

Abstract
Aims
The amino acid sequence of prion protein (PrP) is a key determinant in the transmissibility of prion diseases. While PrP sequence is highly conserved among mammalian species, minor changes in the PrP amino acid sequence may confer alterations in the transmissibility of prion diseases. Classical bovine spongiform encephalopathy (C‐BSE) is the only zoonotic prion strain reported to date causing variant Creutzfeldt‐Jacob disease (vCJD) in humans, although experimental transmission points to atypical L‐BSE and some classical scrapie isolates as also zoonotic. The precise molecular elements in the human PrP sequence that limit the transmissibility of prion strains such as sheep/goat scrapie or cervid chronic wasting disease (CWD) are not well known.
Methods
The transmissibility of a panel of diverse prions from different species was compared in transgenic mice expressing either wild‐type human PrPC (MDE‐HuTg340) or a mutated human PrPC harbouring Val166‐Gln168 amino acid changes (VDQ‐HuTg372) in the β2‐α2 loop instead of Met166‐Glu168 wild‐type variants.
Results
VDQ‐HuTg372 mice were more susceptible to prions than MDE‐HuTg340 mice in a strain‐dependent manner.
Conclusions
Met166‐Glu168 amino acid residues present in wild‐type human PrPC are molecular determinants that limit the propagation of most prion strains assayed in the human PrP context.
Keywords: evolution, prion, PrP, resistance, strain
Met166‐Glu168 amino acid residues present in wild type human PrPC are molecular determinants that limit the propagation of most prion strains assayed in the human PrP context.
![]()
INTRODUCTION
Transmissible spongiform encephalopathies (TSE) or prion diseases are caused by the post‐transcriptional conversion of the cellular form of the prion protein (PrPC) into its accumulative abnormal isoform (PrPSc). 1 PrPSc can be discriminated from PrPC by its detergent insolubility, tendency to aggregate and partial resistance to proteinase K digestion (PK) which produce a truncated fragment (PrPres) mostly used as surrogate marker for prion detection. 1 Prion diversity exists in the form of distinct prion strains that show different features after inoculation of laboratory animals such as survival time, neuroanatomic target areas, patterns of PrPSc deposition in the brain and PrPres biochemical properties. 2 , 3 This variability is thought to be enciphered within different PrPSc conformations. 4 , 5
TSEs affect a large spectrum of mammalian species, including humans [affected by Creutzfeldt‐Jacob disease (CJD) and kuru] and several human food chain species, such as sheep and goats (affected by scrapie), cattle [affected by bovine spongiform encephalopathy (BSE)], cervids [affected by chronic wasting disease (CWD)], and dromedary camels. 6 , 7 , 8 Thus, transmission of animal prions to humans constitutes a great concern for human health. Fortunately, prion transmission between species may be limited as a consequence of the transmission barrier phenomenon. 9 Eventual interspecies transmission of prion strains tends to be an inefficient process that shows partial attack rates and variable, often protracted, survival times. By contrast, intraspecies transmission of prions lacks any transmission barrier and is usually an efficient process showing 100% attack rates, stable survival times and maintenance of the biochemical properties of PrPres. The transmission barrier is mainly driven at the molecular level by the identity between the primary sequences of host PrPC and donor PrPSc but the inoculated prion strain may also influence the outcome. Thus, successful interspecies transmission seems to be determined by the conformational compatibility between host PrPC and the misfolded strain‐specific PrPSc conformers that are present within the infectious prion particles. 5 Given these considerations, “strain barrier” better describes the transmission barrier of a certain prion strain among two different species. Prion strain barriers can be evaluated using mice genetically engineered to express the PrP species to be assayed in the absence of endogenous mouse PrP.
Only one prion strain has ever been reported to overcome the human transmission barrier. Classical BSE (C‐BSE), that started an epidemic in UK cattle during the 1980s and soon extended to other European countries, is the causative agent of variant Creutzfeldt‐Jacob disease (vCJD) in humans as consequence of dietary exposure to C‐BSE contaminated tissues. 10 , 11 , 12 vCJD is responsible for at least 178 deaths in the UK 13 and its estimated prevalence within British population is high, as suggested by the prevalence of vCJD PrPSc in human appendixes (493 cases per one million population). 14 vCJD has only been confirmed in individuals homozygous for Met at residue 129 of human PrP 15 with one exception of heterozygosity (Met/Val) at this codon 16 proving the special importance of this polymorphism for prion transmission in the human PrP context. Another two BSE phenotypes, with pathology and epidemiology different from C‐BSE, were observed after the EU active surveillance implementation in 2001. PrPres biochemical properties from these cases differed from the ones of C‐BSE on Western blot (WB) in terms of the protease‐resistant fragment size and the ratio of glycoforms. These atypical forms were termed L‐BSE or H‐BSE (L‐Low or H‐High) according to the apparent molecular mass of the unglycosylated WB band of the PrPres. 17 , 18 L‐BSE has a higher zoonotic potential than C‐BSE in transgenic mice overexpressing Met129 human PrP variant 19 , 20 , 21 , 22 but no transmission was observed in transgenic mice overexpressing the Val129 human PrP variant. 22 H‐BSE was unsuccessfully transmitted in both models. 19 , 22 As observed in the case of C‐BSE and L‐BSE and the 129 Met/Val dimorphism in human PrP, only one amino acid change may drastically alter the susceptibility to prion strains. Moreover, heterozygosity at 129 codon of human PRNP gene has been associated with prolonged survival in individuals exposed orally to kuru, thus a strong balancing selection at the PRNP 129 locus during the evolution of modern humans has been proposed. 23
The zoonotic potential of other animal prion strains has not yet been definitely proved. However, a study using transgenic mice overexpressing human PrP was done for several scrapie isolates and the transmission efficiency of some of them was comparable to that of C‐BSE. 24 Nevertheless, despite the human alimentary exposure to scrapie, epidemiological studies failed to identify any clear link between scrapie and TSE occurrence in humans. 25 , 26 CWD, which elk, deer, reindeer and moose, have spread extensively throughout North America, 27 has been also detected in South Korean ranched elk, 28 and Norwegian and Finnish wild animals. 29 , 30 , 31 CWD prions are highly infectious and readily transmitted among cervids even through environmental exposure, 32 , 33 causing extraordinarily high prevalence that can exceed 90% in captive deer. 34 Humans, wild animals and livestock species like cattle, swine and sheep are likely exposed to CWD through consumption and/or contact with CWD‐contaminated products/materials. An active surveillance of more than 17,000 US residents revealed that around 20% of them hunt cervids and more than two‐thirds have consumed venison, 35 but a clear risk of developing a TSE through human dietary exposure to CWD has not been found. 36 Experimental transmissions of CWD to transgenic mice expressing human PrP suggest that a strong strain barrier for CWD exists in humans. 37 , 38 , 39
The role of other human PrP residues, apart from the codon 129 polymorphism, in the human transmission barrier for animal prions remains unclear. When the human PrP sequence is compared to the ones of other proven prion susceptible mammals like sheep, cattle, elk or macaque (Figure 1), amino acid changes are observed at the 166 and 168 positions. While humans harbour Met166 and Glu168, most mammal species show Val166 and Gln168 (according to the human amino acid sequence). 166 and 168 amino acid positions are located in the β2‐α2 loop of PrPC. 40 Accumulating evidence suggests that changes in these region are deeply related to alterations in prion strain susceptibility and pathogenicity. 38 , 41 , 42 , 43 , 44 , 45 , 46 For instance, in the sheep PRNP gene, a polymorphism described in the 171 position (equivalent to 168 position in human PrP) was associated either to scrapie susceptibility (Gln168) or resistance (Arg168). 47 Gln to Arg substitution triggers a secondary structure change in the β2‐α2 loop that modifies the connectivity of this region with other PrPC structures. 48
FIGURE 1.

Amino acid comparison of human, gorilla, macaque, american elk, cattle and sheep PrPC amino acid sequences. Only amino acids 88 to 233 (according to human PrP) are included in the comparison for clarity. Deletions are indicated by dashes. Points indicate identical residues. Amino acid numbering is indicated on the right. Species are named on the left. Amino acid changes in 166 and 168 positions (Met/Val and Glu/Gln respectively) are boxed
The conservation of Val166 and Gln168 PrP amino acids in species able to propagate different prion strains supports investigating the role of Val166‐Gln168 to Met166‐Glu168 substitution in the propagation of different prion strains within the human PrPC context. For that purpose, we compare the susceptibility/resistance to a panel of different prion strains of two transgenic mouse models expressing similar levels of wild‐type (MDE‐HuTg340) or Val166‐Gln168 (VDQ‐HuTg372) human PrPC.
MATERIALS AND METHODS
Generation of transgenic mice expressing Val166‐Glu168 amino acid changes of human PrPC
The pMo‐huPrP129M.Xho plasmid previously used for the generation of HuPrP‐Tg340 mouse line 49 has been used as template for directed mutagenesis reactions. pMo‐huPrP129M.Xho plasmid contains the human PrP (Met129 variant) open reading frame inserted in the MoPrP.Xho expression vector. 50 The vector contains the murine wild‐type prnp gene including its promoter, exon 1, exon 2 and 3′‐untranslated sequences excepting intron 2 and the murine PrP ORF. pMo‐huPrP129M.Xho was mutated to generate pMo‐hu166VDQ168‐PrP129M.Xho by using QuikChange II XL kit (Stratagene, CA) with specific oligonucleotides (5′‐GTACTACAGGCCCGTGGATCAGTACAGCAACCAGAAC‐3′ and 5′‐GTTCTGGTTGCTGTACTGATCCACGGGCCTGTAGTAC‐3′) following the procedures described by the manufacturer.
Transgenic mouse lines were generated using the same procedure and mouse colonies previously described for the generation of HuPrP‐Tg340 mouse line. 49 Briefly, transgenes were excised from the expression vector (pMo‐hu166VDQ168‐PrP129M.Xho) by use of the restriction endonuclease NotI, leading to DNA fragments of ~12 kb. NotI digested products were fractionated on a 1% preparative low melting point agarose gel (TopVisionTM, Fermentas Inc.). The gel slice corresponding to 12 kb was excised and digested using β‐agarase I (New England Biolabs) as described by the manufacturer. Purified DNA was resuspended in Tris‐EDTA (10 mM Tris, pH 7.4, 0.1 mM Ethylenediaminetetraacetic acid) at a final concentration of 2–6 ng/mL. Finally, DNA was microinjected into pronuclear‐stage embryos collected from superovulated B6CBAF1 females mated with 129/Ola males carrying a null mutation in their endogenous PrP gene. The homozygous PrP null mouse line used was generated by Manson et al. 51
DNA was extracted from founders’ tail by use of an Extract‐N‐Amp tissue PCR kit (Sigma‐Aldrich) following the manufacturer's instructions. The presence of the human transgene was identified by PCR amplification using specific primers for mouse PrP exon and the human PrP ORF (5′‐CATTCTGCCTTCCTAGTGGTACC‐3′ and 5′‐GTGTTCCATCCTCCAGGCTTC‐3′). muPrP+/− huPrP+/− founders were backcrossed with homozygous PrP null animals (muPrP−/−) 51 to obtain mice homozygosis for the null mutation (muPrP−/− huPrP+/−). The absence of the murine PrP ORF in the transgenic mice generated was confirmed by PCR amplification using specific primers (5′‐TAGATGTCAAGGACCTTCAGCC‐3′ and 5′‐ GTTCCACTGATTATGGGTACC‐3′). Later, muPrP−/− huPrP+/− mice (VDQ‐HuTg372) were used for transmission experiments.
WB analysis of brain PrPC expression in transgenic mice
Whole brains from mouse founders’ lines were homogenised in extraction buffer (0.5% NP‐40, 1% sodium deoxycholate in phosphate‐buffered saline 10 mM pH 7.4, with Complete inhibitor cocktail [Roche]). Samples were precleared by centrifugation at 2,000g for 5 min. Supernatants were mixed with an equal volume of 2× SDS reducing sample loading buffer and boiled for 5 min before loading onto a 12% Bis‐Tris Gel (Criterion XT, BioRad). Protein concentration was measured with PierceTM BCA Protein Assay kit (Thermo Scientific). For immunoblotting experiments, the monoclonal antibody (mAb) Pri308 52 which recognises the111HMAGAAAA118 epitope of the human PrP sequence was used at a concentration of 0.1 µg/mL. Monoclonal antibody recognising β‐actin (Sigma‐Aldrich) was used as loading control at a 1/20.000 dilution. Immunocomplexes were detected incubating the membranes for 1 h with horseradish peroxidase‐conjugated anti‐mouse IgG (GE Healthcare Amersham Biosciences). Immunoblots were developed with enhanced chemiluminescence ECL Select (GE Healthcare Amersham Biosciences). Images were captured using ChemiDoc XRS+System and Image Lab 6.0.1 Software was used for images processing and densitometry analysis. Values were normalised against β‐actin levels.
Prion isolates and inocula preparation
In this work, a panel of field isolates from distinct origin and representing different TSE agents were used (Table 1). As negative control, TSE‐free cattle brain was included on the panel. All inocula were prepared from brain tissues as 10% (w/v) homogenates in 5% glucose.
TABLE 1.
Description of the isolates used in this study
| Isolate | Description and references | Supplier |
|---|---|---|
| sCJD T1 | Type 1 sCJD M129M‐infected case (0.08.02523_001) 53 | BB a |
| sCJD T2/MDE‐HuTg340 | Terminally ill MDE‐HuTg340 mice infected with Type 2 sCJD V129 V‐infected case (BC1452) after two iterative passages | CISA b |
| vCJD | vCJD M129M‐infected case (BC1458) 54 | BHUFA c |
| C‐BSE | Classical BSE natural case from United Kingdom 55 | AHVLA d |
| H‐BSE | Atypical H‐BSE natural case from Poland. Po 45 56 | NVRI e |
| L‐BSE | Atypical L‐BSE natural case from Poland. Po 15 22 | NVRI |
| Scrapie 1 | Naturally scrapie‐infected goat from France (wt; S/P240). Fr−2143 57 | INRA f |
| Scrapie 2 | Naturally scrapie‐infected sheep from France (ARQ/ARQ). Fr‐PS21 24 | INRA |
| Scrapie 2/Tg110 | Terminally ill Bo‐Tg110 mice infected with Scrapie 2 | CISA |
| Scrapie 3 | Naturally scrapie‐infected (ARQ/ARQ) sheep from Italy. It−198–9 58 | ISS g |
| Scrapie 3/Tg110 | Terminally ill Bo‐Tg110 mice infected with Scrapie 3 | CISA |
| CWD | Naturally CWD infected Rocky Mountain elk. #3 | CFIA h |
Basque Biobank. Bilbao. Spain.
Centro de Investigación en Sanidad Animal, Instituto Nacional de Investigación y Tecnología Agraria y Alimentaria. Valdeolmos, Madrid, Spain.
CJD Reference Laboratory. Alcorcón. Spain.
Animal Health and Veterinary Laboratories Agency. New Haw. Addlestone. Surrey, UK.
National Veterinary Research Institute. Pulawy. Poland.
French National Institute for Agricultural Research, Nouzilly, France
Instituto Superiore di Sanitat. Rome. Italy.
National and OIE Reference Laboratory for Scrapie and CWD. Canadian Food Inspection Agency. Ottawa. Ontario, Canada.
Transmission studies
About 6–7 week old mice in groups of 5–9 MDE‐HuTg340 mice (overexpressing human PrPC fourfold the PrP expression level in human brain) 49 and VDQ‐HuTg372 mice (expressing similar levels of PrPC in the brain, the main target for prion propagation) newly generated were inoculated with the above‐mentioned list of prion inocula by intra‐cerebral route with 20 µL of 10% brain homogenate, as previously described. 49 After inoculation, mice were observed daily and their neurological status assessed twice a week. When the progression of the disease was evident or at the end of the lifespan (≈650 days), animals were euthanised for ethical reasons. Once euthanised, necropsy was performed and the brain was harvested for analysis. Half of the brain was fixed by immersion in neutral‐buffered 10% formalin (4% 2‐formaldehyde) and the rest of the tissue was frozen at −20ºC and used for biochemical analysis.
Survival times were calculated as mean ±SD of the days post‐inoculation (dpi) of all the mice scored positive for PrPres. Attack rate was determined as the ratio of PrPres‐positive mice among all the inoculated mice.
WB analysis of brain PrPres in transgenic mice
175 ± 20 mg of frozen brain tissue was homogenised in 5% glucose in distilled water in grinding tubes (Bio‐Rad) adjusted to 10% (w/v) using a TeSeETM Precess 48TM homogenizer (Bio‐Rad) following the manufacturer's instructions. The presence of PrPres in transgenic mice brains was determined by WB using the reagents of the ELISA commercial test (TeSeE, Bio‐Rad). About 10–100 µL of 10% (w/v) brain homogenate was analysed as previously described 56 using 12% Bis‐Tris Gel (Criterion XT, BioRad). For immunoblotting, Sha31 mAb 52 was used at a concentration of 1 µg/mL. Sha31 recognises145‐YEDRYYRE‐152 epitope of the human PrPC sequence. Immunocomplexes were detected as described above for brain PrPC analysis. About 5–50 µL of 10% (w/v) brain homogenate equivalent was loaded per lane in the Western blot.
Paraffin‐embedded tissue blot and histopathological analysis
Paraffin‐embedded tissue blots (PET blot) were conducted as previously described 59 using the Sha31 mAb. 52 Lesion profiles of the brains were done using 4‐µm thick tissue slices stained with haematoxylin and eosin according to the method described by Fraser and Dickinson. 60
Modelling of Val166‐Gln168 human PrPC
In silico human PrP expressing Val166‐Gln168 substitutions based on the prion protein model structures available were generated using Comparative Modelling with Rosetta (RosettaCM) at Robetta Web server on 23 April 2019. (http://robetta.bakerlab.org/). 61 , 62 Briefly, this protocol creates a homology model with given Protein Data Bank files corresponding to one or more template structures. It is used for comparative modelling of target proteins. Figures of the molecular models were obtained with PyMol software. 63
RESULTS
Generation of transgenic mice expressing Val166‐Gln168 human PrPC
The same plasmid vector used to generate the MDE‐HuTg340 mouse line 49 was used to generate the new transgenic mouse lines described in this work. Several mouse lines (founders) hemizygous for the transgene (Val166‐Gln168HuPrP) were obtained. Founder animals also carrying the endogenous murine prnp gene (muPrP+/−, huPrP+/−) were crossed with prnp null mice (muPrP−/−) to obtain transgenic hemizygous lines in a murine prnp null background (muPrP−/−, huPrP+/−). The absence of the murine prnp was determined by PCR using specific primers. Then, hemizygous mice were crossed to produce homozygous animals (muPrP−/−, huPrP+/+). At this point, PrPC expression level was determined in brain homogenates of the founder mice by serial dilution in WB and compared to MDE‐HuTg340 brain PrPC expression levels. From the different founders, the VDQ‐HuTg372 mouse line was selected as it showed comparable brain PrPC levels and similar electrophoretic profile than MDE‐HuTg340 mouse line (Figure 2). VDQ‐HuTg372 mice reached the end of their lifespan with neither evidence of spontaneous prion disease nor behavioural defects such as alterations in reproduction rates or social deficits.
FIGURE 2.
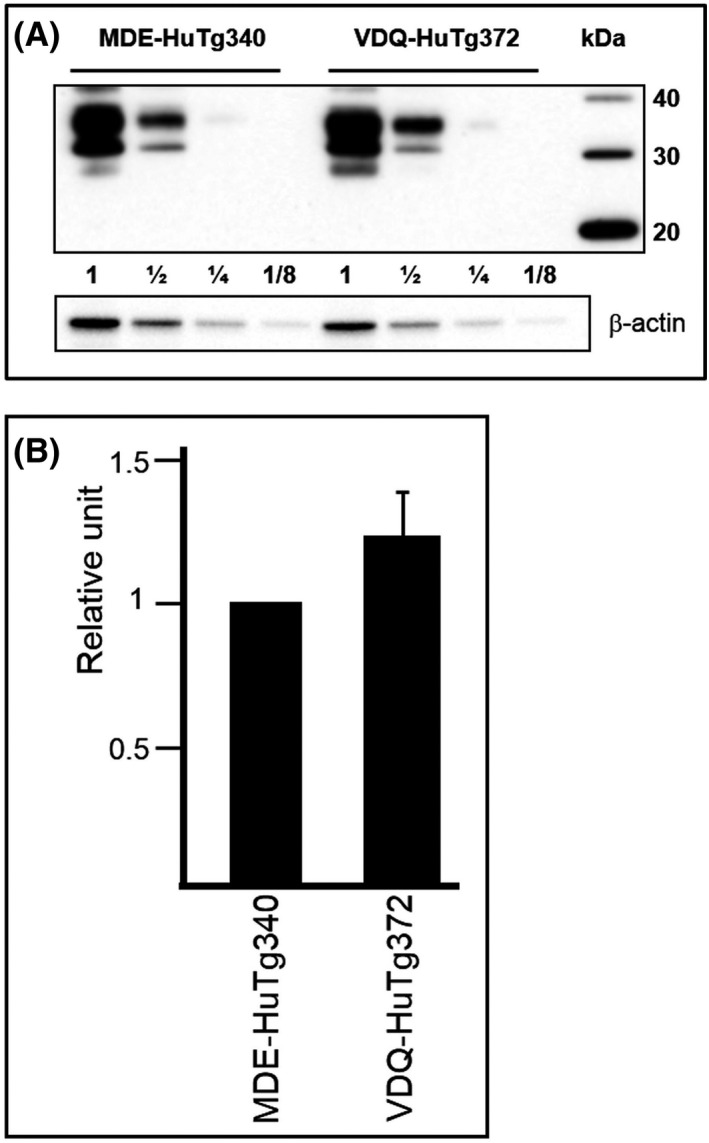
Brain PrPC expression in transgenic mouse lines. (A) Immunoblots of brain PrPC expression in MDE‐HuTg340 in comparison to VDQ‐HuTg372 transgenic mouse line detected with Pri308 mAb. Direct sample (60 micrograms of protein) and 1/2 dilutions were loaded on 12% Bis‐Tris gels. Molecular mass in kDa (MagicMarkTM XP Western Protein Standard) are shown at the right side of the blot. β‐actin was used as loading control. (B) Brain PrPC expression was quantified and normalised against β‐actin levels. Immunoblots illustrates a representative set of three independent experiments and the diagrams illustrates the mean densitometric values from these experiments. Data from MDE‐HuTg340 brains was always standardised as 1 relative unit. Error bar represents the standard deviation of the mean value
Prion resistance/susceptibility of VDQ‐HuTg372 mice compared to MDE‐HuTg340 mice
Once selected, VDQ‐HuTg372 and its control counterpart MDE‐HuTg340 mice were intracranially inoculated with a collection of isolates representative of different prion strains from human, cattle, goat, sheep and elk (Table 2). Since both transgenic lines share the same brain PrPC expression levels, susceptibility/resistance differences among both lines will only be due to the two amino acid substitutions present on VDQ‐HuTg372 mice.
TABLE 2.
Effect of the presence of the 166VDQ168 amino acid residues in the human PrP sequence in the replication of several prion strains from human, bovine, goat, sheep and elk as assayed in MDE‐HuTg340 and VDQ‐HuTg372 mouse models
| Prion origin | Inocula | Passage | Mean survival time in days ± SD, (n/n0) | |
|---|---|---|---|---|
| MDE‐HuTg340 | VDQ‐HuTg372 | |||
| Human | sCJD T1 | 1st | 185 ± 7 (7/7) | 112 ± 10 (6/6) |
| 2 a | 190 ± 8 (5/5) | 113 ± 2 (6/6) | ||
| sCJD T1/VDQ‐HuTg372 | 1st | 196 ± 16 (6/6) | 113 ± 2 (6/6) | |
| sCJD T2/MDE‐HuTg340 | 1st | 469 ± 45 (5/5) | 505 ± 34 (7/7) | |
| vCJD | 1st | 545 ± 146 (5/5) a | 357 ± 28 (7/7) | |
| 2 a | 564 ± 39 (4/4) a | 236 ± 10 (6/6) | ||
| Cattle | H‐BSE | 1st | >650 (0/6) | >650 (0/6) |
| L‐BSE | 1st | 607 ± 13 (7/7) c | 210 ± 13 (6/6) | |
| 2 c | 487 ± 16 (4/4) c | 174 ± 5 (5/5) | ||
| C‐BSE | 1st | >650 (1/8) b | 592 ± 85 (5/5) | |
| 2 b | 633 ± 32 (4/4) b | 328 ± 32 (6/6) | ||
| Goat | Scrapie 1 | 1st | >650 (0/6) | >650 (0/6) |
| Sheep | Scrapie 2 | 1st | >650 (0/6) d | 403 ± 20 (7/7) |
| Scrapie 2/VDQ‐HuTg372 | 1st | >650 (0/6) | 378 ± 53 (6/6) | |
| Scrapie 3 | 1st | >650 (0/6) | >650 (0/6) | |
| Elk | CWD | 1st | >650 (0/6) | 427 (1/7) |
| CWD/HuVDQ‐Tg372 | 1st | 509–594 (2/5) | 236 ± 10 (6/6) | |
Human prion transmission in VDQ‐HuTg372
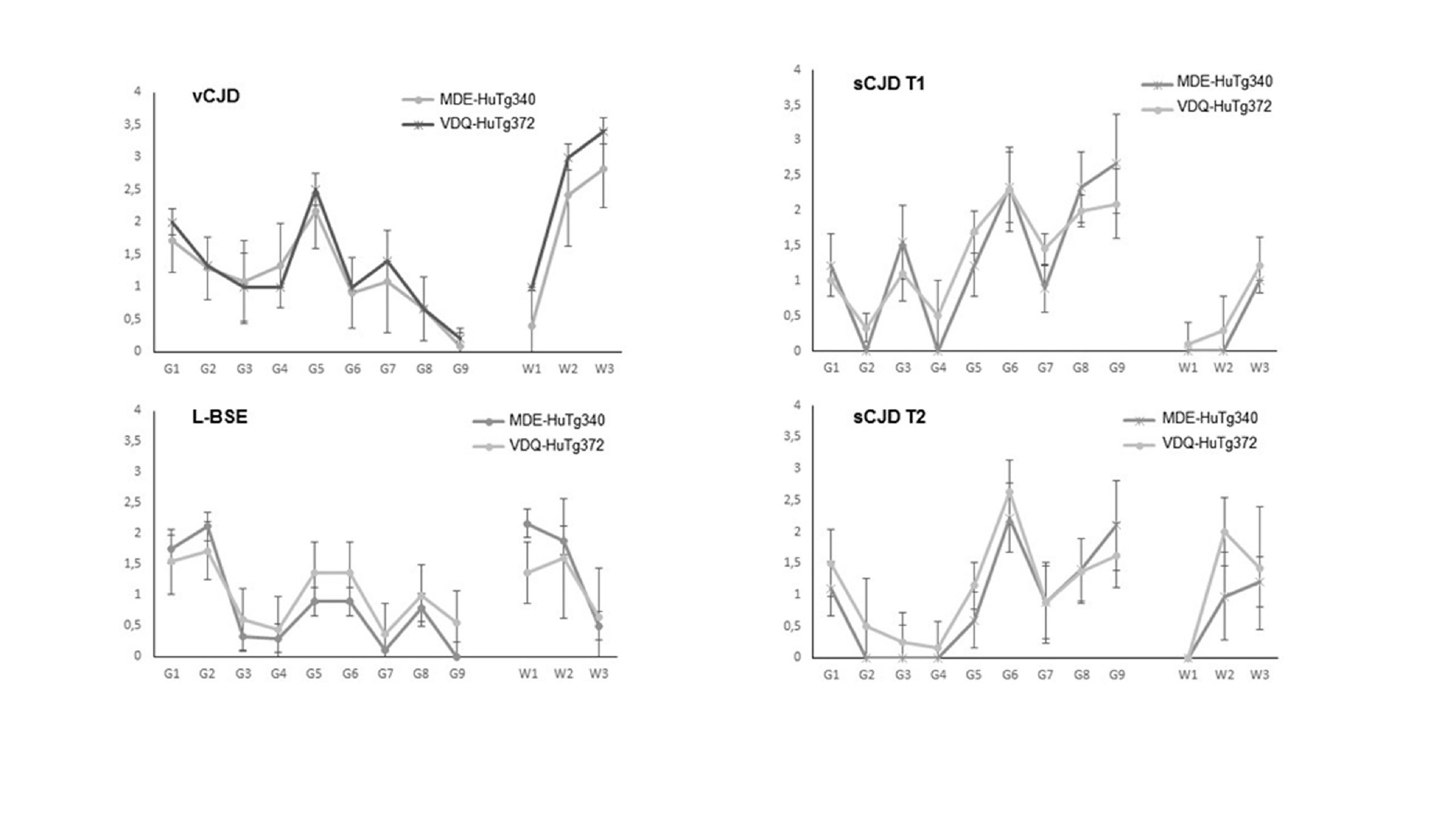
VDQ‐HuTg372 mice and its control counterparts MDE‐HuTg340 mice were inoculated with human prions sporadic CJD (sCJD) and vCJD. VDQ‐HuTg372 mice were readily infected with sCJD T1 prions showing even shorter survival times than MDE‐HuTg340 mice. While VDQ‐Tg372 mice had mean survival times shorter than 120 days, the MDE‐HuTg340 mice had mean survival times around 60 days longer (Table 2). The second passage in VDQ‐HuTg372 mice remained around 120 dpi. However, these extremely fast sCJD T1 prions passaged in VDQ‐HuTg372 did not reduce the sCJD T1 typical mean survival time when inoculated into MDE‐HuTg340 mice (Table 2). Attack rates reached 100% of the inoculated animals in all cases. The sCJD T2 isolate used in this study was previously adapted to MDE‐HuTg340 mice to avoid the transmission barrier due to the codon 129 polymorphism in human PrP (Table 1). VDQ‐HuTg372 showed similar survival times compared to MDE‐HuTg340 mice inoculated with sCJD T2/MDE‐HuTg340, 505 ± 34 and 469 ± 45 dpi respectively (Table 2). Attack rates reached 100% of the inoculated animals in both cases. This difference was found not significant as assessed by Mann‐Whitney's t‐test (p‐value <0.05).
In the case of vCJD prions, VDQ‐HuTg372 mice died at 357 ± 28 days after inoculation showing 100% attack rates, while MDE‐HuTg340 mice showed a much longer survival time of 545 ± 146 dpi and reached 100% attack rates. After the second passage, survival time was maintained for MDE‐HuTg340 mice while it was reduced to 236 ± 10 dpi for VDQ‐HuTg372. Identical brain PrPres electrophoretic signatures were observed in both mouse lines after inoculation with the different human prion strains used in this work (Figure 3A). The sCJD T1 PrPres phenotype is observed for sCJD T2 prion strain in the context of human Met129 genotype as was previously reported. 24 , 64 , 65
FIGURE 3.
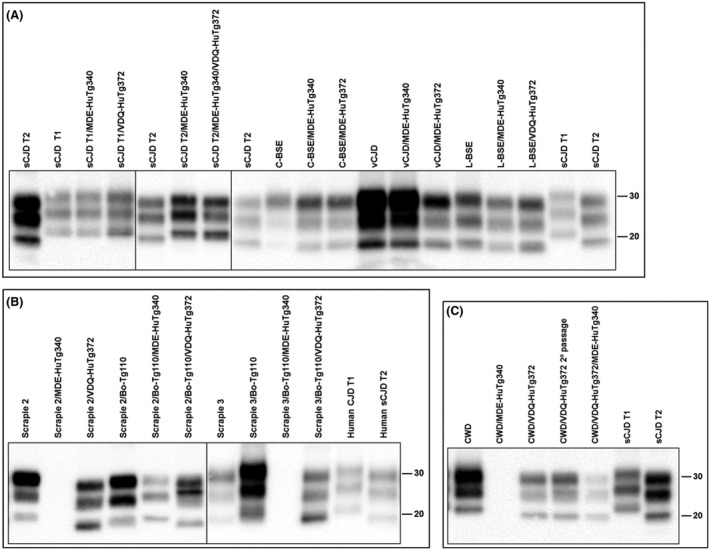
Electrophoretic profiles of PrPres as detected by mAb Sha31 in brain extracts from mice infected with the prion agents indicated in the top. (A) Human and cattle inocula compared with the positive transmissions in MDE‐HuTg340 and VDQ‐HuTg372 transgenic mice. (B) Sheep and sheep‐derived inocula compared with the transmissions in MDE‐HuTg340 and VDQ‐HuTg372 transgenic mice. (C) Elk inoculum compared with the transmissions in MDE‐HuTg340 and VDQ‐HuTg372 transgenic mice. T1 and T2 sCJD from human brain have been included as controls for better comparison. Similar quantities of PrPres were loaded in each lane for better comparison. Molecular mass (in kD) is shown at the right side of the blots
Animal prion transmission in VDQ‐HuTg372 mice
Cattle prion strain C‐BSE, as well as atypical BSE strains L‐BSE and H‐BSE, were inoculated in VDQ‐HuTg372 and MDE‐Tg340 mice. Remarkably for C‐BSE inoculation, only one out of eight MDE‐HuTg340 mice were infected beyond 650 dpi while all VDQ‐HuTg372 mice were positive for brain PrPres after survival times of 592 ± 85 dpi (Table 2). As expected, the second passage showed a 100% attack rate in both models, but in VDQ‐HuTg372 mice the survival time was reduced to 328 ± 32 dpi while MDE‐HuTg340 mice remained at 633 ± 32 dpi (Table 2). Neither VDQ‐HuTg372 nor MDE‐HuTg340 mice were infected with H‐BSE prions. By contrast, L‐BSE was efficiently transmitted in both mouse lines showing 100% attack rates in both cases (Table 2). Again, VDQ‐HuTg372 showed shorter survival times than MDE‐HuTg340 mice, 210 ± 13 (6/6) and 607 ± 13 (7/7) dpi respectively. Second passage in both models resulted in a survival time reduction of around 20%. Identical brain PrPres electrophoretic signatures were observed in both mouse lines after inoculation with the different cattle prion strains used in this work (Figure 3A). Interestingly, the western blot conditions used in this work detected a slightly higher relative mobility in brain PrPres from both mouse lines inoculated with L‐BSE than in the original cattle L‐BSE inoculum. This difference in the L‐BSE migration of PrPres bands in the humanised mice when compared to L‐BSE from cattle could be explained by changes in the electrophoretic mobility due to differences in the amino acid sequence, as was previously described with classical‐BSE after passage in sheep, which also shows a slightly higher mobility than classical‐BSE from cattle. 66
In accordance with previous studies, 24 none of the MDE‐HuTg340 mice inoculated with sheep or goat classical scrapie was scored positive for the disease in the first passage. The same outcome was obtained for VDQ‐HuTg372 mice with isolates Scrapie 1 and 3. Whereas Scrapie 2 infected VDQ‐HuTg372 mice with 100% attack rate and a survival time of 403 ± 20 dpi (Table 2). These prions were then inoculated in MDE‐HuTg340 mice and none of the animals was scored positive for the disease (Table 2). By contrast, second passage in VDQ‐HuTg372 mice resulted in a survival time reduction to 378 ± 53 dpi. Brain‐PrPres from VDQ‐HuTg372 mice inoculated with Scrapie 2 showed an unglycosylated band of 19 kD resembling the human sCJD type 2 PrPres electrophoretic signature (Figure 3B).
Classical scrapie isolates adapted to the bovine PrPC sequence were also inoculated in both models to assess how passage through the bovine species barrier affected classical scrapie zoonotic abilities. Differences were not observed for Scrapie 2, as low attack rates and long survival times were observed in both MDE‐HuTg340 and VDQ‐HuTg372 mice (Table 3). By contrast, bovine‐adapted Scrapie 3 infected just VDQ‐HuTg372 mice showing 100% attack rates and long survival time of 576 ± 37 dpi (Table 3).
TABLE 3.
Comparative transmission of sheep scrapie in MDE‐HuTg340 and VDQ‐HuTg372 mouse models before and after adaptation to cattle‐PrP amino acid sequence
| Prion origin | Inocula | Passage | Mean survival time in days ± SD, (n/n0) | |
|---|---|---|---|---|
| MDE‐HuTg340 | VDQ‐HuTg372 | |||
| Sheep | Scrapie 2 | 1st | >650 (0/6) a | 403 ± 20 (7/7) |
| 2nd | 369,579 (2/6) a | 378 ± 53 (6/6) | ||
| Scrapie 2/Bo‐Tg110 | 1st | 534 (1/5) | 555 (1/5) | |
| Scrapie 3 | 1st | >650 (0/6) | >650 (0/6) | |
| Scrapie 3/Bo‐Tg110 | 1st | >650 (0/6) | 576 ± 37 (6/6) | |
n/n0: diseased, PrPres positive/inoculated animals. Mean survival time is indicated for all mice scored positive for PrPres.
Published in reference. 24
In this case, differences in the brain PrPres signature were observed in MDE‐HuTg340 and VDQ‐HuTg372 mice inoculated with the bovinised scrapie prions (Figure 3B). For Scrapie 2, MDE‐HuTg340 mice showed a PrPres electrophoretic signature resembling type 1 PrPres with a 21 kD unglycosylated band. By contrast, VDQ‐HuTg372 mice showed a PrPres electrophoretic signature characterised by a 19 kD unglycosylated band. Finally, VDQ‐HuTg372 mice inoculated with Scrapie 3 showed an unusual PrPres electrophoretic signature with a 19‐kD predominant unglycosylated band.
Elk CWD prions were unable to infect MDE‐HuTg340 mice (Table 2). By contrast, one out of seven VDQ‐HuTg372 mice was infected showing a 427 dpi survival time. Second passage in VDQ‐HuTg372 mice achieved 100% attack rate and 236 ± 10 dpi survival time. However, these CWD prions adapted to VDQ‐HuTg372 mice were poorly transmitted when inoculated in MDE‐HuTg340 mice as only two out of five animals were scored positive at 509 and 594 dpi respectively.
The brain PrPres electrophoretic signature in VDQ‐HuTg372 or MDE‐HuTg340 mice infected with CWD showed a 19‐kD unglycosylated band resembling type 2 sCJD electrophoretic signature (Figure 4C).
FIGURE 4.
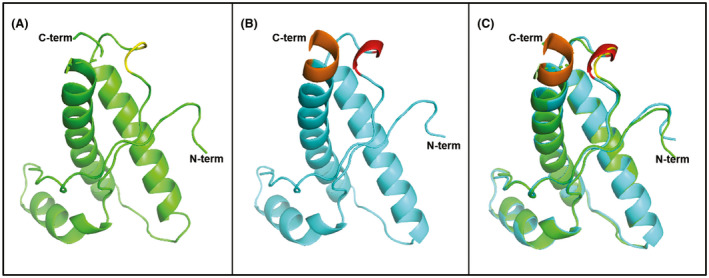
Structural models. (A) Structural Cα backbone of human wt‐PrPC from amino acids 124 to 227 (green). 167–171 positions shown in yellow. (B) Structural Cα backbone of Val166‐Gln168 human PrPC (blue) from amino acids 124 to 227; 167–171 positions shown in red. (C) Structural Cα backbone superposition of 124–227 amino acids of human wt‐PrPC (green) and Val166‐Gln168 human PrPC (blue). Overall folding of the models showing only differences in the carboxy‐terminal region and the β2‐α2 loop. 223–227 amino acid residues in Val166‐Gln168 human PrPC (brown) fold as α‐helix that is not structured in human wt‐PrPC and causing a slight deviation from the straight helix in the α‐helix 3
Comparison of the neuropathological assessment of VDQ‐HuTg372 and MDE‐HuTg340‐infected mice
Vacuolar lesions (Figure S1) and PrPres distribution patterns were compared in VDQ‐HuTg372 and MDE‐HuTg340 mice inoculated with sCJD T1, sCJD T2/MDE‐HuTg340, vCJD and L‐BSE. The same lesion profiles and PrPres distribution patterns were obtained for all strains when compared among the two different mouse models. sCJD prions were characterised by strong PrPres distribution in thalamus and cerebral cortex as well as vacuole accumulation in the superior colliculus, medial thalamus, hippocampus, cerebral cortex and pyramid tract areas in both mouse models as previously described. 22 , 24 vCJD prion transmission showed the histopathological features already reported for C‐BSE‐derived prion transmission, such as granular and strong PrPres distribution, in both mouse lines. 54 Finally, L‐BSE prions were transmitted in MDE‐HuTg340 and VDQ‐Tg372 mice showing unique strain features mainly characterised by a fine non‐granular PrPres distribution which was more intense in the habenular, geniculate and dorsal nuclei of the thalamus and weak vacuole accumulation, more prominent in the cerebellar cortex and cerebellum and mesencephalic tegmentum white matter areas. 22 The only remarkable difference among the two mouse lines was the consistent weak signal intensity in sCJD T1 PrPres pattern in VDQ‐Tg372 mice (data not shown). This could be explained by the extremely short incubation times of around 120 dpi that may not produce big enough PrPSc deposits able to be fully detected by the Pet Blotting technique.
In silico comparative structural analysis
In order to assess if Val166‐Gln168 amino acid changes introduced in VDQ‐Tg372 mice affect the structure of human PrPC, an in silico model was generated and compared with the one of wild‐type human PrPC (Figure 4). Minor changes are observed in the local region of the amino acid changes, which is more structured in Val166‐Gln168 human PrP than in the wild‐type counterpart. Additionally, Val166‐Gln168 human PrP shows a slight deviation from the straight helix axis after residue 220 and enhanced definition of the carboxy‐terminal amino acids of α‐helix 3 when compared to wild‐type human PrP (Figure 4).
DISCUSSION
vCJD cases due to the dietary exposure to the epidemic C‐BSE agent in UK cattle raised concerns about the transmissibility of other animal prions to humans. To date, C‐BSE is the only recognised zoonotic prion. Nevertheless, L‐type BSE seems to propagate with no obvious transmission barrier in transgenic mice expressing Met129 human PrPC 19 , 20 , 21 , 22 but not in mice expressing either Val129 or Met/Val129 PrP variants. 22 In addition, some scrapie isolates were successfully transmitted to humanised mice showing a transmission barrier comparable to that of C‐BSE. 24 Considering this limited susceptibility of human species to prions, we have assessed the transmission features of a collection of prion isolates representative of diverse prion strains in transgenic mice VDQ‐Tg372 expressing a mutant human PrPC containing Val166‐Gln168 amino acid changes. Comparison with MDE‐Tg340 mice that express the same levels of wild‐type human PrPC in brain would elucidate to what extent wild‐type Met166 and Glu168 amino acid residues define the human transmission barrier to prions. Intracranial inoculation was used as the brain is the main target for prion propagation. MDE‐HuTg340 transgenic mice previously described 49 are relevant animal models to assess prion transmission across the human transmission barrier. 22 , 49 , 54 Both transgenic mouse lines originated in such a way that the differences in both models are limited to the amino acid changes at 166 and 168 positions, so the alterations observed in prion propagation are directly related to the referred mutations. Potential differences due to the mixed genetic background of the mouse lines were minimised by inoculating a number of animals (5 to 9) on each experiment. Both amino acid substitutions are included in the β2‐α2 loop, a region previously reported to be of special importance for prion propagation. 38 , 41 , 42 , 43 , 44 , 48 Met166‐Glu168 residues were chosen for mutation because they are together in human PrP while Val166‐Gln168 variants are present in most mammals species like sheep, cattle and elk. 67 The molecular modelling of the human PrP including Val166‐Gln168 amino acid changes do not present substantial changes in the overall PrPC structure although slight differences between both PrP models were found. However, it must be taken into account that the β2‐α2 loop region is generally not well defined in PrP molecular models thus the structural changes produced by mutations on this area may remain underestimated. 40 , 68 , 69 The model shown in this study at least suggests that Val166‐Gln168 substitutions in human PrPC alter α‐helix 3. The alterations in these residues are probably involved in the anchoring of the α‐helix 3 against the β2‐α2 loop and the residues following the first β‐sheet strands previously proposed. 48 , 69 The Y169, F175 and Y218 aromatic cluster present in the wt human PrP could be altered in the Val166‐Gln168 mutant of human PrPC due to modifications in the solvent exposure of Y169. This alteration could allow new interactions with α‐helix 3 as previously proposed for other human PrP mutants associated with disease. 45 , 46
Accelerated propagation of the disease in Val166‐Gln168 human PrPC context
In general terms, the presence of Val166‐Gln168 amino acid changes never delayed the survival time or reduced the attack rates for the prion strains used in this work. Indeed, the prion disease progression was faster in Val166‐Gln168 human PrPC context than in the wild‐type human PrPC one. The only exception was Val166‐Gln168 mice inoculated with human type 2 sCJD. In this case, the disease progression remained unchanged in mice expressing either Val166‐Gln168 human PrPC or wild‐type human PrPC. This effect observed with type 2 sCJD strain suggests that Met166‐Glu168 amino acids are not relevant for the propagation of the type 2 sCJD PrPSc conformers since its mutation does not affect prion propagation. Human strains sCJD type 1 and vCJD and animal strain L‐BSE showed shorter survival times in mutated mice suggesting that Met166‐Glu168 residues somehow increased the resistance to these strains. A mouse model expressing mutant human PrPC Val166‐Gln168‐Asn170‐Thr174 was infected with type 1 sCJD prions showing a 60% increase in the survival time. 38 Thus, evidencing that further modification of the human PrPC β2‐α2 loop region in Ser170‐Asn174 positions creates a significant transmission barrier at least for this prion strain.
Enhanced transmissibility of the infection in Val166‐Gln168 human PrPC context
The reduction of prion transmission barriers in mice expressing the Val166‐Gln168 human PrPC is also observed in terms of attack rates. Partial attack rates observed for C‐BSE prions in mice expressing wild‐type human PrPC were improved to full attack rates in mice expressing Val166‐Gln168 human PrPC. Furthermore, prion strains that were unable to propagate in the wild‐type human PrPC context, like one classical scrapie strain or elk CWD, can propagate in the Val166‐Gln168 human PrPC context. This shows that Met166‐Glu168 residues in wild‐type human PrPC were somehow preventing those strains crossing the human transmission barrier. However, this situation seems again to be strain specific since H‐BSE and other classical scrapie strains were unable to amplify in both animal models despite the amino acid substitutions. As it applies to type 2 sCJD human prions, Met166‐Glu168 residues are not relevant for H‐BSE and certain classical scrapie strains prion propagation. These findings support the idea that different prion PrPSc conformers initially interact and/or convert host PrPC through different regions.
To date, CWD cannot be transmitted to transgenic mice expressing wild‐type human PrPC mice [37, 38, 39, 70, 71 and this work] The inability of CWD to propagate in the wild‐type human PrPC context is abolished in the mouse model expressing Val166‐Gln168 human PrPC even though with a high transmission barrier. This is in agreement with the transmission of CWD in mice expressing mutant human PrPC with Val166‐Gln168‐Asn170‐Thr174 amino acid changes 38 supporting the key role of the β2‐α2 loop in the transmission barrier of CWD. The comparison of Val166‐Gln168 and Val166‐Gln168‐Asn170‐Thr174 mouse models 38 suggest that just Met166‐Glu168 amino acids account for wild type human PrPC resistance against the efficient transmission of CWD. While CWD or certain classical scrapie prions transmitted very efficiently after iterative passage in mice expressing Val166‐Gln168 human PrPC, further transmission of the adapted prions in the wild‐type human PrPC context was inefficient, highlighting the relevance of Met166‐Glu168 amino acid residues as key molecular elements involved in the resistance to propagation of certain classical scrapie or CWD strains.
Biological influence of Met166‐Glu168 amino acid changes
The alterations observed in the propagation of prion strains in Val166‐Gln168 human PrPC context when compared to the wild‐type human PrPC suggest a general but strain‐specific key role of these modifications in the β2‐α2 loop either in the PrPC‐PrPSc heterologous interactions and/or in the PrPSc propagation rates. The higher transmission barrier associated with the wild‐type human PrPC suggests that Met166‐Glu168 amino acid residues could have been selected through evolution as molecular elements enhancing the resistance of human ancestors to circulating prion strains. These prions could affect human ancestors through dietary exposure to prion‐infected tissues either from animals or human cadavers in cannibal rituals. In those cases of dietary exposure to prion‐infected tissues, individuals harbouring Met166‐Glu168 residues in human PrPC putatively would experiment delayed onset of disease and/or poor transmissibility both for intra and interspecies prion transmissions rendering a clear evolutionary advantage over individuals harbouring Val166‐Gln168 amino acid residues in human PrPC.
Prion intraspecies transmission among human ancestors should not be underestimated since ritual cannibalism, besides other kinds of cannibalism, may have formed a driving force in the selection of advantageous human PrP variants, as was previously described for kuru disease 72 and for some prion diseases in the case of Met129Val dimorphism. 22 , 54 , 73 , 74 Such beneficial human PrPC variants may have directed the evolution of modern humans in situations of ancestral prion disease epidemics. 23 Several pieces of evidence support widespread cannibalistic practices in many ancient human populations, for example, anthropic marks on Neanderthal bones 75 and biochemical analysis of fossilised human stools, 76 independently of the funerary or aggressive cause of the cannibalism. Furthermore, non‐human primates evolutionarily related to humans, such as chimpanzees, eat head tissue from hunted prey, due to the high nutritional value of brain. 77 We can speculate that the ancient hunting habits, independent of whether the prey was human or not, exerted pressure for the selection of PrP variants that reduced the susceptibility to circulating prions thus promoting the selection of the Met166‐Glu168 amino acid residues in human PrP sequence. It should be noted that human PrP is the only primate PrP sequence harbouring Met166‐Glu168 amino acid residues. A few highly related primate species such as gorilla, chimpanzee and gibbon harbour only the Met166 residue, 78 suggesting that the Met166 variant was probably obtained earlier than the Glu168 amino acid change during primate evolution.
We can conclude that Met166‐Glu168 amino acids in the human PrP sequence are molecular elements highly involved in the human reduced susceptibility to prion infection although certain prion strains may not be affected by their presence.
ETHICS APPROVAL
Animal experiments were carried out in strict accordance with the recommendations stated in the guidelines of the Code for Methods and Welfare Considerations in Behavioral Research with Animals (Directive 2010/63/EU). All efforts were made to minimise suffering. Experiments were approved by the Committee on the Ethics of Animal Experiments of the author's institution (Instituto Nacional de Investigación y Tecnología Agraria y Alimentaria); Permit Number CEEA 2012/002.
CONFLICT OF INTEREST
There are no conflicts of interest to disclose.
AUTHORS’ CONTRIBUTIONS
The study was conceived by JCE and JMT. JCE, AMM, PAC and JMT performed research. JCE, AMM and JMT analysed data; and JCE, AMM and JMT wrote the paper. All authors read and approved the final manuscript.
FUNDING INFORMATION
This work was supported by grants from the Spanish Ministerio de Economía y Competitividad [AGL2016‐78054‐R (AEI/FEDER, UE), AGL2012‐37988‐C04‐04 and RTA2012‐00004‐00‐00 and fellowship BES‐2010‐040922 to P.A.C.], Fundación La Marató de TV3 (201821‐31) and the Instituto Nacional de Investigación y Tecnología Agraria y Alimentaria (fellowship FPI‐SGIT‐2015‐02 to A.M.M.).
Supporting information
Fig S1
{kind=link}
ACKNOWLEDGEMENTS
We declare no competing financial interests. We thank the following providers of tissues: Basque Biobank (Bilbao, Spain), Animal and Plant Health Agency (New Haw, Addlestone, Surrey U.K.), Biobanco Hospital Universitario Fundación Alcorcón (Alcorcón, Spain), National Veterinary Research Institute (Pulawy, Poland), French National Institute for Agricultural Research (Nouzilly, France) and Instituto Superiore di Sanitat (Rome, Italy).
Espinosa JC, Marín‐Moreno A, Aguilar‐Calvo P Torres JM. Met166‐Glu168 residues in human PrP β2‐α2 loop account for evolutionary resistance to prion infection. Neuropathol Appl Neurobiol. 2021;47:506–518. 10.1111/nan.12676
Juan Carlos Espinosa and Alba Marín‐Moreno are contributed equally to this work.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are in the article.
REFERENCES
- 1. Prusiner SB. Prions. Proc Natl Acad Sci USA. 1998;95:13363‐13383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. DeArmond SJ, Sanchez H, Yehiely F, et al. Selective neuronal targeting in prion disease. Neuron. 1997;19:1337‐1348. [DOI] [PubMed] [Google Scholar]
- 3. Parchi P, Giese A, Capellari S, et al. Classification of sporadic Creutzfeldt‐Jakob disease based on molecular and phenotypic analysis of 300 subjects. Ann Neurol. 1999;46:224‐233. [PubMed] [Google Scholar]
- 4. Peretz D, Williamson RA, Legname G, et al. A change in the conformation of prions accompanies the emergence of a new prion strain. Neuron. 2002;34:921‐932. [DOI] [PubMed] [Google Scholar]
- 5. Collinge J, Clarke AR. A general model of prion strains and their pathogenicity. Science. 2007;318:930‐936. [DOI] [PubMed] [Google Scholar]
- 6. Collins SJ, Lawson VA, Masters CL. Transmissible spongiform encephalopathies. Lancet. 2004;363:51‐61. [DOI] [PubMed] [Google Scholar]
- 7. Aguilar‐Calvo P, Garcia C, Espinosa JC, Andreoletti O, Torres JM. Prion and prion‐like diseases in animals. Virus Res. 2015;207:82‐93. [DOI] [PubMed] [Google Scholar]
- 8. Babelhadj B, Di Bari MA, Pirisinu L, et al. Prion disease in dromedary camels, Algeria. Emerg Infect Dis. 2018;24:1029‐1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Beringue V, Vilotte JL, Laude H. Prion agent diversity and species barrier. Vet Res. 2008;39:47. [DOI] [PubMed] [Google Scholar]
- 10. Collinge J, Sidle KC, Meads J, Ironside J, Hill AF. Molecular analysis of prion strain variation and the aetiology of ‘new variant’ CJD. Nature. 1996;383:685‐690. [DOI] [PubMed] [Google Scholar]
- 11. Hill AF, Desbruslais M, Joiner S, et al. The same prion strain causes vCJD and BSE. Nature. 1997;389:448‐450, 526. [DOI] [PubMed] [Google Scholar]
- 12. Bruce ME, Will RG, Ironside JW, et al. Transmissions to mice indicate that ‘new variant’ CJD is caused by the BSE agent. Nature. 1997;389:498‐501. [DOI] [PubMed] [Google Scholar]
- 13. The National CJD Research & Surveillance Unit Western General Hospital E, EH4 2XU. 26t h ANNUAL REPORT 2017. CREUTZFELDT‐JAKOB DISEASE SURVEILLANCE IN THE UK. Edinburgh: The National CJD Research & Surveillance Unit Western General Hospital, Edinburgh, EH4 2XU. 2017.
- 14. Gill ON, Spencer Y, Richard‐Loendt A, et al. Prevalent abnormal prion protein in human appendixes after bovine spongiform encephalopathy epizootic: large scale survey. BMJ. 2013;347:f5675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Will RG, Zeidler M, Stewart GE, et al. Diagnosis of new variant Creutzfeldt‐Jakob disease. Ann Neurol. 2000;47:575‐582. [PubMed] [Google Scholar]
- 16. Mok T, Jaunmuktane Z, Joiner S, et al. Variant Creutzfeldt‐Jakob disease in a patient with heterozygosity at PRNP Codon 129. N Engl J Med. 2017;376:292‐294. [DOI] [PubMed] [Google Scholar]
- 17. Biacabe AG, Laplanche JL, Ryder S, Baron T. Distinct molecular phenotypes in bovine prion diseases. EMBO Rep. 2004;5:110‐115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Casalone C, Zanusso G, Acutis P, et al. Identification of a second bovine amyloidotic spongiform encephalopathy: molecular similarities with sporadic Creutzfeldt‐Jakob disease. Proc Natl Acad Sci USA. 2004;101:3065‐3070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Beringue V, Herzog L, Reine F, et al. Transmission of atypical bovine prions to mice transgenic for human prion protein. Emerg Infect Dis. 2008;14:1898‐1901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kong Q, Zheng M, Casalone C, et al. Evaluation of the human transmission risk of an atypical bovine spongiform encephalopathy prion strain. J Virol. 2008;82:3697‐3701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wilson R, Dobie K, Hunter N, Casalone C, Baron T, Barron RM. Presence of subclinical infection in gene‐targeted human prion protein transgenic mice exposed to atypical bovine spongiform encephalopathy. J Gen Virol. 2013;94:2819‐2827. [DOI] [PubMed] [Google Scholar]
- 22. Marin‐Moreno A, Hour A, Espinosa JC, et al. Radical change in zoonotic abilities of atypical BSE prion strains as evidenced by crossing of sheep species barrier in transgenic mice. Emerg Infect Dis J. 2020;26:1130‐1139. 10.3201/eid2606.181790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mead S, Stumpf MP, Whitfield J, et al. Balancing selection at the prion protein gene consistent with prehistoric kurulike epidemics. Science. 2003;300:640‐643. [DOI] [PubMed] [Google Scholar]
- 24. Cassard H, Torres JM, Lacroux C, et al. Evidence for zoonotic potential of ovine scrapie prions. Nat Commun. 2014;5:5821. [DOI] [PubMed] [Google Scholar]
- 25. Brown P, Cathala F, Raubertas RF, Gajdusek DC, Castaigne P. The epidemiology of Creutzfeldt‐Jakob disease: conclusion of a 15‐year investigation in France and review of the world literature. Neurology. 1987;37:895‐904. [DOI] [PubMed] [Google Scholar]
- 26. van Duijn CM, Delasnerie‐Laupretre N, Masullo C, et al. Case‐control study of risk factors of Creutzfeldt‐Jakob disease in Europe during 1993–95. European Union (EU) Collaborative Study Group of Creutzfeldt‐Jakob disease (CJD). Lancet. 1998;351:1081‐1085. [DOI] [PubMed] [Google Scholar]
- 27. Miller MW, Fischer JR. The first five (or More) decades of chronic wasting disease: lessons for the five decades to come. Trans N Am Wildl Nat Resour Conf. 2016;81:1–11. [Google Scholar]
- 28. Lee YH, Sohn HJ, Kim MJ, et al. Strain characterization of the Korean CWD cases in 2001 and 2004. J Vet Med Sci. 2013;75:95‐98. [DOI] [PubMed] [Google Scholar]
- 29. Benestad SL, Mitchell G, Simmons M, Ytrehus B, Vikoren T. First case of chronic wasting disease in Europe in a Norwegian free‐ranging reindeer. Vet Res. 2016;47:88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Vikøren T, Våge J, Madslien KI, et al. First detection of chronic wasting disease in a wild red deer (Cervus elaphus) in Europe. J Wildl Dis. 2019;55:970‐972. [PubMed] [Google Scholar]
- 31. ProMED‐Mail . Chronic wasting disease, cervid ‐ FINLAND: first case, moose. 2018; Available from: https://www.promedmail.org/post/5684473
- 32. Mathiason CK, Hays SA, Powers J, et al. Infectious prions in pre‐clinical deer and transmission of chronic wasting disease solely by environmental exposure. PLoS One. 2009;4:e5916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Miller MW, Williams ES, Hobbs NT, Wolfe LL. Environmental sources of prion transmission in mule deer. Emerg Infect Dis. 2004;10:1003‐1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Williams ES, Young S. Spongiform encephalopathies in Cervidae. Rev Sci Tech. 1992;11:551‐567. [DOI] [PubMed] [Google Scholar]
- 35. Abrams JY, Maddox RA, Harvey AR, Schonberger LB, Belay ED. Travel history, hunting, and venison consumption related to prion disease exposure, 2006–2007 FoodNet Population Survey. J Am Diet Assoc. 2011;111:858‐863. [DOI] [PubMed] [Google Scholar]
- 36. Waddell L, Greig J, Mascarenhas M, Otten A, Corrin T, Hierlihy K. Current evidence on the transmissibility of chronic wasting disease prions to humans‐a systematic review. Transbound Emerg Dis. 2018;65:37‐49. [DOI] [PubMed] [Google Scholar]
- 37. Kong Q, Huang S, Zou W, et al. Chronic wasting disease of elk: transmissibility to humans examined by transgenic mouse models. J Neurosci. 2005;25:7944‐7949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kurt TD, Jiang L, Fernandez‐Borges N, et al. Human prion protein sequence elements impede cross‐species chronic wasting disease transmission. J Clin Invest. 2015;125:1485‐1496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wilson R, Plinston C, Hunter N, et al. Chronic wasting disease and atypical forms of bovine spongiform encephalopathy and scrapie are not transmissible to mice expressing wild‐type levels of human prion protein. J Gen Virol. 2012;93:1624‐1629. [DOI] [PubMed] [Google Scholar]
- 40. Zahn R, Liu A, Luhrs T, et al. NMR solution structure of the human prion protein. Proc Natl Acad Sci USA. 2000;97:145‐150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sigurdson CJ, Nilsson KP, Hornemann S, et al. A molecular switch controls interspecies prion disease transmission in mice. J Clin Invest. 2010;120:2590‐2599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Bett C, Fernandez‐Borges N, Kurt TD, et al. Structure of the beta2‐alpha2 loop and interspecies prion transmission. FASEB J. 2012;26:2868‐2876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Billeter M, Riek R, Wider G, Hornemann S, Glockshuber R, Wuthrich K. Prion protein NMR structure and species barrier for prion diseases. Proc Natl Acad Sci USA. 1997;94 7281‐7285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kurt TD, Bett C, Fernandez‐Borges N, et al. Prion transmission prevented by modifying the beta2‐alpha2 loop structure of host PrPC. J Neurosci. 2014;34:1022‐1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Rossetti G, Cong X, Caliandro R, Legname G, Carloni P. Common structural traits across pathogenic mutants of the human prion protein and their implications for familial prion diseases. J Mol Biol. 2011;411:700‐712. [DOI] [PubMed] [Google Scholar]
- 46. Rossetti G, Giachin G, Legname G, Carloni P. Structural facets of disease‐linked human prion protein mutants: a molecular dynamic study. Proteins. 2010;78:3270‐3280. [DOI] [PubMed] [Google Scholar]
- 47. Gonzalez L, Pitarch JL, Martin S, et al. Influence of polymorphisms in the prion protein gene on the pathogenesis and neuropathological phenotype of sheep scrapie after oral infection. J Comp Pathol. 2014;150:57‐70. [DOI] [PubMed] [Google Scholar]
- 48. Soto P, Claflin IA, Al B, Schwab‐McCoy AD, Bartz JC. Cellular prion protein gene polymorphisms linked to differential scrapie susceptibility correlate with distinct residue connectivity between secondary structure elements. J Biomol Struct Dyn. 2020; In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Padilla D, Béringue V, Espinosa JC, et al. Sheep and goat BSE propagate more efficiently than cattle BSE in human PrP transgenic mice. PLoS Pathog. 2011;7:e1001319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Borchelt DR, Davis J, Fischer M, et al. A vector for expressing foreign genes in the brains and hearts of transgenic mice. Genet Anal. 1996;13:159‐163. [DOI] [PubMed] [Google Scholar]
- 51. Manson JC, Clarke AR, Hooper ML, Aitchison L, McConnell I, Hope J. 129/Ola mice carrying a null mutation in PrP that abolishes mRNA production are developmentally normal. Mol Neurobiol. 1994;8:121‐127. [DOI] [PubMed] [Google Scholar]
- 52. Feraudet C, Morel N, Simon S, et al. Screening of 145 Anti‐PrP monoclonal antibodies for their capacity to inhibit PrPSc replication in infected cells. J Biol Chem. 2005;280:11247‐11258. [DOI] [PubMed] [Google Scholar]
- 53. Espinosa JC, Marin‐Moreno A, Aguilar‐Calvo P, Benestad SL, Andreoletti O, Torres JM. Porcine prion protein as a paradigm of limited susceptibility to prion strain propagation. J Infect Dis. 2020; In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Fernandez‐Borges N, Espinosa JC, Marin‐Moreno A, et al. Protective effect of Val129‐PrP against bovine spongiform encephalopathy but not variant Creutzfeldt‐Jakob disease. Emerg Infect Dis. 2017; 23: 1522‐1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Torres JM, Espinosa JC, Aguilar‐Calvo P, et al. Elements modulating the prion species barrier and its passage consequences. PLoS One. 2014;9:e89722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Torres JM, Andreoletti O, Lacroux C, et al. Classical bovine spongiform encephalopathy by transmission of H‐type prion in homologous prion protein context. Emerg Infect Dis. 2011;17:1636‐1644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Aguilar‐Calvo P, Espinosa JC, Pintado B, et al. Role of the goat K222‐PrPC polymorphic variant in prion infection resistance. J Virol. 2014;88:2670‐2676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Vanni I, Migliore S, Cosseddu GM, et al. Isolation of a defective prion mutant from natural scrapie. PLoS Pathog. 2016;12:e1006016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Andreoletti O, Simon S, Lacroux C, et al. PrP(Sc) accumulation in myocytes from sheep incubating natural scrapie. Nat Med. 2004;10:591‐593. [DOI] [PubMed] [Google Scholar]
- 60. Fraser H, Dickinson AG. The sequential development of the brain lesion of scrapie in three strains of mice. J Comp Pathol. 1968;78:301‐311. [DOI] [PubMed] [Google Scholar]
- 61. Song Y, DiMaio F, Wang RY, et al. High‐resolution comparative modeling with RosettaCM. Structure. 2013;21:1735‐1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Raman S, Vernon R, Thompson J, et al. Structure prediction for CASP8 with all‐atom refinement using Rosetta. Proteins. 2009;77(Suppl 9):89‐99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Brunger AT, Adams PD, Clore GM, et al. Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr D Biol Crystallogr. 1998;54:905‐921. [DOI] [PubMed] [Google Scholar]
- 64. Bishop MT, Will RG, Manson JC. Defining sporadic Creutzfeldt‐Jakob disease strains and their transmission properties. Proc Natl Acad Sci USA. 2010;107:12005‐12010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Cassard H, Huor A, Espinosa JC, et al. Prions from sporadic Creutzfeldt‐Jakob disease patients propagate as strain mixtures. MBio 2020;11(3):1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Thuring CM, Erkens JH, Jacobs JG, et al. Discrimination between scrapie and bovine spongiform encephalopathy in sheep by molecular size, immunoreactivity, and glycoprofile of prion protein. J Clin Microbiol. 2004;42:972‐980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Wopfner F, Weidenhofer G, Schneider R, et al. Analysis of 27 mammalian and 9 avian PrPs reveals high conservation of flexible regions of the prion protein. J Mol Biol. 1999;289:1163‐1178. [DOI] [PubMed] [Google Scholar]
- 68. Calzolai L, Lysek DA, Guntert P, et al. NMR structures of three single‐residue variants of the human prion protein. Proc Natl Acad Sci USA. 2000;97:8340‐8345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Caldarulo E, Barducci A, Wuthrich K, Parrinello M. Prion protein beta2‐alpha2 loop conformational landscape. Proc Natl Acad Sci USA. 2017;114:9617‐9622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Sandberg MK, Al‐Doujaily H, Sigurdson CJ, et al. Chronic wasting disease prions are not transmissible to transgenic mice overexpressing human prion protein. J Gen Virol. 2010;91:2651‐2657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Tamguney G, Giles K, Bouzamondo‐Bernstein E, et al. Transmission of elk and deer prions to transgenic mice. J Virol. 2006;80:9104‐9114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Mead S, Whitfield J, Poulter M, et al. A novel protective prion protein variant that colocalizes with kuru exposure. N Engl J Med. 2009;361:2056‐2065. [DOI] [PubMed] [Google Scholar]
- 73. Palmer MS, Dryden AJ, Hughes JT, Collinge J. Homozygous prion protein genotype predisposes to sporadic Creutzfeldt‐ Jakob disease. Nature. 1991;352:340‐342. [DOI] [PubMed] [Google Scholar]
- 74. Wadsworth JD, Asante EA, Desbruslais M, et al. Human prion protein with valine 129 prevents expression of variant CJD phenotype. Science. 2004;306:1793‐1796. [DOI] [PubMed] [Google Scholar]
- 75. Fernandez‐Jalvo Y, Carlos Diez J, Caceres I, Rosell J. Human cannibalism in the early pleistocene of Europe (Gran Dolina, Sierra de Atapuerca, Burgos, Spain). J Hum Evol. 1999;37:591‐622. [DOI] [PubMed] [Google Scholar]
- 76. Marlar RA, Leonard BL, Billman BR, Lambert PM, Marlar JE. Biochemical evidence of cannibalism at a prehistoric Puebloan site in southwestern Colorado. Nature. 2000;407:74‐78. [DOI] [PubMed] [Google Scholar]
- 77. Gilby IC, Wawrzyniak D. Meat eating by wild chimpanzees (Pan troglodytes schweinfurthii): effects of prey age on carcass consumption sequence. Int J Primatol. 2018;39:127‐140. [Google Scholar]
- 78. Schatzl HM, Da Costa M, Taylor L, Cohen FE, Prusiner SB. Prion protein gene variation among primates. J Mol Biol. 1995;245:362‐374. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1
Data Availability Statement
The data that support the findings of this study are in the article.