

Abstract
The stress‐activated protein kinases (SAPKs)/c‐Jun‐N‐terminal‐kinases (JNK) are members of the mitogen‐activated protein kinase family. These kinases are responsible for transducing cellular signals through a phosphorylation‐dependent signaling cascade. JNK activation in immune cells can lead to a range of critical cellular responses that include proliferation, differentiation and apoptosis. MKK4 is a SAPK that can activate both JNK1 and JNK2; however, its role in T‐cell development and function has been controversial. Additionally, loss of either JNK1 or JNK2 has opposing effects in the generation of T‐cell immunity to viral infection and cancer. We used mice with a conditional loss of MKK4 in T cells to investigate the in vivo role of MKK4 in T‐cell development and function during lymphocytic choriomeningitis virus (LCMV) infection. We found no physiologically relevant differences in T‐cell responses or immunity to either acute or chronic LCMV in the absence of MKK4.
Keywords: chronic infection, JNK, LCMV, MAPK, MKK4, T cells
MKK4 is a stress‐activated protein kinase that can activate both JNK1 and JNK2; however, its role in T‐cell development and function has been controversial. We used mice with a conditional loss of MKK4 in T cells to investigate the in vivo role of MKK4 in T‐cell development and function during lymphocytic choriomeningitis virus infection. Contrary to previous reports, we found that T‐cell development and expansion during both acute and chronic viral infection was unperturbed by the loss of MKK4.

Introduction
The stress‐activated protein kinases (SAPKs)/c‐Jun‐N‐terminal‐kinases (JNK) are members of the mitogen‐activated protein kinase (MAPK) family. These kinases are responsible for transducing cellular signals through a phosphorylation‐dependent signaling cascade. JNK activation can lead to a range of critical cellular responses that include proliferation, differentiation and apoptosis. 1 These processes are particularly important in B cells and T cells and, consequently, impact adaptive immunity.
There are three isoforms of JNK (JNK1, JNK2 and JNK3) that are conserved between mice and humans. JNK1 and JNK2 are widely expressed (including the hemopoietic system), while JNK3 is mainly expressed in the brain, heart and testis. 2 The MAPK sequential signaling hierarchy comprises a cascade of activation starting with upstream MKK kinases (MKKKs or MAP3Ks), then MK kinases (MKKs or MAP2Ks) and ending in downstream activation of MAPKs. JNK1 and JNK2 are MAPKs, which can only be phosphorylated by two members of the MAP2Ks called MKK4 and MKK7. 2 Mice with a homozygous germline deletion of the genes that encode MKK4 (Map2k4) or MKK7 (Map2k7) die during gestation at ~E12.5, indicating their critical function during development. 3 , 4 In both cases, embryos displayed liver abnormalities that are consistent with a defect in hematopoiesis in utero. These data suggest non‐redundant functions for MKK4 and MKK7 during hematopoiesis.
MKK7 can only phosphorylate JNKs, whereas MKK4 can additionally activate P38 MAPK. 5 , 6 MKK4 and MKK7 can both directly activate JNKs by phosphorylation but they differ in their preference for phosphorylation site in the T‐P‐Y motif. MKK4 preferentially phosphorylates the Tyr residue and Mkk7 the Thr residue. 7 , 8 Studies have shown that mono‐phosphorylation of the Thr residue by MKK7 alone is sufficient for JNK activation. 5 , 7 , 9 This indicates that MKK7 is important for triggering JNK activation, while the additional phosphorylation of the Tyr residue by MKK4 may only serve to ensure optimal JNK activation. 7 However, one study has found that MKK4 is sufficient for JNK activation. 10 These differences are most likely due to natural variations in the activation stimulus received by any one cell and the availability of JNK substrates.
Consistent with a role for JNK signaling in activation and proliferation, early work on the role of MKK4 and MKK7 in T‐cell biology indicated non‐redundant functions in these processes. 11 , 12 , 13 , 14 However, two of these publications presented conflicting results regarding the function of MKK4. 11 , 13 In support of a role for these MAP2Ks in T‐cell function, loss of both JNK1 and JNK2 in peripheral T cells enhanced their proliferative capacity and IL‐2 production. 3 The potential clinical importance of this has also been shown as a loss of JNK2 provided a gain of function for CD8+ T cells in mouse models of viral infection and cancer. 15 , 16 However, loss of JNK1 alone can also abrogate cellular immunity during acute viral infection. 15 We were interested in the potential non‐redundant functions of the upstream MAP2K that phosphorylates JNK1/2, MKK4, in T‐cell development and function. To this end, we examined mice with a T‐cell specific deletion of the gene that encodes MKK4 (Map2k4) and monitored the development of these T cells. Additionally, we addressed the role of MKK4 in CD8+ T‐cell proliferation and susceptibility to apoptosis in vitro as well as in vivo by infecting MKK4 gene‐targeted animals with an acute or chronic strain of lymphocytic choriomeningitis virus (LCMV).
Results
Thymocytes develop normally in absence of MKK4
Early work examining JNK signaling indicated little or no protein expression of MKK4 in thymocytes. 17 So, we first set out to confirm the expression of MKK4 in thymocytes by comparing protein levels in C57BL/6 mice (WT), control (Map2k4fl/fl) animals and mice that lacked MKK4 specifically in T cells (Map2k4ΔLck). Thymocyte lysates from WT and control animals expressed MKK4 protein levels comparable to those observed in WT brain lysates (Figure 1a). MKK4 protein expression was almost completely abolished in thymocytes harvested from Map2k4Δ Lck mice. A similar expression pattern was observed in peripheral T cells. Furthermore, the expression of MKK7 was unchanged in the absence of MKK4 in peripheral T cells, indicating that there were no compensatory changes in MKK7 expression (Figure 1b). As MKK4 deficiency had been associated with defects in negative selection, 11 we investigated thymocyte development in control and Map2k4ΔLck animals. Loss of MKK4 in thymocytes did not affect the proportions or numbers of T cells in the thymus at any stage of development (Figure 1c–e and Supplementary figure 1a, b). There were also no differences in the proportion or number of thymic T regulatory (Treg) cells (Supplementary figure 1c, d). Nishina et al. 11 had also shown that MKK4‐deficient, double‐positive thymocytes were more sensitive to apoptosis caused by FAS ligation. However, we found that control and Map2k4ΔLck thymocytes displayed identical viability profiles when co‐cultured with sFASL at varying concentrations in vitro (Figure 1f). We also did not observe any proliferative defects when MKK4‐deficient CD8+ T cells were stimulated in vitro with anti‐CD3 and anti‐CD28 (Figure 1g, h) and we did not observe any substantial cell death during in vitro activation (Figure 1i). We also performed western blots on lysates from in vitro stimulated CD8+ T cells and confirmed that the downstream targets of both MKK4 and MKK7, JNK and P38, were both activated (phosphorylated) (Figure 1j). Although MKK7 cannot phosphorylate P38, it has been reported that both MKK3 and MKK6 can perform this function. 18
Figure 1.
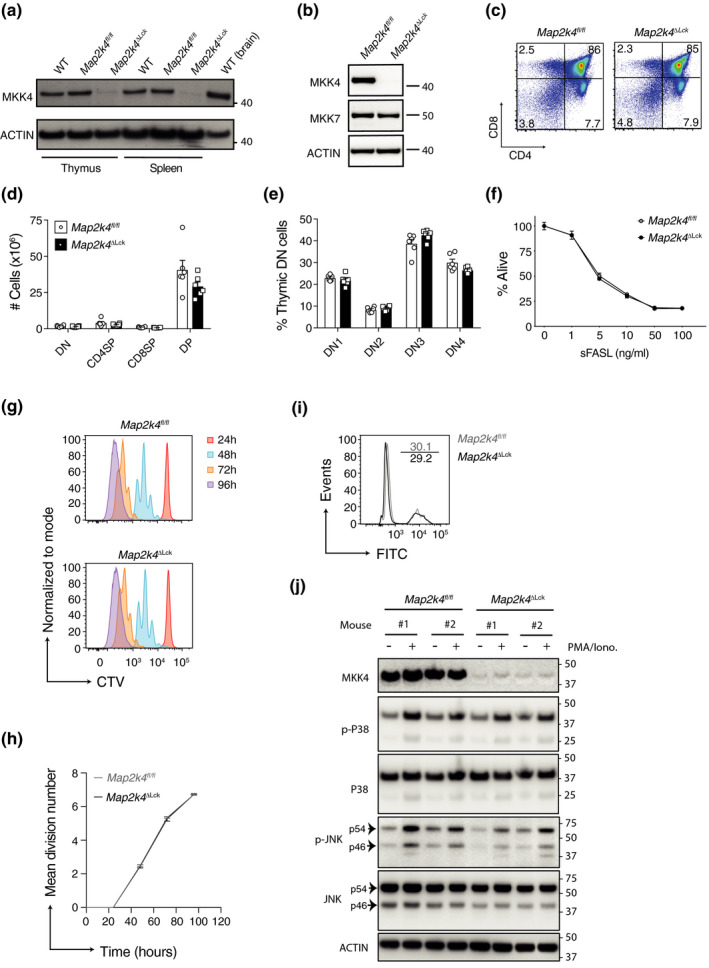
T‐cell development and proliferation occur normally in the absence of MKK4. (a) Immunoblot analysis of MKK4 and ACTIN (loading control) expression in thymocytes (thymus) or flow cytometry sorted splenic CD8+ T cells (spleen). WT (brain) is a positive control. (b) Immunoblot analysis of MKK4 and MKK7 expression in flow cytometry sorted splenic CD8+ T cells. Numbers to the right of the panels represent position of protein size markers (kDa). (c) Representative flow cytometry plots showing thymocytes stained for CD4 and CD8. Percentages of thymocytes in quadrants are indicated. (d) Numbers of each of the thymocyte populations from c. (e) Proportions of DN (double negative; CD4−CD8−) thymocytes at each of the four stages of negative selection. (f) Total thymocytes cultured with oligomerized soluble FasL (sFasL) for 24 h. The mean proportion (± SEM) of thymocytes that are alive, relative to unstimulated, are shown (n = 4 or 5 mice). (g) Representative flow cytometry histograms, illustrating the division of naïve CD8+ T cells sorted using magnetic beads, labeled with CTV and cultured with anti‐CD3 and anti‐CD28 antibodies for 1–4 days (24–96 h). (h) Mean division number at each of the timepoints from experiment in g (n = 3). (i) Naïve CD8+ T cells stained with a fluorescent (FITC) live/dead dye, following 3 days in culture as in g. (j) Immunoblot analysis of cell lysates from magnetically sorted CD8+ T cells that were either left unstimulated or stimulated for 15 min with PMA and ionomycin. Numbers to the right of the panels represent position of protein size markers (kDa). CTV, cell trace violet; h, hours; Iono, Ionomycin; CD4SP, CD4+CD8−; CD8SP, CD4−CD8+; DP, CD4+CD8+; DN1, CD44+CD25−; DN2, CD44+CD25+; DN3, CD44−CD25+; DN4, CD44−CD25−. All experiments were performed at least twice. Graphs show mean and SEM. Each symbol represents one mouse.
Peripheral T‐cell populations were unaffected by loss of MKK4
Coordinated activation and proliferation of both innate and adaptive immune cells is required for the development of a functional immune system. We were interested in examining the role of MKK4‐driven JNK and/ or p38 MAPK activation in the development of a range of immune cells in the periphery. The Lck promoter that drives Cre mediated deletion of Map2k4 in our gene‐targeted animals is active at the DN stage of T‐cell development. 19 We did not observe any differences in the numbers or proportions of total CD4+ or CD8+ T cells (or B cells as a control) in the spleen or pooled LNs of 6–12‐week‐old Map2k4ΔLck animals compared with control mice (Figure 2a, b and Supplementary figure 1e, f). We also did not see any differences in the proportion or number of naïve or memory T‐cell populations, or Treg populations in either the spleen or pLN of Map2k4ΔLck mice compared with control animals (Figure 2c–f and Supplementary figure 1g–i).
Figure 2.
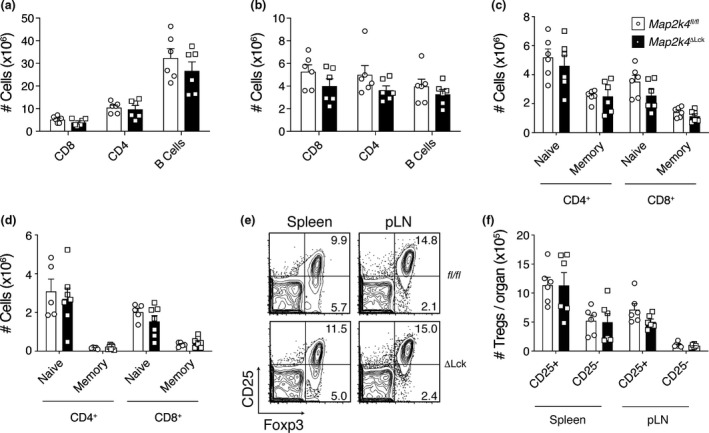
Effect of MKK4 loss on peripheral lymphoid compartments. Numbers of B and T cells in the (a) spleen and (b) pLN. (c, d) Numbers of naïve and memory T cells in the spleen (c) and pLN (d) of naïve animals. (e) Representative flow cytometry plots indicating the proportion of Tregs, gated on CD4+. Percentages of Tregs are indicated in the relevant quadrants. (f) Numbers of CD25− and CD25+ Tregs in the spleen and pLN from the same experiment as in e (n = 6). Graphs show mean and SEM. Data are representative of three independent experiments (a–e) or combined from two independent experiments (f). Each symbol represents one mouse.
MKK4 activity is redundant for induction of the CD8+ T‐cell response to acute and chronic viral infection
JNK1‐deficient mice infected with LCMV had defects in the expansion of a virus‐specific CD8+ T‐cell response. 15 However, JNK2‐deficient mice infected with LCMV generated more virus‐specific CD8+ T cells than WT control mice. 15 As MKK4 is capable of activating both JNK1 and JNK2, we wanted to investigate the effect of MKK4‐deficiency in T cells during LCMV infection. Total numbers of immune cell populations from the spleen were analyzed at the peak of the immunological response to acute LCMV infection, which occurs around day 8 post infection. There were no differences in the numbers of total CD8+ T cells, CD4+ T cells or B cells between MKK4 deficient and control groups (Figure 3a and Supplementary figure 1a). To investigate whether MKK4 was important for the generation of virus‐specific CD8+ T‐cell responses, we analyzed the numbers of these cells in the spleens of LCMV infected mice. The numbers of CD8+ T cells that recognize three immunodominant LCMV epitopes (GP33, GP276, NP396), were similar across gene‐targeted and control mice (Figure 3b and Supplementary figure 2b). As JNK signaling is important for the induction of gene transcription, we also examined the cytokine producing capacity of virus‐specific CD8+ T cells following restimulation with cognate antigens. The ability of virus‐specific T cells to produce TNF and/ or IFNγ was similar across control and Map2k4ΔLck animals (Figure 3c, d and Supplementary figure 2c, d). Although we had not observed any proliferative defect in vitro (Figure 1g, h), previously published in vivo studies had described reduced proliferative potential in MKK4‐deficient peripheral T cells. 12 Using Ki‐67 as a surrogate marker for proliferation, we measured the proportion of CD8+ T cells that had recently divided during acute LCMV infection. Contrary to previous reports, we did not observe any difference in the proportion of CD8+ T cells in Map2k4ΔLck mice that were proliferating, compared with control animals (Figure 3e). We finally confirmed that MKK4 is not required by T cells to control acute LCMV infection by performing viral titers in the lung, liver and spleen, showing that both Map2k4ΔLck animals and controls cleared virus by day 7 post infection (Figure 3f).
Figure 3.
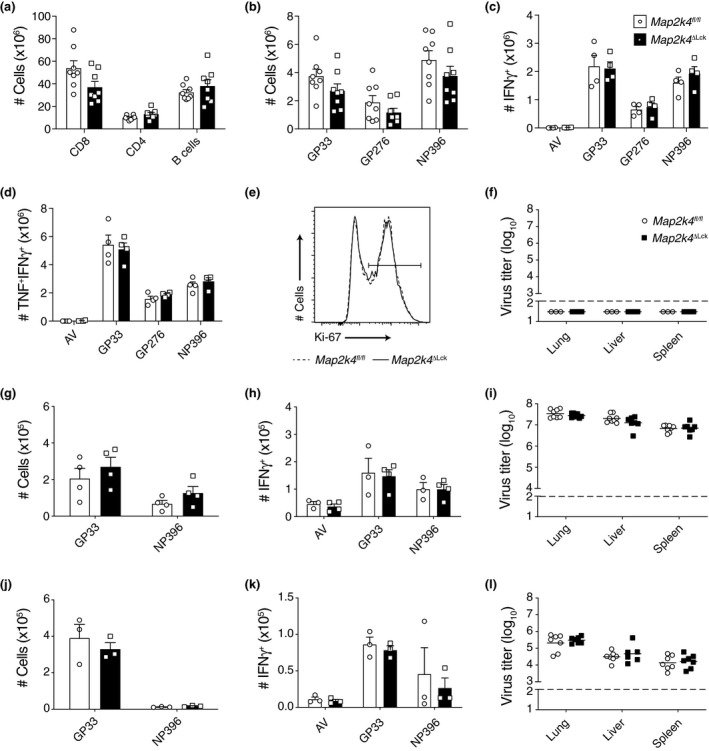
Loss of MKK4 does not perturb the response to viral infection. (a–e) Data generated from animals 8 days post infection with LCMV Armstrong (acute). (a) Numbers of lymphocytes isolated from the spleens of the indicated mice. (b) Numbers of LCMV‐specific CD8+ T cells identified using the relevant tetramers. (c, d) Ex vivo cytokine production by LCMV‐specific CD8+ T cells. Total splenocytes were restimulated with control adenoviral peptide (AV) or the indicated recombinant cognate LCMV peptides. Numbers of CD8+ T cells producing the indicated cytokines after stimulation with the indicated peptides are shown. (e) Representative flow cytometry histogram showing the proportion of ex vivo CD8+ T cells that have recently divided (proliferation). Gated on CD8+. (f) Virus titers in the indicated organs of mice 7 days post infection with LCMV Armstrong. (g–i) Data generated from animals 8 days post infection with LCMV Docile (chronic). (g) Numbers of LCMV‐specific CD8+ T cells identified using the relevant tetramers. (h) Ex vivo cytokine production performed as in c and d. (i) Virus titers in the indicated organs of mice 8 days post infection with LCMV Docile. (j–l) Data generated from animals 35 days post infection with LCMV Docile (chronic). (j) Numbers of LCMV‐specific CD8+ T cells identified using the relevant tetramers. (k) Ex vivo cytokine production performed as in c and d. (l) Virus titers in the indicated organs of mice 35 days post infection with LCMV Docile. Data are pooled from two independent experiments (a, b, i, l) or representative of two independent experiments (c, d, g, h, j, k). Bar graphs show mean and SEM. Viral titers graphs show geometric mean as a horizontal line in each group. The dotted line represents the limit of detection for the viral titer assay. Each symbol represents one mouse.
In contrast to acute LCMV infection, chronic infection with LCMV (docile or clone 13 strains) has been implicated in causing immune exhaustion and functional and numerical attrition of antiviral T cells. 20 , 21 To examine the role of MKK4 signaling in promoting or mitigating these effects we infected Map2k4ΔLck mice with LCMV docile strain and compared their immune response with controls. We found that loss of MKK4 in the T‐cell lineage did not impact the numbers of virus‐specific T cells compared with controls 8 days post infection (Figure 3g and Supplementary figure 2e). There was also no impact on the ability of virus‐specific T cells from Map2k4ΔLck mice to degranulate following ex vivo restimulation, compared with control animals (Figure 3h and Supplementary figure 2f). Unlike infection with acute LCMV, which is cleared from organs by day 7 post infection, LCMV docile remains at a high titer during the protracted chronic infection. Despite the constant SAPK/JNK signaling that occurs through constant T‐cell stimulation seen in chronic LCMV infection, there was no impact on viral titers in Map2k4ΔLck animals compared with controls at day 8 post infection (Figure 3i). We also confirmed that loss of MKK4 did not impact T‐cell responses to LCMV docile or viral control at 35 days post infection in Map2k4ΔLck animals compared with controls (Figure 3j–l and Supplementary figure 2g, h).
Discussion
To inactivate MKK4 in the T‐cell lineage, we used the distal Lck transgene to regulate Cre recombinase expression. 22 This was important as conditional knock out mice that use the proximal Lck promoter to drive Cre expression display a spurious “off‐target” reduction in thymic cellularity and they have aberrant T‐cell development. 23 We observed an almost complete loss of MKK4 protein expression in the thymus and this was maintained in T cells in the periphery of Map2k4ΔLck animals. Contrary to several publications, 11 , 12 , 24 we did not observe any developmental defects in T cells that lacked MKK4. Previous studies reconstituted Rag2−/− animals with Map2k−/− hematopoietic cells. We specifically deleted Map2k4 in the T‐cell lineage during the DN stage of development. It is possible that hematopoietic loss of Map2k4 causes aberrations in early progenitor cells or alternatively loss of Map2k4 across the whole hematopoietic compartment impacts T‐cell development.
We did not observe any proliferative defect, either in vitro or in vivo, in MKK4‐deficient T cells, contradicting some published reports. 11 , 12 However, our data are consistent with a study that showed normal JNK and p38 MAPK phosphorylation in T cells lacking MKK4. 13 This study also found no impact on T‐cell survival or proliferation in the absence of MKK4, concurring with our results. The most important measure of how any one factor affects T‐cell development and function is how T‐cell immunity may be altered during infection with an intracellular pathogen in vivo. Using our Map2k4ΔLck animals, we demonstrated that MKK4 is redundant for the generation of a robust CD8+ T‐cell response during acute LMCV infection. We have additionally shown that loss of MKK4 does not impact the T‐cell response to chronic LCMV infection. Taken together, our data indicate that MKK4 is not required for thymic T‐cell development or emigration of T cells into the periphery. Furthermore, MKK4 is dispensable for the generation of a successful T‐cell response to viral infection.
Methods
Mice and infection with LCMV
The Walter and Eliza Hall Institute Animal Ethics Committee reviewed and approved all animal experiments. Animals used in experiments were 6–12 weeks old. All mice used in our experiments were on a C57BL/6 (H‐2Db) background. Map2k4fl/fl animals have been described previously 25 and Lck‐Cre mice (distal promoter, B6.Cg‐Tg(Lck‐icre)3779Nik/J) were purchased from The Jackson Laboratory (Bar Harbor, ME, USA). Mice were infected with 1 × 103 pfu LCMV Armstrong (acute) or 2 × 106 pfu LCMV Docile (chronic) by intravenous injection into the tail vein. LCMV was propagated on L929 cells. Viral titers were determined by a focus forming assay, using MC57 fibroblast cells, as described previously. 26
Cell culture
For T‐cell viability and proliferation assays, total thymocytes or purified T cells from pooled lymph nodes (pLN; inguinal, cervical, brachial, axillary, mediastinal) were cultured in RPMI 1640 (Thermofisher Scientific, Waltham, USA), supplemented with 10% FCS (Thermofisher Scientific), 50 U mL−1 penicillin (Thermofisher Scientific), 50 μg mL−1 streptomycin (Thermofisher Scientific) and 50 μM 2‐mercaptoethanol (Sigma Aldrich, St Louis, USA). CD8+ T cells were purified using the EasySep Mouse naïve CD8+ T‐cell isolation kit (Stem Cell Technologies, Vancouver, Canada). T cells were stimulated with plate bound anti‐CD3 (10 μg mL−1) and soluble anti‐CD28 (2 μg mL−1) using the eBioscience T‐cell activation kit (Thermofisher Scientific). Alternatively, T cells were cultured with 50 ng mL‐1 PMA (Phorbol 12‐myristate 13‐acetate; Sigma Aldrich) and 1 μM Ionomycin (Sigma Aldrich) for 15 minutes to induce JNK and P38 phosphorylation. Thymocytes were cultured with the indicated concentrations of oligomerized soluble FASL (sFASL; Enzo Life Sciences, Farmingdale, USA) to determine susceptibility to FAS ligation. Proliferation was determined using the CellTrace Violet kit (Invitrogen, Carlsbad, USA). Viability was determined using a flow cytometry based, fixable viability dye (FITC; Invitrogen).
Flow cytometry
Spleen or pooled lymph nodes (pLN; inguinal, cervical, brachial, axillary) were isolated from mice. Red blood cell lysis was performed using ACK buffer (150 mM NH4Cl, 10 mM KHCO3 and 0.1 mM EDTA (all from Sigma Aldrich). 1 × 106 cells were taken for antibody staining and downstream analysis. Specific monoclonal antibodies were purchased from eBioscience (Thermo Fisher Scientific) or BD Biosciences (Franklin Lakes, USA): CD4 APC‐Cy7 (RM4‐5), CD8 BV510 (53‐6.7), CD44 PE (IM7), CD25 PE (PC61), TCRβ APC (H57‐597), Foxp3 APC (FJK‐16), TCRγδ FITC (B1), NK1.1 PE (PK136), CD11b BV510 (M170), CD11c PECy7 (HL3), CD19 PerCP‐Cy5.5 (ID3), Gr1 Alexa700 (Ly6G/Ly6C; RB6‐8C5), MHC‐II FITC (I‐A/I‐E; 2G9) and CD16/32 (2.4G2). H‐2Db restricted LCMV tetramers were purchased from Baylor College Medicine (Houston, USA) and were conjugated to APC: GP33 (KAVYNFATM), GP276 (SGVENPGGYCL) or NP396 (FQPQNGQFI). All flow cytometry data were collected on a Fortessa X20 (BD Biosciences) and analyzed using FlowJo software (Flowjo LLC, Ashland, USA). Ex vivo T‐cell restimulation was performed as described previously 20 using an irrelevant adenoviral peptide (AV; SGPSNTPPEI) or cognate LCMV epitopes with the sequences described above.
Western blots
Cells being analyzed by western blot were strictly kept on ice and were lysed in buffer containing 135 mM NaCl, 20 mM Tris‐HCl (pH 7.5), 1.5 mM Mg2Cl, 1 mM EGTA, 10% glycerol, 1% Triton X‐100, protease and phosphatase inhibitors (Roche, Basel, Switzerland). Lysates were boiled for 5 min in SDS loading buffer and separated using 4–12% Bis‐Tris pre‐cast gels (Thermo Fisher Scientific, Massachusetts, USA). Proteins were transferred onto nitrocellulose membranes (Thermo Fisher Scientific) and detected using primary and secondary antibodies. The following antibodies were used: rabbit anti‐MKK4 (Catalogue # 9152; Cell Signaling Technologies (CST), Danvers, MA, USA), rabbit anti‐β actin (HRP conjugate) (13E5; CST), rabbit anti‐MKK7 (Catalog # 4172; CST), rabbit anti‐p38 total (Catalog # 9212; CST), rabbit anti‐phospho‐p38 (Thr180/Tyr182) (Catalog # 9211; CST), rabbit anti‐SAPK/JNK total (Catalog # 9252; CST), mouse anti‐phospho‐SAPK/JNK (Thr183/Tyr185) (G9; Catalog # 9255; CST). All secondary antibodies used were from Southern Biotech (Birmingham, AL, USA) and were all HRP conjugated.
Statistical analysis
Grouped data are represented as mean ± SD or mean ± SEM as indicated. Prism 6.0d software (Graph Pad Software) was used to perform statistical tests. A t‐test was performed on data that were normally distributed and a Mann‐Whitney U‐test was performed on non‐parametric data. If a statistically significant result was returned, the P‐value is indicated. Where differences between groups did not reach statistical significance, no P‐value is shown.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Author Contributions
SP and MP conceived and designed the research; SP, MD, HS, CA, MH and JC performed research; SP and MP analyzed data; and SP and MP wrote the manuscript.
Funding information
This work was supported by National Health and Medical Research Council Australia (Grants 1147010 to MP, 1039014 to SP, 1133538 to JC), The Silvia and Charles Viertel Senior Medical Research Fellowship (MP), the Victorian State Government Operational Infrastructure Support, and the Independent Research Institutes Infrastructure Support Scheme of the Australian Government National Health and Medical Research Council.
Supporting information
Supplementary Material
Acknowledgments
Map2k4 conditionally gene‐targeted mice were generously provided by Cathy Tournier. We would like to thank Liana Mackiewicz, Carolina Alvarado, Kristy Vella and Merle Dayton for animal husbandry. Daniel Gray, David Vaux, Hamsa Puthalakath and Diana Hansen generously provided reagents. Bruno Helbert, Karen Mackwell and Rainbow Chan performed genotyping.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are included within the manuscript and/or are available from the corresponding author upon reasonable request.
REFERENCES
- 1. Ip YT, Davis RJ. Signal transduction by the c‐Jun N‐terminal kinase (JNK)–from inflammation to development. Curr Opin Cell Biol 1998; 10: 205–219. [DOI] [PubMed] [Google Scholar]
- 2. Haeusgen W, Herdegen T, Waetzig V. The bottleneck of JNK signaling: molecular and functional characteristics of MKK4 and MKK7. Eur J Cell Biol 2011; 90: 536–544. [DOI] [PubMed] [Google Scholar]
- 3. Dong C, Yang DD, Tournier C, et al. JNK is required for effector T‐cell function but not for T‐cell activation. Nature 2000; 405: 91–94. [DOI] [PubMed] [Google Scholar]
- 4. Yang D, Tournier C, Wysk M, et al. Targeted disruption of the MKK4 gene causes embryonic death, inhibition of c‐Jun NH2‐terminal kinase activation, and defects in AP‐1 transcriptional activity. PNAS 1997; 94: 3004–3009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Moriguchi T, Toyoshima F, Masuyama N, Hanafusa H, Gotoh Y, Nishida E. A novel SAPK/JNK kinase, MKK7, stimulated by TNFα and cellular stresses. EMBO J 1997; 16: 7045–7053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lin A, Minden A, Martinetto H, et al. Identification of a dual specificity kinase that activates the Jun kinases and p38‐Mpk2. Science 1995; 268: 286–290. [DOI] [PubMed] [Google Scholar]
- 7. Tournier C, Dong C, Turner TK, Jones SN, Flavell RA, Davis RJ. MKK7 is an essential component of the JNK signal transduction pathway activated by proinflammatory cytokines. Genes Dev 2001; 15: 1419–1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kishimoto H, Nakagawa K, Biological TWJO. Different properties of SEK1 and MKK7 in dual phosphorylation of stress‐induced activated protein kinase SAPK/JNK in embryonic stem cells. ASBMB 2003; 278: 16595–16601. [DOI] [PubMed] [Google Scholar]
- 9. Finch A, Holland P, Cooper J, Saklatvala J, Kracht M. Selective activation of JNK/SAPK by interleukin‐1 in rabbit liver is mediated by MKK7. FEBS Lett 1997; 418: 144–148. [DOI] [PubMed] [Google Scholar]
- 10. Wada T, Nakagawa K, Watanabe T, et al. Impaired synergistic activation of stress‐activated protein kinase SAPK/JNK in mouse embryonic stem cells lacking SEK1/MKK4: different contribution of SEK2/MKK7 isoforms to the synergistic activation. J Biol Chem 2001; 276: 30892–30897. [DOI] [PubMed] [Google Scholar]
- 11. Nishina H, Fischer KD, Radvanyi L, et al. Stress‐signalling kinase Sek1 protects thymocytes from apoptosis mediated by CD95 and CD3. Nature 1997; 385: 350–353. [DOI] [PubMed] [Google Scholar]
- 12. Nishina H, Bachmann M, Oliveira‐dos‐Santos AJ, et al. Impaired CD28‐mediated interleukin 2 production and proliferation in stress kinase SAPK/ERK1 kinase (SEK1)/mitogen‐activated protein kinase kinase 4 (MKK4)‐deficient T lymphocytes. J Exp Med 1997; 186: 941–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Swat W, Fujikawa K, Ganiatsas S, et al. SEK1/MKK4 is required for maintenance of a normal peripheral lymphoid compartment but not for lymphocyte development. Immunity 1998; 8: 625–634. [DOI] [PubMed] [Google Scholar]
- 14. Sasaki T, Wada T, Kishimoto H, et al. The stress kinase mitogen‐activated protein kinase kinase (MKK)7 is a negative regulator of antigen receptor and growth factor receptor‐induced proliferation in hematopoietic cells. J Exp Med 2001; 194: 757–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Arbour N, Naniche D, Homann D, Davis RJ, Flavell RA, Oldstone MBA. c‐Jun NH2‐terminal kinase (JNK)1 and JNK2 signaling pathways have divergent roles in CD8+ T cell‐mediated antiviral immunity. J Exp Med 2002; 195: 801–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Tao J, Gao Y, Li MO, et al. JNK2 negatively regulates CD8+ T cell effector function and anti‐tumor immune response. Eur J Immunol 2007; 37: 818–829. [DOI] [PubMed] [Google Scholar]
- 17. Rincón M, Whitmarsh A, Yang DD, et al. The JNK pathway regulates the in vivo deletion of immature CD4+CD8+ thymocytes. J Exp Med 1998; 188: 1817–1830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Davis RJ. Signal transduction by the JNK group of MAP kinases. Cell 2000; 103: 239–252. [DOI] [PubMed] [Google Scholar]
- 19. Tanigaki K, Tsuji M, Yamamoto N, et al. Regulation of αβ/γδ T cell lineage commitment and peripheral T cell responses by Notch/RBP‐J signaling. Immunity 2004; 20: 611–622. [DOI] [PubMed] [Google Scholar]
- 20. Pellegrini M, Calzascia T, Toe JG, et al. IL‐7 engages multiple mechanisms to overcome chronic viral infection and limit organ pathology. Cell 2011; 144: 601–613. [DOI] [PubMed] [Google Scholar]
- 21. Man K, Gabriel SS, Liao Y, et al. Transcription factor IRF4 promotes CD8+ T cell exhaustion and limits the development of memory‐like T cells during chronic infection. Immunity 2017; 47: 1129–1141.e5. [DOI] [PubMed] [Google Scholar]
- 22. Wang Q, Strong J, Killeen N. Homeostatic competition among T cells revealed by conditional inactivation of the mouse Cd4 gene. J Exp Med 2001; 194: 1721–1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Carow B, Gao Y, Coquet J, Reilly M, Rottenberg ME. lck‐driven Cre expression alters T cell development in the thymus and the frequencies and functions of peripheral T cell subsets. J Immunol 2016; 197: 2261–2268. [DOI] [PubMed] [Google Scholar]
- 24. Nishina H, Radvanyi L, Raju K, Sasaki T, Kozieradzki I, Penninger JM. Impaired TCR‐mediated apoptosis and Bcl‐XL expression in T cells lacking the stress kinase activator SEK1/MKK4. J Immunol 1998; 161: 3416–3420. [PubMed] [Google Scholar]
- 25. Wang X, Nadarajah B, Robinson AC, et al. Targeted deletion of the mitogen‐activated protein kinase kinase 4 gene in the nervous system causes severe brain developmental defects and premature death. Mol Cell Biol 2007; 27: 7935–7946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Battegay M, Cooper S, Althage A, Bänziger J, Hengartner H, Zinkernagel RM. Quantification of lymphocytic choriomeningitis virus with an immunological focus assay in 24‐ or 96‐well plates. J Virol Methods 1991; 33: 191–198. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material
Data Availability Statement
The data that support the findings of this study are included within the manuscript and/or are available from the corresponding author upon reasonable request.