

Here, Soares et al. show an important role for differential binding of TFs to mitotic chromosomes, governed by their electrostatic properties, in defining the temporal order of transcriptional reactivation during mitosis-to-G1 transition. They report that Brn2, an important regulator of neural stem cell identity, associates with condensed chromatin throughout cell division, as assessed by live-cell imaging of proliferating neural stem cells, and ChIP-seq analysis reveals that Brn2 mitotic chromosome binding does not result in sequence-specific interactions prior to mitotic exit, relying mostly on electrostatic forces.
Keywords: Brn2, Ascl1, mitotic bookmarking, transcription, neural stem cell, M-G1 transition
Abstract
During mitosis, chromatin condensation is accompanied by a global arrest of transcription. Recent studies suggest transcriptional reactivation upon mitotic exit occurs in temporally coordinated waves, but the underlying regulatory principles have yet to be elucidated. In particular, the contribution of sequence-specific transcription factors (TFs) remains poorly understood. Here we report that Brn2, an important regulator of neural stem cell identity, associates with condensed chromatin throughout cell division, as assessed by live-cell imaging of proliferating neural stem cells. In contrast, the neuronal fate determinant Ascl1 dissociates from mitotic chromosomes. ChIP-seq analysis reveals that Brn2 mitotic chromosome binding does not result in sequence-specific interactions prior to mitotic exit, relying mostly on electrostatic forces. Nevertheless, surveying active transcription using single-molecule RNA-FISH against immature transcripts reveals differential reactivation kinetics for key targets of Brn2 and Ascl1, with transcription onset detected in early (anaphase) versus late (early G1) phases, respectively. Moreover, by using a mitotic-specific dominant-negative approach, we show that competing with Brn2 binding during mitotic exit reduces the transcription of its target gene Nestin. Our study shows an important role for differential binding of TFs to mitotic chromosomes, governed by their electrostatic properties, in defining the temporal order of transcriptional reactivation during mitosis-to-G1 transition.
During cell division complex cellular changes occur that have a dramatic impact on genome regulation. This is particularly the case in mitosis, as the core transcriptional machinery is heavily inhibited and transcription globally arrested (Festuccia et al. 2017; Palozola et al. 2018a). Condensation of chromatin and the dilution of nuclear proteins occur following nuclear envelope breakdown (NEBD), altering the activities of DNA-binding transcription factors (TFs). Nevertheless, in recent years several TFs have been reported to occupy a fraction of their genomic target sites during mitosis (Kadauke et al. 2012; Caravaca et al. 2013; Deluz et al. 2016; Festuccia et al. 2016; Liu et al. 2017). This process, referred to as “mitotic bookmarking,” provides an attractive model for how gene regulatory information is propagated across cell divisions. TF binding to regulatory regions may therefore mark a subset of genes for efficient reactivation upon mitotic exit (Festuccia et al. 2017; Palozola et al. 2018a). One possibility is that occupancy of regulatory regions by TFs maintains nucleosome positioning, counteracting large chromatin rearrangements characteristic of mitosis, as shown in recent studies (Festuccia et al. 2019; Owens et al. 2019). Nevertheless, the basis and impact of mitotic chromosome binding by TFs to gene regulatory events remain poorly understood.
Distinct types of interactions mediate the association of TFs with DNA. Long-lasting sequence-specific interactions occur via direct binding to specific nucleotide motifs and can be mapped using chromatin immunoprecipitation followed by sequencing (ChIP-seq). In addition, TFs take part in nonspecific interactions with chromatin, mediated primarily by electrostatic forces that do not depend on specific DNA sequences (Suter 2020). These result from the strong electrostatic field surrounding negatively charged DNA (even when in a nucleosome) and are of most importance when chromatin is highly compacted, as it is the case of condensed chromosomes during mitosis or heterochromatic regions in the interphase nucleus (Gebala et al. 2019; Raccaud et al. 2019). Mitotic bookmarking implies site-specific binding (i.e., direct interactions with the specific DNA motifs); however, nonspecific interactions could also contribute to this process (Caravaca et al. 2013). In addition, because many TFs that bind mitotic chromatin are also pioneer TFs (i.e., bind to nucleosomal DNA), the two activities have been often associated in the literature.
Recent studies, performed in a limited number of cell types, suggest reactivation of transcription upon mitotic exit does not occur in bulk during G1, but in sequential waves with distinct kinetics during mitosis-to-G1 (M-G1) transition (Palozola et al. 2018b; Zhang et al. 2019; Kang et al. 2020). How are such distinct temporal profiles of gene reactivation established? Some studies have suggested sequence-specific binding of TFs to mitotic chromatin as the basis for early reactivation of bookmarked genes during M-G1 (Kadauke et al. 2012; Caravaca et al. 2013; Festuccia et al. 2016). However, these observations are still controversial, due to the lack of temporal resolution and sensitivity of cell population assays used to survey active transcription during M-G1. In addition, they also fail to compare TFs with distinct mitotic binding abilities.
Given the view that mitosis (and in particular the M-G1 transition) is a window of opportunity for cell state transitions, recent reports on the interplay between TFs and mitotic chromatin have been performed in the context of pluripotent embryonic stem (ES) cells, which have unusual cell cycle and chromatin features compared with somatic cells. In contrast, no studies have so far investigated mitotic TF binding in neural stem cells, where in vitro models and knowledge of the key cell fate determinants are available.
Members of the class III POU family of TFs (e.g., Brn2; also known as Pou3f2) play diverse functions in neural development, including the acquisition and maintenance of neural progenitor identity. They function by directly binding and regulating a large number of neural stem cell enhancers, often in partnership with Sox2, another TF critical for maintenance of neural stem/progenitor cells (Tanaka et al. 2004; Miyagi et al. 2006; Lodato et al. 2013). Commitment toward the neuronal fate is primarily regulated by basic helix–loop–helix (bHLH) TFs of the proneural family such as Ascl1 (also termed Mash1), which is both necessary and sufficient to activate a full program of neuronal differentiation (Bertrand et al. 2002; Vasconcelos and Castro 2014). Such function during development is reinforced by the extensive use of Ascl1 in reprogramming somatic cells into induced neurons, which has been attributed to its pioneer TF activity. In addition to a pivotal role in triggering neuronal differentiation, Ascl1 is also required for proper proliferation of radial glia (RG) neural stem cells in the embryonic brain and in RG-like cultures growing in nondifferentiating conditions (Castro et al. 2011; Imayoshi et al. 2013). Activation of neuronal differentiation by proneural factors requires (and occurs concomitantly with) the suppression of a progenitor program, of which Sox2 and Brn2 are major regulators (Bylund et al. 2003; Vasconcelos et al. 2016). The balance between these opposing TFs is therefore critical in determining, whether upon cell division, newborn cells maintain their progenitor identity or instead become committed to neuronal differentiation.
Here, we explore the relevance of mitotic bookmarking mechanisms by TFs in neural stem/progenitor cells, focusing on Brn2 and Ascl1, given their critical role in neural stem cell maintenance and differentiation, as well as in neural lineage-specific expression. By combining live-cell imaging, genome-wide location analysis by ChIP-seq, and single-molecule RNA fluorescence in situ hybridization (smRNA-FISH), we addressed how these two pivotal TFs interact with mitotic chromatin and how this impacts their gene regulation functions upon mitotic exit.
Results
Brn2 and Ascl1 display distinct mitotic chromosome binding abilities
Live-cell imaging microscopy has recently emerged as the method of choice for probing the association of TFs with condensed chromosomes, circumventing artifacts caused by chemical fixation (Teves et al. 2016). We applied this imaging method to cultured RG-like neural stem/progenitor cells originated from embryonic mouse telencephalon (herein referred to as NS cells), undergoing proliferative divisions in nondifferentiating conditions. Immunostaining confirms homogeneous Brn2 expression, while Ascl1 displays the characteristic heterogeneous pattern resulting from temporal fluctuations in expression (Supplemental Fig. S1; Castro et al. 2011; Imayoshi et al. 2013). Tagging of endogenous Brn2 and Ascl1 with enhanced green fluorescent protein (eGFP) allowed surveying their dynamics of association with chromatin, without altering expression levels (Fig. 1A,D,G). Strikingly, imaging of Brn2 in NS cells reveals this TF associates with mitotic chromosomes during all stages of mitosis, as shown by strong colocalization of Brn2-eGFP with DNA (Fig. 1B). The degree of colocalization during M-G1 was quantified by determining the level of mitotic chromosome enrichment (MCE; mean fluorescence at the chromosomes divided by total cell fluorescence level) (Fig. 1C). In contrast, the same approach used with Ascl1 reveals clear exclusion of eGFP signal from condensed chromatin since NEBD (i.e., prometaphase), with eGFP signal dispersing within the mixed nucleoplasm and cytoplasm (Fig. 1E). Exclusion from chromatin persisted until completion of cytokinesis (telophase), with colocalization of eGFP with DNA becoming apparent from late telophase onward (+5 min) (Fig. 1E,F).
Figure 1.

Brn2 and Ascl1 display distinct interactions with condensed chromosomes in proliferating NS cells. (A) Adherent cultures of mouse embryonic neural stem/progenitors of ventral telencephalon expressing eGFP-tagged Ascl1 or Brn2 were live imaged undergoing proliferative divisions. (B,E) Time-lapse live-cell imaging of NS cells expressing eGFP-tagged Brn2 (B) and Ascl1 (E) in presence of DNA dye SiR-Hoechst. (C,F) Quantifications of mitotic chromosome enrichment levels (MCEs) at different stages in NS cells expressing Brn2-eGFP (n = 30) and Ascl1-eGFP (n = 24). Data shown as mean ± SD. One-way ANOVA Tukey's multiple test was used to compare MCE levels across different stages. (n.s.) P > 0.05, (*) P ≤ 0.05, (****) P ≤ 0.0001. Not shown in the figure: MCE comparison between TFs at each stage; P ≤ 0.0001. (D,G) Western blot analysis of Brn2-eGFP (D) and Ascl1-eGFP (G) protein expression in lines used in live imaging and their counterparts in parental NS cells. Black arrows mark specific bands corresponding to endogenous or eGFP fusion proteins. Gray arrow marks Brn1, also recognized by the antibody used (see Supplemental Fig. S2 for characterization of Brn2 commercial antibody). (I) Interphase, (PM) prometaphase, (M) metaphase, (A) anaphase, (T) telophase. Scale bars, 10 µm.
Colocalization of Ascl1 with DNA in late telophase (+5′) may be indicative of binding to chromatin or instead result from active import of Ascl1-eGFP into newly formed nuclear envelope, wrapping tightly around decondensing chromosomes. To address this, we started by mapping the Ascl1 nuclear localization signal (NLS). A 23-residue-long sequence N-terminal from the bHLH domain, highly predicted as a bipartite NLS, is both required (when mutated in context of Ascl1-eGFP) and sufficient (when fused to eGFP) to drive nuclear localization (Supplemental Fig. S3). Importantly, the timing of association of Ascl1-eGFP with chromatin in dividing P19 embryonic carcinoma cells, where the analysis of these Ascl1 derivatives was performed, parallels that observed in NS cells (Supplemental Fig. S3D). Strikingly, mutating the Ascl1 NLS in one part, or both parts of the NLS sequence, results in a significant delay (>60 min) or complete inability to colocalize with DNA at mitotic exit, respectively (Supplemental Fig. S3C,D). In conclusion, Brn2 and Ascl1 have contrasting binding patterns throughout mitosis. Colocalization of Ascl1 with DNA in late telophase (+5′) results from its active import into the nuclear envelope rather than its ability to bind to mitotic chromatin prior to nuclear envelope reformation. The observed binding of Brn2 may suggest some role as mitotic bookmarker.
Ascl1 protein is present during M-G1 transition in vivo
TFs can be targeted for degradation during mitosis, imposing another layer of regulation of their potential eviction from mitotic chromatin (Festuccia et al. 2017). While persistence of Brn2 in mitosis has been well documented (Sunabori et al. 2008), we next ascertained whether Ascl1 protein can also be detected during mitosis in an in vivo context. The expression of endogenous Ascl1 during mitosis was characterized by immunohistochemistry in the developing ventral telencephalon at E12.5, a developmental stage when most mitotic events occur in apically dividing progenitors, as seen by phospho-histone H3 staining (Fig. 2A). Most of these are RG that divide asymmetrically to self-renew and originate another more differentiated progenitor (e.g., short neural precursor) (Pilz et al. 2013). Dynamic expression of Ascl1 driven by Hes1 oscillations (Imayoshi et al. 2013) results in heterogeneous expression in neural stem/progenitor cells in both germinal layers (ventricular and subventricular zones [VZs and SVZs]) (Fig. 2B). Ascl1 protein is detectable in 64.9% of apical mitotic cells, which can be segregated by their high or low Ascl1 protein levels. Using DAPI to assess cell cycle phase, it is observed that Ascl1 expression (in both high and low expressing cells) remains constant until anaphase, with fewer (but still significant number of cells) in telophase retaining Ascl1 expression (34.9%) (Fig. 2B–D). Mitotic cells expressing moderate to high levels of Ascl1 were also observed in other VZ (subapically dividing) and SVZ progenitors undergoing mitosis (data not shown). Altogether, these observations suggest that a significant number of neural progenitor cells maintain moderate levels of Ascl1 throughout M phase, although Ascl1 protein levels are reduced upon mitotic exit. We therefore conclude that protein degradation is not the key pathway that limits Ascl1 binding to mitotic chromatin.
Figure 2.

Quantification of Ascl1 expression in apically dividing neural progenitors of ventral telencephalon. (A,B) Cross-sections of mouse telencephalon immunostained for pHH3 and Tuj1 (A) or Ascl1 and Tuj1 (B) together with DAPI staining. Mitotic pHH3+ cells can be found in the ventricular zone (VZ) and subventricular zone (SVZ) in both the lateral and medial ganglionic eminences (LGE and MGE, respectively) of E12.5 ventral telencephalon. (C) The cell cycle stage of apically dividing progenitors, both from LGE and MGE, was assessed using DAPI staining and segregated in cells not expressing (−), expressing low levels of (+), or expressing high levels of (+++) Ascl1 protein. (D) Stacked bar plots showing the percentage of mitotic cells expressing different levels of Ascl1 in prometaphase (PM), metaphase (M), anaphase (A), and telophase (T). Data shown as mean percentage ± SD. Data information: Cross-sections from three mice were used. From each mouse, six consecutive slices were acquired in a total of 1736 cells being quantified. One-way ANOVA Tukey's multiple comparison was used to compare Ascl1 levels between prometaphase and other M-phase stages. (ns) P > 0.05, (*) P ≤ 0.05, (**) P ≤ 0.01. Scale bars:, A,B, 100 µm; C, 5 µm.
eGFP does not hinder Ascl1 mitotic chromosome binding
Ascl1 binds DNA as a heterodimer with ubiquitously expressed bHLH E-proteins (e.g., E47). One possibility could be that the absence of its heterodimeric partner during mitosis hampers Ascl1 binding. However, cotransfecting Ascl1 with E47, or tethering of both proteins via a glycine bridge (Ascl1/E47) (Geoffroy et al. 2009), did not promote its association with mitotic chromosomes (Fig. 3A,D; data not shown). To exclude the possibility that the bulkiness of eGFP (∼27 kDa) hinders the ability of Ascl1 (∼30 kDa) to interact with mitotic chromatin, we performed live-cell imaging analysis of Ascl1/E47 tagged with a small (12-residue-long) tetracysteine tag (TC-Ascl1/E47) (Martin et al. 2005). Strong expression of both TC-Ascl1/E47 and TC-Brn2 can be seen in the interphase nucleus following this labeling method (Fig. 3B,C). In mitotic cells, TC-Ascl1/E47 is found excluded from mitotic chromatin, while TC-Brn2 colocalizes with DNA (Fig. 3B–D). Thus, use of TC-tagged TFs validates previous observations and indicates that the size of eGFP is not the cause of Ascl1 exclusion from mitotic chromatin. Moreover, Ascl1-eGFP was found to be transcriptionally active as judged by its ability to induce neuronal differentiation when expressed in P19 cells or to activate reporter gene expression in transcriptional assays (Supplemental Fig. S4). Altogether, these various lines of evidence indicate Ascl1 does not bind mitotic chromatin. Given previous studies have shown Ascl1 has pioneer TF activity (Wapinski et al. 2013; Raposo et al. 2015), our results clearly indicate these two activities (mitotic chromosome binding and pioneer) can be dissociated and may be deployed differently in reprogramming versus NS cell self-renewal.
Figure 3.
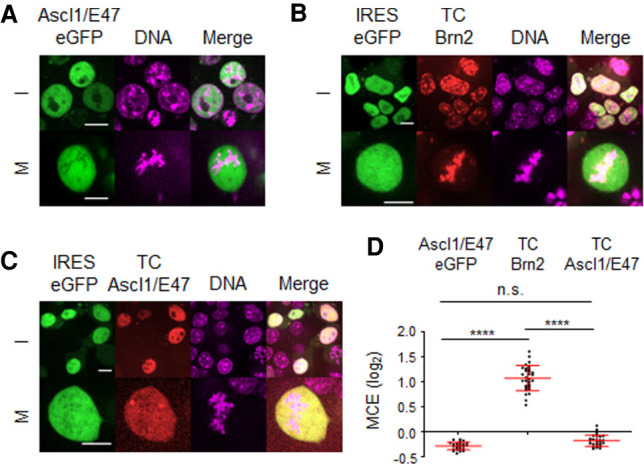
Tetracysteine tag-based live-cell imaging confirms distinct abilities of Ascl1 and Brn2 to interact with mitotic chromosomes. (A–C) Representative captures from live-cell imaging of P19 cells expressing Ascl1/E47-eGFP (A), TC-Brn2-IRES-GFP (B), or TC-Ascl1/E47-IRES-GFP (C) in the presence of DNA dye SiR-Hoechst. (D) Quantifications of mitotic chromosome enrichment levels of all three conditions, measured using cells undergoing metaphase (n = 25 in Ascl1/E47-eGFP; n = 30 in TC-Brn2; n = 26 in TC-Ascl1/E47). Data shown as mean ± SD. (I) Interphase, (M) mitosis. Scale bars, 10µm.
The DNA-binding domain of Brn2 is both required and sufficient for mitotic chromosome binding
Next, we characterized the structural determinants of mitotic chromosome binding of Brn2, using live-cell imaging in transfected P19 cells. Using a truncated Brn2 derivative, we found its structurally conserved DNA-binding domain (DBD), composed of both the POU-specific (POUS) and the POU-homeo (POUH) domains, to be sufficient on its own to associate with mitotic chromosomes (Fig. 4A–C). To further address the contribution of the Brn2 DBD, double point mutations were introduced in residues of either subdomain, which mediate direct contact with DNA as assessed by structural data or in vitro binding assays (Malik et al. 2018). These completely abolish (C311A and R312E) or severely reduce (N406Q and R407G) mitotic chromatin binding (mean MCE = −0.06 and 0.25, respectively), indicating the integrity of both POUS and POUH domains are required (Fig. 4A–C). Of note, although the latter mutant retains some degree of mitotic chromosome binding, this becomes restricted to centromeric and pericentromeric regions (Fig. 4B). Importantly, for each Brn2 derivative analyzed, the heterogeneous expression levels characteristic of transient transfection did not result in significant differences in MCE values across cells (Supplemental Fig. S5A), highlighting the plausibility of our comparisons. In conclusion, the DBD is both required and sufficient to mediate the interaction of Brn2 with condensed chromosomes. Mitotic chromosome binding is likely to be a common feature of POU3F family members, according to live-cell imaging experiments in the context of full-length Oct6 (also known as POU3f1) or DBD-only (Oct6; Brn4; also known as POU3f4) derivatives (Supplemental Fig. S5B,C).
Figure 4.
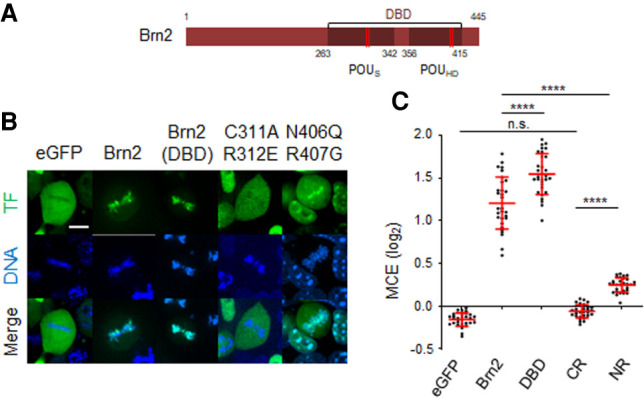
The DNA-binding domain (DBD) of Brn2 is both required and sufficient for mitotic chromosome binding. (A) Representation of Brn2 protein domains to scale, including the DBD with its POUs and POUh domains. Red stripes mark residues involved in sequence-specific binding that were mutated in live-imaging experiments. (B) Representative captures from live-cell imaging of P19 cells expressing eGFP fusion proteins of full-length Brn2, its truncated DBD, or Brn2 derivatives with mutations in residues within the POUs domain (C311A/R312E) or POUh domain (N406Q/R407G). Control eGFP is also shown. Cells were synchronized in metaphase using proTAME and Apcin and live imaged together with DNA staining Hoechst. (C) Quantifications of mitotic chromosome enrichment levels from different Brn2 derivatives. Data shown as mean ± SD (n = 30 for each condition). One-way ANOVA Tukey's multiple comparison test was performed. (n.s.) P > 0.05, (****) P ≤ 0.0001. Scale bar, 10 µm.
Brn2 does not establish sequence-specific interactions with mitotic chromatin
To map the genomic regions occupied by Brn2 in mitosis, we compared the genome-wide binding profile of endogenous Brn2 in NS cells in asynchronous (interphase) and mitotically arrested NS cells using chromatin immunoprecipitation followed by deep sequencing (ChIP-seq). Colchicine incubation followed by shake-off resulted in a mitotic population of NS cells highly enriched in prometaphase (>90%), while maintaining mitotic chromosome binding in live-cell imaging and similar Brn2 expression levels as assessed by western blot analysis (Supplemental Figs. S6A,B, S7A,B). Location analysis in interphase identified a high number (6770) of Brn2 binding events, including within previously characterized neural enhancers driving expression of Nestin and Fabp7 genes (Fig. 5A–C). Gene ontology analysis of genes associated with Brn2 binding showed enrichment for terms related with its proposed function in NS cells, including “somatic” and “neuronal stem cell maintenance” (Supplemental Fig. S6E). A de novo search for enriched DNA motifs present at peak summits found MORE and octamer motifs as main mediators of Brn2 binding (as dimer and monomer, respectively), with a bin-by-bin analysis indicating the MORE motif to be prevalent in the top 1000 peaks (Fig. 5D). Strikingly, sequence-specific binding is drastically reduced in mitosis, with only 85 binding events identified corresponding to genomic regions associated with strong Brn2 binding in interphase (Fig. 5A–C). However, no gene ontology terms were found overrepresented in association with Brn2 peaks found in the mitosis sample (data not shown). Moreover, no mitotic-specific peaks were identified, altogether suggesting the <2% peaks that remain in the mitotic sample may result from contamination with interphase cells. Absence of sequence-specific binding is also in line with the observation that a mutation previously described to abolish Brn2 homodimerization (M414N), a required condition for binding to the MORE motif, does not decrease Brn2 mitotic chromosome binding (Fig. 5E,F; Jerabek et al. 2017). Conversely, the importance of non-sequence-specific binding is suggested by the observation that a single-point mutation in a residue described to interact with the DNA backbone of a MORE motif (Q290A) strongly affects mitotic chromosome binding (Fig. 5E,F; Jauch et al. 2011).
Figure 5.

Brn2 mitotic chromosome binding is not mediated by sequence-specific interactions. (A) Venn diagram depicting the number of genomic regions bound by Brn2 in mitotically synchronized (95% purity sample) or unsynchronized cultures, as determined by ChIP-seq. (B, top panel) ChIP-seq Brn2 binding profile in a large region of chromosome 12 showing multiple peaks found in unsynchronized sample, but no binding in mitosis. (Middle and bottom panels) Brn2 binding was found at expected regions within previously characterized neural enhancers in Nestin and Fabp7 genes, only in the interphase sample. (C) Density plot of ChIP-seq reads from mitotic and interphase samples, mapping to genomic regions centered on Brn2 peak summits found in interphase. Signal intensity represents normalized tag count, ordered by increasing P-values. (D) A bin-by-bin search for enriched DNA-binding motifs centered at Brn2 peak summits identifies the MORE motif as the highest enriched on the top 1000 peaks, while the octamer motif is most enriched in other bins. (E) Representative captures from live-cell imaging of P19 cells expressing eGFP fusion proteins of full-length Brn2, or Brn2 mutants with point mutations that disrupt homodimerization (M414N), predicted to interfere with electrostatic interactions with DNA backbone (Q290A) or in a residue targeted by mitotic phosphorylation that hampers sequence-specific binding and results in phospho-dead (S362A) or phospho-mimetic (S262D) derivatives. Control eGFP is also shown. Cells were synchronized in metaphase using proTAME and Apcin and live imaged together with DNA staining Hoechst. (F) Quantifications of mitotic chromosome enrichment levels from live-imaging experiments shown in E. Data shown as mean ± SD (n = 30 for each condition). One-way ANOVA Tukey's multiple comparison test was performed. (n.s.) P > 0.05, (***) P ≤ 0.001, (****) P ≤ 0.0001. Scale bar, 10 µm.
Phosphorylation of Ser362 impairs Brn2 sequence-specific interactions but is compatible with mitotic chromosome binding
Post-translational modifications of Brn2 during mitosis, in particular phosphorylation, could account for its lack of sequence-specific binding. Western blot analysis revealed a Brn2-specific band more diffused in the mitotic protein extracts (which could be reverted upon phosphatase treatment), confirming Brn2 is a phospho-protein in mitotic NS cells (Supplemental Fig. S7A,B). Of notice, phosphorylation of a conserved serine (Ser362 in Brn2) that inhibits sequence-specific DNA binding of POU proteins was previously shown to occur during G2/M in multiple cell types (Nieto et al. 2007; Sunabori et al. 2008). To test whether Brn2 phosphorylation could indeed abolish sequence-specific binding without compromising mitotic chromosome association, we tested the behavior of a phospho-mimetic derivative (Brn2 S362D). Fluorescence recovery after photobleaching (FRAP) assays reveal that Brn2 S362D displayed similar recovery curves to the Brn2 DBD mutant C311A/R312E, in line with in vitro studies showing Brn2 S362D is devoid of sequence-specific binding (Supplemental Fig. S7C–E). Furthermore, this phospho-mimetic form of Brn2 retains a significant ability to associate with mitotic chromosomes (MCE = 0.7 vs. 1.2 for WT Brn2 vs. 1.3 for its phospho-dead mutant, S362A) (Fig. 5E,F). Thus, mitotic-specific phosphorylation of Brn2 (in particular serine 362) provides a possible molecular basis that conciliates mitotic chromosome binding with the absence of sequence-specific interactions.
Differences in Brn2 and Ascl1 mitotic chromatin binding ability derive from their distinct electrostatic properties
Electrostatic interactions have been proposed to play a major role in mediating the association between TFs and highly compacted DNA (Raccaud et al. 2019). To better understand the contribution of electrostatic forces to the interaction of Brn2 and Ascl1 with mitotic chromatin, we extended our live-cell imaging analysis to cells in interphase. The colocalization of each TF with DNA was characterized by partitioning the cell nucleus in regions of various chromatin densities (heterochromatic, DNA-rich, and DNA-poor) using Hoechst staining as previously described (Fig. 6A; Raccaud et al. 2019). In NS cells, Brn2 was found highly enriched in dense and heterochromatic regions, a property known to correlate with mitotic chromatin binding (Fig. 6B,D; Raccaud et al. 2019). Depletion of Brn2 from DNA-poor regions was also observed in transfected P19 cells. Interestingly, S362D Brn2 (which is devoid of sequence-specific binding but binds mitotic chromatin) presented the same distribution as Brn2, whereas C311A/R312E Brn2 (unable to bind mitotic chromosomes) was evenly distributed across the three domains (Fig. 6C,D). In sharp contrast to Brn2, Ascl1 was not found preferentially at heterochromatic or DNA-dense regions, when compared with DNA-poor regions, in either cell type (Fig. 6B–D). This did not result from an excess of Ascl1 protein over its heterodimeric partners, as a similar result was obtained with Ascl1/E47 tether (Fig. 6C,D). Thus, our results indicate that Brn2 and Ascl1 have very different abilities to associate with highly compacted DNA, indicating the differences observed in mitotic chromosome binding rely on their different electrostatic properties. To further confirm this, we sought to change the electrostatic potential of Ascl1 and assess its impact on the association with mitotic chromatin. Fusion of three highly positively charged SV40 NLS sequences promotes eGFP association with mitotic chromosomes, as assessed by live-cell imaging (mean MCE = 0.37) (Supplemental Fig. S8). This is known to result from an increase in electrostatic interactions, independently of the canonical NLS function (Raccaud et al. 2019). Strikingly, the observed increase in MCE was strongly counteracted by the addition of Ascl1 in fusion with eGFP, even when in the context of an obligatory dimer with E47 (mean MCE = 0.01 and 0.08, respectively). A nearly full recovery of MCE levels was only attained upon fusion of additional NLS sequences (mean MCE = 0.31) (Supplemental Fig. S8), altogether in line with a low electrostatic potential of Ascl1. In conclusion, our results from live-cell imaging in both interphase and mitosis underlie differences of electrostatic properties of Brn2 and Ascl1 and are in line with nonspecific electrostatic interactions being the main determinants for the association of these TFs with mitotic DNA.
Figure 6.
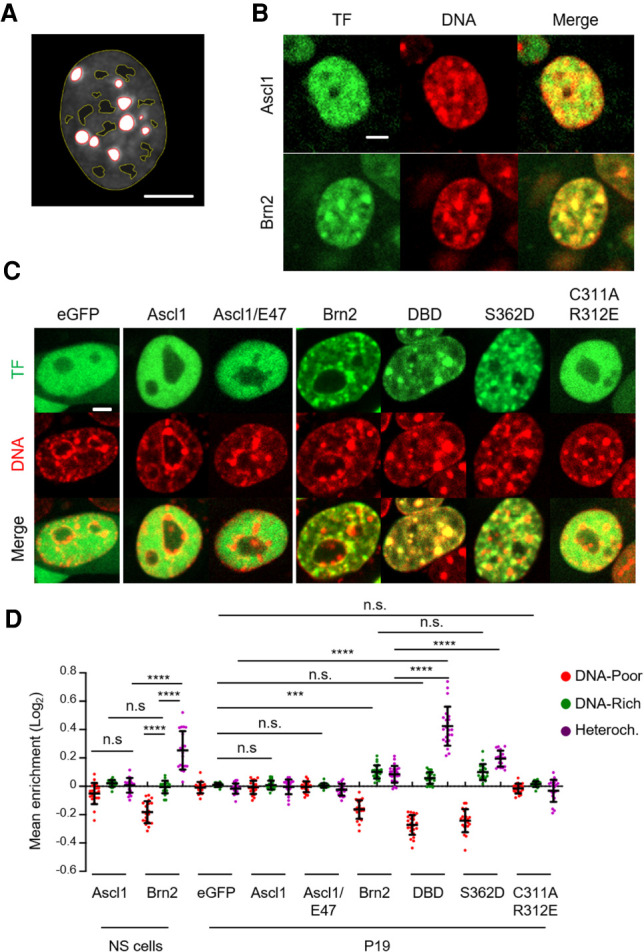
Brn2 (but not Ascl1) associates with highly compacted DNA regions in the interphase nucleus. (A) Image depicting the segmentation of an NS cell nucleus into heterochromatic DNA regions (delineated in red), DNA-poor regions (delineated in yellow), and DNA-rich regions (space in between). (B) Representative captures from live-cell imaging of eGFP-tagged Ascl1- and Brn2-expressing NS cells, stained with Hoechst for segmentation into nuclear regions with different chromatin densities. (C) Representative captures from live-cell imaging of P19 cells expressing variants of Ascl1 and Brn2 eGFP fusion proteins as indicated in the figure, stained with Hoechst for segmentation into nuclear regions with different chromatin densities. (D) Quantification of imaging analyzes shown in B and C. Enrichment levels at different segmented DNA regions are shown as mean ± SD. n = 20 cells for most conditions; n = 24 cells for Brn2 and DBD both in P19 cells. One-way ANOVA Tukey's multiple comparison test was performed. (n.s.) P > 0.05, (***) P ≤ 0.001, (****) P ≤ 0.0001. Scale bars, 5µm.
Targets dependent on Brn2 (Nestin and Fabp7) and Ascl1 (Dll1) have distinct onsets of reactivation during M-G1
A mitotic bookmarking function entails that gene regulatory information is conveyed by TF binding to specific gene regulatory regions. Even in the absence of sequence-specific interactions, retention of Brn2 on mitotic chromatin may increase its local concentration, thereby facilitating how it searches and reactivates target genes during the transition from mitosis to G1 (M-G1). Recent studies suggest the reactivation of a fraction of the transcriptome starts early during M-G1 transition (i.e., late metaphase) (Palozola et al. 2017; Zhang et al. 2019). Thus, Ascl1 dependency on nuclear import may result in a delayed onset of reactivation of its transcriptional program, as compared with Brn2. Proceeding with testing our hypothesis, we next investigated the kinetics of transcriptional reactivation of genes encoding the intermediate filament Nestin and the Notch ligand Dll1, suggested by multiple studies to function as readout of Brn2 and Ascl1, respectively. The requirement of Brn2 for efficient activity of the Nestin neural enhancer was thoroughly documented (Josephson et al. 1998; Sunabori et al. 2008; Lodato et al. 2013). The strict dependency of Dll1 on Ascl1, together with its pattern and kinetics of expression, makes Dll1 a good proxy for Ascl1 activity in neural progenitor cells (Castro et al. 2006; Kageyama et al. 2008).
In order to evaluate the dynamics of transcription during M-G1 transition, we used single-molecule RNA fluorescence in situ hybridization (smRNA-FISH) to simultaneously quantify nascent and mature mRNA in single asynchronous NS cells, using spectrally distinguishable probes against introns or exons. In all interphase NS cells analyzed, exonic signal was much more prevalent than the intronic signal, as expected given the high relative abundance of mature transcripts (Fig. 7A,C). Although many exon foci were found in each interphase cell, particularly for the Nestin probe, colocalization of exonic and intronic probes on DNA (marking primary transcripts) was found to be proportional to the number of loci (zero to four depending on whether in G1 or G2 phase) (Fig. 7A,C). Colocalizing foci from primary transcripts, a readout of active transcription, were often the brightest to be found in the cell. As expected, exon and intron probe signals almost completely disappeared in interphase cells upon RNase treatment (Fig. 7E,F).
Figure 7.

Targets of Brn2 (Nestin) and Ascl1 (Dll1) show distinct kinetics of transcriptional reactivation during M-G1 transition in proliferating NS cells. (A) Representative images of cells in interphase, anaphase, late telophase, and early G1 stained by smRNA-FISH using exonic (FAM) and intronic (Q570) probes for Nestin transcript. Images shown are maximum intensity projections of six optical planes with 0.3-µm Z step intervals, with white arrowheads marking spots of colocalized intron–exon probe signal found on DNA (stained with DAPI) Scale bars, 5 µm. (B) Stacked bar plots showing the fractions of cells containing at least one spot of colocalized intron–exon probe signal found on DNA (green), outside DNA (yellow), or without colocalization spots (red; n = 40 in metaphase; n = 29 in anaphase; n = 37 in telophase, n = 37 in late telophase; n = 32 in early G1; n = 30 in interphase; n = 12 in interphase + RNase). (C) Representative images of cells in interphase, anaphase, late telophase, and early G1 costained by smRNA-FISH using exonic (FAM) and intronic (Q570) probes for Dll1 transcript and by immunocytochemistry for Ascl1 protein. Images shown are maximum intensity projections of six optical planes with 0.3-µm Z step intervals, with white arrowheads marking spots of colocalized intron–exon probe signal found on DNA (stained with DAPI). (D) Stacked bar plots showing the fraction of cells containing at least one spot of colocalized intron/exon probe signal found on DNA (green), outside DNA (yellow), or without colocalization spots (red; n = 32 for metaphase, anaphase, telophase, early G1; n = 21 for late telophase; n = 34 for interphase; n = 11 for interphase + RNase). (E,F) Quantifications of exon probe signal per cell for Nestin (E) and Dll1 (F) transcripts at different stages during M-G1 transition, in experiments described in A–D. Quantification of RNase treated sample is included as control. One-way ANOVA Tukey's multiple comparison test was performed. (n.s.) P > 0.05, (*) P ≤ 0.05, (**) P ≤ 0.01, (***) P ≤ 0.001, (****) P ≤ 0.0001. (M) Metaphase, (A) anaphase, (T) telophase, (Late T) late telophase, (I) interphase. Data shown as mean ± SD. Scale bars, 5 µm (1 µm in high magnification)
When assessing transcription during M-G1 transition, we used coimmunostaining for Ascl1 as a proxy for the timing of nuclear envelope reformation. Telophase cells were defined by having cytokinesis undergoing prior to Ascl1 import, whereas cells in the immediate subsequent step (when Ascl1 import could already be observed) were considered in late telophase (Fig. 7C). In the case of Nestin, a significant fraction of actively transcribing cells was found to increase steadily throughout M-G1 transition, starting as early as anaphase (seven out of 29 cells; 0.24) and until early G1, when the largest number of cells with intron/exon colocalization on DNA was observed (23 out of 32 cells; 0.72) (Fig. 7A,B). Such early reactivation was observed in both Ascl1-positive and -negative cells, in line with Nestin expression being independent of this TF (Supplemental Fig. S9). Active transcription was often observed only in one daughter cell, or in both but in distinct locations, recalling recently found heterogeneity in allele location and 3D interactions at single-cell level during interphase (Finn et al. 2019). At all stages during M-G1 transition, a significant number of intron/exon colocalized foci was observed outside DNA, reaching the highest fraction in telophase (12 out of 37 cells; 0.32) (Fig. 7A,B). However, these were almost never found in interphase, and are likely to represent long-lived unprocessed transcripts resulting from some level of RNA splicing inhibition during mitosis (Shin and Manley 2002) and that diffused away from the transcription site. Moreover, the presence of active Nestin transcription during M-G1 transition is also supported by a concomitant increase in total number of transcripts (exon probe foci) per cell (Fig. 7E). As expected, intron/exon probe colocalization for Nestin transcript almost disappeared upon incubation with transcriptional inhibitor triptolide (Supplemental Fig. S10A–C). This was concomitant with a strong reduction of intron signal (from an average of 10 to three foci per cell), while no increase in exon probe counting was observed throughout M-G1, altogether in line with transcription occurring during this period in control conditions (Supplemental Fig. S10D,E).
When analyzing Dll1 transcription we focused exclusively in Ascl1-expressing cells, given its complete dependency on Ascl1 expression (Casarosa et al. 1999). In contrast to the Nestin gene, smRNA-FISH analysis of Dll1 transcription revealed the absence of actively transcribing cells until late telophase, with a dramatic increase in colocalization of intronic and exonic probes observed in early G1 (29 out of 32 cells; 0.91) (Fig. 7C,D). Quantification of exonic probe foci number revealed no increase during M-G1 transition, in line with Dll1 transcription only restarting in early G1 (Fig. 7F). Finally, we have repeated the same analysis using probes for Nestin and Dll1 transcripts labelled with a different set of fluorophores, and obtained similar results when quantifying exon/intron colocalization events (Supplemental Fig. S11). In conclusion, surveying active transcription with smRNA-FISH shows the onset of activation of Nestin and Dll1 genes occurs at distinct time points during M-G1 transition. This suggests mitotic chromosome binding provides a timing advantage to Brn2 in transcriptional reactivation, as compared with Ascl1. However, not all Brn2 targets are expected to display such an early timing of reactivation, due to additional regulatory constraints. In fact, transcription of Fabp7, another neural stem cell gene that Brn2 regulates via its proximal promoter region (Fig. 5B; Josephson et al. 1998), is also reactivated during mitotic exit but with a later onset relative to Nestin, with significant expression starting in late telophase (Supplemental Fig. S12).
Reactivation of Nestin transcription in early M-G1 is dependent on Brn2
We next sought to understand whether transcriptional reactivation of Nestin during early stages of M-G1 transition depends on Brn2 function. To address this, we designed a mitotic-specific dominant negative of POU3F TFs (DN-Brn2), based on sequence-specific binding competition upon coexpression of a truncated form of Brn2, consisting of its DBD fused to mCherry. Flanking the DBD with nuclear export signal (NES) sequences resulted in its localization outside the nucleus throughout interphase, gaining access to chromatin only during mitosis, upon NEBD. Expression of DN-Brn2 was placed under the control of a tetracycline-inducible promoter in NS cells where endogenous Brn2 was tagged with eGFP (Fig. 8A,B). Upon treatment with the tetracycline analog doxycycline (Dox), expression of DN-Brn2 is found in association with mitotic chromosomes, with its rapid export observed upon nuclear envelope reformation at the end of M-G1 transition (80% exported in 20 min after telophase) (Fig. 8C). The association of DN-Brn2 with condensed chromosomes during metaphase did not alter the association of Brn2-eGFP (MCE = 0.42 vs. 0.46 with or without Dox, respectively, with R2= 0.1175) (Fig. 8D–F). This result implies that DN-Brn2 is unable to compete and prevent Brn2-eGFP association with metaphase chromosomes, in agreement with the importance of non-sequence-specific DNA binding. Accordingly, we detected a linear increase in MCE levels of Brn2, observed in overexpression experiments (Supplemental Fig. S5A). However, such DN-Brn2 is expected to compete with endogenous Brn2 once sequence-specific transcription is initiated (early anaphase) (see Fig. 7A,B). To understand whether reactivation of Nestin transcription during anaphase depends on Brn2 function, we compared the levels of transcripts (using smRNA-FISH) in the presence or absence of DN-Brn2. Strikingly, a significant reduction of Nestin transcripts was observed in the presence of DN-Brn2, as indicated by a decrease in the number of exon probe foci in Dox-treated cells and by their levels remaining unchanged until late telophase (Fig. 8G,H). This finding confirms the requirement of the canonical TF activity of Brn2 in reactivation of Nestin gene during early stages of M-G1 transition, indicating the transcriptional activity of Brn2 is nonlimiting from the start of mitotic exit.
Figure 8.

Transcription reactivation of Nestin gene during M-G1 transition is dependent on Brn2 activity. (A) Experimental strategy whereby a mitotic-specific dominant-negative form of Brn2 (DN-Brn2), composed of its DBD flanked by nuclear export signal (NES) sequences and in fusion with mCherry, was expressed in NS cells under a Dox-inducible promoter. (B) Schematic depicting how DN-Brn2 competes with endogenously expressed POU3F family members for their binding sites in the presence or absence of the nuclear envelope (interphase and mitosis, respectively). (C) Time-lapse live-cell imaging of Dox-treated cells expressing DN-Brn2, from telophase until G1. White arrowheads show association of DN-Brn2 with mitotic DNA (labeled with SiR-Hoechst) during telophase and its cytoplasmic localization after nuclear envelope reformation. Quantification of nuclear export kinetics of DN-Brn2, normalized to interphase levels, is shown below. n = 11 cells. (D) Representative captions from live-cell imaging of DN-Brn2-NS cells in the absence (top) or presence (bottom) of Dox. White arrowheads indicate an example of a metaphase plate in each condition. DNA labeled with SiR-Hoechst. (E) Quantification of MCE of Brn2-eGFP in the presence (+Dox) or absence (−Dox) of DN-Brn2. Mann–Whitney test was performed. (n.s.) P > 0.05. n = 32 for each condition. (F) Correlation analysis between MCE levels of Brn2 and DN-Brn2. (G) Representative images of Dox-treated and -untreated NS cells in telophase stained by smRNA-FISH using an exonic (Q670) probe for Nestin transcript. DNA staining with DAPI. Images shown are maximum intensity projections of 12 optical planes with 0.3-µm Z step intervals. (H) Quantifications of exon probe signal per cell for Nestin transcript at different stages during M-G1 transition. One-way ANOVA Tukey's multiple comparison test was performed. (n.s.) P > 0.05, (**) P ≤ 0.01, (****) P ≤ 0.0001. n = 41 for all untreated cells (−Dox). n = 41 for A with Dox; n = 38 Tel with Dox; n = 41 Late Tel with Dox. (A) Anaphase, (T) telophase, (Late T) late telophase. Data shown as mean ± SD. Scale bars, 5 µm
Discussion
Contrary to the classical view of mitotic chromatin as being transcriptional inert and refractory to TF binding, various studies have in recent years described the ability of certain TFs to associate with mitotic chromatin in dividing cells. However, the molecular basis and functional consequences of such interactions have remained the subject of controversy, with the concept of “mitotic bookmarking by TFs” at the center of it. Recently, genome-wide profiling of transcription through each stage of mitosis revealed that reactivation of the transcriptome occurs in temporally coordinated waves starting early during M-G1 transition (Palozola et al. 2017). These observations raise the question of what is the underlying regulatory logic during this period and how mitotic bookmarking by TFs may be involved in this process. In the present work, we have tackled this question in neural stem cells, a cell type that can be experimentally manipulated and where key TFs are known. Focusing on Brn2 and Ascl1, we investigated how two TFs with distinct functions in neural cell identity interact with mitotic chromatin and how this impacts their transcriptional output during M-G1 transition.
Important mechanistic insights into the temporal pattern of gene reactivation during M-G1 transition have recently started to emerge. Enhancer usage, and the establishment of enhancer–promoter loops, has been observed as early as anaphase (Palozola et al. 2017; Abramo et al. 2019; Zhang et al. 2019). A role for TFs in regulating chromatin accessibility during this period has been recently shown (Festuccia et al. 2019; Friman et al. 2019; Owens et al. 2019); however, a clear link between canonical TF activity and timing of gene reactivation has remained elusive. Addressing this requires the ability to knock down the function of TFs specifically during mitosis, a task made more difficult with Brn2, by the coexpression of redundant POU3f family members in neural stem/progenitor cells (Sugitani et al. 2002). Using a mitotic-specific dominant-negative approach, we were able to overcome these limitations, providing evidence for the need of POU3f TFs in Nestin gene transcription starting from anaphase. This was made possible also by the use of single-cell smRNA-FISH, which provided the sensitivity and temporal resolution that sets aside our study from previous reports using cell population analysis.
In sharp contrast to Brn2 targets, no Dll1 transcription was ever observed before early G1, at which point the concentration of Ascl1 near chromatin increases sharply. Previously, we have shown that activation of Dll1 by Ascl1 requires corecruitment of Brn2 to its neural enhancer, with Ascl1 being rate-limiting given its noncontinuous expression in proliferating NS cells (Castro et al. 2006). Our results indicate that also during M-G1 Ascl1 becomes rate-limiting, with activation of Dll1 occurring when both TFs are available near chromatin.
The concept of mitotic “bookmarking” by TFs as a mechanism to convey gene regulatory information across cell division implies the establishment of sequence-specific interactions with regulatory regions throughout the duration of mitosis/mitotic exit. Our ChIP-seq analysis revealed the absence of sequence-specific binding by Brn2 in cells arrested in prometaphase. We propose phosphorylation of Ser362 as a possible mechanism to impair sequence-specific binding. The equivalent residue in Oct4 is a target of Aurora B kinase during prometaphase, being subsequently dephosphorylated by active protein phosphatase 1 (PP1) during mitotic exit (Shin et al. 2016). Notably, the docking site for PP1 is also conserved in Brn2, with the timing of PP1 activity being compatible with dephosphorylation of Brn2 on time for activation of Nestin gene transcription early in M-G1. In line with these results, we virtually did not detect any ongoing Nestin transcription during metaphase. This suggests reactivation of Nestin does not require the maintenance of low levels transcription throughout mitosis that was recently observed for some genes (Palozola et al. 2017).
The absence of sequence-specific interactions observed with ChIP-seq is in apparent contradiction with mutagenesis of residues in helices 3 of each POUS and POUH subdomain, predicted to establish base-specific interactions with an octamer motif and that impair binding to mitotic chromosomes (Malik et al. 2018). However, these observations can be conciliated, as TFs often rely on the DBD when undergoing an electrostatic-guided one-dimensional (1D) search, and the same residues can switch role from a purely electrostatic interaction with the DNA backbone to a highly specific binding mode (Kalodimos et al. 2004; Suter 2020). Thus, mutagenesis experiments may not always be able to disentangle the different nature of TF–DNA interactions.
We found Brn2 association with mitotic chromatin to be governed mostly by nonspecific electrostatic interactions. This important observation indicates Brn2 does not function as a bona fide mitotic bookmarker in neural stem/progenitor cells. As an alternative, we propose that association of Brn2 with mitotic chromosomes may serve to increase its local concentration near chromatin during M-G1 transition, thereby facilitating its search for target genes. This model was indeed supported by the early timing of reactivation of Nestin transcription during mitotic exit in a Brn2-dependent manner. In contrast, exclusion of Ascl1 from mitotic chromatin is associated with a late reactivation of its target Dll1 in early G1. This occurs concomitant with a sharp increase of Ascl1 concentration near chromatin, which we show requires active nuclear import of TF into the newly reformed nuclear envelope (see model in Fig. 9). Our results support the emerging view that TF association with mitotic chromosomes detected by live-cell imaging is to large extent driven by nonspecific interactions (Festuccia et al. 2019; Raccaud et al. 2019). However, to what extent the lack of sequence-specific binding observed with Brn2 can be extended to other mitotic chromosome binding TFs must be examined in the future. Of note, the Brn2 case finds precedent in a recent study showing that association of Sox2 with mitotic chromatin in ES cells also occurs in the absence of sequence-specific binding (Festuccia et al. 2019).
Figure 9.
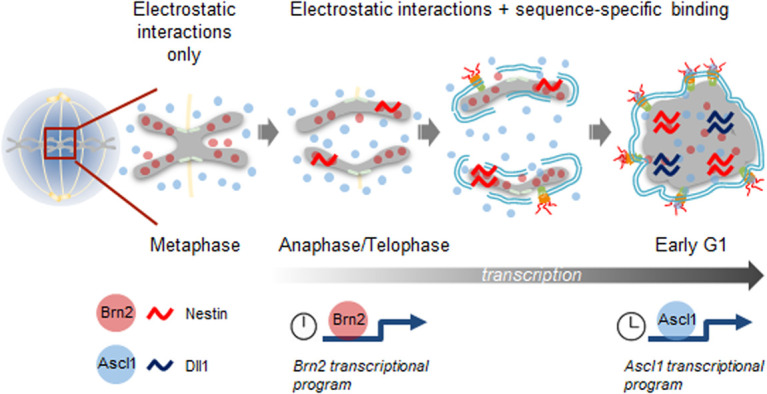
Model for hierarchical reactivation of transcription by Brn2 and Ascl1 in neural stem/progenitor cells undergoing proliferative divisions. Binding of Brn2 to metaphase chromosomes does not involve sequence-specific binding and is mediated by electrostatic interactions. The constant presence near chromatin results in Brn2 inducing the transcriptional reactivation of its target gene Nestin starting from anaphase, a process that requires sequence-specific binding and occurs concomitantly with chromatin decondensation and Brn2 dephosphorylation (events not represented in the figure). In contrast, Ascl1 is excluded from mitotic chromatin, given its low electrostatic potential. Ascl1 concentration in the vicinity of chromatin increases due to nuclear import (upon nuclear envelope reformation), resulting in transcriptional reactivation of its target gene Dll1 in early G1. The different dynamics observed favor an early versus late reactivation of transcriptional targets of Brn2 and Ascl1, respectively.
In contrast to Brn2, Ascl1 exclusion from mitotic chromatin or DNA-rich regions in interphase is suggestive of low electrostatic potential, further supported by its strong ability to counteract the colocalization of SV40 NLS sequences with condensed chromosomes. As a possible consequence, Brn2 and Ascl1 may use different mechanisms when searching the interphase genome for target genes. The presence of Brn2 in the vicinity of mitotic chromatin and interphase DNA-rich regions is in line with Brn2 using more efficient 1D search-based mechanisms to find its target genes. On the contrary, and given its low electrostatic potential, Ascl1 may use preferentially random encountering (3D search), a mechanism highly dependent on protein levels (Suter 2020). This is supported by observations showing Ascl1 cannot induce Dll1 transcription in late telophase, before its highest concentration inside the newly reformed nuclear envelope is reached.
Studies using stem/progenitor cells (including of neural origin) have shown transition through mitosis and G1 phase is crucial for establishing a window of opportunity for executing cell fate decisions (Soufi and Dalton 2016). Understanding the temporal dynamics of transcription during M-G1 transition should cast light into how TFs with a role in cell identity operate. Antagonistic cross-regulatory interactions between pathways promoting progenitor maintenance and neuronal differentiation are well documented. Thus, the intricate balance that regulates cell identity in daughter cells should rely on the kinetics that define how such programs are rewired during mitotic exit. The model we propose describes how the timing of transcriptional reactivation of Brn2 targets may be determined, favoring the re-establishment of the neural stem/progenitor program (Fig. 9). In the case of Ascl1, its expression in neural stem/progenitor cells undergoing proliferative divisions is in apparent contradiction with a well-established ability to promote neuronal commitment and differentiation. The current view is that Ascl1 neurogenic activity is kept at reduced level in a proliferative cell context by mechanisms that may include its (oscillatory) mode of expression and post-translational modifications. We suggest the observed exclusion of Ascl1 from mitotic chromosomes (and delayed transcriptional activity) as yet another layer of regulation to avert premature neuronal differentiation in cycling cells.
Overall, our work results in a model whereby TFs, and their electrostatic properties, play an important role in determining the timing of reactivation of target genes during M-G1 transition. In such a model, TFs occupy distinct hierarchical levels depending on how they interact with mitotic chromatin. It is tempting to speculate that TFs with a bona fide mitotic bookmarking function (i.e., maintain sequence-specific binding throughout mitosis) may be higher in this hierarchy. Emerging genome-wide studies mapping histone marks during M-G1 transition should help dissect further the role of TFs during this period.
Materials and methods
DNA constructs
Expression vectors for eGFP fusions were generated in eGFP N1 (Clontech 6085-1) by subcloning of PCR fragments obtained using primers and template vectors shown in Supplemental Table S1. Mutations of specific residues were introduced by site-directed mutagenesis using oligonucleotides shown in Supplemental Table S2. Plasmids encoding NLS fusion constructs were generated by annealing and subcloning oligonucleotides with complementary sequences (Supplemental Table S3) and BsrGI and XbaI cohesive ends into the C-terminal domain of either eGFP N1, Ascl1-eGFP, or Ascl1/E47-eGFP. All other constructs are described in specific sections below. Constructs generated in this study were confirmed by Sanger sequencing. Full-length Oct6-GFP, DBD (Oct6)-eGFP, and DBD (Brn4)-eGFP expression plasmids were kindly provided by Michael Wegner (Institut fur Biochemie, University Erlangen-Nurnberg).
Establishment, genome editing, and culture of NS lines
NS cell cultures were established from E13.5 mouse Bl6 ventral telencephalon as previously described (Conti et al. 2005) and maintained in mouse NeuroCult basal media supplemented with proliferation supplement (Stem Cell Technologies 05702,), 1% penicillin-streptomycin (Thermo Fisher 15140122), 10 ng/mL EGF (Peprotech 315-09), 10 ng/µL bFGF (Peprotech 100-18B), and 2 µg/mL laminin (Sigma-Aldrich L2020). Cells were kept in T-flasks (Corning, 734-2705) and split every 2–3 d using Accutase (Sigma-Aldrich A6964). For eGFP tagging of Brn2 and Ascl1 by CRISPR/Cas9, template vectors were generated by PCR amplification of both the left and right homology arms of genomic regions and eGFP coding sequence, using primers from Supplemental Table S4, and cloned into pBluescript II KS (+/−) using Gibson assembly (New England Biolabs 174E2611S). For generation of vectors for expression of gRNAs, oligonucleotide pairs (CACCGCGTCCAGTGAACTCAAGCGG, AAACCCGCTTGAGTTCACTGGACGC for Brn2 and CACCGACTTTACCAACTGGTTCTG, AAACCAGAACCAGTTGGTAAAGTC for Ascl1) were cloned into pX330 (Cong et al. 2013) using BbsI. NS cells were electroporated using a Neon nucleofection system. Briefly, cells were rinsed with PBS without Ca2+ and Mg2+ (Biowest L0615-500), dissociated with Accutase, and neutralized with growth medium. Cells were centrifuged at 1000g for 5 min at room temperature, washed with PBS, centrifuged at 1000g for 5 min at room temperature, and resuspended in resuspension buffer at a final density of 1.2 × 106 cells/120 µL. For CRISPR/Cas9 experiments, 1 µg of each plasmid was used in a ratio of 1:1. The cell–DNA mixture was gently mixed and aspirated into the Neon tip (Life Technologies MPK10025), which was inserted into the Neon pipette and subjected to an electric pulse (pulse voltage: 1700 V; pulse width: 20; number of pulses: one). Finally, cells were transferred to a six-well plate containing prewarmed complete NeuroCult medium. Four days later, cultures were enriched for recombined cells (GFP+) using FACSorting and clones originated from single-cells allowed to expand in 96-well plates. The success of genome editing was verified by PCR from genomic DNA and confirmed by Western blot analysis. Briefly, crude cell lysates were diluted in 2× Laemmli buffer (Sigma-Aldrich S3401) and denatured for 5 min at 95°C. Samples were separated in 12% SDS-PAGE gels and transferred to nitrocellulose membranes (GE Healthcare GE10600008) using standard procedures. Blots were probed with primary anti-Ascl1 (1:1000; Abcam, ab211327), anti-Brn2 (1:1000; GeneTex GTX114650), anti-GAPDH (1:1000; Cell Signaling 2118), or anti-histone 3 (1:1000; Cell Signaling 4499) and HRP-conjugated rabbit secondary antibody (Jackson ImmunoResearch 111-035-144).
Live imaging and quantification of mitotic binding
The P19 cell line was cultured in DMEM high-glucose media (BioWest L0101) supplemented with 1% L-glutamine (Thermo Fisher 25030024), 1% penicillin-streptomycin (Thermo Fisher 15140122), and 10% fetal bovine serum heat-inactivated (BioWest S181BH-500). For imaging analysis, P19 cells were transfected on four- or eight-well polymer chambers (ibidi 80426 and 80826, respectively) at 70% confluency, using Lipofectamine 2000 (Life Technologies 11668-019) in the proportion of 1 µg of DNA to 3 µL of lipofectamine (1 µg/well) according to the manufacturer's protocol. Before imaging, P19 cells were moved to phenol red-free DMEM/F12 medium supplemented with FBS (10×). Live-cell imaging of mitotic chromosomes of both NS and P19 cells was achieved by timelapse or snapshot acquisition after cell synchronization in metaphase using 60 µM proTAME (R&D systems I-440-01M) and 200 µM Apcin (R&D Systems I-444-05M) for 2–6 h. Confocal Z-series stacks were acquired at 37°C and 5% CO2, on a Yokogawa CSU-X spinning disk confocal, using a 60× 1.2 NA water immersion objective (488-nm, 561-nm, and 640-nm laser lines were used) and an Andor iXon+ EMCCD camera for image acquisition, respectively. For imaging of NS cells, the same DMEM/F12 was complemented with proliferation supplement, growth factors, and laminin, and cells were grown in same chamber slides. Apcin (100 µM) was used instead for NS cells. For ChIP-seq validation results, Brn2-eGFP NS cells were synchronized with 330 nM colchicine for 10 h. Staining of DNA was done with far-red dye (Lukinavičius et al. 2015), SiR-Hoechst (1:1000; prolonged time lapse), or Hoechst (1:10,000 from 10 mg/mL stock; snapshot acquisition). For imaging of NS cells, higher exposure times (>500 msec) were often required. Postimaging analysis, including background subtraction, was manually performed on time-lapse or snapshot data using Fiji software using DNA staining to threshold chromosome signal. Mitotic chromosome enrichment (MCE) was calculated by dividing the mean fluorescence intensities of TF enrichment at mitotic chromatin and whole cells and then converting data to log2 for visualization, as previously described (Teves et al. 2016). Scatter plots display mean values of MCE of all cells of a given population, in which each dot represents a single quantified cell.
Tetracysteine tag labeling
Ascl1/E47-TC and Brn2-TC expression vectors were generated by PCR amplification using primers shown in Supplemental Table S5 and Ascl1/E47 (Castro et al. 2006) and Brn2 template plasmids (Sugitani et al. 2002). For live imaging, P19 cells were transfected with Ascl1/E47-TC or Brn2-TC expression vectors and synchronized with 60 µM proTAME and 200 µM Apcin. For labelling, cells were washed once with Opti-MEM (Life Technologies 11058-021) and incubated with 2 µM ReAsH (Cayman 19767) for 30 min at 37°C. ReAsH was removed, and cells were washed again with Opti-MEM. A minimum of two 400 µM BAL incubations were carried out for 15 min at 37°C in order to reduce nonspecific ReAsH staining, with Opti-MEM washes in between. BAL was added a third time if cells saw good survival up to that point. After the last BAL incubation, cells were left in live-imaging medium.
ChIP-seq and bioinformatics
Cultures at 70% confluency were incubated with 330 nM colchicine for 10 h. Synchronized NS cells were then gently “shaken off” in order to enrich for the mitotic population. Thereafter, cells were handled in 1.5-mL tubes as typically only around 10 × 106 cells were obtained from seven T-150 flasks of confluent NS cells. The same amount of interphase cells was gathered in parallel by using Accutase on adherent cultures. The purity of our mitotic sample was confirmed in a small sample cytospined (Simport 720-1962) and immunostained with anti-pHH3 (1:500; Merck 06-570) and DAPI. Only mitotic samples with >90% purity were further processed. For chromatin extraction from synchronized and unsynchronized cultures, cells were fixed sequentially with 2 mM di(N-succinimidyl) glutarate (Sigma-Aldrich 80424) and 1% formaldehyde (Sigma-Aldrich F8775) in phosphate-buffered saline and lysed, sonicated, and immunoprecipitated as described (Castro et al. 2011) using rabbit anti-Brn2 antibody (Santa Cruz Biotechnology C-20). Library preparation (from precipitated material and input chromatin as control) was performed using a NEBNext Ultra II DNA library preparation kit (New England Biolabs E7645S) without size selection to maintain sample complexity and according to manufacturer's protocol. Next-generation sequencing was performed at the Instituto Gulbenkian de Ciência (IGC) genomics facility using an Illumina NextSeq500 platform.
Dana analysis was performed in Galaxy (https://usegalaxy.eu). Alignment against mm9 reference genome was done with Bowtie2, format conversion and removal of duplicate reads using SAMTools, and peak-calling using MACS 2.0 (Feng et al. 2012) with a P-value cut-off at 10(−10) using input chromatin as control. Subsampling of the data sets confirmed that peak calling saturation was achieved with ∼80% of sequencing reads. Data visualization was done with the resulting bigwig files at the UCSC genome browser (http://genome.ucsc.edu). Density plots and intersection of data sets were performed using predefined tools available in Galaxy. A de novo DNA motif search was performed using CisFinder with default settings (Sharov and Ko 2009) and gene ontology analysis using GREAT (McLean et al. 2010).
Imaging and DNA colocalization analysis
eGFP fusion constructs were imaged at Z-series stacks of 0.2 µm, acquired at 37°C and 5% CO2, on a Yokogawa CSU-X spinning disk confocal, using a 60× 1.2 NA water immersion objective. An Andor iXon+ EMCCD camera was used to acquire images for the emission of the eGFP (488-nm laser line). The colocalization of transcription factors with different DNA regions was analyzed with an image segmentation pipeline in Fiji. Cell nuclei were identified and segmented by K-means clustering based on the Hoechst signal. Three regions with high, medium, and low Hoechst levels within each nucleus were defined, respectively representing heterochromatic, DNA-dense, and DNA-poor regions. Subsequently, the corresponding eGFP signal intensity in each of the three segmented regions was measured. Data are represented in log2 values in which the intensity of GFP in each DNA region was divided by the whole-cell intensity. At least 20 cells were analyzed per condition. Due to the quality of the Hoechst staining, DNA segmentation by K means clustering resulted in some variability among cells. Thus, only cells yielding a predefined segmentation ratio among the three different regions—heterochromatin (10%–20%), DNA-dense (40%–60%), and DNA-poor (40%)—were included in the final analysis. A max projection of the 20–30 0.2-µm Z slices that were acquired was applied before DNA segmentation.
smRNA-FISH
Custom Stellaris FISH probes were designed against Nestin and Dll1 by using the Stellaris RNA FISH Probe Designer (http://www.biosearchtech.com/stellarisdesigner). Probes were coupled with either FAM, Q570, or Q670 fluorescent reporters (see Supplemental Tables S6–S9). RNA FISH protocol followed the manufacturer's instructions (http://www.biosearch.com/stellarisprotocols). Briefly, NS cells were grown on poly-L lysine-treated (Sigma P8920) coverslips with thickness of #0 (VWR 631.0148). When confluency was reached, cells were fixed for 10 min using RNase-free PBS-diluted 3.7% formaldehyde (Sigma F8775), permeabilized with Triton 0.1% for 5 min, and immunostained with anti-Ascl1 (Abcam ab211327) at 1:500 dilution (1 h at room temperature). At this point, if RNase treatment was intended, permeabilized fixed NS cells were incubated for 20 min with RNase A (Invitrogen 12091-021). NS cells were washed with designated buffers, and coverslips were incubated with the desired custom-made RNA FISH probes (at 1:100 concentration) during 16 h at 37°C in the dark in a humidified chamber. In the next day, cells were washed, stained with DAPI, and mounted in VectaShield mounting medium (Vector Laboratories H-1000). Imaging was performed on a DeltaVision widefield microscope coupled with a high-sensitivity EM-CCD camera using a 100× oil objective (1.4 NA). Images acquired with DeltaVision were deconvolved using Huygens professional version 19.04 (Scientific Volume Imaging) using standard parameters. In control experiments with a transcriptional inhibitor, NS cells were incubated for 1 h with 5 µM triptolide added to fresh media prior to fixation. For DN-Brn2 exon counting, images were acquired using a Leica TCS SP8 confocal microscope equipped with two hybrid detectors for higher sensitivity and required lasers. A 63× (1.4 NA) oil objective was used, coupled with a zoom factor of 3. Images were analyzed using Fiji, and exon and intron signal counting was performed using the find maxima tool, defining the same threshold for all images. A single Z plane was used to confirm colocalization events and max projection of 25 Z stacks (0.3-µm step size) used for exon/intron counting.
Accession numbers
ChIP-seq data sets reported in this study can be accessed under the code E-MTAB-9758 in Array Express.
Supplementary Material
Acknowledgments
We thank Michael Wegner for providing the Oct6-GFP, DBD (Oct6)-eGFP, and DBD (Brn4)-eGFP plasmids. We thank the technical support of Instituto Gulbenkian de Ciência's (IGC) Advanced Imaging Facility (AIF-UIC), which is supported by the national Portuguese funding (PPBI-POCI-01-0145-FEDER-022122), cofinanced by Lisboa Regional Operational Programme (Lisboa 2020), under the Portugal 2020 Partnership Agreement, through the European Regional Development Fund (FEDER) and Fundação para a Ciência e a Tecnologia (FCT; Portugal). In particular, we thank Nuno Pimpão and Gabriel Martins for excellent assistance with imaging acquisition and data analysis. We thank the IGC Genomics Facility for guidance with library synthesis and NGS and the IGC Flow Cytometry Facility. We also acknowledge the support of i3S Scientific Platform Advanced Light Microscopy, member of the national infrastructure PPBI-Portuguese Platform of BioImaging (supported by POCI-01-0145-FEDER-022122). In particular, we thank Maria Azevedo for excellent assistance. This work was funded by grant LISBOA-01-1045-FEDER-029663, cofinanced by Lisboa Regional Operational Programme (Lisboa 2020), under the Portugal 2020 Partnership Agreement through the European Regional Development Fund (FEDER), and by the FCT/Ministério da Ciência, Tecnologia e Ensino Superior (MCTES) through national funds (Programa de Investimentos e Despesas de Desenvolvimento da Administração Central [PIDDAC]) to D.S.C.; FCT doctoral fellowship PD/BD/105999/2014 to M.A.F.S.; EMBO installation grant IG2778 to R.A.O.; and Concurso Estímulo ao Emprego Científico Individual (CEECind) from FCT (CEECIND/01092/2017) to R.A.O.
Author contributions: M.A.F.S. conceived the study; designed the experiments; acquired, analyzed, and interpreted the data; and wrote the draft of the manuscript. D.S.S. conceived and designed the study, and acquired, analyzed, and interpreted the data. V.T. and A.H. acquired, analyzed, and interpreted the data. R.B.B. and S.M.P. contributed unpublished essential reagents and edited the manuscript. R.A.O. and D.S.C. conceived the study, designed the experiments, supervised the study, and wrote the draft of the manuscript.
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.348174.120.
Competing interest statement
S.M.P. is a founder and shareholder of Cellinta Ltd., a cancer gene therapy company. S.M.P. is also an inventor on a University of Edinburgh patent related to NS cell culture methods (WO2005121318A3). The other authors declare no competing interests.
References
- Abramo K, Valton AL, Venev SV, Ozadam H, Fox AN, Dekker J. 2019. A chromosome folding intermediate at the condensin-to-cohesin transition during telophase. Nat Cell Biol 21: 1393–1402. 10.1038/s41556-019-0406-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertrand N, Castro DS, Guillemot F. 2002. Proneural genes and the specification of neural cell types. Nat Rev Neurosci 3: 517–530. 10.1038/nrn874 [DOI] [PubMed] [Google Scholar]
- Bylund M, Andersson E, Novitch BG, Muhr J. 2003. Vertebrate neurogenesis is counteracted by Sox1-3 activity. Nat Neurosci 6: 1162–1168. 10.1038/nn1131 [DOI] [PubMed] [Google Scholar]
- Caravaca JM, Donahue G, Becker JS, He X, Vinson C, Zaret KS. 2013. Bookmarking by specific and nonspecific binding of FoxA1 pioneer factor to mitotic chromosomes. Genes Dev 27: 251–260. 10.1101/gad.206458.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casarosa S, Fode C, Guillemot F. 1999. Mash1 regulates neurogenesis in the ventral telencephalon. Development 126: 525–534. 10.1242/dev.126.3.525 [DOI] [PubMed] [Google Scholar]
- Castro DS, Skowronska-Krawczyk D, Armant O, Donaldson IJ, Parras C, Hunt C, Critchley JA, Nguyen L, Gossler A, Göttgens B, et al. 2006. Proneural bHLH and Brn proteins coregulate a neurogenic program through cooperative binding to a conserved DNA motif. Dev Cell 11: 831–844. 10.1016/j.devcel.2006.10.006 [DOI] [PubMed] [Google Scholar]
- Castro DS, Martynoga B, Parras C, Ramesh V, Pacary E, Johnston C, Drechsel D, Lebel-Potter M, Garcia LG, Hunt C, et al. 2011. A novel function of the proneural factor Ascl1 in progenitor proliferation identified by genome-wide characterization of its targets. Genes Dev 25: 930–945. 10.1101/gad.627811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, et al. 2013. Multiplex genome engineering using CRISPR/Cas systems. Science 339: 819–823. 10.1126/science.1231143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conti L, Pollard SM, Gorba E, Reitano E, Toselli M, Biella G, Sun Y, Sanzone S, Ying Q, Catanneo E, et al. 2005. Niche-independent symmetrical self-renewal of a mammalian tissue stem cell. PLoS Biol 9: e283. 10.1371/journal.pbio.0030283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deluz C, Friman ET, Strebinger D, Benke A, Raccaud M, Callegari A, Leleu M, Manley S, Suter DM. 2016. A role for mitotic bookmarking of SOX2 in pluripotency and differentiation. Genes Dev 30: 2538–2550. 10.1101/gad.289256.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng J, Liu T, Qin B, Zhang Y, Liu XS. 2012. Identifying ChIP-seq enrichment using MACS. Nat Protoc 7: 1728–1740. 10.1038/nprot.2012.101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Festuccia N, Dubois A, Vandormael-Pournin S, Gallego Tejeda E, Mouren A, Bessonnard S, Mueller F, Proux C, Cohen-Tannoudji M, Navarro P. 2016. Mitotic binding of Esrrb marks key regulatory regions of the pluripotency network. Nat Cell Biol 18: 1139–1148. 10.1038/ncb3418 [DOI] [PubMed] [Google Scholar]
- Festuccia N, Gonzalez I, Owens N, Navarro P. 2017. Mitotic bookmarking in development and stem cells. Development 144: 3633–3645. 10.1242/dev.146522 [DOI] [PubMed] [Google Scholar]
- Festuccia N, Owens N, Papadopoulou T, Gonzalez I, Tachtsidi A, Vandoermel-Pournin S, Gallego E, Gutierrez N, Dubois A, Cohen-Tannoudji M, et al. 2019. Transcription factor activity and nucleosome organisation in mitosis. Genome Res 29: 250–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finn EH, Pegoraro G, Brandão HB, Valton AL, Oomen ME, Dekker J, Mirny L, Misteli T. 2019. Extensive heterogeneity and intrinsic variation in spatial genome organization. Cell 176: 1502–1515.e10. 10.1016/j.cell.2019.01.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friman ET, Deluz C, Meireles-Filho ACA, Govindan S, Gardeux V, Deplancke B, Suter DM. 2019. Dynamic regulation of chromatin accessibility by pluripotency transcription factors across the cell cycle. Elife 8: e50087. 10.7554/eLife.50087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gebala M, Johnson S, Narlikar G, Herschlag D. 2019. Ion counting demonstrates a high electrostatic field generated by the nucleosome. Elife 8: e44993. 10.7554/eLife.44993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geoffroy GG, Critchley JA, Castro DS, Ramelli S, Barraclough C, Descombes P, Guillemot F, Raineteau O. 2009. Engineering of dominant active basic helix-loop-helix proteins that are resistant to negative regulation by postnatal central nervous system antineurogenic cues. Stem Cells 27: 847–856. 10.1002/stem.17 [DOI] [PubMed] [Google Scholar]
- Imayoshi I, Isomura A, Harima Y, Kawaguchi K, Kori H, Miyachi H, Fujiwara T, Ishidate F, Kageyama R. 2013. Oscillatory control of factors determining multipotency and fate in mouse neural progenitors. Science 342: 1203–1208. 10.1126/science.1242366 [DOI] [PubMed] [Google Scholar]
- Jauch R, Choo SH, Ng CKL, Kolatkar PR. 2011. Crystal structure of the dimeric Oct6 (POU3f1) POU domain bound to palindromic MORE DNA. Proteins 79: 674–677. 10.1002/prot.22916 [DOI] [PubMed] [Google Scholar]
- Jerabek S, Ng CK, Wu G, Arauzo-Bravo MJ, Kim K, Esch D, Malik V, Chen Y, Velychko S, MacCarthy CM, et al. 2017. Changing POU dimerization preferences converts Oct6 into a pluripotency inducer. EMBO Rep 18: 319–333. 10.15252/embr.201642958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Josephson R, Müller T, Pickel J, Okabe S, Reynolds K, Turner PA, Zimmer A, McKay RDG. 1998. POU transcription factors control expression of CNS stem cell-specific genes. Development 125: 3087–3100. 10.1242/dev.125.16.3087 [DOI] [PubMed] [Google Scholar]
- Kadauke S, Udugama MI, Pawlicki JM, Achtman JC, Jain DP, Cheng Y, Hardison RC, Blobel GA. 2012. Tissue-specific mitotic bookmarking by hematopoietic transcription factor GATA1. Cell 150: 725–737. 10.1016/j.cell.2012.06.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kageyama R, Ohtsuka T, Shimojo H, Imayoshi I. 2008. Dynamic Notch signaling in neural progenitor cells and a revised view of lateral inhibition. Nat Neurosci 11: 1247–1251. 10.1038/nn.2208 [DOI] [PubMed] [Google Scholar]
- Kalodimos CG, Biris N, Bonvin AMJJ, Levandoski MM, Guennuegues M, Boelens R, Kaptein R. 2004. Structure and flexibility adaptation in nonspecific and specific protein-DNA complexes. Science 305: 386–389. 10.1126/science.1097064 [DOI] [PubMed] [Google Scholar]
- Kang H, Shokhirev MN, Xu Z, Chandran S, Dixon JR, Hetzer MW. 2020. Dynamic regulation of histone modifications and long-range chromosomal interactions during postmitotic transcriptional reactivation. Genes Dev 34: 913–930. 10.1101/gad.335794.119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Pelham-Webb B, Di Giammartino DC, Li J, Kim D, Kita K, Saiz N, Garg V, Doane A, Giannakakou P, et al. 2017. Widespread mitotic bookmarking by histone marks and transcription factors in pluripotent stem cells. Cell Rep 19: 1283–1293. 10.1016/j.celrep.2017.04.067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lodato MA, Ng CW, Wamstad JA, Cheng AW, Thai KK, Fraenkel E, Jaenisch R, Boyer LA. 2013. SOX2 co-occupies distal enhancer elements with distinct POU factors in ESCs and NPCs to specify cell state. PLoS Genet 9: e1003288. 10.1371/journal.pgen.1003288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukinavičius G, Blaukopf C, Pershagen E, Schena A, Reymond L, Derivery E, Gonzalez-Gaitan M, D'Este E, Hell SW, Gerlich DW, et al. 2015. SiR-Hoechst is a far-red DNA stain for live-cell nanoscopy. Nat Commun 6: 8497. 10.1038/ncomms9497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malik V, Zimmer D, Jauch R. 2018. Diversity among POU transcription factors in chromatin recognition and cell fate reprogramming. Cell Mol Life Sci 75: 1587–1612. 10.1007/s00018-018-2748-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin BR, Giepmans BNG, Adams SR, Tsien RY. 2005. Mammalian cell-based optimization of the biarsenical-binding tetracysteine motif for improved fluorescence and affinity. Nat Biotechnol 23: 1308–1314. 10.1038/nbt1136 [DOI] [PubMed] [Google Scholar]
- McLean CY, Bristor D, Hiller M, Clarke SL, Schaar BT, Lowe CB, Wenger AM, Bejerano G. 2010. GREAT improves functional interpretation of cis-regulatory regions. Nat Biotechnol 28: 495–501. 10.1038/nbt.1630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyagi S, Nishimoto M, Saito T, Ninomiya M, Sawamoto K, Okano H, Muramatsu M, Oguro H, Iwama A, Okuda A. 2006. The Sox2 regulatory region 2 functions as a neural stem cell-specific enhancer in the telencephalon. J Biol Chem 281: 13374–13381. 10.1074/jbc.M512669200 [DOI] [PubMed] [Google Scholar]
- Nieto L, Joseph G, Stella A, Henri P, Burlet-Schiltz O, Monsarrat B, Clottes E, Erard M. 2007. Differential effects of phosphorylation on DNA binding properties of N Oct-3 Are dictated by protein/DNA complex structures. J Mol Biol 370: 687–700. 10.1016/j.jmb.2007.04.072 [DOI] [PubMed] [Google Scholar]
- Owens N, Papadopoulou T, Festuccia N, Tachtsidi A, Gonzalez I, Dubois A, Vandormael-Pournin S, Nora EP, Bruneau BG, Cohen-Tannoudji M, et al. 2019. CTCF confers local nucleosome resiliency after DNA replication and during mitosis. Elife 8: e47898. 10.7554/eLife.47898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palozola KC, Donahue G, Liu H, Grant GR, Becker JS, Cote A, Yu H, Raj A, Zaret KS. 2017. Mitotic transcription and waves of gene reactivation during mitotic exit. Science 358: 119–122. 10.1126/science.aal4671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palozola KC, Lerner J, Zaret KS. 2018a. A changing paradigm of transcriptional memory propagation through mitosis. Nat Rev Mol Cell Biol 20: 55.– . 10.1038/s41580-018-0077-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palozola KC, Liu H, Nicetto D, Zaret KS. 2018b. Low-level, global transcription during mitosis and dynamic gene reactivation during mitotic exit. Cold Spring Harb Symp Quant Biol 82: 197–205. 10.1101/sqb.2017.82.034280 [DOI] [PubMed] [Google Scholar]
- Pilz GA, Shitamukai A, Reillo I, Pacary E, Schwausch J, Stahl R, Ninkovic J, Snippert HJ, Clevers H, Godinho L, et al. 2013. Amplification of progenitors in the mammalian telencephalon includes a new radial glial cell type. Nat Commun 4: 2125. 10.1038/ncomms3125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raccaud M, Friman ET, Alber AB, Agarwal H, Deluz C, Kuhn T, Gebhardt JCM, Suter DM. 2019. Mitotic chromosome binding predicts transcription factor properties in interphase. Nat Commun 10: 487. 10.1038/s41467-019-08417-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raposo AASF, Vasconcelos FF, Drechsel D, Marie C, Johnston C, Dolle D, Bithell A, Gillotin S, van den Berg DLC, Ettwiller L, et al. 2015. Ascl1 coordinately regulates gene expression and the chromatin landscape during neurogenesis. Cell Rep 10: 1544–1556. 10.1016/j.celrep.2015.02.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharov AA, Ko MSH. 2009. Exhaustive search for over-represented DNA sequence motifs with CisFinder. DNA Res 16: 261–273. 10.1093/dnares/dsp014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin C, Manley JL. 2002. The SR protein SRp38 represses splicing in M phase cells. Cell 111: 407–417. 10.1016/S0092-8674(02)01038-3 [DOI] [PubMed] [Google Scholar]
- Shin J, Kim TW, Kim H, Kim HJ, Suh MY, Lee S, Lee HT, Kwak S, Lee SE, Lee JH, et al. 2016. Aurkb/PP1-mediated resetting of Oct4 during the cell cycle determines the identity of embryonic stem cells. Elife 5: e10877. 10.7554/eLife.10877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soufi A, Dalton S. 2016. Cycling through developmental decisions: how cell cycle dynamics control pluripotency, differentiation and reprogramming. Development 143: 4301–4311. 10.1242/dev.142075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugitani Y, Nakai S, Minowa O, Nishi M, Jishage KI, Kawano H, Mori K, Ogawa M, Noda T. 2002. Brn-1 and Brn-2 share crucial roles in the production and positioning of mouse neocortical neurons. Genes Dev 16: 1760–1765. 10.1101/gad.978002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunabori T, Tokunaga A, Nagai T, Sawamoto K, Okabe M, Miyawaki A, Matsuzaki Y, Miyata T, Okano H. 2008. Cell-cycle-specific nestin expression coordinates with morphological changes in embryonic cortical neural progenitors. J Cell Sci 121: 1204–1212. 10.1242/jcs.025064 [DOI] [PubMed] [Google Scholar]
- Suter DM. 2020. Transcription factors and DNA play hide and seek. Trends Cell Biol 30: 491–500. 10.1016/j.tcb.2020.03.003 [DOI] [PubMed] [Google Scholar]
- Tanaka S, Kamachi Y, Tanouchi A, Hamada H, Jing N, Kondoh H. 2004. Interplay of SOX and POU factors in regulation of the nestin gene in neural primordial cells. Mol Cell Biol 24: 8834–8846. 10.1128/MCB.24.20.8834-8846.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teves SS, An L, Hansen AS, Xie L, Darzacq X, Tjian R. 2016. A dynamic mode of mitotic bookmarking by transcription factors. Elife 5: e22280. 10.7554/eLife.22280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasconcelos FF, Castro DS. 2014. Transcriptional control of vertebrate neurogenesis by the proneural factor Ascl1. Front Cell Neurosci 8: 412. 10.3389/fncel.2014.00412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasconcelos FF, Sessa A, Laranjeira C, Raposo AASF, Teixeira V, Hagey DW, Tomaz DM, Muhr J, Broccoli V, Castro DS. 2016. MyT1 counteracts the neural progenitor program to promote vertebrate neurogenesis. Cell Rep 17: 469–483. 10.1016/j.celrep.2016.09.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wapinski OL, Vierbuchen T, Qu K, Lee QY, Chanda S, Fuentes DR, Giresi PG, Ng YH, Marro S, Neff NF, et al. 2013. Hierarchical mechanisms for direct reprogramming of fibroblasts to neurons. Cell 155: 621–635. 10.1016/j.cell.2013.09.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Emerson DJ, Gilgenast TG, Titus KR, Lan Y, Huang P, Zhang D, Wang H, Keller CA, Giardine B, et al. 2019. Chromatin structure dynamics during the mitosis-to-G1 phase transition. Nature 576: 158–162. 10.1038/s41586-019-1778-y [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.