

In this study, Burgess et al. show that replication of SARS-CoV-2, the agent responsible for the COVID-19 pandemic, and a seasonal human β-coronavirus, HCoV-OC43, can be suppressed by depletion of nuclear RNA methyltransferase METTL3 or cytoplasmic m6A reader proteins YTHDF1 and YTHDF3 and by a highly specific small molecule METTL3 inhibitor. Their findings establish that coronavirus RNAs are m6A-modified and host m6A pathway components control β-coronavirus replication, and illustrate the therapeutic potential of inhibiting METTL3 to restrict coronavirus reproduction.
Keywords: N6-methyladenosine, SARS-CoV-2, HCoV-OC43, coronavirus, virus–host interactions, RNA modification, direct RNA sequencing, nanopore sequencing
Abstract
N6-methyladenosine (m6A) is an abundant internal RNA modification, influencing transcript fate and function in uninfected and virus-infected cells. Installation of m6A by the nuclear RNA methyltransferase METTL3 occurs cotranscriptionally; however, the genomes of some cytoplasmic RNA viruses are also m6A-modified. How the cellular m6A modification machinery impacts coronavirus replication, which occurs exclusively in the cytoplasm, is unknown. Here we show that replication of SARS-CoV-2, the agent responsible for the COVID-19 pandemic, and a seasonal human β-coronavirus HCoV-OC43, can be suppressed by depletion of METTL3 or cytoplasmic m6A reader proteins YTHDF1 and YTHDF3 and by a highly specific small molecule METTL3 inhibitor. Reduction of infectious titer correlates with decreased synthesis of viral RNAs and the essential nucleocapsid (N) protein. Sites of m6A modification on genomic and subgenomic RNAs of both viruses were mapped by methylated RNA immunoprecipitation sequencing (meRIP-seq). Levels of host factors involved in m6A installation, removal, and recognition were unchanged by HCoV-OC43 infection; however, nuclear localization of METTL3 and cytoplasmic m6A readers YTHDF1 and YTHDF2 increased. This establishes that coronavirus RNAs are m6A-modified and host m6A pathway components control β-coronavirus replication. Moreover, it illustrates the therapeutic potential of targeting the m6A pathway to restrict coronavirus reproduction.
Differential RNA modification by N6-methyladenosine (m6A) plays a major role in the control of gene expression, influencing numerous biological processes that impact viral infection biology (Roundtree et al. 2017; Gokhale et al. 2020). m6A is the most abundant internal modification of both coding (Desrosiers et al. 1974; Perry and Kelley 1974) and noncoding RNAs (Alarcón et al. 2015; Patil et al. 2016) and regulates both their biological function and stability (Meyer and Jaffrey 2014). Typically, m6A installation is performed by a nuclear methytransferase (“writer”) complex (METTL3/METTL14/WTAP) that includes the essential methyltransferase-like enzyme 3 (METTL3) catalytic subunit (Bokar et al. 1997; Liu et al. 2014; Wang et al. 2016; Schöller et al. 2018). However, METTL3 is also found at the promoters of specific genes independent of METTL14 and catalyzes cotranscriptional m6A methylation of associated transcripts, an activity required for the maintenance of a leukemic state (Barbieri et al. 2017).
The positions of m6A installation within mRNAs is tightly controlled and the modification is dynamic. The extent of cytoplasmic mRNA m6A demethylation remains controversial; however, at least one nuclear demethylase (“eraser”), ALKBH5, has been identified (Zheng et al. 2013; Ke et al. 2017; Darnell et al. 2018). m6A-modified RNAs are recognized by a number of RNA-binding proteins (“readers”) including nuclear YTHDC1 and three cytoplasmic paralogs YTHDF1, YTHDF2, and YTHDF3 as well as other RNA binding proteins (Lasman et al. 2020; Zaccara and Jaffrey 2020). Whereas it has long been known that many viral mRNAs are m6A-modified, our understanding of the extent to which host components that install, remove, or recognize m6A-modified RNA either enhance or suppress virus infection through varied mechanisms remains in its infancy (Gonzales-van Horn and Sarnow 2017; Tsai et al. 2018; Williams et al. 2019).
Besides shaping host innate immune responses critical for the recognition and response to viral infection, m6A can also directly influence gene expression and reproduction of viruses with nuclear life cycles (Rubio et al. 2018; Williams et al. 2019; Winkler et al. 2019; Gokhale et al. 2020; Kim et al. 2020b; Lu et al. 2020; Price et al. 2020; Shulman and Stern-Ginossar 2020). Several positive-sense, single-stranded RNA viruses that replicate in the cytoplasm have also been shown to contain m6A (Gokhale et al. 2016; Lichinchi et al. 2016; Hao et al. 2019). In the case of hepatitis C virus (HCV), depletion of m6A writers METTL3/14 limited HCV infectious particle production and protein expression without detectably changing viral RNA synthesis (Gokhale et al. 2016; Gonzales-van Horn and Sarnow 2017). Although β-coronaviruses represent a distinct and important RNA virus family (Coronaviridae) that also replicates exclusively in the cytoplasm, how their productive growth cycle might be regulated by the host m6A modification machinery is largely unknown. In addition to widespread circulating β-coronaviruses species associated with mild disease exemplified by HCoV-OC43 (Vijgen et al. 2005), emergent viruses such as SARS-CoV-2 can be far more threatening as evidenced by the COVID-19 global pandemic (Coronaviridae Study Group of the International Committee on Taxonomy of Viruses 2020). Understanding how the host m6A modification machinery influences β-coronavirus reproduction could provide new therapeutic opportunities to interfere with virus replication and spread and possibly reveal differences between pandemic and nonpandemic strains.
Results
Host m6A factors promote HCoV-OC43 replication and spread
To determine whether the cellular m6A modification machinery regulates β-coronavirus replication and identify the host factors involved, a focused RNAi screen was performed. Two distinct siRNAs were used to deplete 14 individual host factors reported to participate in the installation, removal, or recognition of m6A and their efficacy in target depletion validated (Supplemental Fig. S1A). RNAi-treated cultures of MRC-5 normal, diploid human lung fibroblasts in 96-well plates were subsequently infected at low multiplicity with HCoV-OC43, a seasonal, circulating β-coronavirus typically associated with mild respiratory disease in humans. As a measure of virus reproduction and spread, infected cells expressing coronavirus nucleocapsid (N) protein were detected by indirect immunofluorescence and quantified using a high-content imaging platform. Compared with control, nonsilencing siRNA-treated cultures, depletion of select m6A methyltransferase subunits and m6A recognition proteins significantly reduced the percentage of N-expressing cells (Fig. 1A). Both siRNAs specific for the m6A methyltransferase core subunit METTL14 and the catalytic subunit METTL3 reduced the N-positive fraction by ∼30% and 20%, respectively (Fig. 1A). The slightly reduced efficacy observed using METTL3 versus METTL14 siRNA is consistent with METTL3 acting in a catalytic as opposed to stoichiometric fashion and difficulties associated with achieving sufficient enzyme depletion. Only one siRNA for the remaining methyltransferase core subunit WTAP reduced virus replication (Fig. 1A), which was not explained by elevated cytotoxicity (Supplemental Fig. S1B). In contrast, depletion of noncore methyltransferase subunits (RBM15, RBM15B, and ZC3H13), with two different siRNAs each, either did not significantly reduce the percentage of cells that were detectably infected or did so only with a single siRNA (VIRMA) (Fig. 1A). Furthermore, depletion of a different m6A methyltransferase, METTL16, did not detectably alter the infection (Fig. 1A). Thus, HCoV-OC43 reproduction and spread in MRC-5 lung fibroblasts was dependent on a METTL3-containing m6A writer.
Figure 1.

Focused RNA interference screen implicates METTL3 and the YTHDF readers in control of β-coronavirus replication. (A) Human MRC-5 normal lung fibroblasts were transfected with a set of validated siRNAs (two per factor) targeting 14 host components of the m6A pathway and cultured for 72 h before infection with HCoV-OC43 at MOI = 0.001. After 48 h, cells were fixed and the infection assessed by indirect immunofluorescence using an antibody to the viral nucleocapsid protein (N). The percentage of N-positive cells per well was determined using a CellInsight CX7 LZR high-content screening platform. Each assay was performed three times with technical duplicates and normalized to control siRNA treated cells. (B) The impact of siRNA depletion on infectious viral titer was determined by collecting supernatant culture media from A and establishing TCID50 on MRC-5 cells. (C) A549+ACE2 cells were transfected with validated siRNAs targeting METTL3, YTHDF1, YTHDF2, and YTHDF3, either individually or as a mix of single siRNAs to all three YTHDF proteins using siRNA#1 in each case. After 72 h, the cells were infected with icSARS-CoV-2-mNG at MOI = 0.1 for 48 h and then fixed and scored for green fluorescence. The extent of spread was normalized to cells transfected with control siRNA. In each case, an ANOVA test with Dunnett multiple comparison correction was used to establish statistical significance compared with control siRNA. (*) P < 0.033, (**) P < 0.002, (***) P < 0.001.
In addition to host m6A installation components, depletion of specific m6A recognition proteins (readers) reduced the fraction of HCoV-OC43-infected cells. The two siRNAs targeting the nuclear m6A reader YTHDC1 reduced the fraction of N-positive cells by 30%–45% (Fig. 1A). Likewise, both siRNAs against the cytoplasmic m6A readers YTHDF1 and YTHDF3 reduced the N-positive cell number by 40%–50% and 25%–40%, respectively (Fig. 1A). This phenotype was selective for YTHDF1 or YTHDF3 depletion, as only a single siRNA targeting YTHDF2 reduced the N-positive cell number by 13% (Fig. 1A). Additionally, we found that at high multiplicity of infection (MOI), codepletion of YTHDF1, YTHDF2, and YTHDF3 did not detectably reduce overall N protein levels at 24 h beyond the reduction observed from depletion of either YTHDF1 or YTHDF3 alone (Supplemental Fig. S1D). Finally, only a single siRNA specific for the known demethylases ALKBH5 or FTO reduced the fraction that were infected by >25% (Fig. 1A) but, in the case of FTO siRNA #1, might be explained in part by the observed 20% reduction in cell viability (Supplemental Fig. S1B).
To establish whether the decrease in N-positive cells genuinely reflected reduced viral replication, the infectious virus titers from individual RNAi-treated cultures was measured by TCID50 assay. Compared with control, nonsilencing siRNA-treated cultures, depletion of METTL3, YTHDF1, or YTHDF3 reduced virus replication by between threefold and 24-fold (Fig. 1B). Taken together, these results show that the m6A methyltransferase core components METTL3/METTL14 and a subset of m6A reader proteins (YTHDF1, YTHDF3, YTHDC1) are important for HCoV-OC43 reproduction and spread.
SARS-CoV-2 productive replication is antagonized by writer and reader depletion
We next asked whether the host m6A methyltransferase and m6A recognition proteins also impact the reproduction of the recently emerged pandemic β-coronavirus SARS-CoV-2 by performing a similar RNAi screen directed against a more limited set of cellular targets. Because MRC-5 fibroblasts are not sufficiently permissive to study SARS-CoV-2 productive growth, we used human A549 lung carcinoma cells engineered to constitutively express the human ACE2 receptor (A549+ACE2). Additionally, a SARS-CoV-2 reporter virus expressing mNeonGreen (icSARS-CoV-2-mNG) was used to identify infected cells and directly monitor virus reproduction and spread (Xie et al. 2020). Compared with a control, nonsilencing siRNA, the siRNAs against METTL3 reduced the percentage of infected cells by 78% or 81% (Fig. 1C). Likewise, the siRNAs specific for the m6A readers YTHDF2 or YTHDF3 reduced the percentage of mNeonGreen-positive cells by 42%–66% or 75%–76% of the control, respectively (Fig. 1C). In contrast, the YTHDF1 siRNAs initially tested reduced SARS-CoV-2 infection to differing degrees: 68% (YTHDF1 siRNA#1) and 23% (YTHDF1 siRNA#2). Testing of two additional YTHDF1-targeting siRNAs (YTHDF1 siRNA#3 and siRNA#4), however, resulted in a 79% and 89% reduction of SARS-CoV-2 compared with control siRNA, consistent with the more robust inhibition observed using YTHDF1 siRNA#1. Codepletion of all three cellular m6A readers reduced the mNeonGreen-positive cell number by 72% (Fig. 1C). No major impact on cell viability was detected for any siRNA treatment of A549+ACE2 cells (Supplemental Fig. S1D). The apparent greater sensitivity of SARS-CoV-2 to METTL3 and reader protein depletion compared with HCoV-OC43 may be due to biological differences between the two viruses but may also be explained by differences in the cell types or the dynamics of each infection. As exemplified by METTL3 and YTHDF1, we can exclude differences in the efficiency of knockdown between the two cell lines (Supplemental Fig. S1A,E). These data establish that SARS-CoV-2 productive growth is reliant on the major host m6A methyltransferase and cytoplasmic m6A recognition proteins. Thus, a seasonal, circulating human β-coronavirus (HCoV-OC43) and a recently emerged pandemic β-coronavirus (SARS-CoV-2) exhibit similar dependencies upon the host m6A methyltransferase and select m6A reader proteins.
Nuclear accumulation of cytoplasmic m6A readers upon HCoV43 infection
We next examined how the host m6A modification machinery responded to β-coronavirus infection. No substantial change in the overall abundance of the 14 components of the cellular m6A modification machinery evaluated was detected over a 72-h time course in HCoV-OC43-infected MRC-5 fibroblasts (Fig. 2A). However, the subcellular distribution of the m6A readers YTHDF1 and YTHDF3 was altered by infection. While predominantly cytoplasmic in uninfected MRC-5 fibroblasts, YTHDF1 and YTHDF3 both accumulated within the nucleoplasm as evidenced by nucleolar sparing in HCoV-OC43-infected MRC-5 cells (Fig. 2B). Quantitation of nuclear signal intensity in >100 infected cells per condition showed a statistically significant increase in nuclear YTHDF1 and YTHDF3 and, to a lesser extent, METTL3 (Fig. 2C). The distribution of YTHDF2, in contrast, was not detectably altered by infection as it remained predominately cytoplasmic along with the viral N protein (Fig. 2B,C). To evaluate this further using a different methodology, equal numbers of infected and uninfected MRC-5 cells were fractionated into a soluble (cytosolic) fraction and a particulate (nuclear) fraction and probed by immunoblotting (Fig. 2D). Consistent with the indirect immunofluorescence analysis, METTL3 was detected in both fractions (Fig. 2D, lanes 1,2) but was most abundant in the nuclear fraction. Partitioning of METTL3 and YTHDF2 between nuclear and cytosolic fractions was not detectably changed by HCoV-OC43 infection (Fig. 2D, cf. lanes 3,4 and 1,2), and as such, the small increase in nuclear METTL3 detected by indirect immunofluorescence (Fig. 2C) was not recapitulated. In contrast, both YTHDF1 and YTHDF3 were more abundant in the cytoplasmic fraction of uninfected cells but were more abundant in the nuclear fraction in cells infected with HCoV-OC43. Thus, nuclear accumulation of cytoplasmic m6A recognition proteins in response to HCoV-OC43 infection of MRC-5 lung fibroblasts was selective for YTHDF1 and YTHDF3.
Figure 2.

HCoV-OC43 infection results in the nuclear accumulation of METTL3, YTHDF1, and YTHDF2. (A) Lysates from uninfected (UI) or HCoV-OC43-infected MRC-5 cells were prepared at 8, 24, 48, and 72 h postinfection at MOI = 3 and probed by immunoblotting using antibodies to methyltransferase subunits (METTL3, METTL14, WTAP, RBM15, and RBM15B), demethylases (ALKBH5, FTO) and m6A binding proteins (YTHDC1, YTHDF1, YTHDF2, and YTHDF3). Arrowheads denote target bands. (B) Indirect immunofluorescence images of representative uninfected or HCoV-OC43-infected MRC5 cells (24 hpi, MOI = 3) with primary antibodies to METTL3, YTHDF1, YTHDF2, and YTHDF3. Scale bar, 10 µm. (C) Quantitation of nuclear signal for each primary antibody used in panel B after normalization to background fluorescence. Analysis was performed on ≥100 cells/condition and statistical significance determined using an F-test. (****) P ≤ 0.0001, (**) P ≤ 0.0047, (ns) not significant. (D) Uninfected and HCoV-OC43-infected MRC-5 cells (as in B) were rapidly lysed to produce insoluble particulate (nuclear [N]) and soluble (cytosolic [C]) fractions and probed by immunoblotting using antibodies to METTL3, YTHDF1, YTHDF2, and YTHDF3. Lysis and subcellular fractionation efficiency were assessed using antibodies to cytoplasmic β-tubulin and nuclear histone H3.
Redistribution and nuclear accumulation of YTHDF1, YTHDF2, or YTHDF3 was not detected in MRC-5 cells infected with UV-inactivated HCoV-OC43 and much reduced in HCoV-OC43-infected cells treated with the viral RNA synthesis inhibitor remdesivir (Supplemental Fig. S2A,B), indicating that nuclear YTHDF1/3 accumulation is dependent upon virus gene expression and/or the activity of the viral RNA-dependent RNA polymerase. While cytoplasmic YTHDF1/3 were redistributed by HCoV-OC43 infection of MRC-5 cells (Fig. 2B,C), consistent alterations in the subcellular distribution of m6A modification machinery components normally residing within the nucleus was not observed except for a modest reduction in nuclear RBM15 and RBM15B (Supplemental Fig. S3A,B). In contrast, no changes in the subcellular distribution of any components of the host m6A modification machinery were detected in SARS-CoV-2-infected A549+ACE2 cells (Supplemental Fig. S3C) or in Vero E6 cells (data not shown). Whether this reflects differences in how HCoV-OC43 and SARS-CoV-2 impact YTHDF1/3 subcellular distribution, the different inoculum sizes, or cell line-specific differences such as m6A factor abundance remains to be explored.
β-Coronavirus RNAs are m6A-modified
To determine whether m6A installation on cellular transcripts is altered by infection with either SARS-CoV-2 or HCoV-OC43 RNAs, we performed meRIP-seq (Fig. 3) on six biological replicates each of infected and uninfected A549+ACE2 cells (SARS-CoV-2, MOI = 0.1, 48 h) or MRC-5 cells (HCoV-OC43, MOI = 3, 48 h) and observed robust clustering of biological replicates (Supplemental Fig. S4A,B). Strict peak-calling using ExomePeak2 (Meng et al. 2014) identified between 6000 and 11,000 peak regions across data sets (Fig. 3A), all of which were enriched for the canonical DRACH motif associated with sites of m6A installation (Fig. 3B) and primarily clustered toward the boundary between the CDS and 3′ UTR (Fig. 3C), regardless of whether cells were infected or not.
Figure 3.

SARS-CoV-2 and HCoV-OC43 RNAs are m6A-modified. (A) meRIP-seq data sets profiling of SARS-CoV-2-infected A549+ACE2 cells and HCOV-OC43-infected MRC-5 cells were analyzed to determine numbers of significant (Padj < 0.05) peak ranges, enriched over input, in each comparison. (B) Sequence motif analysis of peak regions present in the cellular meRIP-seq data sets show enrichment for the classical RRACH/DRACH motif associated with installation of m6A. (C) Metagene analysis of m6A-peak region distribution across cellular RNAs with annotated 5′ and 3′ untranslated regions (UTR) and coding sequences (CDS). (D) Integration of putative m6A peak ranges (purple) identified by meRIP-seq of SARS-CoV-2-infected A549+ACE2 cells with candidate m6A sites (red) identified through comparative profiling of nanopore direct RNA sequencing (DRS) data sets (SARS-CoV-2-infected A549+ACE2 cells with or without STM2457). The top track shows the normalized coverage for a representative biological replicate of the meRIP-seq paired INPUT (black) and IP (red) data sets (see Supplemental Fig. S4 for additional replicates). To maximize sensitivity and accuracy, comparative DRS analyses were performed using DRUMMER at three different levels: full exome (i.e., all reads), processed exome (i.e., only reads containing the leader sequence), and isoform level (i.e., data sets aligned to the transcriptome rather than genome). The canonical SARS-CoV-2 transcriptome structure is shown below. (E) As in D but for HCOV-OC43-infected MRC-5 cells with or without STM2457.
We next used the virus-infected meRIP-seq data sets to examine whether m6A is installed on SARS-CoV-2 or HCoV-OC43 RNAs. Due to the extremely compact organization of β-coronavirus genomes, we turned to MACS2 (Zhang et al. 2008) for peak-calling and identified 14 (SARS-CoV-2) and six (HCoV-OC43) robust and reproducible peak regions enriched in the IP samples that we posit reflect the presence of one or multiple m6A residues (Fig. 3D,E; Supplemental Fig. S4C,D). It is notable that several peaks were placed in regions represented by the genome length transcripts (gRNA) only, suggesting that both gRNA and subgenomic RNAs (sgRNAs) are m6A-modified. We also compared the SARS-CoV-2 peak regions with the meRIP-seq and miCLIP data from SARS-CoV-2-infected Vero and Huh7 cells reported by Liu and colleagues (Liu et al. 2021). Despite the differences in cell lines and infection conditions, 10 of the meRIP-seq peak regions were found to overlap between the studies and four of our meRIP-seq peaks overlapped with sites identified by miCLIP (Supplemental Fig. S5). The concordance of these independent studies using established human cancer cell lines (A549 lung adenocarcinoma, Huh7 hepatocellular carcinoma) and a nontransformed established African green monkey kidney cell line (Vero) strengthens the conclusion that the genomic and/or subgenomic RNAs of SARS-CoV-2 can contain m6A.
By comparing modified with unmodified RNAs, we have used nanopore direct RNA sequencing (DRS) to identify recurring sequence determination errors that correspond to modified bases and map m6A sites at nucleotide- and transcript-level resolution (Price et al. 2020). Indeed, there was good agreement between sites detected using this DRS approach and peak regions identified by meRIP-seq for polyadenylated RNAs isolated from human adenovirus type 5 (Ad5)-infected cells. To test whether DRS could effectively detect m6A installation on β-coronavirus RNAs, cells were infected with SARS-CoV-2 (A549+ACE2) or HCoV-OC43 (MRC-5) under the same conditions used for meRIP-seq in the presence of STM2457, a new and highly selective small molecule inhibitor of METTL3 activity (Yankova et al. 2021), or a structurally related control compound STM2120 that is >3000-fold less potent as an METTL3 inhibitor as measured by in vitro methylation assay (Supplemental Fig. S6). After subjecting the poly(A) fraction to DRS (Fig. 3; Supplemental Fig. S7A,B), the structures of the SARS-CoV-2 mRNAs were found to be in good agreement with an earlier DRS study (Kim et al. 2020a). Comparative analyses on these data sets using two different informatic approaches, DRUMMER (Price et al. 2020) and ELIGOS2 (Jenjaroenpun et al. 2021), were subsequently performed. Surprisingly, only a single low-confidence site in SARS-CoV-2 that did not match the NNACN motif characteristic of host m6A sites was identified along with two high-confidence sites in HCoV-OC43 (AAACT and GAACT, respectively) that did conform to the degenerate m6A motif (Fig. 3; Supplemental Fig. S7A,B). None of these sites, however, mapped within peak regions identified by meRIP-seq, raising the possibility that they are not genuine m6A installation sites. Thus, unlike the many sites identified by DRS on RNA polymerase II-transcribed adenoviral mRNAs and that were frequently contained within meRIP-seq peak regions, concordance among DRS-detected sites and meRIP-seq peak regions was not observed on β-coronavirus RNAs, which are transcribed by a virus-encoded RNA-dependent RNA polymerase in cytoplasmic structures unique to these viruses.
Small molecule inhibition of METTL3 restricts β-coronavirus replication and spread
Having established that m6A can be detected on β-coronavirus RNAs and that depletion of either METTL3 or individual cytoplasmic m6A reader proteins interferes with productive replication of both viruses, we asked whether selective inhibition of METTL3 catalytic activity using STM2457 could restrict β-coronavirus replication. Following low-multiplicity infection of either MRC-5 lung fibroblasts with HCoV-OC43 or A549+ACE2 lung carcinoma cells with SARS-CoV-2, cultures were treated with vehicle (DMSO), the active METTL3 inhibitor STM2457, or control compound STM2120. As in Figure 1, viral replication and spread was monitored using the high-content imaging platform to score N protein expression (HCoV-OC43) or mNeonGreen fluorescence (icSARS-CoV-2-mNG). Compared with DMSO or STM2120, the higher concentrations of METTL3 inhibitor STM2457 clearly reduced the number of HCoV-OC43-infected MRC-5 cells (Fig. 4A,E) and icSARS-CoV-2-mNG-infected A549+ACE2 cells (Fig. 4B,E). Minor differences in the viability of MRC-5 cells and A549+ACE2 cells were only detected at higher concentrations, indicating that the antiviral activity of STM2457 did not result from general toxicity under these assay conditions (Fig. 4C,D). For HCoV-OC43, the reduction in infected cell number was first evident at 8 µM STM2457 and increased further in a dose-dependent manner up to 30 µM, where a >80% inhibition was observed (Fig. 4A). The half maximal inhibitory concentration (IC50) for STM2457 was ∼21 µM. SARS-CoV-2 reproduction in A549+ACE2 cells was also suppressed by STM2457, with a 10% reduction in mNeonGreen-expressing cells evident at 1.69 µM and increasing in a dose-dependent manner until a >90% reduction at 30 µM (Fig. 4B). The IC50 for STM2457 against SARS-CoV-2 was 16.84 µM. Note that use of two different infection models precludes a direct comparison of the sensitivity of the two viruses to STM2457.
Figure 4.
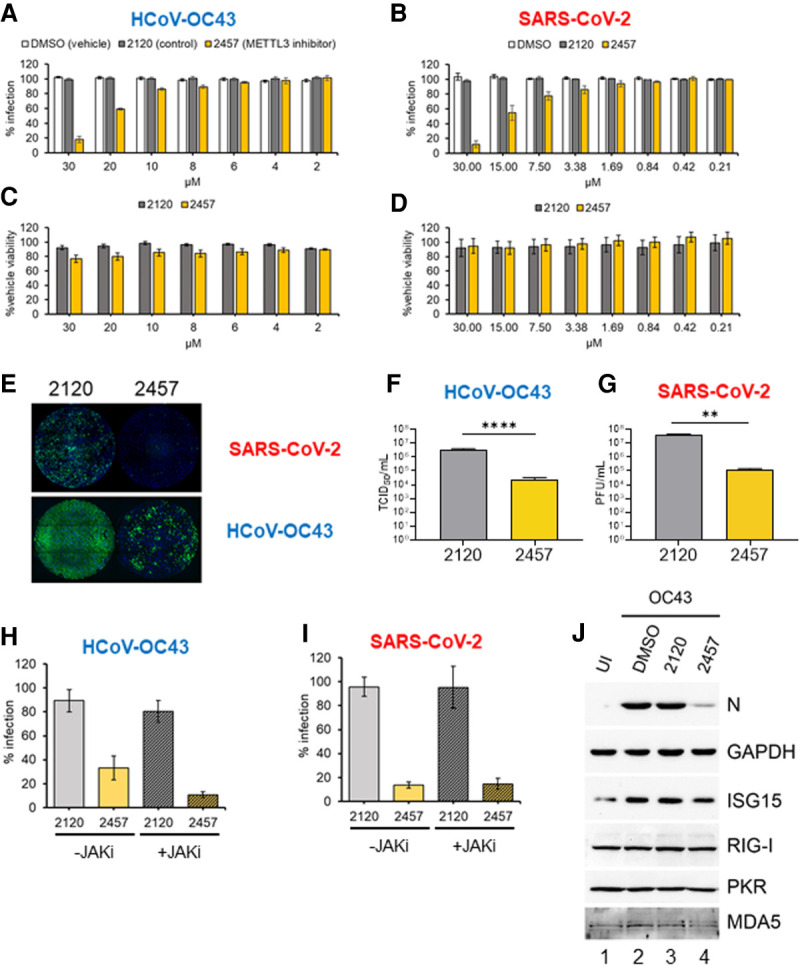
Inhibition of METTL3 activity suppresses β-coronavirus replication. (A) MRC-5 cells were infected with HCoV-OC43 at MOI = 0.001 for 48 h in the presence of METTL3 inhibitor (STM2457, yellow), inactive control compound (STM2120, gray), or vehicle (DMSO, white) at the indicated concentrations. Identification of infected cells by indirect immunofluorescence for nucleocapsid protein was as described in Figure 1A. (B) A549+ACE2 cells were infected with icSARS-CoV-2-mNG at MOI = 0.1 for 48 h, and the percentage of cells infected was established by green fluorescence and normalized to infection of nontreated cells. (C, D) The viability of MRC-5 cells (C) and A549+ACE2 cells (D) in the presence of concentrations of STM2120 or STM2457 used in the infection assays shown in A and B was assessed using a commercial ATP quantitation assay. Cells were maintained at either 33°C or 37°C, respectively, in culture medium containing diluted compound for 48 h prior to lysis. Each experiment was conducted three times with internal duplicates, normalized to DMSO-treated cells processed in parallel and plotted as the mean ± SEM. (E) Representative montages showing wells from the infections quantified in A and B that were treated with 30 µM STM2120 or STM2457 and infected with either icSARS-CoV-2-mNG or HCoV-OC43 as indicated. The signal for the OC43-N antibody and Alexa Fluor 647 secondary antibody is represented in green. (F) Infectious viral titers from MRC-5 cells infected with HCoV-OC43 at MOI = 0.001 treated with 30 µM either STM2120 or STM2457 was determined by TCID50 assay. (G) Infectious virus titers from A549+ACE2 cells infected with icSARS-CoV-2-mNG at MOI = 0.1 and treated with 30 µM either STM2120 or STM2457 was determined by plaque assay. (H,I) MRC-5 and A549+ACE2 cells were infected with OC43 or icSARS-CoV-2-mNG, respectively, as in (A) and (B) in the presence of 30 µM STM2120 or STM2457 and 10 µM JAK inhibitor (pyridone-6) or vehicle control (DMSO) and the percent infected cells quantified. Each experiment was conducted three times with internal duplicates, normalized to DMSO-treated cells processed in parallel, and plotted as the mean ± SEM. (J) Immunoblot analysis of lysates from MRC-5 cells infected with HCoV-OC43 at MOI = 0.001 in the presence of 30 µM STM2120 or STM2457 as in A and collected at 48 hpi and probed for viral N protein, or host ISGs (ISG15, RIG-I, PKR, and MDA5) and GAPDH.
Quantifying virus replication revealed that STM2457 reduced HCoV-OC43 infectious virus production by >100-fold (Fig. 4F) and SARS-CoV-2 infectious virus production by 300-fold (Fig. 4G). This concordance further validates the high-content imaging assay as a measure of viral replication and release. Thus, a selective small molecule METTL3 inhibitor effectively suppresses the productive replication of two human β-coronaviruses, the seasonal HCoV-OC43 and the new pandemic SARS-CoV-2.
Interferon response to β-coronavirus infection unchanged by METTL3 inhibition
We and others have shown that depletion of either METTL3 or METTL14 increases the accumulation of type I interferon (IFN) and antiviral interferon-stimulated gene (ISG) expression in virus-infected cells (Rubio et al. 2018; Winkler et al. 2019; Qiu et al. 2021). To ask whether this might explain the reduction in the β-coronavirus replication and spread when METTL3 catalysis is inhibited using STM2457, cells infected with HCoV-OC43 (Fig. 4H) or SARS-CoV-2 (Fig. 4I) were treated with STM2120 or STM2457 in the presence of a pan-JAK inhibitor to prevent induction of the type I IFN response or vehicle control. This treatment did not prevent the inhibitory effect of STM2457 on spread of either virus but did enhance the replication and spread of human cytomegalovirus (HCMV) in primary fibroblasts (Supplemental Fig. S8A) and blocked the accumulation of phosphorylated STAT1, a JAK substrate, and the interferon-stimulated IFIT2 protein (Supplemental Fig. S8B). Quantitation by RT-qPCR of mRNAs for β-interferon and interferon-stimulated genes IFIT2, ISG15, and OAS3 detected very modest induction upon infection, which was not increased in the presence of STM2457 (Supplemental Fig. S8C). Additionally, immunoblotting of representative IFN-responsive proteins ISG15, RIG-I, PKR, and MDA5 did not show any increase in STM2457-treated cells infected with HCoV-OC43 even though expression of the viral N protein was reduced (Fig. 4J) in accordance with the observed reduction in viral spread (Fig. 4A). Finally, treatment of MRC-5 cells with β-IFN resulted in robust induction of multiple interferon-stimulated proteins, confirming that the pathway is functional in these cells (Supplemental Fig. 8D). These results argue that enhanced type I IFN signaling is not responsible for the antiviral effect of STM2457 against HCoV-OC43 or SARS-CoV-2.
METTL3 inhibition suppresses HCoV-OC43 gene expression
To better understand how STM2457 interferes with HCoV-OC43 replication, MRC-5 cells were infected at high multiplicity, to achieve a synchronous infection, in the presence STM2457 or STM2120, and N protein accumulation was monitored by immunoblotting of lysates collected over a 3-d period (Fig. 5A). This showed a clear reduction in N protein abundance by 24 hpi in the STM2457-treated sample compared with STM2120, although the degree of difference was reduced at both 48 and 72 hpi. However, this delay in viral protein accumulation may account for the reduced spread of newly synthesized virus evident at 48 hpi (Fig. 4A).
Figure 5.

Initial HCoV-OC43 RNA synthesis and protein expression is reduced by inhibition of METTL3 catalysis. (A) Immunoblot analysis of lysates from MRC-5 cells infected with HCoV-OC43 at MOI = 3 in the presence of 30 µM STM2120 or STM2457 collected at 24, 48, or 72 hpi and probed for viral N protein, or host METTL3 and GAPDH. (B) Representative images showing detection of viral N protein or viral dsRNA by indirect immunofluorescence assay (green) in HCoV-OC43-infected MRC-5 cells at 24 hpi in the presence of 30 µM STM2120 or STM2457. Cell nuclei were stained with DAPI. Scale bar, 20 µm. (C). The relative abundance of HCoV-OC43 gRNA (ORF1ab) or sgRNA (N) or host GAPDH at 24 hpi MOI = 3 in the presence of 30 µM STM2120 or STM2457 was determined by RT-qPCR using transcript-specific primers and normalizing to 18S rRNA and plotted as the mean ± SEM (n = 3). (D) MRC-5 cells infected with HCoV-OC43 at MOI = 3 in the presence of 30 µM STM2120 or STM2457 were metabolically pulse-labeled with 35S amino acids for 1 h. Lysates were separated by SDS-PAGE and the fixed, dried gel exposed to film. Migration of molecular weight standards is shown at the left. The arrow at the right indicates migration of virus-encoded N protein. Additionally, the relative levels of total eIF2α and phospho-eIF2α in the same lysates was assessed by immunoblotting. Note that an ultrasensitive enhanced chemiluminescent substrate (SuperSignal West Femto) was required to visualize the very low levels of phospho-eIF2α. (E, left) Cytoplasmic lysates from HCoV-OC43-infected MRC-5 cells treated with STM2120 (gray) or STM2457 (yellow) for 24 h prior to harvest were fractionated over a 10%–50% sucrose gradient, and the absorbance at 254 nm is shown with ribosomal peaks indicated. (Right) The relative abundance of viral N and ORF1a/b RNAs and host GAPDH mRNA in each gradient fraction was determined by RT-qPCR, and the sum of polysomal mRNA (fractions 13–20) under each drug treatment is presented. A representative experiment of two biological replicates in which similar results were obtained is shown.
Some of the earliest steps in the coronavirus life cycle involve the production of negative strand RNA from the incoming positive-strand template and remodeling of the endoplasmic reticulum to form characteristic double-membrane vesicle structures (replication organelles [ROs]) in the cytoplasm that serve as shielded sites of viral RNA synthesis (Kindler et al. 2017; Snijder et al. 2020). This replication strategy results in the accumulation of double-strand RNA (dsRNA) as a replicative intermediate within the cytoplasmic viral ROs. Specific antibodies readily detected viral N protein or dsRNA-containing puncta in HCoV-OC43-infected MRC-5 cells (Fig. 5B) treated with the inactive compound STM2120. In contrast, the METTL3 inhibitor STM2457 suppressed accumulation of both dsRNA in the ROs and N protein accumulation in the cytoplasm. RT-qPCR using transcript-specific primer pairs to distinguish viral gRNA (expressing Orf1ab) and a representative sgRNA (expressing N) further demonstrated a significant reduction of both RNA species in the presence of STM2457, whereas a representative host mRNA (GAPDH) remained similar irrespective of treatment (Fig. 5C). Thus, METTL3 inhibition results in reduced viral RNA accumulation, which in turn is expected to reduce viral protein accumulation.
To monitor the effect of STM2457 on total viral protein synthesis, MRC-5 cells infected with HCoV-OC43 in the presence of either STM2120 or STM2457 were pulsed with 35S-labeled amino acids. After separation of the labeled newly synthesized proteins by SDS-PAGE, a 30% decrease in protein synthesis was evident in the STM2457-treated cells (Fig. 5D, cf. lanes 1 and 2; Supplemental Fig. S9A). Synthesis of multiple host proteins was reduced by infection with HCoV-OC43 likely corresponding to the host shutoff activity of Nsp1 (Schubert et al. 2020; Zhang et al. 2021), and accumulation of abundant viral proteins including N was evident (Fig. 5D, lanes 3,4). Consistent with the immunoblotting data (Fig. 5A), the synthesis rate of N protein was reduced in STM2457-treated cells (Fig. 5A, lane 4) relative to the control (Fig. 5A, lane 3). Additional viral proteins abundantly synthesized in control-treated infected cells were diminished in their synthesis in the presence of STM2457 consistent with a broad inhibitory effect on viral protein synthesis and was not limited to N alone. Furthermore, host shutoff appeared reduced in STM2457-treated cells. Changes in the phosphorylation status of eIF2α, a modification that mediates translational inhibition in response to numerous cell stress conditions including viral infections (Stern-Ginossar et al. 2019), were not observed under any condition (Fig. 5D).
To directly test the impact of STM2457 on viral mRNA translation, we performed a polysome fractionation experiment using RT-qPCR to assess the association of viral mRNAs with ribosomes (Fig. 5E). Remarkably, >80% of mRNAs encoding N were associated with multiple ribosomes (polysome fractions 13–20), and this was reduced to 62% by STM2457 treatment (Fig. 5E; Supplemental Fig. S9B). The full-length gRNA (ORF1a/b) was also very abundant in the polysome fractions and was similarly reduced with STM2457. To ask whether reduced representation on polysomes was selective for coronavirus mRNAs, we probed for GAPDH, a representative cellular transcript. This was less well represented in the polysome fraction, again consistent with the dominance of viral RNAs, but did show a decrease when METTL3 activity was inhibited.
Taken together, these results show that METTL3 activity contributes to both an early step in the β-coronavirus replication cycle that precedes the accumulation of dsRNA within the virus-induced ROs and the translation of viral mRNAs. Consequently, the synthesis of gRNA and sgRNA, and viral N protein expression was reduced by inhibiting METTL3-mediated catalysis. This change in viral RNA accumulation manifested as a reduction in the synthesis of viral polypeptides, including the essential N protein, and thus as a reduction in virus production and spread within the cultures.
Discussion
Our understanding of how internal RNA modification pathways impact the replication and spread of cytoplasmic RNA viruses is limited. Here, we show that the reproduction of two human β-coronaviruses requires the principal cellular m6A methyltransferase METTL3 and two m6A recognition proteins, YTHDF1 and YTHDF3. The catalytic activity of METTL3 is required for efficient synthesis of viral RNAs within the first 24 h of infection and subsequent accumulation of viral proteins. We have also shown by antibody-based RNA capture (meRIP-seq) that viral mRNAs are m6A-modified over multiple regions that contain consensus DRACH m6A acceptor motifs. Recently, Liu et al. (2021) reported eight high-confidence m6A sites on SARS-CoV-2 RNAs using a combination of meRIP-seq and miCLIP. Several of these regions overlap with the meRIP-seq peaks identified in our study but not with the single low-confidence site predicted by comparison of DRS data sets prepared in the presence or absence of a selective METTL3 small molecule inhibitor. A different study implicating METTL3 in the m6A modification of viral RNAs and in regulation of host responses during SARS-CoV-2 infection also appeared while our manuscript was in revision (Liu et al. 2021).
One plausible explanation as to why the DRS methodology, which successfully identified m6A acceptor sites in Ad5 mRNAs (Price et al. 2020), provided little information here is that coronavirus RNAs synthesized by a virus-encoded RNA-dependent RNA polymerase in unique virus-induced cytoplasmic organelles are modified at much lower frequency than either host or Ad5 mRNAs that are transcribed by RNA polymerase II in the nucleus. An earlier DRS study of RNAs isolated from SARS-CoV-2-infected Vero cells compared with synthetic unmodified RNA reported 41 potential sites of base modification, almost half of which mapped to a purine-rich AAGAA motif lacking the canonical AC dinucleotide found in METTL3 substrates (Kim et al. 2020a). Taken together, these findings are consistent with the possibility that β-coronavirus RNAs are infrequently modified and that the requirement for METTL3 catalytic activity most likely reflects the modification of host mRNAs in a manner that is beneficial to the virus. While a broadly up-regulated type-I interferon response was not observed in our study, and can likely be excluded, identification of these host RNAs should be highly informative.
In addition to METTL3, we also found that members of the YTHDF family of m6A readers contribute to β-coronavirus replication and that in MRC-5 cells their subcellular localization can be altered by HCoV-OC43 infection. A recent study characterizing the interactome of SARS-CoV-2 identified YTHDF2, but not YTHDF1 or YTHDF3, associated with viral RNA (Schmidt et al. 2020). Failure to identify YTHDF proteins may reflect their reported low abundance in human cells (Zaccara and Jaffrey 2020). Similarly, several recent genome-wide CRISPR-Cas9 screens failed to implicate YTH domain proteins in the control of β-coronavirus replication (Daniloski et al. 2021; Schneider et al. 2021; Wang et al. 2021; Wei et al. 2021). By their design, these pooled screens include a period of expansion and selection after the targeting vectors are introduced and also present the virus with the mix of cells with different gene disruptions, that together may limit the detection of factors that influence replication and spread rather than cell entry. Transient knockdown of an individual reader presents a more uniform cell population and may be sufficiently short term to minimize compensatory changes in the expression of other factors. It is worth noting, however, that indirect m6A readers from the IGF2BP protein family (Sun et al. 2019) have been identified in multiple SARS-CoV-2 RNA interactome studies, supporting the idea that m6A installation onto viral RNAs influences the RNA-protein interactome (Flynn et al. 2020; Schmidt et al. 2020; Lee et al. 2021). Whether these associations are dependent on RNA methylation by METTL3 needs to be tested.
Collectively, our results reveal that a functioning m6A RNA modification pathway is beneficial to β-coronavirus reproduction and provides a proof of concept that targeting cellular components of this now intensively studied RNA modification pathway could ultimately lead to new therapeutic opportunities to control these important viral pathogens.
Materials and methods
Viruses and cell lines
SARS-CoV-2, isolate USA-WA1/2020 (BEI Resources NR52281, a gift from Dr. Mark Mulligan, New York University Langone Vaccine Center) was amplified once in Vero E6 cells (P1 from the original BEI stock). Briefly, a 90%–95% confluent T175 flask of 1 × 107 Vero E6 cells was infected with 10 μL of the BEI stock in 3 mL of SARS-CoV-2 infection media (DMEM, 2% FBS, 1% NEAA, 10 mM HEPES at pH 7.0) for 1 h. After 1 h, 15 mL of infection media was added to the inoculum and cells were incubated for 72 h at 37°C and 5% CO2. After 72 h, the supernatant was collected and the monolayer frozen and thawed once. Both supernatant and cellular fractions were combined, centrifuged at 1200 rpm for 5 min, and filtered using a 0.22-μm Steriflip filter unit (Millipore). The mNeonGreen expressing SARS-CoV-2 recombinant (icSARS-CoV-2-mNG) based on isolate USA/WA/1/2020 (Xie et al. 2020) was obtained from the UTMB World Reference Center for Emerging Viruses and Arboviruses and similarly amplified by using 50 μL of original stock to inoculate each T175 flask of Vero E6 cells. All experiments with SARS-CoV-2 were performed in the CDC/USDA-approved BSL-3 facility in compliance with the NYU School of Medicine guidelines for biosafety level 3.
HCoV-OC43 was obtained from ATCC (ATCC VR-1558) and propagated in MRC-5 cells. We added 3 × 106 plaque-forming units (pfu) of passage 1 stock to a 90%–95% confluent 10-cm dish of MRC-5 cells in OC43 infection media (DMEM, 2% FBS, penicillin/streptomycin) and incubated it for 4 d at 33°C. The cell monolayer was detached using a cell scraper, collected with the supernatant, and centrifuged at 1000g for 5 min to remove debris. Tissue culture infectious dose 50 (TCID50) was established on MRC-5 cells and working titer (plaque-forming units per milliliter) estimated to be 0.7 TCID50/mL.
A549 cells stably expressing human ACE2 (A549+ACE2 [de Vries et al. 2021]), a kind gift of Adil Mohamed and Meike Dittmann (New York University School of Medicine), were maintained in DMEM, 10% fetal bovine serum, and penicillin/streptomycin at 37 °C with 5% CO2. Cells were supplemented with puromycin (2 μg/mL) every other passage. Vero E6 cells (ATCC CLR-1586) were maintained in DMEM, 10% FBS, 1% nonessential amino acids (NEAA), and penicillin/streptomycin at 37°C with 5% CO2. MRC-5 cells (ATCC CCL-171) were maintained in DMEM, 5% FBS, and penicillin/streptomycin at 37°C with 5% CO2 for a maximum of 24 passages from original stocks.
Cellinsight CX7 LZR high-content screening platform
To monitor virus spread in drug treatment and gene knockdown conditions, cells were seeded in black-walled clear-bottom 96-well plates. For icSARS-CoV-2-mNG infection of A549+ACE2 cells, the next day media was removed and replaced with media containing drugs or vehicle control (DMSO) 2 h prior to infection. Cells were infected in SARS-CoV-2 infection media at MOI = 0.1 in the presence of drugs/vehicle and incubated for 48 h at 37°C. Cells were fixed in a 10% formalin solution for 30 min and permeabilized in 0.5% Triton X-100 in PBS for 15 min prior to DAPI staining and a final PBS wash before analysis. For HCoV-OC43 infections, MRC-5 cells were infected at MOI = 0.001 in OC43 infection media at 33°C. At 48 hpi, cells were fixed in 4% paraformaldehyde for 30 min and permeabilized in 0.5% Triton X-100 in PBS for 15 min before blocking in immunofluorescence blocking buffer (4% FBS in PBS) for 1 h. Infected cells were detected by incubation with anti-OC43-N antibody (1:1000) overnight at 4°C followed by incubation with antimouse Alexa Fluor 647 secondary (Invitrogen A32787) and DAPI for 2 h at room temperature. Plates were imaged using a CellInsight CX7 LZR high-content screening platform by collecting nine images at 4× magnification to cover the entire well. HCS Navigator software was used to quantify cell number by DAPI staining and the percentage infected cells, indicated by either mNeonGreen or Alexa Fluor 647 positivity.
Plaque assays and TCID50 determination
SARS-CoV-2 titers were determined by plaque assay on Vero E6 cells. In brief, 5 × 105 Vero E6 cells/well were seeded in a 12-well plate. The next day, serial 10-fold dilutions of virus in DMEM supplemented with 2% FBS were added to the cells and incubated for 1 h at 37°C. Cells were then overlaid with 0.8% agarose in DMEM + 2% FBS and incubated for 72 h at 37°C. Cells were fixed with 10% formalin, and after agarose plug removal, stained with crystal violet. OC43 virus stock and experiment supernatants were titrated by establishing the tissue culture infectious dose (TCID50) on MRC-5 cells, scoring for CPE after incubation for 7 d at 33°C. Data are presented as mean ± SEM of at least five experiments.
Inhibitor treatments
Small molecule inhibitors STM2120, STM2457, and remdesivir (Medchem Express HY-104077) were reconstituted in DMSO as a 10 mM stock solution and subsequently diluted in appropriate infection media prior to cell treatment. Stocks of the STM compounds were stored at −20°C, whereas remdesivir was stored at −80°C. The pan-Janus protein tyrosine kinase (JAK) inhibitor pyridone 6 (Millipore-Sigma 420099) was reconstituted in DMSO and used at 10 µM.
Cell viability assay
To determine viability, cells were seeded to opaque white 96-well plates. The next day, cells were drug- or vehicle-treated and incubated as in infection experiments (for example, treated-MRC-5 cells were placed for 48 h at 33°C). ATP levels were then assayed using Celltiterglo2.0 (Promega G9242) according to the manufacturer's instructions. Luciferase signal was read on a Perkin Elmer Envision 2103 multilabel reader, and 24-h 1 µM staurosporine treatment used as a positive control for assay sensitivity.
RT-qPCR analysis
Total RNA was isolated from infected cells using TRIzol. For each sample, 500 ng of RNA was subject to cDNA synthesis using qScript XLT (Quanta). Quantitative PCR (qPCR) reactions were conducted using Bio-Rad SsoAdvanced SYBR Green supermix and a Bio-Rad CFX96 real-time system. Primer sequences are detailed in Supplemental Table S1. For each biological replicate, technical duplicates were conducted. mRNA levels relative to 18S rRNA were calculated using the ΔΔCT method, and statistical analyses were performed using GraphPad Prism.
Polysome analysis and metabolic labeling with 35S amino acids
MRC-5 cells (7.2 × 106) were incubated with 100 μg/mL cycloheximide (Sigma C7698) for 10 min at 37°C and 5% CO2 prior to lysis in polysome lysis buffer (20 mM Tris at pH 7.5, 0.05 M KCl, 10 mM MgCl2, 100 μg/mL cycloheximide) containing 1% Triton X-100 and 100 U/mL RiboLock RNase inhibitor (ThermoFisher Scientific, EO0381). After incubation for 10 min on ice, nuclei were pelleted by centrifugation at 20,000g in a tabletop microfuge for 5 min at 4°C. Cytoplasmic lysate was layered onto 10%–50% sucrose gradients (in polysome lysis buffer with 100 μg/mL cycloheximide) in thin-wall polypropylene ultracentrifuge tubes (Beckman Coulter 331372). Gradients were centrifuged at 38,000 rpm for 2.25 h at 4°C in a SW41Ti rotor (Beckman Coulter 331362). Absorbance profiles were produced by pumping the gradients through a flow cell while measuring the absorbance of RNA at 254 nm using a Density Gradient Fractionation System (Brandel, BR-188). RNA was isolated using Trizol and an equal volume from each fraction subject to cDNA synthesis and subsequent RT-qPCR as detailed above. Metabolic labeling of 35S-amino acids was performed as previously described (Burgess and Mohr 2015).
Immunoblotting and cell fractionation
Antibodies and their corresponding dilutions used for immunoblotting are listed in Supplemental Table S2. Immunoblots were visualized using an iBright system CL1000 system (Life Technologies). To generate particulate (nuclear) and soluble (cytoplasmic) fractions, uninfected and HCoV-OC43-infected MRC5 cells were lysed by resuspension in low-salt buffer (20 mM HEPES at pH 7.9, 10% glycerol, 1.5 mM MgCl2, 0.05% NP40, protease inhibitors) and incubated for 5 min on ice before centrifugation at 1000g for 5 min at 4°C. The supernatant (cytosolic fraction) was collected and saved. The nuclear pellet was washed with the same buffer once and resuspended in 1× Laemmli sample buffer and denatured for 5 min at 100°C and saved (nuclear fraction). Lysis and subcellular fractionation efficiency was monitored by blotting for β-tubulin (cytoplasmic) and histone H3 (nuclear).
RNA interference screen
siRNAs used are detailed in Supplemental Table S3. Cells were seeded to clear-bottom, black-walled 96-well plates and transfected the next day using 3 µL/mL Lipofectamine RNAiMax (Life Technologies) at a final siRNA concentration of 20 nM. Three days after transfection, MRC-5 cells were infected with OC43 at MOI = 0.001 and A549+ACE2 cells infected with icSARS-CoV-2-mNG at MOI = 0.1. At 48 hpi, cells were fixed and stained as detailed above prior to infected cell quantification. Knockdown efficiency was established by reproducing transfection conditions in 12-well plates and assaying target protein depletion by immunoblotting where possible or target mRNA depletion by RT-qPCR.
Indirect immunofluorescence
Cells were seeded in chamber well slides and, following infection, fixed in 4% paraformaldehyde for 15 min and permeabilized with 0.2% Triton X-100 in PBS for 15 min. Cells were then incubated with blocking solution (4% FBS in PBS) for 1 h at room temperature and incubated with primary antibodies diluted in blocking solution overnight at 4°C. For the dsRNA primary (J2) antibody, incubation was for 2 h at room temperature. Primary antibody dilutions used are listed in Supplemental Table S2. Cells were washed five times with PBS, incubated with antimouse IgG Alexa Fluor-488 (Invitrogen A11029) or antirabbit IgG Alexa Fluor-555 antibodies (Invitrogen A21429) diluted in blocking solution for 40 min at room temperature, washed five times with PBS, and costained with DAPI in the penultimate wash prior to mounting in fluorescence mounting medium (Dako S302380-2). For m6A factor subcellular localization, cells were imaged using a Zeiss 880 confocal microscope at 63× magnification, and the nuclear signal intensities in uninfected and infected cells (≥100 cells/condition) were quantified using ImageJ (Schneider et al. 2012). Statistical data are presented as the mean ± SEM, as described in the corresponding figure legends, and statistical significance was determined using an F-test with GraphPad Prism 8. P < 0.05 was considered to be statistically significant. For dsRNA and N protein localization following drug treatment, cells were visualized using a Leica DM5000 microscope with a 63× objective using Leica Imaging LAS V4.3 software.
In vitro methyltransferase assay
The enzymatic assay was established to determine IC50 values for the inhibition of RNA methyltransferase activity. The enzyme used was full-length his-tagged METTL3 coexpressed with full length FLAG-tagged METTL14 produced in a baculovirus expression system. The enzyme complex was purified using standard affinity chromatography. Enzymatic reactions were performed at room temperature in 384-well plates using a final reaction volume of 20 µL containing 20 mM TrisCl (pH 7.6), 1 mM DTT, and 0.01% Tween-20. A final concentration of 5 nM ifMETTL3/14 was preincubated with different compound concentrations for 10 min, followed by addition of 0.2 µM final concentration synthetic RNA substrate (5′-UACACUCGAUCUGGACUAAAGCUGCUC-3′) and 0.5 µM final concentration methyl-donor S-adenosyl methionine (SAM). The reaction was incubated for a further 60 min at room temperature, and then quenched by the addition of 40 μL of 7.5% TCA with internal standard. After termination, plates were sealed, centrifuged, and stored at 4°C until analysis. METTL3 activity was assessed using the RapidFire mass spectrometry (RF/MS) platform to measure the S-adenosyl homocysteine (SAH) product. Stopped and stable assay plates were analyzed on the Agilent RF300 integrated autosampler/solid phase extraction (SPE) system coupled to an ABSciex 4000 mass spectrometer for the quantification of the SAH and normalized to the ratio of signal of two internal standards. The mass transition for the product (SAH) was 384.9/135.9 Da. Transitions of the internal standard were used for normalization of matrix effects. IC50 values were calculated based on dilution series of individual compounds. Potency of a compound was measured at varied inhibitor concentrations and normalized to control wells without RNA substrate and without inhibitor (DMSO only).
RNA nucleoside quantification by mass spectrometry
Kasumi-1 cells were grown in RPMI-1640 supplemented with 20% FBS and maintained in culture between 0.3 × 106 and 1.5 × 106 cells/mL. Cells were washed in PBS and resuspended in RPMI-1640 without L-methionine (Thermo Fisher Scientific), 20% FBS, and 30 mg/L L-methionine (99% methyl-13C; 98% methyl-D3) (Cambridge Isotope Laboratories). Cells were plated into round-bottom low-attachment 96-well plates (Corning) at 25,000 cells per well, with compounds, and incubated for 16 h. For analysis, cells were pelleted and snap-frozen at −80°C for 10 min. The plate was then thawed and to each well was added 100 μL of nuclease digest mix containing 62.5 U of benzonase (Sigma Aldrich), 5 U of Antarctic phosphatase (NEB), and 10 mU/µL phosphodiesterase I (Sigma Aldrich) in 20 mM Tris-HCl (pH 8), 20 mM MgCl2, and 100 mM NaCl, and incubated overnight at 37°C. One-hundred microliters of ice-cold 0.1% formic acid containing 1 µg/mL uridine-13C9,15N2 was added to each well and transferred to an AcroPrep Advance 96-well 30-kDa molecular weight cutoff filter plate (Pall Laboratories) and centrifuged at 2000g for 10 min into a fresh 96-well plate. Samples were analyzed on a 4500 triple-quad mass spectrometer (Sciex) fed by a Dionex U3000 standard-flow UPLC (Thermo). Separation was achieved with a gradient of 2%–10% acetonitrile with a constant concentration of 0.1% formic acid on an Acquity UPLC HSS T3 column, 100 Å, 1.8 µm, 2.1 mm × 100 mm, which was held at 15°C. The following transitions were monitored for the specified analytes: adenosine: 268.06 > 135.80; cytidine: 244.06 > 111.90; guanosine: 284.11 > 152.10; uridine: 245.02 > 113.0; heavy uridine: 256.03 > 119.00; m6A: 282.08 > 149.90; and heavy m6A: 286.08 > 153.90.
RNA extraction, meRIP, and sequencing library preparation
The following protocol was adapted from Zeng et al. (2018). Briefly, total RNA was isolated from infected and uninfected cells using Trizol and up to 10 µg was treated with 2 U of DNase I (Roche Diagnostics) for 10 min at 37°C. RNA was fragmented using 10× RNA fragmentation buffer (Thermo Fisher Scientific) in a preheated thermocycler for 6 min at 70°C before stopping the reaction with 0.5 M EDTA and incubating on ice. Following ethanol purification, 10% of fragmented RNA was retained as input while the remainder was subjected to meRIP. Briefly, a 50:50 mix of protein A and protein G beads was washed with 1× IP buffer (150 mM NaCl, 10 mM Tris-HCl at pH 7.5, 0.1% NP40) and tumbled with 5 µg of EpiMark N6-methyladenosine antibody overnight at 4°C. Fragmented RNA was incubated at 4°C with the antibody-bead conjugate for 2 h and subsequently washed once with 1× IP buffer, and twice each with 1× low-salt (50 mM NaCl, 10 mM Tris-HCl at pH 7.5, 0.1% NP40) and 1× high-salt (500 mM NaCl, 10 mM Tris-HCl at ifpH 7.5, 0.1% NP40) IP buffer before eluting in 200 µL of RLT buffer from an RNeasy kit (Qiagen) and extracting according to the manufacturer's protocol before eluting in 14 µL of nuclease-free H20.
meRIP validation was performed by synthesizing cDNA using the high-capacity cDNA reverse transcription kit (Thermo Fisher Scientific) for quantitative real-time PCR using primers targeting GAPDH and SETD7 (Zeng et al. 2018) with a minimum of a 50-fold increase in SETD7 relative to GAPDH required for a meRIP sample to be considered successful.
Sequencing libraries for input and IP fractions were prepared using the SMARTer stranded total RNA-seq kit version 2 (Pico input mammalian, Takara Biosciences) with the following modifications. IP (3.5 µL ) and 100 ng of input were used as starting material, entering the protocol at the addition of first strand cDNA synthesis mix (i.e., skipping fragmentation). For the final PCR step, 12 cycles of amplification were used for input samples and 16 cycles for IP. Purified, indexed libraries were subsequently multiplexed and sequenced in paired-end mode (100 cycles) on an Illumina NovaSeq SP1 flowcell.
meRIP-seq data processing and alignment
Sequence read data sets were trimmed using TrimGalore (https://www.bioinformatics.babraham.ac.uk/projects/trim_galore) to remove adapter sequences, low-quality bases at the 3′ ends, and the first three nucleotides on the R2 read (trim_galore –paired –length 30 –clip_R2 3). This latter step is required to remove additional adapter sequence present at the 5′ end. Processed data sets were competitively aligned using STAR, a splice-aware aligner (Dobin et al. 2013), against a genome index combining the human genome (hg38), the HCoV-OC43 reference genome (NC_006213.1), and the SARS-CoV-2 WA1 strain (MN985325.1). Only uniquely aligned reads were retained (–outFilterMultimapNmax 1) for downstream processing with SAMtools (Li et al. 2009). Here, sorted BAM files were split to provide separate files for each genome sequence and the SARS-CoV-2 and HCoV-OC43 data sets (input and IP) normalized by randomly sampling to produce, respectively, 500,000 and 200,000 unique alignments using reformat.sh from the bbmap package (https://sourceforge.net/projects/bbmap).
To examine the variability between conditions and biological replicates, input and IP data sets were also pseudoaligned against the hg38 transcriptome (protein coding and long noncoding RNAs) using Kallisto (Bray et al. 2016) with subsequent transcript counts converted to gene counts. Variance stabilizing transformation was then used prior to generating sample distance correlation matrices.
meRIP-seq peak-calling
Peak calling for hg38 was performed using ExomePeak2 (Meng et al. 2014) with the following flags (paired_end=true, library_type=1st_strand, consistent_peak=TRUE, genome=“hg38,” peak_calling_mode=”full_tx”), and only consistently detected peaks with Padj < 0.05 were considered true positives. Each data set (comprising six biological replicates of input and IP) was analyzed individually and comparatively (SARS-CoV-2-infected vs. uninfected, and HCoV-OC43-infected vs. uninfected).
Peak calling for SARS-CoV-2 and HCoV-OC43 was performed on sampled BAM files using MACS2 (Zhang et al. 2008) with the following flags (-f BAMPE -g 30000 -q 0.05 -B –call-summits –keep-dup auto –nomodel –extsize 150).
Motif discovery and metagene analysis
Peak ranges derived from the exomePeak2 analyses were processed using the findMotifsGenome.pl from the HOMER package (Heinz et al. 2010) using the following parameters: -mask -size given -norevopp -S 5, -len 5,6,7,8,10,12. Metagene analysis was performed using metaPlotR (https://github.com/olarerin/metaPlotR).
Nanopore direct RNA sequencing
Direct RNA sequencing libraries were generated from 400–1000 ng of poly(A) RNA, isolated using the Dynabeads mRNA purification lit (Invitrogen 61006). Isolated poly(A) RNA was subsequently spiked with 0.5 µL of a synthetic Enolase 2 (ENO2) calibration RNA (Oxford Nanopore Technologies Ltd.) and prepared for sequencing using the SQK-RNA002 edition of the manufacturer's protocol with the following modifications: First strand synthesis was performed using SuperScript IV (Thermo Fisher Scientific) and the incubation time adjusted to 90 min.
Sequencing was performed on a MinION MkIb using R9.4.1 (rev D) flow cells (Oxford Nanopore Technologies Ltd.) for 24–48 h (one library per flowcell), yielding between ∼170,000 and 870,000 reads per data set. Raw fast5 data sets were basecalled using Guppy v3.6.0 (-c rna_r9.4.1_70bps_hac.cfg -r –calib_detect –trim_strategy rna –reverse_sequence true) with only reads in passing filter used for subsequent analyses.
Sequence reads were aligned against either the SARS-CoV-2 (MN985325.1) or HCoV-OC43 (NC_006213.1) genome sequence, using MiniMap2 (-ax splice -k 8 -w 3 -g 30000 -G 30000 -C0 -un –no-end-flt –splice-flank=no), a splice aware aligner (Li 2018), with subsequent parsing through SAMtools and BEDtools (Quinlan and Hall 2010). Subsequent analyses were carried out using either the complete aligned data set (full exome) or a processed data set (processed exome) in which only viral reads containing the SARS-CoV-2 or HCoV-OC43 leader sequence (LS) and poly(A) tail were retained (22%–68% of reads). We additionally performed isoform-level alignment with MiniMap2 (-ax map-ont -L -p 0.99) against both viral transcriptomes (derived from their canonical transcript annotations) and an hg38 transcriptome comprising protein coding and long noncoding RNAs. Here, additional parsing with SAMtools (-F 2324 -q 10) was used to retain only primary alignments (SAM flag 0) with mapping qualities (MapQ) > 0.
RNA modification detection
We used DRUMMER (https://github.com/DepledgeLab/DRUMMER) to perform comparative profiling of DRS data sets. When presented with two data sets aligned against the same genome or transcriptome (e.g., aligned DRS data sets derived from SARS-CoV-2-infected MRC-5 cells treated with either the METTL3 inhibitor STM2457 or noninhibiting control STM2120), DRUMMER identifies individual nucleotides at which basecall error rates differ in a statistically robust manner (Price et al. 2020). Alternative profiling using ELIGOS2 (Jenjaroenpun et al. 2021) was performed using the Full Exome data set with the following flags “–pval 0.05 –oddR 1.5 –esb 0.”
Data visualization
Coverage plots and transcriptome maps were generated using the R Bioconductor packages Gviz (Hahne and Ivanek 2016) and GenomicFeatures (Lawrence et al. 2013). Sequence motif logos were generated using Seq2Logo (Thomsen and Nielsen 2012). Metagene plots were produced by ggplot2 (Wickham 2016).
Data availability
All sequencing data sets associated with this study have been deposited as fastq (Illumina) or fast5 (nanopore) at the European Nucleotide Archive (ENA) under the project accession PRJEB42052.
Supplementary Material
Acknowledgments
We thank our colleagues Mark Mulligan, Adil Mohamed, Maren de Vries, Meike Dittmann, Benjamin E. Nilsson-Payant, and Benjamin tenOever for sharing critical reagents and instrumentation as well as their very generous help with the initial infection studies under extraordinary circumstances. We also thank Zhen Wei for assistance with ExomePeak2 and the New York University (NYU) Langone Health Genome Technology Center (RRID: SCR_017929) and Microscopy Laboratory (RRID: SCR_017934) for access to sequencing and confocal microscopy, respectively. These shared resources are partially supported by a Cancer Center Support Grant (P30CA016087) to the Laura and Isaac Perlmutter Cancer Center. This work was supported by grants from the National Institute of Allergy and Infectious Diseases (AI152543, AI073898, and AI151358) and the National Institute of General Medical Sciences (GM056927), and funding for COVID-related research from NYU Langone Health. R.C.G. was supported by T32 AI100853, and L.T. was supported by T32 AI007180.
Author contributions: H.M.B., D.P.D., L.T., K.P.S., R.C.G., E.I.V., and K.A.S. performed experiments. W.P.B. and A.H. performed compound synthesis and analysis. D.P.D. and J.S.A. performed the Illumina and Nanopore sequencing analysis. H.M.B., D.P.D., M.R.A., T.K., A.C.W., and I.M. designed experiments, discussed the results, analyzed data, and helped H.M.B., A.C.W., and I.M. write the manuscript.
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.348320.121.
Competing interest statement
T.K. is a cofounder of Abcam Plc and Storm Therapeutics Ltd. W.P.B, A.H., and M.R.A. are employees of Storm Therapeutics Ltd, which is developing METTL3 RNA methyltransferase inhibitors.
References
- Alarcón CR, Lee H, Goodarzi H, Halberg N, Tavazoie SF. 2015. N6-methyladenosine marks primary microRNAs for processing. Nature 519: 482–485. 10.1038/nature14281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbieri I, Tzelepis K, Pandolfini L, Shi J, Millán-Zambrano G, Robson SC, Aspris D, Migliori V, Bannister AJ, Han N, et al. 2017. Promoter-bound METTL3 maintains myeloid leukaemia by m6A-dependent translation control. Nature 552: 126–131. 10.1038/nature24678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bokar JA, Shambaugh ME, Polayes D, Matera AG, Rottman FM. 1997. Purification and cDNA cloning of the AdoMet-binding subunit of the human mRNA (N6-adenosine)-methyltransferase. RNA 3: 1233–1247. [PMC free article] [PubMed] [Google Scholar]
- Bray NL, Pimentel H, Melsted P, Pachter L. 2016. Near-optimal probabilistic RNA-seq quantification. Nat Biotechnol 34: 525–527. 10.1038/nbt.3519 [DOI] [PubMed] [Google Scholar]
- Burgess HM, Mohr I. 2015. Cellular 5′′-3′′ mRNA exonuclease Xrn1 controls double-stranded RNA accumulation and anti-viral responses. Cell Host Microbe 17: 332–344. 10.1016/j.chom.2015.02.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coronaviridae Study Group of the International Committee on Taxonomy of Viruses. 2020. The species Severe acute respiratory syndrome-related coronavirus: classifying 2019-nCoV and naming it SARS-CoV-2. Nat Microbiol 5: 536–544. 10.1038/s41564-020-0695-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniloski Z, Jordan TX, Wessels H-H, Hoagland DA, Kasela S, Legut M, Maniatis S, Mimitou EP, Lu L, Geller E, et al. 2021. Identification of required host factors for SARS-CoV-2 infection in human cells. Cell 184: 92–105.e16. 10.1016/j.cell.2020.10.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darnell RB, Ke S, Darnell JE. 2018. Pre-mRNA processing includes N6 methylation of adenosine residues that are retained in mRNA exons and the fallacy of ‘RNA epigenetics’. RNA 24: 262–267. 10.1261/rna.065219.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desrosiers R, Friderici K, Rottman F. 1974. Identification of methylated nucleosides in messenger RNA from Novikoff hepatoma cells. Proc Natl Acad Sci 71: 3971–3975. 10.1073/pnas.71.10.3971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Vries M, Mohamed AS, Prescott RA, Valero-Jimenez AM, Desvignes L, O'Connor R, Steppan C, Devlin JC, Ivanova E, Herrera A, et al. 2021. A comparative analysis of SARS-CoV-2 antivirals characterizes 3CLpro inhibitor PF-00835231 as a potential new treatment for COVID-19. J Virol 95: 1–22. 10.1128/JVI.01819-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. 2013. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29: 15–21. 10.1093/bioinformatics/bts635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flynn RA, Belk JA, Qi Y, Yasumoto Y, Schmitz CO, Mumbach MR, Limaye A, Wei J, Alfajaro MM, Parker KR, et al. 2020. Systematic discovery and functional interrogation of SARS-CoV-2 viral RNA-host protein interactions during infection. bioRxiv 1862: 253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gokhale NS, McIntyre ABR, McFadden MJ, Roder AE, Kennedy EM, Gandara JA, Hopcraft SE, Quicke KM, Vazquez C, Willer J, et al. 2016. N6-methyladenosine in Flaviviridae viral RNA genomes regulates infection. Cell Host Microbe 20: 654–665. 10.1016/j.chom.2016.09.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gokhale NS, McIntyre ABR, Mattocks MD, Holley CL, Lazear HM, Mason CE, Horner SM. 2020. Altered m6A modification of specific cellular transcripts affects Flaviviridae infection. Mol Cell 77: 542–555.e8. 10.1016/j.molcel.2019.11.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzales-van Horn SR, Sarnow P. 2017. Making the mark: the role of adenosine modifications in the life cycle of RNA viruses. Cell Host Microbe 21: 661–669. 10.1016/j.chom.2017.05.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahne F, Ivanek R. 2016. Visualizing genomic data using Gviz and Bioconductor. Methods Mol Biol 1418: 335–351. 10.1007/978-1-4939-3578-9_16 [DOI] [PubMed] [Google Scholar]
- Hao H, Hao S, Chen H, Chen Z, Zhang Y, Wang J, Wang H, Zhang B, Qiu J, Deng F, et al. 2019. N6-methyladenosine modification and METTL3 modulate enterovirus 71 replication. Nucleic Acids Res 47: 362–374. 10.1093/nar/gky1007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, Glass CK. 2010. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell 38: 576–589. 10.1016/j.molcel.2010.05.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenjaroenpun P, Wongsurawat T, Wadley TD, Wassenaar TM, Liu J, Dai Q, Wanchai V, Akel NS, Jamshidi-Parsian A, Franco AT, et al. 2021. Decoding the epitranscriptional landscape from native RNA sequences. Nucleic Acids Res 49: e7. 10.1093/nar/gkaa620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ke S, Pandya-Jones A, Saito Y, Fak JJ, Vågbø CB, Geula S, Hanna JH, Black DL, Darnell JE, Darnell RB. 2017. M6A mRNA modifications are deposited in nascent pre-mRNA and are not required for splicing but do specify cytoplasmic turnover. Genes Dev 31: 990–1006. 10.1101/gad.301036.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Lee J-Y, Yang J-S, Kim JW, Kim VN, Chang H. 2020a. The architecture of SARS-CoV-2 transcriptome. Cell 181: 914–921.e10. 10.1016/j.cell.2020.04.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim GW, Imam H, Khan M, Siddiqui A. 2020b. N6-methyladenosine modification of hepatitis B and C viral RNAs attenuates host innate immunity via RIG-I signaling. J Biol Chem 295: 13123–13133. 10.1074/jbc.RA120.014260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kindler E, Gil-Cruz C, Spanier J, Li Y, Wilhelm J, Rabouw HH, Züst R, Hwang M, V'kovski P, Stalder H, et al. 2017. Early endonuclease-mediated evasion of RNA sensing ensures efficient coronavirus replication. PLoS Pathog 13: e1006195. 10.1371/journal.ppat.1006195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasman L, Krupalnik V, Viukov S, Mor N, Aguilera-Castrejon A, Schneir D, Bayerl J, Mizrahi O, Peles S, Tawil S, et al. 2020. Context-dependent functional compensation between Ythdf m6A reader proteins. Genes Dev 34: 1373–1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence M, Huber W, Pagès H, Aboyoun P, Carlson M, Gentleman R, Morgan MT, Carey VJ. 2013. Software for computing and annotating genomic ranges. PLoS Comp Biol 9: e1003118. 10.1371/journal.pcbi.1003118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Lee Y-S, Choi Y, Son A, Park Y, Lee K-M, Kim J, Kim J-S, Kim VN. 2021. The SARS-CoV-2 RNA interactome. Mol Cell 81: 1–13. 10.1016/j.molcel.2021.04.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. 2018. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34: 3094–3100. 10.1093/bioinformatics/bty191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, 1000 Genome Project Data Processing Subgroup. 2009. The sequence alignment/map format and SAMtools. Bioinformatics 25: 2078–2079. 10.1093/bioinformatics/btp352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lichinchi G, Zhao BS, Wu Y, Lu Z, Qin Y, He C, Rana TM. 2016. Dynamics of human and viral RNA methylation during Zika virus infection. Cell Host Microbe 20: 666–673. 10.1016/j.chom.2016.10.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Yue Y, Han D, Wang X, Fu Y, Zhang L, Jia G, Yu M, Lu Z, Deng X, et al. 2014. A METTL3-METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat Chem Biol 10: 93–95. 10.1038/nchembio.1432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Xu Y-P, Li K, Ye Q, Zhou H-Y, Sun H, Li X, Yu L, Deng Y-Q, Li R-T, et al. 2021. The m6A methylome of SARS-CoV-2 in host cells. Cell Res 31: 404–414. 10.1038/s41422-020-00465-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu M, Zhang Z, Xue M, Zhao BS, Harder O, Li A, Liang X, Gao TZ, Xu Y, Zhou J, et al. 2020. N6-methyladenosine modification enables viral RNA to escape recognition by RNA sensor RIG-I. Nat Microbiol 5: 584–598. 10.1038/s41564-019-0653-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng J, Lu Z, Liu H, Zhang L, Zhang S, Chen Y, Rao MK, Huang Y. 2014. A protocol for RNA methylation differential analysis with MeRIP-seq data and exomePeak R/Bioconductor package. Methods 69: 274–281. 10.1016/j.ymeth.2014.06.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer KD, Jaffrey SR. 2014. The dynamic epitranscriptome: N6-methyladenosine and gene expression control. Nat Rev Mol Cell Biol 15: 313–326. 10.1038/nrm3785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patil DP, Chen C-K, Pickering BF, Chow A, Jackson C, Guttman M, Jaffrey SR. 2016. M6A RNA methylation promotes XIST-mediated transcriptional repression. Nature 537: 369–373. 10.1038/nature19342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry RP, Kelley DE. 1974. Existence of methylated messenger RNA in mouse L cells. Cell 1: 37–42. 10.1016/0092-8674(74)90153-6 [DOI] [Google Scholar]
- Price AM, Hayer KE, McIntyre ABR, Gokhale NS, Abebe JS, Fera Della AN, Mason CE, Horner SM, Wilson AC, Depledge DP, et al. 2020. Direct RNA sequencing reveals m6A modifications on adenovirus RNA are necessary for efficient splicing. Nat Commun 11: 6016–6017. 10.1038/s41467-020-19787-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu W, Zhang Q, Zhang R, Lu Y, Wang X, Tian H, Yang Y, Gu Z, Gao Y, Yang X, et al. 2021. N6-methyladenosine RNA modification suppresses antiviral innate sensing pathways via reshaping double-stranded RNA. Nat Commun 12: 1582–1516. 10.1038/s41467-021-21904-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, Hall IM. 2010. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26: 841–842. 10.1093/bioinformatics/btq033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roundtree IA, Evans ME, Pan T, He C. 2017. Dynamic RNA modifications in gene expression regulation. Cell 169: 1187–1200. 10.1016/j.cell.2017.05.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubio RM, Depledge DP, Bianco C, Thompson L, Mohr I. 2018. RNA m6A modification enzymes shape innate responses to DNA by regulating interferon β. Genes Dev 32: 1472–1484. 10.1101/gad.319475.118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt N, Lareau CA, Keshishian H, Ganskih S, Schneider C, Hennig T, Melanson R, Werner S, Wei Y, Zimmer M, et al. 2020. The SARS-CoV-2 RNA-protein interactome in infected human cells. Nat Microbiol 382: 1708–1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, Eliceiri KW. 2012. NIH image to ImageJ: 25 years of image analysis. Nat Methods 9: 671–675. 10.1038/nmeth.2089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider WM, Luna JM, Hoffmann H-H, Sánchez-Rivera FJ, Leal AA, Ashbrook AW, Le Pen J, Ricardo-Lax I, Michailidis E, Peace A, et al. 2021. Genome-scale identification of SARS-CoV-2 and pan-coronavirus host factor networks. Cell 184: 120–132.e14. 10.1016/j.cell.2020.12.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schöller E, Weichmann F, Treiber T, Ringle S, Treiber N, Flatley A, Feederle R, Bruckmann A, Meister G. 2018. Interactions, localization, and phosphorylation of the m6A generating METTL3-METTL14-WTAP complex. RNA 24: 499–512. 10.1261/rna.064063.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schubert K, Karousis ED, Jomaa A, Scaiola A, Echeverria B, Gurzeler L-A, Leibundgut M, Thiel V, Mühlemann O, Ban N. 2020. SARS-CoV-2 Nsp1 binds the ribosomal mRNA channel to inhibit translation. Nat Struct Mol Biol 27: 959–966. 10.1038/s41594-020-0511-8 [DOI] [PubMed] [Google Scholar]
- Shulman Z, Stern-Ginossar N. 2020. The RNA modification N6-methyladenosine as a novel regulator of the immune system. Nat Immunol 21: 501–512. 10.1038/s41590-020-0650-4 [DOI] [PubMed] [Google Scholar]
- Snijder EJ, Limpens RWAL, de Wilde AH, de Jong AWM, Zevenhoven-Dobbe JC, Maier HJ, Faas FFGA, Koster AJ, Bárcena M. 2020. A unifying structural and functional model of the coronavirus replication organelle: tracking down RNA synthesis. PLoS Biol 18: e3000715. 10.1371/journal.pbio.3000715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern-Ginossar N, Thompson SR, Mathews MB, Mohr I. 2019. Translational control in virus-infected cells. Cold Spring Harb Perspect Biol 11: a033001. 10.1101/cshperspect.a033001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L, Fazal FM, Li P, Broughton JP, Lee B, Tang L, Huang W, Kool ET, Chang HY, Zhang QC. 2019. RNA structure maps across mammalian cellular compartments. Nat Struct Mol Biol 26: 322–330. 10.1038/s41594-019-0200-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomsen MCF, Nielsen M. 2012. Seq2Logo: a method for construction and visualization of amino acid binding motifs and sequence profiles including sequence weighting, pseudo counts and two-sided representation of amino acid enrichment and depletion. Nucleic Acids Res 40: W281–W287. 10.1093/nar/gks469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai K, Courtney DG, Cullen BR. 2018. Addition of m6A to SV40 late mRNAs enhances viral structural gene expression and replication. PLoS Pathog 14: e1006919. 10.1371/journal.ppat.1006919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vijgen L, Keyaerts E, Moës E, Thoelen I, Wollants E, Lemey P, Vandamme A-M, Van Ranst M. 2005. Complete genomic sequence of human coronavirus OC43: molecular clock analysis suggests a relatively recent zoonotic coronavirus transmission event. J Virol 79: 1595–1604. 10.1128/JVI.79.3.1595-1604.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Feng J, Xue Y, Guan Z, Zhang D, Liu Z, Gong Z, Wang Q, Huang J, Tang C, et al. 2016. Structural basis of N6-adenosine methylation by the METTL3-METTL14 complex. Nature 534: 575–578. 10.1038/nature18298 [DOI] [PubMed] [Google Scholar]
- Wang R, Simoneau CR, Kulsuptrakul J, Bouhaddou M, Travisano KA, Hayashi JM, Carlson-Stevermer J, Zengel JR, Richards CM, Fozouni P, et al. 2021. Genetic screens identify host factors for SARS-CoV-2 and common cold coronaviruses. Cell 184: 106–119.e14. 10.1016/j.cell.2020.12.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei J, Alfajaro MM, DeWeirdt PC, Hanna RE, Lu-Culligan WJ, Cai WL, Strine MS, Zhang S-M, Graziano VR, Schmitz CO, et al. 2021. Genome-wide CRISPR screens reveal host factors critical for SARS-CoV-2 infection. Cell 184: 76–91.e13. 10.1016/j.cell.2020.10.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickham H. 2016. ggplot2: elegant graphics for data analysis,2nd ed. Springer-Verlag, New York. [Google Scholar]
- Williams GD, Gokhale NS, Horner SM. 2019. Regulation of viral infection by the RNA modification N6-methyladenosine. Annu Rev Virol 6: 235–253. 10.1146/annurev-virology-092818-015559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkler R, Gillis E, Lasman L, Safra M, Geula S, Soyris C, Nachshon A, Tai-Schmiedel J, Friedman N, Le-Trilling VTK, et al. 2019. m6A modification controls the innate immune response to infection by targeting type I interferons. Nat Immunol 20: 173–182. 10.1038/s41590-018-0275-z [DOI] [PubMed] [Google Scholar]
- Xie X, Muruato A, Lokugamage KG, Narayanan K, Zhang X, Zou J, Liu J, Schindewolf C, Bopp NE, Aguilar PV, et al. 2020. An infectious cDNA clone of SARS-CoV-2. Cell Host Microbe 27: 841–848.e3. 10.1016/j.chom.2020.04.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yankova E, Blackaby W, Albertella M, Rak J, de braekeleer E, Tsagkogeorga G, Pilka ES, Aspris D, Leggate D, Hendrick AG, et al. 2021. Small molecule inhibition of METTL3 as a strategy against myeloid leukaemia. Nature 593: 597–601. 10.1038/s41586-021-03536-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaccara S, Jaffrey SR. 2020. A unified model for the function of YTHDF proteins in regulating m6A-modified mRNA. Cell 181: 1582–1595.e18. 10.1016/j.cell.2020.05.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng Y, Wang S, Gao S, Soares F, Ahmed M, Guo H, Wang M, Hua JT, Guan J, Moran MF, et al. 2018. Refined RIP-seq protocol for epitranscriptome analysis with low input materials. PLoS Biol 16: e2006092. 10.1371/journal.pbio.2006092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nussbaum C, Myers RM, Brown M, Li W, et al. 2008. Model-based analysis of ChIP-seq (MACS). Genome Biol 9: R137–R139. 10.1186/gb-2008-9-9-r137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K, Miorin L, Makio T, Dehghan I, Gao S, Xie Y, Zhong H, Esparza M, Kehrer T, Kumar A, et al. 2021. Nsp1 protein of SARS-CoV-2 disrupts the mRNA export machinery to inhibit host gene expression. Sci Adv 7: eabe7386. 10.1126/sciadv.abe7386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng G, Dahl JA, Niu Y, Fedorcsak P, Huang C-M, Li CJ, Vågbø CB, Shi Y, Wang W-L, Song S-H, et al. 2013. ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Mol Cell 49: 18–29. 10.1016/j.molcel.2012.10.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All sequencing data sets associated with this study have been deposited as fastq (Illumina) or fast5 (nanopore) at the European Nucleotide Archive (ENA) under the project accession PRJEB42052.