

Abstract
Loss of the exocytic Sec1/MUNC18 protein MUNC18‐1 or its target‐SNARE partners SNAP25 and syntaxin‐1 results in rapid, cell‐autonomous and unexplained neurodegeneration, which is independent of their known role in synaptic vesicle exocytosis. cis‐Golgi abnormalities are the earliest cellular phenotypes before degeneration occurs. Here, we investigated whether loss of MUNC18‐1 causes defects in intracellular membrane transport pathways in primary murine neurons that may explain neurodegeneration. Electron, confocal and super resolution microscopy confirmed that loss of MUNC18‐1 expression results in a smaller cis‐Golgi. In addition, we now show that medial‐Golgi and the trans‐Golgi Network are also affected. However, stacking and cisternae ultrastructure of the Golgi were normal. Overall, ultrastructure of null mutant neurons was remarkably normal just hours before cell death occurred. By synchronizing protein trafficking by conditional cargo retention in the endoplasmic reticulum using selective hooks (RUSH) and immunocytochemistry, we show that anterograde Endoplasmic Reticulum‐to‐Golgi and Golgi exit of endogenous and exogenous proteins were normal. In contrast, loss of MUNC18‐1 caused reduced retrograde Cholera Toxin B‐subunit transport from the plasma membrane to the Golgi. In addition, MUNC18‐1‐deficiency resulted in abnormalities in retrograde TrkB trafficking in an antibody uptake assay. We conclude that MUNC18‐1 deficient neurons have normal anterograde but reduced retrograde transport to the Golgi. The impairments in retrograde pathways suggest a role of MUNC18‐1 in endosomal SNARE‐dependent fusion and provide a plausible explanation for the observed Golgi abnormalities and cell death in MUNC18‐1 deficient neurons.
Loss of MUNC18‐1 or the t‐SNAREs SNAP25 and syntaxin‐1 leads to massive, yet unexplained, neurodegeneration. Previous research showed that Golgi abnormalities are the earliest, common phenotype. In this issue, Van Berkel et al. provide an explanation for the mechanism underlying the Golgi abnormalities upon loss of MUNC18‐1. By systematically assessing transport routes to and from the Golgi, they show that retrograde endosome‐to‐Golgi, but not anterograde transport from the Golgi, is disturbed. This imbalance in transport routes provides a plausible explanation for the Golgi abnormalities, and may explain neurodegeneration.
Abbreviations
- CTB
Cholera Toxin Subunit B
- DIV
days in vitro
- E18
embryonic day 18
- EEA1
early endosome antigen 1
- EM
electron microscopy
- ER
endoplasmic reticulum
- KD
knock down
- KO
knock out
- LatB
Latrunculin B
- NGS
normal goat serum
- NPY‐mCherry
Neuropeptide Y fused to mCherry protein
- PM
plasma membrane
- RT
room temperature
- RUSH
retention using selective hooks
- SBP
streptavidin binding protein
- S/M
Sec1/MUNC18
- VSVG‐GFP
vesicular stomatitis virus G fused to green fluorescent protein
- TGN
Trans Golgi Network
- t‐SNARE
Target‐SNARE
- TrkB
tropomyosin receptor kinase B
- WT
wild type
1. INTRODUCTION
Synaptic SNARE complex components are known for their essential role in synaptic vesicle fusion in mammalian neurons (Sudhof, 2013). In addition, loss of the target SNAREs (t‐SNAREs), SNAP25 and syntaxin‐1, or the Sec1/MUNC18 (S/M) protein MUNC18‐1 leads to, yet unexplained, neurodegeneration (Peng et al., 2013; Santos et al., 2017; Vardar et al., 2016; Verhage et al., 2000). Their critical role in viability is independent of their role in synaptic vesicle fusion. Viability, but not synaptic transmission, is fully rescued by expression of non‐neuronal isoforms SNAP23 in Snap25 Knock Out (KO) neurons, syntaxin‐2/3/4 in syntaxin‐1 depleted neurons or MUNC18‐2/3 in Munc18‐1 KO neurons (Delgado‐martı et al., 2007; Peng et al., 2013; Santos et al., 2017). Conversely, several genetic and pharmacological perturbations have been described that block synaptic vesicle fusion and synaptic transmission, but do not cause degeneration (Schiavo et al., 1992; Schoch, 2001; Varoqueaux et al., 2002).
The earliest and shared phenotype among t‐SNAREs and MUNC18‐1 deficient models are abnormalities in Golgi morphology, distinct from apoptosis‐induced Golgi abnormalities (Santos et al., 2017). Expression of non‐neuronal isoforms does not only rescue viability, but also Golgi morphology, suggesting that these two processes are closely connected (Santos et al., 2017). The Golgi is an essential organelle in the (regulated) secretory pathway. Newly synthesized proteins arrive from the endoplasmic reticulum (ER) and move through multiple closely apposed cisternae, allowing posttranslational modifications and subsequent sorting to their final destination (Farquhar & Palade, 1998). This anterograde route is counterbalanced by multiple retrograde trafficking routes to recycle molecules and membrane back to the Golgi (Pavelka & Ellinger, 2008).
Here, we tested the hypothesis that defects in membrane transport pathways could explain the abnormal Golgi morphology, and observed neurodegeneration. Antero‐ and retrograde routes in these pathways were studied using electron, live‐cell, super‐resolution, and confocal microscopy. Our data confirm the previously observed cis‐Golgi abnormalities and extend the phenotype to the Trans Golgi Network (TGN). However, unexpectedly we found that cisternae ultrastructure was normal in Munc18‐1 KO as well as anterograde transport in the secretory pathways. Instead, MUNC18‐1 deficiency led to defects in endosome‐to‐recycling and endosome‐to‐Golgi retrograde pathways. We conclude that loss of Munc18‐1 results in disturbances in retro‐ but not anterograde membrane trafficking pathways. The dysregulation of retrograde trafficking pathways provides a plausible explanation for the previously observed Golgi abnormalities and neurodegeneration.
2. EXPERIMENTAL PROCEDURES
2.1. Animals
Munc18‐1 KO mice were generated as described previously (MGI Cat# 5909101, RRID:MGI:5909101) (Verhage et al., 2000). In short, five exons in Munc18‐1 were targeted by homologous recombination, resulting in complete abolishment of MUNC18‐1 expression. Recombination was performed in 129/SvJ stem cells. After germline transmission, mutant mice were back‐crossed to C57Bl/6J mice for >40 generations, after which 129/SvJ‐derived flanking region contributed <1.5% of the genome, including three genes with passenger mutations from the 129Sv genetic background (Kovačević et al., 2018) Munc18‐1 KO mice are alive until they are born. KO mice were generated by crossing heterozygous mice. Three‐ to eight‐months‐old female pregnant mice were housed in groups in type 2 cages, and had access to food and water ab libitum. On embryonic day 18 (E18) of pregnancy, mice were sacrificed by cervical dislocation and pups were obtained by caesarean section. Because of the limited time of animal suffering (less than a second), no anaesthetics were used. The procedure was executed by trained and qualified personnel. Pups were cooled on ice to minimize suffering and sacrificed by decapitation. KO pups were selected based on the absence of movement, as depletion of MUNC18‐1 results in paralysis. WT pups were arbitrary chosen from WT litters. One E18 pup per litter was used, with a total of 60 pups (30 WT pups, 30 KO pups). All animals were bred and housed according to Institutional and Dutch governmental guidelines. This study was not pre‐registered.
2.2. Neuronal cultures
Cortices were extracted from E18 wild type (WT) and KO embryos and collected in ice‐cold Hanks buffered Salt Solution (Sigma, cat. No. H9394) with 7mM HEPES (Invitrogen, cat. No. 15630‐056). One animal was used for one cell culture. After removal of the meninges, neurons were incubated in Hanks‐HEPES with 0.25% trypsin (Invitrogen, cat. No. T9253) for 20 min at 37°C. Neurons were washed and triturated with fire polished Pasteur pipettes then counted in a Fuchs‐Rosenthal chamber. Neurons were plated in Neurobasal medium supplemented with 2% B‐27 (Invitrogen, cat. No. 11530536), 1.8% HEPES, 0.25% Glutamax (Invitrogen, cat. No. 11574466) and 0.1% Pen/Strep (Invitrogen, cat. No. 11548876). Continental (network) cultures were created by plating WT cortical neurons at 25K/well or Munc18‐1 KO neurons at 75K/well. Neurons were plated on 18mm glass coverslips on a layer of rat glia grown on etched glass coverslips applied with 0.1 mg/ml poly‐d‐lysine and 0.2 mg/ml rat tail collagen (BD Biosciences, cat. No. 354236) solution. For Latrunculin B (LatB) treatment, neurons were plated on a 35mm glass bottom dish. For N‐Cadherin immunostaining, neurons were plated on poly‐L‐ornithine/laminin coated 10mm glass coverslips without glia feeder layer.
2.3. Constructs and lentiviral particles
Constructs encoding pSynapsin‐VSVG‐EGFP, pSynapsin‐NPY‐mCherry, pSynapsin‐ManII‐EGFP (gift Malhotra/Ortega), pSynapsin‐MUNC18WT‐T2A‐CreGFP, and pSynapsin‐MUNC18V263T‐T2A‐CreGFP were subcloned into Lentiviral vectors, and viral particles were produced as described before (Naldini et al., 1996). WT and Munc18‐1 KO neurons were infected with lentiviral particles at 0 days in vitro (DIV). For RUSH experiments, pCMV‐Streptavidin / SBP‐EGFP‐GPI (Addgene #65294, (Boncompain et al., 2012)) and pSynapsin‐ManII‐ECFP were delivered by standard calcium phosphate precipitation transfection at DIV 1, as described previously (Emperador‐Melero et al., 2018).
2.4. Immunocytochemistry
Neuronal cultures were fixed at DIV3 with 3.7% paraformaldehyde (PFA; Electron Microscopy Sciences, cat. No. 15681) then washed three times with Phosphate Buffered Saline pH = 7.4 (PBS). Neurons were permeabilised with 0.5% Triton X‐100, followed by a 30 min incubation in PBS containing 0.1% Triton X‐100 and 2% normal goat serum (NGS) to block a‐specific binding. All antibodies were diluted in NGS. Neurons were stained with primary antibodies for 2 hr at room temperature (RT). The following primary antibodies were used: chicken anti‐MAP2 (1:1,000; Abcam, ab5392, RRID:AB_2138153), mouse anti‐GM130 (1:1,000, Transduction Laboratories, cat. No. 610822, RRID:AB_398141), rat anti‐LAMP1 (1:100, Abcam, ab25245, RRID:AB_449893), rabbit anti‐TGN46 (1:500, Abcam, ab76282, RRID:AB_1524486), rabbit anti‐TrkB (1:500, Millipore, cat. No. 07‐225, RRID:AB_310445), mouse anti‐N‐Cadherin (1:200, Transduction Laboratories, cat. No. 610920, RRID:AB_2077527), rabbit anti‐mCherry (1:1,000, GeneTex, cat. No. GTX128508, RRID:AB_2721247), rabbit anti‐GFP (1:2000, GeneTex, GTX20290, RRID:AB_371415), rabbit anti‐EEA1 (1:50, Cell Signalling, cat. No. 2411S, RRID:AB_2096814). After three washes with PBS, neurons were stained with secondary antibodies Alexa Fluor (1:1,000; Invitrogen) for 1 hr at 20‐25°C. Following three additional washes, coverslips were mounted on microscopic slides with Mowiol‐DABCO.
For surface staining of TrkB, DIV3 neurons were washed twice in Tyrode's solution (2 mM CaCl2, 2.5 mM KCl, 119 mM NaCl, 2mM MgCl2, 20 mM glucose and 25 mM HEPES, pH 7.4) before they were incubated with rabbit anti‐TrkB (1:100, Millipore) diluted in Tyrode's for 15 min at 20‐25°C. After two washes with Tyrode's, neurons were fixed in 3.7% PFA for 20 min and washed three times with PBS. Neurons were blocked in PBS containing 3% bovine serum albumin (BSA; Acros Organics) for 30 min before incubation with Alexa‐647 for 1 hr at 20‐25°C. After three washes with PBS, neurons were permeabilized in 0.25% Triton X‐100 for 5 min and blocked with 3% BSA for 30 min. Neurons were then stained with primary anti‐MAP2 (1:1,000) for 2 hr and secondary Alexa Fluor (1:1,000) for 1 hr. Following three additional washes, coverslips were mounted on microscopic slides with Mowiol‐DABCO.
For retrograde labelling of Cholera Toxin Subunit B (CTB), neurons were incubated with CTB‐488 (100 ng/ml, Thermo Fisher, cat. No. c‐34775) for 15 min at 37°C with 5% CO2 in the dark. After washing the cultures with warm Neurobasal, retrograde trafficking was allowed for another 2 hr before neurons were fixed and stained with normal immunocytochemistry protocol.
Images were made on a Zeiss 510 Meta Confocal microscope (Carl Zeiss) with 40× oil immersion objective (NA = 1.3; Carl Zeiss). Z stacks were acquired with 0.5 or 1 μm intervals. Z stacks were collapsed to maximal projections for image analysis.
STED images were made on a Leica SP8 using 100× oil objective (NA = 1.4). Images were deconvoluted using Huygens Professional software. Line segments were drawn in a single focal plane to plot intensity.
Image analysis was performed in ImageJ software. Golgi roundness was assessed using the ImageJ software build‐in ‘Round’ parameter, which assesses the inverted aspect ratio: .
2.5. Electron microscopy
Neurons were fixed for 90 min at 20‐25°C with 2.5% glutaraldehyde in 0.1M cacodylate buffer, pH 7.4. After washing three times in MilliQ, cells were post‐fixed for 90min at 20‐25°C with 1% OsO4/1% KRu(CN)6. After dehydration through a series of increasing ethanol concentrations, cells were embedded in Epon and polymerised for 48 hr at 60°C. Ultrathin sections (∼80 nm) were cut parallel to the cell monolayer, collected on single‐slot, Formvar‐coated copper grids, and stained in uranyl acetate and lead citrate in Ultrastainer LEICA EM AC20. Somas were arbitrarily selected at low magnification and imaged using a JEOL1010 transmission electron microscope at 40k magnification.
For analysis, the person analysing the images was blinded by obscuring the group assignment for each image, and cisterna width and intercisternae distance were measured in ImageJ software. Each cisterna or the distance between two adjacent cisternae was measured at three locations, and the average was calculated. Next, the average of all cisterna widths (range: 3–7 cisternae, median: 5, per neuron) or intercisternae distance (range: 3–7 distances, median: 5, per neuron) in one neuron was used as one data point.
2.6. Live‐cell imaging
DIV3 neurons were transferred to a NIKON Ti‐Eclipse microscope with a Tokai Hit heating system pre‐warmed to 37°C with 5% CO2. The microscope is equipped with a confocal scanner model A1R+. Time‐lapse recordings were made using resonant scanning mode. For the RUSH system, image acquisition was performed every 7 min for a total of 120 min using a 40×‐oil (NA = 1.3) objective. Biotin (40 μM, Sigma) was added at t = 0. For LatB treatment, imaging was performed every minute for 135 min, with a 60×‐oil (NA = 1.4) with 2× zoom and z‐stacks with 1 μm interval. LatB (20 μM, Sigma) or DMSO (Sigma) as control were added at t = 0. In both assays, neurons were imaged in supplemented Neurobasal.
2.7. TrkB antibody uptake assay
The TrkB antibody uptake assay was adapted from (Carrodus et al., 2014). Neurons were incubated with primary rabbit anti‐TrkB antibody (1:100; Millipore) diluted in Neurobasal medium at 37°C with 5% CO2 for 1 hr to promote TrkB internalization. After two washes with warm supplemented Neurobasal, neurons were fixed in 3.7% PFA. Cultures were washed in PBS and incubated in 3% bovine serum albumin (BSA) in PBS for 30 min to block a‐specific binding. Subsequently, surface TrkB was stained with Alexa‐488 (1:1,000; Invitrogen) for 1 hr at 20‐25°C. After three washing steps in PBS, neurons were incubated for 18 hr with secondary antibody conjugated with alkaline phosphatase, anti‐ rabbit‐HRP (111‐055‐003; 1:100; Jackson ImmunoResearch) diluted in 3% BSA to ensure all remaining surface bound anti‐TrkB was blocked prior to the permeabilisation steps detecting internalized TrkB. Cultures were washed with PBS and post fixed with 3.7% PFA. Next, neurons were permeabilised with 0.5% Triton X‐100 for 5 min and blocked in 3% BSA for 30 min. Internalized TrkB was stained with Alexa‐647 (1:1,000; Invitrogen), diluted in 3% BSA along with primary chicken anti‐MAP2 antibody (1:500; Abcam) for 1.5 hr at 20‐25°C. After three additional washes, neurons were stained against MAP2 with Alexa‐546 (1:1,000; Invitrogen) for 1 hr at 20‐25°C. Coverslips were mounted on microscopic slides with Mowiol‐DABCO and imaged with 40X oil immersion objective (NA = 1.3). Z stacks were acquired with 0.5 μm intervals and collapsed to maximal projections. ImageJ software was used to generate particle masks to analyse puncta area, size, and count. The intensity of internalized TrkB was calculated by subtracting surface intensity from the internalized pool using, taking the mean intensity per pixel using ImageJ software.
2.8. Statistical analysis
Statistical analysis and graphing were performed using GraphPad Prism versions 5 and 6 and MATLAB (MathWorks Inc., v2017a). Normality was assessed using the D’Agostino & Pearson normality test. No test for outliers was performed. Parametric tests were used whenever assumptions of homoscedasticity and normality were met. Otherwise, non‐parametric tests were used. For the Golgi roundness parameter, non‐parametric were used regardless normality to avoid potential bias of nonlinearity. When comparing more than two groups, One‐way ANOVA (assumptions met) or Kruskal–Wallis test (assumptions not met) with post‐hoc Dunn's comparisons test was performed. All statistical tests were two‐tailed. An error probability level of p < .05 was accepted as statistically significant. All cellular data is represented in Tukey boxplots, showing the minimum and maximum values (ends of whiskers), interquartile range (length of box) and median (line through box). Columns and dots represent individual litters and neurons, respectively. Sample size is indicated in Figure legends (N = number of neurons/number of independent cell cultures). No sample calculation was performed. Except for EM analysis, no blinding of groups was performed. No exclusion criteria were pre‐determined.
3. RESULTS
3.1. Loss of MUNC18‐1 leads to cis‐Golgi and TGN abnormalities, but normal alignment
Previously, we have shown that loss of MUNC18‐1 results in a smaller and rounder cis‐Golgi (Santos et al., 2017). Here, we confirmed that the GM130 (cis‐Golgi marker) positive area was ~33% smaller and ~50% rounder in DIV 3 Munc18‐1 KO neurons, whereas the mean GM130 intensity was not significantly different (Figure 1c–e). Staining for TGN46, a TGN marker, revealed a 30% smaller area in Munc18‐1 KO neurons, but the shape of the TGN46 positive area was normal, as quantified with the roundness parameter (Figure 1f,g).
Figure 1.
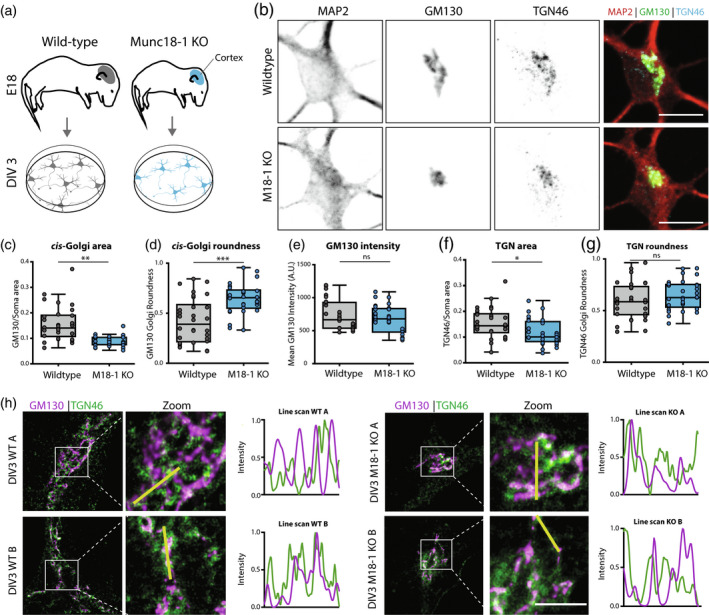
Munc18‐1 KO neurons have a smaller cis and trans Golgi complex. (a) Cortical neurons from E18 WT and Munc18‐1 KO mice were plated on glia feeder layers. At DIV3, cultures were fixed and stained for Golgi markers. (b) Typical example of WT and Munc18‐1 KO neurons, stained for MAP2 (dendritic marker), GM130 (cis‐Golgi marker) and TGN46 (TGN marker). Scale bar is 10μm. (c) The GM130‐positive area was 33% smaller in Munc18‐1 KO neurons compared to WT neurons (p < .0001, Mann–Whitney U test). N: WT = 28/3, KO = 30/3. (d) The GM130‐positive area of Munc18‐1 KO neurons was 50% rounder (p = .0001, Mann–Whitney U test). N: WT = 28/3, KO = 30/3. (e) Average intensity of GM130 was comparable between Munc18‐1 KO and WT controls (ns p = .27, unpaired T test). N: WT = 28/3, KO = 30/3. (f) Munc18‐1 KO neurons presented a 30% smaller TGN compared to WT neurons (p = .01, unpaired T test). N: WT = 30/3, KO = 29/3. (g) TGN roundness is not affected in Munc18‐1 KO neurons (ns p = .22, Mann–Whitney U test). N: WT = 30/3, KO = 29/3. (h) Representative STED images and intensity line scans of DIV 3 WT and Munc18‐1 KO neurons, stained for GM130 (cis‐Golgi marker) and TGN46 (TGN marker). Scale bar is 2μm. Data is represented in Tukey boxplots. Columns and dots represent individual litters and neurons, respectively. N = number of neurons/number of independent cell cultures
Super‐resolution STED imaging was performed to visualize the Golgi abnormalities at higher resolution. Also using STED, GM130, and TGN46‐positive areas were smaller in Munc18‐1 KO neurons (Figure 1h), but the alignment of the GM130 positive areas and TGN46 positive areas was comparable between WT and Munc18‐1 KO. Together, these data confirm cis‐Golgi abnormalities in Munc18‐1 KO neurons and extend this to the TGN, but indicate a normal spatial alignment of the two compartments.
3.2. Cisternae width and inter‐cisterna distance are normal in Munc18‐1 KO neurons
Golgi stack defects, such as dilated cisternae, are frequently observed in mutants that show trafficking defects (Bailey Blackburn et al., 2016; Emperador‐Melero et al., 2018). To test for such possible defects, cortical neurons in culture at DIV 1–3 were imaged using electron microscopy (EM) (Figure 2a). DIV3 is the latest possible time point, since approximately 50% of Munc18‐1 KO neurons has died at this stage and at DIV4 almost all neurons have died (Santos et al., 2017). From DIV 1 onwards, the total Golgi area was smaller in Munc18‐1 KO neurons. At high magnification (50k), individual cisternae had a normal appearance in Munc18‐1 KO neurons (Figure 2b). Cisternae width and inter‐cisternae distance in DIV3 neurons were similar in Munc18‐1 KO and WT neurons (Figure 2c). The overall morphology and ultrastructure of Munc18‐1 KO neurons were remarkably normal, given the fact that these neurons will die in the next hours (Santos et al., 2017). No signs of neurodegeneration were observed, for example, membrane blebbing or ruptures, abundant vacuoles or aggregates, or DNA condensation. Ultrastructure of all organelles were indistinguishable from organelles in WT neurons. Together, EM confirms the smaller Golgi area in Munc18‐1 KO neurons, in line with the confocal and STED imaging. However, the overall morphology and ultrastructure, including Golgi cisterna morphology, are normal in Munc18‐1 KO neurons.
Figure 2.
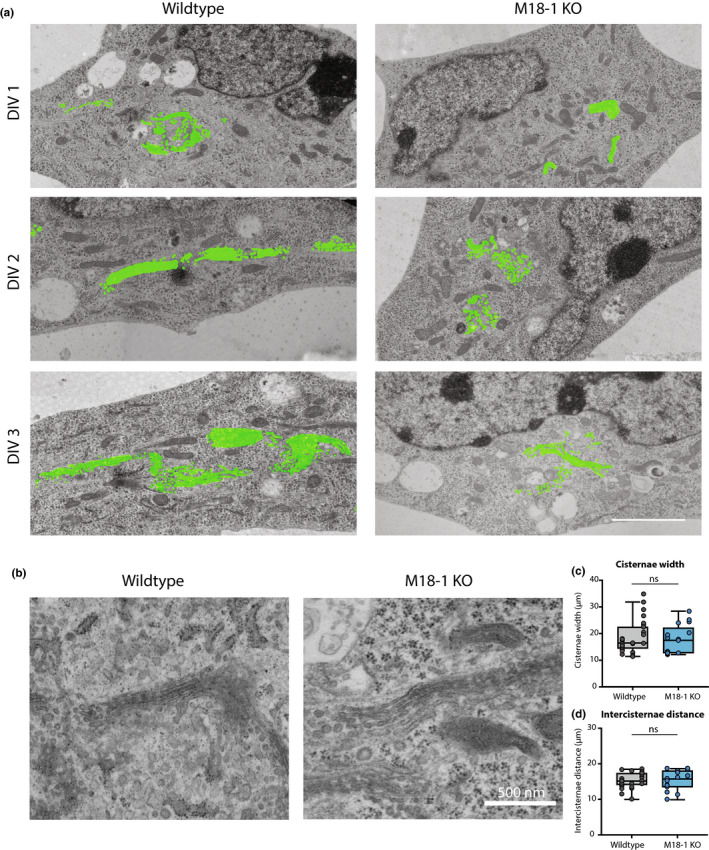
Less Golgi membrane, but normal cisternae structure in Munc18‐1 KO neurons. (a) Typical examples of EM images of WT and Munc18‐1 KO neurons fixed at DIV 1, 2 and 3. Golgi membrane is highlighted in green. Scale bar 2.5μm. (b) High magnification EM images of Golgi apparatus in WT and Munc18‐1 KO neurons at DIV 3. Scale bar 500 nm. (c) Average width of Golgi cisternae was not different between WT and Munc18‐1 KO neurons (ns p = .49, Mann–Whitney U test). N: WT = 29/3, KO = 17/3. (d) Munc18‐1 KO neurons did not differ in distance between cisternae (ns p = .97, unpaired T test). N: WT = 29/3, KO = 17/2. Mean and individual neurons as dots are shown. N = number of neurons/number of independent cell cultures
3.3. Actin depolymerisation in WT neurons phenocopies Golgi abnormalities in Munc18‐1 KO neurons
MUNC18‐1 is known to regulate F‐actin networks (Pons‐Vizcarra et al., 2019; Toonen et al., 2006). F‐actin is involved in maintaining Golgi morphology (Dippold et al., 2009) and actin depolymerisation by LatB leads to a condensed Golgi (Lázaro‐Diéguez et al., 2006), similar to the phenotype observed in Munc18‐1 KO neurons. To test if actin dysregulation causes the Munc18‐1 KO Golgi phenotype, WT and Munc18‐1 KO neurons were treated with LatB (20µM). ManII‐GFP was used as a live marker for the medial‐Golgi. In Munc18‐1 KO neurons, the ManII‐GFP positive area was ~50% smaller, as expected (Figure 3b,c). LatB treatment in WT neurons resulted in reduction in the ManII‐GFP positive area, cumulating in a ~45% total reduction 135 min after LatB addition. However, in Munc18‐1 KO neurons, LatB had no effect on the ManII‐GFP positive area. Hence, actin depolymerisation in WT neurons phenocopies the Munc18‐1 KO Golgi phenotype, but does not further reduce Golgi area in Munc18‐1 KO neurons, suggesting that dysregulation of F‐actin may indeed contribute to Golgi abnormalities in Munc18‐1 KO neurons.
Figure 3.
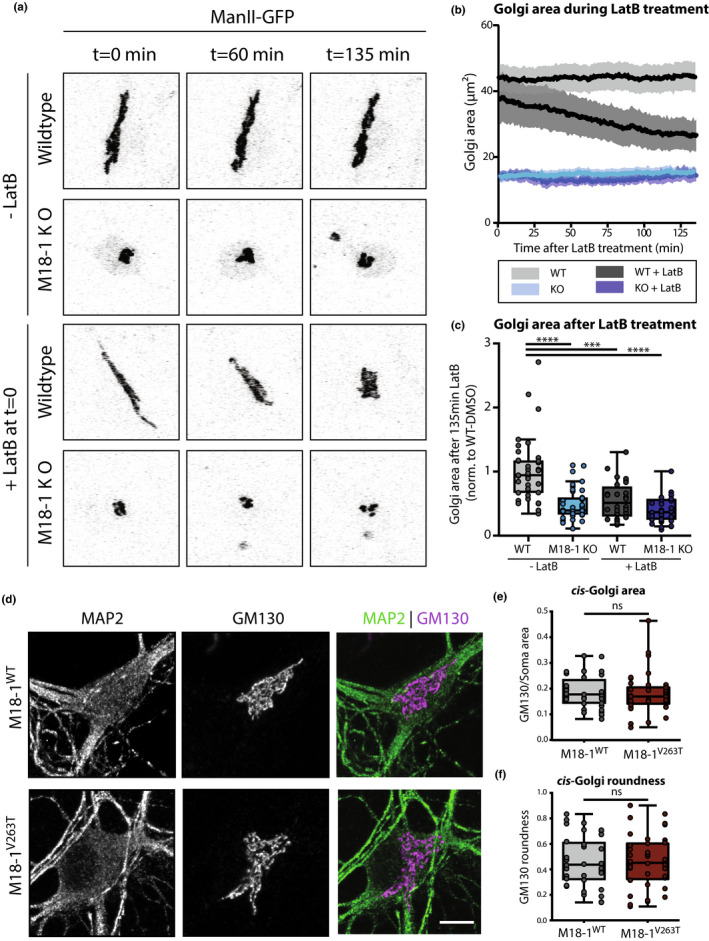
Actin depolymerization reduces Golgi area in WT but not in Munc18‐1 KO neurons. (a) WT and Munc18‐1 KO neurons were infected with ManII‐GFP (Golgi marker) and treated with LatB at DIV3. Golgi morphology was imaged for 135 min. (b) Golgi area in WT and Munc18‐1 KO neurons with and without LatB treatment over time. (c) Average Golgi area in WT and Munc18‐1 KO neurons with and without LatB treatment after 135 min of treatment. Data were normalized to the respective batch average of WT‐DMSO condition. The groups were significantly different from each other (p < .0001, Kruskal–Wallis test). Post hoc multiple comparisons revealed that WT Golgi area was significantly larger than untreated Munc18‐1 KO, LatB‐treated WT and LatB‐treated KO neurons, whereas these latter groups were not significantly different from each other ( post‐hoc Dunn's comparisons test). N: WT + DMSO=37/3, KO + DMSO=40/3, WT + LatB=28/3, KO + LatB=32/3. (d) Typical examples of Munc18‐1 KO neurons rescued with M18‐1WT and M18‐1V263T at DIV14 stained for MAP2 (dendritic marker) and GM130 (cis‐Golgi marker). Scale bar is 10 μm. (e) GM130‐positive area in M18‐1V263T was not different from M18‐1WT neurons (ns p = .51, Mann–Whitney U test). N: WT = 36/3, KO = 40/3. (f) GM130‐positive area roundness was not affected in M18‐1V263T neurons (ns p = .87, Mann–Whitney test). (g) N: WT = 36/3, KO = 40/3. (h) Data is represented in Tukey boxplots. Columns and dots represent individual litters and neurons, respectively. N = number of neurons/number of independent cell cultures
To test this possibility further, a MUNC18‐1 mutant (V263T) was expressed in Munc18‐1 KO neurons known to rescue secretion and Syntaxin‐1 targeting, but not F‐actin dysregulation (Pons‐Vizcarra et al., 2019). Munc18‐1 KO neurons were infected with MUNC18‐1WT or MUNC18‐1V263T and cis‐Golgi morphology was examined at DIV14 (Figure 3d). MUNC18‐1V263T neurons showed normal survival. GM130‐positive area and roundness were not affected by the V263T mutation (Figure 3e,f). Together, these data show that MUNC18‐1’s role in F‐actin regulation is not causally related to its function in maintaining Golgi morphology.
3.4. Endogenous proteins of the secretory pathway do not accumulate in the Golgi of Munc18‐1 KO neurons
To understand whether the abnormal Golgi morphology affects Golgi function, we examined the expression levels and localisation of two proteins that are trafficked through the Golgi: cell‐adhesion protein N‐Cadherin and Neurotrophin receptor Tropomyosin Receptor Kinase B (TrkB) (Figure 4. A‐B) (Klein et al., 1991; Wiertz et al., 2011). In Munc18‐1 KO neurons, the N‐Cadherin staining intensity in the TGN46‐positive area was comparable to WT neurons (Figure 4c), while the TrkB intensity was ~% lower (Figure 4d). However, these differences between N‐Cadherin and TrkB staining were not specific for the Golgi marker areas. The total staining intensity in the dendrites and soma was also similar for N‐Cadherin, and also ~55% reduced for TrkB (Figure 4e,f). Hence, TrkB levels are reduced in Munc18‐1 KO neurons, not specifically in the Golgi and no evidence for specific Golgi retention was obtained for either marker.
Figure 4.
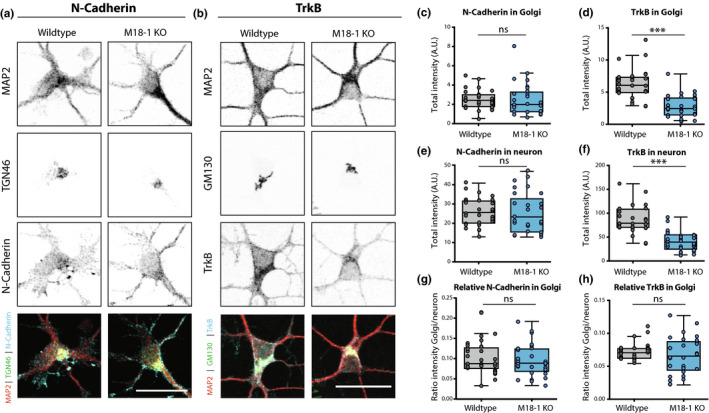
Endogenous markers of the secretory pathway do not accumulate in the Golgi of Munc18‐1 KO neurons. (a) Typical example of WT and Munc18‐1 KO neurons at DIV3 stained for MAP2 (dendritic marker), TGN46 (Trans Golgi Network Marker) and N‐Cadherin. Scale bar is 25μm. (b) Typical example of WT and Munc18‐1 KO neurons at DIV3 stained for MAP2 (dendritic marker), GM130 (cis‐Golgi marker) and TrkB. Scale bar is 25μm. (c) N‐Cadherin intensity in Golgi was not different between WT and Munc18‐1 KO (ns p = .24, Mann–Whitney test). N: WT = 30/3, KO = 29/3. (d) TrkB intensity in Golgi was ~55% lower in Munc18‐1 KO (p < .0001, Mann–Whitney test). N: WT = 28/3, KO = 30/3. (e) Munc18‐1 KO neurons did not differ in total N‐Cadherin intensity (ns p = .82, unpaired T test). N: WT = 30/3, KO = 29/3. (f) Total TrkB intensity in Munc18‐1 KO neurons was ~55% lower (p < .0001, unpaired T test). N: WT = 28/3, KO = 30/3. (g) Relative N‐Cadherin distribution in the Golgi, measured by N‐Cadherin intensity in Golgi (c) divided by total N‐Cadherin intensity (e), was not different in Munc18‐1 KO neurons (ns p = .65, Mann–Whitney test). N: WT = 30/3, KO = 29/3. (h) Relative TrkB distribution in the Golgi, measured by TrkB intensity in Golgi (d) divided by total TrkB intensity (f), was not different in Munc18‐1 KO neurons (ns p = .36, Mann–Whitney test). N: WT = 28/3, KO = 30/3. Data is represented in Tukey boxplots. Columns and dots represent individual litters and neurons, respectively. N = number of neurons/number of independent cell cultures
To confirm this conclusion quantitatively, TrkB staining intensity in GM130‐positive area was divided by the staining intensity in the total neuron. For both N‐Cadherin and TrkB, no differences were found in the relative levels in the GM130‐positive areas compared to the rest of the neuron (Figure 4g,h). Together, these data show that although some proteins show lower expression levels, no evidence was obtained for specific Golgi retention, suggesting that Golgi function is not affected by its smaller size and altered shape in Munc18‐1 KO neurons.
3.5. ER‐to‐Golgi and Golgi exit cargo trafficking is not affected upon MUNC18‐1 loss
To examine whether specific secretory pathways are disturbed in Munc18‐1 KO neurons, we over‐expressed cargo proteins for the constitutive (Vesicular stomatitis virus G fused to green fluorescent protein; VSVG‐GFP) and regulated (Neuropeptide Y fused to mCherry protein; NPY‐mCherry) pathway (Horton & Ehlers, 2003; Persoon et al., 2018). Expression levels of VSVG‐GFP and NPY‐mCherry were 60% and 55% lower in the GM130‐positive area of Munc18‐1 KO neurons, respectively (Figure 5c,d). Quantification of expression in dendrites and soma showed a similar reduction in VSVG‐GFP (~60%, Figure 5e) and NPY‐mCherry levels (~45%, Figure 5f) in Munc18‐1 KO neurons. Hence, similar to the situation for endogenous cargo (Figure 4), the reduced expression of heterologous cargo in the Golgi area appears to be a consequence of overall lower expression levels, rather than Golgi‐retention.
Figure 5.
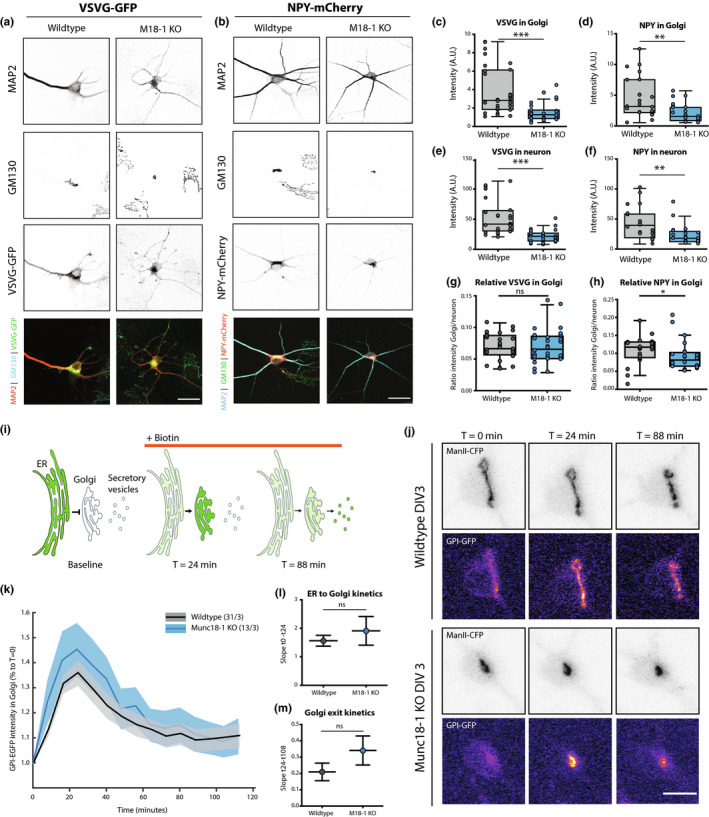
Anterograde transport through the Golgi is unaffected in Munc18‐1 KO neurons. (a) Typical example of WT and Munc18‐1 KO neurons over‐expressing VSVG‐GFP at DIV3, stained for MAP2 (dendritic marker), cis‐Golgi (GM130) and GFP. Scale bar is 25μm. (b) Typical example of WT and Munc18‐1 KO neurons over‐expressing NPY‐mCherry at DIV 3, stained for MAP2 (dendritic marker), cis‐Golgi (GM130) and mCherry. Scale bar is 25μm. (c) VSVG‐GFP intensity in the Golgi of Munc18‐1 KO neurons was ~60% lower (p < .0001, Mann–Whitney test). N: WT = 28/3, KO = 28/3. (d) Munc18‐1 KO neurons showed a ~55% reduction in NPY‐mCherry intensity in the Golgi area (p = .001, Mann–Whitney test). N: WT = 23/3, KO = 24/3. (e) VSVG‐GFP intensity in neuron was ~60% lower in Munc18‐1 KO neurons (p < .0001, Mann–Whitney test). N: WT = 28/3, KO = 28/3. (f) NPY‐mCherry intensity in Munc18‐1 KO neurons was ~45% lower (p = .0024, Mann–Whitney test). N: WT = 23/3, KO = 24/3. (g) Relative VSVG‐GFP distribution in the Golgi, measured by VSVG‐GFP intensity in Golgi (c) divided by total VSVG‐GFP intensity (E), was not different in Munc18‐1 KO neurons (ns p = .58, Mann–Whitney test). N: WT = 28/3, KO = 28/3. (h) Relative NPY‐mCherry distribution in the Golgi, measured by NPY‐mCherry intensity in Golgi (d) divided by total NPY‐mCherry intensity (f), was 15% lower in Munc18‐1 KO neurons (ns p = .026, Mann–Whitney test). N: WT = 23/3, KO = 24/3. Data is represented in Tukey boxplots. Columns and dots represent individual litters and neurons, respectively. (i) Cartoon representing RUSH assay. Under baseline conditions, GPI‐GFP‐SBP is bound to KDEL‐streptavidin and therefore retained in the ER. Upon biotin administration, the binding between SBP and streptavidin is reversed, thereby releasing the GPI‐GFP cargo into the secretory pathway. (j) Representative images of WT and Munc18‐1 KO DIV3 neurons expressing Golgi marker ManII‐CFP and GPI‐GFP. Biotin was added at T = 0. Scalebar is 10μm. (k) GPI‐GFP intensity in the Golgi of WT and Munc18‐1 KO neurons over time after biotin administration. Shown are mean and SEM. N: WT = 31/3, KO = 13/3. (l) GPI transport to the Golgi, measured as the fitted slope between t = 0 and the highest intensity, t = 24min, was not significantly different between WT and Munc18‐1 KO neurons (p = .42, F = 0.633, DFn = 1, DFd = 172, linear regression). N: WT = 31/3, KO = 13/3. (m) No differences were observed in Golgi exit, measured as the fitted slope between t = 24 and t = 112, between the two conditions (p = .19, F = 1.656, DFn = 1, DFd = 480, linear regression). N: WT = 31/3, KO = 13/3. N = number of neurons/number of independent cell cultures
To confirm this conclusion again quantitatively, relative expression levels overlapping with GM130 staining, were divided by the fluorescence intensity in the total neuron. For VSVG‐GFP, the relative levels in the Golgi were not different in Munc18‐1 KO neurons (Figure 5g) and ~15% lower for NPY‐mCherry (Figure 5h). Hence, overall expression levels of markers for the constitutive and regulated secretory pathway are lower in Munc18‐1 KO neurons, but no evidence was observed for Golgi retention of either marker.
The retention using selective hooks (RUSH) system was used to examine cargo trafficking in live neurons (Boncompain et al., 2012). GPI‐GFP, a marker of the constitutive secretory pathway, fused to Streptavidin binding protein (SBP) and the ER target peptide sequence KDEL fused to Streptavidin were expressed in Munc18‐1 KO and WT neurons. Under baseline conditions, SBP and Streptavidin bind, trapping GPI‐GFP in the ER. Biotin administration reverses the binding, allowing GPI‐GFP to travel through the secretory pathway (Figure 5i,j). Before adding biotin, GPI‐GFP was similarly localised in WT and Munc18‐1 KO neurons (Figure 5j, T = 0 min). Addition of biotin resulted in rapid translocation of GPI‐GFP to ManII‐CFP positive Golgi areas, reaching a maximum after 24 min (Figure 5k). No differences in the kinetics of ER to Golgi transport were detected between WT and Munc18‐1 KO neurons (Figure 5k,l). Subsequent Golgi exit resulted in decreasing GPI‐GFP intensity in the ManII‐CFP areas. Munc18‐1 KO neurons performed equally well in Golgi exit as WT neurons (Figure 5k–m). Taken together, these data indicate that the altered Golgi size and morphology in Munc18‐1 KO neurons does not affect ER‐to‐Golgi transport and Golgi exit, suggesting that anterograde trafficking in the two main secretory routes is normal in the absence of Munc18‐1.
3.6. Retrograde Golgi pathways are affected in Munc18‐1 KO neurons
Retrograde trafficking pathways from the endolysosomal system to the Golgi are important to replenish essential proteins and lipids to the Golgi, which is essential for survival (Bonifacino & Rojas, 2006). First, we examined the morphology of the endolysosomal pathway. LAMP1 is an integral component of lysosomes, and often used as a marker for lysosomes (Figure 6a) (Chen et al., 1988). LAMP1‐positive area and intensity in the soma were unaffected in Munc18‐1 KO neurons (Figure 6b,c). Staining for early endosome antigen 1 (EEA1), a widely used marker for early endosomes, showed a punctate pattern in soma and dendrites (Figure 6d). MUNC18‐1 depletion did not affect the number of EEA1 puncta nor the size of EEA1‐positive puncta (Figure 6e,f). These data indicate that, unlike the Golgi, endolysosomal morphology is normal upon deletion of MUNC18‐1 expression.
Figure 6.

Munc18‐1 KO neurons show reduced endosome‐to‐Golgi retrograde transport of Cholera Toxin B. (a) Typical example of WT and Munc18‐1 KO neurons at DIV3 stained for MAP2 (dendritic marker) and LAMP1 (lysosomes). Scale bar is 25 μm. (b) The relative LAMP1 area, measured as the LAMP1‐positive area over soma area, was not changed in Munc18‐1 KO neurons (ns p = .41, Mann–Whitney test). N: WT = 25/3. KO = 26/3. (c) No differences were observed in LAMP1 intensity between both conditions (ns p = .81, unpaired T test). N: WT = 25/3. KO = 26/3. (d) Typical example of WT and Munc18‐1 KO neurons at DIV 3 stained for MAP2 (dendritic marker) and EAA1 (early endosomes). Scale bar is 25 μm. (e) Number of EEA1‐positive puncta per μm2 was unchanged in Munc18‐1 KO neurons (ns p = .34, unpaired T test). N: WT = 30/3, KO = 29/3. (f) The size of EEA1‐positive puncta was comparable between the two conditions (ns p = .42, Mann–Whitney test). N: WT = 30/3, KO = 29/3. (g) Cartoon representing the CTB assay. After a 15‐min incubation of CTB, uninternalized CTB is washed out. Internalized CTB is allowed to retrogradely traffic to the Golgi for 2 hr. (h) Typical examples of WT and Munc18‐1 KO neurons fixed after the CTB assay. Neurons were stained for MAP2 (dendritic marker) and GM130&TGN46 (cis‐Golgi and TGN markers). (i) CTB intensity in Golgi was ~45% lower in Munc18‐1 KO neurons (p < .0001, Mann–Whitney test). N: WT = 35/3, KO = 35/3. (j) Total CTB internalized in Munc18‐1 KO neurons was ~30% lower (p < .0001, Mann–Whitney test). N: WT = 35/3, KO = 35/3. (k) Relative CTB distribution in the Golgi, measured by CTB intensity in Golgi (I) divided by total CTB intensity (J), was 20% lower in Munc18‐1 KO neurons (p = .004, unpaired T test). N: WT = 35/3, KO = 35/3. Data are represented in Tukey boxplots. Columns and dots represent individual litters and neurons, respectively. N = number of neurons/number of independent cell cultures
Cholera Toxin Subunit B (CTB) fused to Alexa‐488 was used to investigate retrograde transport (Figure 6g). CTB travels from the plasma membrane (PM) to early endosomes, recycling endosomes and the Golgi (Matsudaira et al., 2015; Wang et al., 2016; Wernick et al., 2010). After two hours of CTB incubation, the majority of the CTB was translocated from the PM to GM130/TGN46‐positive areas in the soma (Figure 6h). CTB intensity in these Golgi areas was ~45% lower in Munc18‐1 KO neurons than in WT neurons (Figure 6i). Total CTB‐Alexa‐488 internalized in Munc18‐1 KO neurons showed a ~30% reduction (Figure 6j). The relative distribution of CTB in the GM130/TGN46 positive areas, as calculated by the ratio of CTB‐Alexa‐488 fluorescence in GM130/TGN46‐positive areas over total CTB fluorescence, was ~20% lower in Munc18‐1 KO neurons (Figure 6k). Hence, retrograde transport of CTB to the Golgi is impaired in Munc18‐1 KO neurons.
3.7. Abnormal retrograde TrkB pathways in Munc18‐1 KO neurons
To further investigate the impaired retrograde pathways, we examined the retrograde endosomal pathway of TrkB using live labelling with a TrkB antibody (Barford et al., 2017; Budzinska et al., 2020; Carrodus et al., 2014). First, TrkB surface expression was assessed in fixed neurons prior to permeabilization. Mean TrkB surface labelling was similar in WT and Munc18‐1 KO neurons (Figure 7a,b). Next, live neurons were incubated with a TrkB antibody for 60 min at 37°C. To distinguish surface from internalized TrkB, both fractions were stained with different secondary antibodies (Figure 7c). In both WT and Munc18‐1 KO neurons a punctate pattern of internalized TrkB was observed (Figure 7d). The mean TrkB punctum intensity in Munc18‐1 KO neurons was ~30% and ~35% lower in the soma and dendrites, respectively (Figure 7e,f). Conversely, the number of TrkB puncta was ~20% higher in the soma of Munc18‐1 KO neurons, but not different in neurites (Figure 7g,h). Puncta size was increased by 15% and 10% in soma and dendrites, respectively (Figure 7i,j). The total pool of internalized TrkB, measured by multiplying punctum intensity by total puncta area, remained unaffected in Munc18‐1 KO neurons during the one‐hour antibody incubation (Figure 7k). These data together show that endocytosed TrkB puncta were bigger and contained less TrkB in Munc18‐1 KO neurons. Moreover the density of TrkB puncta was higher in the soma, but not in dendrites, while the total pool of internalized TrkB was unaffected. Together with the impaired retrograde CTB transport, we conclude that retrograde pathways are affected by MUNC18‐1 deficiency.
Figure 7.
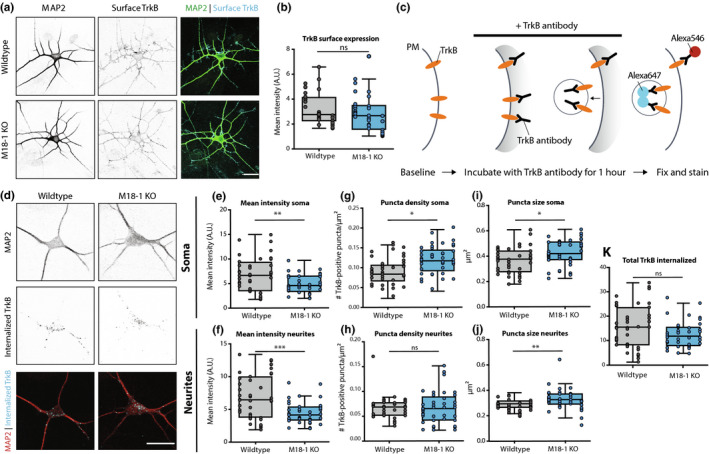
Retrograde TrkB transport is affected in Munc18‐1 KO neurons. (a) Typical example of WT and Munc18‐1 KO neurons at DIV3 stained for MAP2 (dendritic marker) and surface TrkB. Scale bar is 25 μm. (b) Average TrkB surface levels were not affected in Munc18‐1 KO neurons (ns p = .23, Mann–Whitney test). N: WT = 35/3. KO = 30/3. (c) Cartoon representing TrkB antibody uptake assay. Neurons were incubated with TrkB antibody for one hour, before they were fixed and stained. Remaining surface TrkB was stained with Alexa546, whereas internalized TrkB was stained with Alexa647. (d) Typical examples of WT and Munc18‐1 KO neurons fixed after the TrkB antibody uptake assay and stained for MAP2 (dendritic marker) and internalized TrkB (Alexa647). (e) Mean TrkB puncta intensity in the soma of Munc18‐1 KO neurons was ~30% lower (p = .003, unpaired T test). N: WT = 41/4. KO = 41/4. (f) Mean TrkB puncta intensity in neurites was ~35% lower in Munc18‐1 KO neurons (p = .0007, Mann–Whitney test). N: WT = 41/4. KO = 41/4. (g)Munc18‐1 KO neurons showed an ~20% increase in TrkB puncta density in the soma (p = .01, Mann–Whitney test). N: WT = 41/4. KO = 41/4. (h) TrkB puncta density was not different in neurites of Munc18‐1 KO neurons (ns p = .12, Mann–Whitney test). N: WT = 41/4. KO = 41/4. (i) Average TrkB puncta size in the soma of Munc18‐1 KO neurons was ~15% bigger (p = .01, unpaired T test). N: WT = 41/4. KO = 41/4. (j) In neurites of Munc18‐1 KO neurons, average TrkB puncta size was ~10% bigger (p = .12, Mann–Whitney test). N: WT = 41/4. KO = 41/4. (k) Total pool of internalized TrkB was not different between WT and Munc18‐1 KO neurons (p = .07, unpaired T test). N: WT = 41/4. KO = 41/4. Data are represented in Tukey boxplots. Columns and dots represent individual litters and neurons, respectively. N = number of neurons/number of independent cell cultures
4. DISCUSSION
In this study, we investigated the impact of MUNC18‐1 loss in cellular trafficking routes. We show that in absence of MUNC18‐1, neurons have a smaller cis‐, medial‐Golgi and TGN, while Golgi stack ultrastructure was normal. ER‐to‐Golgi and Golgi exit of markers in the constitutive and regulated secretory pathway were unaffected. Retrograde trafficking to the Golgi, however, was impaired.
4.1. Munc18‐1 KO neurons transform from healthy to death within hours
Munc18‐1 KO neurons at DIV3, that is, during the steepest part of the survival curve, and hours before most neurons have died (Santos et al., 2017), show a strikingly normal morphology when examined at the ultrastructural level. No cellular signs of degeneration, as described in previous studies on cell death (e.g. Taatjes et al., 2008), were observed. Hence, Munc18‐1 KO neurons transform from morphologically normal, healthy neurons to cell death within hours, with only a smaller Golgi/TGN as a morphological indicator of the nearing cell death. In addition, reduced expression of several, but not all proteins was detected at this stage (Figures 4 and 5), which is a common feature in other cell death models (e.g. Patel et al., 2002). Cell death in other cell types is known to also occur within hours, e.g. at a rate of 5% per hour (Wolbers et al., 2004), but such high rates are induced by lethal cytotoxic compounds or radiation (Forcina et al., 2017; Jessel et al., 2002; Wolbers et al., 2004). In conclusion, the transformation from normal, healthy neurons to death is remarkably fast in Munc18‐1 KO neurons. The cell autonomous defects caused by MUNC18‐1 loss, and probably also syntaxin‐1 or SNAP25 loss, trigger cell death with exceptionally fast kinetics, comparable to cytotoxic poisoning.
Reduced expression of proteins in Munc18‐1 KO neurons was not only observed in the present study. It was previously shown that depletion of MUNC18‐1 also leads to a 70% reduction in syntaxin‐1 levels (Toonen et al., 2005). Yet it is unlikely that reduced syntaxin‐1 levels account for the observed phenotypes, as STX1A KO or STX1B heterozygous mice show normal survival (Gerber et al., 2008; Wu et al., 2015). In addition, viability and Golgi morphology, but not syntaxin‐1 expression levels, are rescued upon MUNC18‐3 over‐expression in Munc18‐1 KO neurons (Santos & Wierda, 2016). Thus, the partial loss of syntaxin‐1 expression cannot explain the observed phenotypes in Munc18‐1 KO neurons.
4.2. MUNC18‐1 deficiency leads to atypical Golgi condensation without affecting Golgi functions
The earliest phenotype detected upon loss of MUNC18‐1 is a smaller and rounder Golgi area, suggesting condensation of the Golgi complex. However, Golgi stacking and ultrastructure cisternae morphology were not different. In addition, the average intensity of a Golgi‐resident marker was not increased in Munc18‐1 KO neurons. This is strikingly different from classical Golgi condensation phenotypes, where the round Golgi morphology is accompanied by swollen cisternae and/or accumulation of Golgi resident markers (Bard et al., 2003; Dippold et al., 2009; Lázaro‐Diéguez et al., 2006; Young et al., 2005). The clear absence of ultrastructure abnormalities places MUNC18‐1‐induced Golgi abnormalities in a novel category, distinct from typical Golgi condensation associated with cell death.
Golgi morphology and function are tightly linked. A previously published RNAi screen showed that 75% of the proteins that regulate Golgi morphology, also played essential roles in Golgi function (Chia et al., 2012). Since loss of MUNC18‐1 results in a severe reduction of Golgi membrane, it was anticipated that Golgi function would be disturbed. Remarkably, anterograde trafficking and Golgi exit were normal for multiple cargo markers in the constitutive and regulated secretory pathway in Munc18‐1 KO neurons. In addition, it is unlikely that glycosylation, one of the main functions of the Golgi, is affected, since glycosylation of TrkB is essential for its correct localisation in the cell (Mutoh et al., 2000; Watson et al., 1999) and TrkB distribution was normal in Munc18‐1 KO neurons. Hence, the atypical Golgi condensation upon MUNC18‐1 depletion does not affect Golgi‐intrinsic functions and Golgi‐export.
4.3. MUNC18‐1’s role in actin regulation does not explain Golgi abnormalities
The characteristic elongated morphology of the Golgi is maintained by a complex interplay between lipids and the actin cytoskeleton (Dippold et al., 2009). MUNC18‐1 has an established role in controlling actin organization via a hydrophobic residue in β‐sheet 10 (Pons‐Vizcarra et al., 2019). We showed that depolymerisation of actin in WT neurons by LatB treatment phenocopied the smaller and rounder Golgi area observed upon MUNC18‐1 loss, indicating that actin defects might explain the observed Golgi abnormalities. However, Golgi morphology was restored upon expression of a MUNC18‐1 mutant that fails to support a normal actin organization. Moreover previous studies investigating Golgi abnormalities upon actin depolymerisation reported perforation/fragmentation and severe swelling of Golgi cisternae (Lázaro‐Diéguez et al., 2006), and defects in VSVG transport (Dippold et al., 2009). This is in contrast with the observations in Munc18‐1 KO neurons in the present study; cisternae morphology was normal and VSVG transport was not affected. Hence, it is unlikely that defects in the actin cytoskeleton explain the Golgi abnormalities in Munc18‐1 KO neurons.
4.4. A potential role for MUNC18‐1 in endosomal SNARE‐dependent fusion
While anterograde transport was unaffected, two independent assays uncovered retrograde trafficking defects in Munc18‐1 KO neurons. First, retrograde transport of CTB from the PM to the Golgi was reduced in Munc18‐1 KO neurons. Second, retrograde TrkB trafficking in the soma was disturbed. Upon endocytosis, CTB and TrkB are both sorted to the endocytic pathway in Rab7‐positive organelles and transported back to the soma (Bhattacharyya et al., 1997; Ehlers et al., 1995; Wang et al., 2016). These organelles, referred to as signalling endosomes, engage interactors to enable downstream signalling pathways (Delcroix et al., 2003; Villarroel‐Campos et al., 2018). Once in the soma, the targeting of CTB and TrkB diverge. TrkB is either directed to lysosomes or to the plasma membrane for recycling (Barford et al., 2017), whereas CTB is targeted to the TGN and Golgi (Emperador‐Melero et al., 2018; Ragnarson et al., 1998). For both assays, it is unlikely that the defects are explained by impaired endocytosis: for CTB retrograde transport to the Golgi was impaired after normalizing for the total internalized CTB pool (Figure 6k), and in the TrkB assay the total pool of internalized TrkB was unaffected (Figure 7k). Hence, we conclude that the retrograde pathway defects occur after the initial endocytosis of CTB/TrkB. Indeed, perturbations in retrograde transport of signalling endosomes in Endophilin triple KO neurons or inhibition of dynein motor proteins result in similar cellular phenotypes and reduced survival, albeit less pronounced, as presented in this study (Budzinska et al., 2020; Burk et al., 2017). Thus, defects in retrograde transport of signalling endosomes‐to‐Golgi and/or ‐to‐recycling provide a plausible explanation for the observed abnormalities in CTB transport and retrograde TrkB trafficking.
These data suggest a role for MUNC18‐1 in endosomal transport in neurons. Since MUNC18‐1 is known for its function in catalysing SNARE‐dependent fusion, a defective fusion reaction in this pathway is the most plausible explanation for the observed abnormalities. Endosome‐to‐recycling and endosome‐to‐Golgi retrograde trafficking rely on multiple SNARE‐dependent fusion reactions (Mallard et al., 2002). In non‐neuronal cells, other dedicated S/M proteins have been described to operate in these fusion reactions, especially mVps45 (Tellam et al., 1997) and possibly also mSly1 (Laufman et al., 2009). VPS45 localises to the Golgi complex and peri‐Golgi structures, and is involved in homotypic late‐endosomal and endosome‐to‐Golgi fusion (Gengyo‐Ando et al., 2007; Rahajeng et al., 2009; Roccisana et al., 2013; Tellam et al., 1997). Knockdown (KD) of VPS45 in non‐neuronal cells produced strikingly similar cellular abnormalities (Rahajeng et al., 2009) as observed in the current study. In a comparable antibody uptake assay, using β1‐Integrin receptor internalisation instead of TrkB, VPS45 KD cells showed accumulation of endocytosed vesicles in the soma (Rahajeng et al., 2009). In agreement to our study, receptor internalization and early endosome formation were not affected (Rahajeng et al., 2009). In addition, KD of hVPS45 led to condensation of the cis‐Golgi and TGN (Rahajeng et al., 2009). In neurons, little is known about the function of mVps45. MUNC18‐1 may be involved in the same fusion reactions in neurons as mVps45 is in non‐neuronal cells, or in a distinct, neuron‐specific, retrograde pathway. Alternatively, its loss may indirectly affect the equilibrium in cellular levels or availability of other SNAREs and SNARE‐organisers in retrograde cellular trafficking, causing the observed retrograde pathway defects and Golgi abnormalities.
4.5. Defects in retrograde pathways can explain degeneration in Munc18‐1 KO brains
The impairments in endosomal fusion might not only provide an explanation for the Golgi abnormalities, but also for the fact that Munc18‐1 KO neurons die. Neuronal survival is critically regulated by neurotrophic signalling pathways (Reichardt, 2006). The present study showed a) reduced total levels of the neurotrophic receptor TrkB in Munc18‐1 KO neurons and b) disturbed retrograde trafficking of TrkB. Since surface and internalized TrkB levels were normal in Munc18‐1 KO neurons, the reduced total TrkB levels indicates reduced TrkB synthesis, increased turnover or depletion of intracellular (reserve) pools. This reduction in TrkB levels is, however, not sufficient to explain cell death, as TrkB heterozygous mice are viabile (Klein et al., 1993; Minichiello & Klein, 1996) despite reduced cellular TrkB levels to a similar extent as observed here. In contrast, correct retrograde signalling of TrkB is critical for neuronal survival (Burk et al., 2017; Reichardt, 2006). Comparable abnormalities in TrkB‐containing endosomes have been reported upon perturbations of the retrograde pathways, leading to defective neurotrophic signalling and reduced neuronal survival (Budzinska et al., 2020; Wan et al., 2008). In addition, enlarged signalling endosomes have directly been linked to neurodegenerative processes (Xu et al., 2016). It was previously shown that administration of BDNF to Munc18‐1 KO neurons delays the degeneration (Heeroma et al., 2004), strengthening the notion that neurotrophic signalling is involved in MUNC18‐1‐dependent viability. Together, this suggests that defective retrograde pathways in Munc18‐1 KO neurons result in several intracellular impairments, including neurotrophic signalling, ultimately leading to cell death.
5. CONFLICT OF INTEREST STATEMENT
The authors declare no competing financial interests.
ACKNOWLEDGMENTS
This work was supported by the European Union ERC Advanced Grant 322966 to M.V. and Horizon 2020 grant COSYN (RIA grant agreement no 610307, to M.V.) and by NWO Gravitation program BRAINSCAPES (NWO: 024.004.012). The authors thank Desiree Schut and Lisa Laan for preparing glia feeders and culturing neurons; Robbert Zalm for virus production and cloning; Joost Hoetjes and Joke Wortel for breeding and genotyping mutant mice. This study was posted as a preprint at BioRxiv: https://www.biorxiv.org/content/10.1101/2020.05.12.090811v1.
All experiments were conducted in compliance with the ARRIVE guidelines.
van Berkel AA, Santos TC, Shaweis H, van Weering JRT, Toonen RF, Verhage M. Loss of MUNC18‐1 leads to retrograde transport defects in neurons. J Neurochem.2021;157:450–466. 10.1111/jnc.15256
REFERENCES
- Bailey Blackburn, J. , Pokrovskaya, I. , Fisher, P. , Ungar, D. , & Lupashin, V. V. (2016). COG complex complexities: detailed characterization of a complete set of HEK293T cells lacking individual COG subunits. Front Cell Dev Biol, 4, 23. 10.3389/fcell.2016.00023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bard, F. , Mazelin, L. , Péchoux‐Longin, C. , Malhotra, V. , & Jurdic, P. (2003). Src regulates Golgi structure and KDEL receptor‐dependent retrograde transport to the endoplasmic reticulum. Journal of Biological Chemistry, 278, 46601–46606. 10.1074/jbc.M302221200 [DOI] [PubMed] [Google Scholar]
- Barford, K. , Deppmann, C. , & Winckler, B. (2017). The neurotrophin receptor signaling endosome: where trafficking meets signaling. Dev Neurobiol, 77, 405–418. 10.1002/dneu.22427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharyya, A. , Watson, F. L. , Bradlee, T. A. , Pomeroy, S. L. , Stiles, C. D. , & Segal, R. A. (1997). Trk receptors function as rapid retrograde signal carriers in the adult nervous system. Journal of Neuroscience, 17, 7007–7016. 10.1523/JNEUROSCI.17-18-07007.1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boncompain, G. , Divoux, S. , Gareil, N. , De, F. H. , Lescure, A. , Latreche, L. , Mercanti, V. , Jollivet, F. , Raposo, G. , & Perez, F. (2012). Synchronization of secretory protein traffic in populations of cells. Nature Methods, 9(5), 493–498. 10.1038/nmeth.1928 [DOI] [PubMed] [Google Scholar]
- Bonifacino, J. S. , & Rojas, R. (2006). Retrograde transport from endosomes to the trans‐Golgi network. Nature Reviews Molecular Cell Biology, 7, 568–579. 10.1038/nrm1985 [DOI] [PubMed] [Google Scholar]
- Budzinska, M. I. , Villarroel‐Campos, D. , Golding, M. , Weston, A. , Collinson, L. , Snijders, A. P. , & Schiavo, G. (2020). PTPN23 binds the dynein adaptor BICD1 and is required for endocytic sorting of neurotrophin receptors. Journal of Cell Science, 133(6), jcs242412. 10.1242/jcs.242412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burk, K. , Murdoch, J. D. , Freytag, S. , Koenig, M. , Bharat, V. , Markworth, R. , Burkhardt, S. , Fischer, A. , & Dean, C. (2017). EndophilinAs regulate endosomal sorting of BDNF‐TrkB to mediate survival signaling in hippocampal neurons. Scientific Reports, 7, 2149. 10.1038/s41598-017-02202-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrodus, N. L. , Teng, K.‐S.‐L. , Munro, K. M. , Kennedy, M. J. , & Gunnersen, J. M. (2014). Differential labeling of cell‐surface and internalized proteins after antibody feeding of live cultured neurons. Journal of Visualized Experiments, 84, e51139. 10.3791/51139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, J. , Cha, Y. , Yuksel, K. , Gracy, R. , & August, J. (1988). Isolation and sequencing of a cDNA clone encoding lysosomal membrane glycoprotein mouse LAMP‐1. Journal of Biological Chemistry, 263, 8754–8758. [PubMed] [Google Scholar]
- Chia, J. , Goh, G. , Racine, V. , Ng, S. , Kumar, P. , & Bard, F. (2012). RNAi screening reveals a large signaling network controlling the Golgi apparatus in human cells. Molecular Systems Biology, 8, 1–33. 10.1038/msb.2012.59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delcroix, J. D. , Valletta, J. S. , Wu, C. , Hunt, S. J. , Kowal, A. S. , & Mobley, W. C. (2003). NGF signaling in sensory neurons: evidence that early endosomes carry NGF retrograde signals. Neuron, 39, 69–84. 10.1016/S0896-6273(03)00397-0 [DOI] [PubMed] [Google Scholar]
- Delgado‐martı, I. , Nehring, R. B. , & Sørensen, J. B. (2007). Differential abilities of SNAP‐25 homologs to support neuronal function. Journal of Neuroscience, 27, 9380–9391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dippold, H. C. , Ng, M. M. , Farber‐Katz, S. E. , Lee, S. K. , Kerr, M. L. , Peterman, M. C. , Sim, R. , Wiharto, P. A. , Galbraith, K. A. , Madhavarapu, S. , Fuchs, G. J. , Meerloo, T. , Farquhar, M. G. , Zhou, H. , & Field, S. J. (2009). GOLPH3 bridges phosphatidylinositol‐4‐ phosphate and actomyosin to stretch and shape the Golgi to promote budding. Cell, 139, 337–351. 10.1016/j.cell.2009.07.052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehlers, M. D. , Kaplan, D. R. , Price, D. L. , & Koliatsos, V. E. (1995). NGF‐stimulated retrograde transport of trkA in the mammalian nervous system. Journal of Cell Biology, 130, 149–156. 10.1083/jcb.130.1.149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emperador‐Melero, J. , Huson, V. , van Weering, J. , Bollmann, C. , Fischer von Mollard, G. , Toonen, R. F. , & Verhage, M. (2018). Vti1a/b regulate synaptic vesicle and dense core vesicle secretion via protein sorting at the Golgi. Nature Communications, 9, 3421. 10.1038/s41467-018-05699-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farquhar, M. G. , & Palade, G. E. (1998). The Golgi apparatus: 100 years of progress and controversy. Trends in Cell Biology, 8, 2–10. 10.1016/S0962-8924(97)01187-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forcina, G. C. , Conlon, M. , Wells, A. , Cao, J. Y. , & Dixon, S. J. (2017). Systematic quantification of population cell death kinetics in mammalian cells. Cell Systems, 4, 600–610.e6. 10.1016/j.cels.2017.05.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gengyo‐Ando, K. , Kuroyanagi, H. , Kobayashi, T. , Murate, M. , Fujimoto, K. , Okabe, S. , & Mitani, S. (2007). The SM protein VPS‐45 is required for RAB‐5‐dependent endocytic transport in Caenorhabditis elegans . EMBO Reports, 8, 152–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerber, S. H. , Rah, J.‐C. , Min, S.‐W. , Liu, X. , de Wit, H. , Dulubova, I. , Meyer, A. C. , Rizo, J. , Arancillo, M. , Hammer, R. E. , Verhage, M. , Rosenmund, C. , & Südhof, T. C. (2008). Conformational switch of syntaxin‐1 controls synaptic vesicle fusion. Science, 321, 1507–1510. 10.1126/science.1163174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heeroma, J. H. , Roelandse, M. , Wierda, K. , Van, A. K. I. , Toonen, R. F. G. , Hensbroek, R. A. , Brussaard, A. , Matus, A. , & Verhage, M. (2004). Trophic support delays but does not prevent cell‐intrinsic degeneration of neurons deficient for munc18‐1. European Journal of Neuroscience, 20(3), 623–634. 10.1111/j.1460-9568.2004.03503.x [DOI] [PubMed] [Google Scholar]
- Horton, A. C. , & Ehlers, M. D. (2003). Dual Modes of endoplasmic reticulum‐to‐Golgi transport in dendrites revealed by live‐cell imaging. Journal of Neuroscience, 23, 6188–6199. 10.1523/JNEUROSCI.23-15-06188.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jessel, R. , Haertel, S. , Socaciu, C. , Tykhonova, S. , & Diehl, H. A. (2002). Kinetics of apoptotic markers in exogeneously induced apoptosis of EL4 cells. Journal of Cellular and Molecular Medicine, 6, 82–92. 10.1111/j.1582-4934.2002.tb00313.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein, R. , Nanduri, V. , Jing, S. A. , Lamballe, F. , Tapley, P. , Bryant, S. , Cordon‐Cardo, C. , Jones, K. R. , Reichardt, L. F. , & Barbacid, M. (1991). The trkB tyrosine protein kinase is a receptor for brain‐derived neurotrophic factor and neurotrophin‐3. Cell, 66, 395–403. 10.1016/0092-8674(91)90628-C [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein, R. , Smeyne, R. J. , Wurst, W. , Long, L. K. , Auerbach, B. A. , Joyner, A. L. , & Barbacid, M. (1993). Targeted disruption of the trkB neurotrophin receptor gene results in nervous system lesions and neonatal death. Cell, 75, 113–122. 10.1016/S0092-8674(05)80088-1 [DOI] [PubMed] [Google Scholar]
- Kovačević, J. , Maroteaux, G. , Schut, D. , Loos, M. , Dubey, M. , Pitsch, J. , Remmelink, E. , Koopmans, B. , Crowley, J. , Cornelisse, L. N. , Sullivan, P. F. , Schoch, S. , Toonen, R. F. , Stiedl, O. , & Verhage, M. (2018). Protein instability, haploinsufficiency, and cortical hyper‐excitability underlie STXBP1 encephalopathy. Brain, 141(5), 1350–1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laufman, O. , Kedan, A. , Hong, W. , & Lev, S. (2009). Direct interaction between the COG complex and the SM protein, Sly1, is required for Golgi SNARE pairing. EMBO Journal, 28, 2006–2017. 10.1038/emboj.2009.168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lázaro‐Diéguez, F. , Jiménez, N. , Barth, H. , Koster, A. J. , Renau‐Piqueras, J. , Llopis, J. L. , Burger, K. N. J. J. , & Egea, G. (2006). Actin filaments are involved in the maintenance of Golgi cisternae morphology and intra‐Golgi pH. Cell Motility and the Cytoskeleton, 63, 778–791. 10.1002/cm.20161 [DOI] [PubMed] [Google Scholar]
- Mallard, F. , Tang, B. L. , Galli, T. , Tenza, D. , Saint‐Pol, A. , Yue, X. , Antony, C. , Hong, W. , Goud, B. , & Johannes, L. (2002). Early/recycling endosomes‐to‐TGN transport involves two SNARE complexes and a Rab6 isoform. Journal of Cell Biology, 156, 653–664. 10.1083/jcb.200110081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsudaira, T. , Niki, T. , Taguchi, T. , & Arai, H. (2015). Transport of the cholera toxin B‐subunit from recycling endosomes to the Golgi requires clathrin and AP‐1. Journal of Cell Science, 128, 3131–3142. 10.1242/jcs.172171 [DOI] [PubMed] [Google Scholar]
- Minichiello, L. , & Klein, R. (1996). TrkB and TrkC neurotrophin receptors cooperate in promoting survival of hippocampal and cerebellar granule neurons. Genes & Development, 10, 2849–2858. 10.1101/gad.10.22.2849 [DOI] [PubMed] [Google Scholar]
- Mutoh, T. , Hamano, T. , Tokuda, A. , & Kuriyama, M. (2000). Unglycosylated Trk protein does not co‐localize nor associate with ganglioside GM1 in stable clone of PC12 cells overexpressing Trk (PCtrk cells). Glycoconjugate Journal, 17, 233–237. [DOI] [PubMed] [Google Scholar]
- Naldini, L. , Blomer, U. , Gallay, P. , Ory, D. , Mulligan, R. , Gage, F. , Verma, I. , & Trono, D. (1996). In vivo gene delivery and stable transduction of nondividing cells by a lentiviral vector. Science, 272, 263–267. 10.1126/science.272.5259.263 [DOI] [PubMed] [Google Scholar]
- Patel, J. , McLeod, L. E. , Vries, R. G. J. , Flynn, A. , Wang, X. , & Proud, C. G. (2002). Cellular stresses profoundly inhibit protein synthesis and modulate the states of phosphorylation of multiple translation factors. European Journal of Biochemistry, 269, 3076–3085. 10.1046/j.1432-1033.2002.02992.x [DOI] [PubMed] [Google Scholar]
- Pavelka, M. , & Ellinger, A. (2008). Retrograde plasma membrane‐to‐Golgi apparatus transport. The Golgi Apparatus: State of the Art 110 Years after Camillo Golgi’s Discovery (pp. 459–474). Springer‐Verlag Wien. [Google Scholar]
- Peng, L. , Liu, H. , Ruan, H. , Tepp, W. H. , Stoothoff, W. H. , Brown, R. H. , Johnson, E. A. , Yao, W. , Zhang, S. , & Dong, M. (2013). Cytotoxicity of botulinum neurotoxins reveals a direct role of syntaxin 1 and SNAP25 in neuron survival. Nature Communications, 4, 1412–1472. 10.1038/ncomms2462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persoon, C. M. , Moro, A. , Nassal, J. P. , Farina, M. , Broeke, J. H. , Arora, S. , Dominguez, N. , Weering, J. R. , Toonen, R. F. , & Verhage, M. (2018). Pool size estimations for dense‐core vesicles in mammalian <scp>CNS</scp> neurons. EMBO Journal, 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pons‐Vizcarra, M. , Kurps, J. , Tawfik, B. , Sørensen, J. B. , van Weering, J. R. T. , & Verhage, M. (2019). MUNC18‐1 regulates the submembrane F‐actin network, independently of syntaxin1 targeting, via hydrophobicity in β‐sheet 10. Journal of Cell Science, 132, jcs234674. 10.1242/jcs.234674 [DOI] [PubMed] [Google Scholar]
- Ragnarson, B. , Ornung, G. , Ottersen, O. P. , Grant, G. , & Ulfhake, B. (1998). Ultrastructural detection of neuronally transported choleragenoid by postembedding immunocytochemistry in freeze‐substituted Lowicryl HM20 embedded tissue. Journal of Neuroscience Methods, 80, 129–136. 10.1016/S0165-0270(97)00206-9 [DOI] [PubMed] [Google Scholar]
- Rahajeng, J. , Caplan, S. , & Naslavsky, N. (2010). Common and distinct roles for the binding partners Rabenosyn‐5 and Vps45 in the regulation of endocytic trafficking in mammalian cells. Experimental Cell Research, 316(5), 859–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reichardt, L. F. (2006). Neurotrophin‐regulated signalling pathways. Philosophical Transactions of the Royal Society of London. Series B, Biological Sciences, 361, 1545–1564. 10.1098/rstb.2006.1894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roccisana, J. , Sadler, J. B. A. , Bryant, N. J. , & Gould, G. W. (2013). Sorting of GLUT4 into its insulin‐sensitive store requires the Sec1/Munc18 protein mVps45. Molecular Biology of the Cell, 24, 2389–2397. 10.1091/mbc.e13-01-0011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos, T. , & Wierda, K. (2016) Early Golgi abnormalities and neurodegeneration upon loss of presynaptic proteins. [DOI] [PMC free article] [PubMed]
- Santos, T. C. , Wierda, K. , Broeke, J. H. , Toonen, R. F. , & Verhage, M. (2017). Early Golgi abnormalities and neurodegeneration upon loss of presynaptic proteins Munc18‐1, Syntaxin‐1, or SNAP‐25. Journal of Neuroscience, 37, 4525–4539. 10.1523/JNEUROSCI.3352-16.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiavo, G. , Poulain, B. , Rossetto, O. , Benfenati, F. , Tauc, L. , & Montecucco, C. (1992). Tetanus toxin is a zinc protein and its inhibition of neurotransmitter release and protease activity depend on zinc. EMBO Journal, 11, 3577–3583. 10.1002/j.1460-2075.1992.tb05441.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoch, S. (2001). SNARE function analyzed in synaptobrevin/VAMP knockout mice. Science, 294, 1117–1122. 10.1126/science.1064335 [DOI] [PubMed] [Google Scholar]
- Sudhof, T. C. (2013) Neurotransmitter Release : The Last Millisecond in the Life of a Synaptic Vesicle, 675–690. [DOI] [PMC free article] [PubMed]
- Taatjes, D. J. , Sobel, B. E. , & Budd, R. C. (2008). Morphological and cytochemical determination of cell death by apoptosis. Histochemistry and Cell Biology, 129, 33–43. 10.1007/s00418-007-0356-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tellam, J. T. , James, D. E. , Stevens, T. H. , & Piper, R. C. (1997). Identification of a mammalian Golgi Sec1p‐like protein, mVps45. Journal of Biological Chemistry, 272, 6187–6193. 10.1074/jbc.272.10.6187 [DOI] [PubMed] [Google Scholar]
- Toonen, R. F. G. , De, V. K. J. , Zalm, R. , & Su, T. C. (2005). Munc18‐1 stabilizes syntaxin 1, but is not essential for syntaxin 1 targeting and SNARE complex formation. Journal of Neurochemistry, 93(6), 1393–1400. [DOI] [PubMed] [Google Scholar]
- Toonen, R. F. , Kochubey, O. , De, W. H. , Gulyas‐kovacs, A. , Konijnenburg, B. , Sørensen, J. B. , Klingauf, J. , & Verhage, M. (2006). Dissecting docking and tethering of secretory vesicles at the target membrane. The EMBO Journal, 25(16), 3725–3737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vardar, G. , Chang, S. , Arancillo, M. , Wu, Y. , Trimbuch, T. , & Rosenmund, C. (2016). Distinct functions of syntaxin‐1 in neuronal maintenance, synaptic vesicle docking, and fusion in mouse neurons. Journal of Neuroscience, 36, 7911–7924. 10.1523/JNEUROSCI.1314-16.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varoqueaux, F. , Sigler, A. , Rhee, J. , Brose, N. , Enk, C. , Reim, K. , & Rosenmund, C. (2002). Total arrest of spontaneous and evoked synaptic transmission but normal synaptogenesis in the absence of Munc13‐mediated vesicle priming. Proceedings of the National Academy of Sciences, 99(13), 9037–9042. 10.1073/pnas.122623799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhage, M. , Maia, A. S. , Plomp, J. J. , Brussaard, A. B. , Heeroma, J. H. , Vermeer, H. , Toonen, R. F. , Hammer, R. E. , van den Berg, T. K. , Missler, M. , Geuze, H. J. , & Sudhof, T. C. (2000). Synaptic assembly of the brain in the absence of neurotransmitter secretion. Science, 287, 864–869. 10.1126/science.287.5454.864 [DOI] [PubMed] [Google Scholar]
- Villarroel‐Campos, D. , Schiavo, G. , & Lazo, O. M. (2018). The many disguises of the signalling endosome. FEBS Letters, 592, 3615–3632. 10.1002/1873-3468.13235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan, J. , Cheung, A. Y. , Fu, W. Y. , Wu, C. , Zhang, M. , Mobley, W. C. , Cheung, Z. H. , & Ip, N. Y. (2008). Endophilin B1 as a novel regulator of nerve growth factor/TrkA trafficking and neurite outgrowth. Journal of Neuroscience, 28, 9002–9012. 10.1523/JNEUROSCI.0767-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, T. , Martin, S. , Nguyen, T. H. , Harper, C. B. , Gormal, R. S. , Martínez‐Mármol, R. , Karunanithi, S. , Coulson, E. J. , Glass, N. R. , Cooper‐White, J. J. , van Swinderen, B. , & Meunier, F. A. (2016). Flux of signalling endosomes undergoing axonal retrograde transport is encoded by presynaptic activity and TrkB. Nature Communications, 7, 12976. 10.1038/ncomms12976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson, F. L. , Porcionatto, M. A. , Bhattacharyya, A. , Stiles, C. D. , & Segal, R. A. (1999). TrkA glycosylation regulates receptor localization and activity. Journal of Neurobiology, 39, 323–336. [DOI] [PubMed] [Google Scholar]
- Wernick, N. L. B. , Chinnapen, D.‐J.‐F. , Cho, J. A. , & Lencer, W. I. (2010). Cholera toxin: an intracellular journey into the cytosol by way of the endoplasmic reticulum. Toxins, 2, 310–325. 10.3390/toxins2030310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiertz, R. W. F. , Marani, E. , & Rutten, W. L. C. (2011). Neural cell–cell and cell–substrate adhesion through N‐cadherin, N‐CAM and L1. Journal of Neural Engineering, 8, 46004. 10.1088/1741-2560/8/4/046004 [DOI] [PubMed] [Google Scholar]
- Wolbers, F. , Buijtenhuijs, P. , Haanen, C. , & Vermes, I. (2004). Apoptotic cell death kinetics in vitro depend on the cell types and the inducers used. Apoptosis, 9, 385–392. 10.1023/B:APPT.0000025816.16399.7a [DOI] [PubMed] [Google Scholar]
- Wu, Y.‐J. , Tejero, R. , Arancillo, M. , Vardar, G. , Korotkova, T. , Kintscher, M. , Schmitz, D. , Ponomarenko, A. , Tabares, L. , & Rosenmund, C. (2015). Syntaxin 1B is important for mouse postnatal survival and proper synaptic function at the mouse neuromuscular junctions. Journal of Neurophysiology, 114, 2404–2417. 10.1152/jn.00577.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, W. , Weissmiller, A. M. , White, J. A. , Fang, F. , Wang, X. , Wu, Y. , Pearn, M. L. , Zhao, X. , Sawa, M. , Chen, S. , Gunawardena, S. , Ding, J. , Mobley, W. C. , & Wu, C. (2016). Amyloid precursor protein‐mediated endocytic pathway disruption induces axonal dysfunction and neurodegeneration. Journal of Clinical Investigation, 126, 1815–1833. 10.1172/JCI82409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young, J. , Stauber, T. , del Nery, E. , Vernos, I. , Pepperkok, R. , & Nilsson, T. (2005). Regulation of microtubule‐dependent recycyling at the trans‐golgi network by Rab6A and Rab6A’. Molecular Biology of the Cell, 16, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]