

Abstract
Synucleinopathies such as Parkinson's disease (PD) are hallmarked by α‐synuclein (α‐syn) pathology and neuroinflammation. This neuroinflammation involves activated microglia with increased secretion of interleukin‐1β (IL‐1β). The main driver of IL‐1β secretion from microglia is the NLRP3 inflammasome. A critical link between microglial NLRP3 inflammasome activation and the progression of both α‐syn pathology and dopaminergic neurodegeneration has been identified in various PD models in vivo. α‐Syn is known to activate the microglial NLRP3 inflammasome in murine models, but its relationship to this inflammasome in human microglia has not been established. In this study, IL‐1β secretion from primary mouse microglia induced by α‐syn fibrils was dependent on NLRP3 inflammasome assembly and caspase‐1 activity, as previously reported. We show that exposure of primary human microglia to α‐syn fibrils also resulted in significant IL‐1β secretion that was dependent on inflammasome assembly and involved the recruitment of caspase‐1 protein to inflammasome scaffolds as visualized with superresolution microscopy. While canonical IL‐1β secretion was clearly dependent on caspase‐1 enzymatic activity, this activity was less clearly involved for α‐syn‐induced IL‐1β secretion from human microglia. This work presents similarities between primary human and mouse microglia in the mechanisms of activation of the NLRP3 inflammasome by α‐syn, but also highlights evidence to suggest that there may be a difference in the requirement for caspase‐1 activity in IL‐1β output. The data represent a novel characterization of PD‐related NLRP3 inflammasome activation in primary human microglia and further implicate this mechanism in the pathology underlying PD.
Keywords: caspase‐1, NLRP3 inflammasome, Parkinson's disease, primary human microglia, STED, α‐synuclein
Main points
Primary human microglia undergo canonical inflammasome activation involving caspase‐1
α‐Syn fibrils activate the NLRP3 inflammasome in human microglia
α‐Syn‐induced inflammasomes recruit caspase‐1; its activity may not be required for IL‐1β yield
1. INTRODUCTION
The neuropathology of Parkinson's disease (PD) involves both the formation of intraneuronal inclusions known as Lewy bodies (LBs) and Lewy neurites (LNs), rich in aggregates of the protein α‐synuclein (α‐syn), and the degradation of dopaminergic neurons (Glass, Saijo, Winner, Marchetto, & Gage, 2010; Heneka, Kummer, & Latz, 2014; Spillantini, 1999; Q. S. Zhang, Heng, Yuan, & Chen, 2017; W. Zhang et al., 2005). Pathology occurs in various brain regions but is most prominent in the substantia nigra pars compacta (SNpc) region of the midbrain (Braak et al., 2003; Dauer & Przedborski, 2003; Jellinger, 2018; Sgobio et al., 2019). The SNpc, notably, is the brain region in which the density of microglia is the highest (Béraud & Maguire‐Zeiss, 2012; Mastroeni et al., 2009; Rogers, Mastroeni, Leonard, Joyce, & Grover, 2007). In PD, as in other neurodegenerative diseases, neuronal loss is invariably associated with a neuroinflammatory response that includes activation of the innate immune system, hallmarked by the presence of activated microglia which assemble around the degenerating neurons (Béraud & Maguire‐Zeiss, 2012); (Rogers et al., 2007). A major role in the perpetuation of this innate immune activation is played by the pro‐inflammatory cytokine interleukin‐1β (IL‐1β), which is secreted in the central nervous system (CNS) primarily by microglia acting as immune effectors (Blum‐Degen et al., 1995; Griffin, Liu, Li, Mrak, & Barger, 2006; Mendiola & Cardona, 2018; Wang et al., 2016). Mutations leading to the upregulation of IL‐1β gene expression have been linked to increased risk for PD (McGeer, Yasojima, & McGeer, 2002; Surendranathan, Rowe, & O'Brien, 2015; Wahner, Sinsheimer, Bronstein, & Ritz, 2007), and the concentration of IL‐1β protein is increased in the striatal dopaminergic tissue and cerebrospinal fluid of PD patients (Blum‐Degen et al., 1995; Daniele et al., 2015; Mogi et al., 1994).
The canonical mechanism for IL‐1β release from microglia involves activation of the NLRP3 inflammasome (Latz, Xiao, & Stutz, 2013; Mendiola & Cardona, 2018; Place & Kanneganti, 2018), which is a multimolecular scaffold whose primary function is to sense, amplify, and broadcast pro‐inflammatory signals from one cell to another by driving cytokine secretion. The three traditionally accepted component proteins of the NLRP3 inflammasome are: (1) the intracellular pattern recognition receptor NACHT domain‐, leucine‐rich repeat (LRR)‐, and pyrin domain‐containing protein 3 (NLRP3), (2) the small adaptor molecule known as apoptosis‐associated speck‐like protein containing a caspase activation and recruitment domain (CARD), or ASC, and (3) the cysteine‐aspartate protease‐1 (caspase‐1) (Boucher et al., 2018; Heneka et al., 2014; Latz et al., 2013; Schroder & Tschopp, 2010).
NLRP3 and ASC protein levels are upregulated in the nigral microglia of PD patients and in the striatal microglia of mouse models of parkinsonism (Gordon et al., 2018). This implies that the NLRP3 inflammasome upstream of IL‐1β secretion may be a key agent in PD pathology in vivo. Furthermore, microglial NLRP3 inflammasome activation has been shown to be critical for both dopaminergic neurodegeneration and the spreading of α‐syn aggregates in several murine parkinsonism models, where inhibition of NLRP3 activation by MCC950, and thus blockade of inflammasome assembly, or knockdown of NLRP3 or caspase‐1 expression ameliorates both of these aspects of pathology (Gordon et al., 2018; E. Lee et al., 2019; Martinez et al., 2017; Yan et al., 2015). However, the relationship of dopaminergic neurodegeneration and α‐syn pathology to NLRP3 inflammasome activation awaits further elucidation, particularly in human‐specific models.
One mechanistic link between α‐syn pathology and NLRP3 inflammasome activation is that α‐syn aggregates are known to activate the inflammasome in microglia from in vivo mouse models of parkinsonism to induce IL‐1β production in a delayed fashion (Gordon et al., 2018; Plotegher et al., 2017). A similar inflammasome‐activating effect of α‐syn has been demonstrated for primary human peripheral monocytes and the THP‐1 cell model (Codolo et al., 2013); (Freeman et al., 2013). The important question of how α‐syn might influence NLRP3 inflammasome activation in human brain microglia to contribute to PD pathology, however, has not been investigated previously. In this study, we hypothesized that α‐syn fibrils can activate the NLRP3 inflammasome in primary human microglia, inducing inflammasome‐dependent, caspase‐1‐mediated IL‐1β production. We tested this hypothesis with a combination of biochemical techniques and STED superresolution microscopy that provided novel insight into the nature and protein composition of α‐syn‐induced NLRP3 inflammasome complexes in primary human microglia.
2. MATERIALS AND METHODS
2.1. Isolation and culture of primary microglia from human brain tissue
Adult primary human microglia were isolated from postmortem brain specimens by density gradient centrifugation essentially as previously described by de Groot, Hulshof, Hoozemans, and Veerhuis (2001). Corpus callosum or subventricular cortical white matter tissue specimens were acquired from rapid autopsy according to the standard protocols of the Netherlands Brain Bank (Ravid & Swaab, 1993), with informed donor consent having been obtained from either patients or next of kin during life. In addition, normal cortical tissue not needed for diagnostic purposes was acquired in cooperation with the VU University Medical Center Department of Neurosurgery from patients undergoing focal cortical resection for medication‐refractory epilepsy, also with informed donor consent. The use of primary human tissue for in vitro experiments was in compliance with the Declaration of Helsinki. All tissue was collected in Dulbecco's modified Eagle medium [DMEM] supplemented with 0.1% gentamycin. The isolated microglia were cultured in medium comprising DMEM and Ham F10 (1:1) supplemented with 10% v/v heat inactivated fetal bovine serum (Hyclone, Thermo Fisher Scientific), a mixture of 100 IU/ml penicillin and 50 μg/ml streptomycin (Gibco), and 0.5% l‐glutamine. For experimentation, microglia were seeded in 24‐ or 48‐well uncoated culture plates (Corning Costar) and incubated at 37°with 5% CO2. Then, 24 hr after isolation, the microglia were treated with 25 ng/ml granulocyte macrophage colony stimulating factor (recombinant human GM‐CSF, Immunotools) to allow for better adherence and proliferation, after which the medium was replaced with fresh culture medium approximately every 72 hr. Microglia were utilized in experiments between days 6–10 postisolation, and the results of each experiment as indicated in figures represent an individual microglial culture from an individual patient.
2.2. Isolation and culture of primary microglia from mouse brain tissue
Primary mouse microglia cells were derived from mixed gender cultures of C57BL/6J mouse brains (postnatal days P0–P2). Cerebral cortices were stripped of the meninges and mechanically dissociated as described by Russo et al. (2015). The cell suspension obtained from the two brains was plated on poly‐l‐lysine (0.1 mg/ml, Sigma‐Aldrich)‐coated T‐75 flask and cultivated in DMEM (Gibco by Thermo Fischer Scientific, Breda, the Netherlands), supplemented with 10% heat‐inactivated fetal bovine serum (FBS; Gibco) and 1% penicillin/ streptomycin (P/S; Gibco). The next day, the cells were washed three times with DPBS (Gibco) to remove cellular debris and cultured as reported by Scheiblich et al. (2017). After 8–10 days in culture, weakly attached mature microglia were shaken loose from the astrocytic monolayer with a repetition of the harvesting procedure every 2–3 days, up to three times.
2.3. Cell culture and reagents
The human monocyte‐like leukemia cell line THP‐1 was obtained from ATCC (Rockville, MD, CLS Cat# 300356/p804_THP‐1, RRID:CVCL_0006). The cells were cultured in Roswell Park Memorial Institute (RPMI) 1640 medium with GlutaMAX (Gibco) supplemented with 10% heat inactivated FBS (Hyclone, Thermo Fisher Scientific, Breda, the Netherlands) and a mixture of 100 IU/ml penicillin and 50 μg/ml streptomycin (Gibco). The THP‐1 cells were differentiated by the addition to culture medium upon seeding in 24‐ or 48‐well uncoated culture plates (Corning Costar, Amsterdam, the Netherlands) of 100 ng/ml phorbol 12‐myristate 13‐acetate (PMA, Sigma‐Aldrich, Mechelen, Belgium) for 72 hr prior to exposure. Lipopolysaccharide (LPS) from Escherichia coli O55:B5 was from Sigma‐Aldrich (catalog no. L‐2880). Recombinant human α‐syn was overexpressed and purified in monomeric form from E. coli and then aggregated into fibrils (250 mM nominal concentration of monomers to form 3615 μg/ml aqueous fibril stock solution) as previously reported (Codolo et al., 2013). No endotoxin contamination was detectable in the α‐Syn monomer and fibril preparations, as determined with a limulus amoebocyte lysate (LAL) assay. Fibrils were not sonicated prior to application in culture. α‐Syn monomers, upon thawing, were centrifuged through an Amicon 100 kDa filter (Sigma‐Aldrich, Mechelen, Belgium) to remove any spontaneously formed aggregates from solution. The NLRP3 inhibitor MCC950 (CP‐456773, CRID3) was from Sigma‐Aldrich (Mechelen, Belgium), and the caspase‐1 inhibitor Z‐YVAD‐FMK was from PromoCell GmbH (Heidelberg, Germany).
2.4. Exposure conditions
PMA‐differentiated THP‐1 cells in 24‐ (450,000 cells/well) or 48‐well (225,000 cells/well) culture plates were washed once with PBS to remove serum proteins left over from culture medium. Cells were exposed to LPS (50 ng/ml) as a time‐matched NLRP3 inflammasome activation control stimulus or to various concentrations of α‐syn fibrils or filtered monomers up to 289.2 μg/ml (nominal concentration) in serum‐free medium for 18–24 hr. For canonical activation, THP‐1 cells were exposed to LPS (50 ng/ml) for 3.5 hr followed by the addition of nigericin (10 μM, Adipogen, Epalinges, Switzerland) for 30 min. GM‐CSF‐treated primary human microglia in 24‐ or 48‐well plates after 6–10 days in culture were also washed once with PBS and were exposed to LPS (20 ng/ml) or the same concentration ranges of α‐syn fibrils or filtered monomers in serum‐free medium for 18–24 hr. For canonical activation, primary human microglia were exposed to LPS (20 ng/ml) for 3.5 hr followed by the addition of nigericin (10 μM) for 30 min. In inhibition experiments, MCC950 (1 μM) or Z‐YVAD‐FMK (10 μM) were administered at the time of addition of the initial stimulus (priming) and left in the system for the duration of the exposure. After cell exposure, supernatants were collected immediately and centrifuged at 0.8× g for 5 min at room temperature to remove any cell debris. Cell lysates were collected by scraping with a pipet tip in lysis buffer (0.5% NP‐40 in PBS with protease inhibitor cocktail [cOmplete mini, EDTA‐free, Roche, Woerden, the Netherlands]) on ice, and cell debris was removed via centrifugation at 21,000× g for 10 min at 4°C. Cleared supernatants and cell lysates were stored at −20°C until testing.
2.5. ELISA analysis
The concentration of IL‐1β in culture supernatants was determined with a sandwich enzyme‐linked immunosorbent assay (ELISA) specific for the detection of human IL‐1β (PeliKine compact kit, Sanquin, Amsterdam, the Netherlands).
2.6. Thioflavin T aggregation assay
Then, 20, 10, and 5 μl aqueous stock α‐syn fibrils (3,615 μg/ml) and 20 μl filtered monomers (250 μM, 3,615 μg/ml) were added to 10 μl 2 mM thioflavin T in HEPES buffer (20 mM HEPES, 150 mM NaCl, pH 7.2), in each well of a 96‐well black fluorescence plate. HEPES buffer was added to a final volume of 100 μl per well, the plate was shaken, and the fluorescence was measured at 37°C with a Tecan Spark plate reader (Tecan GmbH, Grödig, Austria) at an excitation wavelength of 440 nm and an emission wavelength of 485 nm.
2.7. Western blot analysis
Supernatants and lysates were collected as described for ELISA. For western blotting, samples were denatured with heating in lithium dodecyl sulfate (LDS)‐containing loading buffer and reducing agent containing 500 mM dithiothreitol (DTT) for 5 min @ 95°C (except for native α‐syn aggregates, which were subjected to electrophoresis without heating or denaturation to maintain aggregate structure) and separated by polyacrylamide gel electrophoresis (PAGE) on a 4–12% Bis‐Tris gel in MES running buffer (NuPAGE, Thermo Fisher Scientific, Breda, the Netherlands). After PAGE, proteins were transferred to polyvinylidene fluoride (PVDF) membranes which were then blocked with 5% milk and 0.5% Tween in PBS and then immunolabeled. Detection was performed using enhanced chemiluminescence (ECL Western Blotting Substrate, Pierce, Breda, the Netherlands) with a ChemiDoc imaging system (Biorad). β‐actin was used as a loading control for cell lysate western blots. Primary antibodies included: anti‐α‐syn polyclonal antibody (MJFR1, 1:1,000 dilution, Abcam, Cambridge, United Kingdom), anti‐IL‐1β polyclonal antibody (anti‐human IL‐1β/IL‐1F2, R and D Systems Cat# AF‐201‐NA, RRID:AB_354387, 1:2,000 dilution), and anti‐β‐actin (Sigma‐Aldrich Cat# A3853, RRID:AB_262137, 1:10,000 dilution, Sigma‐Aldrich, Mechelen, Belgium). Secondary antibodies included goat anti‐mouse polyclonal antibody with horseradish peroxidase (HRP) conjugate (Agilent Cat# P0447, RRID:AB_2617137, 1:1,500 dilution) and rabbit anti‐goat polyclonal antibody with HRP conjugate (Agilent Cat# P0449, RRID:AB_2617143, 1:2,000 dilution).
2.8. Viability assays
The lactate dehydrogenase leakage assay (LDH, Pierce, Breda, the Netherlands), to assess membrane integrity, and the methyl tetrazolium assay (MTT, Sigma M‐2128, Mechelen, Belgium), to assess mitochondrial function, were performed according to manufacturer's protocols. For the MTT assay, the supernatants were first removed from the cell culture and 0.25 mg/ml MTT in complete cell culture medium was added. After 2 hr at 37°C, the MTT solution was removed and 100 μl DMSO was added to each well. The plate was shaken for 1 min and absorbance was measured at a wavelength of 540 nm.
2.9. Confocal and STED superresolution microscopy
Postisolation primary human microglia or PMA‐treated THP‐1‐derived macrophage‐like cells were allowed to adhere to 12 mm round borosilicate glass coverslips (Menzel #1.5, Thermo Fisher Scientific, Breda, the Netherlands) in separate wells of a 24‐well culture plate. No coating or treatment of the coverslips was required. After exposure of the cells, supernatants were collected for ELISA analysis of IL‐1β to confirm inflammasome activation and the cells were washed 3× with PBS, fixed for 10 min at room temperature with 4% paraformaldehyde in PBS with gentle shaking, and washed another 3× with PBS. Nonspecific binding sites were blocked with 10% goat serum/0.1% Triton X‐100 with azide (Life Technologies) diluted 1:1 (to 5%/0.05%) in PBS for at least 30 min with gentle shaking. After blocking, cells were incubated without washing with primary antibodies in dilution buffer (PBS with 1% bovine serum albumin [BSA]) either at room temperature for 1 hr or at 4°C overnight, then washed 3× with PBS and incubated with secondary antibodies in dilution buffer. As a final labeling step, cells were incubated with labeled primary antibody for caspase‐1 in dilution buffer. Cells were washed 3× with PBS after each antibody incubation. Primary antibodies included: anti‐NLRP3/NALP3 monoclonal antibody (Cryo‐2, AdipoGen Cat# AG‐20B‐0014, RRID:AB_2490202, 1:200 dilution), anti‐ASC polyclonal antibody (AL 177, AdipoGen Cat# AG‐25B‐0006, RRID:AB_2490440, 1:200 dilution), anti‐α‐syn polyclonal antibody (MJFR1, Abcam Cat# ab138501, RRID:AB_2537217, 1:200 dilution), anti‐p20‐caspase‐1 (AA 145‐170) (Alexa Fluor 488) polyclonal antibody (Bioss Primary Conjugated Antibodies, 1:100 dilution). Secondary antibodies included: Abberior STAR 580 [Abberior Cat# 2‐0002‐005‐1, RRID:AB_2620153] and Abberior STAR 635p (Abberior GmbH, 1:100 dilution). Nuclear counterstaining was performed with DAPI dihydrochloride (Thermo Fisher Scientific Cat# D1306, RRID:AB_2629482), diluted to 300 nM in PBS and f‐actin counterstaining with Alexa Fluor 546 phalloidin diluted 1:1,000 in PBS. Images were acquired with a Leica TCS SP8 STED 3× confocal scanning laser microscope equipped with 3 STED lasers (confocal 40×, STED 100× objective; NA 1.4; immersion in oil) and analyzed with the ImageJ software (Fiji, RRID:SCR_002285). Three dimensional (3D) rendering of ImageJ‐generated Z‐stacks was acquired using the Imaris software (Imaris, RRID:SCR_007370).
2.10. Transmission electron microscopy
α‐Syn fibrils: α‐Syn fibrils resuspended in PBS were adsorbed onto a carbon‐coated copper grid and were then negative‐stained with 0.05% uranyl acetate solution. Transmission electron microscopy (TEM) micrographs were taken with a Tecnai‐12 electron microscope (Philips‐FEI) at the EM Facility of University of Padua.
2.11. Mouse primary microglia
Cells were fixed and processed for electron microscopy 24 hr after treatment with α‐syn fibrils. Mouse primary microglia cells were fixed for 1 hr at room temperature with freshly prepared 2.5% (v/v) glutaraldehyde in 0.1 M sodium cacodylate (pH 7.4). After washing with 0.1 M sodium cacodylate, cells were postfixed in 1% OsO4, 1.5% K4Fe(CN)6 in 0.1 M sodium cacodylate pH 7.4, stained with 0.5% uranyl acetate, dehydrated in ethanol and embedded in Embed 812. Thin sections were imaged on a Tecnai‐12 electron microscope (Philips‐FEI) at the EM Facility of University of Padua.
2.12. Atomic force microscopy
Atomic force microscopy (AFM) imaging of α‐syn fibrils was performed as previously described (Plotegher, Greggio, Bisaglia, & Bubacco, 2014) in a “PeakForce tapping” mode with Scanasyst‐Air probes (Bruker, Mannheim, Germany) on a Nanoscope V system equipped with a Multimode head and a type‐E piezoelectric scanner (Bruker, Mannheim, Germany). Ten microliters of sample were deposited on freshly cleaved mica (RubyRed Mica Sheets, Electron Microscopy Sciences, Fort Washington, USA) and left to adsorb for 5 min at room temperature (∼20°C). The mica surface was then rinsed with ∼500 μl of MilliQ H2O (Millipore Simplicity) at the same temperature and dried with dry nitrogen.
2.13. Statistical analysis
Two‐way ANOVA with Bonferroni multiple testing correction was performed with individual experimental replicates in GraphPad Prism v.7. Graphs presented as means and standard deviations per experiment (unless otherwise indicated in figure legend) with experiments differentiated by color.
3. RESULTS
3.1. Primary human microglia are competent for canonical NLRP3 inflammasome activation
Canonical NLRP3 inflammasome activation, involving the recruitment of NLRP3, ASC, and caspase‐1 and resulting in IL‐1β secretion, can be induced in primary mouse and nonhuman primate microglia by priming with bacterial LPS followed by treatment with the K+/H+ antiporter nigericin (Burm et al., 2015; Venegas et al., 2017). The NLRP3 inflammasome can be activated by nigericin with or without LPS priming in PMA‐differentiated, human macrophage‐like THP‐1 cells, which are frequently used as surrogate microglia (Freeman et al., 2013; Man et al., 2014; Netea et al., 2010; Nurmi et al., 2013). However, canonical activation of the NLRP3 inflammasome leading to the secretion of IL‐1β has not been documented previously for primary human microglia. We confirmed that the THP‐1 model secreted IL‐1β in response to PMA differentiation (72 h) and nigericin treatment (30 min) alone (Figure 1a). Primary human microglia were also competent to secrete IL‐1β upon exposure to nigericin for 30 min, but only after priming by LPS (3.5 hr, Figure 1b,c). Secretion of mature p17 IL‐1β upon canonical activation was confirmed by western blot in combined and concentrated supernatants of both THP‐1 cells (not shown) and microglia (Figure 1d). The p17 IL‐1β band was not detectable in supernatants collected from individual experiments, presumably due to unavoidably limited overall protein levels in microglia experiments.
FIGURE 1.
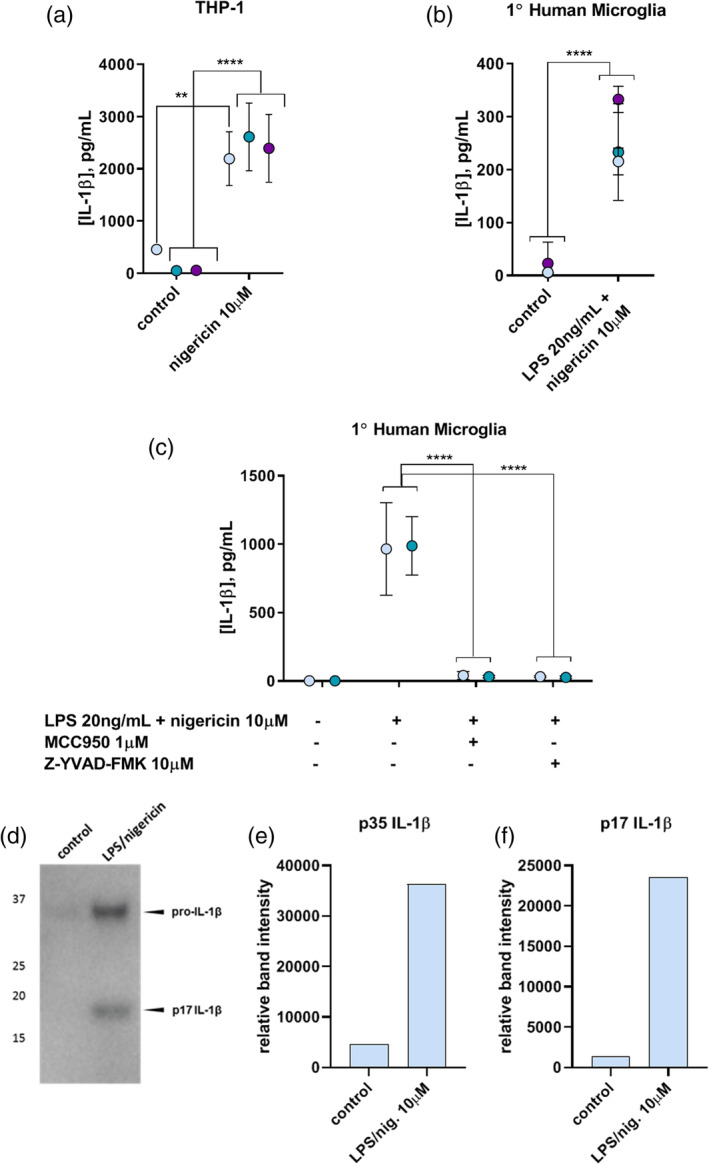
(a) IL‐1β in culture supernatants of PMA‐differentiated THP‐1 cells upon canonical inflammasome activation with nigericin alone (10 μM, 30 min) in the absence of LPS priming, as measured by ELISA. (b) IL‐1β in culture supernatants of primary human microglia upon LPS priming (20 ng/ml, 3.5 hr) followed by nigericin activation (10 μM, 30 min). (c) Inhibition of canonical NLRP3 inflammasome activation in primary human microglia by inhibiting NLRP3 with MCC950 or caspase‐1 activity with Z‐YVAD‐FMK. ** = p = .001; 2 or 3 experiments, n = 3. Colors indicate individual experiments. (d) IL‐1β western blot of combined, concentrated supernatants from several primary human microglia experiments showing secretion of p35 pro‐IL‐1β and the mature p17 form upon canonical inflammasome activation with LPS priming (20 ng/ml, 3.5 hr) followed by nigericin activation (10 μM, 30 min). (e,f) Quantification of relative p35 (e) and p17 (f) IL‐1β band intensities between no treatment (control) and canonical inflammasome activation in primary human microglia [Color figure can be viewed at wileyonlinelibrary.com]
To ensure that the IL‐1β maturation induced by LPS and nigericin from primary human microglia was indeed dependent on NLRP3 inflammasome activation, inhibitors of various steps in the canonical inflammasome signaling pathway were employed. Since downstream IL‐1β processing relies on effective NLRP3 inflammasome scaffold formation to recruit caspase‐1, the NLRP3 inhibitor MCC950, also known as CRID3 (Coll et al., 2015; Coll, Robertson, Butler, Cooper, & O'Neill, 2011; Gordon et al., 2018), was used to block this process. One micrometer MCC950 concomitant with LPS priming prior to nigericin activation of primary human microglia prevented the secretion of IL‐1β, suggesting that IL‐1β production in response to these stimuli is indeed NLRP3 dependent (Figure 1c). The caspase‐1 requirement for this IL‐1β secretion was confirmed with co‐administration of the caspase‐1 inhibitor Z‐YVAD‐FMK together with LPS priming prior to nigericin activation (Figure 1c). These results demonstrate that primary human microglia, like primary human monocytes and mouse and nonhuman primate microglia (Burm et al., 2015), are competent for canonical NLRP3 inflammasome assembly and activation involving caspase‐1 processing to elicit the production of IL‐1β.
3.2. Characterization of α‐syn fibrils and monomers
To validate the structures of fibrillar and monomeric α‐syn species prior to cell treatment, α‐syn fibrils and monomers were tested on western blot under nondenaturing conditions (Figure 2a). α‐Syn fibrils and monomers were also examined with the thioflavin T assay to determine the extent of fibrillar structure in fibril fractions as compared to monomer fractions (Figure 2b). Aggregates of α‐syn were indeed largely fibrillar and monomers monomeric, and there was no detectable spontaneous fibrillization of monomers under assay conditions. The fibrillar nature of the α‐syn was further confirmed with TEM (Figure 2c) and AFM (Figure 2d).
FIGURE 2.
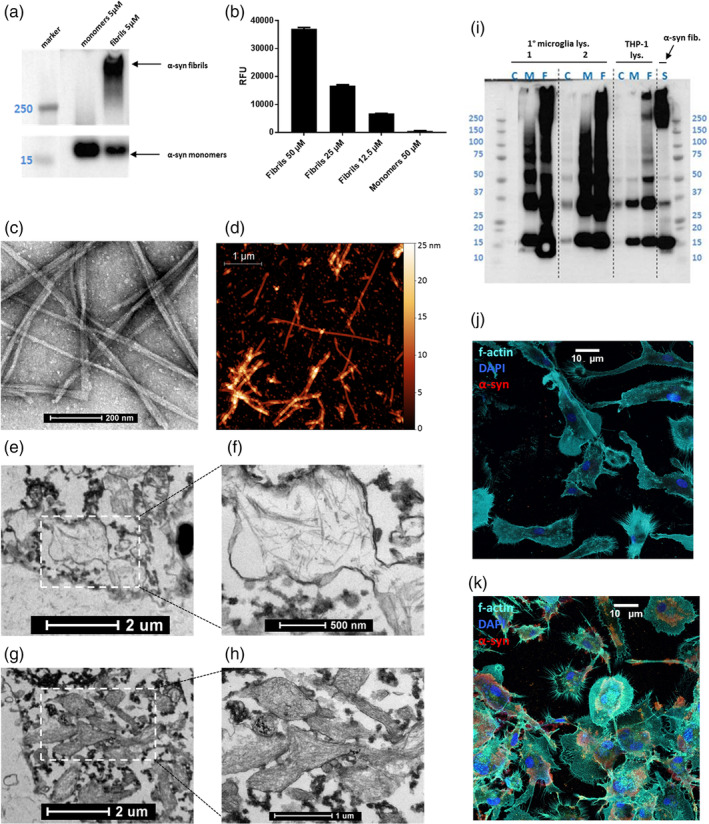
(a) α‐Syn fibrils and monomers as characterized by western blot with MJFR1 anti‐α‐syn antibody. Spliced for clarity to juxtapose lanes of interest. (b) Thioflavin T assay of α‐syn fibrils (12.5‐50 μM gradient) and monomers (50 μM). X‐axis in RFU, relative fluorescence units. (c) α‐Syn fibrils as visualized with TEM. Scale bar = 200 nm. (d) α‐Syn fibrils as visualized with AFM (courtesy of Dr. Marco Brucale, CNR ISMN). Scale bar = 1 μm. Representative experiments. (e,g) Primary mouse microglia with internalized α‐syn, visualized with TEM. Scale bars = 2 μm. (f,h) zooms of indicated regions of interest from e and g. Scale bars = 500 nm (f) and 1 μm (h). (i) Western blot with MJFR1 anti‐α‐syn antibody of lysates of primary human microglia from two patients (1 and 2) and lysates of THP‐1 cells after overnight exposure to 72.3 μg/ml α‐syn monomers or fibrils in comparison to 72.3 μg/ml α‐syn fibrils applied directly to gel. C, untreated control lysate; F, fibril‐treated lysate; M, monomer‐treated lysate, S, α‐syn “standard” (fibrils as applied to cells). (j,k) Confocal fluorescence microscopy stitched tile overview of primary human microglia, either untreated (j) or exposed to 72.3 μg/ml α‐syn overnight (k) (scale bar = 10 μm). All cell types tested are competent for the uptake of α‐syn fibrils [Color figure can be viewed at wileyonlinelibrary.com]
3.3. Exogenous α‐syn is internalized by primary mouse and human microglia
Primary mouse microglia (Plotegher et al., 2014) and primary human monocytes (Codolo et al., 2013) as well as THP‐1 cells (Freeman et al., 2013) have been reported previously to internalize α‐syn fibrils. This was confirmed in primary mouse microglia using TEM (Figure 2e–h ) and in the THP‐1 model and primary human microglia with western blot analysis of cell lysates (Figure 2i). The capability of primary human microglia to take up fibrillar α‐syn was further explored using confocal microscopy (Figure 2j,k). In the primary human microglia, the internalized α‐syn localized to the paranuclear area (Figure 2k).
3.4. α‐Syn fibrils, but not monomers, induce IL‐1β secretion from primary mouse and human microglia
Primary mouse microglia responded to α‐syn fibrils as expected by secreting IL‐1β into culture supernatants in a dose‐dependent fashion, with somewhat higher levels of IL‐1β detected at 24 hr exposure time than at 16 hr (Figure 3a–b). Monomers up to 361.5 μg/ml did not elicit any IL‐1β secretion from mouse microglia. These results are consistent with previous observations in primary mouse microglia (Russo et al., 2015). NLRP3 inflammasome involvement in this process is evident upon observation with immunofluorescent confocal microscopy, which shows the overlap of the three component proteins in primary mouse microglia after 16 hr treatment with 144.6 μg/ml α‐syn (Figure 3c).
FIGURE 3.

(a) 16 hr or (b) 24 hr exposure of primary mouse microglia to α‐syn fibrils and monomers (144.6 or 361.5 μg/ml nominal concentration). Fibrils induce IL‐1β secretion from the microglia whereas monomers do not. Points indicate mean of three replicates from independent microglia cultures generated from ≥6 mice each. (c) Confocal microscopic overview of NLRP3 (gray), ASC (red), and caspase‐1 (green) overlap illustrating inflammasome complex formation in primary mouse microglia treated for 16 hr with 144.6 μg/ml α‐syn fibrils. (d) Overnight exposure of PMA‐primed THP‐1 cells to a concentration gradient of α‐syn fibrils (fib., 72.3–289.2 μg/ml nominal concentration) induces dose‐dependent IL‐1β secretion. α‐Syn monomers (mon., 72.3–289.2 μg/ml) also induce some IL‐1β production from these cells at higher concentrations. **** = p <.0001; three experiments, n = 3. Colors indicate individual experiments. (e) Overnight exposure of primary human microglia to the same concentration gradient of α‐syn fibrils also induces dose‐dependent IL‐1β secretion. α‐Syn monomers, however, do not induce IL‐1β production at any concentration tested. ** = p <.01; 3 experiments, n = 3. Colors indicate individual experiments. (f) IL‐1β western blot of combined, concentrated supernatants from several primary human microglia experiments showing secretion of p35 pro‐IL‐1β and the mature p17 form upon noncanonical LPS‐mediated NLRP3 inflammasome activation (20 ng/ml overnight) or α‐syn‐mediated NLRP3 inflammasome activation (72.3 μg/ml overnight). (g–i) quantification of p35 (g), p26 (h), and p17 (i) IL‐1β bands from western blot (f) after overnight treatment of primary human microglia with 20 ng/ml LPS or 72.3 μg/ml α‐syn [Color figure can be viewed at wileyonlinelibrary.com]
LPS on its own has been shown to induce IL‐1β secretion via noncanonical NLRP3 inflammasome activation from THP‐1 cells and primary human peripheral monocytes after internalization of the endotoxin following either long‐term (16 hr) exposure or direct transfection (Baker et al., 2015; Vigano et al., 2015). Primary human microglia responded to overnight exposure to LPS by secreting IL‐1β, as did the THP‐1 model (Figure 3d,e). This secretion was diminished in the presence of MCC950 (Figure 4d–f), indicating NLRP3 inflammasome involvement, so LPS was utilized as an exposure time‐matched positive control inflammasome activation stimulus in experiments with α‐syn in human cells.
FIGURE 4.
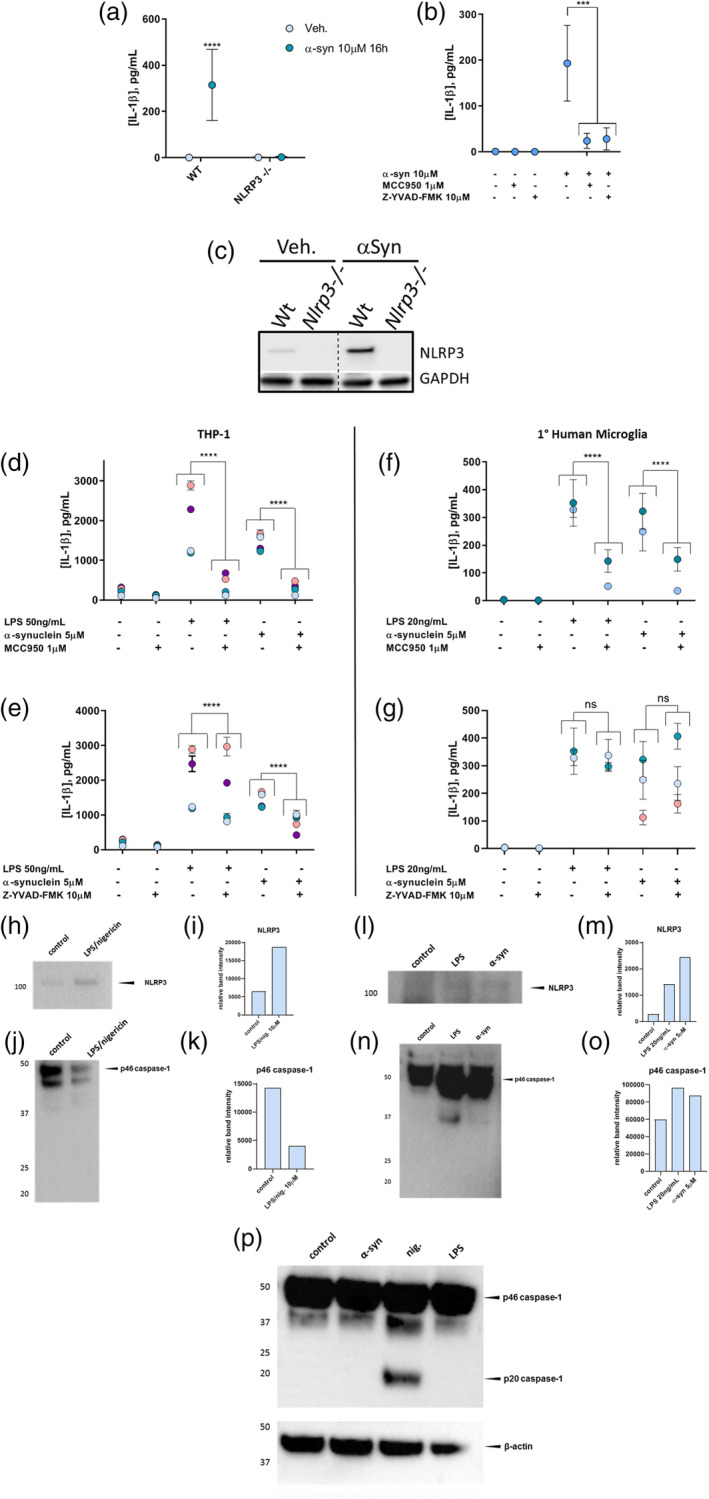
(a) Primary microglia from WT mice show an IL‐1β secretion response to 16 hr exposure to 144.6 μg/ml α‐syn, while microglia from NLRP3−/− mice do not. (b) Il‐1β secretion from WT primary mouse microglia induced by exposure to 144.6 μg/ml α‐syn for 16 hr is abrogated by inhibition of either NLRP3 with MCC950 or caspase‐1 activity with Z‐YVAD‐FMK (YVAD). (c) Primary microglia from WT mice demonstrate an increase in NLRP3 protein levels in cell lysates as detected on western blot in response to the same treatment as in Figure 4a, while microglia from NLRP3−/− mice do not. Spliced to juxtapose NLRP3 bands directly. (d) MCC950 Inhibition of LPS or α‐syn‐induced IL‐1β secretion by the THP‐1 model. Four experiments, n = 2 or 3. (e) LPS‐ or α‐syn‐induced IL‐1β secretion from THP‐1 cells is partially dependent on caspase‐1 activity. Four experiments, n = 2 or 3. (f) MCC950 Inhibition of LPS‐ or α‐syn‐induced IL‐1β secretion by primary human microglia. Two experiments, n = 3. (g) LPS‐ or α‐syn‐induced IL‐1β secretion from primary human microglia is independent of caspase‐1 activity. 2 (LPS) or 3 (α‐syn) experiments, n = 3. Colors indicate individual experiments. (h,l) NLRP3 western blots of combined, concentrated supernatants of several primary human microglia experiments after canonical (h) or LPS‐ or α‐syn‐mediated (l) NLRP3 inflammasome activation. (i,m) Quantification of NLRP3 bands shown in h (I, canonical activation) and l (m, LPS‐ or α‐syn‐mediated activation). (j,n): Caspase‐1 western blots of combined, concentrated supernatants of several primary human microglia experiments after canonical (j) or LPS‐ or α‐syn‐mediated (n) NLRP3 inflammasome activation. (k,o) Quantification of caspase‐1 bands shown in j (k, canonical activation) and n (o, LPS‐ or α‐syn‐mediated activation). (p) Caspase‐1 western blot of lysates of THP‐1 cells after canonical, LPS‐, or α‐syn‐mediated NLRP3 inflammasome activation versus β‐actin loading control. ns, not significant, **** = p <.0001 [Color figure can be viewed at wileyonlinelibrary.com]
THP‐1 cells responded to a concentration gradient of α‐syn fibrils by releasing IL‐1β into the culture medium in a dose‐dependent fashion (Figure 3d). A similar dose‐dependent secretion pattern was elicited from primary human microglia with the same range of α‐syn fibril concentrations (Figure 3e). While the THP‐1 model showed a modest response to α‐syn monomers at higher test concentrations, the monomers were unable to induce IL‐1β secretion from the human microglia, like the mouse microglia, at any test concentration. This suggests a fibril‐specific effect on IL‐1β secretion from both primary human and mouse microglia, and also indicates that the observed result is not due to endotoxin contamination of the α‐syn. Cleaved p17 IL‐1β was detected by western blot in combined, concentrated supernatants of both LPS‐treated and α‐syn‐treated microglia (Figure 3f), implying NLRP3 inflammasome involvement in IL‐1β processing. Further experiments with α‐syn were conducted using a concentration of 72.3 μg/ml, as this exhibited a robust and significant effect on IL‐1β secretion from primary human microglia.
3.5. α‐Syn‐induced IL‐1β secretion from primary mouse microglia is dependent on NLRP3 and on caspase‐1 activity
Primary microglia from wild type mice responded to 16 hr exposure to α‐syn fibrils by secreting IL‐1β (Figure 4a) and increasing NLRP3 protein production (Figure 4c). Primary microglia from NLRP3−/− mice, on the other hand, showed no response to the same treatment, highlighting the dependence of this mechanism on the NLRP3 protein. NLRP3 inhibition by MCC950 blocked α‐syn‐induced IL‐1β production from wild type primary mouse microglia, as did inhibition of caspase‐1 activity with Z‐YVAD‐FMK (Figure 4b). These findings suggest that IL‐1β secretion by primary mouse microglia in response to α‐syn fibrils is dependent on the assembly of the NLRP3 inflammasome and activation of caspase‐1, supporting previous observations by others (Plotegher et al., 2014).
3.6. α‐Syn‐induced IL‐1β secretion from primary human microglia is dependent on NLRP3 inflammasome assembly but may be independent of caspase‐1 activity
Consistent with the MCC950‐mediated inhibition of positive control inflammasome activation with LPS/nigericin observed in human cells (Figure 1) and α‐syn‐induced inflammasome activation in primary mouse microglia (Figure 4c), 1 μM MCC950 in conjunction with either LPS or α‐syn fibril administration overnight attenuated the production of IL‐1β taking place in response to either activation stimulus in THP‐1 cells (Figure 4d) and primary human microglia (Figure 4f). This suggests that IL‐1β secretion by both THP‐1 cells and primary human microglia in response to either LPS or α‐syn after overnight exposure is in large part dependent upon NLRP3 inflammasome scaffold formation.
We next examined the caspase‐1 dependence of α‐syn‐induced IL‐1β secretion from the THP‐1 model and primary human microglia. Since we had observed that α‐syn‐stimulated IL‐1β secretion from mouse microglia could be abrogated with the caspase‐1 inhibitor Z‐YVAD‐FMK (Figures 1 and 4b), we expected that this would also be the case for human microglia. Inhibiting caspase‐1 activity with Z‐YVAD‐FMK only partially blocked the release of IL‐1β induced by α‐syn in the THP‐1 model (Figure 4e). Surprisingly, caspase‐1 inhibition appeared to be unable to block IL‐1β secretion from primary human microglia in these experiments (Figure 4g). This suggests that α‐syn‐induced IL‐1β secretion from primary human microglia, unlike primary mouse microglia, may be independent of caspase‐1 activity and thus the canonical pathway of inflammasome activation.
To gain more clarity regarding the role of caspase‐1 in the maturation of IL‐1β in α‐syn‐treated microglia, we checked for the presence of the cleaved p20 caspase‐1 band in combined, concentrated supernatants of primary microglia after canonical, noncanonical, and α‐syn‐mediated NLRP3 inflammasome activation. No p20 caspase‐1 was detected in microglial supernatants after any type of activation despite clear bands indicating the presence of relatively high amounts of p46 and p33 pro‐caspase‐1 (Figure 4j,n). To ensure that the antibody utilized was indeed able to detect the p20 caspase‐1 band, we examined lysates from THP‐1 cells activated in the same ways. While activation with LPS or α‐syn showed no p20 caspase‐1, this band was clearly evident upon canonical activation with nigericin (Figure 4p).
3.7. NLRP3 inflammasome complexes including caspase‐1 protein are formed in primary human microglia upon activation by α‐syn
Consistent with the IL‐1β secretion observed upon treatment of THP‐1 cells and primary human microglia with fibrillar α‐syn, NLRP3 inflammasome complexes were visible in both cell types upon exposure to α‐syn with STED microscopy using fluorescently labeled antibodies specifically detecting the inflammasome components NLRP3, ASC, and caspase‐1, as well as α‐syn (Figure 5a–d [THP‐1] and e–g [primary human microglia]). NLRP3, ASC, and caspase‐1 proteins were clearly present in α‐syn‐induced NLRP3 inflammasome complexes in both cell types (Figure 5a,b,e). Though caspase‐1 enzymatic activity appeared not to be required for α‐syn‐induced IL‐1β secretion from primary human microglia as shown in Figure 4g, caspase‐1 protein was clearly associated with α‐syn‐induced NLRP3 inflammasome complexes in both cell types (Figure 5). This observation implies that caspase‐1 is activated by virtue of its recruitment to the scaffold (Boucher et al., 2018).
FIGURE 5.

(a) STED zoom of α‐syn‐induced inflammasome complex showing canonical inflammasome components NLRP3, ASC, and caspase‐1 in the THP‐1 model. (b) 3D surface rendering of the composite image detailing the orientation of NLRP3, ASC, and caspase‐1 in the inflammasome complex (THP‐1). NLRP3 (yellow); ASC (red); caspase‐1 (green). Scale bar = 1 μM. See Figure S1 for 3D animation of this structure. (c) STED zoom of α‐syn‐induced inflammasome complex showing the localization of α‐syn relative to inflammasome components NLRP3 and caspase‐1 in the THP‐1 model. Scale bar = 1 μM. (d) 3D surface rendering of the composite image detailing the orientation of NLRP3, α‐syn, and caspase‐1 in the inflammasome complex (THP‐1). NLRP3 (yellow); α‐syn (red); caspase‐1 (green). See Figure S2 for 3D animation of this structure. (e) STED zoom of α‐syn‐induced inflammasome complex showing canonical inflammasome components NLRP3, ASC, and caspase‐1 in the primary human microglia model. NLRP3 (magenta); ASC (red); caspase‐1 (green). Scale bar = 1 μM. (f) STED zoom of α‐syn‐induced inflammasome complex showing the localization of α‐syn relative to inflammasome components NLRP3 and caspase‐1 in the primary human microglia model. NLRP3 (yellow); α‐syn (red); caspase‐1 (green); DAPI (nuclear counterstain, blue); phalloidin (f‐actin counterstain, cyan). Scale bar = 1 μM. (g) 3D surface rendering of the composite image detailing the orientation of NLRP3, α‐syn, and caspase‐1 in the inflammasome complex along with f‐actin (primary human microglia). NLRP3 (yellow); α‐syn (red); caspase‐1 (green); f‐actin (phalloidin, cyan). See Figure S3 for 3D animation of this structure [Color figure can be viewed at wileyonlinelibrary.com]
f‐Actin was evident in the inflammasome complexes in primary human microglia (Figure 5f,g). This is not unprecedented; the presence of f‐actin associated with NLRP3 inflammasomes has been reported by others for mouse macrophages activated by ATP, nigericin, or asbestos (Burger, Fickentscher, de Moerloose, & Brandt, 2016; MacPherson, Westbom, Kogan, & Shukla, 2017).
α‐Syn internalized by both THP‐1 cells (Figure 5c,d) and primary human microglia (Figure 5f,g) also appeared to be associated with inflammasome structures, with the inflammasome situated within a network of α‐syn fibrils while some of the α‐syn was incorporated into the inflammasome scaffold. Further detail regarding inflammasome protein orientation and the positioning of the α‐syn in and around the inflammasome can be more clearly visualized in the video files in the accompanying Supporting Information (Figures [Link], [Link]).
4. DISCUSSION
The findings of this study provide evidence in a human‐specific system for the role of the microglial NLRP3 inflammasome in the underlying mechanisms of PD. They reveal (i) that primary human microglia are susceptible to canonical NLRP3 inflammasome‐mediated, caspase‐1‐dependent IL‐1β secretion induced by established positive control stimuli, (ii) that α‐syn fibrils activate the NLRP3 inflammasome in primary human microglia to release mature IL‐1β, (iii) that despite caspase‐1 recruitment to the α‐syn‐induced inflammasome scaffold in primary human microglia, its caspase activity may not be required for IL‐1β processing, and (iv) that NLRP3 inflammasome structures induced by α‐syn appear to associate with the α‐syn itself.
Canonical inflammasome activation by the positive control priming/activation stimulus combination of LPS and nigericin has been previously demonstrated for primary mouse and nonhuman primate microglia (Venegas et al., 2017); (Burm et al., 2015). The current study established that primary human microglia are also competent to undergo this type of inflammasome activation, as expected. Moreover, IL‐1β was released in a dose‐dependent manner into supernatants from both primary mouse and human microglia and THP‐1 cells after overnight exposure to a concentration gradient of α‐syn fibrils. Monomers had some effect on IL‐1β secretion from THP‐1 cells, but elicited no cytokine secretion from primary human or mouse microglia, suggesting a fibril‐specific effect on the primary microglia of both species. Conversion of a portion of pro‐IL‐1β to the 17 kDa mature form of the cytokine was confirmed by western blot for each activator in combined, concentrated supernatants of both primary human microglia and THP‐1 cells. Our observations regarding IL‐1β secretion are coherent between primary mouse and human microglia and are consistent with what has been reported by others for primary human monocytes, primary mouse microglia, and the THP‐1 model in response to α‐syn‐mediated activation of the NLRP3 inflammasome (Codolo et al., 2013; Freeman et al., 2013; Plotegher et al., 2014).
Canonical, LPS‐mediated, and α‐syn‐mediated IL‐1β production by primary human and mouse microglia were all significantly diminished upon NLRP3 inhibition by MCC950. This implies that these modes of IL‐1β production are, as expected, highly dependent on NLRP3 inflammasome scaffold assembly in both species. Furthermore, primary microglia derived from NLRP3−/− mice were unable to respond to α‐syn exposure by secreting IL‐1β, as opposed to microglia from wild‐type mice (Figure 4a,b), highlighting the upstream requirement of the NLRP3 protein for IL‐1β processing induced by α‐syn.
Results of primary human microglia experiments with caspase‐1 inhibition by Z‐YVAD‐FMK revealed that canonical inflammasome stimulation, involving short‐term LPS priming followed by nigericin activation, was clearly dependent on caspase‐1 enzymatic activity as expected. This confirms the functionality of caspase‐1 in inflammasome activation in human microglia. On the other hand, both LPS‐ and α‐syn‐mediated IL‐1β secretion were much less dependent on caspase‐1 activity in macrophage‐like THP‐1 cells and, unexpectedly, appeared to be independent of caspase‐1 activity in primary human microglia. This observation stands in stark contrast to the clear requirement for caspase‐1 in α‐syn‐mediated inflammasome activation in primary mouse microglia, a contrast which indicates that the potential difference in caspase‐1 requirement between activation stimuli in human microglia cannot be accounted for by the time frame of activation (4 hr for canonical activation vs. overnight for LPS‐ and α‐syn‐mediated activation). Since Z‐YVAD‐FMK is largely specific for caspase‐1 (Bian et al., 2009; Lipinska et al., 2014; Takasu et al., 2016), our results suggest that alternative caspases to caspase‐1 could be involved in the IL‐1β processing mechanism induced by α‐syn in human microglia. Noncanonical NLRP3 inflammasome activation induced by 16 hr exposure to LPS has been reported in human monocytes to be mediated by inflammatory caspases‐4 and ‐5, for which caspase‐11 is the homologous caspase in mice (Baker et al., 2015; Lagrange et al., 2018; Schmid‐Burgk et al., 2015; Vigano et al., 2015). The typically apoptotic caspase‐8 has been implicated in the processing of IL‐1β in murine macrophages (Antonopoulos et al., 2013); (Maelfait et al., 2008). Caspase‐8 is also involved in Salmonella typhimurium‐induced NLRC4 inflammasome activation, which was shown to also recruit NLRP3 to its scaffold, in THP‐1 cells (Man et al., 2014). The results presented here suggest that α‐syn‐mediated NLRP3 inflammasome activation leading to IL‐1β secretion from primary human microglia may be caspase‐1‐independent, but more work remains to be done to confirm whether this is indeed the case, and also to clarify which other caspases may be involved. This is a topic of ongoing study.
Despite the apparent lack of requirement for caspase‐1 enzymatic activity in α‐syn‐mediated IL‐1β secretion from primary human microglia based on the experiments presented here, STED microscopy revealed that the caspase‐1 protein is clearly recruited to α‐syn‐induced NLRP3 inflammasomes. This recruitment suggests that caspase‐1 undergoes activation upon inflammasome assembly, even if that activity is not critical for IL‐1β processing in α‐syn‐mediated inflammasome activation, since caspase‐1 activation occurs upon its proximity‐induced dimerization and autocatalytic cleavage on the inflammasome scaffold (Boucher et al., 2018). The tetrameric p20/p10 cleavage product of caspase‐1, previously considered to be the active caspase species responsible for downstream IL‐1β maturation (Boucher et al., 2018; Thornberry et al., 1992), was more recently shown to be an inactive byproduct of the higher molecular weight active species p33/p10 (Boucher et al., 2018). Breakdown of the p20/p10 tetramer, unstable upon its release from the inflammasome scaffold, serves as an intrinsic mechanism for the cessation of caspase‐1 activity (Boucher et al., 2018). In the present study, the visibility on western blot of higher molecular weight species but lack of detectable p20 caspase‐1 may indicate that the caspase remains active in human microglia under the conditions tested. However, this remains speculative, and more work needs to be done to characterize the duration of caspase‐1 activity as it has been reported to differ as a function of inflammasome size and cell type (Boucher et al., 2018).
If, as we suggest, caspase‐1 is active upon NLRP3 inflammasome activation in primary human microglia, but this activity is not required for IL‐1β processing, it follows that other substrates for the caspase should be present. Wang et al. (2016) reported that α‐syn itself is a substrate for caspase‐1, being cleaved at aspartate residue 121 to yield a truncated, aggregation‐prone form of α‐syn that subsequently perpetuates further caspase‐1 activation through the inflammasome. It may be that in the conditions presented here, caspase‐1 activity is diverted to α‐syn truncation, leaving the task of IL‐1β processing to other caspases.
NLRP3 inflammasome activation associated with extensive dopaminergic neuronal death has been specifically identified as being microglial in origin in both PD patients and numerous murine models of parkinsonism (Gordon et al., 2018; E. Lee et al., 2019). The exact trigger(s) of NLRP3 inflammasome activation in human microglia in vivo and the mechanism by which this activation has a detrimental effect on dopaminergic neurons remain to be identified. It is known that α‐syn is readily released by neurons and that microglia have the highest rate of clearance of this neuron‐derived α‐syn (H. J. Lee, Suk, Bae, & Lee, 2008). The present study shows that α‐syn can induce inflammasome activation upon uptake into primary human microglia. Thus, it may be that neuronal α‐syn contributes to activation of the NLRP3 inflammasome in PD patients in vivo, leading to caspase activation, IL‐1β production and signaling, and the propagation of innate immune activity, contributing to neurodegeneration.
LPS is often used as an inflammasome priming agent prior to cell activation with protein aggregates such as α‐syn (Freeman et al., 2013) or amyloid‐β (Halle et al., 2008; Venegas et al., 2017). However, the induction of IL‐1β secretion by α‐syn was previously shown to take place, albeit to a somewhat lesser extent, in the absence of a separate LPS priming step in primary human monocytes (Plotegher et al., 2014) and primary mouse microglia (Gordon et al., 2018). Treatment of primary cortical mouse microglia with aggregated recombinant human α‐syn has been reported to lead to the strong (240‐fold) upregulation of IL‐1β mRNA from baseline expression (Béraud & Maguire‐Zeiss, 2012), suggesting an inflammasome priming effect of the α‐syn on its own. Although in the present study, shorter‐term (4 hr) canonical NLRP3 inflammasome activation did indeed require LPS priming prior to a secondary activation step with nigericin, NLRP3 inflammasome‐mediated production of IL‐1β by primary mouse, and human microglia upon overnight exposure to α‐syn required no separate LPS priming of the microglia. The longer exposure time frame for α‐syn‐mediated NLRP3 inflammasome activation in THP‐1 cells and primary human microglia is consistent with the kinetics previously reported for α‐syn activation of primary mouse microglia, which were attributed to the time requirement for cellular uptake of the α‐syn (Gordon et al., 2018). Since it is improbable under physiological conditions that sufficient LPS would be present in the brains of PD patients to directly prime the microglia, but probable that there would be long‐term exposure of microglia to α‐syn, we consider the activation scheme tested here to be a more accurate representation of in vivo PD conditions than the inclusion of LPS as a priming agent for this mode of inflammasome activation. Alternatively, it may be that endogenous ligands of Toll‐like receptor 4 (TLR4), the receptor to which LPS binds to exert its downstream signaling effects, serve to prime microglia for subsequent activation by α‐syn in vivo. High mobility group box 1 (HMGB1), biglycan, and heat shock protein 70 (Hsp70) all have been identified as endogenous TLR4 ligands in the brain (Gou et al., 2020; Martín‐Hernández et al., 2018; Xie et al., 2020).
The results of this study demonstrate for the first time the canonical NLRP3 inflammasome competence of primary human microglia, similar to that previously reported for primary mouse and nonhuman primate microglia as well as human peripheral macrophages. Moreover, they reveal the ability of PD‐relevant α‐syn fibrils to activate the NLRP3 inflammasome in primary human microglia to induce IL‐1β processing. Our work suggests that this α‐syn‐mediated IL‐1β production may take place in a caspase‐1 activity‐independent manner that differs from α‐syn‐mediated activation of primary mouse microglia, implying the likelihood of alternative caspase involvement in this process in human microglia. Taken together, these observations serve to implicate the NLRP3 inflammasome as a contributor to PD pathology in a human‐specific microglia system. The data reported here are subject to the usual limitations of in vitro human cell studies and in vivo animal models in terms of extrapolating results from experimental models to authentic disease conditions in patients. However, they allow for a direct comparison of the mouse model to primary human microglia, revealing parallels between models and species in terms of mechanisms of NLRP3 inflammasome activation as well as highlighting important downstream divergence in pathways, particularly with regard to caspase involvement in α‐syn‐induced inflammasome signaling. By casting doubt on the requirement for caspase‐1 but supporting that for NLRP3 in α‐syn‐mediated inflammasome activation in human microglia, these findings lend further credence to currently ongoing clinical research into NLRP3 as a therapeutic target for PD.
Furthermore, the unique possibility of generating an NLRP3−/− primary microglia culture from the in vivo mouse paradigm in this work provided the opportunity to single out the specific contribution of the NLRP3 inflammasome to pathology in murine models of parkinsonism. Considering the crucial role of NLRP3 inflammasome activation in the spreading of α‐syn pathology and in dopaminergic neurodegeneration that has been documented by others through previous research in such models (Gordon et al., 2018; E. Lee et al., 2019; Martinez et al., 2017; Yan et al., 2015), this comparison is very valuable in the rationalization of PD‐related inflammasome studies in human systems.
CONFLICT OF INTEREST
The authors declare that they have no conflict of interest.
Supporting information
Figure S1 Supporting information
Figure S2 Supporting information
Figure S3 Supporting information
ACKNOWLEDGMENTS
The authors wish to thank Marc van Eik and Iris Gombert for contributing to experimental data on human cells, Prof. Dr. Michael Heneka and Dr. Róisín McManus of the German Center for Neurodegenerative Diseases (DZNE, Bonn, Germany) for providing essential mouse microglial culture materials and protocols, Dr. Marko Popovic and Evelien Timmermans‐Huisman of the VUmc Amsterdam Department of Anatomy and Neurosciences for assistance with obtaining and analyzing confocal and STED images and video, Dr. Marco Brucale of the Consiglio Nazionale delle Ricerche, Istituto per lo Studio dei Materiali Nanostrutturati (CNR‐ISMN, Bologna, Italy) for providing AFM imaging, and Dr. Hans Baaijen and Dr. Sander Idema of the VUmc Amsterdam Department of Neurosurgery for the tissue acquired upon focal cortical resection procedures. The work presented is part of the EU Joint Programme‐Neurodegenerative Disease Research (JPND) project “InCure.” The project is supported through the following funding organizations under the aegis of JPND: France, Agence National de la Recherche; Germany, Federal Ministry of Education and Research (BMBF; funding code 01ED1505); Italy, Ministry of Education, Universities and Research; Netherlands, ZonMw (ZonMw; funding code 733051051); Sweden, Swedish Research Council.
Pike AF, Varanita T, Herrebout MAC, et al. α‐Synuclein evokes NLRP3 inflammasome‐mediated IL‐1β secretion from primary human microglia. Glia. 2021;69:1413–1428. 10.1002/glia.23970
Funding information ZonMw, Grant/Award Number: 733051051; Swedish Research Council; Ministry of Education, Universities and Research; Federal Ministry of Education and Research, Grant/Award Number: 01ED1505; Agence National de la Recherche
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- Antonopoulos, C. , Russo, H. M. , El Sanadi, C. , Martin, B. N. , Li, X. , Kaiser, W. J. , … Dubyak, G. R. (2013). Caspase‐8 as an effector and regulator of NLRP3 Inflammasome signaling. The Journal of Biological Chemistry, 290(33), 20167–20184. 10.1074/jbc.M115.652321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker, P. J. , Boucher, D. , Bierschenk, D. , Tebartz, C. , Whitney, P. G. , D'Silva, D. B. , … Masters, S. L. (2015). NLRP3 inflammasome activation downstream of cytoplasmic LPS recognition by both caspase‐4 and caspase‐5. European Journal of Immunology, 45(10), 2918–2926. 10.1002/eji.201545655 [DOI] [PubMed] [Google Scholar]
- Béraud, D. , & Maguire‐Zeiss, K. A. (2012). Misfolded α‐synuclein and toll‐like receptors: Therapeutic targets for Parkinson's disease. Parkinsonism & Related Disorders, 18, S17–S20. 10.1016/s1353-8020(11)70008-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bian, Z.‐M. , Elner, S. G. , & Elner, V. M. (2009). Dual Involvement of Caspase‐4 in Inflammatory and ER Stress‐Induced Apoptotic Responses in Human Retinal Pigment Epithelial Cells. Investigative Opthalmology & Visual Science, 50(12), 6006. 10.1167/iovs.09-3628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blum‐Degen, D. , Muller, T. , Kuhn, W. , Gerlach, M. , Przuntek, H. , & Riederer, P. (1995). Interleukin‐1 beta and interleukin‐6 are elevated in the cerebrospinal fluid of Alzheimer's and de novo Parkinson's disease patients. Neuroscience Letters, 202(1–2), 17–20. [DOI] [PubMed] [Google Scholar]
- Boucher, D. , Monteleone, M. , Coll, R. C. , Chen, K. W. , Ross, C. M. , Teo, J. L. , … Schroder, K. (2018). Caspase‐1 self‐cleavage is an intrinsic mechanism to terminate inflammasome activity. The Journal of Experimental Medicine, 215(3), 827–840. 10.1084/jem.20172222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braak, H. , Del Tredici, K. , Rub, U. , de Vos, R. A. , Jansen Steur, E. N. , & Braak, E. (2003). Staging of brain pathology related to sporadic Parkinson's disease. Neurobiology of Aging, 24(2), 197–211. [DOI] [PubMed] [Google Scholar]
- Burger, D. , Fickentscher, C. , de Moerloose, P. , & Brandt, K. J. (2016). F‐actin dampens NLRP3 inflammasome activity via flightless‐I and LRRFIP2. Scientific Reports, 6, 29834. 10.1038/srep29834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burm, S. M. , Zuiderwijk‐Sick, E. A. , t Jong, A. E. , van der Putten, C. , Veth, J. , Kondova, I. , & Bajramovic, J. J. (2015). Inflammasome‐induced IL‐1beta secretion in microglia is characterized by delayed kinetics and is only partially dependent on inflammatory caspases. The Journal of Neuroscience, 35(2), 678–687. 10.1523/JNEUROSCI.2510-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Codolo, G. , Plotegher, N. , Pozzobon, T. , Brucale, M. , Tessari, I. , Bubacco, L. , & de Bernard, M. (2013). Triggering of inflammasome by aggregated alpha‐synuclein, an inflammatory response in synucleinopathies. PLoS One, 8(1), e55375. 10.1371/journal.pone.0055375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coll, R. C. , Robertson, A. , Butler, M. , Cooper, M. , & O'Neill, L. A. (2011). The cytokine release inhibitory drug CRID3 targets ASC oligomerisation in the NLRP3 and AIM2 inflammasomes. PLoS One, 6(12), e29539. 10.1371/journal.pone.0029539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coll, R. C. , Robertson, A. A. , Chae, J. J. , Higgins, S. C. , Munoz‐Planillo, R. , Inserra, M. C. , … O'Neill, L. A. (2015). A small‐molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nature Medicine, 21(3), 248–255. 10.1038/nm.3806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniele, S. G. , Beraud, D. , Davenport, C. , Cheng, K. , Yin, H. , & Maguire‐Zeiss, K. A. (2015). Activation of MyD88‐dependent TLR1/2 signaling by misfolded alpha‐synuclein, a protein linked to neurodegenerative disorders. Science Signaling, 8(376), ra45. 10.1126/scisignal.2005965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dauer, W. , & Przedborski, S. (2003). Parkinson's disease: Mechanisms and models. Neuron, 39(6), 889–909. [DOI] [PubMed] [Google Scholar]
- de Groot, C. J. , Hulshof, S. , Hoozemans, J. J. , & Veerhuis, R. (2001). Establishment of microglial cell cultures derived from postmortem human adult brain tissue: Immunophenotypical and functional characterization. Microscopy Research and Technique, 54(1), 34–39. 10.1002/jemt.1118 [DOI] [PubMed] [Google Scholar]
- Freeman, D. , Cedillos, R. , Choyke, S. , Lukic, Z. , McGuire, K. , Marvin, S. , … Campbell, E. M. (2013). Alpha‐synuclein induces lysosomal rupture and cathepsin dependent reactive oxygen species following endocytosis. PLoS One, 8(4), e62143. 10.1371/journal.pone.0062143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glass, C. K. , Saijo, K. , Winner, B. , Marchetto, M. C. , & Gage, F. H. (2010). Mechanisms underlying inflammation in neurodegeneration. Cell, 140(6), 918–934. 10.1016/j.cell.2010.02.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon, R. , Albornoz, E. A. , Christie, D. C. , Langley, M. R. , Kumar, V. , Mantovani, S. , … Woodruff, T. M. (2018). Inflammasome inhibition prevents alpha‐synuclein pathology and dopaminergic neurodegeneration in mice. Science Translational Medicine, 10(465), eaah4066. 10.1126/scitranslmed.aah4066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gou, X. , Ying, J. , Yue, Y. , Qiu, X. , Hu, P. , Qu, Y. , … Mu, D. (2020). The roles of high mobility group box 1 in cerebral ischemic injury. Frontiers in Cellular Neuroscience, 14, 600280. 10.3389/fncel.2020.600280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin, W. S. , Liu, L. , Li, Y. , Mrak, R. E. , & Barger, S. W. (2006). Interleukin‐1 mediates Alzheimer and Lewy body pathologies. Journal of Neuroinflammation, 3, 5. 10.1186/1742-2094-3-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halle, A. , Hornung, V. , Petzold, G. C. , Stewart, C. R. , Monks, B. G. , Reinheckel, T. , … Golenbock, D. T. (2008). The NALP3 inflammasome is involved in the innate immune response to amyloid‐beta. Nature Immunology, 9(8), 857–865. 10.1038/ni.1636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heneka, M. T. , Kummer, M. P. , & Latz, E. (2014). Innate immune activation in neurodegenerative disease. Nature Reviews Immunology, 14(7), 463–477. 10.1038/nri3705 [DOI] [PubMed] [Google Scholar]
- Jellinger, K. A. (2018). Dementia with Lewy bodies and Parkinson's disease‐dementia: Current concepts and controversies. Journal of Neural Transmission (Vienna), 125(4), 615–650. 10.1007/s00702-017-1821-9 [DOI] [PubMed] [Google Scholar]
- Lagrange, B. , Benaoudia, S. , Wallet, P. , Magnotti, F. , Provost, A. , Michal, F. , … Henry, T. (2018). Human caspase‐4 detects tetra‐acylated LPS and cytosolic Francisella and functions differently from murine caspase‐11. Nature Communications, 9(1), 242. 10.1038/s41467-017-02682-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latz, E. , Xiao, T. S. , & Stutz, A. (2013). Activation and regulation of the inflammasomes. Nature Reviews. Immunology, 13(6), 397–411. 10.1038/nri3452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, E. , Hwang, I. , Park, S. , Hong, S. , Hwang, B. , Cho, Y. , … Yu, J. W. (2019). MPTP‐driven NLRP3 inflammasome activation in microglia plays a central role in dopaminergic neurodegeneration. Cell Death and Differentiation, 26(2), 213–228. 10.1038/s41418-018-0124-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, H. J. , Suk, J. E. , Bae, E. J. , & Lee, S. J. (2008). Clearance and deposition of extracellular alpha‐synuclein aggregates in microglia. Biochemical and Biophysical Research Communications, 372(3), 423–428. 10.1016/j.bbrc.2008.05.045 [DOI] [PubMed] [Google Scholar]
- Lipinska, K. , Malone, K. E. , Moerland, M. , & Kluft, C. (2014). Applying caspase‐1 inhibitors for inflammasome assays in human whole blood. Journal of Immunological Methods, 411, 66–69. 10.1016/j.jim.2014.05.018. [DOI] [PubMed] [Google Scholar]
- MacPherson, M. , Westbom, C. , Kogan, H. , & Shukla, A. (2017). Actin polymerization plays a significant role in asbestos‐induced inflammasome activation in mesothelial cells in vitro. Histochemistry and Cell Biology, 147(5), 595–604. 10.1007/s00418-016-1530-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maelfait, J. , Vercammen, E. , Janssens, S. , Schotte, P. , Haegman, M. , Magez, S. , & Beyaert, R. (2008). Stimulation of toll‐like receptor 3 and 4 induces interleukin‐1beta maturation by caspase‐8. The Journal of Experimental Medicine, 205(9), 1967–1973. 10.1084/jem.20071632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Man, S. M. , Hopkins, L. J. , Nugent, E. , Cox, S. , Gluck, I. M. , Tourlomousis, P. , … Bryant, C. E. (2014). Inflammasome activation causes dual recruitment of NLRC4 and NLRP3 to the same macromolecular complex. Proceedings of the National Academy of Sciences of the United States of America, 111(20), 7403–7408. 10.1073/pnas.1402911111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez, E. M. , Young, A. L. , Patankar, Y. R. , Berwin, B. L. , Wang, L. , von Herrmann, K. M. , … Havrda, M. C. (2017). Editor's highlight: Nlrp3 is required for inflammatory changes and Nigral cell loss resulting from chronic Intragastric rotenone exposure in mice. Toxicological Sciences, 159(1), 64–75. 10.1093/toxsci/kfx117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martín‐Hernández, D. , Caso, J. R. , Javier Meana, J. , Callado, L. F. , Madrigal, J. , … Leza, J. C. (2018). Intracellular inflammatory and antioxidant pathways in postmortem frontal cortex of subjects with major depression: Effect of antidepressants. Journal of Neuroinflammation, 15(1), 251. 10.1186/s12974-018-1294-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mastroeni, D. , Grover, A. , Leonard, B. , Joyce, J. N. , Coleman, P. D. , Kozik, B. , … Rogers, J. (2009). Microglial responses to dopamine in a cell culture model of Parkinson's disease. Neurobiology of Aging, 30(11), 1805–1817. 10.1016/j.neurobiolaging.2008.01.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGeer, P. L. , Yasojima, K. , & McGeer, E. G. (2002). Association of interleukin‐1 beta polymorphisms with idiopathic Parkinson's disease. Neuroscience Letters, 326(1), 67–69. [DOI] [PubMed] [Google Scholar]
- Mendiola, A. S. , & Cardona, A. E. (2018). The IL‐1beta phenomena in neuroinflammatory diseases. Journal of Neural Transmission (Vienna), 125(5), 781–795. 10.1007/s00702-017-1732-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mogi, M. , Harada, M. , Kondo, T. , Riederer, P. , Inagaki, H. , Minami, M. , & Nagatsu, T. (1994). Interleukin‐1 beta, interleukin‐6, epidermal growth factor and transforming growth factor‐alpha are elevated in the brain from parkinsonian patients. Neuroscience Letters, 180(2), 147–150. [DOI] [PubMed] [Google Scholar]
- Netea, M. G. , Simon, A. , van de Veerdonk, F. , Kullberg, B. J. , Van der Meer, J. W. , & Joosten, L. A. (2010). IL‐1beta processing in host defense: Beyond the inflammasomes. PLoS Pathogens, 6(2), e1000661. 10.1371/journal.ppat.1000661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nurmi, K. , Virkanen, J. , Rajamaki, K. , Niemi, K. , Kovanen, P. T. , & Eklund, K. K. (2013). Ethanol inhibits activation of NLRP3 and AIM2 inflammasomes in human macrophages—A novel anti‐inflammatory action of alcohol. PLoS One, 8(11), e78537. 10.1371/journal.pone.0078537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Place, D. E. , & Kanneganti, T. D. (2018). Recent advances in inflammasome biology. Current Opinion in Immunology, 50, 32–38. 10.1016/j.coi.2017.10.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plotegher, N. , Berti, G. , Ferrari, E. , Tessari, I. , Zanetti, M. , Lunelli, L. , … Bubacco, L. (2017). DOPAL derived alpha‐synuclein oligomers impair synaptic vesicles physiological function. Scientific Reports, 7, 40699. 10.1038/srep40699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plotegher, N. , Greggio, E. , Bisaglia, M. , & Bubacco, L. (2014). Biophysical groundwork as a hinge to unravel the biology of alpha‐synuclein aggregation and toxicity. Quarterly Reviews of Biophysics, 47(1), 1–48. 10.1017/S0033583513000097 [DOI] [PubMed] [Google Scholar]
- Ravid, R. , & Swaab, D. F. (1993). The Netherlands brain bank—A clinico‐pathological link in aging and dementia research. Journal of Neural Transmission. Supplementum, 39, 143–153. [PubMed] [Google Scholar]
- Rogers, J. , Mastroeni, D. , Leonard, B. , Joyce, J. , & Grover, A. (2007). Neuroinflammation in Alzheimer's disease and Parkinson's disease: Are microglia pathogenic in either disorder? International Review of Neurobiology, 82, 235–246. 10.1016/s0074-7742(07)82012-5 [DOI] [PubMed] [Google Scholar]
- Russo, I. , Berti, G. , Plotegher, N. , Bernardo, G. , Filograna, R. , Bubacco, L. , & Greggio, E. (2015). Leucine‐rich repeat kinase 2 positively regulates inflammation and down‐regulates NF‐kappaB p50 signaling in cultured microglia cells. Journal of Neuroinflammation, 12, 230. 10.1186/s12974-015-0449-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheiblich, H. , Schlutter, A. , Golenbock, D. T. , Latz, E. , Martinez‐Martinez, P. , & Heneka, M. T. (2017). Activation of the NLRP3 inflammasome in microglia: The role of ceramide. Journal of Neurochemistry, 143(5), 534–550. 10.1111/jnc.14225 [DOI] [PubMed] [Google Scholar]
- Schmid‐Burgk, J. L. , Gaidt, M. M. , Schmidt, T. , Ebert, T. S. , Bartok, E. , & Hornung, V. (2015). Caspase‐4 mediates non‐canonical activation of the NLRP3 inflammasome in human myeloid cells. European Journal of Immunology, 45(10), 2911–2917. 10.1002/eji.201545523 [DOI] [PubMed] [Google Scholar]
- Schroder, K. , & Tschopp, J. (2010). The inflammasomes. Cell, 140(6), 821–832. 10.1016/j.cell.2010.01.040 [DOI] [PubMed] [Google Scholar]
- Sgobio, C. , Sun, L. , Ding, J. , Herms, J. , Lovinger, D. M. , & Cai, H. (2019). Unbalanced calcium channel activity underlies selective vulnerability of nigrostriatal dopaminergic terminals in parkinsonian mice. Scientific Reports, 9(1), 4857. 10.1038/s41598-019-41091-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spillantini, M. G. (1999). Parkinson's disease, dementia with Lewy bodies and multiple system atrophy are alpha‐synucleinopathies. Parkinsonism & Related Disorders, 5(4), 157–162. [DOI] [PubMed] [Google Scholar]
- Surendranathan, A. , Rowe, J. B. , & O'Brien, J. T. (2015). Neuroinflammation in Lewy body dementia. Parkinsonism & Related Disorders, 21(12), 1398–1406. 10.1016/j.parkreldis.2015.10.009 [DOI] [PubMed] [Google Scholar]
- Takasu, A. , Masui, A. , Hamada, M. , Imai, T. , Iwai, S. , & Yura, Y. (2016). Immunogenic cell death by oncolytic herpes simplex virus type 1 in squamous cell carcinoma cells. Cancer Gene Therapy, 23(4), 107–113. 10.1038/cgt.2016.8. [DOI] [PubMed] [Google Scholar]
- Thornberry, N. A. , Bull, H. G. , Calaycay, J. R. , Chapman, K. T. , Howard, A. D. , Kostura, M. J. , … Tocci, M. J. (1992). A novel heterodimeric cysteine protease is required for interleukin‐1βprocessing in monocytes. Nature, 356(6372), 768–774. 10.1038/356768a0. [DOI] [PubMed] [Google Scholar]
- Venegas, C. , Kumar, S. , Franklin, B. S. , Dierkes, T. , Brinkschulte, R. , Tejera, D. , … Heneka, M. T. (2017). Microglia‐derived ASC specks cross‐seed amyloid‐beta in Alzheimer's disease. Nature, 552(7685), 355–361. 10.1038/nature25158 [DOI] [PubMed] [Google Scholar]
- Vigano, E. , Diamond, C. E. , Spreafico, R. , Balachander, A. , Sobota, R. M. , & Mortellaro, A. (2015). Human caspase‐4 and caspase‐5 regulate the one‐step non‐canonical inflammasome activation in monocytes. Nature Communications, 6, 8761. 10.1038/ncomms9761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahner, A. D. , Sinsheimer, J. S. , Bronstein, J. M. , & Ritz, B. (2007). Inflammatory cytokine gene polymorphisms and increased risk of Parkinson disease. Archives of Neurology, 64(6), 836–840. 10.1001/archneur.64.6.836 [DOI] [PubMed] [Google Scholar]
- Wang, W. , Nguyen, L. T. , Burlak, C. , Chegini, F. , Guo, F. , Chataway, T. , … Hoang, Q. Q. (2016). Caspase‐1 causes truncation and aggregation of the Parkinson's disease‐associated protein alpha‐synuclein. Proceedings of the National Academy of Sciences of the United States of America, 113(34), 9587–9592. 10.1073/pnas.1610099113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie, Y. , Peng, J. , Pang, J. , Guo, K. , Zhang, L. , Yin, S. , … Jiang, Y. (2020). Biglycan regulates neuroinflammation by promoting M1 microglial activation in early brain injury after experimental subarachnoid hemorrhage. Journal of Neurochemistry, 152(3), 368–380. 10.1111/jnc.14926 [DOI] [PubMed] [Google Scholar]
- Yan, Y. , Jiang, W. , Liu, L. , Wang, X. , Ding, C. , Tian, Z. , & Zhou, R. (2015). Dopamine controls systemic inflammation through inhibition of NLRP3 inflammasome. Cell, 160(1–2), 62–73. 10.1016/j.cell.2014.11.047 [DOI] [PubMed] [Google Scholar]
- Zhang, Q. S. , Heng, Y. , Yuan, Y. H. , & Chen, N. H. (2017). Pathological alpha‐synuclein exacerbates the progression of Parkinson's disease through microglial activation. Toxicology Letters, 265, 30–37. 10.1016/j.toxlet.2016.11.002 [DOI] [PubMed] [Google Scholar]
- Zhang, W. , Wang, T. , Pei, Z. , Miller, D. S. , Wu, X. , Block, M. L. , … Zhang, J. (2005). Aggregated alpha‐synuclein activates microglia: A process leading to disease progression in Parkinson's disease. The FASEB Journal, 19(6), 533–542. 10.1096/fj.04-2751com [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Supporting information
Figure S2 Supporting information
Figure S3 Supporting information
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.