

Abstract
Following a request from the European Commission, the EFSA Panel on Nutrition, Novel Foods and Food Allergens (NDA) was asked to deliver an opinion on the safety of calcidiol monohydrate as a novel food (NF) pursuant to Regulation (EU) 2015/2283, including its bioavailability as a metabolite of vitamin D3 when added for nutritional purposes to food supplements. The NF is produced chemically. It is proposed in food supplements up to 10 μg/day for individuals ≥ 11 years of age, including pregnant and lactating women and up to 5 μg/day in 3‐ to 10‐year‐old children. The production process, composition, specifications and stability of the NF do not raise safety concerns. Animal and human data indicate efficient absorption. The NF contains a fraction of nanoparticles, which are fat soluble and unlikely to reach systemic distribution. There are no concerns regarding genotoxicity. Human adult studies do not raise safety concerns. Combined intake estimates of calcidiol from the NF and calcidiol and vitamin D from the diet were below the tolerable upper intake level (UL) for vitamin D for subjects above 11 years of age. The achieved mean serum 25(OH)D concentration in adults supplemented with 10 μg NF per day remained below 200 nmol/L. The Panel concludes that the NF is safe under the proposed conditions of use and use levels for individuals ≥ 11 years old, including pregnant and lactating women. The applicant did not provide data on the bioavailability and safety of the NF in children. The combined intake estimation in children (3–10 years) is close to the UL for vitamin D. Therefore, the Panel could not conclude on the safety of consumption of the NF in children (3–10 years) at the proposed daily intake. The NF is a bioavailable source of the biologically active metabolite of vitamin D, i.e. 1,25‐dihydroxyvitamin D.
Keywords: Novel Foods, calcidiol monohydrate, food supplement, safety, bioavailability, vitamin D
1. Introduction
1.1. Background as provided by the European Commission
On 16 May 2018, the company DSM Nutritional Products Ltd. submitted a request to the European Commission in accordance with Article 10 of Regulation (EU) 2015/2283 to place on the Union market calcidiol monohydrate produced by chemical synthesis as a novel food and to be added to the list of vitamin D forms specified in Annex II of Directive 2002/46/EC as a source of vitamin D.
The novel food is intended for use in food supplements targeting the general healthy population (including pregnant and lactating women), excluding children below the age of three. The proposed use level is 10 μg per day for children above 11 years of age and adults. For children from 3 to 10 years of age, the proposed use level is 5 μg per day.
The applicant has also requested data protection under Article 26 of Regulation (EU) 2015/2283.
1.2. Terms of Reference as provided by the European Commission
In accordance with Article 29(l)(a) of Regulation (EC) No 178/2002, the European Commission asks the European Food Safety Authority to provide a scientific opinion:
-
–
by carrying out the assessment for calcidiol monohydrate produced by chemical synthesis as a novel food in accordance with Article 10(3) of Regulation (EU) 2015/2283, and
-
–
following the outcome of the assessment of the safety of calcidiol monohydrate produced by chemical synthesis as a novel food, by carrying out the assessment of the bioavailability of calcidiol monohydrate as a metabolite of vitamin D3 when it is added for nutritional purposes to food supplements.
The European Commission asks the European Food Safety Authority to evaluate and inform the Commission as to whether and if so, to what extent, the requirements of Article 26(2)(c) of Regulation (EU) 2015/2283 are fulfilled in elaborating its opinion on calcidiol monohydrate regarding the proprietary data for which the applicant is requesting data protection.
1.3. Information on existing evaluations and authorisations
Vitamin D
-
–
The EFSA NDA Panel issued a scientific opinion on the dietary reference values (DRVs) for vitamin D (EFSA NDA Panel, 2016b ), which is the generic term for vitamin D 2 (ergocalciferol) and vitamin D 3 (cholecalciferol).1 In this opinion, the Panel set an adequate intake (AI) of 10 μg/day for infants aged 7–11 months, and of 15 μg/day under conditions of assumed minimal cutaneous vitamin D synthesis for the other population groups.2,3
-
–
The EFSA NDA Panel (2012) revised the previously set tolerable upper intake levels (ULs) for vitamin D (D2 and D3) (SCF, 2003). Based on two studies in men consuming vitamin D 3, the Panel proposed a UL of 100 μg/day vitamin D for adults. Based on two studies in pregnant or lactating women consuming vitamin D3 or vitamin D2 and D3, the Panel established that the UL of 100 μg/day for adults also applies to pregnant and lactating women. The Panel considered that ‘there is no reason to believe that adolescents in the phase of rapid bone formation and growth have a lower tolerance for vitamin D compared to adults’, thus extended to adolescents aged 11 to 17 years the UL of 100 μg/day set for adults. Taking into account their smaller body size, a UL of 50 μg/day was selected for children aged 1 to 10 years (EFSA NDA Panel, 2012). In a subsequent opinion considering vitamin D2 and D3, the Panel also kept the UL previously set by EFSA of 25 μg/day for infants aged up to 6 months and set a UL of 35 μg/day for infants 6–12 months (EFSA NDA Panel, 2018).
-
–
Regarding authorisations of ‘vitamin D substances’ in food, at the time of the adoption of this opinion, Directive 2002/46/EC on food supplements includes the following forms which may be used in the manufacture of food supplements as a source of vitamin D: vitamin D3 (‘cholecalciferol’) and vitamin D2 (‘ergocalciferol’).
25‐Hydroxycholecalciferol (calcidiol) monohydrate
-
–
Regarding consumption in humans:
-
o
‘Calcifediol’ (calcidiol) is used in the European Union (EU) as a human medicinal product, approved at EU Member State level4 and in some countries outside the EU.
-
o
-
–
Regarding consumption in animals:
-
o
Following the submission of a dossier by DSM Nutritional Products Ltd. (i.e. also the NF applicant), the EFSA FEEDAP Panel issued an opinion on the safety for target species, consumers, users and the environment and on the efficacy of the product of trade name ‘Hy•D (calcifediol)’ (25‐hydroxycholecalciferol monohydrate or 25(OH)D monohydrate, CAS No 63 283‐36-3) (EFSA, 2005). The FEEDAP Panel was able to set upper tolerable limits and maximum content of 25(OH)D in feed for chickens for fattening, laying hens and turkey. The product was approved as a feed additive (EC Number E 670a) in the EU according to Commission Regulation 1443/2006. When assessing the safety for the human consumer, the FEEDAP Panel suggested a provisional UL for 25(OH)D3 (10 μg/day in adults, 5 μg/day in children). This was estimated using a ‘biological activity factor’ relative to vitamin D3 of 5 applied to previous ULs for vitamin D of 50 μg/day in adults and 25 μg/day in children up to the age of 11 (European Commission, 2002; Institute of Medicine, 1997), i.e. before the update of the ULs for vitamin D by the EFSA NDA Panel (2012). The FEEDAP Panel considered that this ‘biological activity factor’ represented a ‘conservative approach’, considering data in rat (Blunt et al., 1968; Reeve et al., 1982) and chicken (related to calcium absorption, bone ash, plasma calcium, tibia ash and body weight). The ‘provisional ULs’ were confirmed by the FEEDAP Panel as provisional ULs in the following opinion of 2009 regarding the extended use of 25‐hydroxycholecalciferol monohydrate in the feed of poultry and pigs (EFSA, 2009). The FEEDAP Panel concluded that the exposure resulting from such use, at the proposed maximum doses, would not present a risk for the consumer. An assessment on the renewal of the authorisation of this product as a feed additive was ongoing at the time of the adoption of the present opinion of the NDA Panel.
-
o
Outside of the EU, 25‐hydroxycholecalciferol is generally recognized as safe (GRAS) for the feeding of broiler chicken in the United States5 and has been registered for the feeding of food producing animals (swine and poultry) in Latin America, Asia, Oceania and Africa.
-
o
2. Data and methodologies
2.1. Data
The safety assessment of this NF is based on data supplied in the application and information submitted by the applicant following EFSA requests for supplementary information.
During the assessment, the Panel identified additional data which were not included in the application.
Administrative and scientific requirements for NF applications referred to in Article 10 of Regulation (EU) 2015/2283 are listed in the Commission Implementing Regulation (EU) 2017/24696.
A common and structured format on the presentation of NF applications is described in the EFSA guidance on the preparation and presentation of a NF application.7 As indicated in this guidance, it is the duty of the applicant to provide all of the available (proprietary, confidential and published) scientific data, including both data in favour and not in favour of supporting the safety of the proposed NF.
This NF application includes a request for protection of proprietary data in accordance with Article 26 of Regulation (EU) 2015/2283. The data requested by the applicant to be protected comprise:
-
o
Annex 1 to Annex 16 of the dossier: Master data Calcifediol and product specifications; ADME studies comparing calcidiol and vitamin D3 (cholecalciferol) (Beck, 2016; Beck et al., 2017; Beck and Punler, 2017); Toxicity studies (Weber and Arcelin, 2004; Weber and Schulz, 2005; Thiel et al., 2007, 2014; Wöhrle and Sokolowski, 2013; Remus and Verbaan, 2016; Remus and Verspeek‐Rip, 2016); Human studies (Wittwer, 2015 and Kunz et al., 2016)
-
o
The analytical reports ‘Report DSM_EFSA_Calcifediol’ and its respective annexes (including detailed information on the particle size and dissolution kinetics and supporting annexes with a selection of representative micrographs, the addendum analytical report and respective annexes).
2.2. Methodologies
The assessment follows the methodology set out in the EFSA guidance on NF applications (EFSA NDA Panel, 2016a) and the principles described in the relevant existing guidance documents from the EFSA Scientific Committee. The legal provisions for the assessment are laid down in Article 11 of Regulation (EU) 2015/2283 and in Article 7 of the Commission Implementing Regulation (EU) 2017/2469.
This assessment concerns only risks that might be associated with consumption of the NF under the proposed conditions of use and is not an assessment of the efficacy of the NF with regard to any claimed benefit.
The evaluation of bioavailability of the NF, calcidiol monohydrate (25‐hydroxycholecalciferol monohydrate) in comparison with vitamin D3 was conducted in line with the principles contained in the ‘Guidance on safety evaluation of sources of nutrients and bioavailability of nutrient from the sources’ (EFSA ANS Panel, 2018). Regarding bioavailability, this guidance ‘aims to consider the relative bioavailability of the nutrient from the source compared under identical experimental conditions with the bioavailability of the nutrient in forms that are already permitted for use’. It is noted that the NF is a source of the biologically active form of vitamin D (i.e. 1,25‐dihydroxyvitamin D) and not a source of the nutrient ‘vitamin D’ as such.
3. Assessment
3.1. Introduction
The NF which is the subject of the application is calcidiol monohydrate (25‐hydroxycholecalciferol monohydrate, called ‘Calcidiol’ by the applicant).
The NF is produced from cholestatrienol by chemical synthesis and thus it falls within the class ‘chemical substances’ of the EFSA Guidance on novel food.7 The NF falls under the category ‘ix) Vitamins, minerals and other substances used in accordance with Directive 2002/46/EC, Regulation (EC) No 1925/2006 or Regulation (EU) No 609/2013’.
The NF is intended to be used in food supplements. The target population is adults including pregnant and lactating women, and children above 3 years of age.
The Panel notes that the NF contains the monohydrate form of the major circulating metabolite of vitamin D3 in the body and is a source of 1,25‐dihydroxyvitamin D, the biologically active form of vitamin D.
The applicant intends to market the NF as a diluted form called ‘0.25% w/w’ or ‘Calcidiol 0.25% SD/S’. The Panel notes that this formulation contains values in the range of 0.250–0.275% w/w of calcidiol (anhydrous), hence that 0.25% w/w is only the lower bound of the content range.
In view of the above, and for consistency with the naming in the dossier submitted and in the mandate received, these two products are further called in the opinion the NF (‘Calcidiol’) and ‘0.25% w/w formulation’ with inverted commas, while calcidiol is the non‐hydrated form which is referred to oral consumption of 25‐hydroxyvitamin D in general and 25(OH)D refers to the serum concentration of the molecule (in line with the previous scientific opinions of the NDA Panel on vitamin D).
3.2. Identity of the NF
The applicant has provided the following information on the identity of the product.
Table 1.
Chemical identity of the NF provided by the applicant
| Chemical substance | |
|---|---|
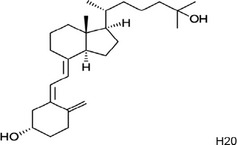
| Chemical name according to IUPAC | (1S,3Z)‐3‐[(2E)‐2‐[(1R,3αS,7αR)‐1‐[(2R)‐6‐hydroxy‐6‐methylheptan‐2‐yl]‐7α‐methyl‐2,3,3α,5,6,7‐hexahydro‐1H‐inden‐4‐ylidene]ethylidene]‐4‐methylidenecyclohexan‐1‐ol; hydrate |
| Synonyms, trade names, abbreviations | Common synonyms: 25‐Hydroxycholecalciferol monohydrate; Calcifediol, 25‐OH‐D3, 25(OH)D3; 25‐hydroxy vitamin D3Synonyms found in various reports: HyD®, Calcifediol, photoconversion HD3 crystal FG, photoconversion 25‐OH‐D3 crystal FG |
| CAS Number: | 63283‐36‐3 (Calcifediol monohydrate) |
| Empirical formula: | C27H44O2.H2O |
| Structural formula (as proposed by applicant): |
|
| Molecular weight | 418.7 g/mol |
The identity of the NF has been demonstrated by infrared (IR) and high‐performance liquid chromatography–ultraviolet detection (HPLC‐UV, 254 nm) by comparison with an authentic specimen.
The NF has a particulate nature and the morphology and number‐based size distribution of the particles have been characterised by means of transmission electron microscopy‐energy dispersive X‐ray analysis (TEM‐EDX), in accordance with the ISO norm 21363:2020, and scanning electron microscopy (SEM). The NF is a polydisperse material composed of small irregular particles with a median length, width and thickness of 541 nm, 333 nm and 264 nm, respectively. The D108 of the length, width and thickness are 201 nm, 144 nm and 108 nm, respectively. The NF contains a fraction of nanoparticles (the minimum length, width and thickness being 111 nm, 76 nm and 19 nm, respectively).
The Panel notes that the applicant has addressed the requirements and provided additional information in line with the up‐to‐date state of the science (as reflected in the draft EFSA Guidance on Technical requirements for regulated food and feed product applications to establish the presence of small particles including nanoparticles (EFSA Scientific Committee, 2020 ‐currently under finalisation9).
3.3. Production process
According to the information provided, the NF is produced according to Good Manufacturing Practice (GMP) conditions and Hazard Analysis Critical Control Points (HACCP) principles.
The first step of the production process of the NF is a yeast fermentation utilising a genetically engineered strain of Saccharomyces cerevisiae. This yeast is categorised by EFSA as a microorganism with Qualified Presumption of Safety (QPS) status (EFSA BIOHAZ Panel, 2013). The applicant provided a detailed description of the genetic modification steps applied to the parental strain of S. cerevisiae to obtain the derivative strain used to produce the NF.
The yeast fermentation results in a mixture of sterols, with trienol being the major sterol obtained. After the fermentation, purification and several chemical steps follow, resulting in a crude NF after crystallisation.
The chemical steps include saponification and extraction, where the trienol is isolated from the biomass. This is followed by a hydroxylation step to separate the trienol from the other sterols. Trienol is then epoxidised and subsequently reduced to give 25‐hydroxydehydrocholesterol (Hy‐DHC).
A photoreaction follows, to obtain a mixture of 25‐hydroxy‐previtamin D3, 25‐hydroxy‐tachysterol and small amounts of 25‐hydroxy‐lumisterol. Thereafter, the 25‐hydroxy‐previtamin D3 is thermally isomerised to ‘Calcidiol’ and recrystallised to obtain the NF of the required purity.
The applicant intends to market the NF as ‘0.25% w/w’ (see Section 3.1), containing 0.25–0.275% w/w of calcidiol (anhydrous), in a matrix consisting of several constituents complying with EU Regulation 1333/2008 in order to provide a more stable product.
Upon EFSA's request, the applicant provided additional data on the production process steps, including clarifications on the microorganism used in the fermentation step and absence of viable cells. The applicant provided evidence on the absence of DNA in the NF and absence of viable cells from the final product and data to demonstrate the validity of the method applied, in accordance with the EFSA Guidance on the characterisation of microorganisms used as feed additives or as production organisms (EFSA FEEDAP Panel, 2018).
The Panel considers that the production process is sufficiently described and does not raise safety concerns.
3.4. Compositional data
The NF consists of calcidiol monohydrate. In order to confirm that the manufacturing process is reproducible and adequate to produce the NF on a commercial scale and in accordance with the specifications, the applicant provided analytical information for four batches (from which 2 consecutive and 2 non‐consecutive batches) of the NF (see Table 2).
Table 2.
Batch‐to‐batch analysis of the NF
| Parameter | Method of analysis | Batch number | |||
|---|---|---|---|---|---|
| #1 | #2 | #3 | #4 | ||
| Appearance | Visual | White | White | White | White |
| Identity | Infrared spectrometry | Corresponds to specifications | Corresponds to specifications | Corresponds to specifications | Corresponds to specifications |
| Calcidiolj 25(OH)D3.H2O | HPLC/UVa | 100.6% | 101.3% | 101.1% | 102.2% |
| ∆22‐25(OH)D3 | HPLC/UV | 0.2% | 0.2% | 0.3% | 0.2% |
| 25(OH) lumisterol(f) | HPLC‐MSc | 0.14% | 0.14% | 0.1% | 0.1% |
| pre‐25(OH)D3 (g) | HPLC‐MS | 0.14% | 0.14% | 0.1% | 0.1% |
| iso‐25(OH) Tachysterol(h) | HPLC‐MS | 0.14% | 0.14% | 0.1% | 0.1% |
| trans‐Vitamin D3 (i) | HPLC‐MS | 0.14% | 0.14% | 0.1% | 0.1% |
| Other impurities | HPLC‐MS | 0.09% | 0.09% | 0.10% | 0.10% |
| Water content | Karl Fisher | 4.4% | 4.4% | 4.9% | 4.5% |
| Acetone mg/kg | GC‐HS‐FIDd | 840 | 821 | 789 | 840 |
| Isopropanol mg/kg | GC‐HS‐FID | NDb | NDb | NDb | NDb |
| Heavy metals | |||||
| Arsenic mg/kg | ICP‐MSe | < 1.0 | < 1.0 | < 1.0 | < 1.0 |
| Lead mg/kg | ICP‐MS | < 1.0 | < 1.0 | < 1.0 | < 1.0 |
| Mercury mg/kg | ICP‐MS | < 0.1 | < 0.1 | < 0.1 | < 0.1 |
| Cadmium mg/kg | ICP‐MS | < 0.5 | < 0.5 | < 0.5 | < 0.5 |
HPLC/UV: High performance liquid chromatography/Ultraviolet Spectroscopy.
ND: Not Detectable.
HPLC–MS: High‐performance liquid chromatography/mass spectrometry.
GC‐HS‐FID: Gas Chromatography with Headspace‐Sampler and Flame Ionisation Detector.
ICP‐MS: Inductively coupled plasma/mass spectrometry.
Impurity: 9β,10α‐cholesta‐5,7‐diene‐3β,25‐diol.
Impurity: Cholesta‐5,7‐diene‐3β,25‐ diol.
Impurity: (6E)‐9,10‐secocholesta‐5(10),6,8‐triene‐3β,25‐diol (iso‐ 25(OH).
Impurity: (5E,7E)‐9,10‐secocholesta‐5,7,10(19)‐triene‐3β,25‐diol.
Values exceeding 100% are due to technical variabilities.
The NF has been analysed according to the specifications and methods for calcifediol as described in United States and/or European Pharmacopeia.
The applicant states that all the ingredients used in the commercial ‘0.25% w/w’ preparation of the NF (sodium ascorbate, d,l‐alpha‐tocopherol, medium chain triglycerides, sucrose, modified starch and silicon dioxide) are in compliance with the Food Additive Regulation (EC) No 1333/200810. Each batch of the NF included in the preparation is tested for all the parameters listed in the specifications of the NF, prior to the manufacture of the commercial preparation. In response to the request of additional information from EFSA, the applicant provided the certificate of analysis of four independent batches of the ‘0.25% w/w’ commercial preparation.
Information was provided on the accreditation of the laboratories that conducted the analyses presented in the application.
The Panel considers that the information provided on the composition is sufficient for characterising the NF.
3.4.1. Stability
The applicant used for the stability tests the NF and the ‘0.25% w/w’ preparation.
The applicant performed stability tests with three independently produced batches of the crystalline NF. The tests were carried out at 5°C for a period of 12 months and at 25°C, at 60% relative humidity (RH) for a period of 6 months. The batches were kept in closed aluminium bottles.
The stability tests with the food preparation ‘Calcifediol 0.25% SD/S’ (i.e. ‘0.25% w/w’) were performed under the following conditions: temperature at 15°C and at 25°C, RH 60% for 18 months and at 40 °C, RH 75% for six months. As expected, stability decreased at higher temperatures (e.g. calcidiol, starting from an output value of 0.271%, after 6 months at storage < 15°C resulted in 0.268% and after 6 months at 40°C, 0.257%).
Upon EFSA's request, the applicant provided additional data up to 36 months at 15 and 25°C for both the NF and the 0.25% w/w preparation. The following parameters were tested for the NF: appearance, identity, assay, water content and the various impurities. Appearance, colour, dispersibility at 20°C, loss on drying and assay were tested for the preparation (results not reported, confidential).
The Panel considers that the data provided sufficient information with respect to the stability of the NF.
3.5. Specifications
The applicant describes the NF appearance as white to almost white crystals. The specifications of the NF are indicated in Table 3.
Table 3.
Specifications of the NF
| Parameter | Specification |
|---|---|
| 25(OH)D3.H2O | 97.0–100% |
| Total related substances: | ≤ 1.5% |
| ∆22‐25(OH)D3 | ≤ 0.5% |
| Lumisterol(a) | ≤ 0.5% |
| pre‐25(OH)D3 (b) | ≤ 0.5% |
| Tachysterol(c) | ≤ 0.5% |
| trans‐Vitamin D3 (d) | ≤ 0.5% |
| Other impurities | ≤ 0.10% |
| Water content | 3.8–5.0% |
| Acetone (mg/kg) | ≤ 1,000 |
| Isopropanol (mg/kg) | ≤ 500 |
| Heavy metals | |
| Arsenic (mg/kg) | ≤ 1.0 |
| Lead (mg/kg) | ≤ 1.0 |
| Cadmium (mg/kg) | ≤ 0.5 |
| Mercury (mg/kg) | ≤ 0.1 |
9β,10α‐Cholesta‐5,7‐diene‐3β,25‐diol (25(OH)).
Cholesta‐5,7‐diene‐3β,25‐diol.
(6E)‐9,10‐Secocholesta‐5(10),6,8‐triene‐3β,25‐diol (iso‐25(OH)).
(5E,7E)‐9,10‐Secocholesta‐5,7,10(19)‐triene‐3β,25‐diol.
Upon EFSA's request, the applicant clarified that the parameters for the NF (e.g. the impurities) are tested according to the European and US Pharmacopeia (EP and USP). In addition to the parameters from the monographs of these pharmacopeia, the applicant considered key parameters to ensure consistent manufacturing process and quality (e.g. residual solvents, heavy metals).
The applicant did not provide microbiological specifications for the NF. Instead, microbiological specifications were provided for the 0.25% w/w preparation (Table 4).
Table 4.
Proposed microbiological criteria for the ‘0.25% w/w’ preparation of the NF
| Parameter | Specification |
|---|---|
| Microbiological | |
| TAMC | ≤ 103 CFU/g |
| TYMC | ≤ 102 CFU/g |
| Enterobacteria | < 10 CFU/g |
| Escherichia coli | Not detected in 10 g |
| Salmonella spp. | Not detected in 25 g |
| Staphylococcus aureus | Not detected in 10 g |
| Pseudomonas aeruginosa | Not detected in 10 g |
TAMC: total aerobic microbial count; TYMC: total yeast and mould count; CFU: colony forming units.
The applicant's rationale for not including microbiological criteria in the specifications of the NF is that the pristine crystalline ‘Calcidiol’ (the NF) will not be commercialised as such, but will always be formulated into a nutrient preparation as defined in the preamble of Annex III of Regulation (EU) No 1333/2008.
The Panel considered the microbiological results for the 0.25% w/w preparation to be within acceptable limits. When considering the production process, the provided batch testing, the specifications proposed for the 0.25% w/w preparation, and the proposed uses, the Panel considers that microbiological specifications are not needed for the NF.
The 0.25% w/w preparation containing the NF is a fine, free flowing powder with an off‐white to yellowish colour containing calcidiol (according to a Calcifediol anhydrous assay) in the range of 0.25–0.275% w/w. The applicant also specified the presence of DL‐alpha‐tocopherol and sodium ascorbate in the commercial preparation of the NF.
The Panel considers that the information provided on the specifications of the NF is sufficient and does not raise safety concerns.
3.6. History of use of the NF and/or of its source
The NF (25‐hydroxycholecalciferol monohydrate) has no history of use in the EU for the proposed use as a food supplement.
As indicated in Section 1.3 ‘Information on existing evaluations and authorisations’, 25‐hydroxycholecalciferol monohydrate is approved as a feed additive in the EU (EFSA 2005, 2009) and in some countries outside the EU. Hence, 25‐hydroxycholecalciferol may be present in foods of animal origin (see Section 3.7.3).
In addition, calcidiol may be used in human medicinal products in the EU. The Eudravigilance database indicates that cases of hypervitaminosis D have been observed in subjects that had consumed this medicinal product, at doses (when reported) higher than the daily intake of the NF proposed by the applicant (see Section 3.7.2), e.g. about 266–270 μg/day.4
3.7. Proposed uses and use levels and anticipated intake
3.7.1. Target population
The target population of the NF as proposed by the applicant is adults including pregnant and lactating women, and children except those below the age of 3 years.
3.7.2. Proposed uses and use levels
The applicant intends to market the NF as a new substance to be used in the manufacture of food supplements as an alternative to the already authorised forms of vitamin D, i.e. vitamin D3 (cholecalciferol) and vitamin D2 (ergocalciferol) in accordance with Directive 2002/46/EC). The applicant intends to market the NF as a preparation containing 0.25–0275% w/w calcidiol.
The proposed maximum daily intake is 10 μg of the NF per day for children aged 11 years and above, as well as for adults including pregnant and lactating women. For children of age 3–10 years, the proposed maximum daily intake of the NF is 5 μg/day.
3.7.3. Combined intake from the NF and other sources
Vitamin D intake from the diet (D2 or D3), sun exposure and calcidiol intake from some foods of animal origin (EFSA NDA Panel, 2016b) may contribute to total serum 25(OH)D concentrations in the population. Hence, serum 25(OH)D concentrations (for which results from specific studies are discussed in Section 3.9) may be considered as the outcome of the combined influence of the intake of vitamin D and of calcidiol and of sun exposure. Cashman et al. (2012) showed that the range of mean serum 25(OH)D concentrations in 14 European studies in children and adult populations, measured following the protocol of the Vitamin D Standardization Program, was 38.3–65 nmol/L (EFSA NDA Panel, 2016b).
In foods, calcidiol may be present as 25‐hydroxycholecalciferol (25(OH)D3) or 25‐hydroxyergocalciferol (25(OH)D2). Contents of 25‐hydroxycholecalciferol per 100 g have been reported in milk and butter,11 eggs,12 fish,13 meat products14 and offal.15 Contents of 25‐hydroxyergocalciferol per 100 g have been reported in whole milk and butter,16 and in some meat products and offal.17
The FEEDAP Panel (EFSA 2009) presented a scenario for the daily exposure of the consumer to 25(OH)D3 from consumption of pig and poultry containing 25(OH)D3 concentrations that result from feeding of animals with feed containing the highest recommended 25(OH)D3 concentrations. The scenario included daily consumption of muscle from pig and poultry, liver from piglets, kidney from poultry, skin/fat from chicken and eggs, with portion sizes based on consumption data from a scientific cooperation among EU MS (European Commission, 2004). The estimated total intake for the consumer of such products was 2.44 μg 25(OH)D3/day.
A combined intake of 25‐hydroxycholecalciferol from the NF and from the background diet according to this calculation of the FEEDAP Panel could theoretically amount to 12.44 μg/day for adults and children aged 11 years and above, and to 7.44 μg/day for children aged 3–10 years.
3.8. Absorption, distribution, metabolism and excretion (ADME)
The applicant provided a narrative review by Borel et al. (2015) which reports in particular on a study in healthy men (20–35 year), in which the total amount of radioactivity recovered in plasma was higher after [3H]25(OH)D3 oral administration than after [3H]vitamin D3 intake (means 6 h post‐dose were 47–49% of total radioactive dose given vs about 13%, respectively). A higher percentage of radioactivity was found in the chylomicron fraction after ingestion of [3H]vitamin D3 than after ingestion of [3H]25(OH)D3 (means of 18% vs about 5–6%, respectively) (Compston et al., 1981). The data suggest that absorption of vitamin D3 occurs via chylomicrons, while that of 25(OH)D3 occurs via the portal route.
Vitamin D from the diet or from cutaneous synthesis is hydroxylated in the liver to 25(OH)D, which is hydroxylated primarily in the kidneys to the biologically active metabolite 1,25‐dihydroxyvitamin D (Section 1.3). Of the two metabolites of vitamin D, the major circulating form is 25(OH)D, with a longer mean half‐life, of about 13–15 days. 25(OH)D is taken up from the blood into tissues, including the adipose tissue, muscle and liver for storage. The two main pathways of degradation are the C23 lactone pathway and the C24 oxidation pathway. Vitamin D metabolites 25(OH)D and 1,25(OH)2D in the body are degraded in an oxidative pathway involving stepwise side‐chain modifications by the actions of CYP24A1 (24‐hydroxylase): their catabolism involves inactivation by 24‐hydroxylation, which gives rise initially to 24,25(OH)2D and to 1,24,25(OH)3D (i.e. 1,24,25‐trihydroxyvitamin D, then leading to calcitroic acid). After several steps, one of the final products of the C24 oxidation pathway, i.e. calcitroic acid, is excreted, mainly in the bile and thus in the faeces. Human CYP24A1 also catalyses, although to a lesser extent, the 23‐hydroxylation of both 25(OH)D and 1,25(OH)2D leading, in sequential steps, to 25(OH)D‐26,23‐lactone and 1,25(OH)2D‐26,23‐lactone, respectively (EFSA NDA Panel, 2016b).
In addition, the Panel notes that the NF is composed of small particles and contains a fraction of nanoparticles (see Section 3.2), which are insoluble in water. The applicant provided an in vitro study simulating human gastrointestinal (GI) digestion, which showed that these particles do not quickly dissolve in the GI tract,18 implying that a fraction may reach the human intestine as particles. However, if the NF is absorbed at least partly as small particles or nanoparticles, they are expected to partition and quickly solubilise into the lipophilic compartments, suggesting that systemic distribution of particles is unlikely to occur.
3.8.1. Data on bioavailability in humans
Taking into account the methodological approach of the EFSA guidance on bioavailability (Section 2.2) and the particular nature of the NF (Section 1.3), the Panel compared the effect of oral calcidiol and oral vitamin D3 (which is an authorised source of vitamin D according to Directive 2002/46/EC) on serum/plasma 25(OH)D concentration (the accepted biomarker of vitamin D status).
In a study on patients requiring treatment with vitamin D for a number of conditions e.g. deficiency, X‐linked hypophosphataemic rickets or osteomalacia, osteoporosis and hypoparathyroidism, and healthy volunteers, a higher bioavailability was shown of oral 25(OH)D3 (for at least six weeks) than of vitamin D2 and D3 (for at least 4 months). Consumption of 25(OH)D3 in daily dosage of 50 and 100 μg produced mean 25(OH)D concentrations (measured by a radio‐competitive assay) which were respectively 5 and 6 times higher than with vitamin D (Stamp et al., 1977).
In another study discussed by the applicant, on three healthy volunteers who received orally 14C‐vitamin D3 and 3H‐25(OH)D3, plasma maximum radioactivity after single dose administration was higher and was reached faster with calcidiol (6–10 h post‐dose) than with vitamin D3 (8–10 h post‐dose) (Haddad and Rojanasathit, 1976).
The applicant provided six parallel‐design, human intervention studies (Barger‐Lux et al., 1998; Bischoff‐Ferrari et al., 2012; Jetter et al., 2014; Cashman et al., 2012; Navarro‐Valverde et al., 2016; Wittwer, 2015; Vaes et al., 2018; Kunz et al., 2016).19 They investigated the effectiveness of (daily) oral doses of vitamin D3 or calcidiol in raising serum 25(OH)D in men and/or not‐pregnant and not‐lactating women, with or without considering background vitamin D intake or sun exposure, at latitudes relevant for Europe (37–51°N), and generally excluding users of vitamin D supplements. Among them, four of the six studies were funded by the applicant, either with sponsorship or study materials (Cashman et al., 2012; Bischoff‐Ferrari et al., 2012; Jetter et al., 2014; Wittwer, 2015; Vaes et al., 2018; and Kunz et al., 2016) (see Section 3.10.5). The applicant developed a commercial preparation of the NF to be used in the clinical trials. Upon EFSA's request, the applicant clarified that the products used in the clinical studies have the same composition as the ‘0.25 % w/w formulation’ (see Sections 3.1, 3.3 and 3.4), i.e. the commercial preparation for which analysis on 4 batches were provided in this application (data not shown, see explanations in Section 3.4).
Upon request from EFSA, the applicant provided the outcome of a literature search on calcidiol or calcidiol monohydrate given as a supplement or treatment to humans (healthy or with a disease, all ages including preterm infants, with single or repeated doses), in Scopus and Chemical Abstracts in March 2013 and with a particular focus on children, pregnant and lactating women, in Pubmed in April 2021. From this, no additional paper was found that the applicant would consider relevant for this dossier.
A detailed description of the six studies is presented in Appendix A and they are also addressed in Sections 3.9 and 3.10.5. Parameters possibly influencing exposure or the level of confidence in the reported values for exposure and the biomarker are also included, i.e. whether compliance was checked, latitude and season of the study, whether the dose administered was analytically checked, the conditions of consumption e.g. with a meal or not, the form of the supplement i.e. capsules or drops, the background vitamin D intake, whether or not vitamin D supplement users or those going on sunny holidays were excluded, and details of the analytical method and quality control to measure serum 25(OH)D.
Daily amounts of oral calcidiol ranged from 5 to 50 μg/day. All studies investigated generally healthy adults aged more than 50 years, except one study in younger adults (20–37 years) (Barger‐Lux et al., 1998). Study samples ranged between 20 and 116 subjects. Duration of supplementation ranged from 4 to 52 weeks. Baseline mean serum 25(OH)D concentration by study groups ranged from about 30 to 67 nmol/L. Mean body mass index (BMI) in the groups was about 25–28 kg/m2.
Among the limitations of some of these studies, the Panel notes e.g.:
Two were open‐label (Barger‐Lux et al., 1998; Navarro‐Valverde et al., 2016).
Vitamin D intake from the background diet was assessed in only two studies (Cashman et al., 2012; Wittwer, 2015 (Study report – proprietary unpublished data) published in Vaes et al., 2018).
Check of the compliance was sometimes not mentioned (Navarro‐Valverde et al., 2016).
Analytical check of the doses of oral 25(OH)D administered was sometimes not mentioned (Barger‐Lux et al., 1998; Navarro‐Valverde et al., 2016; Unpublished proprietary study report Kunz et al., 2016).
One study did not use the same analytical method to measure baseline and final serum 25(OH)D concentrations (Unpublished proprietary study report Wittwer, 2015 published in Vaes et al., 2018).
Furthermore, comparisons between studies were complicated by the fact that the season when the trial took place was either unspecified, corresponded to winter (hence to low cutaneous vitamin D synthesis) or covered sunny seasons, so that the level of endogenous cutaneous vitamin D synthesis, influencing serum 25(OH)D concentrations, was probably not similar between studies.
Taking the characteristics and possible limitations of these studies into account, the Panel considers that these human intervention studies showed that oral administration of 25(OH)D3 in adults increases serum 25(OH)D concentration more than vitamin D. In one study with a low risk of bias for the measurement of serum 25(OH)D (Cashman et al., 2012), the increase from baseline was up to 5 times higher with 20 μg/day oral 25(OH)D (a dose twice as much as the proposed daily intake of the NF) than with the same amount of vitamin D3. The Panel concludes that altogether the studies show that the NF at the proposed daily intake is bioavailable in adults as it increases serum 25(OH)D concentration and hence vitamin D status, and is a source of the biologically active form of vitamin D (1,25(OH)2D).
The Panel notes that no data were provided on the bioavailability of the NF in children aged 3 years or more or in pregnant or lactating women.
3.8.2. Data on bioavailability in animals
The applicant submitted two studies in rodents, in which the metabolism of ‘Calcidiol’ ([14C]‐Calcifediol) and vitamin D3 (14C‐Cholecalciferol) were compared (Beck et al., 2017; Beck and Punler, 2017, unpublished study reports).
The Panel notes that, due to the high doses tested in animals and as human data is available, these studies are not informative for the present assessment.
3.8.3. Conclusion on ADME
The Panel considers that the potential of oral calcidiol to increase serum 25(OH)D concentration in comparison to vitamin D3 has been consistently demonstrated in the literature on adult humans provided by the applicant. In four intervention studies, this mean increase is higher than the one observed with oral administration of vitamin D3 at similar dose (20 μg/day, which is twice the proposed daily intake of the NF). In one of the two intervention studies with a low risk of bias for the measurement of serum 25(OH)D, this increase from baseline was up to 5 times higher with 20 μg/day oral 25(OH)D than with the same dose of vitamin D3. No such comparison is available at the proposed daily intakes of the NF in adults or children.
The Panel notes that there is considerable variability in the achieved increases in 25(OH)D with calcidiol in comparison to vitamin D3, depending on the dose and experimental conditions in the various studies. It is known that factors such as vitamin D status (i.e. 25(OH)D concentrations) at baseline, endogenous synthesis by exposure to UV‐radiation, body composition and also polymorphism of genes encoding proteins involved in vitamin D synthesis, transport and metabolism may influence serum 25(OH)D concentrations (EFSA NDA Panel, 2016b).
3.9. Nutritional information
The applicant provided a compositional analysis of the NF (Section 3.4). The quantity of the commercial preparation ‘0.25% w/w’ indicated by the applicant is 4 mg in order to reach the proposed daily NF intake of 10 μg/day. Other components of the commercial preparation (dl‐alpha‐tocopherol, sodium ascorbate, carbohydrates and fats) are present only in small amounts that are not nutritionally relevant.
In the following, the outcome of the combined influence of the intake of vitamin D, calcidiol and sun exposure (Section 3.7.3) in relation to (i) the current ULs for vitamin D and (ii) reference values for serum 25(OH)D concentrations, will be considered.
-
i
Considerations with regard to ULs for vitamin D (D 2 and D 3 )
A combined intake of calcidiol from the NF and from foods of animal origin originated from animals fed with calcidiol has been estimated to be 12.44 μg/day (rounded to 12.5 μg/day) for adults and children aged 11 years and above, and to 7.44 μg/day (rounded to 7.5 μg/day) for children aged 3–10 years (Section 3.7.3). Also, based on dietary intake data for vitamin D that have been collected for adults in 14 European countries and for infants and children in 11 European countries by EFSA (EFSA NDA Panel, 2012), mean/median dietary intake of vitamin D has been estimated to range between 1.1 to 8.2 μg/day in adults, from 1.4 to 5.6 μg/day in children aged 1–13 years, and from 1.6 to 4.0 μg/day in children aged about 11–18 years. Among high consumers (95th percentile), intake in adults was up to 16 μg/day from foods. In high consumers (90th or 95th percentile according to surveys) of children and adolescents, the highest intake from foods was 11.9 and 7.7 μg/day, respectively (EFSA NDA Panel, 2012).
The DRVs, including the ULs set for vitamin D by the Panel were based on data for vitamin D2 and D3 only and did not cover data on oral calcidiol consumption (EFSA NDA Panel, 2012, 2016b, 2018) (Section 1.3). Also, a systematic review of data, assessing the extent to which oral calcidiol is more bioavailable than oral vitamin D3 in all population groups and dietary context was outside the remit of this opinion and the data provided by the applicant do not permit this question to be answered for the proposed daily intake of 5 or 10 μg/day. Thus, as a theoretical calculation for this opinion, the NDA Panel used the factor of 5 set by the FEEDAP Panel (EFSA, 2005) (see Section 1.3) to convert calcidiol to vitamin D.
Following this approach, 5 and 10 μg calcidiol from the NF (included in the commercial preparation at 0.25% w/w) would theoretically correspond to a vitamin D intake of 25 and 50 μg, respectively (27.5 and 55 μg if the NF is added to the supplement at 0.275%). Values of 25 (or 27.5) and 50 (or 55) μg/day for, respectively, the age ranges 3–10 years and 11 years and above are below the current ULs for vitamin D (D2 and D3) for children 1–10 years (50 μg/day) and for adults (including pregnant and lactating women) and children 11–17 years (100 μg/day). These values would however be largely above the DRVs for vitamin D (adequate intakes) set as 15 μg/day of vitamin D (D2 and D3) for healthy individuals over 1 year of age including pregnant and lactating women under the condition of assumed minimal cutaneous vitamin D synthesis.
The combined intake of calcidiol from the NF and the background diet of 12.5 (for adults and children ≥ 11 years) and 7.5 μg/day (for children 3–10 years) mentioned above would translate into a theoretical vitamin D intake of 62.5 and 37.5 μg/day, respectively. Added to the intake of vitamin D from the background diet, this would sum up for adults, when using the P95 intake of vitamin D from the diet (16 μg/day, see above) to 78.5 μg/day. This theoretical calculation leads to a result below the current UL for vitamin D (D2 and D3) set for adults (100 μg/day).
For children ≥ 11 years, the combined intake of calcidiol from the NF and from the background diet added to the background intake of vitamin D would sum up, using the P95 intake of vitamin D from the diet, to 70.2 μg/day (62.5 + 7.7 μg/day). This theoretical calculation leads to a result below the current UL for vitamin D (D2 and D3) set for this age group (100 μg/day).
For children 3–10 years, the combined intake of calcidiol from the NF and from the background diet added to the background intake of vitamin D would add up, using the P95 intake of vitamin D from the diet, to 49.4 μg/day (37.5 + 11.9 μg/day). This result is close to the UL for vitamin D (D2 and D3) for this age group (50 μg/day) and the UL would be exceeded if the concentration of the NF in the commercial preparation to be used in supplements was at 0.275%.
Table 5 provides an overview of the exposure to vitamin D from different sources separately and combined, and the ULs established by EFSA for the target population of this NF (EFSA NDA Panel, 2012).
Table 5.
Total vitamin D intake (μg/day) resulting from combined intake of calcidiol and vitamin D from the background diet and from the NF at the maximum use levels as proposed by the applicant
| Population group | Combined intake of calcidiol from the NF and background dieta | Theoretical combined vitamin D intake (as calcidiol) from the NF and background dietb | Highest intake of vitamin D from foods in the background diet (EFSA NDA Panel, 2012)c | Total combined intake of vitamin D from calcidiol and the background dietd | UL (μg/day) EFSA NDA Panel (2012) |
|---|---|---|---|---|---|
| Children 3–10 years old | 7.5 | 37.5 | 11.9 | 49.4 | 50 |
| Adolescents (children ≥ 11 years old) | 12.5 | 62.5 | 7.7 | 70.2 | 100 |
| Adults (f) | 12.5 | 62.5 | 16 | 78.5 | 100 |
Resulting from the combined intake of calcidiol (25‐hydroxycholecalciferol) from the NF (5 or 10 μg/day) and from the background diet according to the refined calculation of the FEEDAP Panel (2.44 μg/day) (EFSA, 2009).
Resulting from the Combined intake of calcidiol from the NF and foods from animal origin by using the factor of 5 between calcidiol and vitamin D set by the FEEDAP Panel (EFSA, 2005).
Intake of vitamin D from foods in the background diet in high consumers, P95th intake for adults and adolescents, P90th for children (according to surveys) (EFSA NDA Panel, 2012).
Resulting from the sum of the theoretical combined vitamin D intake (as calcidiol) from the NF + background diet and the highest intake of vitamin D from foods in the background diet (EFSA NDA Panel, 2012).
-
ii
Considerations with regard to achieved serum 25(OH)D concentrations
Regarding reference values for serum 25(OH)D concentrations, the Panel had considered that a serum 25(OH) concentration above 220 nmol/L may lead to hypercalcaemia (EFSA NDA Panel, 2016b). Later, in its revision of the UL for infants, the Panel considered that a serum 25(OH) concentration of 200 nmol/L or below is unlikely to pose a risk of adverse health outcomes (such as hypercalciuria, hypercalcaemia, nephrocalcinosis, or abnormal growth) in healthy infants, highlighting however that this should not be regarded as a cut‐off for toxicity in infants.The Panel noted that ‘high’ 25(OH)D concentration is not an adverse health outcome per se, but that it can be considered as a surrogate endpoint (EFSA NDA Panel, 2018).
Hence, in the present opinion, the Panel considered data from six human intervention studies in adults provided by the applicant previously described (Section 3.8 and Appendix A).19 The Panel paid attention specifically to achieved serum 25(OH)D concentrations after oral daily calcidiol supplementation, generally without concomitant consumption of vitamin D supplements or high cutaneous synthesis of vitamin D via sun exposure, in comparison to the serum 25(OH)D concentrations of 200 nmol/L mentioned above.
The Panel notes that, in the intervention studies provided by the applicant, supplemental calcidiol intake of 10 or 5 μg/day in adult populations raised mean 25(OH)D concentrations to values exceeding the serum 25(OH)D concentration of 50 nmol/L previously considered by the Panel as the ‘suitable target value’ when setting an adequate intake for vitamin D (EFSA NDA Panel, 2016b), but remaining below 200 nmol/L.
The Panel also notes that, with a consumption of up to 15 or 20 μg/day calcidiol (generally without concomitant consumption of vitamin D supplements or high cutaneous synthesis of vitamin D via sun exposure, but in addition to background diet of vitamin D, reported in only two studies), mean serum 25(OH)D concentration remained below 200 nmol/L in the studies in adults provided by the applicant. However, in some of the studies in which subjects received calcidiol at 20 μg/day, results showed that some individuals may achieve concentration above 200 nmol/L.
No data were available in children aged 3 years and above or in pregnant or lactating women with regard to serum 25(OH)D concentrations.
3.9.1. Conclusion on nutritional information
Noting that the UL for vitamin D in high consumers aged 3–10 years could be exceeded, the Panel could not conclude for this age group.
Considering the available evidence, the target population, proposed uses and use levels, the theoretical combined intake of calcidiol from the NF and foods of animal origin (Section 3.7), and the bioavailability of the NF (Section 3.8), the Panel considers that the consumption of the NF is not nutritionally disadvantageous.
3.10. Toxicological information
Since the NF is of particulate nature and contains a fraction of nanoparticles, the adequacy of the toxicological studies for assessing the hazards of the fraction of small particles was assessed as reflected in the Guidance on risk assessment of the application of nanoscience and nanotechnologies in the food and feed chain adopted by the EFSA Scientific Committee in 2018 and the draft EFSA Guidance on Technical requirements for regulated food and feed product applications to establish the presence of small particles including nanoparticles (currently under finalisation9). The applicant provided evidence that the batches tested were representative of the NF in terms of particle size distribution. For the 90‐day oral toxicity study, the NF was administered by mixing it as powder in the diet, which represents the realistic way of exposure of consumers (as required by the EFSA Guidance on the application of nanoscience and nanotechnologies in the food and feed chain, EFSA Scientific Committee, 2018). Following absorption, the NF, if taken up as nanoparticles, is expected to partition and quickly solubilise into the lipophilic compartments, suggesting that systemic distribution of particles is unlikely to occur. Taking all this into account, the available toxicological studies are considered adequate for the assessment.
3.10.1. Genotoxicity
In their opinion on ‘Hy•D’ (‘Calcidiol’, based on 25‐hydroxylcholecalciferol/25‐hydroxy‐pre‐cholecalciferol) as feed additive in accordance with Council Directive 70/524/EEC) adopted in 2005, the EFSA FEEDAP Panel concluded that, based on the studies considered (acute toxicity in mice and rats, repeated dose sub‐chronic toxicity in rats, two mutagenicity studies, and reproduction studies both in rat and rabbit), there was no reason to suspect that the NF is genotoxic (EFSA, 2005) (see Section 1.3). In a further opinion on calcidiol monohydrate, published in 2009, the EFSA FEEDAP Panel confirmed their previous conclusion which was corroborated by an additional negative in vitro chromosomal aberration assay (EFSA, 2009) (see section 1.3).
For the present opinion, the applicant submitted a series of new genotoxicity tests carried out with the NF (proprietary data). Table 6 provides a summary of the genotoxicity tests submitted.
Table 6.
Summary of genotoxicity tests with the NF ‘Calcidiol’
| Study | Concentrations | Result | Reference |
|---|---|---|---|
| Plate incorporation test (experiment 1) Pre‐incubation test (experiment 2) Salmonella Typhimurium GLP, OECD 471, Batch WICSP1098B, purity 98.3% | 3, 10, 33, 100, 333, 1,000, 2,500 μg/plate ± S9 10, 33, 100, 333, 1,000, 2,500, 5,000 μg/plate ± S9 | Negative Negative | Wöhrle and Sokolowski (2013) (unpublished) |
| In vitro CA with human lymphocytes GLP, OECD 473, Batch WICS‐R185, purity 97.3% | 2, 3.5, 5.7, 6.1, 10.7 18.7, 32.7 and 57.1 μg/mL, ± S9 | Negative | Weber and Schulz (2005) (unpublished) |
| In vitro gene mutation L5178Y cells, GLP, OECD 490, DSM047117, Batch crystalline WICSP1131B, purity 96.9% | ≤ 7.5 (with +S9) and ≤ 25 μg/mL (‐S9), incubation time 3 h; ≤ 5 μg/mL, 24 h incubation time | Negative Negative | Remus and Verspeek‐Rip (2016) (unpublished) |
| In vivo MN test, 5 rats per group, GLP, OECD 474, GLP, Batch WICSP1131B, purity 96.9% | Main study: 10, 25 and 50 mg/kg bw (MTD) | Negative | Remus and Verbaan (2016) (unpublished) |
CA: chromosomal aberration; GLP: Good Laboratory Practice; MN: micronucleus; MTD: Maximum Tolerated Dose; OECD: Organisation for Economic Co‐Operation and Development; bw: body weight.
In an OECD Test No 471 and GLP compliant reverse mutation assay, Salmonella Typhimurium TA1535, TA1537, TA98, TA100 and Escherichia coli WP2 uvrA were tested (in triplicate) in two independent investigations at concentrations of 3, 10, 33, 100, 333, 1,000, 2,500 and 5,000 μg/plate (first experiment) and at concentrations of 10, 33, 100, 333, 1,000, 2,500 and 5,000 μg/plate (second experiment) of the NF (Batch WICSP113B, purity 96.9%), with and without metabolic activation. No toxicity (reduction in number of revertants) was seen at any of the concentrations tested. No mutagenicity (increase in number of revertants compared to negative control) was seen at any concentration with any strain with or without metabolic activation (Wöhrle and Sokolowski, 2013, unpublished study report).
The Panel concludes that the NF is not mutagenic in the S. typhimurium/E. coli reverse mutation assay.
The NF (Batch WICS‐R185, purity 97.3%) was investigated in an OECD Test No 473 and GLP compliant in vitro chromosomal aberration (CA) assay with human lymphocytes in two independent experiments at concentrations of 2, 3.5, 5.7, 6.1, 10.7 18.7, 32.7 and 57.1 μg/mL (at higher doses cytotoxicity was observed) with and without metabolic activation (Weber and Schulz, 2005, unpublished study report). While, with the positive controls (ethylmethane sulfonate (EMS) and cyclophosphamide (CP)), significantly increased numbers of structural CAs were observed both without and with metabolic activation (S9), no effects were seen in the treatment groups. The Panel concludes that the NF is not clastogenic in vitro in human lymphocytes.
The NF (‘Calcidiol’ Batch WICSP1131B, purity 96.9%) was tested in an OECD Test No 490 and GLP compliant in vitro mutagenicity test (induction of forward mutations in TK locus) with L5178Y mouse lymphoma cells, with and without metabolic activation (Remus and Verspeek‐Rip, 2016, unpublished study report). In a first experiment at concentrations of up to of 7.5 (with S9) and 25 μg/mL (without S9), and incubation time of 3 h, relative growth (RTG) was 14% and 13% with and without S9, respectively. In a second experiment the NF was incubated up to a concentration of 5 μg/mL without S9 for 24 h and an RTG of 28% resulted. MMS and CP, that served as positive controls, increased mutation frequencies. ‘Calcidiol’ did not induce increased mutations under the conditions of test described. The Panel concludes that the NF is not mutagenic in vitro in murine lymphoma cells.
In addition, the NF (Batch WICSP1131B, 96.9% purity) was tested in an OECD Test No 474 and GLP compliant in vivo micronucleus (MN) assay in the bone marrow of rats (Remus and Verbaan, 2016, unpublished study report). Groups of 5 male rats were gavaged twice (at 0 and 24 h) with vehicle control and with 10, 25 or 50 mg/kg body weight (bw) of ‘Calcidiol’ and once with 20 mg/kg of CP as a positive control. Bone marrow was collected 48 h after the first dose. No increase in the mean frequency of micronucleated polychromatic erythrocytes (PCE) was observed at any dose level as compared to the control, whereas CP induced statistically significant increase in the number of micronucleated PCEs. At doses of 25 mg ‘Calcidiol’/kg bw and higher, and with CP, decreased PCE/NCE ratios were observed, and at 50 mg/kg (the only dose where such parameters were investigated) also changes in haematologic parameters were observed, proving toxic effects on erythropoiesis and being indicative of the test item and/or its metabolites reaching the target organ. The Panel concludes that the NF is not clastogenic or aneugenic in the bone marrow of male rats up to a dose of 50 mg/kg (the maximum tolerated dose).
Overall, the Panel concludes that the genotoxicity studies submitted confirm the previous conclusion of the FEEDAP Panel (EFSA, 2005, 2009) that there is no concern for the NF regarding genotoxicity.
3.10.2. Acute toxicity
The applicant has provided an acute toxicity study in rat (Weber and Arcelin, 2004, unpublished study report).
The Panel considers that, in general, acute toxicity studies are not pertinent for the safety assessment of NFs.
3.10.3. Subacute and subchronic toxicity
The applicant refers to a study in the public domain that investigated toxicity effects of vitamin D3 and 25‐hydroxyvitamin D3. Shepard and DeLuca (1980) gavaged groups of 10 male rats with 0.25, 2.5, 25, 250 or 2,500 μg vitamin D3 per day and a second set of 10 male rats with 0.185, 1.85, 18.5, 185 and 1,850 μg 25‐hydroxy vitamin D3 per day. The animals were dosed daily for 14 days.
Animals given vitamin D3 started to lose weight at doses of 25 μg/day and above, and 9 out of 10 animals died at the highest doses. No animals died in the 25‐hydroxyvitamin D3 group, though, again, the animals lost weight in the two highest groups. Changes in calcium and phosphorus concentrations were seen in the highest dosed animals. These animals were ‘generally in poor health’ and had mottled and greyish kidneys, suggesting calcification. The authors suggest that the toxicity results from a gross exaggeration of normal function of vitamin D, induced by the presence of high concentrations of these precursors, although they also point out that high concentration of 25(OH) D3 can substitute directly for 1,25(OH)2D3. Given the high doses used, and the high toxicity, no conclusions can be drawn from this study.
In an OECD 408 and GLP‐compliant 90‐day study, Thiel et al. (2014) (unpublished study report) added 25‐hydroxyvitamin D3 (NF formulations) (containing 25‐hydroxy vitamin D3 12.5 mg, dl‐alpha‐tocopherol 76.5 mg, coconut oil 18 mg, modified food starch 709 mg, maltodextrin 149 mg, sodium ascorbate 25 mg, silicon dioxide 10 mg) to the diets of groups of 20 (10 male and 10 female) Wistar rats, yielding final dose levels of 0, 7, 20, 60 and 180 μg ‘Calcidiol’/kg bw per day. Five animals per sex in the control and high dose group had an additional 28 days recovery period at the end of the study. Clinical signs and functional observations were evaluated in week 12–13, body weight, food and water consumption daily during treatment period, ophthalmoscopy before treatment and in week 13, clinical pathology in weeks 4, 8, end of treatment and end of recovery, and macroscopy and histopathology at the end of treatment and recovery.
No clinical signs or abnormalities were noted during the observation period. Motor activity was similar between treated and control groups. No toxicologically significant changes in food consumption, body weight or body weight gain and no treatment‐related changes in ophthalmology or haematology were observed. As for possible effects at the first site of contact, histopathological examination of the mesenteric lymph nodes, duodenum, jejunum, ileum, Peyer's Patches, cecum, colon and rectum of treated rats showed no differences compared to control animals. Therefore, no evidence of local toxicity from the nanofraction of particles of the NF was found.
An increase in water consumption, a decrease in pH, higher calcium concentration and excretion, higher inorganic phosphate concentration and excretion and higher calcium creatinine ratios in urine were observed at all tested doses and time points. Most of these changes returned to normal after the recovery period.
In blood, increased Ca concentrations were observed in both sexes at all dose levels. These were not completely reversed after the recovery period. Increased inorganic P was observed in both sexes at the highest dose. Increased sodium concentrations (by 1–2%, not in a dose‐response manner) were observed in females at all doses of the NF. Increases in adrenal weight and adrenal to body weight ratio were noted for females treated at 4.5 and 13.4 mg/kg at the end of the treatment period, changes were reversed at the end of the recovery period. Macroscopic analysis of organs upon necropsy did not reveal toxicologically relevant alterations. In addition to other microscopic findings observed at the end of the study, pyelonephritis and hyperplasia of kidney urothelium and hypertrophy in interstitial cells of ovaries were observed. Mineralisation in the renal pelvis of the kidneys was observed in both sexes at 20, 60 and 180 μg/kg bw per day and also in 4 female animals at 7 μg/kg bw per day (notably also in two female animals of the control group mineralisation was seen). There was a dose‐response relationship regarding the number of affected animals and the grade of the mineralisation. The effect was classified by the study investigators as ‘minimal’ to ‘slight’, with the exception of the effect reported for one male animal of the highest dose which was considered ‘moderate’. The localisation of the background mineralisation differed from the test item‐related mineralisation in that the mineralisation was present mainly in the pelvic urothelium and the area where papillary epithelium and pelvic urothelium connect, whereas the background mineralisation was mainly present in the medulla. The hyperplasia of the urothelium and the pyelonephritis were found in one or maximum two animals per group and of minimal to slight grade. These effects were considered to be likely triggered by the deposition of minerals which was mainly present in the pelvic urothelium. Mineralisation remained present in most animals after the 28‐recovery period. Mineralisation was not found in any other organ than kidney except very mild mineralisation in the lung (vascular) and gut (Peyer's patches) and this was also seen in a similar proportion of the control animals and at the same quality/location. Thus, the mineralisation in lung and gut are considered not treatment‐related.
The Panel considers the effects observed in relation to the kidney to be treatment‐related and caused by the known biological effects of 1,25‐dihydroxycholecalciferol, the active form of vitamin D3, for which calcidiol is the precursor, on calcium and phosphorus metabolism. The effects were observed at all doses. The Panel concludes that no no observed adverse effect level (NOAEL) can be derived from this study.
3.10.4. Chronic toxicity, carcinogenicity, reproductive and developmental toxicity
Chronic toxicity, carcinogenicity and reproductive studies have not been provided by the applicant. The NDA Panel considers that such studies are not necessary for the evaluation of the NF.
3.10.5. Human data
In line with previous assessments (EFSA NDA Panel, 2012, 2016b, 2018), this section describes data on urinary calcium/hypercalciuria, serum calcium/hypercalcaemia and adverse events. These data were reported in the six human intervention studies investigating oral daily doses of vitamin D3 or calcidiol in men and/or not‐pregnant and not‐lactating women (Sections 3.8 and 3.8). The applicant developed a commercial preparation of the NF to be used in the clinical trials. Upon EFSA's request, the applicant clarified that the products used in the clinical studies have the same composition as the ‘0.25 % w/w formulation’ (see Sections 3.1, 3.3 and 3.4), i.e. the commercial preparation for which analysis on 4 batches were provided in this application (data not shown, see explanations in Section 3.4). Although the studies provided by the applicant were primary efficacy or pharmacokinetic studies, the Panel addresses below possible occurrence of adverse outcomes, with a special focus on parameters related to hypercalciuria and hypercalcaemia as possible indicators of excessive vitamin D intake (see Section 3.9).
Four of the six human intervention studies report on urinary calcium (Appendix A).
In the unpublished study report by Wittwer (2015) and the publication Vaes et al. (2018), calcium (Ca) and creatinine (creat) concentrations were measured in morning spot urine and expressed as Ca/creat‐ratio. At baseline, there was no significant difference in urinary Ca concentration between the groups supplemented with either 20 μg vitamin D3 or 5, 10 or 15 μg 25(OH)D3/day (p = 0.29), with an overall mean baseline value of 129 mg Ca/g creat (95% CI 107–151). Over the 24‐week intervention period, the treatment per time interaction was non‐significant (p = 0.95). There was also no significant difference in urinary Ca/creat‐ratios between the groups after the 24 weeks of supplementation. On average, all treatment groups remained below the reference value of 220 mg Ca/g creat or within the reference range used in this study (which corresponds to a Ca/creat‐ratio of 0.62 mmol/mmol). Although the wide CIs are indicative for individuals exceeding this reference, individuals exceeding the reference values were in all groups at different time points including at baseline.
In the study of Bischoff‐Ferrari et al. (2012) and Jetter et al. (2014), there was no significant difference in urinary Ca excretion assessed as Ca/creat ratio (mean ± SE) in spot urine between the groups receiving either vitamin D3 or 25(OH)D3, considering all time points over 4 months (0.41 ± 0.03 vs 0.36 ± 0.03, p = 0.37) or at the end of follow‐up (0.33 ± 0.06 vs 0.33 ± 0.06, p = 0.98). Units are assumed by the NDA Panel to be mmol/mmol, although the information was not provided in the publications, and no reference range was indicated by the authors.
In the study by Navarro‐Valverde et al. (2016), mean (± SD), the baseline level for Ca/creat‐ratio (mmol/mmol) for the combined study group was 0.1 ± 0.03 and within the range of 0.08–0.3 considered as reference range in this study. The daily administration of calcidiol oral drops (20 μg/day) or equal amounts of vitamin D3 resulted in significantly higher Ca/creat‐ratios (mmol/mmol) at six months (0.19 ± 0.04 vs 0.13 ± 0.03) and at 12 months (0.27 ± 0.06 vs. 0.17 ± 0.03) in the calcidiol group. The Panel assumes that the Ca/creat‐ratio was determined in spot urine and the units to be mg/mg (EFSA NDA Panel, 2018), although the information was not provided in the publication.
In the study by Kunz et al. (2016) (unpublished study report), fasting 2‐h morning urine Ca/creat‐ratios (mg/mg) showed no change over the 6 months course of the study and no significant differences between the groups receiving either 20 μg vitamin D3 or 10, 15 or 20 μg per day of 25(OH)D3. Calcium and creatinine were analysed in 24 h urine at baseline and visit 14 (day 182). Higher mean 24 h‐urine calcium levels were seen in the 15 and 20 μg (but not in the 10 μg) 25(OH)D3 groups compared with 20 μg vitamin D but stayed within ‘a range of no concern’ defined as < 300 mg/24 h by the authors, although in all groups some individuals exceeded this value at certain time points including at baseline.
Regarding serum calcium, reference ranges for serum calcium or hypercalcaemia were defined in all studies (in different ways), except one (Navarro‐Valverde et al., 2016) (see Appendix A for details). Numerical values of serum calcium were similar between groups receiving 20 μg/day of vitamin D3 or oral calcidiol (Navarro‐Valverde et al., 2016). In the other five studies, serum calcium remained below the reference range or the value defining hypercalcaemia in groups receiving oral calcidiol.
Regarding adverse events in general, either they were not measured or not reported (Barger‐Lux et al., 1998; Bischoff‐Ferrari et al., 2012; Jetter et al., 2014; Navarro‐Valverde et al., 2016) or none were observed (Cashman et al., 2012) or their number was not significantly different between the groups (Wittwer, 2015;/Vaes et al., 2018) or they were considered as not related to the study products (Wittwer, 2015; Kunz et al., 2016; Vaes et al., 2018) (see Appendix A for details).
The Panel notes that, in the studies provided by the applicant, urinary calcium excretion or serum calcium were not primary outcomes. Urinary calcium excretion was mainly measured as Ca/creat‐ratio in spot urine for which low sensitivity and specificity to diagnose hypercalciuria, compared to 24‐h urinary calcium measurement, has been reported (Jones et al., 2012). Nevertheless, the Panel considers that in the studies provided by the applicant, no hypercalciuria or hypercalcaemia was observed, at the doses tested including at the daily intake of the NF proposed by the applicant, and there was no increased number of adverse events in the groups receiving oral calcidiol compared to those receiving vitamin D3. The Panel considers that the data regarding urinary calcium, serum calcium and adverse events in general in the human intervention studies provided by the applicant in adults do not raise safety concerns. No data on urinary calcium, serum calcium or possible adverse events were provided in children aged 3 years and over or in pregnant or lactating women consuming the NF at the proposed daily intake.
3.11. Allergenicity
The Panel considers that, owing to the absence of protein, the NF is unlikely to trigger allergic reactions in the target population under the proposed conditions of use.
4. Discussion
The NF, which is the subject of the application, is calcidiol monohydrate (25‐hydroxycholecalciferol monohydrate), referred to as ‘Calcidiol’ by the applicant. The NF contains the monohydrate form of the major circulating metabolite of vitamin D3 in the body and is a source of 1,25‐dihydroxyvitamin D, the biologically active form of vitamin D.
The NF is intended to be used in food supplements at 10 μg/day for individuals aged 11 years and above, and 5 μg/day for children of age 3–10 years. The target population is adults including pregnant and lactating women, and children above 3 years of age. The applicant intends to market the NF as a diluted form, called ‘0.25% w/w’ or ‘Calcidiol 0.25% SD/S’ by the applicant, that contains values in the range of 0.250–0.275% w/w of calcidiol (anhydrous).
The NF is produced by chemical synthesis and there are no safety issues associated with the levels of reaction by‐products, residual solvents or heavy metals in the NF.
The NF is of particulate nature, contains a fraction of nanoparticles, and these particles may reach the human intestine as such. However, if the NF is absorbed at least partly as nanoparticles, they are expected to partition and quickly solubilise into the lipophilic compartments, suggesting that systemic distribution of particles is unlikely to occur.
Regarding bioavailability, the potential of supplementation with calcidiol (25‐hydroxycholecalciferol) to increase serum 25(OH)D concentration in comparison to vitamin D3, which is an authorised source of vitamin D according to Directive 2002/46/EC, has been demonstrated in studies among adults provided by the applicant. These data indicate efficient absorption.
Regarding achieved serum 25(OH)D, the studies providing supplemental calcidiol intake of 10 or 5 μg/day in various adult populations did not raise mean 25(OH)D concentrations above 107 and 52.2 nmol/L respectively. These concentrations exceed the serum 25(OH)D of 50 nmol/L which was considered as the ‘suitable target value’ by the Panel when setting an adequate intake (AI) for vitamin D (EFSA NDA Panel, 2016b), but are in the range of concentrations (i.e. below 200 nmol/L) unlikely to pose a risk of adverse health outcomes such as hypercalciuria, hypercalcaemia or nephrocalcinosis (EFSA NDA Panel 2012, 2016b, 2018).
Regarding safety, no NOAEL can be derived from the experimental animal toxicological data provided by the applicant. In the human studies in adults provided by the applicant, no hypercalciuria or hypercalcaemia was observed, at the daily intake of the NF proposed by the applicant, and no increased number of adverse events compared to supplementation with vitamin D3 was observed.
Conservative highest vitamin D intake estimates were obtained from a theoretical approach, combining intake of calcidiol from the NF with intake of calcidiol and of vitamin D from other dietary sources, and considering calcidiol to be 5‐fold more potent than vitamin D3 (factor set by the FEEDAP Panel based on data in rats and chicken (EFSA, 2005)). These intake estimates were below the upper levels for vitamin D (D2 and D3) (i.e. 100 μg/day) as established by the NDA Panel for individuals above 10 years old (EFSA NDA Panel, 2012).
No data were provided by the applicant to assess the bioavailability (i.e. the impact on serum 25(OH)D concentrations) and safety of the consumption of the NF by pregnant women or lactating women. However, the Panel notes that the estimated exposure mentioned above for adults are below the UL for vitamin D, and that the achieved mean serum 25(OH)D concentration when (non‐pregnant non‐lactating) adults are supplemented with 10 μg/day NF remains below 200 nmol/L.
The Panel also notes the uncertainty regarding the calculated combined exposures to vitamin D of the general population, given the fact that the range of foods fortified with vitamin D as well as food supplements containing a high dose of vitamin D has increased over the years (including since the vitamin D intake data used in the calculations for this opinion were obtained). The Panel notes that, depending on the latitude and the time of the year, an additional uncertainty is represented by the endogenous cutaneous vitamin D synthesis (impacting on serum 25(OH)D concentrations).
No data were provided by the applicant to assess the bioavailability and safety of the consumption of the NF by children. For children 3–10 years, the combined intake of calcidiol from the NF and from the background diet, added to the background intake of vitamin D, would sum up, using the P95 intake of vitamin D from the diet to 49.4 μg/day. The Panel notes that the intake estimation is close to the UL for vitamin D (D2 and D3) for this age group (50 μg/day). The Panel also notes that this value might be exceeded if the NF is added to the supplement at a concentration above 0.25%.
5. Conclusions
The Panel concludes that the NF, ‘Calcidiol’, i.e. calcidiol monohydrate (25‐hydroxycholecalciferol monohydrate), is safe under the proposed conditions of use and use levels, (up to 10 μg/day) for children ≥ 11 years old and adults, including pregnant and lactating women.
The Panel notes that, in children, for high consumers, the combined intake of the NF (5 μg/day) and of calcidiol from the background diet, added to the background intake of vitamin D, would approach the UL for vitamin D (D2 and D3) in children of age 3–10 years old. Furthermore, the applicant proposes to add the NF to food supplements as a preparation containing 0.25% to 0.275% w/w of calcidiol. This could result in the UL for vitamin D in children of this age being exceeded. Given the uncertainties, the Panel could not conclude on the safety of consumption of the NF in children of 3–10 years at the proposed daily intake.
The Panel also concludes that the NF is a bioavailable source of the biologically active metabolite of vitamin D, i.e. 1,25‐dihydroxyvitamin D.
5.1. Protection of Proprietary data in accordance with Article 26 of Regulation (EU) 2015/2283
The Panel could not have reached the conclusion on the safety of the NF under the proposed conditions of use without the data claimed as proprietary by the applicant (master data and product specifications, ADME studies, toxicity studies, human studies and the analytical reports including the annexes).
Steps taken by EFSA
Letter from the European Commission to the European Food Safety Authority with the request for a scientific opinion on the safety of Calcidiol produced by chemical synthesis Ref. Ares (2018) 6458095, dated 14 December 2018.
On 14/12/2018, a valid application on Calcidiol, which was submitted by DSM Nutritional Products Ltd., was made available to EFSA by the European Commission through the Commission e‐submission portal (NF 2018/0402) and the scientific evaluation procedure was initiated.
On 17/05/2019 and 22/11/2019, EFSA requested the applicant to provide additional information to accompany the application and the scientific evaluation was suspended.
On 30/09/2019 and 24/04/2020 additional information was provided by the applicant and the scientific evaluation was restarted.
On 18/05/2020, 18/12/2020, 17/02/2021, 31/03/2021 and 07/05/2021, EFSA requested the applicant to provide clarifications on the information provided.
On 27/11/2020, 28/01/2021, 11/03/2021, 20/04/2021 and 10/05/2021 additional information were provided by the applicant through the Commission e‐submission portal and the scientific evaluation was restarted.
During its meeting on 25/05/2021, the NDA Panel, having evaluated the data, adopted a scientific opinion on the safety of calcidiol monohydrate produced by chemical synthesis as a novel food pursuant to Regulation (EU) 2015/2283.
Abbreviations
- 1,25(OH)2D
1,25‐dihydroxy‐vitamin D
- 1,25(OH)2D2
1,25‐dihydroxy‐ergocalciferol
- 1,25(OH)2D3
1,25‐dihydroxy‐cholecalciferol
- 25(OH)D3
25‐hydroxyvitamin D3
- 25(OH)D
25‐hydroxyvitamin D
- ADME
absorption, distribution, metabolism and excretion
- AI
adequate intake
- BIOHAZ
Panel on Biological Hazards
- BMI
body mass index
- bw
body weight
- Ca/creat
calcium/creatinine
- CA
chromosomal aberration
- CAS
Chemical Abstracts Service
- creat
creatinine
- CFU
colony forming unit
- CI
confidence interval
- CP
cyclophosphamide
- DBP
vitamin D‐binding protein
- DRV
dietary reference value
- EMS
ethylmethane sulfonate
- EDX
energy dispersive X‐ray spectroscopy
- EP
European Pharmacopeia
- FAIM
Food Additive Intake Model
- FEEDAP
Panel on Additives and Products or Substances used in Animal Feed
- GC‐HS-FID
gas chromatography with headspace‐sampler and flame ionisation detector
- GI
gastrointestinal
- GLP
Good Laboratory Practice
- GMP
Good Manufacturing Practice
- GRAS
generally recognized as safe
- HACCP
Hazard Analysis Critical Control Points
- HPLC
high‐performance liquid chromatography
- HPLC–UV
high‐performance liquid chromatography–ultraviolet spectroscopy
- HPLC–MS
high‐performance liquid chromatography–mass spectrometry
- Hy‐DHC
25‐hydroxydehydrocholesterol
- ICP/MS
inductively coupled mass‐mass spectrometry
- IR
infrared spectroscopy
- ISO
International Standard Organization
- IUPAC
International Union for Pure and Applied Chemistry
- LC–MS/MS
liquid chromatography with tandem mass spectrometry
- MMS
methyl methanesulfonate
- MN
micronucleus
- MS
Member State
- MTD
Maximum Tolerated Dose
- NDA
Panel on Nutrition, Novel Foods and Food Allergens
- NF
novel food
- NOAEL
no observed adverse effect level
- NCE
normochromatic erythrocytes
- OECD
Organisation for Economic Co‐operation and Development
- PCE
polychromatic erythrocytes
- QPS
Qualified Presumption of Safety
- RH
relative humidity
- RTG
relative growth
- RCT
randomised clinical trial
- SCF
Scientific Committee for Food
- SD
standard deviation
- SE
standard error
- SEM
scanning electron microscopy
- TAMC
total aerobic microbial count
- TEM
transmission electron microscopy
- TLC
thin‐layer chromatography
- TYMC
total yeast and mould count
- TEM
transmission electron microscopy
- UL
tolerable upper intake level
- UV
ultraviolet
- USP
United States pharmacopeia
- Vitamin D2
ergocalciferol
- Vitamin D3
cholecalciferol
- w/w
weight for weight
Appendix A – Vitamin D3 and Calcifediol (calcidiol) human data
1.
Comparison serum response to Vitamin D and calcifediol supplementation in adults
| Reference Study design and characteristics | Population Population size Compliance checked? | Study site Latitude Season | Treatment and dose Substance | Duration Dose analytical check Condition of consumption | Baseline 25(OH)D nmol/L | Final serum 25(OH)D nmol/L | Background vit D intake | Vitamin D supplement users or those going on sunny holidays excluded | BMI kg/m2 | Analytical method 3 | Final serum 25(OH)D > 200 nmol/L? Urinary calcium Serum calcium Adverse events |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Bischoff‐Ferrari et al. (2012) Jetter et al. (2014) Parallel design, intervention study Subjects, investigators, study physician, and nurses aware of the daily/weekly intervention regimen. But they were blinded to the type of intervention. | Healthy post‐menopausal women 50–70 years, with baseline 25(OH)D of 20–60 nmol/L and BMI of 18–29 kg/m2 Bischoff‐Ferrari: n = 20 (5 per randomised group). Not reported in the 4 groups as randomised: groups of daily and weekly doses (D3 or 25(OH)D) merged by the authors for the analysis. Compliance checked. | Zurich, Switzerland 47°N, Jan–July | D3 Group of 20 μg daily combined with group of 140 μg weekly25(OH)D3 Group of 20 μg daily combined with group of 140 μg weekly | 15 week Analytical check of the doses in the capsules. Capsules consumed with breakfast | Mean ± SD 35.4 ± 9 (whole D3 groups, Bischoff‐Ferrari et al., 2012) 30.2 ± 3.9 (group with daily D3, Jetter et al., 2014)Mean ± SD 30.7 ± 10.2 (whole 25(OH)D groups, Bischoff‐Ferrari et al., 2012) 32.7 ± 9.9) in group with daily 25(OH)D (Jetter et al., 2014) | 77 (4) Mean (SE) (whole D3 groups, Bischoff‐Ferrari et al., 2012) Final maximal serum 25(OH)D: 82.8 ± 37.8 nmol/L in those receiving daily D3 (geometric mean and CV, Jetter et al., 2014)174 (4) Mean (SE) (whole 25(OH)D groups, Bischoff‐Ferrari et al., 2012) Final maximal serum 25(OH)D: 183 ± 36.8 nmol/L in those receiving daily 25(OH)D (geometric mean and CV, Jetter et al., 2014) | Not reported | Yes | Mean ± SD D3 daily: 25.46 ± 4.47 D3 weekly: 25.52 ± 2.37 25(OH)D daily: 24.90 ± 3.20 25(OH)D weekly: 21.59 ± 2.49 | HPLC‐MS/MS, validated in an international NIST/NIH quality assurance comparison |
|
| Cashman et al. (2012) Parallel design, Randomised double‐blind, placebo‐controlled intervention study | Healthy adults, ≥ 50 y n = 58 (2 dropouts, 56 completers) Compliance checked. | Cork, Ireland, 51°N, Jan‐April | Placebo | 10 w Analytical check of the doses in the capsules Conditions of consumption not reported | 42.7± 12.6 (mean ± SD) | 41.2 ± 11.1 (mean ± SD) | 6.5 (2.9–7.9) μg/d (mean, IQR) | Yes | Mean ± SD 28.3 ± 4.8 | ELISA, quality control via the Vitamin D External Quality Assessment Scheme |
|
| 20 μg/day D3 | 49.7 ± 16.2 (mean ± SD) | 69.0 ± 8.7 (mean ± SD) | 7.6 (2.9–5.4) μg/d (mean, IQR) | ||||||||
| 7 μg/day 25(OH)D3 | 42.5 ± 8.9 (mean ± SD) | 70.7 ±9.9 (mean ± SD) | 5.1 (2.8–6.6) μg/d (mean, IQR) | ||||||||
| 20 μg/day 25(OH)D3 | 38.2 ± 9.9 (mean ± SD) | 134.6 ±26.0 (mean ± SD) | 4.4 (3.7–6.1) μg/d (mean, IQR) | ||||||||
| Navarro‐Valverde et al. (2016) Parallel design, open‐label, randomised convenience intervention study | Healthy post‐menopausal osteopenic women, average age 67 y, all with serum 25(OH)D < 50 nmol/L n = 40 Compliance not checked. | Córdoba, Spain, 36.7°N Period of start/end of study not reported | 20 μg/day D3 | 12 m No analytical check of the doses (drops for both daily doses, ampoules for both weekly doses). Conditions of consumption not reported | 40.5± 4.7 | 86.2 ± 23.7 | Not reported | Not reported | Mean ± SD 26.4 ±4 | Automatic online solid phase extraction coupled with HPLC and UV detection |
|
| 20 μg/day 25(0H)D3 | 37.2 ± 4.2 | 188.0 ± 24.0 | |||||||||
| 266 μg 25(OH)D3 once every week | 38.0 ± 3.7 | 233.0 ± 81.2 | |||||||||
| 266 μg 25(OH)D3 once every two weeks | 39.5 ± 4 | 210.5 ± 22.2 | |||||||||
| Kunz et al. (2016), (unpublished) Parallel design, randomised, double‐blind intervention study | Healthy subjects, age > 50 years n = 93 randomised (91 received the products, 4 drop outs, 87 completers) Compliance checked | Leatherhead, UK 51.3 °N Period of start/end of study not reported in Kunz et al. (2016). Nov–May (stated by applicant) | 20 μg/day D3 | 6 m (+ 6m follow‐up without consumption of the capsules) No analytical check of the dose in the capsules reported. Capsules consumed before breakfast | Shown on figure, mean probably around 47 nmol/L (extracted from figure) | Increase of + 38.7 nmol/L (read on figure: mean final serum 25(OH)D of about 80 nmol/L) | Not assessed | Yes | Inclusion: 20–32 (mean: 26.2) | In‐house HPLC/MS/MS method 1 | – No final mean serum 25(OH)D above 200 nmol/L. – Urinary Ca (24 h urine or fasting 2 h urine, depending on the time points): – ‘Range of no concern’ defined as urinary Ca < 300 mg/24 h – Urinary Ca/creat. ratio remained stable during supplementation (24 h: group 10 μg 25(OH)D3: 95% CI = –0.0016, 0.0022], p = 0.7216; group 15 μg 25(OH)D3: 95% CI = –0.0005, 0.0032], p = 0.1586; group 20 μg 25(OH)D3: 95% CI = [0.0004, 0.0042], p = 0.0164) – Mean baseline urinary Ca (groups: D3, 10 μg, 15 μg and 20 μg 25(OH)D3): 158.73, 165.60, 188.47 and 159.14 mg/24 h – Final urinary Ca: 161.66, 160.69, 225.49, 202.71 mg/24 h – Higher 24‐h final mean urinary Ca in groups with 15 and 20 μg 25(OH)D. – in all groups some individuals exceeded this value at baseline and end of study – Serum Ca within reference levels (2.12 – 2.52 mmol/L), remained stable during supplementation and was not different between groups. – 482 AEs (e.g. headaches, reported infections, respiratory complaints) reported by 88 subjects during 1 y. No adverse event was assessed by the authors as being related to the products. There were 136 AEs in the 20 μg 25(OH)D group, 129 AEs in the 15 μg 25(OH)D group, 109 AEs in the D3 group, and 108 in the 10 μg 25(OH)D group. –Seven subjects had ‘serious’ AEs (one death, angina pectoris, back injury and fractured ribs, large bowel obstruction, probable urinary tract infection, breast cancer). The authors considered that no serious AE was related to the products. |
| 10 μg/day 25(OH)D3 | Increase of + 50.1 nmol/L (read on figure: mean final serum 25(OH)D of about 100 nmol/L) | ||||||||||
| 15 μg/day 25(OH)D3 | Increase of + 75.5 nmol/L (read on figure: mean final serum 25(OH)D of about 120 nmol/L) | ||||||||||
| 20 μg/day 25(OH)D3 | Increase of + 97.4 nmol/L (read on figure: mean final serum 25(OH)D of about 150 nmol/L) | ||||||||||
| Barger‐Lux et al., (1998) Parallel design, open‐label, intervention study. Comparisons across compounds are not randomised: subjects randomised for the dose but not the form administered (i.e. dependant on scheduling consideration and subject availability). | Healthy young men; 20–37 y n = 116 Compliance checked. | Omaha, USA 41.3 °N Jan‐Apr | 25 μg/day D3 | 8 w Analytical check of the doses in the capsules. Consumed at bedtime | 67 ± 252 (mean ± SD) average over all 9 groups (groups receiving 1,25(OH)2D not shown) | Increase of + 28.6 (final 25(OH)D* about 100 nmol/L) | Not reported | Yes | Mean ± SD 25.7 ± 3.2 | Protein‐binding assay | – Final serum 25(OH)D not reported (as an indication,* calculations of possible final 25(OH)D by EFSA, assuming a baseline of 67 nmol/L). – Urinary Ca: not measured – Serum Ca: measured. Values above the upper limit of the reference range for this paper (2.55 mmol/L) only in the groups receiving 1,25(OH)2D3. – Adverse events not measured. |
| 250 μg/day D3 | Increase of + 146.1 (final 25(OH)D* about 213 nmol/L) | ||||||||||
| 1250 μg/day D3 | Increase of + 643.0 (final 25(OH)D* about 710 nmol/L) | ||||||||||
| 10 μg/day 25(OH)D3 | 4 w (i.e. shorter duration than D3 groups) No analytical check of the doses in the capsules. Consumed at bedtime. | Increase of + 40.0 (final 25(OH)D* about 107 nmol/L) | |||||||||
| 20 μg/day 25(OH)D3 | Increase of + 76.1 (NB: assuming baseline of 67 nmol/L, final 25(OH)D about 143 nmol/L) | ||||||||||
| 50 μg/day 25(OH)D3 | Increase of + 206.4 (NB: assuming baseline of 67 nmol/L, final 25(OH)D about 273 nmol/L) | ||||||||||
| Wittwer, 2015 (unpublished) Vaes et al., 2018; Parallel, randomised, double‐blind intervention study | Subjects aged 65 y and older (mean 79 y), Serum 25(OH)D of 25–50 nmol/L Including frail subjects (Wittwer, 2015). n = 60 randomised (1 did not start due to violation of eligibility criteria), 5 drop‐outs, 54 completers, 3 with major protocol violation, i.e. increased sun exposure and non‐compliance) Compliance checked. | Wageningen, The Netherlands 51° N August‐April | 20 μg/day D3 | 24 w Analytical check of the doses in the capsules Capsules consumed with a standardised breakfast | Mean (SD) (unadjusted): 37.7 (7.0) | Model‐predicted mean (95%CI),‐model adjusted for baseline concentration and BMI): 71.6 (63.2–80.0) | mean (SD): 3.7 (1.2) | Yes | Inclusion: 20–35 (mean 26.8) | Full study report (Wittwer, 2015): analytical method not reported (Vaes et al., 2018) – At screening: Isotope dilution‐online solid phase extraction liquid chromatography‐tandem mass spectrometry (IDXLC‐MS/MS) – end of study: LC/MS/MS (in‐house method of the applicant, presented as validated) | – No individual with final serum 25(OH)D > 200 nmol/L (Wittwer, 2015). – No differences in serum Ca or urine Ca/creatinine ratios (Ca/creat) in morning spot urine, between groups at baseline and study end (Wittwer, 2015). – On average, all groups with urinary Ca/creat ratio remaining below the reference range of < 220 mg/g creatinine (0.62 mmol/mmol). – Also 23 subjects > 220 mg/g at one or more timepoints: considered as not clinically relevant by the authors(baseline: p = 0.29 CI 95%; study end: p = 0.95) No hypercalcemia observed (no serum Ca > 2.6 mmol/L). 76 AEs in 39 subjects (‘infections and infestation’, ‘metabolism and nutrition disorders’, ‘psychiatric disorders’, ‘nervous system disorders’, ‘cardiac disorders’, ‘vascular disorders’, ‘respiratory, thoracis and mediastinal disorders, gastrointestinal disorders, skin and subcutaneous tissue disorder, musculoskeletal and connective tissue disorders, renal and urinary disorders, general disorders and administration side conditions’, ‘injury, poisoning and procedural complications’). Among them: 19 AEs in 7 subjects receiving 5 μg 25(0H)D3; 17 AEs in 11 subjects receiving 10 μg 25(OH)D3, 18 AEs in 9 subjects receiving 15 μg 25(0H)D3. Also, 8 SAEs in 6 objects. Number of AEs and SAEs not significantly different between groups. None of the AEs or SAEs led to study discontinuation or changes in supplementation. Wittwer (2015): 8 AEs reported as being related to the study product. Vaes et al., 2018: no AE due to compound according to Ethics Committee. |
| 5 μg/day 25(OH)D3 | Mean (SD) (unadjusted): 43.4 (15.8) | Model‐predicted mean (95%CI),‐model adjusted for baseline concentration and BMI): 52.2 (44.4–60.2) | Mean (SD): 4.2 (1.6); | ||||||||
| 10 μg/day 25(OH)D3 | Mean (SD) (unadjusted): 38.3 (10.5) | Model‐predicted mean (95%CI),‐model adjusted for baseline concentration and BMI): 88.7 (81.4–96.1) | Mean (SD): 3.3 (1.3) | ||||||||
| 15 μg/day 25(OH)D3 | Mean (SD) (unadjusted): 38.6 (12.9) | Model‐predicted mean (95%CI),‐model adjusted for baseline concentration and BMI): 109.9 (102.5–117.2) | Mean (SD): 3.5 (1.5) |
AE: adverse event, BMI: Body mass index, Ca: calcium, Ca/create: calcium/creatinine ratio, CI: Confidence interval, IQR: interquartile range, m: months, HPLC: high‐performance liquid chromatography, LC‐MS/MS: liquid chromatography–mass spectrometry, NIST: National Institute of Standards and Technology; SAE: serious adverse event: SE: standard error, SD: Standard deviation, w: weeks.
Calculated by EFSA assuming a baseline of 67 nmol/L.
According to a communication from the applicant, the method is validated (the method is not described in the study report).
Value above the ‘suitable target value’ of 50 nmol/L considered by the Panel for serum 25(OH)D when setting AIs for vitamin D (EFSA NDA Panel, 2016b).
Regarding analytical methods, previous discussions on LC‐MS/MS and HPLC method, the vitamin D external quality assessment scheme (DEQAS), the US National Institute of Standards and Technology (NIST) and the vitamin D Standardisation Program (VDSP) present in previous EFSA opinions on dietary reference values for vitamin D and tolerable upper intake level for vitamin D in infants (EFSA NDA Panel, 2016b, 2018) were taken into account.
Suggested citation: EFSA NDA Panel (EFSA Panel on Nutrition, Novel Foods and Food Allergens) , Turck D, Castenmiller J, De Henauw S, Hirsch‐Ernst KI, Kearney J, Maciuk A, Mangelsdorf I, McArdle HJ, Naska A, Peláez C, Pentieva K, Siani A, Thies F, Tsabouri S, Vinceti M, Cubadda F, Frenzel T, Heinonen M, Marchelli R, Neuhauser‐Berthold M, Poulsen M, Prieto Maradona M, Schlatter JR, van Loveren H, Dumas C, Roldán‐Torres R, Steinkellner H and Knutsen HK, 2021. Scientific Opinion on the safety of calcidiol monohydrate produced by chemical synthesis as a novel food pursuant to Regulation (EU) 2015/2283. EFSA Journal 2021;19(7):6660, 30 pp. 10.2903/j.efsa.2021.6660
Requestor: European Commission
Question number: EFSA‐Q‐2018‐00514
Panel members: Dominique Turck, Jacqueline Castenmiller, Stefaan De Henauw, Karen Ildico Hirsch‐Ernst, John Kearney, Helle Katrine Knutsen, Alexandre Maciuk, Inge Mangelsdorf, Harry J McArdle, Androniki Naska, Carmen Pelaez, Kristina Pentieva, Alfonso Siani, Frank Thies, Sophia Tsabouri and Marco Vinceti.
Declarations of interest: The declarations of interest of all scientific experts active in EFSA's work are available at https://ess.efsa.europa.eu/doi/doiweb/doisearch.
Acknowledgements: The Panel wishes to thank the following for the support provided to this scientific output: the former member of the EFSA Novel Foods Working Group Yolanda Sanz for the contribution provided to the development of the opinion until 3 January 2020, the members of the EFSA SCER Cross‐cutting WG nanotechnologies and EFSA staff members Gabriela Precup and Patricia Romero Fernández.
Adopted: 25 May 2021
Notes
For conversion between μg and International Units (IU) of vitamin D intake: 1 μg = 40 IU and 0.025 μg = 1 IU. For conversion between nmol/L and ng/mL for serum 25(OH)D concentration: 2.5 nmol/L = 1 ng/mL.
In the presence of endogenous cutaneous vitamin D synthesis, the requirement for dietary vitamin D is lower or may be even zero.
Vitamin D from the diet or from cutaneous synthesis following UV irradiation is hydroxylated in the liver to 25‐hydroxyvitamin D or 25(OH)D, also named calcidiol or calcifediol (i.e. 25(OH)D2 or 25(OH)D3), which is the accepted biomarker of vitamin D status (EFSA NDA Panel, 2016b). The 25(OH)D is hydroxylated primarily in the kidneys to the biologically active metabolite 1,25‐dihydroxyvitamin D (calcitriol, i.e. 1,25(OH)2D3 or 1,25(OH)2D2). Vitamin D, 25(OH)D and 1,25(OH)2D are transported in the blood mainly by the vitamin D‐binding protein (DBP).
Commission Implementing Regulation (EU) 2017/2469 of 20 December 2017 laying down administrative and scientific requirements for applications referred to in Article 10 of Regulation (EU) 2015/2283 of the European Parliament and of the Council on novel foods. OJ L 351, 30.12.2017, pp. 64–71.
EFSA NDA Panel (EFSA Panel on Dietetic Products, Nutrition and Allergies), Turck D, Bresson J‐L, Burlingame B, Dean T, Fairweather‐Tait S, Heinonen M, Hirsch‐Ernst KI, Mangelsdorf I, McArdle HJ, Naska A, Neuhäuser‐Berthold M, Nowicka G, Pentieva K, Sanz Y, Siani A, Sjödin A, Stern M, Tomé D, Vinceti M, Willatts P, Engel K‐H, Marchelli R, Pöting A, Poulsen M, Salminen S, Schlatter J, Arcella D, Gelbmann W, de Sesmaisons‐Lecarré A, Verhagen H and van Loveren H, 2016. Guidance on the preparation and presentation of an application for authorisation of a novel food in the context of Regulation (EU) 2015/2283. EFSA Journal 2016;14(11):4594, 24 pp. https://doi.org/10.2903/j.efsa.2016.4594
The portion of particles with sizes smaller than this value is 10%.
Regulation (EC) No 1333/2008 of the European Parliament and of the Council of 16 December 2008 on food additives, https://eur-lex.europa.eu/legal-content/EN/TXT/?uri=CELEX%3A02008R1333-20201223
E.g. of 4.2 ng in semi‐skimmed milk to 96 ng in butter, mean values (Jakobsen and Saxholt, 2009).
Ranging from 0.5 to 1.2 µg in egg yolks (Ovesen et al., 2003).
0.11 µg in raw salmon flesh and 0.22 µg in raw trout (Ovesen et al., 2003).
0.07–0.14 µg in pork cuts and up to 0.34 µg in pork rind, mean values (Clausen et al., 2003).
0.48 µg in pork liver, Mattila et al., 1995; 0.51–0.98 µg in kidney of cows (Ovesen et al., 2003).
Mean: 3.1 and 58 ng, respectively (Jakobsen and Saxholt, 2009).
Not detectable contents up to 0.17 µg/100 g in beef liver, (Mattila et al., 1995).
According to the definition of the Guidance on risk assessment of the application of nanoscience and nanotechnologies in the food and feed chain: Part 1, human and animal health.
References
- Barger‐Lux MJ, Heaney RP, Dowell S and Holick MF, 1998. Vitamin D and its major metabolites: serum levels after graded oral dosing in healthy men. Osteoporosis International, 8, 222–230. [DOI] [PubMed] [Google Scholar]
- Beck M, 2016. Comparison of 25(0H)D concentration increases in human serum/plasma following oral supplementation of calcifediol (25‐hydroxyvitamin D3) or cholecalciferol (vitamin D3). DSM Proprietary unpublished data.
- Beck M and Punler M, 2017. The Comparative Metabolism of [14C]‐Calcifediol and [14C]‐Cholecalciferol Following Single Oral Administration in the Bile Duct Cannulated Male Han Wistar Rat. (Study conducted at Charles River Laboratories, Edinburgh, UK; Study No 172566; Report No. 37799). DSM Proprietary unpublished data.
- Beck M, Punler M, Real Sesma V, O'Neill J, Chenal E and Etheve S, 2017. The Comparative Metabolism of [14C]‐Calcifediol and [14C]‐Cholecalciferol Following Multiple Oral Administration in the Intact Male Han Wistar Rat. (Study conducted at Charles River Laboratories, Edinburgh, UK; Study No 172451; Report No. 37794). DSM Proprietary unpublished data.
- Bischoff‐Ferrari HA, Dawson‐Hughes B, Stöcklin E and Egli A, 2012. Oral Supplementation with 25(OH)D3 versus Vitamin D3: effects on 25(OH) levels, lower extremity function, blood pressure and markers of innate immunity. Journal of Bone and Mineral Research, 27, 160–169. [DOI] [PubMed] [Google Scholar]
- Blunt JW, Tanaka Y and DeLuca HF, 1968. The biological activity of 25‐hydroxycholecalciferol, a metabolites of vitamin D3. Proceedings of the National Academy of Sciences of the United States of America, 61, 1503–1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borel P, Caillaud D and Cano NJ, 2015. Vitamin D Bioavailability: State of the Art. Critical Reviews in Food Science and Nutrition, 55, 1193–1205. [DOI] [PubMed] [Google Scholar]
- Cashman KD, Seamans KM, Lucey AJ and Hill TR, 2012. Relative effectiveness of oral 25‐hydroxyvitamin D3 and vitamin D3 in raising wintertime serum 25‐hydroxyvitamin D in older adults. The American Journal of Clinical Nutrition, 95, 1350–1356. [DOI] [PubMed] [Google Scholar]
- Clausen I, Jakobsen J, Leth T and Ovesen L, 2003. Vitamin D3 and 25‐hydroxyvitamin D3 in raw and cooked pork cuts. Journal of Food Composition and Analysis, 16, 575–585. [Google Scholar]
- Commission Regulation (EC) No 1333/2008 of the European Parliament and of the Council of 16 December 2008 on food additives. Available online: https://eur-lex.europa.eu/legal-content/EN/TXT/?uri=CELEX%3A02008R1333-20201223
- Commission Regulation (EU) No 609/2013 of the European Parliament and of the Council of 12 June 2013 on food intended for infants and young children, food for special medical purposes, and total diet replacement for weight control and repealing Council Directive 92/52/EEC, Commission Directives 96/8/EC, 1999/21/EC, 2006/125/EC and 2006/141/EC, Directive 2009/39/EC of the European Parliament and of the Council and Commission Regulations (EC) No 41/2009 and (EC) No 953/2009. OJ L, 181, 35–56.
- Compston JE, Merrett AL, Hammett FG and Magill P, 1981. Comparison of the appearance of radiolabelled vitamin D3 and 25‐hydroxy‐vitamin D3 in the chylomicron fraction of plasma after oral administration in man. Clinical Science, (Lond) 60, 241–243. [DOI] [PubMed] [Google Scholar]
- Directive 2002/46/EC of the European Parliament and of the Council of 10 June 2002 on the approximation of the laws of the Member States relating to food supplements. Official Journal‐European Communities Legislation, 45, 51–57.
- EFSA (European Food Safety Authority), 2005. Opinion of the Scientific Panel on additives and products or substances used in animal feed (FEEDAP) on the evaluation of safety and efficacy of “Hy•D” (calcifediol), based on 25‐hydroxylcholecalciferol/25‐hydroxy‐pre‐cholecalciferol, as feed additive in accordance with Council Directive 70/524/EEC. EFSA Journal 2005;3(7):224, 35 pp. 10.2903/j.efsa.2005.224 [DOI] [Google Scholar]
- EFSA (European Food Safety Authority), 2009. Opinion of the Scientific Panel on additives and products or substances used in animal feed (FEEDAP) on the safety and efficacy of 25‐hydroxycholecalciferol as a feed additive for poultry and pigs. EFSA Journal 2009;7(2):969, 32 pp. 10.2903/j.efsa.2009.969 [DOI] [Google Scholar]
- EFSA ANS Panel (EFSA Panel on Food Additives and Nutrient Sources added to Food), Younes M, Aggett P, Aguilar F, Crebelli R, Dusemund B Filipicč M, Frutos MJ, Galtier P, Gundert‐Remy U, Kuhnle GG, Lambré C, Leblanc J‐C, Lillegaard IT, Moldeus P, Mortensen A, Oskarsson A, Stankovic I, Waalkens‐Berendsen I, Woutersen RA, Wright M, Di Domenico A, Fairweather‐Tait S, McArdle H, Smeraldi C and Gott D, 2018. Guidance on safety evaluation of sources of nutrients and bioavailability of nutrient from the sources. EFSA Journal 2018;16(6):5294, 35 pp. 10.2903/j.efsa.2018.5294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- EFSA BIOHAZ Panel (Panel on Biological Hazards), 2013. Scientific Opinion on the maintenance of the list of QPS biological agents intentionally added to food and feed (2013 update). EFSA Journal 2013;11(11):3449, 107 pp. 10.2903/j.efsa.2013.3449 [DOI] [Google Scholar]
- EFSA FEEDAP Panel (Panel on Additives and Products or Substances used in Animal Feed), 2018. Guidance on the characterisation of microorganisms used as feed additives or as production organisms. EFSA Journal 2018;16(3):5206, 24 pp. 10.2903/j.efsa.2018.5206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- EFSA NDA Panel (EFSA Panel on Dietetic Products, Nutrition and Allergies), 2012. Scientific Opinion on the Tolerable Upper Intake Level of Vitamin D. EFSA Journal 2012;10(7):2813, 45 pp. 10.2903/j.efsa.2012.2813 [DOI] [Google Scholar]
- EFSA NDA Panel (EFSA Panel on Dietetic Products, Nutrition and Allergies), 2016a. Guidance on the preparation and presentation of an application for authorisation of a novel food in the context of Regulation (EU) 2015/2283. EFSA Journal 2016;14(11):4594, 24 pp. 10.2903/j.efsa.2016.4594 [DOI] [Google Scholar]
- EFSA NDA Panel (EFSA Panel on Dietetic Products, Nutrition and Allergies), 2016b. Scientific opinion on dietary reference values for vitamin D. EFSA Journal 2016;14(10):e04547, 145 pp. 10.2903/j.efsa.2016.4547 [DOI] [Google Scholar]
- EFSA NDA Panel (EFSA Panel on Dietetic Products, Nutrition and Allergies), 2018. Scientific opinion on the update of the tolerable upper intake level for vitamin D for infants. EFSA Journal 2018;16(8):5365, 118 pp. 10.2903/j.efsa.2018.5365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- EFSA Scientific Committee , 2020. Public consultation on the draft EFSA Guidance on technical requirements for regulated food and feed product applications to establish the presence of small particles including nanoparticles. Available online: https://www.efsa.europa.eu/en/consultations/call/public-consultation-draft-efsaguidance-technical-requirements [DOI] [PMC free article] [PubMed]
- EFSA Scientific Committee , Hardy A, Benford D, Halldorsson T, Jeger MJ, Knutsen HK, More S, Naegeli H, Noteborn H, Ockleford C, Ricci A, Rychen G, Schlatter JR, Silano V, Solecki R, Turck D, Younes M, Chaudhry Q, Cubadda F, Gott D, Oomen A, Weigel S, Karamitrou M, Schoonjans R and Mortensen A, 2018. Guidance on risk assessment of the application of nanoscience and nanotechnologies in the food and feed chain: part 1, human and animal health. EFSA Journal 2018;16(7):5327, 95 pp. 10.2903/j.efsa.2018.5327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- European Commission , 2002. Scientific Committee on Food. Opinion on the Tolerable Upper intake level of vitamin D. Available online: http://europa.eu.int/comm/food/fs/sc/scf/index_en.html
- European Commission , 2004. Reports on Tasks for Scientific Cooperation (SCOOP) Task 3.2.11. Assessment of the dietary exposures to arsenic, cadmium, lead and mercury of the population of the EU Member States Directorate General Health and Consumer Protection.
- Haddad JG and Rojanasathit S, 1976. Acute administration of 25‐hydroxycholecalciferol in man. The Journal of Clinical Endocrinology and Metabolism, 42, 284–290. [DOI] [PubMed] [Google Scholar]
- Institute of Medicine , 1997. Dietary Reference Intakes for Calcium, Phosphorus, Magnesium, Vitamin D, and Fluoride. National Academy Press, Washington, DC. [PubMed] [Google Scholar]
- Jakobsen J and Saxholt E, 2009. Vitamin D metabolites in bovine milk and butter. Journal of Food Composition and Analysis, 22, 472–478. [Google Scholar]
- Jetter A, Egli A, Dawson‐Hughes B, Staehelin HB, Stoecklin E, Goessl R, Henschkowski J and Bischoff‐Ferrari HA, 2014. Pharmacokinetics of oral vitamin D3 and calcifediol. Bone, 59, 1419. [PubMed] [Google Scholar]
- Jones AN, Shafer MM, Keuler NS, Crone EM and Hansen KE, 2012. Fasting and postprandial spot urine calcium‐to‐creatinine ratios do not detect hypercalciuria. Osteoporosis International, 23, 553–562. 10.1007/s00198-011-1580-7. Epub 2011 Feb 24. PMID: 21347742; PMCID: PMC3295234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunz I, Beck M, Schoop R and Etheve S, 2016. Leatherhead Final Study Report: Response of serum 25‐hydroxyvitamin D to different doses of Catcifediol 0.25 SD/S compared to vitamin D3 supplementation: A randomized, controlled, double blind, tong term pharmacokinetic study. DSM Proprietary unpublished data.
- Mattila PH, Piironen VI, Uusi‐Rauva EJ and Koivistoinen PE, 1995. Contents of cholecalciferol, ergocalciferol, and their 25‐hydroxylated metabolites in milk products and raw meat and liver as determined by HPLC. Journal of Agricultural and Food Chemistry, 43, 2394–2399. [Google Scholar]
- Navarro‐Valverde C, Sosa‐Henriquez M, Alhambra‐Exposito MR and Quesada‐Gomez JM, 2016. Vitamin D3 and calcidiol are not equipotent. The Journal of Steroid Biochemistry and Molecular Biology, 164, 205–208. [DOI] [PubMed] [Google Scholar]
- Ovesen L, Brot C and Jakobsen J, 2003. Food contents and biological activity of 25‐hydroxyvitamin D: a vitamin D metabolite to be reckoned with? Annals of Nutrition and Metabolism, 47, 107–113. [DOI] [PubMed] [Google Scholar]
- Reeve LE, Jorgensen NA and DeLuca HF, 1982. Vitamin D compounds in cow's milk. The Journal of Nutrition, 112, 667–672. [DOI] [PubMed] [Google Scholar]
- Remus T and Verbaan I, 2016. Micronucleus test in bone marrow cells of the rat with DSM047117. (Study conducted at WIL Research Europe B.V., 5231 DD's Hertogenbosch, The Netherlands; WIL study number 511353). DSM Proprietary unpublished data.
- Remus T and Verspeek‐Rip C, 2016. Evaluation of the mutagenic activity of DSM047117 in an in vitro mammalian cell gene mutation test with L5178Y mouse lymphoma cells (Study conducted at WIL Research Europe B.V., 5231 DD's Hertogenbosch, The Netherlands; WIL study number 511352). DSM Proprietary unpublished data.
- SCF (Scientific Committee on Food), 2003. Opinion on the Tolerable Upper Intake Level of vitamin D., 35 pp.
- Shepard RM and DeLuca HF, 1980. Plasma concentration of vitamin D3 and its metabolites in the rat as influenced by vitamin D3 or 25‐hydroxyvitamin D3 intakes. Archives of Biochemistry and Biophysics, 202, 43–53. [DOI] [PubMed] [Google Scholar]
- Stamp TC, Haddad JG and Twigg CA, 1977. Comparison of oral 25‐hydroxycholecalciferol, vitamin D, and ultraviolet light as determinants of circulating 25‐hydroxyvitamin D. Lancet, 1, 1341–1343. [DOI] [PubMed] [Google Scholar]
- Thiel A, Scase K, Hofmann P, Köhnlein M and Martland M, 2007. 3‐Month Oral Toxicity Study with Rovimix® D3‐500 by Dietary Administration Followed by a 4‐Week Recovery Period in Wistar Rats (Study conducted at Notox BV, Hambakenwetering 7, 5231 DD's‐ Hertogenbosch, The Netherlands, Notox Study 458257). DSM Proprietary unpublished data.
- Thiel A, van Otterdijk F, Etheve S, Schierle J and Hard G, 2014. 90 day oral Toxicity study with DSM047117 by Dietary administration in the rat followed by a 28 day recovery period (Study performed at: WIL Research Europe B.V., 5231 DD s. Hertogenbosch, The Netherlands, WIL Study Number: Project 503200). DSM Proprietary unpublished data. [Google Scholar]
- Vaes AMM, Tieland M, de Regt MF, Wittwer J, van Loon LJC and de Groot L, 2018. Dose‐response effects of supplementation with calcifediol on serum 25‐hydroxyvitamin D status and its metabolites: a randomized controlled trial in older adults. Clinical Nutrition, 37, 808–814. 10.1016/j.clnu.2017.03.029 [DOI] [PubMed] [Google Scholar]
- Weber E and Arcelin G, 2004. Calcifediol: Acute Oral Toxicity Study in Rats. (‘Study performed at RCC Ltd, Wölferstr”. 4, CH‐4414 Füllinsdorf, Switzerland; RCC Project Number 844765). DSM Proprietary unpublished data.
- Weber E and Schulz M, 2005. Chromosome Aberration Test in Human Lymphocytes in vitro with Calcifediol. DSM Proprietary unpublished data.
- Wittwer J, 2015. Dose finding study in physically non‐frail and (pre‐)frail elderly to measure 25(OH) Vitamin D levels after supplementation with HY.D Calcifediol 25 SD/S and Vitamin D3 (D‐Dose study) DSM Proprietary unpublished data.
- Wöhrle T and Sokolowski A, 2013. DSM047117: Salmonella typhimurium and Escherichia coli reverse mutation assay. (Study conducted at Harlan CCR; D‐64380 Rossdorf; Harlan CCR study Number 1533500). DSM Proprietary unpublished data.