbamazepine‐protein conjugates in healthy volunteers) and PICME II (Pharmacokinetic Investigation into the formation of Carbamazepine Metabolites and carbamazepine–protein conjugates in epilepsy patients). Both studies were granted clinical trial authorisation by the Medicines and Healthcare products Regulatory Agency in the UK (EudraCT: 2012–004700‐35 and EudraCT: 2013–002743‐28). Ethical approval for PICME I was granted by the North West Research Ethics Committee—Greater Manchester (ethics reference: 12/NW/0780). Ethical approval for PICME II was granted by the North West Research Ethics Committee—Haydock (ethics reference: 13/NW/0503).
2.2. Study population
Healthy male adult volunteers were recruited at the Royal Liverpool and Broadgreen University Hospitals NHS Trust, UK, as part of the PICME I clinical study from September 2013 to November 2013. The PICME II clinical study was conducted at the Walton Centre NHS Foundation Trust, UK, from December 2013 to September 2014. Two groups of patient volunteers were recruited as part of the PICME II study: an autoinduction group and a maintenance group. The autoinduction group consisted of patients with epilepsy who were newly initiated on CBZ treatment. The maintenance group consisted of patients with epilepsy who had been established on a stable dose of CBZ for at least 4 weeks. Detailed inclusion and exclusion criteria are available in the supplementary material.
2.3. Dosing and sampling schedules
In PICME I, healthy volunteers were administered a single oral dose of 400 mg immediate release CBZ (Novartis Pharmaceuticals UK Ltd., London, UK). Blood samples (5 mL) were taken predose and at 0.25, 0.5, 1, 2, 4, 6, 8, 12, 24, 48 and 72 hours after drug administration. CBZ was administered by study clinicians and nurses, with dosing times recorded in case report forms.
In PICME II, patients with epilepsy in the autoinduction group, who were newly prescribed CBZ, received a single immediate release oral dose of 200 mg (Novartis Pharmaceuticals UK Ltd., London, UK). Blood samples (5 mL) were taken predose and at 1, 2, 4, 6, 8 and 24 hours after drug administration. Subjects were discharged and self‐administered 200 mg CBZ once daily at home. On day 14, subjects returned for further blood sampling predose and then at 1, 2, 4, 6 and 8 hours postdose. After this visit, the dose of CBZ was increased to 200 mg twice daily. On day 28, blood samples (5 mL) were collected at the same time points as on day 14. The dose of CBZ was increased to 200 mg in the morning and 400 mg at night. On day 42, blood samples (5 mL) were collected at the time points given above. This dose titration regimen is standard practice for initiation of CBZ therapy at the Walton Centre NHS Foundation Trust.
Participants in the maintenance group of the PICME II clinical trial underwent sparse PK sampling. Blood samples (5 mL) were collected during routine epilepsy clinic appointments. Postdose timing was calculated by asking the subject the timing and dosage of the previous dose of self‐administered CBZ. All PK blood samples were immediately centrifuged at 1500 g for 10 minutes at 4°C. Aliquots of plasma (300 μL) were stored at −80°C until further analysis. Subjects in the maintenance group obtained their CBZ prescription from the community so it was not possible to determine all the manufacturers.
2.4. Plasma drug and metabolite assays methods
CBZ and metabolites in plasma samples were assayed by liquid chromatography–tandem mass spectrometry using the method reported in Yip et al. 5 The calibration ranges for the analytes were as follows: CBZ, 0.33–15.95 μg/mL; CBZE, 0.10–6.30 μg/mL; 2OH‐CBZ, 0.92–118.18 ng/mL; and 3OH‐CBZ, 4.73–605.28 ng/mL. The intraday coefficients of variation for the analytes were as follows: CBZ, 3.7%; CBZE, 3.0%; 2OH‐CBZ, 4.7%; and 3OH‐CBZ, 3.4%. The interday coefficients of variation for each analyte were: CBZ, 6.5%; CBZE, 6.5%; 2OH‐CBZ, 5.5%; and 3OH‐CBZ, 4.0%.
2.5. DNA extraction procedure
Blood samples (10 mL) for genetic analyses were collected from all participants at their first study visit and stored at −80°C. DNA was extracted from 4–5 mL of whole blood, thawed at room temperature, using a Chemagic magnetic separation module (Chemagen Biopolymer‐Technologie AG, Baeweiler, Germany) according to the manufacturer's protocol. The concentration of DNA was measured using a NanoDrop microvolume spectrophotometer (Thermo Scientific, Wilmington, USA). The extracted DNA was stored in aliquots at 4°C.
2.6. Selection of genetic polymorphisms
The effect of genetic variation on the PK of CBZ was investigated by analysis of single nucleotide polymorphisms (SNPs). The selection of genes and SNPs (Table 2) was based upon the known enzymology of CBZ's metabolism, 4 , 5 , 7 , 46 efflux transporter activity for CBZ and CBZE, 47 , 48 expected minor allele frequencies in the study population (>5%), and a review of previous polymorphism association studies (Table 1). P450 oxidoreductase (POR) supports reactions for all P450 enzymes and its most common variant (POR*28) has been associated with altered activity of CYPs such as CYP2C9 49 and CYP3A. 50 A total of 20 SNPs were chosen in 11 genes (Table 2).
TABLE 2.
Selection of genes and single nucleotide polymorphisms for genotyping studies
| SNP ID | Gene | Nucleotide change/allele | Potential change in function | SNP typing platform | References |
|---|---|---|---|---|---|
| rs1045642 | ABCB1 | 3435C > T | Reduced expression | Taqman | 35 |
| rs1128503 | ABCB1 | 1236 T > C | Inconclusive | MassArray | 35 |
| rs2032582 | ABCB1 | 2677G > T | Inconclusive | MassArray | 35 |
| rs2273697 | ABCC2 | 1249G > A | Increased activity | MassArray | 36 |
| rs3740066 | ABCC2 | 3972C > T | Inconclusive | MassArray | 36 |
| rs717620 | ABCC2 | –24C > T | Reduced activity | Taqman | 36 |
| rs28365062 | UGT2B7 | 735A > G | Increased activity | MassArray | 37 |
| rs28365063 | UGT2B7 | 372A > G | Increased activity | MassArray | 29 |
| rs7438135 | UGT2B7 | –900G > A | Increase activity | MassArray | 38 |
| rs1051740 | EPHX1 | 337 T > C | Increased activity | MassArray | 39 |
| rs2234922 | EPHX1 | 416A > G | Reduced activity | MassArray | 39 |
| rs11572080 | CYP2C8 | *3 | Inconclusive | MassArray | 40 |
| rs1058930 | CYP2C8 | *4 | Reduced activity | MassArray | 40 |
| rs4244285 | CYP2C19 | *2 | Loss of function | MassArray | 41 |
| rs192154563 | CYP2C19 | *17 | Increase activity | MassArray | 41 |
| rs3745274 | CYP2B6 | *6 | Reduced activity | Taqman | 42 |
| rs2242480 | CYP3A4 | *1G | Increased activity | MassArray | 27 |
| rs776746 | CYP3A5 | *3 | Loss of function | MassArray | 43 |
| rs2333227 | MPO | –463G > A | Reduced activity | Taqman | 44 |
| rs1057868 | POR | *28 | Increased activity | Taqman | 45 |
2.7. Analysis of single nucleotide polymorphisms
SNPs were typed using either the Agena MassArray iPLEX platform (Agena Inc, San Diego, CA, USA) or manufacturer‐validated TaqMan real‐time PCR SNP genotyping assays (Life Technologies, Paisley, UK) according to the manufacturer's protocols (Table 2). The genotype frequencies of each SNP were examined for deviation from Hardy–Weinberg equilibrium (HWE) using the χ2 test.
2.8. Population PK modelling
Equations 1–9 are the final ordinary differential equations and associated (re)parameterisations for the structural PK model applied to concentration data from simultaneous assays of CBZ and the 3 metabolites in plasma. A schematic of the final model is given in Figure 2.
| (1) |
| (2) |
| (3) |
| (4) |
| (5) |
Where:
| (6) |
| (7) |
| (8) |
| (9) |
FIGURE 2.
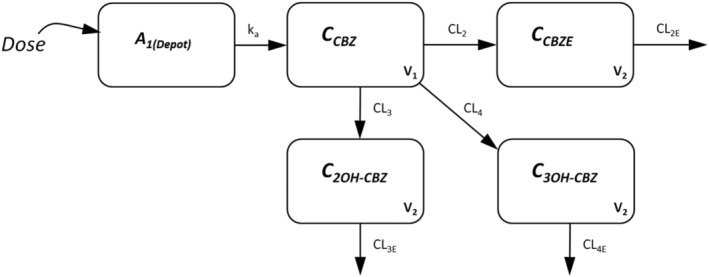
Schematic of the pharmacokinetic model for carbamazepine
A 1‐compartment PK model with first‐order absorption and elimination was chosen for CBZ based on visual inspection of the raw data and previously published analyses of the PK of CBZ. Exploratory analyses involved 1‐compartment fitting to parent CBZ data alone to provide initial estimates for the final fitting. To account for CBZE, 2OH‐CBZ and 3OH‐CBZ metabolite production, the total clearance of CBZ (CLTOT) was partitioned into CL2, CL3, and CL4, respectively, via the FM1 parameter (Equations 6–8) and under the assumptions that production of the 3 observed metabolites accounted for approximately all the primary elimination of CBZ, and that CL3 equals CL4. 3 , 4 , 51 CBZE, 2OH‐CBZ and 3OH‐CBZ were all subject to first‐order elimination governed by CL2E, CL3E and CL4E parameters, respectively, with the same volume of distribution (V2) estimated for the 3 metabolites. To ensure identifiability of the model, FM1 was fixed to a value of 0.64 according to prior mass balance information regarding the metabolic fate of CBZ (i.e. 36% of parent CBZ is primarily converted to CBZE), 51 , 52 and, in addition, the absorption rate constant for CBZ (KA) was constrained to be greater than the elimination rate constant for CBZ by estimating a KAstar parameter and the use of Equation 9. Assuming the same volume of distribution was also necessary for the identifiability of the model and was deemed reasonable given the metabolites are isomerically similar and have similar physicochemical properties. The structural identifiability of the model was confirmed (7 PK parameters estimated: KAstar, CLTOT, CL2E, CL3E, CL4E, V1, V2; 4 analytes/states observed) using the DAISY software package, 53 and in the absence of IV data, estimates of CL and V parameters in the model are apparent in nature, i.e. CL/F, V/F etc. A schematic of the final model is provided in Figure 2.
Population PK analysis of the data for CBZ and its metabolites was performed with NONMEM (version 7.3, ICON plc, South County Business Park, Leopardstown, Dublin, Republic of Ireland), using the first‐order conditional estimation method (with interaction). Interindividual variability (assuming a log‐normal distribution across the population) was included in each of the structural model parameters, while residual error was accounted for using an exponential error model, with a separate residual error variance estimated for each of the 4 plasma analytes. In addition, to achieve shorter computational run times and improved model stability, the closed form analytical solutions of the model's differential equations were derived and implemented for model fitting, with multiple dosing super‐positioning (according to each individual dosing record) implemented via the DOWHILE/ILOOP functionality in NONMEM 7.4 abbreviated code. Subjects in the PICME II maintenance group were modelled with an assumed dosing record of 8 weeks BID dosing (at each individual's specific dose level) to replicate steady state conditions. The analytical solutions of the model equations are provided in the supplementary information.
The known auto‐induction effect of CBZ, was incorporated in the base model as a binary, categorical induction status effect on CLTOT and CL3E. Data from PICME I and the first visit of the PICME II autoinduction group were categorised as having come from a noninduced period while data from the second visit of the PICME II autoinduction group and the PICME II maintenance group were categorised as from an induced period (i.e. a binary, noncontinuous time function)—reflecting the length of CBZ dosing up to the point of PK observation, with the latter groups having received at least 14 days daily dosing of CBZ, which would be expected to increase clearance. More sophisticated models for autoinduction as a continuous function of time (and drug exposure, taking the form of e.g. an autoinduction enzyme turnover model used previously for rifampicin were attempted in exploratory analysis, but failed to minimise successfully and/or give acceptable parameter estimate precision. 54 We believe that this is at least in part due to the dataset lacking information on a continuous range of levels of induction over time among the patients, which a turnover autoinduction model might need for its parameters to be sufficiently well estimated: patient PK in this study was observed under the more binary condition of either showing the effects of induction after several weeks dosing or not. Exploratory analyses went on to test the induction effect on CLTOT, FM1 and metabolite clearances (CL2E,3E,4E) with the final base model retaining the effect on CLTOT and CL3E, expressions for which are given in Equations 10 and 11, with IND as a dummy variable equal to 1 or 0 for an induced or noninduced period datapoint, θx the fixed effect values for the parameter or the induction effect in question, and ηx the log interindividual variability random effect component, drawn from a normal distribution with mean of 0 and variance ωx 2. Data from PICME I and PICME II autoinduction visit 1 were IND = 0, while data from PICME II autoinduction visit 2 and PICME II maintenance were IND = 1. Further equations for covariate models are expressed in typical value format, omitting the random effect component for clarity.
| (10) |
| (11) |
Exploratory analyses investigating incorporation of interoccasion variability as a random effect in the model failed to produce acceptable fittings, despite the potential for this to be required to describe the PICME II autoinduction group data in particular, where patients' PK was monitored on 2 occasions.
Age and weight were investigated as continuous covariates in the model on CLTOT, V1 and metabolite clearances (CL2E,3E,4E) using expressions of the form in Equation 12, where individual covariate values (covi) were normalised to the population means (45.8 years for age or 80.8 kg for weight).
| (12) |
The daily dose of CBZ (DOSE) was investigated as a covariate on CLTOT in a similar manner (Equation 13, where 400 mg was the most common daily dose) to allow, for example, for a potentially greater CLTOT in patients on higher doses due to greater induction of metabolism.
| (13) |
Smoking status, and whether or not a patient was administered a specific comedication, were examined as categorical covariate effects on CLTOT using an expression of the form in Equation 14, with MED as an indicator equal to 1 or 0 if the comedication was present or absent, respectively. Comedications investigated included valproate, phenytoin, phenobarbital, clobazam, lamotrigine, levetiracetam, zonisamide, topiramate, lacosamide, pregabalin, perampanel, omeprazole, atorvastatin, pravastatin and simvastatin.
| (14) |
The potential effects of the SNPs genotyped in the subject population were generally examined as categorical covariate effects on CLTOT, using an expression of the form in Equation 15 with 2 θCOV parameters (2 extra degrees of freedom) to allow characterisation of heterozygous mutant, vs homozygous mutant, vs wild type status. SNPHOM and SNPHET are dummy variables equal to 1 or 0 to record the genotype status as yes/no, respectively, with both equal to 0 for wild‐type individuals.
| (15) |
The exceptions to equation 14 among the SNPs investigated were the EPHXTC and EPHXAG genotypes (Table 2), which were investigated for potential effects on CL2E as these genes are specifically associated with the metabolic clearance of CBZE.
SNP covariate effects were also examined for effect of homozygous mutant only vs combined wild type/heterozygous mutant group and for either homozygous or heterozygous mutant vs wild type. These analyses required only a single θCOV degree of freedom parameter:
| (16) |
Covariate model selection adopted a standard forward addition/backward deletion approach. 55 The covariates were initially examined in univariate analyses as single additions to the base model. They were deemed statistically significant (P < .05) based on a drop in objective function value compared to the base model of 3.8 or 6.0 (or greater) for 1 or 2 degrees of freedom, respectively. In terms of extra covariate effect parameters added to the model, −2 degrees of freedom were required, e.g. for genotype HOM vs HET vs WT covariate models. Final covariate model selection via backward deletion adopted a statistical significance criterion of P < .01, i.e. changes in objective function value of 6.6 or 9.2 for 1 or 2 degrees of freedom, respectively.
Standard goodness‐of‐fit plots (observed concentrations vs population‐ and individual‐predicted concentrations, as residual plots) and a prediction‐corrected visual predictive check (VPC, based on 1000 simulations of the dataset population) were used to evaluate the final model fitting to the dataset, with parametric standard errors (expressed as percentage relative standard errors) used to assess the precision of model parameter estimates.
2.9. Nomenclature of targets and ligands
Key protein targets and ligands are hyperlinked to corresponding entries in http://www.guidetopharmacology.com, 56 and are permanently archived in the Concise Guide to Pharmacology 2019/20. 57
3. RESULTS
3.1. Subject demographics and genetic polymorphisms
In total, 80 subjects were recruited into the studies (Table 3). Rich PK sampling data were obtained from 8 healthy volunteers who completed the PICME I study. Rich PK sampling data were also obtained from 3 patients with a new diagnosis of epilepsy who completed dose titration of CBZ as part of the autoinduction group of PICME II. Eighty sparse PK samples were collected from 69 patients who were recruited to the maintenance group of PICME II. In total, 248 sets of drug and metabolite assays were used in the analysis.
TABLE 3.
Demographic and medication data for subjects included in population PK analysis. Data are expressed as mean values ± standard deviation (ranges)
| PICME I: Healthy volunteer | PICME II: Autoinduction group | PICME II: Maintenance group | |
|---|---|---|---|
| Numbers of subjects | 8 | 3 | 69 |
| Sets of drug/metabolite assays | 96 | 75 | 77 |
| Gender (%): | |||
| Male | 8 (100%) | 1 (33.3%) | 37 (53.6%) |
| Female | 0 | 2 (66.6%) | 32 (46.4%) |
| Age (y) | 27.4 ± 3.5 (21.7–32.8) | 46.7 ± 4.4 (43.5–51.7) | 47 ± 14.3 (25.9–75.8) |
| Total body weight (kg) | 84.6 ± 11.2 (69.8–99.4) | 95.2 ± 26.4 (73.1–124.5) | 79.7 ± 18.8 (39.0–140.5) |
| Smoker (%): | |||
| Yes | 0 (0%) | 0 (0%) | 20 (29.0%) |
| No | 8 (100%) | 3 (100%) | 49 (71.0%) |
| Sparse PK sampling time points (minutes) | NA | NA | 337.5 ± 230.9 (70–1030) |
| CBZ dose (mg/d) | 400‐mg single dose | 200 mg once daily (days 1–14) | 819 mg ± 367 (range: 200–1600 mg) |
| 200 mg twice daily (days 15–28) | |||
| 200 mg AM and 400 mg PM (days 29–42) | |||
| CBZ therapy without concomitant AEDs or CYP3A4 inducer/inhibitor (%) | 8 (100%) | 2 (66.6%) | 16 (23.2%) |
| Concomitant with 1 AED (%) | 0 | 0 | 26 (37.7%) |
| Concomitant with 2 AEDs (%) | 0 | 0 | 16 (23.2%) |
| Concomitant with 3 AEDs (%) | 0 | 0 | 5 (7.2%) |
| Concomitant with 1 CYP3A4 inducer/inhibitor (%) | 0 | 1 (33.3%) | 12 (17.4%) |
| Concomitant with 2 CYP3A4 inducer/inhibitors (%) | 0 | 0 | 3 (4.3%) |
| Number of subjects for each AED: | |||
| Levetiracetam | 0 | 0 | 26 |
| Clobazam | 0 | 0 | 18 |
| Lamotrigine | 0 | 0 | 5 |
| Zonisamide | 0 | 0 | 5 |
| Phenytoin | 0 | 0 | 4 |
| Topirmate | 0 | 0 | 4 |
| Pregabalin | 0 | 0 | 4 |
| Valproate | 0 | 0 | 3 |
| Lacosamide | 0 | 0 | 3 |
| Perampanel | 0 | 0 | 1 |
| Phenobarbital | 0 | 0 | 1 |
| Number of subjects for each CYP3A4 | |||
| Inducer/inhibitor: | |||
| Statin | 0 | 1 | 12 |
| Omeprazole | 0 | 0 | 6 |
time points applicable only to sparse PK sampling group; AED: antiepileptic drug; CBZ: carbamazepine, PK: pharmacokinetic.
The distribution of the 20 SNPs amongst these subjects is recorded in Table 4. No subjects were carriers for CYP2C19*17 (rs192154563). Each genotype frequency was consistent with HWE, and minor allele frequencies ranged from 4 to 55% in keeping with polymorphism frequencies reported in the literature. No demographic covariates were significant in the final PK model.
TABLE 4.
Distribution of single nucleotide polymorphisms (SNPs) amongst study subjects (n = 80)
| SNP ID | Gene | Allele | Genotype frequency (%) | Minor allele frequency | Hardy–Weinberg equilibrium (P) | ||
|---|---|---|---|---|---|---|---|
| Wild type | Heterozygote | Homozygote | |||||
| rs1045642 | ABCB1 | 3435C > T | 14 (17.5%) | 44 (55%) | 22 (27.5%) | 0.55 | .32 |
| rs1128503 | ABCB1 | 1236 T > C | 14 (17.5%) | 42 (52.5%) | 24 (30%) | 0.56 | .55 |
| rs2032582 | ABCB1 | 2677G > T | 24 (30.0%) | 39 (48.8%) | 17** (21.2%) | 0.46 | .88 |
| rs2273697 | ABCC2 | 1249G > A | 49 (61.3%) | 29 (36.3%) | 2 (2.4%) | 0.21 | .34 |
| rs3740066 | ABCC2 | 3972C > T | 25 (31.3%) | 42 (52.5%) | 13 (16.2%) | 0.43 | .51 |
| rs717620 | ABCC2 | –24C > T | 50 (62.5%) | 26 (32.5%) | 4 (5.0%) | 0.21 | .80 |
| rs28365062 | UGT2B7 | 735A > G | 62 (77.5%) | 18 (22.5%) | 0 (.0%) | 0.11 | .26 |
| rs28365063 | UGT2B7 | 372A > G | 56 (70.0%) | 24 (30.0%) | 0 (.0%) | 0.15 | .11 |
| rs7438135 | UGT2B7 | –900G > A | 27 (33.8%) | 39 (48.7%) | 14 (7.5%) | 0.42 | .99 |
| rs1051740 | EPHX1 | 337 T > C | 36 (45.0%) | 37 (46.3%) | 7 (8.7%) | 0.32 | .56 |
| rs2234922 | EPHX1 | 416A > G | 53 (66.3%) | 24 (30.0%) | 3 (3.7%) | 0.19 | .89 |
| rs11572080 | CYP2C8 | *3 | 62 (77.5%) | 16 (20.0%) | 2 (2.5%) | 0.13 | .44 |
| rs1058930 | CYP2C8 | *4 | 70 (87.5%) | 10 (12.5%) | 0 (.0%) | 0.06 | .55 |
| rs4244285 | CYP2C19 | *2 | 57 (71.3%) | 19 (23.7%) | 4 (5.0%) | 0.17 | .17 |
| rs192154563 | CYP2C19 | *17 | 80 (100%) | 0 (0.0%) | 0 (.0%) | 0.00 | NA |
| rs3745274 | CYP2B6 | *6 | 52 (65.0%) | 26 (32.5%) | 2 (2.5%) | 0.19 | .55 |
| rs2242480 | CYP3A4 | *1G | 71 (88.8%) | 9 (11.2%) | 0 (.0%) | 0.06 | .59 |
| rs776746 | CYP3A5 | *3 | 73 (91.3%) | 7 (8.7%) | 0 (.0%) | 0.04 | .68 |
| rs2333227 | MPO | –643G > A | 12 (15%) | 41 (51.3%) | 27 (33.7%) | 0.59 | .58 |
| rs1057868 | POR | *28 | 40 (50.0%) | 27 (33.8%) | 13 (16.2%) | 0.33 | .53 |
included 3 TA subjects, NA: not applicable.
3.2. Base model and covariate fitting
As illustrated by the VPC and diagnostic plots (Figures 3 and 4), the PK profiles for CBZ and its 3 assayed metabolites, and their variability, were adequately described by the applied final model. VPC plots stratified according to the study group (PICME I, PICME II autoinduction, and PICME II maintenance) are provided in the Supplementary Material with plots of raw data and mean observed profiles where applicable. There was, however, some apparent underprediction of 2OH‐CBZ concentrations at time points later than 24 h. These 48 and 72 h timepoints all originate from the 8 patients of the PICME I study group, with the majority of the exposure dataset coming from the 72 patients of PICME II (autoinduction and maintenance); it is feasible therefore that the apparent poor fit at 48 h and 72 h for 2‐OH reflects that the overall population fitting is weighted to the description of the 90% of overall subjects in the PICME II study groups, where a higher CL3E is more typical, as accounted for at least in part by the 1.7‐fold increased induction effect on CL3E retained in the final model. The result would be a higher population CL3E estimate than is ideal to describe the late timepoints of PICME I profiles; however, with the PICME II subjects in turn having no 48‐ and 72‐h observations to balance out the VPC plot at the later timepoints, the apparent underprediction can be accounted for then as a result of the compromise resulting from the combined analysis of the complete dataset.
FIGURE 3.
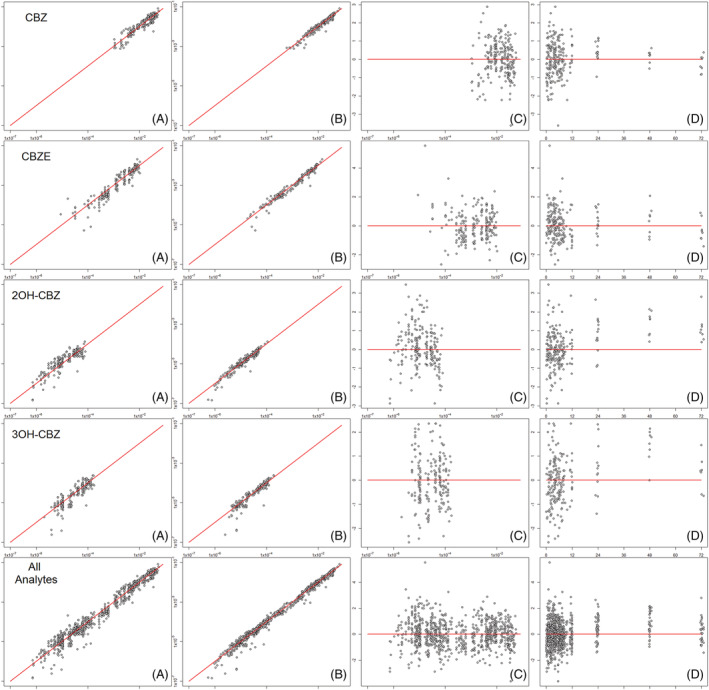
Goodness of fit diagnostic plots for of carbamazepine and metabolites (as labelled by row): (A) Observed concentrations (mM) vs population predicted concentrations (mM); (B) Observed concentrations (mM) vs individual predicted concentrations (mM); (C) Conditional weighted residuals vs population predicted concentrations (mM); (D) Conditional weighted residuals vs time after dose (h)
FIGURE 4.
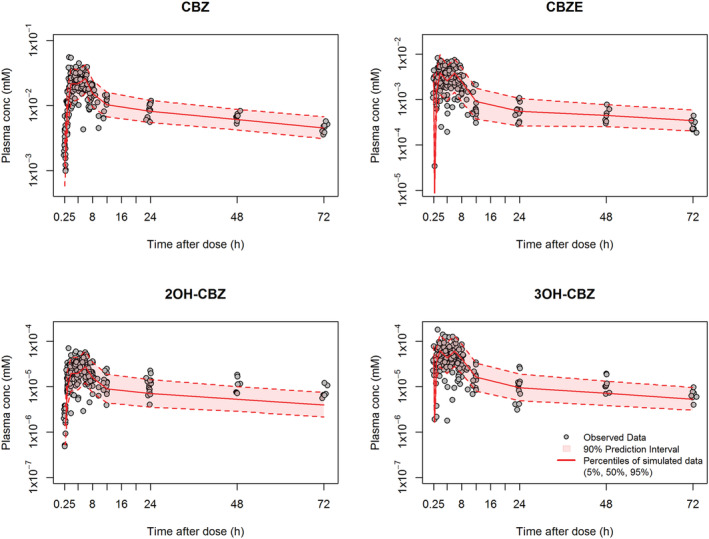
Prediction‐corrected visual predictive check for the final pharmacokinetic model fitting for each of the analytes. 90% prediction interval (broken line) and median population prediction (continuous line) determined from 1000 simulations for CBZ with the covariate values of those individuals used in the model building process. CBZ: carbamazepine; CBZE: carbamazepine‐10,11‐epoxide; 2OH‐CBZ: 2‐hydroxy‐carbamazepine; 3OH‐CBZ ‐ 3‐hydroxy‐carbamazepine
The population mean estimates for CLTOT, V1 and KA of CBZ are provided in Table 5. The CLTOT value in our model was 1.96 L/h which falls in the centre of previously published CLTOT values which range between 1.15 and 3.58 L/h. 10 , 11 , 13 , 14 The CL value for CBZE in our model was 9.71 L/h. In a previous population PK model, which included CBZE the CL estimate for CBZE in a 70 ‐kg patient prescribed 400 mg CBZ was 28 L/h. 58 A significant part of the discrepancy can be explained by the assumed conversion ratio between CBZ and CBZE. Our model assumes that 0.36 of CBZ is converted to CBZE 51 , 59 whilst the model of Jiao et al. assumes that 0.65 of CBZ is converted to CBZE.
TABLE 5.
Final model parameters
| Parameter | Estimated value [%RSE] (EPS‐shrinkage %) | % Interindividual variability [%RSE] (ETA‐shrinkage %) | ||
|---|---|---|---|---|
| CLTOT (L/h) | 1.96 | [18] | 25.3 | [23] (20) |
| FM1 | 0.64 | (fixed) | 6.3 | [62] (65) |
| CL2E (L/h) | 9.71 | [5] | 27.9 | [16] (29) |
| CL3E (L/h) | 726 | [25] | 42.3 | [16] (14) |
| CL4E (L/h) | 543 | [6] | 34.5 | [14] (23) |
| KASTAR (h−1) | 0.409 | [24] | 75.3 | [19] (63) |
| V1 (L) | 161 | [7.0] | 13.6 | [27] (22) |
| V2 (L) | 39.2 | [45] | 149.7 | [30] (64) |
| CLTOT induction effect | 1.73 | [19] | ‐ | ‐ |
| CL3E induction effect | 1.63 | [27] | ‐ | ‐ |
| Cov.Eff.(DOSE on CLTOT) | 1.36 | [10] | ‐ | ‐ |
| Cov.Eff.(EPHXAGHOM on CL2E) | 0.502 | [14] | ||
| Cov.Eff.(PHT on CLTOT) | 1.93 | [25] | ‐ | ‐ |
| CBZ resid err.Sd | 0.221 | [10] (12) | ‐ | ‐ |
| CBZE resid err.Sd | 0.310 | [6.0] (12) | ‐ | ‐ |
| 2OH‐CBZ resid err.Sd | 0.297 | [8.0] (13) | ‐ | ‐ |
| 3OH‐CBZ resid err.Sd | 0.362 | [12.0] (11) | ‐ | ‐ |
Cov.eff: covariate effect; FM: metabolite fraction; PHT: phenytoin; %RSE: percentage relative standard error
A summary of relevant computational runs for building the final covariate model is given in Table 6, with the final model including the effects of total daily dose (DGRP), EPHX1‐416G/G (homozygous mutant) genotype and coadministration of phenytoin as significant covariates on CBZ total clearance. None of the other SNP genotypes investigated showed a statistically significant effect on clearance of either CBZ or CBZE during the backward deletion component of model selection. A table of changes in objective function following incorporation of the various covariates investigated during model building is provided in the Supplementary Material.
TABLE 6.
Covariate model building
| Covariate model | Objective function value | Difference in objective function value |
|---|---|---|
| Base model | −14 739.0 | |
| DOSE on CLTOT | −14 804.3 | −65.3 |
| EPHXAGHOM on CL2E | −14 747.7 | −8.7 |
| PHT on CLTOT | −14 744.3 | −5.3 |
| DOSE on CLTOT & EPHXAGHOM on CL2E | −14 813.0 | −8.6 |
| DOSE & PHT on CLTOT | −14 810.2 | −5.9 |
| DOSE & PHT on CLTOT & EPHXAGHOM on CL2E | −14 819.2 | −6.2 |
PHT: phenytoin
The final model equation for an individual subject's CLTOT and CL3E are given in equations 17 and 18:
| (17) |
| (18) |
4. DISCUSSION
A population PK model was developed to investigate the effects of SNPs in genes involved with metabolism and efflux transport of CBZ on the plasma PK of CBZ and 3 metabolites in healthy volunteers and epilepsy patients. The influence of demographic and clinical characteristics of the patients on the clearance of CBZ was also examined. The model revealed that the EPHX1‐416G/G genotype was a significant covariate for the clearance of CBZE. Concomitant therapy with phenytoin and the total daily dose of CBZ were also significant covariates for the clearance of CBZ. However, none of the other SNPs investigated exerted a demonstrable effect on CBZ PK.
The gene EPHX1 encodes microsomal epoxide hydrolase, which catalyses hydrolysis of the electrophilic CBZE to DiOH‐CBZ. 6 The SNP EPHX1 c.416A > G is associated with reduced epoxide hydrolase activity. 39 Three subjects in our model possessed the EPHX1‐416G/G genotype. When compared with EPHX1‐416G/A and ‐416A/A genotype subjects, their clearance of CBZE was reduced by approximately 50%. CBZE retains anticonvulsant activity but elevated plasma levels, secondary to concurrent brivaracetam therapy, have been associated with reduced tolerability to CBZ treatment and an increase in adverse events such as blurred vision, dizziness and fatigue. 60 Brivaracetam inhibits microsomal epoxide hydrolase and led to a rise in CBZE levels by a median of 83.9% (range 57.9–341.7%), which highlights that a 50% reduction in clearance that has been associated with EPHX ‐416G/G genotype may also be clinically relevant. One previous investigation reported that patients with the EPHX c.416A > G SNP had higher adjusted plasma CBZ levels 30 whilst another identified lower adjusted levels of DiOH‐CBZ 27 in patients receiving CBZ. However, 3 studies were unable to detect differences between plasma levels of either CBZ or its metabolites in patients with the EPHX1 c. 416A > G polymorphism. 26 , 29 , 31
A second SNP in EPHX1, namely c.337 T > C, was not associated with altered clearance of CBZE in our model. A study in Han Chinese patients with CBZ‐induced SJS/TEN found a significant association between EPHX1 c.337 T > C polymorphisms and the development of SJS/TEN. 61 The authors postulate that the polymorphism may contribute to the risk of CBZ‐induced SJS/TEN by increasing the plasma concentration of CBZE. The same SNP in EPHX1 was associated with increased levels of CBZE in 1 study 24 but no differences were detected in the current study or in studies by others. 26 , 27 , 30 In our model, SNPs in genes of CYPs, other enzymes (including UGT2B7, generally regarded as a detoxification enzyme) and drug transporters were not significant covariates of the PK of CBZ.
The influence of phenytoin therapy on CBZ's metabolism has been recognised in several other population PK models. 11 , 12 , 13 , 15 Phenytoin increases the metabolism of many drugs, including immunosuppressants, 62 chemotherapeutic agents 63 and antiretroviral drugs, 64 through induction of multiple CYP isoforms and upregulation of P‐gp. 65 Patients who are coprescribed CBZ with phenytoin potentially require larger doses of CBZ to maintain plasma levels.
The total daily dose of CBZ was another significant covariate in the population PK model. All subjects in the study were prescribed controlled release formulations of CBZ. There was a positive correlation between total daily dose and clearance of the drug. The effect of dose on clearance may be explained by a reduction in bioavailability and increase in clearance through greater autoinduction at higher doses. 66 Several other population PK models of CBZ have reported that the total daily dose is a significant covariate. 10 , 67 , 68 , 69 , 70
Concomitant treatments with sodium valproate, 11 , 12 , 14 , 69 phenobarbital 11 , 12 , 13 , 67 , 69 , 71 and felbamate 13 have been reported to be significant covariates in population PK models of CBZ. None of those antiepileptic drugs (AEDs) achieved a significant effect in the current model. This is most likely to have been due to the small numbers of subjects receiving any of these drugs. No other AEDs were significant in our model. Concomitant treatment with omeprazole (n = 6) and statins (n = 13) were also investigated as covariates in the population PK model. Omeprazole is a proton‐pump inhibitor that is associated with drug–drug interactions secondary to its inhibition of CYP2C19 and CYP3A4. 72 Statins have been reported to inhibit CYP enzymes, 73 and they act as substrates of the organic anion transporters OATP1B1 and OATP1B3 and of P‐gp. 74 Neither was found to affect metabolism of CBZ significantly.
There is considerable uncertainty regarding the influence of genetic variation on metabolism of CBZ, with conflicting results from several studies (Table 1). Up to 1/3 of patients with epilepsy do not respond to AED therapy and the transporter hypothesis proposes that over expression of efflux transporters such as ABCB1 in the blood–brain barrier limits access of AEDs to the epileptic focus. 75 Earlier studies have reported the requirement for higher CBZ maintenance doses, 24 increased clearance of CBZ 29 as well as both higher 32 and lower 33 plasma levels of CBZ in patients with SNPs in ABCB1 and ABCC2. In our population PK model 6 SNPs in the genes ABCB1 and ABCC2, which both encode drug efflux transporters, did not demonstrate a significant effect on the PK of CBZ.
CYP3A4 is the major CYP isoform responsible for oxidative metabolism (aromatic hydroxylation and 10,11‐epoxidation) of CBZ. 4 , 76 The allele CYP3A4*1G has been associated with lower serum levels of CBZ and CBZE in Chinese patients with epilepsy. 27 However, 3 other analyses were not able to replicate this association, 24 , 25 , 30 and our own population PK model did not detect a significant association between SNPs in CYP isoforms and clearance of CBZ. It is possible that the multiple enzymatic pathways involved in CBZ metabolism, 5 and the multiplicity of CYP isoforms catalysing each of those pathways, 4 , 5 may compensate for the altered functioning of the gene for a particular enzyme, thereby limiting the effect on total drug clearance of individual SNPs. Furthermore, most of the subjects in our study who carried a variant were heterozygotes; their functional allele will have limited the consequences of the polymorphism. Collectively, the effect of the PK gene variants investigated in this study might be relatively small, and thus much larger sample sizes would be required to detect the effect.
HLA‐B*15:02 and HLA‐A*31:01 are recognised as biomarkers for susceptibility to CBZ‐induced hypersensitivity reactions. 16 However, carriage of these HLA alone may be insufficient to trigger a hypersensitivity reaction. A recent experimental study has demonstrated that it is CBZE, and not CBZ, which binds to the HLA‐B*15:02 molecule and alters its peptide‐binding motif for B*15:02‐restricted peptides. 22 It is possible that susceptibility to CBZ hypersensitivity reactions is dependent on the presence of specific immunogenetic factors, such as carriage of a risk HLA genotype, and combinations of allelic enzyme variants in the metabolic pathways of CBZ that generate above threshold levels of immunoreactive metabolites. For example, 1 study reported that patients required higher dosages of CBZ only if they possessed a combination of SNPs in both UGT2B7 and ABCC2. 24 Similarly, combined HLA and CYP2C9*3 pharmacogenetic screening for phenytoin hypersensitivity improved the sensitivity and specificity of predictive testing. 20
We have previously reported that chemically reactive metabolites of CBZ, namely CBZE and arene oxides, are able to form covalent adducts with human serum albumin. 5 We hypothesise that reduced clearance of CBZE in subjects with the EPHX1‐416G/G genotype leads to greater formation of covalent protein adducts, ultimately resulting in an increased frequency of immune‐mediated hypersensitivity reactions, especially in those expressing the HLA risk allele.
Limitations of the current study include the relatively small number of subjects and the limited number of elderly patients. Consequently, the power of the study might have been insufficient to detect significant covariates with small effects or covariates affecting extremes of age only. In addition, several P450 isoforms known to be significant contributors to CBZ hydroxylation, such as CYP1A2, 4 were not investigated as part of the present analyses. The CYP1A2 (c.‐163C > A) genotype has been shown to affect CBZ's PK in children. 70 Finally, the recent discovery of 856 SNPs in human CYP3A4, 77 which codes the principal monoxygenase catalysing CBZ 10,11‐epoxidation, 76 indicates PK might be influenced by many more genetic polymorphisms than were included in this or any other study. To make significant progress, future studies should attempt to characterise the genetic polymorphisms on a much larger scale in patients with CBZ hypersensitivity reactions in order to determine the influence of many more variants on metabolism pathways within a relevant, pathological, context.
In conclusion, a population PK model has been developed for CBZ which successfully incorporates certain clinical parameters of adult epilepsy patients. We identified carriage of the EPHX1‐416G/G genotype as being a significant covariate of CBZE clearance and concomitant treatment with phenytoin and CBZ dose as significant covariates in affecting the PK of CBZ.
COMPETING INTERESTS
M.P. receives research funding from various organisations including the MRC, NIHR, EU Commission and Health Education England. He has also received partnership funding for the following: MRC Clinical Pharmacology Training Scheme (cofunded by MRC and Roche, UCB, Eli Lilly and Novartis); a PhD studentship jointly funded by EPSRC and Astra Zeneca; and grant funding from VistaGen Therapeutics. He has also unrestricted educational grant support for the UK Pharmacogenetics and Stratified Medicine Network from Bristol‐Myers Squibb and UCB. He has developed an HLA genotyping panel with MC Diagnostics, but does not benefit financially from this. None of the funding declared above has been used for the current paper.
CONTRIBUTORS
Study concept and design: V.L.M.Y., B.K.P., A.G.M. and M.P. Acquisition of Data: V.L.M.Y., X.M., J.L.M., D.F.C. and A.G.M. Model Development: V.L.M.Y. and H.P. Analysis or interpretation of Data: All authors. Drafting of the manuscript: V.L.M.Y., H.P. and J.L.M. Critical revision of the manuscript for important intellectual content: All authors.
Supporting information
Table S1 Inclusion and exclusion criteria for PICME I and PICME II clinical studies
Table S2 Objective function changes for selected covariate runs
Figure S1 VPC for the final PK model for each analyte in PICME I
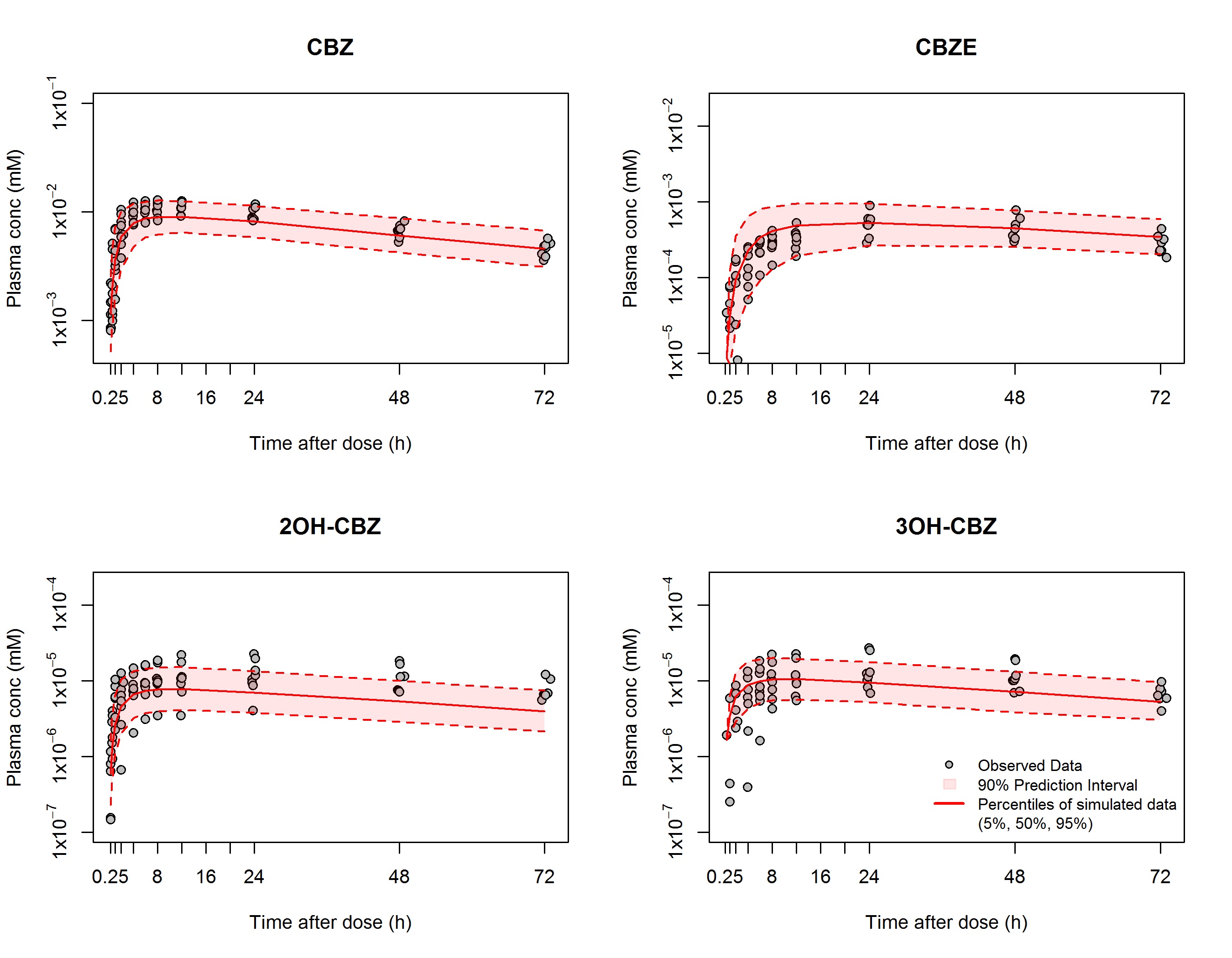{kind=link}
Figure S2 VPC for the final PK model for each analyte in PICME II (autoinduction group)
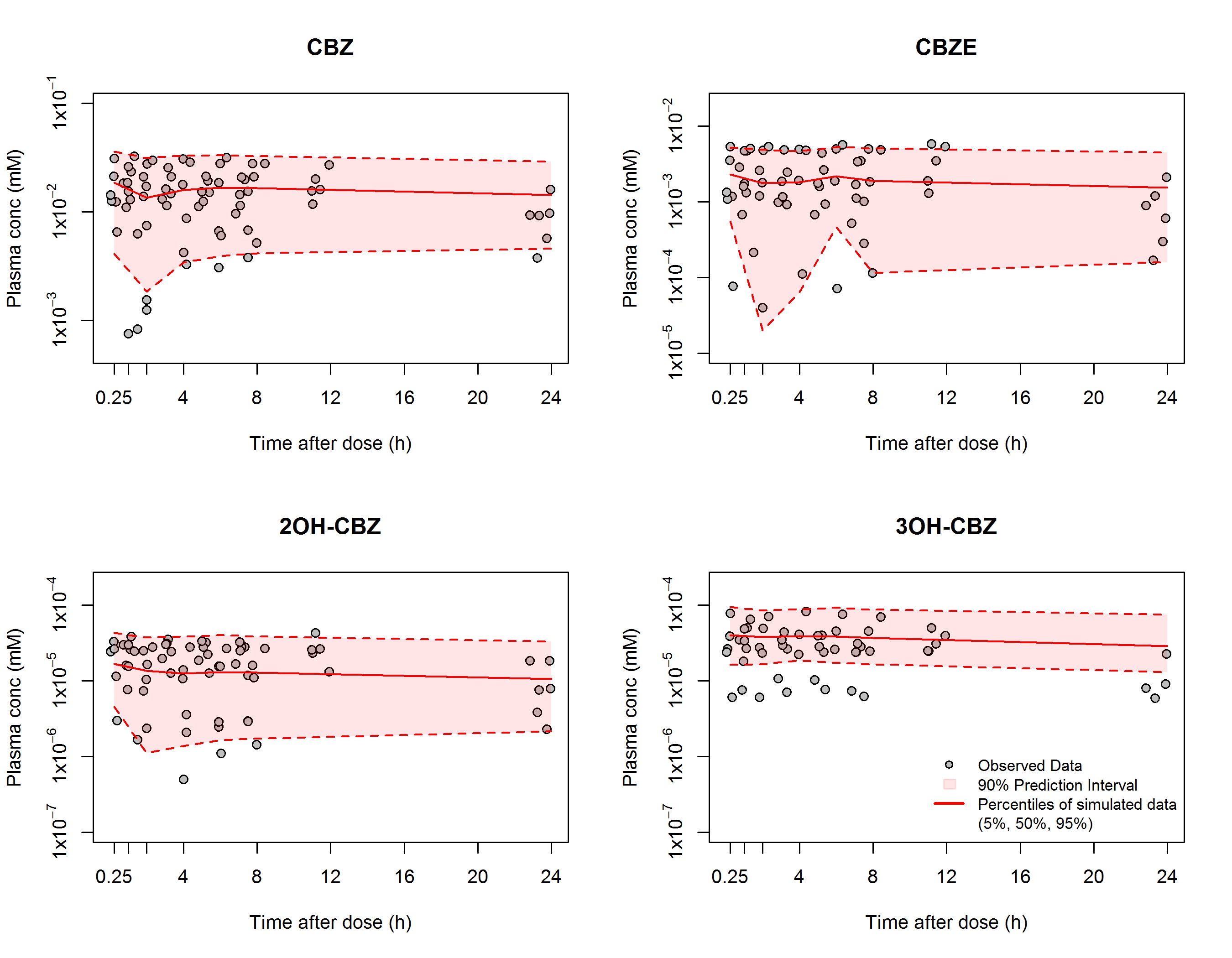{kind=link}
Figure S3 VPC for the final PK model for each analyte in PICME II (maintenance group)
{kind=link}
Figure S4 Raw concentration data for all analytes according to study group
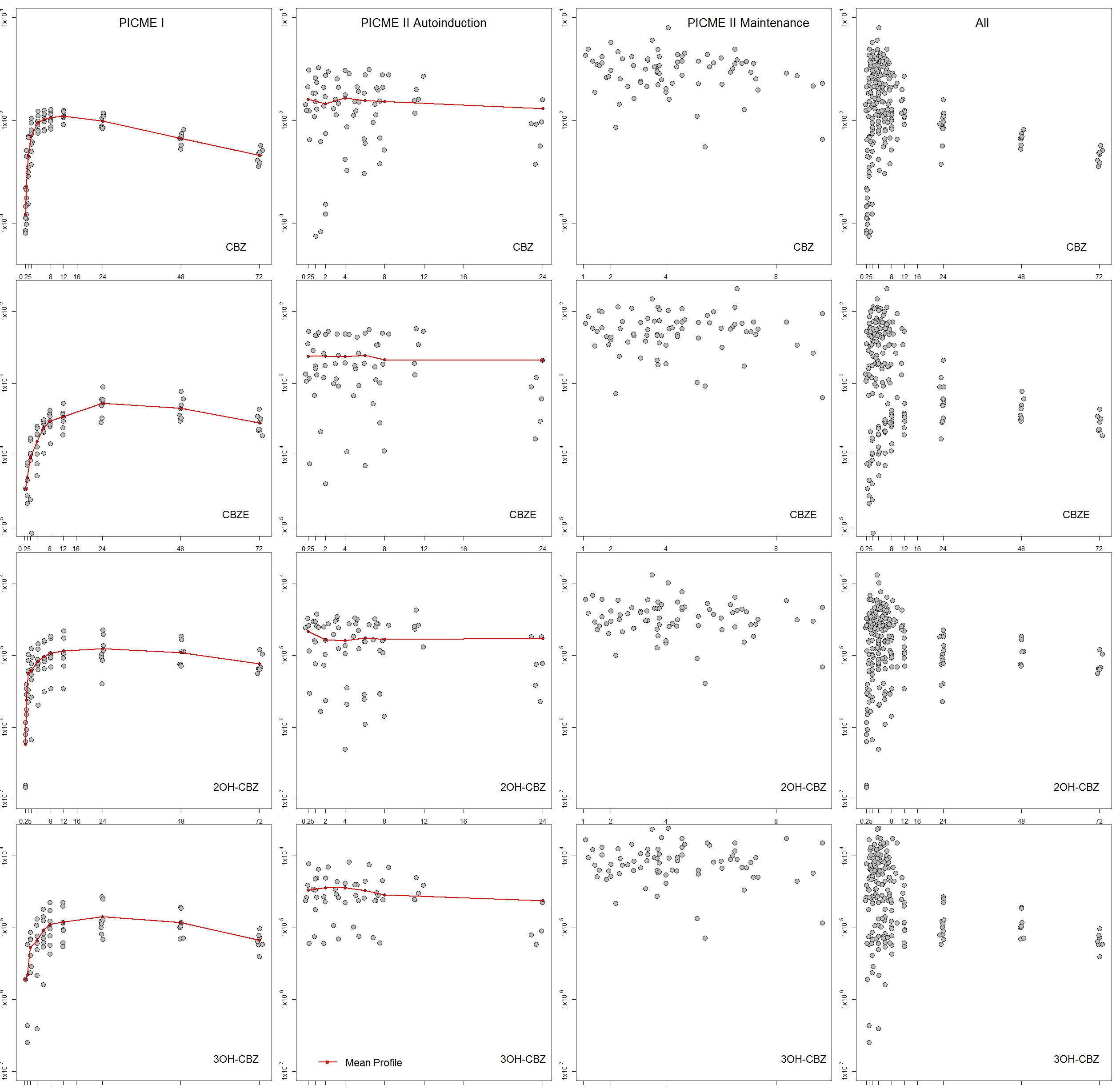{kind=link}
Figure S5 VPC for the final PK model for each analyte in all study groups
{kind=link}
Data S1 Closed form analytical solution for differential equations of structural model
Data S2 Coefficients of exponential terms
Data S3 Analytical solutions for structural model states as functions of dose and time
ACKNOWLEDGEMENTS
Dr R. Mills from ICON Development Solutions (Manchester) for input into the clinical trial design. Prof L Aarons and Dr K Ogungbenro from the Centre of Applied Pharmacokinetic Research at the University of Manchester for input into the population‐PK model. V.L.M.Y. was an MRC Clinical Training Fellow supported by the North West England Medical Research Council Fellowship Scheme in Clinical Pharmacology and Therapeutics, which is funded by the Medical Research Council (grant number G1000417/94909), ICON plc, GlaxoSmithKline, AstraZeneca, and the Medicines Evaluation Unit Ltd. M.P. was Director of this Fellowship programme. This work was also supported by the European Union's Seventh Framework Programme FP7/2007‐2013 under grant agreement number 238132 and the Medical Research Council Centre for Drug Safety Science (Grant code MR/L006758/1). M.P. is a NIHR Emeritus Senior Investigator and has received funding from the NHS Chair of Pharmacogenetics (from the UK Department of Health). North West England Medical Research Council Fellowship Scheme in Clinical Pharmacology and Therapeutics, which is funded by the Medical Research Council, ICON plc, GlaxoSmithKline, AstraZeneca and Medicines Evaluation Unit Ltd, Grant/Award Number: G1000417/94909; European Union's Seventh Framework Programme FP7/2007–2013, Grant Award/Number: 238132; Medical Research Council Centre for Drug Safety Science, Grant Award/Number: MR/L006758/1.
Yip VLM, Pertinez H, Meng X, et al. Evaluation of clinical and genetic factors in the population pharmacokinetics of carbamazepine. Br J Clin Pharmacol. 2021;87:2572–2588. 10.1111/bcp.14667
The authors confirm that the Principal Investigator for this paper is Vincent Yip and that he had direct clinical responsibility for patients.
Clinical trial registration numbers: ISRCTN00131154. ISRCTN62125126.
Funding information FP7 Health, Grant/Award Number: 238132; ICON plc, GlaxoSmithKline, AstraZeneca, and the Medicines Evaluation Unit Ltd. M.P; Medical Research Council, Grant/Award Numbers: G1000417/94909, MR/L006758/1; North West England Medical Research Council Fellowship Scheme in Clinical Pharmacology and Therapeutics
Contributor Information
Vincent L.M. Yip, Email: vyip@liverpool.ac.uk.
Henry Pertinez, Email: vyip@liverpool.ac.uk.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- 1. Marson AG, Al‐Kharusi AM, Alwaidh M, et al. The SANAD study of effectiveness of carbamazepine, gabapentin, lamotrigine, oxcarbazepine, or topiramate for treatment of partial epilepsy: an unblinded randomised controlled trial. The Lancet. 2007;369(9566):1000‐1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Pearce RE, Lu W, Wang Y, Uetrecht JP, Correia MA, Leeder JS. Pathways of carbamazepine bioactivation in vitro. III. The role of human cytochrome P450 enzymes in the formation of 2,3‐Dihydroxycarbamazepine. Drug Metab Dispos. 2008;36(8):1637‐1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Pearce RE, Uetrecht JP, Leeder JS. Pathways of carbamazepine bioactivation in vitro: ii. The role of human cytochrome P450 enzymes in the formation of 2‐hydroxyiminostilbene. Drug Metab Dispos. 2005;33(12):1819‐1826. [DOI] [PubMed] [Google Scholar]
- 4. Pearce RE, Vakkalagadda GR, Leeder JS. Pathways of carbamazepine bioactivation in vitro I. characterization of human cytochromes P450 responsible for the formation of 2‐ and 3‐Hydroxylated metabolites. Drug Metab Dispos. 2002;30(11):1170‐1179. [DOI] [PubMed] [Google Scholar]
- 5. Yip VLM, Meng X, Maggs JL, et al. Mass spectrometric characterization of circulating covalent protein adducts derived from epoxide metabolites of carbamazepine in patients. Chem Res Toxicol. 2017;30(7):1419‐1435. [DOI] [PubMed] [Google Scholar]
- 6. Thorn CF, Klein TE, Altman RB, et al. PharmGKB summary: carbamazepine pathway. Pharmacogenet Genomics. 2011;21(12):906‐910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Staines AG, Coughtrie MWH, Burchell B. N‐Glucuronidation of carbamazepine in human tissues is mediated by UGT2B7. J Pharmacol Exp Ther. 2004;311(3):1131‐1137. [DOI] [PubMed] [Google Scholar]
- 8. Lu W, Uetrecht JP. Peroxidase‐mediated bioactivation of hydroxylated metabolites of carbamazepine and phenytoin. Drug Metab Dispos. 2008;36(8):1624‐1636. [DOI] [PubMed] [Google Scholar]
- 9. Schulz M, Iwersen‐Bergmann S, Andresen H, Schmoldt A. Therapeutic and toxic blood concentrations of nearly 1,000 drugs and other xenobiotics. Crit Care. 2012;16(4):R136. 10.1186/cc11441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Milovanovic JR, Jankovic SM. Factors influencing carbamazepine pharmacokinetics in children and adults: population pharmacokinetic analysis. Int J Clin Pharmacol Ther. 2011;49(7):428‐436. [DOI] [PubMed] [Google Scholar]
- 11. Jiao Z, Zhong MK, Shi XJ, Hu M, Zhang JH. Population pharmacokinetics of carbamazepine in Chinese epilepsy patients. Ther Drug Monit. 2003;25(3):279‐286. [DOI] [PubMed] [Google Scholar]
- 12. Gray AL, Botha JH, Miller R. A model for the determination of carbamazepine clearance in children on mono‐ and polytherapy. Eur J Clin Pharmacol. 1998;54(4):359‐362. [DOI] [PubMed] [Google Scholar]
- 13. Graves NM, Brundage RC, Wen Y, et al. Population pharmacokinetics of carbamazepine in adults with epilepsy. Pharmacotherapy. 1998;18(2):273‐281. [PubMed] [Google Scholar]
- 14. Jankovic SM, Jovanovic D, Milovanovic JR. Pharmacokinetic modeling of carbamazepine based on clinical data from Serbian epileptic patients. Methods Find Exp Clin Pharmacol. 2008;30(9):707‐713. [DOI] [PubMed] [Google Scholar]
- 15. Punyawudho B, Ramsay ER, Brundage RC, Macias FM, Collins JF, Birnbaum AK. Population pharmacokinetics of carbamazepine in elderly patients. Ther Drug Monit. 2012;34(2):176‐181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Yip VL, Marson AG, Jorgensen AL, Pirmohamed M, Alfirevic A. HLA genotype and carbamazepine‐induced cutaneous adverse drug reactions: A systematic review. Clin Pharmacol Ther. 2012;92(6):757‐765. [DOI] [PubMed] [Google Scholar]
- 17. Locharernkul C, Loplumlert J, Limotai C, et al. Carbamazepine and phenytoin induced Stevens‐Johnson syndrome is associated with HLA‐B*1502 allele in Thai population. Epilepsia. 2008;49(12):2087‐2091. [DOI] [PubMed] [Google Scholar]
- 18. Hung SI, Chung WH, Liu ZS, et al. Common risk allele in aromatic antiepileptic‐drug induced Stevens‐Johnson syndrome and toxic epidermal necrolysis in Han Chinese. Pharmacogenomics. 2010;11(3):349‐356. [DOI] [PubMed] [Google Scholar]
- 19. Chung W‐H, Chang W‐C, Lee Y‐S, et al. Genetic variants associated with phenytoin‐related severe cutaneous adverse reactions. JAMA. 2014;312(5):525‐534. [DOI] [PubMed] [Google Scholar]
- 20. Su S‐C, Chen C‐B, Chang W‐C, et al. HLA alleles and CYP2C9*3 as predictors of phenytoin hypersensitivity in east Asians. Clinical Pharmacol Ther. 2019;105(2):476‐485. [DOI] [PubMed] [Google Scholar]
- 21. Knowles SR, Uetrecht J, Shear NH. Idiosyncratic drug reactions: the reactive metabolite syndromes. The Lancet. 2000;356(9241):1587‐1591. [DOI] [PubMed] [Google Scholar]
- 22. Simper GS, Hò GGT, Celik AA, et al. Carbamazepine‐mediated adverse drug reactions: CBZ‐10, 11‐epoxide but not carbamazepine induces the alteration of peptides presented by HLA‐B15: 02. J Immunol Res. 2018;2018:5086503. 10.1155/2018/5086503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Jaramillo NM, Galindo IF, Vazquez AO, Cook HJ, A LL, Lopez ML. Pharmacogenetic potential biomarkers for carbamazepine adverse drug reactions and clinical response. Drug Metabol Drug Interact. 2014;29(2):67‐79. [DOI] [PubMed] [Google Scholar]
- 24. Ma CL, Jiao Z, Wu XY, Hong Z, Wu ZY, Zhong MK. Association between PK/PD‐involved gene polymorphisms and carbamazepine‐individualized therapy. Pharmacogenomics. 2015;16(13):1499‐1512. [DOI] [PubMed] [Google Scholar]
- 25. Wang P, Yin T, Ma HY, et al. Effects of CYP3A4/5 and ABCB1 genetic polymorphisms on carbamazepine metabolism and transport in Chinese patients with epilepsy treated with carbamazepine in monotherapy and bitherapy. Epilepsy Res. 2015;117:52‐57. [DOI] [PubMed] [Google Scholar]
- 26. Caruso A, Bellia C, Pivetti A, et al. Effects of EPHX1 and CYP3A4 polymorphisms on carbamazepine metabolism in epileptic patients. Pharmgenomics Pers Med. 2014;7:117‐120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zhu X, Yun W, Sun X, Qiu F, Zhao L, Guo Y. Effects of major transporter and metabolizing enzyme gene polymorphisms on carbamazepine metabolism in Chinese patients with epilepsy. Pharmacogenomics. 2014;15(15):1867‐1879. [DOI] [PubMed] [Google Scholar]
- 28. Panomvana D, Traiyawong T, Towanabut S. Effect of CYP3A5 genotypes on the pharmacokinetics of carbamazepine when used as monotherapy or co‐administered with phenytoin, phenobarbital or valproic acid in Thai patients. J Pharm Pharm Sci. 2013;16(4):502‐510. [DOI] [PubMed] [Google Scholar]
- 29. Puranik YG, Birnbaum AK, Marino SE, et al. Association of carbamazepine major metabolism and transport pathway gene polymorphisms and pharmacokinetics in patients with epilepsy. Pharmacogenomics. 2013;14(1):35‐45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yun W, Zhang F, Hu C, et al. Effects of EPHX1, SCN1A and CYP3A4 genetic polymorphisms on plasma carbamazepine concentrations and pharmacoresistance in Chinese patients with epilepsy. Epilepsy Res. 2013;107(3):231‐237. [DOI] [PubMed] [Google Scholar]
- 31. Hung CC, Chang WL, Ho JL, et al. Association of polymorphisms in EPHX1, UGT2B7, ABCB1, ABCC2, SCN1A and SCN2A genes with carbamazepine therapy optimization. Pharmacogenomics. 2012;13(2):159‐169. [DOI] [PubMed] [Google Scholar]
- 32. Sterjev Z, Trencevska GK, Cvetkovska E, et al. The association of C3435T single‐nucleotide polymorphism, Pgp‐glycoprotein gene expression levels and carbamazepine maintenance dose in patients with epilepsy. Neuropsychiatr Dis Treat. 2012;8:191‐196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Meng H, Guo G, Ren J, Zhou H, Ge Y, Guo Y. Effects of ABCB1 polymorphisms on plasma carbamazepine concentrations and pharmacoresistance in Chinese patients with epilepsy. Epilepsy Behav. 2011;21(1):27‐30. [DOI] [PubMed] [Google Scholar]
- 34. Park PW, Seo YH, Ahn JY, Kim KA, Park JY. Effect of CYP3A5*3 genotype on serum carbamazepine concentrations at steady‐state in Korean epileptic patients. J Clin Pharm Ther. 2009;34(5):569‐574. [DOI] [PubMed] [Google Scholar]
- 35. Hodges LM, Markova SM, Chinn LW, et al. Very important pharmacogene summary: ABCB1 (MDR1, P‐glycoprotein). Pharmacogenet Genomics. 2011;21(3):152‐161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bruhn O, Cascorbi I. Polymorphisms of the drug transporters ABCB1, ABCG2, ABCC2 and ABCC3 and their impact on drug bioavailability and clinical relevance. Expert Opin Drug Metab Toxicol. 2014;10(10):1337‐1354. [DOI] [PubMed] [Google Scholar]
- 37. Kwara A, Lartey M, Boamah I, et al. Interindividual variability in pharmacokinetics of generic nucleoside reverse transcriptase inhibitors in TB/HIV‐coinfected Ghanaian patients: UGT2B7*1c is associated with faster zidovudine clearance and glucuronidation. J Clin Pharmacol. 2009;49(9):1079‐1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Matic M, Norman E, Rane A, et al. Effect of UGT2B7‐900G>A (−842G>A; rs7438135) on morphine glucuronidation in preterm newborns: results from a pilot cohort. Pharmacogenomics. 2014;15(12):1589‐1597. [DOI] [PubMed] [Google Scholar]
- 39. Nakajima Y, Saito Y, Shiseki K, et al. Haplotype structures of EPHX1 and their effects on the metabolism of carbamazepine‐10,11‐epoxide in Japanese epileptic patients. Eur J Clin Pharmacol. 2005;61(1):25‐34. [DOI] [PubMed] [Google Scholar]
- 40. Aquilante CL, Niemi M, Gong L, Altman RB, Klein TE. PharmGKB summary: very important pharmacogene information for cytochrome P450, family 2, subfamily C, polypeptide 8. Pharmacogenet Genomics. 2013;23(12):721‐728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Scott SA, Sangkuhl K, Shuldiner AR, et al. PharmGKB summary: very important pharmacogene information for cytochrome P450, family 2, subfamily C, polypeptide 19. Pharmacogenet Genomics. 2012;22(2):159‐165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Thorn CF, Lamba JK, Lamba V, Klein TE, Altman RB. PharmGKB summary: very important pharmacogene information for CYP2B6. Pharmacogenet Genomics. 2010;20(8):520‐523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lamba J, Hebert JM, Schuetz EG, Klein TE, Altman RB. PharmGKB summary: very important pharmacogene information for CYP3A5. Pharmacogenet Genomics. 2012;22(7):555‐558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hansson M, Olsson I, Nauseef WM. Biosynthesis, processing, and sorting of human myeloperoxidase. Arch Biochem Biophys. 2006;445(2):214‐224. [DOI] [PubMed] [Google Scholar]
- 45. Lunde I, Bremer S, Midtvedt K, et al. The influence of CYP3A, PPARA, and POR genetic variants on the pharmacokinetics of tacrolimus and cyclosporine in renal transplant recipients. Eur J Clin Pharmacol. 2014;70(6):685‐693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Miyata‐Nozaka Y, Zain SM, Taguchi M, Shigeyama M, Isobe T, Hanioka N. Carbamazepine 10,11‐epoxidation in human liver microsomes: influence of the CYP3A5*3 polymorphism. Pharmazie. 2017;72(12):747‐750. [DOI] [PubMed] [Google Scholar]
- 47. Zhang C, Zuo Z, Kwan P, Baum L. In vitro transport profile of carbamazepine, oxcarbazepine, eslicarbazepine acetate, and their active metabolites by human P‐glycoprotein. Epilepsia. 2011;52(10):1894‐1904. [DOI] [PubMed] [Google Scholar]
- 48. Shen XM, Cheng J. Effects of MDR1 (C3435T) polymorphism on resistance, uptake, and efflux to antiepileptic drugs DNA. Cell Biol. 2019;38(3):250‐255. [DOI] [PubMed] [Google Scholar]
- 49. Hatta FH, Aklillu E. P450 (cytochrome) oxidoreductase gene (POR) common variant (POR*28) significantly alters CYP2C9 activity in Swedish, but not in Korean healthy subjects. Omics: A J Integ Biol. 2015;19(12):777‐781. [DOI] [PubMed] [Google Scholar]
- 50. Elens L, Nieuweboer AJ, Clarke SJ, et al. Impact of POR*28 on the clinical pharmacokinetics of CYP3A phenotyping probes midazolam and erythromycin. Pharmacogenet Genomics. 2013;23(3):148‐155. [DOI] [PubMed] [Google Scholar]
- 51. Spina E. Carbamazepine: Chemistry, Biotransformation and Pharmacokinetics. In: Levy RH, ed. Antiepileptic Drugs. 5th ed. Philadelphia, PA, USA: Wolters Kluwer; 2002:236‐246. [Google Scholar]
- 52. Bourgeois BF, Wad N. Individual and combined antiepileptic and neurotoxic activity of carbamazepine and carbamazepine‐10,11‐epoxide in mice. J Pharmacol Exp Ther. 1984;231(2):411‐415. [PubMed] [Google Scholar]
- 53. Bellu G, Saccomani MP, Audoly S, D'Angio L. DAISY: a new software tool to test global identifiability of biological and physiological systems. Comput Methods Programs Biomed. 2007;88(1):52‐61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Svensson RJ, Aarnoutse RE, Diacon AH, et al. A population pharmacokinetic model incorporating Saturable pharmacokinetics and autoinduction for high rifampicin doses. Clin Pharmacol Ther. 2018;103(4):674‐683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Hutmacher MM, Kowalski KG. Covariate selection in pharmacometric analyses: a review of methods. Br J Clin Pharmacol. 2015;79(1):132‐147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Harding SD, Sharman JL, Faccenda E, et al. The IUPHAR/BPS guide to PHARMACOLOGY in 2018: updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucleic Acids Res. 2018;46(D1):D1091‐d106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Alexander SPH, Kelly E, Mathie A, et al. THE CONCISE GUIDE TO PHARMACOLOGY 2019/20: Introduction and other protein targets. Br J Pharmacol. 2019;176(S1):S1‐S20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Jiao Z, Shi XJ, Zhao ZG, Zhong MK. Population pharmacokinetic modeling of steady state clearance of carbamazepine and its epoxide metabolite from sparse routine clinical data. J Clin Pharm Ther. 2004;29(3):247‐256. [DOI] [PubMed] [Google Scholar]
- 59. Garnett WR, Levy B, McLean AM, et al. Pharmacokinetic evaluation of twice‐daily extended‐release carbamazepine (CBZ) and four‐times‐daily immediate‐release CBZ in patients with epilepsy. Epilepsia. 1998;39(3):274‐279. [DOI] [PubMed] [Google Scholar]
- 60. Steinhoff BJ, Bacher M, Blickhan M, et al. Is brivaracetam‐induced elevation of carbamazepine‐epoxide levels common and clinically relevant? — A case series. Epilepsy Res. 2020;159:106236. 10.1016/j.eplepsyres.2019.106236 [DOI] [PubMed] [Google Scholar]
- 61. He XJ, Jian LY, He XL, et al. Association of ABCB1, CYP3A4, EPHX1, FAS, SCN1A, MICA, and BAG6 polymorphisms with the risk of carbamazepine‐induced Stevens‐Johnson syndrome/toxic epidermal necrolysis in Chinese Han patients with epilepsy. Epilepsia. 2014;55(8):1301‐1306. [DOI] [PubMed] [Google Scholar]
- 62. D'Souza MJ, Pollock SH, Solomon HM. Cyclosporine‐phenytoin interaction. Drug Metab Dispos. 1988;16(2):256‐258. [PubMed] [Google Scholar]
- 63. Pursche S, Schleyer E, von Bonin M, et al. Influence of enzyme‐inducing antiepileptic drugs on trough level of imatinib in glioblastoma patients. Curr Clin Pharmacol. 2008;3(3):198‐203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Lim ML, Min SS, Eron JJ, et al. Coadministration of lopinavir/ritonavir and phenytoin results in two‐way drug interaction through cytochrome P‐450 induction. J Acquir Immune Defic Syndr. 2004;36(5):1034‐1040. [DOI] [PubMed] [Google Scholar]
- 65. Brodie MJ, Mintzer S, Pack AM, Gidal BE, Vecht CJ, Schmidt D. Enzyme induction with antiepileptic drugs: cause for concern? Epilepsia. 2013;54(1):11‐27. [DOI] [PubMed] [Google Scholar]
- 66. Kudriakova TB, Sirota LA, Rozova GI, Gorkov VA. Autoinduction and steady‐state pharmacokinetics of carbamazepine and its major metabolites. Br J Clin Pharmacol. 1992;33(6):611‐615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Delgado Iribarnegaray MF, Santo Bueldga D, Garcia Sanchez MJ, Otero MJ, Falcao AC, Dominguez‐Gil A. Carbamazepine population pharmacokinetics in children: mixed‐effect models. Ther Drug Monit. 1997;19(2):132‐139. [DOI] [PubMed] [Google Scholar]
- 68. Reith DM, Hooper WD, Parke J, Charles B. Population pharmacokinetic modeling of steady state carbamazepine clearance in children, adolescents, and adults. J Pharmacokinet Pharmacodyn. 2001;28(1):79‐92. [DOI] [PubMed] [Google Scholar]
- 69. Vucicevic K, Miljkovic B, Velickovic R, Pokrajac M, Mrhar A, Grabnar I. Population pharmacokinetic model of carbamazepine derived from routine therapeutic drug monitoring data. Ther Drug Monit. 2007;29(6):781‐788. [DOI] [PubMed] [Google Scholar]
- 70. Djordjevic N, Milovanovic DD, Radovanovic M, et al. CYP1A2 genotype affects carbamazepine pharmacokinetics in children with epilepsy. Eur J Clin Pharmacol. 2016;72(4):439‐445. [DOI] [PubMed] [Google Scholar]
- 71. Chan E, Lee HS, Hue SS. Population pharmacokinetics of carbamazepine in Singapore epileptic patients. Br J Clin Pharmacol. 2001;51(6):567‐576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Shirasaka Y, Sager JE, Lutz JD, Davis C, Isoherranen N. Inhibition of CYP2C19 and CYP3A4 by omeprazole metabolites and their contribution to drug‐drug interactions. Drug Metab Dispos. 2013;41(7):1414‐1424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Transon C, Leemann T, Dayer P. In vitro comparative inhibition profiles of major human drug metabolising cytochrome P450 isozymes (CYP2C9, CYP2D6 and CYP3A4) by HMG‐CoA reductase inhibitors. Eur J Clin Pharmacol. 1996;50(3):209‐215. [DOI] [PubMed] [Google Scholar]
- 74. Lennernas H, Fager G. Pharmacodynamics and pharmacokinetics of the HMG‐CoA reductase inhibitors. Similarities and differences. Clin Pharmacokinet. 1997;32(5):403‐425. [DOI] [PubMed] [Google Scholar]
- 75. Aronica E, Sisodiya SM, Gorter JA. Cerebral expression of drug transporters in epilepsy. Adv Drug Deliv Rev. 2012;64(10):919‐929. [DOI] [PubMed] [Google Scholar]
- 76. Kerr BM, Thummel KE, Wurden CJ, et al. Human liver carbamazepine metabolism. Role of CYP3A4 and CYP2C8 in 10,11‐epoxide formation. Biochem Pharmacol. 1994;47(11):1969‐1979. [DOI] [PubMed] [Google Scholar]
- 77. Guttman Y, Nudel A, Kerem Z. Polymorphism in cytochrome P450 3A4 is ethnicity related. Front Genet. 2019;10:224. 10.3389/fgene.2019.00224 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 Inclusion and exclusion criteria for PICME I and PICME II clinical studies
Table S2 Objective function changes for selected covariate runs
Figure S1 VPC for the final PK model for each analyte in PICME I
Figure S2 VPC for the final PK model for each analyte in PICME II (autoinduction group)
Figure S3 VPC for the final PK model for each analyte in PICME II (maintenance group)
Figure S4 Raw concentration data for all analytes according to study group
Figure S5 VPC for the final PK model for each analyte in all study groups
Data S1 Closed form analytical solution for differential equations of structural model
Data S2 Coefficients of exponential terms
Data S3 Analytical solutions for structural model states as functions of dose and time
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.