

Abstract
Charcot‐Marie‐Tooth disease (CMT) is one of the most common inherited neurodegenerative disorders with an increasing number of CMT‐associated variants identified as causative factors, however, there has been no effective therapy for CMT to date. Aminoacyl‐tRNA synthetases (aaRS) are essential enzymes in translation by charging amino acids onto their cognate tRNAs during protein synthesis. Dominant monoallelic variants of aaRSs have been largely implicated in CMT. Some aaRSs variants affect enzymatic activity, demonstrating a loss‐of‐function property. In contrast, loss of aminoacylation activity is neither necessary nor sufficient for some aaRSs variants to cause CMT. Instead, accumulating evidence from CMT patient samples, animal genetic studies or protein conformational analysis has pinpointed toxic gain‐of‐function of aaRSs variants in CMT, suggesting complicated mechanisms underlying the pathogenesis of CMT. In this review, we summarize the latest advances in studies on CMT‐linked aaRSs, with a particular focus on their functions. The current challenges, future direction and the promising candidates for potential treatment of CMT are also discussed.

Keywords: aminoacyl‐tRNA synthetases, animal model, charcot‐marie‐tooth disease, mutation, pathogenesis
Variants of some aminoacyl‐tRNA synthetases (aaRS) are strongly associated with Charcot–Marie–Tooth disease (CMT), one of the most common inherited neuromuscular disorders. Some variants affect enzymatic activity, showing loss‐of‐function effects. In contrast, evidence from CMT patient samples, animal genetic studies or protein conformational analysis has confirmed toxic gain‐of‐function of some aaRSs variants in CMT outside of aminoacylation. In this review, we will discuss the latest advances in studies on CMT‐associated aaRSs, with a particular focus on the complicated mechanisms in the pathogenesis of CMT.

Abbreviations
- aaRS(s)
aminoacyl‐tRNA synthetase(s)
- AlaRS
alanyl‐tRNA synthetase
- CMT
Charcot–Marie–Tooth disease
- CMT1
demyelinating CMT type 1
- CMT2
axonal CMT type 2
- CMT2N
CMT type 2N
- CMT2U
CMT type 2U
- CMT2W
CMT type 2W
- dHMN
distal hereditary motor neuronopathy
- dHMN‐V
dHMN type V
- DI‐CMT
dominant intermediate CMT
- DI‐CMTC
DI‐CMT type C
- EMAPII
endothelial monocyte‐activating polypeptide II
- EPRS
glutamyl‐prolyl‐tRNA synthetase
- GlyRS
glycyl‐tRNA synthetase
- HDAC6
histone deacetylase 6
- HDX
hydrogen‐deuterium exchange
- HisRS
histidyl‐tRNA synthetase
- iNPCs
induced neuronal progenitor cells
- MetRS
methionyl‐tRNA synthetase
- MS
mass spectrometry
- MSC
multi‐synthetase complex
- Nrp1
neuropilin‐1
- PBMCs
peripheral blood mononuclear cells
- SAXS
small‐angle X‐ray scattering
- Trk
tropomyosin receptor kinase
- TrkR(s)
Trk receptor(s)
- TrpRS
tryptophanyl‐tRNA synthetase
- TyrRS
tyrosyl‐tRNA synthetase
- VE‐cadherin
vascular endothelial‐cadherin
- VEGF
vascular endothelial growth factor
- WT
wild type
1. INTRODUCTION
Charcot–Marie–Tooth disease (CMT) is one of the most common inherited neuromuscular disorders with an estimated prevalence of 1/2,500 worldwide (Rossor et al., 2016; Skre, 1974). The major clinical manifestation of CMT is the degeneration of both motor and sensory peripheral nerves, leading to a loss of muscle tissue and touch sensation in bodily extremities (Patzko & Shy, 2011).
Based on electrophysiological and histopathological criteria, CMT is divided into two major groups: demyelinating CMT type 1 (CMT1) and axonal CMT type 2 (CMT2). CMT1 is the most prevalent type presented as demyelinating peripheral neuropathy, which manifests as markedly decreased nerve‐conduction velocities. In contrast, CMT2 accounts for approximately 20% of cases characterized by pathological axonal loss at nerve biopsy and has relatively normal conduction velocities (Pareyson & Marchesi, 2009). Beyond this clear classification, rapidly increasing knowledge of this disease has led to the recognition of other forms of CMT, such as a dominant intermediate CMT subtype (DI‐CMT) with features of both CMT1 and CMT2, and a pure motor form, distal hereditary motor neuronopathy (dHMN), characterized by the sparing of sensory nerves upon examination. Further subdivisions of these CMT types are mainly based on causative genes and variants.
In typical cases, CMT onset often occurs in the first two decades of life, and the disease processes slowly without affecting life expectancy (Laura et al., 2019; Pareyson & Marchesi, 2009). However, the age of onset, disease course and severity vary greatly based on the CMT subtype, causative genes and types of variants. Despite significant advances in the genetic diagnosis of CMT using next‐generation sequencing technology, no effective therapies have thus far been developed.
To date, over 100 CMT‐associated genes have been identified as causative factors (Laura et al., 2019). Among them, five genes (GARS, YARS, AARS, HARS and WARS) encode aminoacyl‐tRNA synthetases (aaRSs) including glycyl‐, tyrosyl‐, alanyl‐, histidyl‐ and tryptophanyl‐tRNA synthetases (GlyRS, TyrRS, AlaRS, HisRS and TrpRS, respectively) (Antonellis et al., 2003; Jordanova et al., 2006; Latour et al., 2010; Tsai et al., 2017; Vester et al., 2013), which is the largest family implicated in CMT and highlights the vital importance of aaRSs in the pathogenesis of CMT.
The aaRS family comprises ubiquitously expressed enzymes that are involved in the translation of the genetic code by charging amino acids onto their cognate tRNAs during protein synthesis (Ling et al., 2009). Based on structural features, 20 canonical aaRSs are divided into two classes that differ in the architecture of their active sites for adenylate synthesis. Class I aaRSs contain a typical Rossmann‐fold utilized for nucleotide binding and two well‐conserved signature sequences (HIGH and KMSKS), whereas class II aaRSs are less conserved and include three signature motifs (Motifs 1‐3) within a seven‐stranded β‐sheet and three flanking α‐helices. Among the five known CMT‐associated aaRSs, TryRS and TrpRS belong to class I, and the other three (GlyRS, AlaRS and HisRS) belong to class II.
Intriguingly, variants of these tRNA synthetase genes are strongly associated with CMT, but not all variants affect the aminoacylation activities of the enzymes. More strikingly, CMT‐like neuropathy phenotypes in animal models generated by some variants cannot be rescued by overexpression of wild type (WT) proteins, suggesting that CMT is very likely linked to toxic gain‐of‐function associated with the variants themselves. In this review, we discuss the latest advances in studies on aaRSs in CMT, particularly focusing on their roles in the pathogenesis of the disease.
2. GARS VARIANTS
GlyRS is a class II aaRS with a conserved catalytic domain composed of a central antiparallel β‐sheet flanked with α‐helices and three conserved sequence motifs (Xie et al., 2007), followed by an anticodon domain. The N‐terminal extension contains a specific appended helix‐turn‐helix structural motif named the WHEP domain, which derives from three of the five WHEP‐containing proteins TrpRS, HisRS and EPRS (Guo et al., 2010) (Figure 1a). GlyRS was the first tRNA synthetase implicated in CMT (Antonellis et al., 2003). To date, more than twenty missense variants of GARS have been discovered in patients with CMT2D (OMIM# 601472), an autosomal‐dominant axonal subtype of CMT, or dHMN type V (dHMN‐V, OMIM# 600794), a subtype of dHMN with upper‐limb predominance (Motley et al., 2010) (Table 1). Among them, most variants are located in the catalytic domain, one variant is in the WHEP domain and the remaining two are in the anticodon domain (Figure 1a and b). Interestingly, several variants (GARS E71G, GARS L129P, GARS G240R, GARS I280F, GARS H418R, GARS G526R and GARS G598A) are closely associated with the disease; however, some affect aminoacylation activity, while others do not (Griffin et al., 2014) (Table 1). For example, GARS E71G segregates with CMT2D in large families but has WT‐like aminoacylation activity (Antonellis et al., 2003, 2006; Nangle et al., 2007; Niehues et al., 2015). Given the differential enzymatic activities of GlyRS variants, it is not surprising for scientists to seek out other disease‐associated functions of these variants that are distinct from the canonical aminoacylation functions.
FIGURE 1.
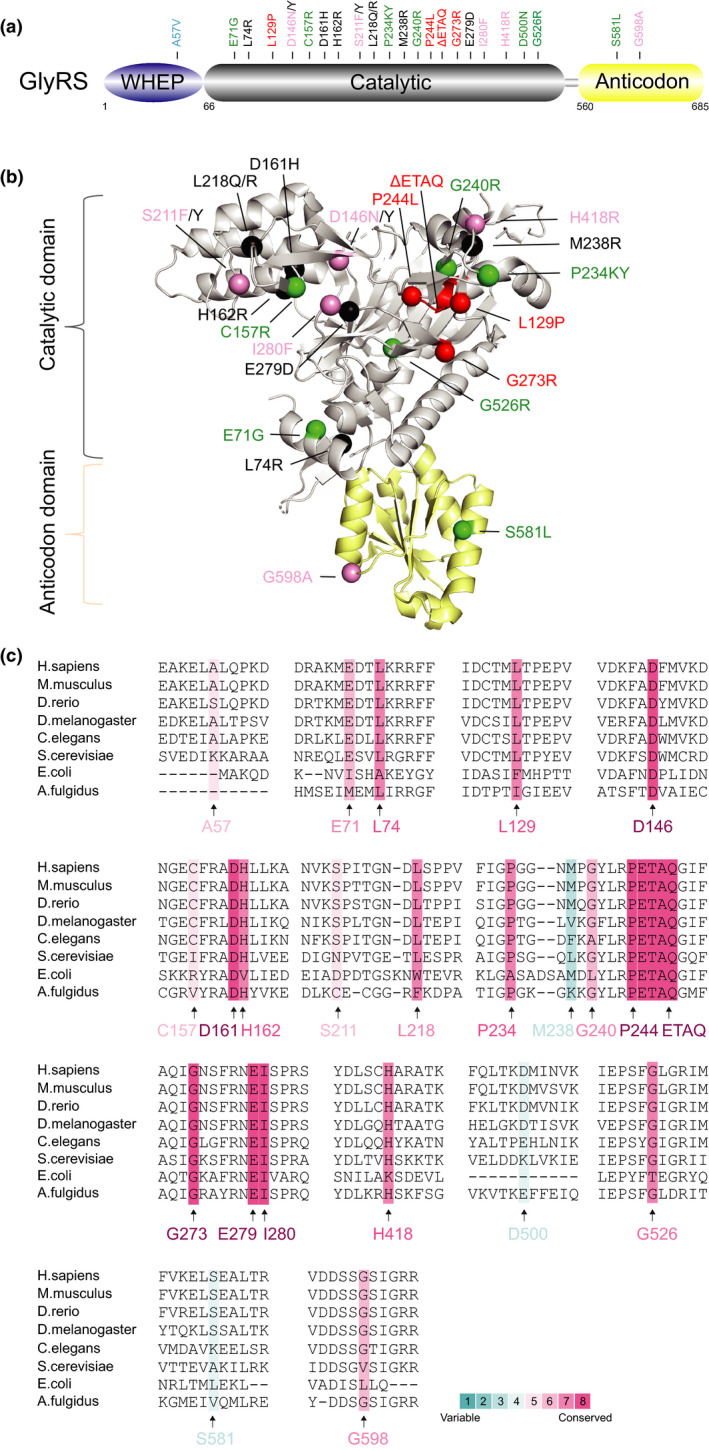
Distribution and conservation of CMT‐associated variant sites in human GlyRS. (a) Functional domains of GlyRS including a WHEP domain (in purple), a catalytic domain (in grey) and an anticodon domain (in yellow). (b) The crystal structure of human GlyRS (PDB entry 2PME). CMT variant sites in either schematic diagram (a) or crystal structure (b) are indicated with different colors based on enzymatic activity of each variant. CMT variants with WT‐like enzymatic activity (fully active) are colored in green; variants with the activity ≥1/2 are labeled in blue; variants with the activity <1/2 are indicated in pink; variants with no activity (inactive) are colored in red; variants with activity undetermined are indicated in black. The priority of enzymatic activity displayed here is ranked based on aminoacylation assays in patient sample > animal models > in vitro using purified human enzyme > yeast orthologs. (c) Evolutionary conservation analysis of CMT‐linked GlyRS across archaea, bacteria and eukaryotes. Sequence alignment of each GlyRS variant site is indicated by the color intensity, with blue representing variable and red representing conserved
TABLE 1.
Effect of CMT variants on aminoacylation activity and conformational change of aaRSs
| aaRS | Variants | Segregation* | Age at onset | Aminoacylation activity** | Phenotype | Conformational change | References | |
|---|---|---|---|---|---|---|---|---|
| Yeast† | Animal model‡ | |||||||
| GARS | A57V | P1 in F1 | 12 y | k cat/K m: 1/2 (tRNA) | NA | ND | ND | (Griffin et al., 2014; Rohkamm et al., 2007) |
| E71G | P17 in F1 | 17.7 y b |
Normal (Drosophila); Normal (in vitro) |
Viable | Neuronal phenotypes [trans‐OE, Drosophila] | Little change versus. WT | (Antonellis et al., 2003, 2006; Nangle et al., 2007; Niehues et al., 2015; Qin et al., 2014; Sivakumar et al., 2005) | |
| L74R | P6 in F1 | 13.3 y b | ND | ND | ND | ND | (Yu et al., 2018) | |
| L129P | P5 in F1 | 16.9 y b | Loss (in vitro) | Reduced | ND | HDX increase d,e,f : 37% | (Antonellis et al., 2003, 2006; He et al., 2011; Nangle et al., 2007; Sivakumar et al., 2005) | |
| D146N | P2 in F1 | 15 y b | k cat/K m: 1/14 (tRNA) | Reduced | ND | ND | (Griffin et al., 2014; Lee et al., 2012) | |
| D146Y | P1 in F1 | 3 mo | ND | ND | ND | ND | (Liao et al., 2015) | |
|
C157R /C201R M |
‐‐ | ‐‐ |
Insignificant reduced (± mouse); Reduced (+/+mouse) |
ND | Neuropathy phenotypes in ±; postnatal death in +/+ [KI, mouse] | ND | (Achilli et al., 2009) | |
| D161H | P3 in F1 | 17 y c | ND | ND | ND | ND | (Nan et al., 2019) | |
|
H162R /H218R Mt |
P2 in F1 | Early 20s | ND | ND | ND | ND | (Boczonadi et al., 2018) | |
| S211F | P4 in F2 from 2C | 8 y c | k cat/K m: 1/4,400 (tRNA) | NA | ND | ND | (Griffin et al., 2014; Lee et al., 2012; Sun et al., 2017) | |
| S211Y | P2 in F1 | 11 y c | ND | ND | ND | ND | (Yalcouye et al., 2019) | |
| L218Q | P1 in F1 | Toddler | ND | ND | ND | ND | (Kawakami et al., 2014) | |
| L218R | P4 in F1 | 8.7 mo c | ND | ND | ND | ND | (Chung et al., 2018) | |
|
P234KY /P278KY M |
– | – |
Insignificant reduced (±mouse); Normal (in vitro) k cat/K m: 1/500 (tRNA) |
Viable or Lethal |
Neuropathy phenotypes in ±; embryonic lethal in +/+ [KI, mouse]; Neuronal phenotypes [trans‐OE, Drosophila] |
HDX increase d,e,f : 15% | (Grice et al., 2015; He et al., 2015; Morelli et al., 2019; Nangle et al., 2007; Seburn et al., 2006; Stum et al., 2011) | |
| M238R | P1 in F1 | 2 y | ND | ND | ND | ND | (Liao et al., 2015) | |
| G240R | P8 in F1 a | 23 y b |
Normal (Drosophila); Loss (in vitro); k cat/K m: 1/110 (tRNA) |
Viable | Neuronal phenotypes [trans‐OE, Drosophila] | HDX increase d,e,f : 30% | (Antonellis et al., 2003, 2006; He et al., 2011; Niehues et al., 2015; Sivakumar et al., 2005) | |
| P244L | P1 in F1 | Adolescence | k cat/K m: undetectable | Lethal | ND | ND | (Abe & Hayasaka, 2009; Griffin et al., 2014) | |
| ΔETAQ | P1 in F1 | 13 mo | k cat/K m: 1/11000 (tRNA) | Lethal | Neuropathy phenotypes in ± [microinj, mouse] | ND | (Morelli et al., 2019) | |
| G273R | P2 in F2 | 11.5 y b | ND | Lethal | ND | ND | (Lee et al., 2019) | |
| E279D | P8 in F1 | Adolescence | ND | ND | ND | ND | (Sun et al., 2015) | |
| I280F | P4 in F2 from 2C | 22.5 y b | k cat/K m: 1/1,700 (tRNA) | Viable | ND | ND | (Griffin et al., 2014; James et al., 2006; Klein et al., 2014) | |
| H418R | P8 in F1 | 26 y b |
Reduced (in vitro); k cat/K m: 1/16 (tRNA) |
Lethal | ND | ND | (Antonellis et al., 2006; Griffin et al., 2014; Nangle et al., 2007; Sivakumar et al., 2005) | |
| D500N | P6 in F1 | 21.7 y b | Normal (in vitro) | NA | ND | ND | (Del Bo et al., 2006; Griffin et al., 2014; Nangle et al., 2007) | |
| G526R | P18 in F4 from 2C | 21.4 y c |
Normal (Drosophila); Loss (in vitro); k cat/K m: 1/44,000 (tRNA) |
Lethal | Neuronal phenotypes [trans‐OE, Drosophila] |
Little change versus. WT; Rg g : 15.6%; HDX increase d,e,f : 16% |
(Antonellis et al., 2003, 2006; Dubourg et al., 2006; He et al., 2011; Niehues et al., 2015; Xie et al., 2007) | |
| S581L | P1 in F1 | 4 y |
Normal (in vitro); k cat/K m: 1/2 (tRNA) |
NA | ND |
Little change versus. WT; HDX increase d,e,f : 18% |
(Cader et al., 2007; Griffin et al., 2014; He et al., 2011; James et al., 2006; Nangle et al., 2007) | |
| G598A | P3 in F2 from 2C | Infantile | k cat/K m: 1/180 (tRNA) | Viable | ND | HDX increase d,e,f : 22% | (Eskuri et al., 2012; Griffin et al., 2014; He et al., 2011; James et al., 2006; Stum et al., 2011) | |
| YARS | G41R | P15 in F1 | 1st and 2nd decades |
Reduced (Drosophila); Loss (in vitro) |
Lethal | Neuronal phenotypes [trans‐OE, Drosophila] |
Rg: 5.2% RH h : 6.9% HDX increase d,e,f : 1.3% |
(Gonzaga‐Jauregui et al., 2015; Jordanova et al., 2003, 2006; Storkebaum et al., 2009) |
| D81I | P1 in F1 | 23 y | ND | ND | ND | ND | (Hyun et al., 2014) | |
| Δ153−156 | P1 in F1 | ND |
Reduced (Drosophila); Reduced (in vitro) |
Reduced | Neuronal phenotypes [trans‐OE, Drosophila] |
Rg: −2.4% RH: −13.8% HDX increase d,e,f : none |
(Jordanova et al., 2006; Storkebaum et al., 2009) | |
| E196K | P38 in F1 | 7–59 y |
Normal (Drosophila); Normal (in vitro) |
Viable | Neuronal phenotypes [trans‐OE, Drosophila] |
Rg: 5.8% RH: 10.3% HDX increase d,e,f : 10.8% |
(Froelich & First, 2011; Gonzaga‐Jauregui et al., 2015; Jordanova et al., 2003, 2006; Storkebaum et al., 2009) | |
| E196Q | P7 in F1 | ND | ND | Reduced | ND | ND | (Gonzaga‐Jauregui et al., 2015) | |
| AARS | N71Y | P7 in F1 | 31.5 y c |
Loss (in vitro); k cat/K m: 1/4,130 (tRNA) |
Lethal | ND | ND | (Lin et al., 2011; McLaughlin et al., 2012) |
| G102R | P5 in F1 | 34 y c | ND | Lethal | ND | ND | (Motley et al., 2015) | |
| R326W | P11 in F1 | 35.8 y c | ND | Lethal | Abnormal embryos [microinj, Zebrafish] | ND | (Weterman et al., 2018) | |
| R329H | P66 in F8 from 4C | 25.0 y c |
Loss (in vitro); k cat/K m: 1/50 (tRNA) |
Lethal | ND | ND | (Bansagi et al., 2015; Latour et al., 2010; McLaughlin et al., 2012) | |
| R337K | P6 in F1 | 19.7 y c | k cat/K m: 3.87 (tRNA) | Enhanced | Abnormal embryos [microinj, Zebrafish] | ND | (McLaughlin et al., 2012; Weterman et al., 2018) | |
| S627L | P4 in F1 | 30 y c | ND | Reduced | Abnormal embryos [microinj, Zebrafish] | ND | (Weterman et al., 2018) | |
| E688G | P3 in F1 | Early onset | ND | ND | ND | ND | (Bansagi et al., 2015) | |
| E778A | P4 in F1 | ND |
Normal (in vitro); k cat/K m: 1/1.4 (tRNA) |
Viable | ND | ND | (McLaughlin et al., 2012) | |
| D893N | P4 in F1 | 28.5 y c | ND | ND | ND | ND | (Zhao et al., 2012) | |
| HARS | T132I | P10 in F1 | 23.7 y c |
Loss (in vitro); k cat/K m: undetectable |
Lethal | ND |
Rg: 2.3% DH h : 4% HDX decrease d,e,f : 4% |
(Blocquel et al., 2019; Safka Brozkova et al., 2015) |
| P134H | P5 in F1 | Childhood |
Normal (patient); Reduced (in vitro); k cat/K m: 1/83 (His); 1/25 (ATP) |
Lethal | ND |
Rg: 5.2% DH: 18% HDX increase d,e,f : 6% |
(Blocquel et al., 2019; Safka Brozkova et al., 2015) | |
| R137Q | P1 in F1 | 49 y | ND | Lethal | Neurotoxicity [trans‐OE, C. elegans] | ND | (Vester et al., 2013) | |
| V155G | P5 in F1 | 2nd decade |
Reduced (in vitro); k cat/K m: 1/2.2 (tRNA); 1/166.7 (His); 1/342.1 (ATP) |
Viable | ND | ND | (Abbott et al., 2018) | |
| D175E | P4 in F1 | 25.5 y c |
Reduced (in vitro); k cat/K m: 1/2,000 (His); 1/62 (ATP) |
Reduced | ND |
Rg: 2.3% DH: 20% HDX decrease d,e,f : 5% |
(Blocquel et al., 2019; Safka Brozkova et al., 2015) | |
| Y330C | P2 in F1 | Childhood |
Reduced (in vitro); k cat/K m: 1/4.1 (tRNA); 1/125 (His); 1/477.9 (ATP) |
Reduced | ND | ND | (Abbott et al., 2018) | |
| S356N | P1 in F1 | 10 y |
Reduced (in vitro); k cat/K m: 1/4.6 (tRNA); 1/12.5 (His); 1/866.7 (ATP) |
Reduced | ND | ND | (Abbott et al., 2018) | |
| D364Y | P9 in F1 | 21.9 y c |
Normal (in vitro); k cat/K m: 1/50 (His); 1/1.2 (ATP) |
Lethal | Neurotoxicity [trans‐OE, C. elegans] |
Rg: 4.1% DH: 16% HDX increase d,e,f : 3% |
(Blocquel et al., 2019; Safka Brozkova et al., 2015) | |
| WARS | H257R | P12 in F3 from 2C | 11.8 y c | Reduced (in vitro) | Enhanced †† | ND | ND | (Tsai et al., 2017) |
| D314G | P5 in F1 | 17.8 y b | ND | ND | ND | ND | (Wang et al., 2019) | |
| MARS | A397T | P1 in F1 | < 1 y | ND | Lethal | ND | ND | (Gillespie et al., 2019) |
| R618C | P3 in F1 | 56 y c | ND | Lethal | ND | ND | (Gonzalez et al., 2013) | |
| R737W | P2 in F1 | 12 y c | ND | ND | ND | ND | (Sagi‐Dain et al., 2018) | |
| P800T | P5 in F3 from 2C | 34.3 y c | ND | ND | ND | ND | (Hirano et al., 2016; Hyun et al., 2014; Nam et al., 2016) | |
M, mouse; +/+, homozygous; ±, heterozygous; Mt, mitochondria; His, histidine; versus. versus; WT, wild type; Ref., references; ND, not determined.
This variant was identified in 2 families with information in one family unknown.
Average age at onset.
Average age at onset of available individual(s) except those deceased individuals, asymptomatic individuals, affected individuals with age at onset unknown or ambiguous, or alive individuals but refused to have a clinical evaluation.
Difference of radius of gyration (Rg, Å) for each variant relative to WT enzyme based on SAXS analysis. The value is calculated using the formula of (Rg Vriant‐Rg WT)/Rg WT × 100%, with a positive value indicating size increase and a negative value indicating size decrease, respectively.
Difference of hydrodynamic radius (RH, nm) or diameter (DH, nm) of each variant relative to WT enzyme based on the switchSENSE® Technology. The value is calculated using the formula of (RH or DH Variant‐ RH or DH WT)/RH or DH WT × 100%, with a positive value indicating a larger protein size and a negative value indicating a smaller protein size, respectively.
Family segregation of genetic variants. P, person(s) carrying the variant; F, family(ies); C, countries.
Aminoacylation activity of each variant is ranked based on different assays from in vivo to in vitro charging to in vitro kinetics. In in vivo assays, the types of models are shown in parentheses. Enzymatic activity in both in vivo and in vitro assays is shown with ‘Normal’ indicating WT‐like activity, ‘Loss’ indicating inactive, and ‘Reduced’ indicating activity between ‘Normal’ and ‘Loss’. For kinetic analysis, catalytic rate constant/Michaelis constant (k cat/K m) is shown in each variant relative to WT for different substrates including tRNA, amino acid or ATP shown in parentheses; ‘undetectable’ indicates values that are below the limits of kinetic detection.
Growth of yeast strain containing variant form compared with WT strain. NA, not applicable because of the variant that could not be modeled in yeast ortholog.
Growth of yeast strains containing human variant relative to human WT gene.
Phenotypes of each variant in animal models with the genetic manipulation of species shown in square brackets. trans‐OE, transgenic over‐expression; KI, knockin; microinj, microinjection.
Average increase/decrease in deuterium incorporation for each variant relative to WT enzyme after 1 hd or at 1,000 se or after 10,000 sf of exchange based on HDX analysis. h, hour; s, seconds.
2.1. Animal models of GlyRS‐linked CMT
Among all CMT‐associated aaRSs, GlyRS is well‐established and the most studied in animal models. In Drosophila models, GlyRS variants significantly reduce the levels of newly synthesized proteins, and this translational defect is not attributed to altered tRNA aminoacylation because overexpression of Drosophila gars fails to rescue impaired protein translation (Niehues et al., 2015). Additionally, the enzymatic activity in the tissues of mice carrying heterozygous Gars Nmf249/+ and Gars C201R/+ variants (corresponding to GARS P234KY and GARS C157R in human) was not significantly decreased compared with that in WT animals (Achilli et al., 2009; Seburn et al., 2006). Notably, overexpression of WT GlyRS could not improve the neuropathy phenotypes in either Gars Nmf249/+ or Gars C201R/+ mice (Motley et al., 2011), and the same conclusion regarding another GARS G240R variant was reached with Drosophila CMT models, which showed no phenotypic rescue upon overexpression of the WT proteins (Niehues et al., 2015).
Recently, one group identified a de novo GARS variant (GARS ΔETAQ) in a single patient with severe peripheral neuropathy, and this allele was further introduced into a mouse model (Morelli et al., 2019). This mouse model carrying a human disease allele displays primary features of peripheral neuropathy. Strikingly, the allele‐specific knockdown of GARS ΔETAQ using RNAi prevents the neuropathy phenotypes in mice, and the same efficacy is confirmed in Gars Nmf249/+ mice, both before and after onset. These findings provide important proof‐of‐concept for virally delivered RNAi‐based gene therapy for treating CMT2D; however, whether this approach can be applied to other CMT‐causing single‐base pair variants requires additional research. Of note, in this study, re‐evaluation of in vitro kinetic properties and yeast models for the P234KY allele supported a loss‐of‐function effect (Morelli et al., 2019), contrary to previous reports (Table 1) (Nangle et al., 2007; Seburn et al., 2006; Stum et al., 2011). Such discrepancies in activity assays may reflect different sensitivities of experimental settings.
Given the well‐characterized Gars Nmf249/+ in mice, one group further modeled the P234KY variant in Drosophila (gars P234KY). Ubiquitous expression of gars P234KY was shown to severely affect fly fitness, with no adults emerging, whereas pan‐neuronal expression caused late pupal lethality, suggesting that toxicity may manifest or derive from tissues beyond the nervous system (Ermanoska et al., 2014). Indeed, muscular expression of gars P234KY in flies induces significant neuronal defects, and the same is true for the gars G240R variant, suggesting that the pathology has a noncell autonomous contribution (Grice et al., 2015). Interestingly, the neuronal toxicity of gars P234KY is dependent on the WHEP domain since its removal from gars P234KY abrogates neuromuscular and survival defects, revealing a clear dominant toxic gain‐of‐function mechanism for variants that may contribute to the pathology of CMT2D (Grice et al., 2015).
In contrast, mice carrying homozygous GARS variants, such as those of Gars Nmf249/Nmf249 and Gars C201R/C201R, are not viable or even undergo early death, and perinatal lethality can be rescued by overexpression of the WT proteins (Achilli et al., 2009; Seburn et al., 2006). Likewise, a homozygous gars variant (gars P98L) in Drosophila, although not the cause of CMT, leads to defects in dendritic and axonal terminal arborization, which can be fully rescued by transgenic expression of WT human GARS (Chihara et al., 2007). This finding provides evidence that human GlyRS has equivalent functions in Drosophila. However, the defects in fly models could be only partially rescued by the human GARS E71G variant, whereas the GARS L129P variant did not show any rescue capability, indicating that human CMT2D variants do have loss‐of‐function properties and may affect the normal function of GlyRS to different degrees. Thus, based on studies from animal models, different GARS variants may cause the same disease via distinct mechanisms, with some variants having toxic gain‐of‐function effects and others demonstrating undefined loss‐of‐function properties.
2.2. Conformational changes in CMT‐linked GARS variants
To date, crystal structures of three GARS variants, GARS G526R, GARS S581L and GARS E71G, have been solved; however, few conformational changes were observed compared with the WT proteins (Cader et al., 2007; Qin et al., 2014; Xie et al., 2007) (Table 1). Nonetheless, different CMT‐causing variants have distinct effects on dimer formation. For example, GARS D500N, GARS G526R and GARS S581L strengthen the capacity of dimer formation, whereas GARS L129P and GARS G240R disrupt dimer formation (Nangle et al., 2007). Because the dimeric form of GlyRS is essential for aminoacylation, both GARS L129P and GARS G240R show a loss of enzyme activity, while GARS D500N and GARS S581L demonstrate full aminoacylation activity compared to that of the WT enzyme. However, despite an enhanced dimer formation capability, GARS G526R has abolished aminoacylation activity (Nangle et al., 2007). Therefore, considering this together with another fully active variant (GARS E71G), almost half of these CMT‐causing variants are active, further supporting the conclusion that CMT is not simply caused by a deficiency in aminoacylation. Furthermore, this finding also raises the possibility that CMT‐causing variants may disrupt an unknown function of GlyRS, leading to a toxic gain‐of‐function that is associated with only GlyRS variants.
Considering these data, the same research group further explored five GARS variants (GARS L129P, GARS G240R, GARS G526R, GARS S581L and GARS G598A) in solution utilizing hydrogen‐deuterium exchange (HDX) analysis monitored by mass spectrometry (MS) (He et al., 2011). Interestingly, despite their differential effects on dimerization and aminoacylation activity (Griffin et al., 2014; Nangle et al., 2007), all five variants exhibit the same neomorphic conformational opening that is mostly buried in WT GlyRS (He et al., 2011). This conformational change also occurs in the mouse carrying the GARS P234KY variant, which has a similar neomorphic structural opening. Further small‐angle X‐ray scattering (SAXS) analysis of the GARS G526R variant confirmed that this neomorphic structural opening is associated with an unknown physiological function of GlyRS that is suppressed by the WHEP domain. Perhaps for this reason, these CMT‐causing variants might disrupt WHEP‐mediated suppression, resulting in gain‐of‐function phenotypes. Whether the induced conformational changes confer the variants with the ability to interact with other proteins needs to be further addressed.
It is important to note that, based on conservation analysis (Figure 1c), some variant sites such as E71 and G526 in GARS, show less divergence in the evolutionary progression to humans, suggesting essential roles of these sites during evolution; however, based on the above findings, variations in these sites do not significantly affect enzymatic activity, further supporting non‐canonical activities outside of aminoacylation imparted by these variants. Other essential functions of aaRSs beyond translation have been well documented (Guo & Schimmel, 2013; Guo et al., 2010).
2.3. Interaction partners of GARS variants
In fact, several interaction partners of GARS variants as well as the molecular mechanisms underlying the interactions have been uncovered. Variant, but not WT, GlyRS are capable of binding the neuropilin‐1 (Nrp1) receptor, and such an aberrant interaction competitively interferes with the binding of the cognate ligand vascular endothelial growth factor (VEGF) to Nrp1, thereby contributing to motor defects in CMT2D (He et al., 2015). More importantly, aberrant signaling of GlyRS/Nrp1/VEGF is observable in the neural tissues of Gars Nmf249/+ mice, and VEGF overexpression can partially rescue motor defects in Gars Nmf249/+ mice. The same GlyRS‐Nrp1 interaction also occurs in lymphocytes from CMT2D patients with the GARS L129P variant. This work demonstrates that CMT arises from the neomorphic activity of misfolded GlyRS interacting with susceptible signaling targets independent of aminoacylation. Nonetheless, not all GlyRS variants contribute to the disease through the GlyRS‐Nrp1 interaction. For example, the GARS ΔETAQ variant does not have a strong interaction with Nrp1, suggesting distinct mechanisms for different variants (Morelli et al., 2019). The possibility of Nrp1‐interacting GlyRS variants interacting with other extracellular and/or intercellular targets cannot be ruled out.
In addition to Nrp1, tropomyosin receptor kinase (Trk) receptors (TrkRs) were subsequently shown to interact with CMT2D‐linked GlyRS variants (GARS CMT2D). Unlike Nrp1/VEGF signaling, which targets motor axons, TrkR signaling specifically acts on sensory axons. Variant, but not WT, GlyRS misactivates Trk signaling by binding to multiple TrkRs, leading to defective differentiation and development of sensory neurons (Sleigh et al., 2017). Interestingly, Gars C201R/+ mice exhibited nonprogressive perturbation of sensory neuronal fate during early stages of development, in line with the clinical features of some CMT2D patients who present subtle and undiagnosed sensory symptoms in advance of motor deficits (Sleigh et al., 2017). This finding also explains the absence of sensory defects in patients with dHMN‐V who present predominantly with motor degeneration.
The aberrant interaction of GlyRS with Nrp1 or TrkR depends on the extracellular domains of Nrp1 or TrkR. One intracellular partner, histone deacetylase 6 (HDAC6), was recently identified and surprisingly shown to aberrantly interact with almost all GARS CMT2D variants in reach (GARS E71G, GARS L129P, GARS S211F, GARS G240R, GARS E279D, GARS H418R, GARS G526R, GARS S581L and GARS G598A), and this aberrant crosstalk was shown to functionally stimulate the deacetylase activity of HDAC6 on α‐tubulin (Mo et al., 2018). Deacetylation of α‐tubulin further causes axonal transport deficits prior to disease onset in Gars Nmf249/+ mice, with specific targeting at peripheral nerves rather than the brain or spinal cord. More essentially, the defective phenotypes can be rescued by the HDAC6 inhibitor, and the same is true for Gars C201R/+ mice from another study (Benoy et al., 2018). Although abnormal GlyRS‐HDAC6 interactions have been identified in many human GARS CMT2D variants, the differential degrees of interplay among variants is closely associated with distinct clinical manifestations among CMT2D patients. For example, the GARS S581L and GARS G598A variants trigger the strongest HDAC6 interaction among all variants, of which the GARS G598A variant can also interact with Nrp1, concordant with unfavorable clinical features of infantile onset and extreme severity for patients harboring the GARS G598A variant (Mo et al., 2018). For the other human GARS CMT2D variants with relatively weak HDAC6 interactions, the possibility of alternative pathogenic mechanisms in CMT2D cannot be excluded.
2.4. Mitochondrial role of GlyRS in CMT
It is worth noting that GlyRS is one of two aaRSs (the other is lysyl‐tRNA synthetase) encoding both cytosolic and mitochondrial forms; however, little is known about the mitochondrial role of GlyRS and how it affects the phenotypes of diseases such as CMT.
A recent study identified a novel disease‐associated dominant variant of GARS H162R (reported as GARS H216R because of inclusion of the mitochondrial target sequence) in patients presenting with typical clinical and electrophysiological signs of dHMN‐V (Boczonadi et al., 2018). Indeed, GlyRS is present in RNA granules in mitochondria and is involved in mitochondrial translation; downregulation of GARS results in defective mitochondrial translation in neuronal cells and myoblasts but not in fibroblasts, indicating a tissue‐specific feature (Boczonadi et al., 2018). Interestingly, reduced mitochondrial respiration and decreased calcium uptake are found in induced neuronal progenitor cells (iNPCs) generated from patient carrying the GARS H162R variant, suggesting that this neuropathy‐associated variant leads to a complicated alteration of mitochondrial function in neurons. In Gars C201R mice, mitochondrial dysfunction is found in only the sciatic nerve, while the other five highly metabolic tissues display no mitochondrial defects, further supporting a tissue‐specific mechanism in vivo. This study, together with the complex interactions of GlyRS exemplified above, suggests that more cellular compartments are involved in aaRS‐linked CMT in addition to the cytoplasm and mitochondria.
3. YARS VARIANTS
Similar to other class I aaRSs, TyrRS has a Rossmann‐fold domain that consists of mostly parallel β‐strands connected by α‐helices as the catalytic domain (Blocquel et al., 2017); however, both TyrRS and TrpRS function as homodimers, while the remaining class I aaRSs are functional monomers. In addition to an N‐terminal catalytic domain and an anticodon domain, the C‐terminal fragment of human TyrRS is highly homologous to endothelial monocyte‐activating polypeptide II (EMAPII) (Kao et al., 1992), a known cytokine that is dispensable for aminoacylation by TyrRS (Figure 2a). To date, five catalytic domain variants of YARS have been reported in DI‐CMT type C (DI‐CMTC or CMTDIC, OMIM# 608323) (Gonzaga‐Jauregui et al., 2015; Hyun et al., 2014; Jordanova et al., 2006) (Table 1 and Figure 2a and b); three variants (YARS G41R, YARS E196K and YARS E196Q) segregate with DI‐CMTC in large families, whereas the remaining two variants (YARS D81I and YARS Δ153‐156) have been found in only one patient each. Notably, the highly conserved YARS G41R and YARS Δ153‐156 variants (Figure 2c) are defective in their ability to activate tyrosine, whereas the YARS E196K variant does not affect the formation of the tyrosyl‐adenylate intermediate but rather enhances the catalytic rate compared with that achieved with the WT enzyme (Froelich & First, 2011). In Drosophila models, the same conclusion of transgenic YARS E196K variant overexpression severely impairing motor performance but having normal enzymatic activity is reached (Storkebaum et al., 2009). In contrast, the YARS G41R and YARS Δ153‐156 variants induce less toxicity in flies than the YARS E196K variant despite the severely decreased enzymatic activities of the YARS G41R and YARS Δ153‐156 variants in Drosophila. Furthermore, Drosophila in which the yars is inhibited by 50% display normal motor performance. These findings indicate that the loss of aminoacylation activity is neither necessary nor sufficient to cause peripheral neuropathy, suggesting that TyrRS‐linked neurodegeneration results from a gain‐of‐function of TyrRS variants separate from aminoacylation.
FIGURE 2.
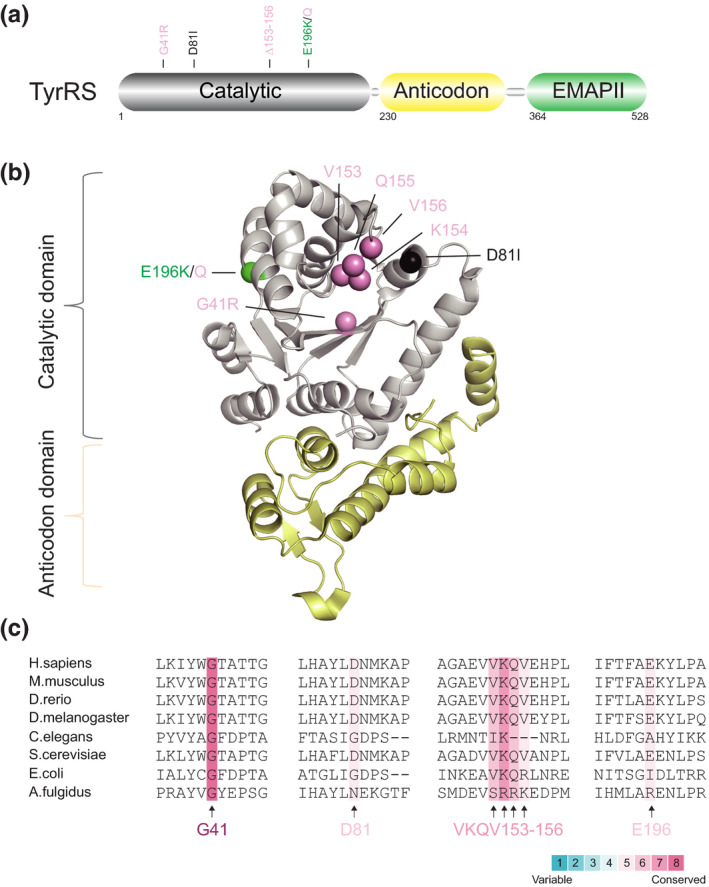
Distribution and conservation of CMT‐associated variant sites in human TyrRS. (a) Functional domains of TyrRS including a catalytic domain (in grey), an anticodon domain (in yellow) and an EMAPII domain (in green). (b) The crystal structure of human TyrRS without the C‐terminal EMAPII domain (PDB entry 1N3L). CMT variant sites in either schematic diagram (a) or crystal structure (b) are indicated with different colors based on enzymatic activity of each variant with the same priority ranking as Figure 1b. (c) Evolutionary conservation analysis of CMT‐linked TyrRS across species. Sequence alignment of each variant site is indicated by the color intensity, with blue representing variable and red representing conserved
To further pinpoint the functions gained from YARS variants, Xiang‐lei Yang and her colleagues uncovered conformational changes induced by the YARS G41R and YARS E196K variants; the YARS Δ153‐156 variant did not induce conformational change but did expose the same area that was opened by the YARS G41R and YARS E196K variants (Blocquel et al., 2017). In addition, all three variants had the same enhanced binding affinity for TRIM28. These data suggest that DI‐CMTC, like CMT2D, is caused by a specific protein structure change that allows the generation of an alternative stable conformation for potential pathological interactions. The fact that the YARS Δ153‐156 variant lacks conformational change but aberrantly interacts with TRIM28 suggests that residues 153–156 act as a structural blocker to prevent aberrant interactions of the WT enzyme (Blocquel et al., 2017).
Nevertheless, the abnormal TyrRS‐TRIM28 interaction should not be the only dysregulated pathway for DI‐CMTC variants since the TRIM28 ortholog is not found in Drosophila, suggesting alternative pathological mechanisms for CMT‐like phenotypes in fly models. Indeed, TyrRS can translocate to the nucleus for protection from DNA damage under oxidative stress, and this beneficial effect of nuclear TyrRS is achieved by activation of the transcription factor E2F1 (Wei et al., 2014). Further studies verified significant increases in the expression levels of E2F1‐regulated target genes in peripheral blood mononuclear cells (PBMCs) from patients with DI‐CMTC carrying the YARS G41R and YARS E196K variants (Bervoets et al., 2019). Remarkably, inhibition of nuclear TyrRS using pharmacological or genetic approaches suppresses the hallmark phenotypes of CMT in Drosophila, highlighting the importance of nuclear TyrRS variant for CMT neuropathology; however, the involvement of other cellular compartments or molecules mediating the toxicities outside the nucleus cannot be excluded.
Collectively, despite completely different molecular architectures for catalysis of class I and class II aaRSs (e.g., TyrRS and GlyRS), both types of architecture may predispose the enzymes to conformational perturbations that permit aberrant interactions. The nuclear involvement of TyrRS CMT neurotoxicity could also have essential implications for other CMT‐associated aaRSs.
4. AARS VARIANTS
AlaRS is the third aaRS known to be involved in CMT. Unlike other CMT‐associated aaRSs, AlaRS does not have an anticodon domain but rather has an editing domain and a C‐terminal domain C‐Ala (Figure 3a). To date, nine AARS variants leading to dominant axonal CMT type 2N (CMT2N, OMIM# 613287) have been reported worldwide (Bansagi et al., 2015; Latour et al., 2010; Lin et al., 2011; McLaughlin et al., 2012; Motley et al., 2015; Weterman et al., 2018; Zhao et al., 2012) (Table 1). Among them, five variants (AARS N71Y, AARS G102R, AARS R326W, AARS R329H and AARS R337K) are located in the N‐terminal catalytic domain, two (AARS S627L and AARS E688G) are located in the editing domain and the final two (AARS E778A and AARS D893N) are located in the C‐Ala domain (Figure 3a and b). Of these variants, the AARS R329H variant was first discovered to segregate with CMT2N in two unrelated French families (Latour et al., 2010) and was then identified in a large Australian family (McLaughlin et al., 2012) and in a cohort of patients from one Irish and four British families (Bansagi et al., 2015), thus representing a recurrent variant worldwide. Yeast complementation assay results revealed impaired enzyme functions of the AARS R329H variant as well as the AARS N71Y, AARS G102R, AARS R326W and AARS S627L variants (McLaughlin et al., 2012; Motley et al., 2015; Weterman et al., 2018). In contrast, the cellular growth of yeast was not shown to be affected by the AARS E778A variant compared to that of the WT enzyme (McLaughlin et al., 2012), and the AARS R337K variant even improved yeast cell growth and showed a nearly 4‐fold increase in tRNA charging activity (Weterman et al., 2018). Interestingly, although different enzyme functions are caused by the AARS R326W, AARS R337K and AARS S627L variants (Table 1), equivalent neural developmental toxicities were observed in the embryos of zebrafish after microinjections of human variant mRNAs, suggesting that the abnormal phenotypes in zebrafish are dominant‐negative or toxic gains of function (Weterman et al., 2018).
FIGURE 3.
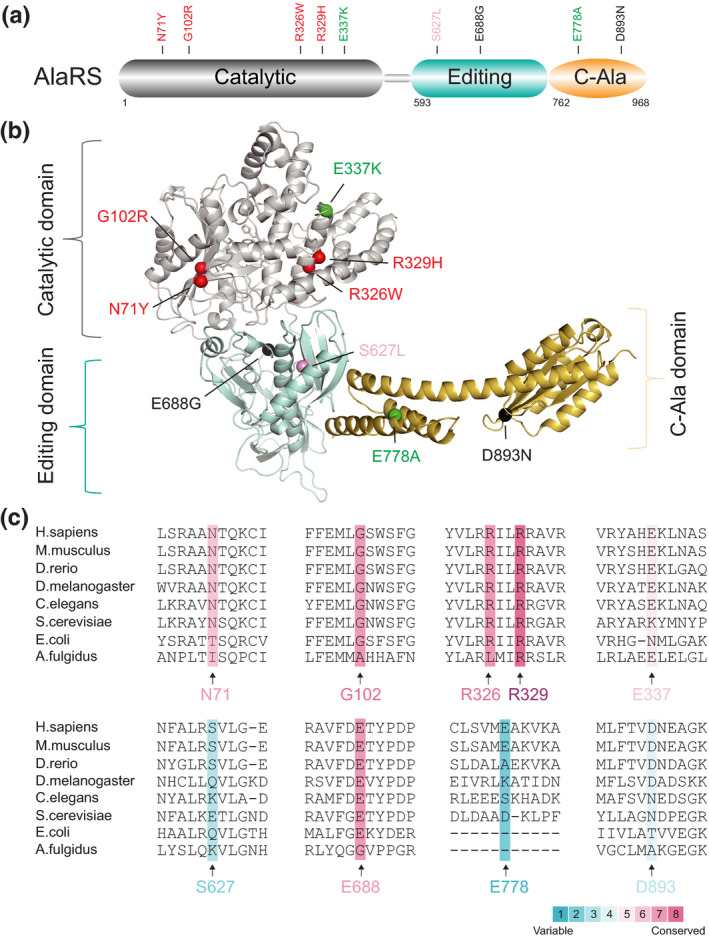
Distribution and conservation of CMT‐associated variant sites in human AlaRS. (a) Functional domains of AlaRS including a catalytic domain (in grey), an editing domain (in palegreen) and a C‐Ala domain (in orange). (b) The structure model of AlaRS was generated by the protein structure homology‐modelling server SWISS‐MODEL (Waterhouse et al., 2018). Human AlaRS catalytic domain (PDB entry 5KNN) and C‐Ala domain (PDB entry 5T5S) were further docked into the model and adjusted manually. CMT variant sites in either schematic diagram (a) or structure model (b) are indicated with different colors based on enzymatic activity of each variant with the same priority ranking as Figure 1b. (c) Evolutionary conservation analysis of CMT‐linked AlaRS across species. Sequence alignment of each variant site is indicated by the color intensity, with blue representing variable and red representing conserved
Other variants in AARS, including AARS D893N from a Chinese family (Zhao et al., 2012) and AARS E688G from an Irish family (Bansagi et al., 2015), were identified; however, their impacts on enzymatic activity and phenotypes in animal models need to be verified in the future.
As mentioned above, AlaRS includes an editing domain wherein two human CMT‐linked AlaRS variants (AARS S627L and AARS E688G) are located (Figure 3a and b). In mice, a missense variant at A734E in the editing domain of murine AlaRS (Aars A734E) can cause cell‐lethal accumulation of misfolded protein in neurons, leading to severe neurodegenerative phenotypes (Lee et al., 2006). Although the A734E variant in Aars is recessive, this finding, to some extent, pinpoints the fundamental roles of the editing activity of AlaRS for maintaining the accurate processing of genetic information and provides insight into novel mechanisms underlying human neurodegenerative diseases, such as AlaRS‐linked CMT.
Interestingly, the C‐Ala domain was once termed the dimerization domain because it provides contacts for dimerization in archaeal AlaRS (Naganuma et al., 2009). Over evolutionary time, the C‐Ala domain has been completely dispensable for aminoacylation but has developed distinct roles in higher organisms (Sun et al., 2016). Such a functional evolution of the C‐Ala domain allows AlaRS to be the single exception among the 19 other human aaRSs with no new motif or domain additions (Guo, Schimmel, et al., 2010). Perhaps for this reason, the AARS E778A variant in the C‐Ala domain shows WT‐like catalytic activity (McLaughlin et al., 2012), implying that nonenzymatic gain‐of‐function mechanisms underlie the pathogenesis of CMT2N.
5. HARS VARIANTS
HisRS has a domain structure identical to that of GlyRS, as it consists of a WHEP domain, a catalytic domain and an anticodon binding domain (Figure 4a). HARS variants have been successively identified in dominant axonal CMT type 2W (CMT2W, OMIM# 616625) (Laura et al., 2019), with all variants being located in the catalytic domain (Figure 4a and b). Of eight variants, seven result in a loss of function as determined by yeast complementation assays, and neurotoxicity has been successfully recapitulated in transgenic C. elegans models of HARS R137Q and HARS D364Y variants (Abbott et al., 2018; Safka Brozkova et al., 2015; Vester et al., 2013). However, the discrepancies between yeast model phenotype and aminoacylation activities in vitro (e.g., the HARS D364Y variant is lethal in yeast cells but has normal charging activity in vitro) (Table 1), as well as the causal link between these variants and CMT2W, remain unclear.
FIGURE 4.
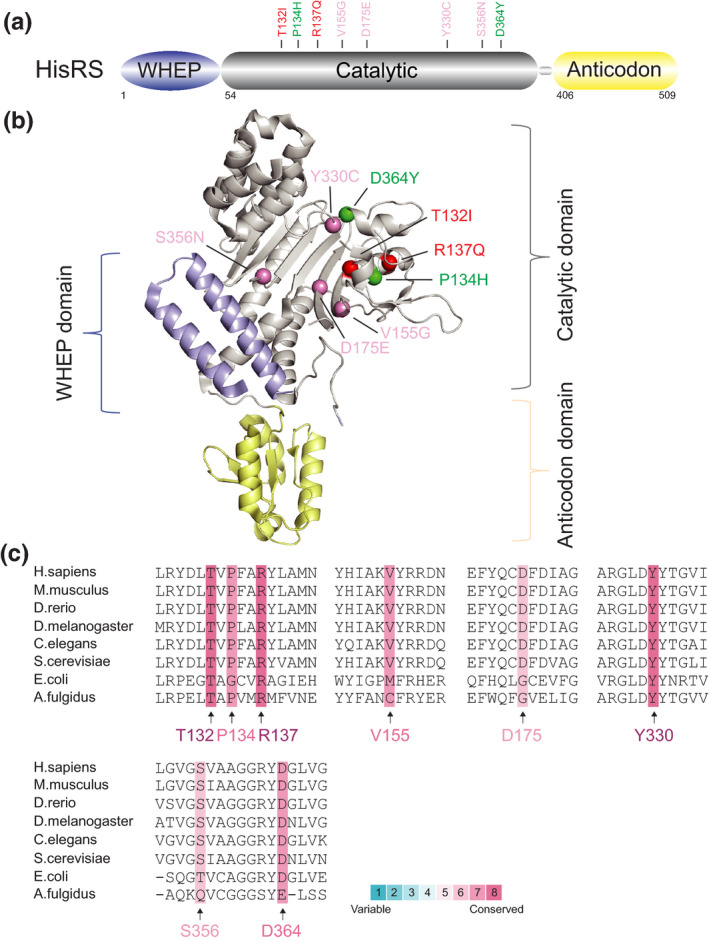
Distribution and conservation of CMT‐associated variant sites in human HisRS. (a) Functional domains of TyrRS including a WHEP domain (in purple), a catalytic domain (in grey) and an anticodon domain (in yellow). (b) The crystal structure of human HisRS (PDB entry 6O76). CMT variant sites in either schematic diagram (a) or crystal structure (b) are indicated with different colors based on enzymatic activity of each variant with the same priority ranking as Figure 1b. (c) Evolutionary conservation analysis of CMT‐linked HisRS across species. Sequence alignment of each variant site is indicated by the color intensity, with blue representing variable and red representing conserved
Based on these considerations, a recent study further investigated charged tRNAs in actual patients with CMT using assays enabling the detection of aminoacylation within the human cell environment. Unexpectedly, no differences in the aminoacylation levels of tRNAHis and other tRNAs were observed between patients carrying the HARS P134H variant and their unaffected family members. Additionally, no correlation between the enzymatic activities of variants causing CMT2W and disease severity were found (Blocquel et al., 2019). Further biochemical and biophysical analyses of HisRS variants (HARS T132I, HARS P134H, HARS D175E and HARS D364Y) demonstrated a conformational change that opens the dimerization interface of HisRS and likely exposes neomorphic surfaces that may mediate aberrant interactions with CMT2W‐causing variants. This work ruled out a correlation between enzymatic activity and disease severity, but instead showed a link between the degree of variant‐induced structural opening and disease severity, strongly supporting that HARS‐linked CMT disease is in fact not simply caused by a loss‐of‐function mechanism or a dominant‐negative effect of the variants but actually by open conformation‐driven gain‐of‐function mechanisms. The nonenzymatic functions gained by the HARS P134H and HARS D364Y variants in CMT are also rationalized by conservation analysis which showed highly conserved P134 and D364 sites with full enzymatic activity (Figure 4c).
It is worth noting that both GlyRS and HisRS contain a WHEP domain, which is indispensable for the neuronal toxicity caused by the gars P234KY variant in Drosophila (Grice et al., 2015). WHEP‐mediated suppression has also been shown to be a gain of function in the GARS G526R variant (He et al., 2011). In the case of HisRS, the conformational changes induced by HisRS variants strengthen the interactions between the WHEP domain and the C‐terminal part of the catalytic domain, which may help to open the dimerization interface (Blocquel et al., 2019). Interestingly, removal of the WHEP domain mostly had no effect on the aminoacylation activities of the WHEP‐containing aaRSs (Guo, Schimmel, et al., 2010), suggesting a potential relevance of the WHEP domain for CMT.
6. WARS VARIANTS
WARS encodes the human cytosolic TrpRS, which is composed of a WHEP domain, a catalytic domain and an anticodon binding domain (Figure 5a–c). WARS variants have just recently been identified in CMT, and the research on TrpRS in CMT is thus very limited.
FIGURE 5.
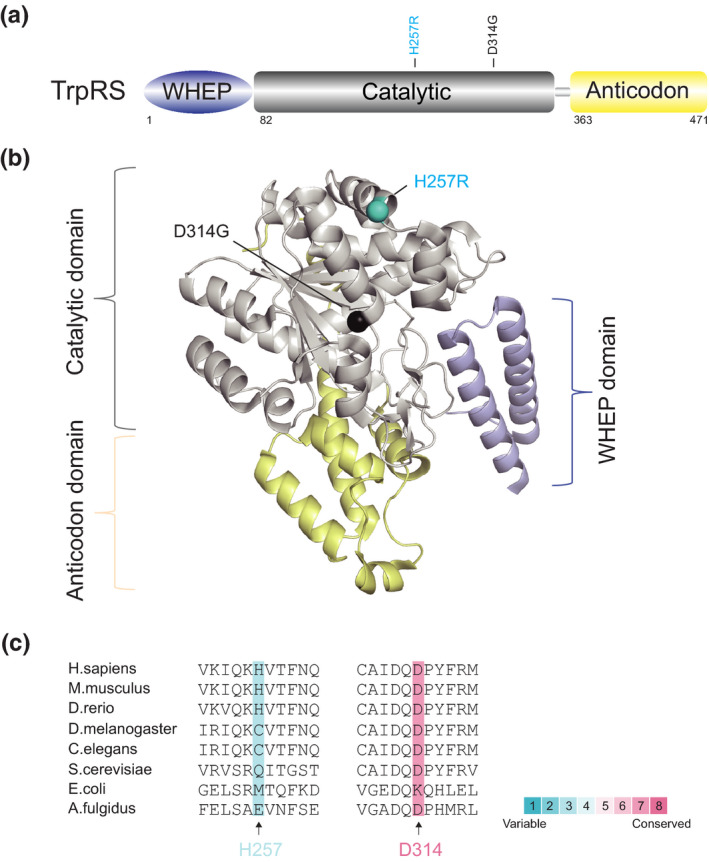
Distribution and conservation of CMT‐associated variant sites in human TrpRS. (a) Functional domains of TrpRS including a WHEP domain (in purple), a catalytic domain (in grey) and an anticodon domain (in yellow). (b) The crystal structure of human TrpRS (PDB entry 1R6T). CMT variant sites in either schematic diagram (a) or crystal structure (b) are indicated with different colors based on enzymatic activity of each variant with the same priority ranking as Figure 1b. (c) Evolutionary conservation analysis of CMT‐linked TrpRS across species. Sequence alignment of each variant site is indicated by the color intensity, with blue representing variable and red representing conserved
The first missense variant of WARS (WARS H257R) was identified in two unrelated Taiwanese pedigrees with autosomal‐dominant dHMN and later found in one additional dHMN family of European ancestry in Belgium (Tsai et al., 2017). This recurrent catalytic domain variant of WARS does not affect protein dimerization but does have a damaging and dominant‐negative effect on the enzymatic activity of TrpRS, resulting in disturbed protein synthesis and defective cell viability (Tsai et al., 2017). Furthermore, the variant, but not the WT, TrpRS inhibits neurite outgrowth in primary motor neurons and leads to neurite degeneration. Interestingly, in contrast to human WT TrpRS, which fails to complement wrs1 deficiency in yeast models, the WARS H257R variant can partially complement wrs1 deficiency in yeast cells, suggesting that the WARS H257R variant may change the structure of human TrpRS, enhancing the catalytic ability of the TrpRS variant toward yeast tRNA. Notably, the variant proteins exhibit enabled and enhanced binding to vascular endothelial‐cadherin (VE‐cadherin), leading to an enhanced angiostatic effect. This result is very similar to the previous finding that an aberrant GlyRS‐Nrp1 interaction interferes with the binding of VEGF, thus causing motor defects (He et al., 2015). In this regard, axonal neuropathy is very likely to be closely associated with the angiogenesis pathway, but this nonenzymatic function needs to be further investigated.
Very recently, another novel heterozygous WARS variant (WARS D314G) was reported in a Chinese family presenting with a mild‐to‐moderate and late‐onset phenotype of dHMN (Wang et al., 2019). Further structural model analysis predicted that this catalytic domain variant might have an impact on the recognition, binding and activation of tryptophan; however, other functional assays are lacking.
7. OTHER PUTATIVE NEUROPATHY‐ASSOCIATED VARIANTS
7.1. MARS variants
Unlike other CMT‐linked aaRSs, methionyl‐tRNA synthetase (MetRS) functions as a monomer and associates with the multi‐synthetase complex (MSC), which is anchored by the N‐terminal GST domain. MetRS also has a conserved class I catalytic domain and an anticodon domain, followed by a C‐terminal‐appended WHEP domain with an unclear function. Four MARS variants have thus far been linked to CMT type 2U (CMT2U, # OMIM 616280) (Table 1). Three variants are located in the anticodon binding domain, and one is in the catalytic domain. The monoallelic variant of R618C in the MARS (MARS R618C) was first described in a family with two affected patients who presented with late‐onset CMT2U and one unaffected member (Gonzalez et al., 2013). The MARS R618C variant is nonfunctional in yeast models, suggesting a loss‐of‐function feature; however, the potential mechanisms and contributions of this variant in CMT are less well characterized (Gonzalez et al., 2013). Subsequently, the MARS P800T variant was identified in a Korean family with late‐onset CMT2U (Hyun et al., 2014), and then in another Japanese family with late‐onset CMT2U (Hirano et al., 2016) and a Korean family with CMT2U but with a relatively early onset (Nam et al., 2016), suggesting that this recurrent variant causes variability and diversity of the CMT phenotype. Recently, two novel, likely disease‐associated missense variants, MARS R737W and MARS A397T, were reported in a 13‐year‐old girl with CMT2U (Sagi‐Dain et al., 2018) and in an 11‐year‐old girl with early‐onset CMT2U (Gillespie et al., 2019), respectively. These findings enrich the spectrum of the MARS variants in CMT2U, but disease‐associated function of the variants is unclear, and the human genetic evidence for MARS variants in CMT remains unequivocal.
7.2. NARS variants
In addition to CMT, aaRSs variants have been frequently implicated in other neuropathies. For example, biallelic NARS variants were identified in 7 affected patients with recessive microcephaly from three unrelated families (Wang et al., 2020). Another study described de novo dominant heterozygous and biallelic recessive variants in the NARS in 32 individuals from 21 families, presenting with multiple neurodevelopmental defects (Manole et al., 2020). Interestingly, functional data indicated that genotypes with dominant heterozygous NARS variants produce a toxic gain‐of‐function, whereas the homozygous recessive variants probably induce a partial loss of function (Manole et al., 2020; Wang et al., 2020). Although neuropathies other than CMT are outside the scope of this review, these studies do shed light on the complex pathogenic mechanisms of aaRSs in neuropathies.
8. SUMMARY AND PROSPECTS
Despite the identification of an increasing number of causative genes, CMT remains incurable. A more accurate classification and deeper understanding of CMT remain great challenges to scientists.
One major challenge in this field is understanding the pathogenic commonalities among different aaRS‐linked CMT subtypes. Some aaRS variants may cause CMT through a loss of function (either through haplo‐insufficiency or a dominant‐negative effect). Many CMT‐linked aaRS variant sites, such as D146, P244, ETAQ, G273 and I280 in GARS (Figure 1c), G41 in YARS (Figure 2c), G102 and R329 in AARS (Figure 3c), and T132, R137 and Y330 in HARS (Figure 4c), are evolutionarily highly conserved, suggesting their important roles, and variations in them do have loss‐of‐function properties in vitro or in yeast models (Table 1). In addition, phenotypes caused by recessive homozygous variants or those beyond CMT can be rescued by WT proteins in animal models (Achilli et al., 2009; Chihara et al., 2007; Seburn et al., 2006), suggesting that some variants result in undefined loss of function. Nevertheless, the specific mechanism by which aminoacylation deficiency causes peripheral neuropathy remains unknown.
Many lines of evidence have confirmed the toxic functions gained from aaRS variants, and this discovery may serve as a shared disease‐causing mechanism for aaRS‐associated CMT. For example, different CMT‐linked variants of GlyRS, HisRS and TyrRS lead to shared neomorphic structural opening which allows aberrant interactions with membrane receptors or intracellular proteins, thereby interfering with certain signaling pathways and trafficking in motor and sensory neurons (Blocquel et al., 2017, 2019; He et al., 2011, 2015). This shared property may facilitate the identification of new therapies by targeting these opened sites in all aaRS‐linked CMT subtypes. In addition, both CMT2D and DI‐CMTC models in Drosophila share common genetic modifiers with nuclear localization (Ermanoska et al., 2014). This finding implies that the nuclear involvement of CMT‐linked aaRSs, such as TyrRS (Bervoets et al., 2019), may very likely be another shared pathogenic mechanism, but this topic needs further exploration. Last, except for TyrRS and AlaRS, three CMT‐linked aaRSs (GlyRS, HisRS and TrpRS) and one putative CMT‐associated MetRS contain a specific appended WHEP domain, which mostly does not affect the enzymatic activities of their aaRSs (Guo, Schimmel, et al., 2010) but does show a close association with aaRS‐linked CMT (Blocquel et al., 2019; Grice et al., 2015; He et al., 2011), implying a potential shared pathogenic mechanism. Further studies on the specific role of the WHEP domain in CMT are of great interest. Nonetheless, we cannot rule out the possibility that both functional losses and functional gains are simultaneously involved in the pathogenic mechanisms in certain CMT forms, although the underlying factors remain unknown.
Furthermore, multiple aaRS variants have been identified in CMT, but only a few have been tested in animal models or patient cells. To date, the CMT‐causing GlyRS is the only one that has been successfully recapitulated in mouse models, while the functions of the other aaRS variants have been determined in only yeast strains, which are apparently insufficient to illustrate the true regulatory functions of aaRSs under physiological conditions. The discrepant aminoacylation levels of the HARS P134H variant in yeast models and CMT patients suggests that the pathogenic mechanisms caused by aaRS variants in CMT are highly context‐dependent. Furthermore, simple experimental models of aaRS variants established in flies, fish and worms also require re‐evaluation in mammalian animal models or patient cells. As such, detailed analysis of a higher model system will be critical for addressing this issue, and it will help to not only distinguish the contributions of each aaRS variant to CMT, but also to determine the pathogenic commonalities among different aaRS‐associated CMT subtypes to further advance therapeutic interventions.
Last but not least, neither ideal biomarkers nor therapeutic targets are currently available for slowly progressive CMT, although we do have some promising candidates to target in the diagnosis and treatment of CMT, such as Nrp‐1, TRIM28 and nuclear TyrRS. Deeply understanding why peripheral nerves are predominantly affected in aaRS‐linked CMT and how they work within the human cell environment will hopefully lay the foundation for the precise stratification and targeted treatment of CMT in the future.
CONFLICTS OF INTEREST
The authors report no conflicts of interest.
AUTHORS’ CONTRIBUTIONS
H.Z. and L.S. wrote the paper and made the figures. Z‐W.Z. and L.S. designed the review.
ACKNOWLEDGMENT
This work was supported by the National Natural Science Foundation of China (No. 31971147) and Shenzhen Science and Technology Innovation Commission (No. JCYJ20190807155011406) given to L.S.; Natural Science Foundation of Guangdong Province (No. 2019A1515010881) and Shenzhen Science and Technology Innovation Commission (No. JCYJ20190807154407467) given to Z‐W.Z.; Natural Science Foundation of Yunnan Province (No. 2019FB089) and the Fundamental Research Funds for the Central Universities (No. 3332018130) given to H.Z.
Zhang H, Zhou Z‐W, Sun L. Aminoacyl‐tRNA synthetases in Charcot–Marie–Tooth disease: A gain or a loss?. J Neurochem.2021;157:351–369. 10.1111/jnc.15249
Contributor Information
Zhong‐Wei Zhou, Email: zhouzhw6@mail.sysu.edu.cn.
Litao Sun, Email: sunlt@mail.sysu.edu.cn.
REFERENCES
- Abbott, J. A. , Meyer‐Schuman, R. , Lupo, V. , Feely, S. , Mademan, I. , Oprescu, S. N. , Griffin, L. B. , Alberti, M. A. , Casasnovas, C. , Aharoni, S. , Basel‐Vanagaite, L. , Züchner, S. , De Jonghe, P. , Baets, J. , Shy, M. E. , Espinós, C. , Demeler, B. , Antonellis, A. , & Francklyn, C. (2018). Substrate interaction defects in histidyl‐tRNA synthetase linked to dominant axonal peripheral neuropathy. Human Mutation, 39, 415–432. 10.1002/humu.23380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abe, A. , & Hayasaka, K. (2009). The GARS gene is rarely mutated in Japanese patients with Charcot‐Marie‐Tooth neuropathy. Journal of Human Genetics, 54, 310–312. 10.1038/jhg.2009.25 [DOI] [PubMed] [Google Scholar]
- Achilli, F. , Bros‐Facer, V. , Williams, H. P. , Banks, G. T. , AlQatari, M. , Chia, R. , Tucci, V. , Groves, M. , Nickols, C. D. , Seburn, K. L. , Kendall, R. , Cader, M. Z. , Talbot, K. , van Minnen, J. , Burgess, R. W. , Brandner, S. , Martin, J. E. , Koltzenburg, M. , Greensmith, L. , … Fisher, E. M. C. (2009). An ENU‐induced mutation in mouse glycyl‐tRNA synthetase (GARS) causes peripheral sensory and motor phenotypes creating a model of Charcot‐Marie‐Tooth type 2D peripheral neuropathy. Disease Models & Mechanisms, 2, 359–373. 10.1242/dmm.002527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonellis, A. , Ellsworth, R. E. , Sambuughin, N. , Puls, I. , Abel, A. , Lee‐Lin, S.‐Q. , Jordanova, A. , Kremensky, I. , Christodoulou, K. , Middleton, L. T. , Sivakumar, K. , Ionasescu, V. , Funalot, B. , Vance, J. M. , Goldfarb, L. G. , Fischbeck, K. H. , & Green, E. D. (2003). Glycyl tRNA synthetase mutations in Charcot‐Marie‐Tooth disease type 2D and distal spinal muscular atrophy type V. American Journal of Human Genetics, 72, 1293–1299. 10.1086/375039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonellis, A. , Lee‐Lin, S.‐Q. , Wasterlain, A. , Leo, P. , Quezado, M. , Goldfarb, L. G. , Myung, K. , Burgess, S. , Fischbeck, K. H. , & Green, E. D. (2006). Functional analyses of glycyl‐tRNA synthetase mutations suggest a key role for tRNA‐charging enzymes in peripheral axons. Journal of Neuroscience, 26, 10397–10406. 10.1523/JNEUROSCI.1671-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bansagi, B. , Antoniadi, T. , Burton‐Jones, S. , Murphy, S. M. , McHugh, J. , Alexander, M. , Wells, R. , Davies, J. , Hilton‐Jones, D. , Lochmüller, H. , Chinnery, P. , & Horvath, R. (2015). Genotype/phenotype correlations in AARS‐related neuropathy in a cohort of patients from the United Kingdom and Ireland. Journal of Neurology, 262, 1899–1908. 10.1007/s00415-015-7778-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benoy, V. , Van Helleputte, L. , Prior, R. , d’Ydewalle, C. , Haeck, W. , Geens, N. , Scheveneels, W. , Schevenels, B. , Cader, M. Z. , Talbot, K. , Kozikowski, A. P. , Vanden Berghe, P. , Van Damme, P. , Robberecht, W. , & Van Den Bosch, L. (2018). HDAC6 is a therapeutic target in mutant GARS‐induced Charcot‐Marie‐Tooth disease. Brain, 141, 673–687. 10.1093/brain/awx375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bervoets, S. , Wei, N. A. , Erfurth, M.‐L. , Yusein‐Myashkova, S. , Ermanoska, B. , Mateiu, L. , Asselbergh, B. , Blocquel, D. , Kakad, P. , Penserga, T. , Thomas, F. P. , Guergueltcheva, V. , Tournev, I. , Godenschwege, T. , Jordanova, A. , & Yang, X.‐L. (2019). Transcriptional dysregulation by a nucleus‐localized aminoacyl‐tRNA synthetase associated with Charcot‐Marie‐Tooth neuropathy. Nature Communications, 10, 5045. 10.1038/s41467-019-12909-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blocquel, D. , Li, S. , Wei, N. A. , Daub, H. , Sajish, M. , Erfurth, M.‐L. , Kooi, G. , Zhou, J. , Bai, G. E. , Schimmel, P. , Jordanova, A. , & Yang, X.‐L. (2017). Alternative stable conformation capable of protein misinteraction links tRNA synthetase to peripheral neuropathy. Nucleic Acids Research, 45, 8091–8104. 10.1093/nar/gkx455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blocquel, D. , Sun, L. , Matuszek, Z. , Li, S. , Weber, T. , Kuhle, B. , Kooi, G. , Wei, N. A. , Baets, J. , Pan, T. , Schimmel, P. , & Yang, X.‐L. (2019). CMT disease severity correlates with mutation‐induced open conformation of histidyl‐tRNA synthetase, not aminoacylation loss, in patient cells. Proceedings of the National Academy of Sciences, 116, 19440–19448. 10.1073/pnas.1908288116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boczonadi, V. , Meyer, K. , Gonczarowska‐Jorge, H. , Griffin, H. , Roos, A. , Bartsakoulia, M. , Bansagi, B. , Ricci, G. , Palinkas, F. , Zahedi, R. P. , Bruni, F. , Kaspar, B. , Lochmüller, H. , Boycott, K. M. , Müller, J. S. , & Horvath, R. (2018). Mutations in glycyl‐tRNA synthetase impair mitochondrial metabolism in neurons. Human Molecular Genetics, 27, 2187–2204. 10.1093/hmg/ddy127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cader, M. Z. , Ren, J. , James, P. A. , Bird, L. E. , Talbot, K. , & Stammers, D. K. (2007). Crystal structure of human wildtype and S581L‐mutant glycyl‐tRNA synthetase, an enzyme underlying distal spinal muscular atrophy. FEBS Letters, 581, 2959–2964. 10.1016/j.febslet.2007.05.046 [DOI] [PubMed] [Google Scholar]
- Chihara, T. , Luginbuhl, D. , & Luo, L. (2007). Cytoplasmic and mitochondrial protein translation in axonal and dendritic terminal arborization. Nature Neuroscience, 10, 828–837. 10.1038/nn1910 [DOI] [PubMed] [Google Scholar]
- Chung, P. , Northrup, H. , Azmath, M. , Mosquera, R. A. , Moody, S. , & Yadav, A. (2018). Glycyl tRNA Synthetase (GARS) gene variant causes distal hereditary motor neuropathy V. Case Rep Pediatr, 2018, 8516285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Bo, R. , Locatelli, F. , Corti, S. , Scarlato, M. , Ghezzi, S. , Prelle, A. , Fagiolari, G. , Moggio, M. , Carpo, M. , Bresolin, N. , & Comi, G. P. (2006). Coexistence of CMT‐2D and distal SMA‐V phenotypes in an Italian family with a GARS gene mutation. Neurology, 66, 752–754. 10.1212/01.wnl.0000201275.18875.ac [DOI] [PubMed] [Google Scholar]
- Dubourg, O. , Azzedine, H. , Yaou, R. B. , Pouget, J. , Barois, A. , Meininger, V. , Bouteiller, D. , Ruberg, M. , Brice, A. , & LeGuern, E. (2006). The G526R glycyl‐tRNA synthetase gene mutation in distal hereditary motor neuropathy type V. Neurology, 66, 1721–1726. 10.1212/01.wnl.0000218304.02715.04 [DOI] [PubMed] [Google Scholar]
- Ermanoska, B. , Motley, W. W. , Leitão‐Gonçalves, R. , Asselbergh, B. , Lee, L. T. H. , De Rijk, P. , Sleegers, K. , Ooms, T. , Godenschwege, T. A. , Timmerman, V. , Fischbeck, K. H. , & Jordanova, A. (2014). CMT‐associated mutations in glycyl‐ and tyrosyl‐tRNA synthetases exhibit similar pattern of toxicity and share common genetic modifiers in Drosophila. Neurobiology of Diseases, 68, 180–189. 10.1016/j.nbd.2014.04.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eskuri, J. M. , Stanley, C. M. , Moore, S. A. , & Mathews, K. D. (2012). Infantile onset CMT2D/dSMA V in monozygotic twins due to a mutation in the anticodon‐binding domain of GARS. Journal of the Peripheral Nervous System, 17, 132–134. 10.1111/j.1529-8027.2012.00370.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Froelich, C. A. , & First, E. A. (2011). Dominant Intermediate Charcot‐Marie‐Tooth disorder is not due to a catalytic defect in tyrosyl‐tRNA synthetase. Biochemistry, 50, 7132–7145. 10.1021/bi200989h [DOI] [PubMed] [Google Scholar]
- Gillespie, M. K. , McMillan, H. J. , Kernohan, K. D. , Pena, I. A. , Meyer‐Schuman, R. , Antonellis, A. , & Boycott, K. M. (2019). A novel mutation in MARS in a patient with charcot‐marie‐tooth disease, axonal, Type 2U with congenital onset. Journal of Neuromuscular Diseases, 6, 333–339. 10.3233/JND-190404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzaga‐Jauregui, C. , Harel, T. , Gambin, T. , Kousi, M. , Griffin, L. B. , Francescatto, L. , Ozes, B. , Karaca, E. , Jhangiani, S. N. , Bainbridge, M. N. , Lawson, K. S. , Pehlivan, D. , Okamoto, Y. , Withers, M. , Mancias, P. , Slavotinek, A. , Reitnauer, P. J. , Goksungur, M. T. , Shy, M. , & Lupski J. R. (2015). Exome sequence analysis suggests that genetic burden contributes to phenotypic variability and complex neuropathy. Cell Reports, 12(7), 1169–1183. 10.1016/j.celrep.2015.07.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez, M. , McLaughlin, H. , Houlden, H. , Guo, M. , Yo‐Tsen, L. , Hadjivassilious, M. , Speziani, F. , Yang, X. ‐L. , Antonellis, A. , Reilly, M. M. , & Züchner, S. (2013). Exome sequencing identifies a significant variant in methionyl‐tRNA synthetase (MARS) in a family with late‐onset CMT2. Journal of Neurology, Neurosurgery & Psychiatry, 84(11), 1247–1249. 10.1136/jnnp-2013-305049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grice, S. J. , Sleigh, J. N. , Motley, W. W. , Liu, J. L. , Burgess, R. W. , Talbot, K. , & Cader, M. Z. (2015). Dominant, toxic gain‐of‐function mutations in gars lead to non‐cell autonomous neuropathology. Human Molecular Genetics, 24, 4397–4406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin, L. B. , Sakaguchi, R. , McGuigan, D. , Gonzalez, M. A. , Searby, C. , Zuchner, S. , Hou, Y. M. , & Antonellis, A. (2014). Impaired function is a common feature of neuropathy‐associated glycyl‐tRNA synthetase mutations. Human Mutation, 35, 1363–1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo, M. , & Schimmel, P. (2013). Essential nontranslational functions of tRNA synthetases. Nature Chemical Biology, 9, 145–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo, M. , Schimmel, P. , & Yang, X. L. (2010). Functional expansion of human tRNA synthetases achieved by structural inventions. FEBS Letters, 584, 434–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo, M. , Yang, X. L. , & Schimmel, P. (2010). New functions of aminoacyl‐tRNA synthetases beyond translation. Nature Reviews Molecular Cell Biology, 11, 668–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He, W. , Bai, G. , Zhou, H. , Wei, N. , White, N. M. , Lauer, J. , Liu, H. , Shi, Y. , Dumitru, C. D. , Lettieri, K. , Shubayev, V. , Jordanova, A. , Guergueltcheva, V. , Griffin, P. R. , Burgess, R. W. , Pfaff, S. L. , & Yang, X. ‐L. (2015). CMT2D neuropathy is linked to the neomorphic binding activity of glycyl‐tRNA synthetase. Nature, 526(7575), 710–714. 10.1038/nature15510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He, W. , Zhang, H. M. , Chong, Y. E. , Guo, M. , Marshall, A. G. , & Yang, X. L. (2011). Dispersed disease‐causing neomorphic mutations on a single protein promote the same localized conformational opening. Proceedings of the National Academy of Sciences of the United States of America, 108, 12307–12312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirano, M. , Oka, N. , Hashiguchi, A. , Ueno, S. , Sakamoto, H. , Takashima, H. , Higuchi, Y. , Kusunoki, S. , & Nakamura, Y. (2016). Histopathological features of a patient with Charcot‐Marie‐Tooth disease type 2U/AD‐CMTax‐MARS. Journal of the Peripheral Nervous System, 21, 370–374. [DOI] [PubMed] [Google Scholar]
- Hyun, Y. S. , Park, H. J. , Heo, S. H. , Yoon, B. R. , Nam, S. H. , Kim, S. B. , Park, C. I. , Choi, B. O. , & Chung, K. W. (2014). Rare variants in methionyl‐ and tyrosyl‐tRNA synthetase genes in late‐onset autosomal dominant Charcot‐Marie‐Tooth neuropathy. Clinical Genetics, 86, 592–594. [DOI] [PubMed] [Google Scholar]
- James, P. A. , Cader, M. Z. , Muntoni, F. , Childs, A. M. , Crow, Y. J. , & Talbot, K. (2006). Severe childhood SMA and axonal CMT due to anticodon binding domain mutations in the GARS gene. Neurology, 67, 1710–1712. 10.1212/01.wnl.0000242619.52335.bc [DOI] [PubMed] [Google Scholar]
- Jordanova, A. , Irobi, J. , Thomas, F. P. , Van Dijck, P. , Meerschaert, K. , Dewil, M. , Dierick, I. , Jacobs, A. N. , De Vriendt, E. , Guergueltcheva, V. , Rao, C. V. , Tournev, I. , Gondim, F. A. A. , D'Hooghe, M. , Van Gerwen, V. , Callaerts, P. , Van Den Bosch, L. , Timmermans, J.‐P. , Robberecht, W. , … Timmerman, V. (2006). Disrupted function and axonal distribution of mutant tyrosyl‐tRNA synthetase in dominant intermediate Charcot‐Marie‐Tooth neuropathy. Nature Genetics, 38, 197–202. 10.1038/ng1727 [DOI] [PubMed] [Google Scholar]
- Jordanova, A. , Thomas, F. P. , Guergueltcheva, V. , Tournev, I. , Gondim, F. A. A. , Ishpekova, B. , De Vriendt, E. , Jacobs, A. N. , Litvinenko, I. , Ivanova, N. , Buzhov, B. , De Jonghe, P. , Kremensky, I. , & Timmerman, V. (2003). Dominant intermediate Charcot‐Marie‐Tooth type C maps to chromosome 1p34‐p35. American Journal of Human Genetics, 73, 1423–1430. 10.1086/379792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kao, J. , Ryan, J. , Brett, G. , Chen, J. , Shen, H. , Fan, Y. G. , Godman, G. , Familletti, P. C. , Wang, F. , & Pan, Y. C. (1992). Endothelial monocyte‐activating polypeptide II. A novel tumor‐derived polypeptide that activates host‐response mechanisms. Journal of Biological Chemistry, 267, 20239–20247. [PubMed] [Google Scholar]
- Kawakami, N. , Komatsu, K. , Yamashita, H. , Uemura, K. , Oka, N. , Takashima, H. , & Takahashi, R. (2014). A novel mutation in glycyl‐tRNA synthetase caused Charcot‐Marie‐Tooth disease type 2D with facial and respiratory muscle involvement. Rinsho Shinkeigaku, 54, 911–915. 10.5692/clinicalneurol.54.911 [DOI] [PubMed] [Google Scholar]
- Klein, C. J. , Middha, S. , Duan, X. , Wu, Y. , Litchy, W. J. , Gu, W. , Dyck, P. J. B. , Gavrilova, R. H. , Smith, D. I. , Kocher, J.‐P. , & Dyck, P. J. (2014). Application of whole exome sequencing in undiagnosed inherited polyneuropathies. Journal of Neurology, Neurosurgery and Psychiatry, 85, 1265–1272. 10.1136/jnnp-2013-306740 [DOI] [PubMed] [Google Scholar]
- Latour, P. , Thauvin‐Robinet, C. , Baudelet‐Méry, C. , Soichot, P. , Cusin, V. , Faivre, L. , Locatelli, M.‐C. , Mayençon, M. , Sarcey, A. , Broussolle, E. , Camu, W. , David, A. , & Rousson, R. (2010). A major determinant for binding and aminoacylation of tRNA(Ala) in cytoplasmic Alanyl‐tRNA synthetase is mutated in dominant axonal Charcot‐Marie‐Tooth disease. American Journal of Human Genetics, 86, 77–82. 10.1016/j.ajhg.2009.12.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laura, M. , Pipis, M. , Rossor, A. M. , & Reilly, M. M. (2019). Charcot‐Marie‐Tooth disease and related disorders: An evolving landscape. Current Opinion in Neurology, 32, 641–650. 10.1097/WCO.0000000000000735 [DOI] [PubMed] [Google Scholar]
- Lee, D. C. , Meyer‐Schuman, R. , Bacon, C. , Shy, M. E. , Antonellis, A. , & Scherer, S. S. (2019). A recurrent GARS mutation causes distal hereditary motor neuropathy. Journal of the Peripheral Nervous System, 24, 320–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, H. J. , Park, J. , Nakhro, K. , Park, J. M. , Hur, Y. M. , Choi, B. O. , & Chung, K. W. (2012). Two novel mutations of GARS in Korean families with distal hereditary motor neuropathy type V. Journal of the Peripheral Nervous System, 17, 418–421. [DOI] [PubMed] [Google Scholar]
- Lee, J. W. , Beebe, K. , Nangle, L. A. , Jang, J. , Longo‐Guess, C. M. , Cook, S. A. , Davisson, M. T. , Sundberg, J. P. , Schimmel, P. , & Ackerman, S. L. (2006). Editing‐defective tRNA synthetase causes protein misfolding and neurodegeneration. Nature, 443, 50–55. 10.1038/nature05096 [DOI] [PubMed] [Google Scholar]
- Liao, Y. C. , Liu, Y. T. , Tsai, P. C. , Chang, C. C. , Huang, Y. H. , Soong, B. W. , & Lee, Y. C. (2015). Two novel de novo GARS mutations cause early‐onset axonal charcot‐marie‐tooth disease. PLoS One, 10, e0133423. 10.1371/journal.pone.0133423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, K. P. , Soong, B. W. , Yang, C. C. , Huang, L. W. , Chang, M. H. , Lee, I. H. , Antonellis, A. , & Lee, Y. C. (2011). The mutational spectrum in a cohort of Charcot‐Marie‐Tooth disease type 2 among the Han Chinese in Taiwan. PLoS One, 6, e29393. 10.1371/journal.pone.0029393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling, J. , Reynolds, N. , & Ibba, M. (2009). Aminoacyl‐tRNA synthesis and translational quality control. Annual Review of Microbiology, 63, 61–78. 10.1146/annurev.micro.091208.073210 [DOI] [PubMed] [Google Scholar]
- Manole, A. , Efthymiou, S. , O’Connor, E. , Mendes, M. I. , Jennings, M. , Maroofian, R. , Davagnanam, I. , Mankad, K. , Lopez, M. R. , Salpietro, V. , Harripaul, R. , Badalato, L. , Walia, J. , Francklyn, C. S. , Athanasiou‐Fragkouli, A. , Sullivan, R. , Desai, S. , Baranano, K. , Zafar, F. , … Houlden, H. (2020). De novo and Bi‐allelic pathogenic variants in NARS1 cause neurodevelopmental delay due to toxic gain‐of‐function and partial loss‐of‐function effects. American Journal of Human Genetics, 107, 311–324. 10.1016/j.ajhg.2020.06.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaughlin, H. M. , Sakaguchi, R. , Giblin, W. , Wilson, T. E. , Biesecker, L. , Lupski, J. R. , Talbot, K. , Vance, J. M. , Züchner, S. , Lee, Y.‐C. , Kennerson, M. , Hou, Y. ‐M. , Nicholson, G. , & Antonellis, A. , (2012). A Recurrent loss‐of‐function alanyl‐tRNA synthetase (AARS ) mutation in patients with charcot‐marie‐tooth disease type 2N (CMT2N). Human Mutation, 33(1), 244–253. 10.1002/humu.21635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mo, Z. , Zhao, X. , Liu, H. , Hu, Q. , Chen, X.‐Q. , Pham, J. , Wei, N. A. , Liu, Z. E. , Zhou, J. , Burgess, R. W. , Pfaff, S. L. , Caskey, C. T. , Wu, C. , Bai, G. E. , & Yang, X.‐L. (2018). Aberrant GlyRS‐HDAC6 interaction linked to axonal transport deficits in Charcot‐Marie‐Tooth neuropathy. Nature Communications, 9, 1007. 10.1038/s41467-018-03461-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morelli, K. H. , Griffin, L. B. , Pyne, N. K. , Wallace, L. M. , Fowler, A. M. , Oprescu, S. N. , Takase, R. , Wei, N. A. , Meyer‐Schuman, R. , Mellacheruvu, D. , Kitzman, J. O. , Kocen, S. G. , Hines, T. J. , Spaulding, E. L. , Lupski, J. R. , Nesvizhskii, A. , Mancias, P. , Butler, I. J. , Yang, X.‐L. , … Burgess, R. W. (2019). Allele‐specific RNA interference prevents neuropathy in Charcot‐Marie‐Tooth disease type 2D mouse models. Journal of Clinical Investigation, 129, 5568–5583. 10.1172/JCI130600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motley, W. W. , Griffin, L. B. , Mademan, I. , Baets, J. , De Vriendt, E. , De Jonghe, P. , Antonellis, A. , Jordanova, A. , & Scherer, S. S. (2015). A novel AARS mutation in a family with dominant myeloneuropathy. Neurology, 84, 2040–2047. 10.1212/WNL.0000000000001583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motley, W. W. , Seburn, K. L. , Nawaz, M. H. , Miers, K. E. , Cheng, J. , Antonellis, A. , Green, E. D. , Talbot, K. , Yang, X.‐L. , Fischbeck, K. H. , & Burgess, R. W. (2011). Charcot‐Marie‐Tooth‐linked mutant GARS is toxic to peripheral neurons independent of wild‐type GARS levels. PLoS Genetics, 7, e1002399. 10.1371/journal.pgen.1002399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motley, W. W. , Talbot, K. , & Fischbeck, K. H. (2010). GARS axonopathy: Not every neuron's cup of tRNA. Trends in Neurosciences, 33, 59–66. 10.1016/j.tins.2009.11.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naganuma, M. , Sekine, S. , Fukunaga, R. , & Yokoyama, S. (2009). Unique protein architecture of alanyl‐tRNA synthetase for aminoacylation, editing, and dimerization. Proceedings of the National Academy of Sciences, 106, 8489–8494. 10.1073/pnas.0901572106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nam, S. H. , Hong, Y. B. , Hyun, Y. S. , da Nam, E. , Kwak, G. , Hwang, S. H. , Choi, B. O. , & Chung, K. W. (2016). Identification of genetic causes of inherited peripheral neuropathies by targeted gene panel sequencing. Molecules and Cells, 39, 382–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nan, H. , Takaki, R. , Hata, T. , Ichinose, Y. , Tsuchiya, M. , Koh, K. , & Takiyama, Y. (2019). Novel GARS mutation presenting as autosomal dominant intermediate Charcot‐Marie‐Tooth disease. Journal of the Peripheral Nervous System, 24, 156–160. [DOI] [PubMed] [Google Scholar]
- Nangle, L. A. , Zhang, W. , Xie, W. , Yang, X. L. , & Schimmel, P. (2007). Charcot‐Marie‐Tooth disease‐associated mutant tRNA synthetases linked to altered dimer interface and neurite distribution defect. Proceedings of the National Academy of Sciences, 104, 11239–11244. 10.1073/pnas.0705055104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niehues, S. , Bussmann, J. , Steffes, G. , Erdmann, I. , Köhrer, C. , Sun, L. , Wagner, M. , Schäfer, K. , Wang, G. , Koerdt, S. N. , Stum, M. , Jaiswal, S. , RajBhandary, U. L. , Thomas, U. , Aberle, H. , Burgess, R. W. , Yang, X.‐L. , Dieterich, D. , & Storkebaum, E. (2015). Impaired protein translation in Drosophila models for Charcot‐Marie‐Tooth neuropathy caused by mutant tRNA synthetases. Nature Communications, 6, 7520. 10.1038/ncomms8520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pareyson, D. , & Marchesi, C. (2009). Diagnosis, natural history, and management of Charcot‐Marie‐Tooth disease. The Lancet Neurology, 8, 654–667. 10.1016/S1474-4422(09)70110-3 [DOI] [PubMed] [Google Scholar]
- Patzko, A. , & Shy, M. E. (2011). Update on Charcot‐Marie‐Tooth disease. Current Neurology and Neuroscience Reports, 11, 78–88. 10.1007/s11910-010-0158-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin, X. , Hao, Z. , Tian, Q. , Zhang, Z. , Zhou, C. , & Xie, W. (2014). Cocrystal structures of glycyl‐tRNA synthetase in complex with tRNA suggest multiple conformational states in glycylation. Journal of Biological Chemistry, 289, 20359–20369. 10.1074/jbc.M114.557249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohkamm, B. , Reilly, M. M. , Lochmüller, H. , Schlotter‐Weigel, B. , Barisic, N. , Schöls, L. , Nicholson, G. , Pareyson, D. , Laurà, M. , Janecke, A. R. , Miltenberger‐Miltenyi, G. , John, E. , Fischer, C. , Grill, F. , Wakeling, W. , Davis, M. , Pieber, T. R. , & Auer‐Grumbach, M. (2007). Further evidence for genetic heterogeneity of distal HMN type V, CMT2 with predominant hand involvement and Silver syndrome. Journal of the Neurological Sciences, 263, 100–106. 10.1016/j.jns.2007.06.047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossor, A. M. , Tomaselli, P. J. , & Reilly, M. M. (2016). Recent advances in the genetic neuropathies. Current Opinion in Neurology, 29, 537–548. 10.1097/WCO.0000000000000373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Safka Brozkova, D. , Deconinck, T. , Beth Griffin, L. , Ferbert, A. , Haberlova, J. , Mazanec, R. , Lassuthova, P. , Roth, C. , Pilunthanakul, T. , Rautenstrauss, B. , Janecke, A. R. , Zavadakova, P. , Chrast, R. , Rivolta, C. , Zuchner, S. , Antonellis, A. , Beg, A. A. , De Jonghe, P. , Senderek, J. , & Baets, J. (2015). Loss of function mutations inHARScause a spectrum of inherited peripheral neuropathies. Brain, 138(8), 2161–2172. 10.1093/brain/awv158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sagi‐Dain, L. , Shemer, L. , Zelnik, N. , Zoabi, Y. , Orit, S. , Adir, V. , Schif, A. , & Peleg, A. (2018). Whole‐exome sequencing reveals a novel missense mutation in the MARS gene related to a rare Charcot‐Marie‐Tooth neuropathy type 2U. Journal of the Peripheral Nervous System, 23, 138–142. [DOI] [PubMed] [Google Scholar]
- Seburn, K. L. , Nangle, L. A. , Cox, G. A. , Schimmel, P. , & Burgess, R. W. (2006). An active dominant mutation of glycyl‐tRNA synthetase causes neuropathy in a Charcot‐Marie‐Tooth 2D mouse model. Neuron, 51, 715–726. 10.1016/j.neuron.2006.08.027 [DOI] [PubMed] [Google Scholar]
- Sivakumar, K. , Kyriakides, T. , Puls, I. , Nicholson, G. A. , Funalot, B. , Antonellis, A. , Sambuughin, N. , Christodoulou, K. , Beggs, J. L. , Zamba‐Papanicolaou, E. , Ionasescu, V. , Dalakas, M. C. , Green, E. D. , Fischbeck, K. H. , & Goldfarb, L. G. (2005). Phenotypic spectrum of disorders associated with glycyl‐tRNA synthetase mutations. Brain, 128, 2304–2314. 10.1093/brain/awh590 [DOI] [PubMed] [Google Scholar]
- Skre, H. (1974). Genetic and clinical aspects of Charcot‐Marie‐Tooth's disease. Clinical Genetics, 6, 98–118. 10.1111/j.1399-0004.1974.tb00638.x [DOI] [PubMed] [Google Scholar]
- Sleigh, J. N. , Dawes, J. M. , West, S. J. , Wei, N. , Spaulding, E. L. , Gómez‐Martín, A. , Zhang, Q. , Burgess, R. W. , Cader, M. Z. , Talbot, K. , Yang, X. L. , Bennett, D. L. , & Schiavo G. (2017). Trk receptor signaling and sensory neuron fate are perturbed in human neuropathy caused by Gars mutations. Proceedings of the National Academy of Sciences, 114(16), E3324–E3333. 10.1073/pnas.1614557114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storkebaum, E. , Leitão‐Gonçalves, R. , Godenschwege, T. , Nangle, L. , Mejia, M. , Bosmans, I. , Ooms, T. , Jacobs, A. , Van Dijck, P. , Yang, X. L. , Schimmel, P. , Norga, K. , Timmerman, V. , Callaerts, P. , & Jordanova, A. (2009). Dominant mutations in the tyrosyl‐tRNA synthetase gene recapitulate inDrosophilafeatures of human Charcot–Marie–Tooth neuropathy. Proceedings of the National Academy of Sciences, 106(28), 11782–11787. 10.1073/pnas.0905339106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stum, M. , McLaughlin, H. M. , Kleinbrink, E. L. , Miers, K. E. , Ackerman, S. L. , Seburn, K. L. , Antonellis, A. , & Burgess, R. W. (2011). An assessment of mechanisms underlying peripheral axonal degeneration caused by aminoacyl‐tRNA synthetase mutations. Molecular and Cellular Neurosciences, 46, 432–443. 10.1016/j.mcn.2010.11.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun, A. , Liu, X. , Zheng, M. , Sun, Q. , Huang, Y. , & Fan, D. (2015). A novel mutation of the glycyl‐tRNA synthetase (GARS) gene associated with Charcot‐Marie‐Tooth type 2D in a Chinese family. Neurological Research, 37, 782–787. 10.1179/1743132815Y.0000000055 [DOI] [PubMed] [Google Scholar]
- Sun, B. , Chen, Z. , Ling, L. , Yang, F. , & Huang, X. (2017). Clinical and genetic spectra of Charcot‐Marie‐Tooth disease in Chinese Han patients. Journal of the Peripheral Nervous System, 22, 13–18. 10.1111/jns.12195 [DOI] [PubMed] [Google Scholar]
- Sun, L. , Song, Y. , Blocquel, D. , Yang, X. L. , & Schimmel, P. (2016). Two crystal structures reveal design for repurposing the C‐Ala domain of human AlaRS. Proceedings of the National Academy of Sciences, 113, 14300–14305. 10.1073/pnas.1617316113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai, P.‐C. , Soong, B.‐W. , Mademan, I. , Huang, Y.‐H. , Liu, C.‐R. , Hsiao, C.‐T. , Wu, H.‐T. , Liu, T.‐T. , Liu, Y.‐T. , Tseng, Y.‐T. , Lin, K.‐P. , Yang, U.‐C. , Chung, K. W. , Choi, B.‐O. , Nicholson, G. A. , Kennerson, M. L. , Chan, C.‐C. , De Jonghe, P. , Cheng, T.‐H. , … Lee, Y.‐C. (2017). A recurrent WARS mutation is a novel cause of autosomal dominant distal hereditary motor neuropathy. Brain, 140, 1252–1266. 10.1093/brain/awx058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vester, A. , Velez‐Ruiz, G. , McLaughlin, H. M. , NISC Comparative Sequencing Program , Lupski, J. R. , Talbot, K. , Vance, J. M. , Züchner, S. , Roda, R. H. , Fischbeck, K. H. , Biesecker, L. G. , Nicholson, G. , Beg, A. A. , & Antonellis, A. (2013). A loss‐of‐function variant in the human histidyl‐tRNA synthetase (HARS) gene is veurotoxic in vivo. Human Mutation, 34(1), 191–199. 10.1002/humu.22210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, B. , Li, X. , Huang, S. , Zhao, H. , Liu, J. , Hu, Z. , Lin, Z. , Liu, L. , Xie, Y. , Jin, Q. , Zhao, H. , Tang, B. , Niu, Q. , & Zhang R. (2019). A novel WARS mutation (p.Asp314Gly) identified in a Chinese distal hereditary motor neuropathy family. Clinical Genetics, 96(2), 176–182. 10.1111/cge.13563 [DOI] [PubMed] [Google Scholar]
- Wang, L. U. , Li, Z. , Sievert, D. , Smith, D. E. C. , Mendes, M. I. , Chen, D. Y. , Stanley, V. , Ghosh, S. , Wang, Y. , Kara, M. , Aslanger, A. D. , Rosti, R. O. , Houlden, H. , Salomons, G. S. , & Gleeson, J. G. (2020). Loss of NARS1 impairs progenitor proliferation in cortical brain organoids and leads to microcephaly. Nature Communications, 11, 4038. 10.1038/s41467-020-17454-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterhouse, A. , Bertoni, M. , Bienert, S. , Studer, G. , Tauriello, G. , Gumienny, R. , Heer, F. T. , de Beer, T. A. P. , Rempfer, C. , Bordoli, L. , Lepore, R. , & Schwede, T. (2018). SWISS‐MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Research, 46, W296–W303. 10.1093/nar/gky427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei, N. A. , Shi, Y. I. , Truong, L. N. , Fisch, K. M. , Xu, T. , Gardiner, E. , Fu, G. , Hsu, Y.‐S. , Kishi, S. , Su, A. I. , Wu, X. , & Yang, X.‐L. (2014). Oxidative stress diverts tRNA synthetase to nucleus for protection against DNA damage. Molecular Cell, 56, 323–332. 10.1016/j.molcel.2014.09.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weterman, M. A. J. , Kuo, M. , Kenter, S. B. , Gordillo, S. , Karjosukarso, D. W. , Takase, R. , Bronk, M. , Oprescu, S. , van Ruissen, F. , Witteveen, R. J. W. , Bienfait, H. M. E. , Breuning, M. , Verhamme, C. , Hou, Y.‐M. , de Visser, M. , Antonellis, A. , & Baas, F. (2018). Hypermorphic and hypomorphic AARS alleles in patients with CMT2N expand clinical and molecular heterogeneities. Human Molecular Genetics, 27, 4036–4050. 10.1093/hmg/ddy290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie, W. , Nangle, L. A. , Zhang, W. , Schimmel, P. , & Yang, X. L. (2007). Long‐range structural effects of a Charcot‐Marie‐Tooth disease‐causing mutation in human glycyl‐tRNA synthetase. Proceedings of the National Academy of Sciences, 104, 9976–9981. 10.1073/pnas.0703908104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yalcouyé, A. , Diallo, S. H. , Coulibaly, T. , Cissé, L. , Diallo, S. , Samassékou, O. , Diarra, S. , Coulibaly, D. , Keita, M. , Guinto, C. O. , Fischbeck, K. , Landouré, G. , & H3Africa Consortium . (2019). A novel mutation in the GARS gene in a Malian family with Charcot‐Marie‐Tooth disease. Molecular Genetics & Genomic Medicine, 7(7), 10.1002/mgg3.782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu, X. , Chen, B. , Tang, H. , Li, W. , Fu, Y. , Zhang, Z. , & Yan, Y. (2018). A novel mutation of GARS in a Chinese family with distal hereditary motor neuropathy type V. Frontiers in Neurology, 9, 571. 10.3389/fneur.2018.00571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao, Z. , Hashiguchi, A. , Hu, J. , Sakiyama, Y. , Okamoto, Y. , Tokunaga, S. , Zhu, L. , Shen, H. , & Takashima, H. (2012). Alanyl‐tRNA synthetase mutation in a family with dominant distal hereditary motor neuropathy. Neurology, 78, 1644–1649. 10.1212/WNL.0b013e3182574f8f [DOI] [PMC free article] [PubMed] [Google Scholar]