

Abstract
Astrocytes regulate synaptic communication and are essential for proper brain functioning. In Alzheimer's disease (AD) astrocytes become reactive, which is characterized by an increased expression of intermediate filament proteins and cellular hypertrophy. Reactive astrocytes are found in close association with amyloid‐beta (Aβ) deposits. Synaptic communication and neuronal network function could be directly modulated by reactive astrocytes, potentially contributing to cognitive decline in AD. In this review, we focus on reactive astrocytes as treatment targets in AD in the APPswePS1dE9 AD mouse model, a widely used model to study amyloidosis and gliosis. We first give an overview of the model; that is, how it was generated, which cells express the transgenes, and the effect of its genetic background on Aβ pathology. Subsequently, to determine whether modifying reactive astrocytes in AD could influence pathogenesis and cognition, we review studies using this mouse model in which interventions were directly targeted at reactive astrocytes or had an indirect effect on reactive astrocytes. Overall, studies specifically targeting astrocytes to reduce astrogliosis showed beneficial effects on cognition, which indicates that targeting astrocytes should be included in developing novel therapies for AD.
Keywords: AD mouse model, Alzheimer's disease, amyloid‐beta, APPswePS1dE9, reactive astrocytes
MAIN POINTS
The APPswePS1dE9 mouse displays extensive amyloid‐induced reactive astrogliosis.
Reactive astrocytes play a pivotal role in AD pathogenesis as they affect synaptic function.
Targeting reactive astrocytes may help to prevent cognitive decline.
1. INTRODUCTION
Alzheimer's disease (AD) is a prime example of a central nervous system (CNS) disease with a clear presence of reactive astrocytes. AD is the most common form of dementia and is neuropathologically characterized by extracellular deposits of amyloid‐beta (Aβ) and intracellular neurofibrillary tangles, consisting of hyperphosphorylated tau (Selkoe, 1991). Moreover, Aβ plaques are invaded and surrounded by activated microglia (Itagaki, Mcgeer, & Akiyama, 1989; Yin et al., 2017), and reactive astrocytes (Kamphuis et al., 2014; Kato et al., 1998; Simpson et al., 2010). Over the past decade, there has been an increasing insight into the effect of reactive astrogliosis on neural functioning. Therefore, it is opportune to review reactive astrocytes in relation to cognitive function in a widely used AD mouse model: the APPswePS1dE9 mouse (Box 1).
BOX 1. The APPswePS1dE9 mouse model.
APPswePS1dE9 mice overexpress two transgenes; humanized amyloid precursor protein (hAPP) harboring the Swedish mutation (K595N/M596L; Johnston, O'Neill, Lannfelt, Winblad, & Cowburn, 1994; Mullan et al., 1992) and human PS1 with a deletion of exon 9 (PS1dE9) (Perez‐Tur et al., 1995). The APPswePS1dE9 mice were made by co‐injection of two plasmids into a single cell embryo derived from F2 hybrids of C57BL/6J and C3H/HeJ mice (Figure 1d). Each plasmid contained one of the two transgenes, either humanized APP or human PS1dE9, under the control of a mouse prion protein (PrP) promoter element (the MoPrP.Xho vector). The two transgenes co‐integrated and co‐segregated as a single locus (Jankowsky et al., 2001). For coinjection of both transgenes, the MoPrP.Xho vector was used. Within this modified prion protein (MoPrP) vector, an Xho I restriction site replaced the open reading frame (ORF) of the third exon, removing the coding sequence of PrP. The transgene sequences, containing a start and stop codon, were cloned into the vector at this Xho I site. The main advantage of the use of the MoPrP.Xho vector was that the ORF is located within an exon, and can, therefore, be easily replaced by a recombinant ORF. Furthermore, as the MoPrP.Xho vector is small by itself, large cDNA fragments can be introduced (Borchelt, Davis, et al., 1996; Jankowsky et al., 2001). The expression of the transgenes is driven by the mouse PrP promoter, which was thought to lead to a predominant neuronal expression of the transgenes (Borchelt, Davis, et al., 1996; Jankowsky et al., 2001). However, it is known that in addition to the nervous system, the promoter also drives the expression of transgenes in other organs, such as heart and kidneys (Borchelt, Davis, et al., 1996; Esquerda‐Canals, Montoliu‐Gaya, Güell‐Bosch, & Villegas, 2017; Jankowsky et al., 2007). Although transgene expression in heart and kidneys probably would not affect the pathological conditions in the brain, it might be associated with the premature death of APPswePS1dE9 mice (Section 3.5).
Multiple lines of mice were derived from embryos with the coinjected vectors (Jankowsky et al., 2001). The mice of line 85 were deposited at the Jackson Laboratories (Jankowsky et al., 2004). In this review, we focus on APPswePS1dE9 mice on a C57BL/6J × C3H/HeJ genetic background, MMRRC Stock No: 34829 (formerly Jackson Lab Stock No: 004462) and APPswePS1dE9 mice on a congenic C57BL/6J genetic background MMRRC Stock No: 34832 (formerly Jackson Lab Stock No: 005864).
The first increase in soluble Aβ42 levels has been observed in 3‐month‐old APPswePS1dE9 mice (Van Tijn et al., 2012) and amyloid plaques in 4‐month‐old mice (Garcia‐Alloza et al., 2006; Ruan, Kang, Pei, & Le, 2009). Impaired synaptic plasticity has been detected as early as 3.5 months in the cortex (Shemer et al., 2006). A decrease in presynaptic and postsynaptic terminals has been observed by 4 months of age in the hippocampus (Hong et al., 2016). Cognitive impairments have been observed as early as 3 months of age, in a contextual fear memory task and 6‐month‐old APPswePS1dE9 mice in the Morris water maze spatial memory task (Végh et al., 2014). Astrogliosis develops around the age of 6 months, based on the increased expression of GFAP (Ruan et al., 2009; Van Tijn et al., 2012; Végh et al., 2014).
Astrocytes are essential for proper brain functioning (Box 2; Kettenmann & Verkhratsky, 2008; Verkhratsky & Nedergaard, 2018). They are actively involved in synaptic communication (Araque et al., 2014; Araque, Parpura, Sanzgiri, & Haydon, 1998) and play a key role in maintaining ion, neurotransmitter (Kirischuk, Parpura, & Verkhratsky, 2012), and energy homeostasis in the CNS (Allaman, Bélanger, & Magistretti, 2011; Verkhratsky & Nedergaard, 2018). Perisynaptic astrocyte protrusions closely interact with synapses and together they form the tripartite synapse (Araque, Parpura, Sanzgiri, & Haydon, 1999). In the reactive state, astrocytes show cellular hypertrophy, which is likely to affect these protrusions. Besides, there is a change in the intermediate filament (IF) cytoskeleton, due to an increase in the expression of the IF proteins glial fibrillary acidic protein (GFAP; the classical marker for reactive astrocytes; Box 3), vimentin, and nestin (Hol & Pekny, 2015; Pekny et al., 2016). The changes associated with the reactive state can directly impact synaptic transmission and neuronal circuit activity, thereby potentially contributing to the cognitive problems observed in AD (Chung, Welsh, Barres, & Stevens, 2015; Dossi, Vasile, & Rouach, 2018; Jo et al., 2014; Kuchibhotla et al., 2008; Ortinski et al., 2010; Osborn, Kamphuis, Wadman, & Hol, 2016; Peters et al., 2009; Wu, Guo, Gearing, & Chen, 2012; Xing, Yang, Cui, & Chen, 2019).
BOX 2. Physiological functions of astrocytes.
Astrocytes are involved in numerous physiological functions, including, but not limited to, maintaining molecular homeostasis of the CNS, recycling of neurotransmitters, supplying energy substrates to neurons, and maintaining the blood–brain barrier (BBB) (for a comprehensive review see Verkhratsky & Nedergaard, 2018).
Astrocyte processes are in close contact with neurons, forming the so‐called tripartite synapse (Araque et al., 1999), and with the vasculature, forming the gliovascular unit (Giaume, Koulakoff, Roux, Holcman, & Rouach, 2010). Astrocytes interact with synapses, respond to signals of surrounding neurons, and can, in turn, modulate synaptic function. They can regulate synaptic transmission by buffering extracellular potassium, releasing gliotransmitters, for example, ATP, glutamate, or GABA, and taking up neurotransmitters from the extracellular environment, establishing tight and dynamic neuron–glia interactions (Shrivastava et al., 2013; Steinhäuser, Seifert, & Bedner, 2012). As part of the gliovascular unit, astrocytes form a link between the vasculature and neurons. Astroglial endfeet are involved in the regulation of BBB function, supplying nutrients, removal of toxic waste products, and maintenance of ionic balance around synapses (De Bock, Leybaert, & Giaume, 2017).
Even though astrocytes occupy individual domains (Bushong, Martone, Jones, & Ellisman, 2002), they are coupled via gap junctions (Giaume et al., 2010). Connexins (Cxs), specifically Cx30 and Cx43, are the proteins that form transmembrane hemichannels (HCs) or gap junctions between astrocytes. The intercellular gap junctions enable the diffusion of small molecules through the astrocyte network and play an important role in the metabolic support of neurons (Rouach, Koulakoff, Abudara, Willecke, & Giaume, 2008) and in potassium homeostasis (Bazzigaluppi, Weisspapir, Stefanovic, Leybaert, & Carlen, 2017; Wallraff et al., 2006). As a hemichannel, connexins also form a pore between the cytoplasm and the extracellular space (Yi et al., 2016).
Astrocytes have an active role in CNS physiology, and astrocyte dysfunction has been implicated in various CNS disorders. In AD, astrocytes undergo morphological, molecular, and functional changes, a process known as astrogliosis.
BOX 3. GFAP.
Glial fibrillary acidic protein (GFAP) is the characteristic intermediate filament (IF) protein in astrocytes. Astrocytes express 10 different isoforms of GFAP, which form together with vimentin and nestin the cytoskeleton of astrocytes (Hol & Pekny, 2015; Pekny et al., 2016). GFAP expression is highly upregulated in disease. Reactive astrocytes are observed in various brain pathologies, for example, epilepsy, neurodegenerative diseases, ischemia, infection, and brain trauma. An increased GFAP expression and changed morphology are the most used markers for reactive astrocytes (Escartin, Guillemaud, & Carrillo‐de Sauvage, 2019; Hol & Pekny, 2015; Pekny et al., 2016), also evident from the studies listed in Tables 1 and 2 of this review. Although some functional changes after the loss of GFAP have been described (Hughes, Maguire, McMinn, Scholz, & Sutherland, 2004; Liedtke et al., 1996), the link between increased GFAP expression and functional changes in reactive astrocytes remain largely unstudied. Even though GFAP is a valid and valuable marker for reactive astrocytes, there are some remaining concerns and limitations. GFAP‐immunostainings only show the IF cytoskeleton, which is about 10% of the astrocyte surface, and therefore cannot be used to determine cellular hypertrophy. Cortical astrocytes have lower expression levels of GFAP in comparison to hippocampal astrocytes, and it remains to be determined whether these astrocytes respond with a similar GFAP increase in response to AD pathology. In conclusion, by only studying GFAP, early changes and subpopulation specific changes of reactive astrocytes are missed in AD. Alternative and AD specific reactive astrocyte markers should be determined to fully understand the pathological changes in AD astrocytes.
To understand how the changes in reactive astrocytes contribute to cognitive decline in AD, it is crucial to study the alterations in neuron–glia interactions during AD pathogenesis, as these can directly affect neuronal functioning. In this systematic review, we focus on the APPswePS1dE9 mouse. This is a widely used AD mouse model, which excellently mimics amyloidosis and gliosis. Of note, in the scientific literature, this model is often referred to as APP/PS1 mice. Since the abbreviation APP/PS1 is also used to refer to other AD mouse models with different APP and PS1 mutations and driven by different promoters, we will use the full name APPswePS1dE9 throughout the review. We will first introduce the amyloid hypothesis, on which the model is based, and review the characteristics and the merit of the APPswePS1dE9 mouse model for AD research. We then systematically review studies that describe interventions that directly or indirectly affected reactive astrocytes in the APPswePS1dE9 AD mouse model, to determine whether modifying reactive astrocytes in AD could influence pathogenesis and cognition.
2. THE AMYLOID HYPOTHESIS AND THE APPSWEPS1DE9 MOUSE MODEL
Several AD mouse models, including the APPswePS1dE9 model, are based on the amyloid‐cascade hypothesis. Even though the debate on the exact etiology of AD is still strongly ongoing (De Strooper & Karran, 2016; Selkoe & Hardy, 2016), the amyloid hypothesis (also known as the Aβ hypothesis or amyloid cascade hypothesis) has been a main hypothesis in the AD field for almost 30 years (Hardy & Allsop, 1991; Hardy & Selkoe, 2002; Selkoe, 1991; Selkoe & Hardy, 2016). The hypothesis places Aβ, a 39–43 amino acid long peptide proteolytically cleaved from the amyloid precursors protein (APP; Figure 1a), at the beginning of the disease process. The amyloid hypothesis states that the overproduction of Aβ and its aggregation in plaques are the first events in AD pathogenesis, eventually resulting in the development of neurofibrillary tangles, loss of synapses, and neuronal death (Hardy & Allsop, 1991; Hardy & Selkoe, 2002; Selkoe, 1991). In the 1990s, AD‐causative mutations were observed in APP and in genes that were later found to be components of γ‐secretase that cleaves APP, that is, presenilin 1 (PS1), and presenilin 2 (PS2; Chartier‐Harlin et al., 1991; Goate et al., 1991; Levy‐lahad et al., 1995; Mullan et al., 1992; Rogaev et al., 1995; Sherrington et al., 1995). These genetic findings point into the direction of an enhanced processing of APP and production of Aβ, and support the hypothesis of a primary involvement of Aβ in the disease process. A major concern with the amyloid hypothesis is the poor correlation between the development of cognitive impairment and Aβ plaque burden (Nelson et al., 2012). Still, mouse models based on mutations in APP, PS1, and PS2 are valuable tools to study specific parts of the AD pathogenesis.
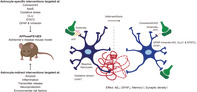
FIGURE 1.
An overview of the amyloidogenic and non‐amyloidogenic pathway, the generation of the APPswePS1dE9 mouse model, and gliosis. (a) Alternative processing of APP by α‐, β‐, and/or γ‐secretases resulting in amyloidogenic and non‐amyloidogenic cleavage. (b) Humanized mouse APP sequence (the white blocks indicate the human‐specific amino acids that were introduced in the mouse sequence), containing the Swedish mutation (yellow block). (c) PS1 is a transmembrane protein and part of the γ‐secretase complex. The PS1dE9 mutation results in the deletion of exon 9. (d) The APPswe and PS1dE9 transgenes were integrated in MoPrP vectors between exon 2 and 3; the coding sequence of PrP was completely removed, as described in Borchelt et al. (1996) and Jankowsky et al. (2001). The two vectors were co‐injected into a single cell embryo derived from F2 hybrids of C57BL/6J and C3H/HeJ mice: C3/B6HeJ mice. This resulted in the generation of double transgenic APPswePS1dE9 mice, in which the expression of both the APP and the PS1 gene is driven by the mouse prion protein promoter. (e) Integration of the transgenes occurred at chromosome 9 between the Arpp21 and Pdcd6ip genes (Jackson et al., 2015). (f) From the age of 6 months, reactive astrocytes (GFAP, green) and activated microglia (Iba1, red) surrounding Aβ plaques (6E10, blue) were detected, scale bar: 50 μm, adjusted from original picture in Orre et al. (2014). Aβ, amyloid‐beta; APP, amyloid precursor protein; ε‐site, epsilon cleavage site; GFAP, glial fibrillary acidic protein; Iba1, ionized calcium‐binding adapter molecule 1; MoPrP, modified prion protein; PrP, prion protein; PS1, presenilin 1; sAPPα, soluble amyloid precursor protein‐α; sAPPβ, soluble amyloid precursor protein‐β. Based on Jackson et al. (2015) and Jankowsky et al. (2001, 2007). Scientific illustration toolkits from Motifolio were used to generate parts of this figure
Over the years, the amyloid hypothesis has been refined and now also includes the important role of Aβ oligomers, which is the most pathogenic and toxic form of Aβ (Selkoe & Hardy, 2016). Of the various forms of Aβ (monomers, oligomers, fibrils), it is oligomeric Aβ that disrupt synaptic function (Walsh et al., 2002). In several AD mouse models, based on genetic mutations in APP, PS1, and PS2, an impairment in synaptic transmission has been detected before fibrillar plaques become apparent (Larson, Lynch, Games, & Seubert, 1999; Moechars et al., 1999; Végh et al., 2014).
In this review, we focus on APPswePS1dE9 mice (Box 1), which overexpress two transgenes: humanized amyloid precursor protein (hAPP) harboring the Swedish mutation (K595N/M596L, Figure 1b; Johnston et al., 1994; Mullan et al., 1992) and human PS1 with a deletion of exon 9 (PS1dE9, Figure 1c; Perez‐Tur et al., 1995). Both mutations cause early‐onset AD in humans (Mullan et al., 1992; Perez‐Tur et al., 1995) and are known to change APP processing, resulting in an enhanced production of Aβ (Citron et al., 1992; Perez‐Tur et al., 1995).
2.1. Amyloid precursor protein, start of the amyloidogenic pathway
AD‐causative mutations were found in the APP gene, leading to an enhanced processing of the APP protein into Aβ. Even though its central place in the amyloid hypothesis, the normal function of APP and its cleaved fragments is largely unknown. APP is a ubiquitously expressed Type I membrane glycoprotein with only a small intracellular domain (Nalivaeva & Turner, 2013; Selkoe et al., 1996). In the brain, APP regulates the cell cycle progression of fetal neural stem cells (Joo et al., 2010) and stimulates neural cell migration, thereby playing an important role in brain development. In the adult brain, APP is involved in long‐term potentiation (LTP; Weyer et al., 2011) and maintenance of calcium homeostasis (Octave, Pierrot, Ferrao Santos, Nalivaeva, & Turner, 2013).
APP can be processed by different proteolytic enzymes, that is, α‐, β‐, and γ‐secretases, resulting in different products (Figure 1a). Via the non‐amyloidogenic pathway, APP is sequentially cleaved by α‐ and y‐secretase, resulting in a soluble amyloid precursor protein‐α (sAPPα), an extracellular P3, and an intracellular APP C‐terminal domain (AICD) fragment. The amyloidogenic pathway yields sAPPβ, Aβ, and AICD due to cleavage by β‐ and γ‐secretases (Hall & Roberson, 2012; Salminen et al., 2013). The cleavage of APP by β‐secretase results in the formation of sAPPβ and an intramembrane β‐carboxyl‐terminal fragment (β‐CTF). The formation of Aβ40 or Aβ42 starts with ε‐cleavage, followed by γ‐cleavage of the β‐CTF (Takami et al., 2009). Products of the primary ε‐cleavage, Aβ49, and AICD50‐99 or Aβ48 and AICD49‐99, are sequential cleaved resulting in Aβ40 and Aβ42, respectively. The two major pathways are Aβ49 ➔ Aβ46 ➔ Aβ43 ➔ Aβ40 and Aβ48 ➔ Aβ45 ➔ Aβ42 (tripeptide hypothesis; Kakuda et al., 2006; Szaruga et al., 2017; Takami et al., 2009). Over the years, also other proteolytic products of APP have been implicated in AD. In 2004, Cao and Südhof (2004) proposed a role for AICD in the regulation of gene transcription. Although others have shown that the effect on downstream targets is weak and might be indirect (Hébert et al., 2006). Thus, the role of AICD as a transcriptional regulator remains controversial (for a review, Bukhari et al., 2017). Also, the extracellular N‐terminal fragment of APP (N‐APP) produced after cleavage of APP by β‐secretase, has been shown to mediate neurodegeneration. This outcome is distinct from effects mediated by Aβ peptides (Nikolaev, McLaughlin, O'Leary, & Tessier‐Lavigne, 2009).
APP is differentially spliced, resulting in several isoforms. The most common and most stable isoforms are APP695, APP751, and APP770, with the number referring to the number of amino acids (Johnston et al., 1994). In the brain, APP695 is mainly expressed in neurons (Johnston et al., 1994; Nalivaeva & Turner, 2013). In neuronal cell lines, it has been shown that sAPPβ, Aβ, and AICD are preferentially formed from APP695 (Belyaev et al., 2010). The Kunitz‐type protease inhibitor sequence in APP770 and APP751 promotes cleavage by α‐secretase, not yielding Aβ. APP695 lacks this sequence, making it the most amyloidogenic APP isoform (Nalivaeva & Turner, 2013).
2.2. APPswePS1dE9 mouse: Swedish mutation in APP results in an increased Aβ production
For the generation of the APPswePS1dE9 model, humanized mouse APP695 was used (Jankowsky et al., 2007). Human and mouse APP sequences differ in 17 amino acids, three of which are positioned within the Aβ‐encoding part. In humanized mouse APP695, the mouse‐specific amino acids in the Aβ‐encoding sequence were replaced by the human‐specific amino acids (Figure 1b). Thus, glycine at position 5 in the Aβ domain was replaced by arginine, phenylalanine at position 10 by tyrosine, and arginine at position 13 by histidine. The last replacement causes Aβ to be more susceptible to zinc‐mediated aggregation (Jankowsky et al., 2007; Nalivaeva & Turner, 2013). Importantly, the produced Aβ peptide in the mouse will be entirely human (Mellott et al., 2017), which is essential for testing therapies targeted at human Aβ.
The humanized APP sequence of the APPswePS1dE9 mice contains the Swedish mutation. Several mutations in APP have been linked to familial AD (FAD; https://www.alzforum.org/mutations). The humanized APP sequence of the APPswePS1dE9 mice contains the Swedish mutation. The Swedish mutation (APPswe) was discovered in a Swedish family with early‐onset AD. Two pedigrees showed a double missense mutation in exon 16 of APP, which results in the replacement of the lysine residue at position 595 in APP695 by asparagine (K595N) and the methionine residue at position 596 by leucine (M596L). The mutated codons are positioned just before the N‐terminal side of the Aβ domain (Figure 1b). Due to the position of these mutations, β‐secretase can cleave APP more efficiently. The level of Aβ in cells expressing APPswe is 6–8 times higher than in cells expressing wild‐type APP (Citron et al., 1992). Processing of wild‐type APP by β‐secretase occurs in early endosomes (Rajendran et al., 2006; Sannerud et al., 2011; Sannerud & Annaert, 2009), whereas APPswe is likely processed by β‐secretase in vesicles on their way to the cell surface (Haass et al., 1995). In short, the APPswe mutation affects APP processing towards an increased Aβ production.
2.3. Presenilin 1, function in the amyloidogenic pathway
Presenilin 1 (PS1) and 2 (PS2) are transmembrane proteins that undergo endoproteolytic processing. This yields large N‐terminal and smaller C‐terminal fragments (NTFs and CTFs) (Hardy, 1997), which accumulate equally in the brain (Lee et al., 1997). In contrast to the holoprotein, the NTFs and CTFs are stable protein fragments forming complexes that represent the active form of PSs (Xia, 2000).
PS1 is crucial for normal development as PS1−/− mice die late in embryogenesis or at birth (Shen, 2014; Xia, 2000). PSs also play an essential role in synaptic function and neuronal survival in adult mice, since age‐dependent synapse and neuron loss has been observed in a conditional double knockout mouse. These PS1−/−/PS2−/− mice showed memory and behavioral impairments (reviewed by Shen, 2014). In PS1‐deficient neurons, accumulation of AICD fragments and loss of Aβ production have been observed (De Strooper et al., 1998). These studies revealed that PS1 is essential for the proteolytic function of γ‐secretase (De Strooper et al., 1998; Wolfe et al., 1999). We now know that PS1 or PS2 form the catalytic active subunit of the γ‐secretase intramembrane protease complexes, which also contain Nicastrin (Nct), anterior pharynx defective 1 (Aph‐1), and presenilin‐enhancer‐2 (Pen‐2; De Strooper, 2003; Francis et al., 2002; Goutte, Tsunozaki, Hale, & Priess, 2002; Yu et al., 2000).
Diverse molecular effects are induced by the different mutations in PSs. Mutations in PS1 and PS2 increase the ratio of Aβ42:Aβ40 (Borchelt et al., 1996). Consistent with this observation, Aβ deposition was observed earlier in transgenic mice in which APPswe was co‐expressed with mutant PS1 compared to mice only expressing APPswe (Borchelt et al., 1997). The largest change in the Aβ42:Aβ40 ratio was observed when APPswe was co‐expressed with PS1 lacking exon 9, which shifted the ratio from 1:3 to 1:0.75 (Jankowsky et al., 2004). This change in the Aβ42:Aβ40 ratio could be caused by the subcellular distribution of PSs. PS1 and PS2 have distinct subcellular locations, contributing to substrate specificity. Whereas PS1 is broadly distributed in the cell, PS2 is directed to late endosomes/lysosomes. The strongest increase in the Aβ42:Aβ40 ratio was induced by mutations in PS1 when it changed the localization of PS1 to late endosomes/lysosomes. FAD‐associated mutations can affect the subcellular location of PS1 and overall increase the Aβ42:Aβ40 ratio, predominantly affecting the intracellular fraction and Aβ40 formation (Sannerud et al., 2016). Overall the mutations appear to decrease the C‐ to N‐terminal cleavage of APP, resulting in longer, more hydrophobic, and self‐aggregating Aβ peptides (Selkoe & Hardy, 2016).
When modulating PS1, it has to be noted that PSs/γ‐secretase not only cleave APP, but that they target numerous type I integral membrane proteins, such as Notch1, ErbB4, E‐cadherin, CD44, and LDL‐receptor related protein (LRP; De Strooper, 2003; Duggan & McCarthy, 2016; Wakabayashi & De Strooper, 2008). These changes could also contribute to AD pathogenesis.
2.4. APPswePS1dE9 mouse: PS1 Exon 9 deletion results in an increased Aβ production
Mutations in PS1 and PS2, are the main cause of FAD (Jankowsky et al., 2004) and are linked to early‐onset AD (Levy‐lahad et al., 1995; Rogaev et al., 1995; Sherrington et al., 1995). Together, they represent approximately 75% of all known FAD mutations (https://www.alzforum.org/ mutations). For the generation of the APPswePS1dE9 model, human PS1 with a deletion of exon 9 (PS1dE9) was used (Jankowsky et al., 2007). The deletion of exon 9 causes early‐onset FAD (Lee et al., 1997; Sherrington et al., 1995). The PS1dE9 mutation is a point mutation (G to T) in the splice site for exon 9, which breaks up the consensus sequence for the splice acceptor site, resulting in the (in‐frame) deletion of exon 9 (Hardy, 1997; Steiner et al., 1999).
As this is the domain in which PS processing normally occurs, PS1dE9 ultimately results in the accumulation of the uncleavable PS holoprotein. However, the pathological function of PS1dE9 is independent of its defect to undergo proteolytic processing, but rather due to a missense translation that is the result of the aberrant exon 8/10 splice junction leading to S290C and an increase in Aβ42. Correcting the S290C change prevented the threefold increase in Aβ42 production (Steiner et al., 1999). Thus, the PS1dE9 variant affects APP processing towards an increased Aβ42 production. How the PS1dE9 mutation results in an increased production of Aβ42 remains unclear. A hypothesis is that this mutation results in disturbed transport of PS1 from the endoplasmic reticulum (Kim et al., 2007) and that this change in the subcellular location of PS1 may affect the ratio of Aβ42:Aβ40 formation (Section 2.3).
The PS1dE9 mutation is associated with atypical Aβ pathology and motor symptoms. In the cortex of several AD patients with this mutation large, loose, “cotton wool” Aβ deposits and few dense core plaques were detected (Crook et al., 1998; Mann et al., 2001; Smith et al., 2001). However, not all patients with the PS1dE9 mutation display this atypical pathology (Hiltunen et al., 2000; Ishii et al., 1997). In PS1dE9 transgenic mice, this atypical pathology is not observed and APPswePS1dE9 mice display characteristic neuritic plaques (Jankowsky et al., 2004).
3. CHARACTERIZATION OF THE APPSWEPS1DE9 MOUSE MODEL
The discovery of AD‐causative mutations in the APP, PS1, and PS2 genes contributed to the generation of various transgenic AD mouse models (Elder, Gama Sosa, & De Gasperi, 2010). Around 100 models are now available to study AD pathogenesis (https://www.alzforum.org/research-models). Several are based on the amyloid‐cascade hypothesis (APPswePS1dE9, PDAPP, J20, and 5xFAD), but other models are based on mutations in tau, Trem2, or ApoE (3xTG, Trem2 R47H knock‐in, ApoE4 knock‐in). The APPswePS1dE9 mouse model, which is the focus of this review, was generated in 2001 (Jankowsky et al., 2001) and has since been widely used.
3.1. Cell‐type‐specific expression of the transgenes
In APPswePS1dE9 mice, the expression of the transgenes is driven by the mouse PrP promoter (Box 1, Figure 1d), which was thought to lead to a predominant neuronal expression of the transgenes (Borchelt, Davis, et al., 1996; Jankowsky et al., 2001).
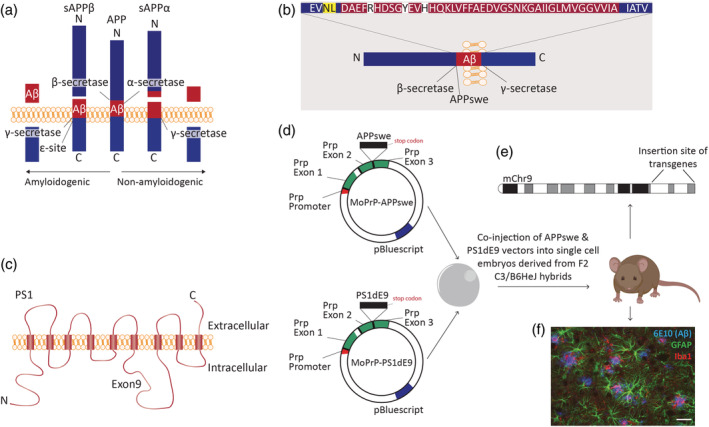
However, by comparing expression levels of PrP in different cell types in the brain, the highest expression is found in astrocytes and endothelial cells, followed by neurons, oligodendrocyte progenitor cells (OPCs), and oligodendrocytes. The expression of the Prp gene in microglia is low (Zhang et al., 2014). Although it is mentioned in the original manuscript that there is MoPrP.Xho‐driven transgene expression in astrocytes, it is not further discussed (Borchelt, Davis, et al., 1996). APP and PS1 are endogenously expressed by astrocytes and microglia of wild‐type mice (Figure 2a; Orre, Kamphuis, Osborn, Jansen, et al., 2014; Tasic et al., 2018; Zhang et al., 2014). We observed expression of the APP and PS1 transgenes in cortical astrocytes (Figure 2b,d) and microglia (Figure 2c,e) isolated from 15‐ to 18‐month‐old APPswePS1dE9 mice (unpublished data, Willem Kamphuis and Tamar Smit). This shows that the transgenes are actively transcribed in glia, and therefore might also directly change glia function in the APPswePS1dE9 mice.
FIGURE 2.
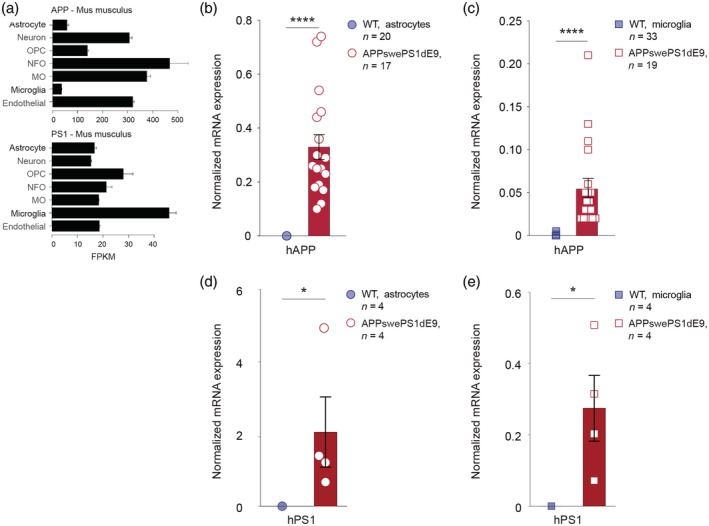
Endogenous and transgenic APP and PS1 expression in astrocytes and microglia. (a). Expression levels of endogenous APP and PS1, modified from https://web.stanford.edu/group/barres_lab/brain_rnaseq.html. Cells were isolated from the cortices of P7 mice (Zhang et al., 2014). (b,c) Cortical astrocytes and microglia were isolated from 15‐ to 18‐month‐old WT (blue) and APPswePS1dE9 mice (red) by FACS procedures, described in detail by Orre et al. (2014). RNA was isolated using TRIsure and cDNA was generated following the manufacturer's instructions (Quantitect – Qiagen). Resulting cDNA served as a template in real‐time qPCR assays (SYBR Green PCR Master Mix; Applied Biosystems), as described by Kamphuis et al. (2015). To determine expression levels of the humanized APP (hAPP) transgene the following primers were used: FW: TGAACCATTTCAACCGAGCTG and REV: GTGGGTACCTCCAGAGCC. Transcript levels were normalized to HPRT and GAPDH levels. (b) Normalized mRNA expression of hAPP is higher in astrocytes of APPswePS1dE9 mice (n = 17 mice) compared to WT (n = 20 mice, the level of hAPP was below the limit of detection in 17 samples). (c) Normalized mRNA expression of hAPP is higher in microglia of APPswePS1dE9 mice (n = 19 mice) compared to WT (n = 33 mice, the level of hAPP was below the limit of detection in 29 samples). (d,e) Cortical astrocytes and microglia were isolated from 4‐month‐old wild‐type (blue) and APPswePS1dE9 mice (red) by MACS procedures, adapted from protocol by Orre, Kamphuis, Osborn, Melief, et al. (2014). To determine expression levels of the hPS1 transgene the following primers were used: FW: GAGGACAACCACCTGAGCAA and REV: ATCTTGCTCCACCACCTGC. Transcript levels were normalized to HPRT and GAPDH levels. (d) Normalized mRNA expression of hPS1 is higher in astrocytes of APPswePS1dE9 mice (n = 4 mice) compared to WT (n = 4 mice, the level of hPS1 was below the limit of detection in three samples). (e) Normalized mRNA expression of hPS1 is higher in microglia of APPswePS1dE9 mice (n = 4 mice) compared to WT (n = 4 mice, the level of hPS1 was below the limit of detection in three samples). FACS, fluorescence‐activated cell sorting; FPKM, fragments per kilobase million; GAPDH, glyceraldehyde‐3‐phosphate dehydrogenase; hAPP, humanized amyloid precursor protein; HPRT, hypoxanthine phosphoribosyltransferase; hPS1, human presenilin 1; MACS, magnetic‐activated cell sorting; MO, myelinating oligodendrocyte; NFO, newly formed oligodendrocyte; OPC, oligodendrocyte progenitor cell; PS1, presenilin 1; WT, wild‐type. *p < .05, ****p < .0001, Mann–Whitney test
3.2. Influence of genetic background on AD pathogenesis
The genetic background of mice can affect behavior and the timing of the formation of plaques (Hyman & Tanzi, 2019; Jackson et al., 2015; Neuner, Heuer, Huentelman, O'Connell, & Kaczorowski, 2019), and therefore should always be mentioned in manuscripts and taken into account when designing experiments. The original APPswePS1dE9 mouse was generated in F2 hybrids of C57BL/6J and C3H/HeJ (B6C3) mice. This mouse line has also been backcrossed to C57BL/6J, as this background is favorable for behavioral experiments to test memory and cognition.
The partial C3H/HeJ background is especially relevant concerning neuroinflammation and reactive gliosis research in this AD mouse model, as the C3H/HeJ mice have a mutation the in Toll‐like receptor‐4 gene (Tlr4). Normally, peripheral administration of the bacterial endotoxin lipopolysaccharide (LPS) induces a microglia response and reactive astrocytes. However, a spontaneous mutation in the Lps locus (defective LPS response: Lpsd/d) makes C3H/HeJ mice resistant to LPS (Barber, Perera, & Vogel, 1995; Poltorak et al., 1998). C57BL/6J mice do not have this mutation (Lpsn/n allele) and therefore have a normal LPS response. This Lps gene was later identified as the Tlr4 gene, and a single mutation in the third exon of the Tlr4 gene is responsible for the LPS resistance. Aside from their inability to respond to LPS, C3H/HeJ mice are developmentally and immunologically normal (Poltorak et al., 1998). Maintaining APPswePS1dE9 mice on a B6C3 background will result in a mixed litter with different Tlr4 genotypes. Mice can be homozygous (Lpsd/d), heterozygous (Lpsd/n), or wild‐type (Lpsn/n) for the Tlr4 mutation (Tahara et al., 2006). This is of importance for AD research, as TLR4 (lpsd/d) mediates the Aβ‐induced activation of microglia. Indeed microglia cultures of C3H/HeJ mice exposed to Aβ show a reduced production of nitrite and IL‐6 in comparison to microglia from wild‐type C3H/HeN mice (which do not have a mutation in the TLR4 gene) (Walter et al., 2007). TLR4 signaling is involved in the clearance and uptake of Aβ (Tahara et al., 2006) and studies indicate that this Tlr4 mutation increases the Aβ load in APPswePS1dE9mice, without affecting APP and PS1 levels (Song et al., 2011; Tahara et al., 2006). Moreover, the Lpsd/d‐APPswePS1dE9 mice are more vulnerable to cognitive deficits compared to Lpsn/n‐APPswePS1dE9 of the same age, as assessed by the Morris water maze (Song et al., 2011). Therefore, it is important to indicate the specific strain and for B6C3 mice the specific Lps genotype, when reporting results, as the background strain of APPswePS1dE9 mice affects amyloidosis, microglia activation, and cognition (Song et al., 2011; Tahara et al., 2006). Unfortunately, this information is not always specified in the scientific literature.
The Jackson Laboratory provides two strains of the APPswePS1dE9 mice, one on a mixed B6C3 background (B6C3‐Tg(APPswe, PSEN1dE9)85Dbo/Mmjax; Stock No: 34829) and one on a C57BL/6J congenic background (B6.Cg‐Tg(APPswe, PSEN1dE9)85Dbo/Mmjax; Stock No: 34832). For the studies selected in part 4 of this review, we indicate the background used when specified.
3.3. Neuropathological, neurobiological, and behavioral characteristics
Transgenic mouse models of AD with a mutation only in APP are characterized by a relatively slow accumulation of Aβ, whereas mice with mutations in both APP and PS1 accumulate Aβ faster, develop plaques earlier in life, and show behavioral impairments at younger ages (Jankowsky & Zheng, 2017). APPswePS1dE9 mice develop the main pathological hallmarks observed in AD patients, including aggregation of Aβ in plaques, reactive astrocytes, neuroinflammation, and cognitive impairments (Malm, Koistinaho, & Kanninen, 2011; Xiong et al., 2011). In the hippocampus of 12‐month‐old APPswePS1dE9 mice, even tau hyperphosphorylation is increased compared to wild‐type controls (Sui, Zhang, Dong, Xu, & Sun, 2019; Zhu, Xu, Sun, Zhu, & Sui, 2017). However, it is important to highlight that tau pathology in the form of neurofibrillary tangles is not observed in APPswePS1dE9 mice and other AD mouse models, except when the animals also express human mutant tau (Duyckaerts, Potier, & Delatour, 2008; Moreno‐Gonzalez, Estrada, Sanchez‐Mejias, & Soto, 2013).
In 3‐month‐old APPswePS1dE9 mice, an increase in soluble Aβ42 is detected in the posterior cerebral cortex (Van Tijn et al., 2012). Plaques are detected in 4‐month‐old mice in the (frontal) cortex and hippocampus (Garcia‐Alloza et al., 2006; Ruan et al., 2009) or in the cortex and hippocampus of 6‐month‐old mice (Jankowsky et al., 2004). In general, the Aβ pathology and glial activation progress during aging (Garcia‐Alloza et al., 2006; Kamphuis, Orre, Kooijman, Dahmen, & Hol, 2012; Orre, Kamphuis, Osborn, Jansen, et al., 2014; Ruan et al., 2009; Wirz et al., 2013). The age at which Aβ plaques are detected is depending on the method used, the sex of the animals, and the genetic background. For instance, the studies by Tahara et al. (2006) and Song et al. (2011) showed that in APPswePS1dE9 mice on a mixed B6C3 background, the Aβ burden is increased in LPSd/d and LPSd/n animals compared to LPSn/n mice (Song et al., 2011; Tahara et al., 2006). Also, sex influences the Aβ pathology in APPSwePS1dE9 mice (Jiao et al., 2016). The study by Jiao et al. (2016) shows that there are sex differences in other AD pathology markers as well. In 12‐month‐old female APPswePS1dE9 mice higher levels of the astroglial protein GFAP, the microglial marker CD45, and proinflammatory cytokines are found together with an increased Aβ burden and a lower level of synaptic markers compared to male mice (Jiao et al., 2016). These studies indicate that the genetic background and sex are important factors that should be considered when designing studies.
Plaque‐associated reactive astrocytes and activated microglia are detected in APPswePS1dE9 mice from the age of 6 months (Ruan et al., 2009; Van Tijn et al., 2012; Végh et al., 2014), making it a good model to study the development of both amyloidosis and gliosis.
Electrophysiologically, the first changes are observed at the onset of Aβ pathology. Impaired synaptic plasticity is detected as early as 3.5 months in the cortex of APPswePS1dE9 mice (Shemer et al., 2006). In the hippocampus, no major defects in glutamatergic transmission or plasticity are found in 3‐month‐old APPswePS1dE9 mice. Only the induction of transient LTP is affected, in an age‐independent manner (Volianskis et al., 2010). Loss of synapses or neurons could mediate impairment in synaptic plasticity. While some studies indicate a decrease in the number of presynaptic and postsynaptic terminals in the hippocampus as early as 4 months (Hong et al., 2016) or 7 months (Ding et al., 2008; Woo et al., 2015), others found no changes in synaptic density in the cortex at 3 or 6–7 months (Shemer et al., 2006; Van Tijn et al., 2012) or even an increase in the number of synaptic contacts in the CA1 region of the hippocampus of 12‐month‐old APPswePS1dE9 mice (West, Bach, Søderman, & Jensen, 2009).
Cognitive impairments are first observed in 3‐month‐old APPswePS1dE9 mice in a contextual fear memory task and in 6‐month‐old mice in the Morris water maze spatial memory task (Végh et al., 2014). In other studies, impairments have been found in a spatial working memory task in 4‐month‐old APPswePS1dE9 mice (Park et al., 2006), and 6‐month‐old APPswePS1dE9 mice made more mistakes and showed longer latencies in the radial arm water maze (RAWM) test compared to age‐matched wild‐type mice (Xiong et al., 2011). Whereas another study found no differences in spatial learning between wild‐type and APPswePS1dE9 mice at the age of 7 months, the performance of APPswePS1dE9 mice in the RAWM was impaired at the age of 12–13 months (Volianskis et al., 2010). The first changes in cognition are detected by hippocampal‐dependent cognitive tests, including the contextual fear memory task, of which the performance inversely correlates with regions in which Aβ pathology was found.
3.4. Reactive astrocytes
Due to their active and crucial function in the brain, changes in astrocytes in pathological conditions are no longer considered as a nonspecific or passive secondary reaction (Escartin et al., 2019). A variety of pathological stimuli can induce morphological, molecular, and functional changes in astrocytes (Escartin et al., 2019; Hol & Pekny, 2015; Pekny et al., 2016; Escartin et al., 2021). Different terms, including (astro)gliosis, astrocyte activation, reactive astrocytes, and astrocyte reactivity are used in literature to describe these astroglial changes (for a comprehensive review, Escartin et al., 2019, 2021). Detrimental effects on brain function can be mediated by the loss of homeostatic functions and/or gain of functions related to the reactive state of astrocytes (Osborn et al., 2016). Nevertheless, beneficial functions of reactive astrocytes have also been found. Reactive astrocytes form a glial scar to restrict damage, repair the blood–brain barrier, and provide energy substrates (Buffo, Rolando, & Ceruti, 2010; Sofroniew, 2015). Preventing the reactive astrocytes‐associated increase in GFAP (sometimes together with vimentin) can exacerbate inflammation and pathology associated with autoimmune encephalomyelitis (Liedtke, Edelmann, Chiu, Kucherlapati, & Raine, 1998), stroke (Li et al., 2008; Liu et al., 2014), and even Aβ accumulation (Kraft et al., 2013), although the latter could not be repeated by us in a similar experiment (Kamphuis et al., 2015).
Plaque‐associated reactive astrocytes are detected in APPswePS1dE9 mice at the age of 6 months, based on the increased expression of GFAP (Ruan et al., 2009; Van Tijn et al., 2012; Végh et al., 2014). Despite the widespread presence of reactive astrocytes in APPswePS1dE9 mice at late stages of AD pathology, no evidence has been found for proliferation of astrocytes (Kamphuis et al., 2012). At the age of 15–18 months, isolated cortical astrocytes revealed a proinflammatory phenotype and a reduced expression of genes involved in neuronal support and communication (Orre, Kamphuis, Osborn, Jansen, et al., 2014). The expression of immunoproteasome subunits is increased in astrocytes during ageing, starting in 9‐ to 12‐month‐old APPswePS1dE9 mice (Orre et al., 2013). Elevated resting intracellular calcium levels and more frequent calcium transients were found in cortical astrocytes after the development of senile plaques (6–8 months). These astrocytic calcium transients originated near plaques, occurred synchronously over longer distances, and were uncoupled from neuronal activity (Kuchibhotla, Lattarulo, Hyman, & Bacskai, 2009). When astrocytes become reactive they enhance the release of various gliotransmitters, including GABA, glutamate, and ATP (Jo et al., 2014; Yi et al., 2016). The astroglial GABA immunoreactivity is inversely correlated with the distance from amyloid plaques (Jo et al., 2014). These findings indicate that there are clear morphological, molecular, and functional changes in reactive astrocytes in APPswePS1dE9 mice.
3.5. Advantages and limitations
Although no animal model fully mimics the pathology and cognitive impairments seen in human AD patients, AD mouse models have been invaluable in AD research. Each AD (mouse) model has its strengths and limitations (Sasaguri et al., 2017). AD mouse models enable investigating astrocyte function in an intact system and studying behavioral changes. However, it should be taken into account that there are species differences between mouse and human astrocytes at a molecular and functional level (Oberheim et al., 2009; Zhou et al., 2020). The characterization of astrocytes acutely isolated from human brain tissue (Zhang et al., 2016) and induced pluripotent stem cell (iPSC)‐derived astrocytes help to address these species differences (Mungenast, Siegert, & Tsai, 2016; Sullivan & Young‐Pearse, 2017). Validation of these findings in a complete organism, to assess its effect on AD pathology and on memory and cognition, remains an essential step in therapy development for AD. This makes AD mouse models extremely valuable.
A major advantage of the APPswePS1dE9 mouse model is that it enables efficient crossing with other mice, as the transgenes were incorporated at the same chromosomal location and co‐segregate as a single locus (Jankowsky et al., 2001). APPswePS1dE9 mice develop the main pathological hallmarks as observed in AD patients; age‐related aggregation of Aβ in plaques, neuroinflammation, reactive gliosis, and cognitive impairments (Malm et al., 2011; Wirz et al., 2013; Xiong et al., 2011). Moreover, due to the expression of both APPswe and PS1dE9, the mice develop Aβ pathology at an early age (Borchelt et al., 1997). However, as the APPswePS1dE9 mice overexpress mutant transgenes from an early age (embryonic), it might affect brain development and function. If and how overexpression of humanized APPswe and human PS1dE9 affects endogenous APP‐ and/or PS1‐processes beyond Aβ production (Sections 2.1 and 2.3) and influence AD pathogenesis remains largely unstudied. Finally, it must be noted that there are no AD patients with mutations in both APP and PS1. While the APP and PS1 mutations used in the APPswePS1dE9 mouse model are linked to FAD (Mullan et al., 1992; Perez‐Tur et al., 1995), FAD accounts for <5% of all AD patients (Malm et al., 2011).
In AD patients, the earliest pathological changes are observed in the medial temporal lobe (entorhinal cortex and hippocampus; Braak & Braak, 1991). This spatial and temporal expression is not mimicked by the transgene expression that is driven by the PrP promoter. However, also in the APPswePS1dE9 mice, Aβ pathology is detected from an early age in the cortex and hippocampus (Garcia‐Alloza et al., 2006; Ruan et al., 2009; Van Tijn et al., 2012). In AD knock‐in models, in which the endogenous mouse APP gene is humanized, the target gene is under the control of its native promoter. While this leaves the temporal, spatial, and gene expression level of APP undisturbed (Guo et al., 2013; Saito et al., 2014), these models also do not exhibit tau pathology. Another potential disadvantage of these knock‐in models is the less severe pathology development and minor behavioral changes (Sasaguri et al., 2017).
Variable premature death rates, between 10% and 35%, have been reported for APPswePS1dE9 mice. Although cardiomyocyte contractile dysfunction was found in APPswePS1dE9 mice, this was not linked to the mortality rate (Turdi et al., 2009). Overall, gross pathological abnormalities were not observed in these mice (Xiong et al., 2011). One study using female mice indicates that before the age of 6 months, about 35% of the APPswePS1dE9 mice kept as a hemizygote on a C57BL/6J congenic background died. Seizures might be a factor accounting for the premature death of APPswePS1dE9 mice (Minkeviciene et al., 2009; Xiong et al., 2011). Indeed, patch–clamp electrophysiology identified that a depolarized resting membrane potential contributed to the increased neuronal network excitability observed in this transgenic line (Minkeviciene et al., 2009). The mortality rate seems to be highly variable, as in our group it is around 10% for APPswePS1dE9 mice at 6 months of age (personal communication Willem Kamphuis). Whether the variable mortality rate is due to the specific background used of the APPswePS1dE9 mice, the environment in which the mice are housed including the type of caging, noise levels, food, or social housing is not clear.
Despite its limitations, the APPswePS1de9 mouse model is overall a good model to study amyloidosis and the development of reactive astrocytes and its consequences, which forms the focus of this review.
4. SYSTEMATIC REVIEW: REACTIVE ASTROCYTES AS A TREATMENT TARGET IN THE APPSWEPS1DE9 MOUSE MODEL
Astrocytes display a reactive phenotype in the CNS of AD patients and APPswePS1dE9 mice (Section 3.4); meaning hypertrophic soma and a pronounced upregulation of the intermediate filament cytoskeleton. How this affects astrocyte normal function (Box 2) and astrocyte–neuron interactions is just beginning to be unraveled.
To determine how intervening with reactive astrocytes could affect AD pathogenesis and cognition, we have systematically reviewed studies on the APPswePS1dE9 mouse model in which astrocytes were targeted by either direct or indirect interventions. Besides the effect on astrocytes, we also included the effect on Aβ burden, microglia activation, synaptic density, and memory. When indicated in the study, we included the specific APPswePS1dE9 strain purchased, including genetic background, and which sex was used. The review is based on a PubMed search, updated last on April 2, 2021, with the following search terms: ((((Alzheimer*) AND mouse model*) NOT (5xFAD OR 2xKI OR 3xTg OR Tg2576 OR TgCRND8 OR TASTPM OR ARTE10 OR EAE OR NL‐F OR v717I OR J20 OR JNPL3 OR P301L OR P301S OR PS19 OR APP23 OR PDAPP OR TAPP)) AND (reactive astrocyt* OR astrocytosis OR astrogliosis OR gliosis)) AND (“2001”[Date ‐ Publication]: “3000”[Date ‐ Publication]). We only included articles written in English. This search term yielded 314 research articles (Figure 3). We screened the title and abstract of these research articles and selected studies referring to an APP/PS1 model or an unspecified AD mouse model. Selecting the studies that found an effect of astrocytes and/or gliosis or used an astrocyte‐specific intervention resulted in the inclusion of 101 articles. After screening the full text, 55 articles were selected that used the APPswePS1dE9 mouse model. In nine studies, astrocytes were directly targeted in APPswePS1dE9 mice either by blocking the negative functional changes of reactive astrocytes, by inducing gene expression in astrocytes, or by preventing astrocytes to become reactive (Table 1). We found 46 studies in which astrocytes were indirectly affected by various interventions. We divided these studies into five categories based on the main target of the intervention: Aβ, inflammation, transmitter release, neuroprotection, or environmental risk factors (Table 2). Altogether the results from the studies indicate that targeting astrocytes in AD should be included in developing novel therapies.
FIGURE 3.
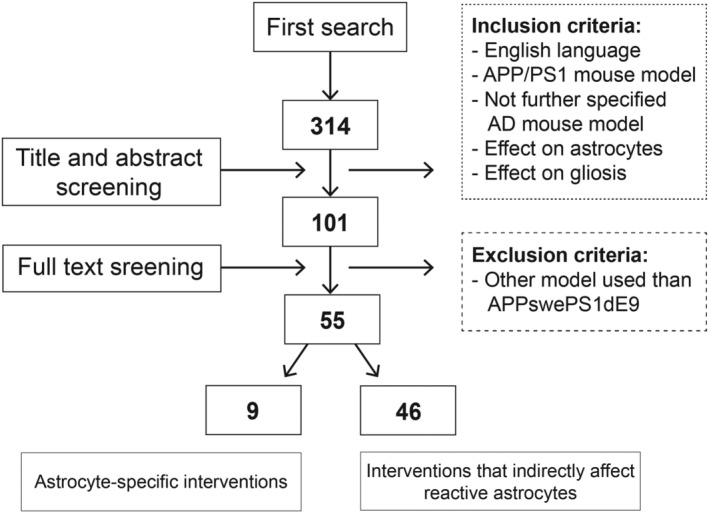
Flowchart of study selection. Refer to Section 4 for the search terms used in the first search
TABLE 1.
Astrocyte‐specific interventions in APPswePS1dE9 mice
| Intervention | Target | Age | Sex | Background | Amyloid‐beta | Astrocytes | Microglia | Synaptic density | Memory | References |
|---|---|---|---|---|---|---|---|---|---|---|
| Gfap‐Cx43 −/− ‐APPswePS1dE9 a | Cx43 | 9 mo | F/M | – | – |
ATP release↓(ATPlite) Glutamate release↓ (CE‐LIF) GABA release↔ (CE‐LIF) |
– | Hip: RTN‐3 dystrophic neurites↓ (IHC) | – | Yi et al. (2016) |
| Gfap‐Cx43 −/− ‐APPswePS1dE9 b | Cx43 | 12 mo | – | BALB/c × C57BL/6NHsd × 129S7/SvEvBrd‐Hprtb‐m2 | Cx and Hip: Aβ number↔ (IHC, 6E10) |
Cx and Hip: GFAP number↓ (IHC) GFAP, GLAST and MEGF10↓ (qPCR and WB) |
– | Hip: Syn↔, PSD95↑ (WB) |
MWM↑ NOR↑ |
Ren, Zhang, and Wang (2018) |
|
GFAP‐iE3‐APPswePS1dE9 c Induced from birth |
ApoE | 6 mo1 or 9 mo2 | F/M | C57BL/6J × C3H/HeJ |
Cx: (in)soluble Aβ 40, Aβ 42 ↔ (ELISA) Cx: Aβ↔ (IHC) |
Cx and Hip: GFAP↓ 2 (IHC) | Cx and Hip: Iba1↔ 2 (IHC) | Cx: Syn↔ 2, PSD95↑ 2 (WB) | – | Liu et al. (2017) |
|
GFAP‐iE4‐APPswePS1dE9 c Induced from birth1,2 or 6 mo3 |
ApoE | 6 mo1, 9 mo2 or 9 mo3 | F/M | C57BL/6J × C3H/HeJ |
Cx: (in)soluble Aβ 40, Aβ 42 ↑ 1,2 (ELISA) Cx: (in)soluble Aβ 40, Aβ 42 ↔ 3 (ELISA) Cx and Hip: Aβ↑ 1,2 (IHC) Cx and Hip: Aβ↔ 3 (IHC) |
Cx and Hip: GFAP↑ 1,2 Cx and Hip: GFAP↔ 3 (IHC) |
Cx and Hip: Iba1↑ 2 (IHC) | Cx: Syn↔ 2, PSD95↔ (WB) | – | Liu et al. (2017) |
| AAV‐GFAP104‐DTR injection in hippocampus at 10–12 mo | Oxidative stress | 11–13 mo | F/M | C57BL/6J | – | Hip: GFAP intensity↑ (IHC) | Hip: Iba1 area↑ (IHC) | NeuN number↓ (IHC) | NPR↓ | Chun et al. (2020) |
| Intracerebroventricular injection of AAV‐GFAP‐CLU at 2 d | Clusterin | 8 mo | F/M | C57BL/6J × C3H/HeJ |
Cx: (in)soluble Aβ 40, Aβ 42 ↓ (ELISA) Cx and Hip: Aβ area↓ (IHC, X‐34) |
Cx: GFAP intensity↓ (IHC) Cx: Gfap↓ (qPCR) |
Cx: Iba1 intensity↓ (IHC) | Cx and Hip: Dystrophic neurites↓ (IHC) | – | Wojtas et al. (2020) |
| AAV‐gfaABC1D‐SOCS3 injection in hippocampus at 3–4 mo1 or injection at 15 mo2 | STAT3 | 9–10 mo1 or 16 mo2 | M | C57BL/6J × C3H/HeJ |
Hip: Aβ number↓ 1 (IHC, BAM10) Hip: Aβ 40, Aβ 42 ↔ 1 (ELISA) |
Hip: GFAP intensity↓ (IHC and WB) Vim intensity↓ 1 (IHC) |
Hip: Iba1 number per plaque↔ 1 (IHC) | – | MWM↑ 1 | Ceyzériat et al. (2018) |
|
Cx43‐STAT3 −/− ‐APPswePS1dE9 b Induced from 6 wk |
STAT3 | 8 mo | F/M | C57BL/6N |
Aβ area↓ (IHC, IC16, and ThS) Soluble Aβ 40, Aβ 42 ↓ (ELISA) |
Cx and Hip: GFAP area↔ (IHC) Peri‐plaque astroglial volume↑ |
Cx and Hip: Iba1 area↔ (IHC) Number of microglia branches and junctions↑ |
– | MWM↑ | Reichenbach et al. (2019) |
| APPswePS1dE9 c ‐GFAP −/− vimentin −/− | Intermediate filament proteins | 4 mo1, 8 mo2 and 12 mo3 | – | B6C3 × C57BL × 129SV × 129Ola |
Cx and Hip: Aβ area↑ 2,3 (IHC, HJ3.4 and X‐34) Cx: insoluble Aβ 40, Aβ 42 ↔ 1 Cx: Insoluble Aβ 40 ↑ 3 , Aβ 42 ↑ 2 (ELISA) |
Plaque‐astrocyte overlap↓ 3 GFAP number↔ 3 (IHC) |
Plaque‐microglia overlap↑ Iba1 number↑ 2 (IHC) Iba1↑ 3 (qPCR) |
ECx: RTN‐3↑ 3 (IHC) | – | Kraft et al. (2013) |
| APPswePS1dE9 c ‐GFAP −/− vimentin −/−1 and APPswePS1dE9 b ‐GFAP −/−2 | Intermediate filament protein(s) | 6 mo, 9 mo, and 15 mo | – | C57BL/6 × 129Sv × 129Ola | Aβ area↔ (IHC, 6E10) |
Plaque‐astrocyte overlap↓ Vim area↑ 2 (IHC) |
Iba1 number↔ (IHC) | – | – | Kamphuis et al. (2015) |
Abbreviations: General—Specified interventions or ages indicated with 1–3. When not indicated: same effect for all interventions or ages. When a specific group is not indicated: read‐out was not determined for that group; ↑, Increased or improved (Memory) compared to control of same age; ↓, decreased or impaired (Memory); ↔, no (significant) difference; –, not studied; d, day(s); mo, month(s); wk, week(s). Intervention and Target—Different APPswePS1dE9 strains indicated with a–c. aAPPswePS1dE9 mice, specific strain not indicated; bMMRRC Stock No: 34832‐JAX–C57BL/6J; cMMRRC Stock No: 34829‐JAX–C57BL/6J × C3H/HeJ. AAV, adeno‐associated virus‐based vectors; ApoE, apolipoprotein E; CLU, clusterin; Cx43, Connexin43; DTR, diphtheria toxin receptor; gfaABC1D, promoter to mediate astrocyte‐specific expression; GFAP, glial fibrillary acidic protein; iE3, inducible human ApoE3 expression; iE4, inducible human ApoE4 expression; SOCS3, suppressor of cytokine signaling 3; STAT3, signal transducer and activator of transcription 3. Age —at read‐out. Sex—Female (F), Male (M), or both (F/M). –, not indicated. Background: Genetic background of mice used in the study. x, Crossed with. Brain Regions—Cx, cortex; ECx, entorhinal cortex; Hip, hippocampus. Techniques—ATPlite, Luciferin‐luciferase bioluminescence assay; CE‐LIF, capillary electrophoresis with laser‐induced fluorescence; ELISA, enzyme‐linked immunosorbent assay; IHC, immunohistochemistry; MWM, Morris water maze; NOR, novel object recognition; NPR, novel place recognition; qPCR, quantitative polymerase chain reaction; WB, western blot. Genes, proteins, and transmitters—6E10, Aβ1–16 antibody; Aβ, amyloid‐beta; ATP, adenosine triphosphate; BAM10, Aβ1–40 antibody; GABA, gamma‐aminobutyric acid; GFAP, glial fibrillary acidic protein; GLAST, glutamate–aspartate transporter; HJ3.4, Aβ1–13 antibody; Iba1, ionized calcium‐binding adapter molecule 1; IC16, Aβ1‐16 antibody; MEGF10, multiple EGF‐like domains 10; PSD95, postsynaptic density protein 95; RTN‐3, reticulon‐3, marker of neuritic dystrophy; Syn, synaptophysin; ThS, thioflavin‐S; Vim, vimentin; X‐34 dye, fluorescent amyloid‐specific dye.
TABLE 2.
Interventions in APPswePS1dE9 mice indirectly affecting reactive astrocytes
| Intervention | Target | Age | Sex | Background | Amyloid‐beta | Astrocytes | Microglia | Synaptic density | Memory | Reference |
|---|---|---|---|---|---|---|---|---|---|---|
| Amyloid‐beta | ||||||||||
| Monthly immunization targeting Aβ42 from 3 mo | Aβ42 | 15‐16 mo | – | C57BL/6J × C3H/HeJ |
Cx and Hip: Aβ 42 area↓ (IHC) Aβ 42 ↓ (ELISA) |
Cx and Hip: GFAP↓ (IHC) | – | – | – | Qu et al. (2007) |
| Daily oral administration of Tannic acid from 6 mo | β‐secretase | 12 mo | F/M | C57BL/6J × C3H/HeJ† |
CC, ECx, and Hip: Aβ number and size↓ (IHC, 4G8) Wb: Aβ 40, Aβ 42 ↓ (ELISA) |
CC, ECx, and Hip: GFAP area↓ (IHC) | CC, ECx, and Hip: Iba1 area↓ (IHC) | – |
NOR↑ MWM↑ |
Mori et al. (2012) |
| c‐fms‐ACE 10/10 ‐APPswePS1dE9 c | Aβ42 | 5 mo1, 7 mo2, 8.5 mo3, 11–12 mo4 or 13 mo5 | F/M | C57BL/6 |
Cx and Hip: Aβ↓ 3 (IHC, 6E10 and ThS) Soluble Aβ 40 ↓ 2 Soluble Aβ 42 ↓ 1,2,5 (ELISA) |
GFAP area and number↓ 2,5 (IHC) | – | – | BM↑ 4 | Bernstein et al. (2014) |
| Weekly injection with low (L) or high (H) doses of IVIG 1, Oli‐Nabs 2 or Blue‐Nabs 3 | Aβ42 oligomers | 8–9 mo | M | C57BL/6J |
Aβ↓ 2H,3H (IHC, 6E10) Insoluble Aβ 40 ↓ 3H, ↔ 1,2,3L, Aβ 42 ↓ 3H, ↔ 1,2,3L Soluble Aβ 40 ↓, Aβ 42 ↓ H,2L, ↔ 1L,3L (ELISA) |
Cx: GFAP↓ 2,3H, ↔ 1,3L Hip: GFAP area↓ 2, ↔ 1,3 (IHC) |
Cx and Hip: Iba1↓ 2,3H, ↔ 1,3L (IHC) | Syn↑ 2H,3H, ↔ 1,2L,3L (WB) |
MWM↑ 2L,3H, ↔ 1,2H,3L NOR↑ |
Wang et al. (2016) |
| Bone marrow transplantation of WT1 or c‐fms‐ACE 10/10 2 mice at 2 mo | Aβ42 | 9 mo | F/M | C57BL/6J |
Cx: Aβ area↓ 2 (IHC, 6E10) Cx: Soluble Aβ 42 ↓ 2 (ELISA) |
Cx: GFAP area↓ 2, number↓ (IHC) | – | – | – | Koronyo‐Hamaoui et al. (2020) |
| Oral administration of BPN‐15606 from 3 mo1 or 6 mo2 | γ‐secretase | 6 mo1 or 9 mo2 | F | C57BL/6J × C3H/HeJ |
Cx: Aβ number and size↓ 1 (IHC, 82E1) Hip: Aβ size↓ 1 Cx and Hip: Aβ↔ 2 (IHC) Insoluble Aβ 38, Aβ 40 and Aβ 42 ↔ Soluble Aβ 38, Aβ 40 ↔ and Aβ 42 ↓ 1, ↔ 2 (ELISA) |
Cx and Hip: GFAP area and size↔ |
Cx and Hip: Iba1 area and number↓ 1, ↔ 2 (IHC) |
– | MWM↑ 1, ↔ 2 | Prikhodko et al. (2020) |
| Injection of IDOL ASO at 3 mo and 6 mo | Aβ | 8–9 mo | M | C57BL/6J |
Aβ area↓ (IHC, 82E1 and X‐34) (In)soluble Aβ 40, Aβ 42 ↓ (ELISA) |
GFAP area↓ (IHC) |
Iba1 intensity↔ Iba1 soma size↓ (IHC) |
– |
MWM↑ Contextual and cued FC↔ |
Gao et al. (2020) |
| Ad libitum access to Gemfibrozil 1 or Wy14643 2 via drinking water | Aβ | 8–10 mo | F/M | – |
Aβ↓ (IHC, 4G8) Cx and Hip: (in)soluble Aβ 40, Aβ 42 ↓ (ELISA) |
Cx and Hip: GFAP↑ (IHC and WB) | Cx and Hip: Iba1↑ (IHC and WB) | Cx and Hip: Syn and PSD95↑ (WB) |
MWM↑ Open Field test↑ |
Luo et al. (2020) |
| Gal‐3 +/− ‐APPswePS1dE9 c | Aβ | 7 mo | F/M | C57BL/6J × 129/Sv × SJL |
Hip: Aβ↓ (IHC) Aβ oligomers↓ (WB) |
Hip: GFAP↓ (IHC) | Hip: Iba1↓ (IHC) | – | MWM↑ | Tao et al. (2020) |
| Daily oral intake of Gallic Acid from 12 mo | α/β‐secretase | 18 mo | M | C57BL/6J | Cx, ECx, and Hip: Aβ area and number↓ (IHC, 4G8) | Cx, ECx, and Hip: GFAP area↓ (IHC) | Cx, ECx, and Hip: Iba1 area↓ (IHC) | – |
NOR↑ RAWM↑ YM↑ |
Mori et al. (2020) |
| Inflammation | ||||||||||
| Daily oral intake of PDTC from 9 mo | NF‐κB | 16 mo | – | C57BL/6J × C3H/HeJ † |
Cx: Aβ area↔ (IHC, pan‐Aβ, and ThS) Hip: (in)soluble Aβ 40, Aβ 42 ↔ (ELISA) |
Cx: GFAP intensity↔ (IHC) Cx: GLT‐1↑ (WB) |
Cx: CD11b↔ Cx: CD45 intensity↔ (IHC) |
– | MWM↑ | Malm et al. (2007) |
| LPS d/d ‐APPswePS1dE9 | Innate immune response | 13–15 mo | – | C57BL/6J × C3H/HeJ | – |
Cx: GFAP area↑ (IHC) GFAP↑ (WB) |
Cx: CD11b area↑ (IHC) CD11b↔ (WB) CD45↔ (IHC and WB) |
– | – | Jin, Kim, Maxwell, Li, and Fukuchi (2008) |
| Daily injection of PDTC | NF‐κB | 7–12 mo | – | C57BL/6J × C3H/HeJ | Cx and Hip: Aβ 40 ↔, Aβ 42 ↑ (ELISA) | Cx and Hip: GFAP↓ (IHC and WB) | – | – | – | Zhang et al. (2009) |
| Daily injection of Valproic acid from 7 mo | Inflammation/neuroprotection | 8 mo | – | C57BL/6J × C3H/HeJ |
Cx and Hip: Aβ area and number↓ (IHC, 4G8) Soluble Aβ 40, Aβ 42 ↓ (ELISA) |
Hip: GFAP number↓ (IHC) | Hip: Iba1 number↓ (IHC) | – | MWM↑ | Xuan et al. (2015) |
| Immunization with GA 1, Mo BM 2 or GA and Mo BM 3 from 10 mo | Myelin‐derived antigens/inflammation | 12 mo | M | C57BL/6J |
Cx and Hip: Aβ area↓ (IHC, 4G8, and ThS) Aβ 40, Aβ 42 ↓ (ELISA) |
Cx: GFAP area and number↓ (IHC) | – | Hip: VGluT1↑ (IHC) | BM↑ | Koronyo et al. (2015) |
| TRPA1 −/− ‐APPswePS1dE9 c | Inflammation/Calcium signaling | 8 mo | – | C57BL/6J | Aβ oligomers, monomers↓ (WB) | GFAP↑ (WB) | – | – |
MWM↑ YM↑ |
Lee et al. (2016) |
| NFATc2 −/− ‐APPswePS1dE9 c | Cytokines | 8 mo | M | C57BL/6J × 129X1/SvJ | Cx: Aβ intensity↔ (IHC, 4G8 and Campbell‐Switzer staining) | Cx: GFAP intensity↓ (IHC) | Cx: Iba1↔ and CD68 intensity↓ (IHC) | Hip: PSD95↓ and Syn↔ (WB) | – | Manocha et al. (2017) |
| Early postnatal antibiotic treatment for 1 wk from P14 | Inflammation | 6.5 mo | M | C57BL/6J × C3H/HeJ |
Aβ area↓ (IHC, 3D6) (In)soluble Aβ 40, Aβ 42 ↔ (ELISA) |
GFAP number↓ (IHC) | Iba1 number↓ (IHC) | – | – | Minter et al. (2017) |
| Daily administration of Humulus Japonicus from 5 mo | Oxidative stress/inflammation | 7.5 mo | F/M | C57BL/6J × C3H/HeJ | Cx: Aβ area↓ (IHC, BAM10) | Cx: GFAP area↓ (IHC) | Cx: Iba1 number↓ (IHC) | – |
NOR↑ YM↑ |
Park et al. (2017) |
| CCR3 −/− APPswePS1dE9 b | CCR3 | 12 mo | M | C57BL/6J × – | Cx and Hip: Aβ number↓ (IHC, 6E10) | Cx and Hip: GFAP number↓ (IHC) |
Cx and Hip: Iba1 number↓ (IHC) |
Hip: Drebrin and PSD95↑ (WB) Hip: Synapse number↑ (EM) |
MWM↑ | Zhu et al. (2017) |
| Weekly immunization with anti‐CD49d 1 or IgG control2 from 9 mo | Proinflammatory microglia | 10 mo | F | C57BL/6J |
Cx: (In)soluble Aβ 40, Aβ 42 ↔ (WB) Wb: Aβ↔ (IHC, 4G8) |
Cx: GFAP↓ 1, ↔ 2 (WB) Wb: GFAP↓ 1, ↔ 2 (IHC) |
Wb: CD68 and Iba1↓ (IHC) | Cx: Syn↔ and PSD95↓ (WB) | – | Manocha, Ghatak, Puig, and Combs (2018) |
|
ApoA‐I −/− ‐APPswePS1dE9 c KO was compared to hemizygous group |
ApoA‐I | 12 mo | F/M | C57BL/6J |
Cx: Aβ↑ (IHC) Hip: Aβ↔ (IHC) Hemisphere: (In)soluble Aβ 40, Aβ 42 ↔ (ELISA) |
Cx and Hip: GFAP area↑ (IHC) | – | – | Contextual and cued FC↔ | Button et al. (2019) |
| Daily oral administration of UA for 13 d from 7 mo | Inflammation | 7.5 mo | F | C57BL/6J × C3H/HeJ | Cx and Hip: Aβ 40, Aβ 42 number and area↓ (IHC) | Cx and Hip: GFAP number↓ (IHC) | Cx and Hip: Iba1 number↓ (IHC) | – | MWM↑ | Gong et al. (2019) |
| Two daily injections of YM344031 from 10 mo | CCR3 | 12 mo | M | C57BL/6J × C3H/HeJ | Hip and NCx: Aβ number↓ (IHC) | Cx: GFAP number↓ (IHC) | Cx: Iba1 number↓ (IHC) | Hip: Syn, drebrin and PSD95↑(WB) | MWM↑ | Sui et al. (2019 ) |
| pdMCAO 1 or sham‐operated 2 daily injection of ONO‐8713 for 14 d from 4.5 mo | EP1 receptor | 5 mo | M | C57BL/6J × C3H/HeJ | Cx: Aβ 40, Aβ 42 ↔ (ELISA) |
Cx: GFAP↔ Hip: GFAP↓ 2, ↔ 1 (IHC) |
Cx: Iba1↓ 1, ↔ 2 Hip: Iba1↔ (IHC) |
– | PAT↑ 1, ↔ 2 | Mendes et al. (2020) |
| Weekly injection of GA from 20 mo | Myelin‐derived antigens/Inflammation | 22 mo | F/M | C57BL/6J | Cx, ECx, and Hip: Aβ area↓ (IHC, 6E10) | Cx, ECx, and Hip: GFAP area↓ (IHC) | Cx, ECx, and Hip: Iba1 area↓ (IHC) |
ECx: PSD95↑ Hip and Wb: PSD95↔ (IHC) Syn↑ (MS) |
BM↑ | Doustar et al. (2020) |
| Transmitter release | ||||||||||
| Daily oral intake of selegiline for 3 d1, 1 wk2 or 3 wk3 from 10 mo | MAO‐B | 10–12 mo | F/M | C57BL/6J and C3H/HeJ | Aβ↔ 2 (WB) | GABA within GFAP positive area↓ 1 (IHC) | – | – |
MWM↔ 3 PAT↑ 2 |
Jo et al. (2014) |
| Boldine in drinking water from 6 mo | Hemichannel | 9 mo | F/M | C57BL/6J | Cx and Hip: Aβ area↔ (IHC) |
GFAP↔ (WB) d‐serine and GABA release↔ ATP and glutamate release↓ (CE‐LIF) |
Iba1↔ (WB) | Hip: RTN‐3 dystrophic neurites↓ (IHC) | – | Yi et al. (2017) |
| Daily oral intake of selegiline for 3 d1, 4 wk2 or KDS2010 from 8 mo3 | MAO‐B | 8 mo1, 9 mo2, or 13 mo3 | F/M | C57BL/6J × C3H/HeJ | – |
GABA within GFAP positive are↓ 1,3, ↔ 2 GFAP↓ 3 (IHC) |
– | – |
PAT↑ 1,3 , ↔ 2 MWM↑ 3 |
Park et al. (2019) |
| Neuroprotection | ||||||||||
|
Daily injection of Tetrahydrohyperforin (IDN5706) from 12 mo |
Inflammation, oxidative activity, and monoamines/neurotransmitter uptake | 13 mo | M | C57BL/6J × C3H/HeJ |
Hip: Aβ area↔ (IHC, Aβ1‐17) Aβ area↓ (IHC, ThS) |
Hip: GFAP number↔ Hip: GFAP intensity soma and soma size↓ (IHC) |
– | – | MWM↑ | Cerpa et al. (2010) |
| Daily administration of peroxisome proliferator WY 1 or 4‐PB 2 via drinking water from 7 mo | Peroxisomes | 9 mo | F/M | C57BL/6J × C3H/HeJ | Aβ area↓ (IHC, 4G8 and ThS) |
Cx: GFAP area↔ 1, ↓ 2 Hip: GFAP area↓ (IHC) |
Cx: CD11b area↔ 1, ↓ 2 Hip: CD11b area↓ (IHC) |
Hip: Syn, VAMP1/2 and VGlut1 ↔ GluR2, PSD95 and NR2B↑ (WB) |
MWM↑ NOR↔ |
Inestrosa et al. (2013) |
| Biweekly injection of recombinant NOIs vaccine from 2 to 3 mo | Neurite outgrowth | 5 mo | F/M | C57BL/6J | Cx and Hip: Aβ area and number↓ (IHC, 6E10) Hemisphere: Aβ↓ (WB, 6E10) | Cx and Hip: GFAP area↓ (IHC) | – | – | MWM↑ | Zhang et al. (2013) |
|
Endo‐B1 −/− APPswePS1dE9 c |
Apoptosis, autophagy, and mitochondrial function | 6 mo1 or 12 mo2 | M | C57BL/6J |
Cx and DG: Aβ area and number↑ 1 (IHC, 6E10) Soluble Aβ 40, Aβ 42 ↑ 1 (Luminex assay) |
Cx and Hip: GFAP area↑ 1 (IHC) |
– | CA3: Syn↓ (IHC) | MWM↓ | Wang et al. (2015) |
| Cofilin +/‐ ‐APPswePS1dE9 a | Filamentous actin | 7 mo | F/M | – × 129 5/SvEvBrd × C57BL/6J | – | Hip: GFAP intensity↓ (IHC) | – | Hip: PSD95 and Syn intensity↑ (IHC) |
Contextual FC↑ Cued FC↔ |
Woo et al. (2015) |
| Perinatal choline‐supplemented diet | Choline | 6 mo1, 9 mo2 or 12 mo3 | F/M | C57BL/6J × C3H/HeJ |
Hip: Aβ40, Aβ42 area↓2,3 Hip: Aβ 40, Aβ 42 number↓ 2,3 (IHC) Soluble Aβ↓ 2F,3M, ↔ 3F,2M (WB, 6E10) Soluble Aβ 40, Aβ 42 ↔ 1,2M,3F, ↓ 2F,3M (ELISA) |
GFAP↓ 2F , ↔ 3F,2M,3M (WB) | – | – | – | Mellott et al. (2017) |
| Daily injection of ASS234 from 10 wk | AChE/MAO | 4.5 mo | M | – |
Cx: Aβ↓ Hip: Aβ↔ (IHC, ThS) |
Cx: GFAP number↓ (IHC) | Cx: Iba1 number↓ (IHC) | – | – | Serrano et al. (2017) |
| Daily injection of liraglutide 1 or palm 11 ‐PrRP31 2 from 7 to 8 mo | GLP‐1 | 9–10 mo | M | C57BL/6J |
CX: Aβ area↔ Hip: Aβ area↔ 1, ↓ 2 (IHC) |
Cx: GFAP area↓ Hip: GFAP area↔ 1, ↓ 2 (IHC) |
Cx: Iba1 area↔ Hip: Iba1 area↔ 1, ↓ 2 (IHC) |
Hip: Syn↑ 2 (WB) | – | Holubová et al. (2019) |
| Pyk2 −/− ‐APPswePS1dE9 c | Pyk2 | 12 mo | F/M | C57BL/6J | Hip: Aβ area↔ (IHC) | Hip: GFAP area↓ (IHC) | Hip: Iba1 area↔ (IHC) | DG: PSD95 and SV2A area↑ (IHC) |
MWM↑ NOR↑ PAT↑ |
Salazar et al. (2019) |
| Oral administration of WBQ5187 from 6 mo | Multiple targets | 9 mo | M | C57BL/6J |
Aβ area↓ (IHC) Soluble Aβ 40, Aβ 42 ↓ (ELISA) |
GFAP↓ (IHC) | Iba1↓ (IHC) | – | MWM↑ | Wang et al. (2019) |
| Daily injections of liraglutide 1 or DA‐JC1 2 from 10 to 12 mo | GLP‐1 | 11–13 mo | F | C57BL/6J × C3H/HeJ | CA1, Cx, and DG: Aβ area↓ (IHC) | Hip: GFAP area↓ 2,↔ 1 (IHC) | Hip: Iba1 area↓ (IHC) | – | – | Salles et al. (2020) |
| Daily leptin injection for 1 wk from 3 mo1 or 12 mo2 | Neurogenesis | 3 mo or 12 mo | M | C57BL/6J | Hip: Aβ number↓ (IHC, 6E10) |
Hip: Gfap↔ 1, ↓ 2 (qPCR) SGZ: BrdU+GFAP+↔1, ↓2 SVZ: BrdU+GFAP+↔ (IHC) |
Hip: Iba1 number↓ (IHC) | – | – | Calió et al. (2021) |
| Environmental risk factors | ||||||||||
| Restraint stress for 21 d, 6 hrs a day from 4 mo | Stress | 5 mo | M | C57BL/6J | (In)soluble Aβ 40, Aβ 42 ↔ (ELISA) | Cx and Hip: GFAP area↑ (IHC) | – | – | – | Perez‐Nievas et al. (2011) |
| Five days a week: Low1 or high2 dose cigarette smoke exposure from 3 mo | Smoking | 7 mo | F/M | C57BL/6J × C3H/HeJ |
Cx: Aβ↔ 1, ↑ 2 Hip: Aβ area↑ Cx and Hip: Aβ number↔ 1, ↑ 2 (IHC, 4G8) Cx and Hip: Aβ area↔ 1, ↑ 2 Subiculum: Aβ area↔ (IHC, ThS) |
Cx and Hip: GFAP area↔ 1, ↑ 2 (IHC) | Cx and Hip: Iba1 area↔ 1, ↑ 2 (IHC) | – | – | Moreno‐Gonzalez et al. (2013) |
| Voluntary wheel running from 5 mo | Exercise | 7.5 mo | – | C57BL/6J × C3H/HeJ |
Cx: Aβ area↔ (IHC, 4G8, and ThS) Cx: Aβ number↓ (IHC, ThS) Hip: Aβ area↓ (IHC, A11, 4G8 and ThS) Hip: Aβ number↓ (IHC, ThS) |
Cx and Hip: GFAP intensity↓ (IHC) | – | – | MWM↑ | Tapia‐Rojas, Aranguiz, Varela‐Nallar, and Inestrosa (2016) |
|
Social housing: Social isolation1 Social contact with one2 or social contact with five3 from 6 mo |
Social interaction | 12 mo | M | C57BL/6J |
Hip: Aβ area and number↔ (IHC, ThS and 6E10) Aβ 40, Aβ 42 ↔ (ELISA) |
Hip: GFAP number↑ 3 (IHC) | – |
Syn, SNAP‐25, PSD95 and GluN2B↑ 3 GluA2 and GluN2A↔ (WB) Hip: Dendritic spine density↔ 1,2,↑ 3 (IHC) |
MWM↔1,2, ↑ 3 | Liang, Yang, Zhang, and Hao (2019) |
| Early life stress by limited nesting and bedding from P2‐9 | Early life stress | 4 mo1 or 10 mo2 | M | C57BL/6J | – |
ECx and Hip: GFAP area↔ (IHC) Hip: Aldh1l1, Aqp4, GFAP, GLAST, GLT1, GluS, Vim↔, Fasn↑ 1 (qPCR) |
– | – | – | Abbink et al. (2020) |
Abbreviations: General—Specified interventions or ages indicated with 1–5. When not indicated: the same effect for all interventions or ages. When a specific group is not indicated: read‐out was not determined for that group; ↑, increased or improved (Memory) compared to the control group of the same age; ↓, decreased or impaired (Memory); ↔, no (significant) difference; –, not studied; d, day(s); mo, month(s); wk, week(s). Intervention and target—Different APPswePS1dE9 strains indicated witha–c. aAPPswePS1dE9 mice, specific strain not indicated; bMMRRC Stock No: 34829‐JAX–C57BL/6J × C3H/HeJ; cMMRRC Stock No: 34832‐JAX—C57BL/6J. 4‐PB, 4‐phenylbutyrate: activates peroxisome proliferation; AChE, acetylcholinesterase; ApoA‐I, apolipoprotein A1: primary component of high‐density lipoproteins; ASS234, multipotent acetyl and butyrylcholinesterase/monoamine oxidase A–B inhibitor; Blue‐Nabs, naturally occurring autoantibodies against Aβ oligomers purified by Cibacron Blue; Boldine, inhibitor of hemichannel activity; BPN‐15606, γ‐secretase modulator; CCR3, C‐C chemokine receptor 3; CD49d, antibody against α4‐integrin; c‐fms‐ACE10/10, mouse line overexpressing angiotensin‐converting enzyme (ACE) under the control of the c‐fms promoter: resulting in ACE expression in myelomonocytic lineage cells; Cofilin, filamentous‐actin‐severing protein; DA‐JC1, Glucagon‐like peptide‐1 (GLP‐1)/glucagon‐dependent insulinotropic polypeptide dual agonist; Endo‐B1, endophilin‐B1 (Bif‐1: Bax‐interacting factor 1); GA, Glatiramer acetate: weak agonist of myelin‐derived antigens. Gal‐3, Galactin‐3: Aβ oligomerization and Aβ toxicity; Gemfibrozil, peroxisome proliferator‐activated receptor agonist; GLP‐1, glucagon‐like peptide‐1; IDOL ASO, inducible degrader of low‐density lipoprotein receptor (IDOL) antisense oligonucleotide (ASO); IVIG, intravenous immunoglobulin; KDS2020, reversible monoamine oxidase‐B inhibitor; Liraglutide, glucagon‐like peptide‐1 (GLP‐1) receptor analogue; LPSd/d, mouse line expressing a spontaneous mutation in the lipopolysaccharide (LPS) locus: resulting in a defective LPS response; MAO‐A‐B, monoamine oxidase‐A and B; MoBM, bone marrow‐derived CD115+ monocytes; NFATc2, nuclear factor of activated T cells, isoform c2; NOI, neurite outgrowth inhibitor; Oli‐Nabs, naturally occurring autoantibodies against Aβ oligomers purified by Aβ42 oligomers; ONO‐8713, Selective prostaglandin receptor 1 (EP1) antagonist; Palm‐PrRP, prolactin‐release peptide (PrRP) palmitoylated at the N‐terminus; pdMCAO, permanent middle cerebral artery occlusion; PDTC, pyrrolidine dithiocarbamate; NF‐κB, nuclear factor‐κB inhibitor; Pyk2, (PTK2B) Protein tyrosine kinase 2β; Selegiline, monoamine oxidase‐B inhibitor; Tannic acid, anti‐amyloidogenic polyphenol (flavonoid); TRPA1, transient receptor potential ankyrin 1: cation channel; UA, urolithin A; gut‐microbial metabolite of ellagic acid; WBQ5187, Quinolone‐benzofuran derivative; WT, wild‐type; WY, Wy‐14643 peroxisome proliferator‐activated receptor agonist: activators of peroxisome proliferators receptors agonist; YM344031, C–C chemokine receptor 3 antagonist. Age —at read‐out. Sex—female (F), male (M), or both (F/M). –, not indicated. Background: Genetic background of mice used in the study. x, Crossed with. †MMRRC Stock No: 34829‐JAX–C57BL/6J × C3H/HeJ crossed with C57BL/6J. Brain regions—CA, cornu ammonis; CC, cingulate cortex; Cx, cortex; DG, dentate gyrus; ECx, entorhinal cortex; Hip, Hippocampus; NCx, neocortex; SGZ, subgranular zone of the hippocampal dentate gyrus; SVZ, subventricular zone; Wb, Whole brain. Techniques—BM, Barnes Maze; BrdU, 5‐bromo‐2′‐deoxyuridine; CE‐LIF, capillary electrophoresis with laser‐induced fluorescence; ELISA, enzyme‐linked immunosorbent assay; EM, electron microscopy; FC, fear conditioning; IHC, immunohistochemistry; MS, mass spectrometry; MWM, Morris water maze; NOR, novel object recognition; PAT, passive avoidance test; RAWM, radial arm water maze; qPCR, quantitative polymerase chain reaction; WB, western blot; YM, Y‐Maze test. Genes, proteins, and transmitters—3D6, Aβ1–5 antibody. 4G8, Aβ17–24 antibody; 6E10, Aβ1–16 antibody; 82E1, Soluble and fibrillar Aβ; A11, oligomer antibody; Aβ, amyloid‐beta; Aldh1I1, 10‐formyltetrahydrofolate dehydrogenase; Aqp4, aquaporin‐4; ATP, adenosine triphosphate; BAM10, Aβ1‐40 antibody; CD, cluster of differentiation molecule; Fasn, fatty acid synthase; GABA, gamma‐aminobutyric acid; GFAP, glial fibrillary acidic protein; GLAST, glutamate–aspartate transporter; GLT‐1, glutamate transporter 1; GluA2, glutamate receptor, ionotropic, AMPA2 (alpha 2); GluN2A, glutamate receptor, ionotropic, NMDA2A (epsilon 1); GluN2B, glutamate receptor, ionotropic, NMDA2B (epsilon 2); GluR2, glutamate receptor 2; GluS, glutamate synthetase; Iba1, ionized calcium‐binding adapter molecule 1; NR2B, N‐methyl d‐aspartate receptor subtype 2B; PSD95, postsynaptic density protein 95; RTN‐3, reticulon‐3, marker of neuritic dystrophy; SNAP‐25, synaptosomal nerve‐associated protein 25; SV2A, synaptic vesicle glycoprotein 2A; Syn, synaptophysin; ThS, thioflavin‐S; VAMP1, vesicle‐associated membrane protein 1; VGlut, vesicular glutamate transporter; Vim, Vimentin; X‐34 dye, fluorescent amyloid‐specific dye.
4.1. Interventions directly affecting reactive astrocytes
The following studies applied various astrocyte‐specific interventions in APPswePS1dE9 mice to unravel the contribution of reactive astrocytes to AD pathogenesis (Table 1). Studies with a similar approach to target reactive astrocytes are discussed together in the following sections.
4.1.1. Intervening with connexins in reactive astrocytes
Connexin30 (Cx30) and Cx43 are the proteins that form gap junctions between astrocytes or form transmembrane hemichannels (HCs), which are pores between the cytoplasm and the extracellular milieu of astrocytes. Reactive astrocytes, that surround Aβ plaques, upregulate the expression of Cx30 and Cx43 (Yi et al., 2016). Regardless of their distance to plaques, the gap junction communication between APPswePS1dE9 astrocytes, however, was similar to that in wild‐type astrocytes. In the hippocampus of APPswePS1dE9 mice, the HCs composed of Cx43 were chronically open, indicating that specifically the hemichannel function is affected in AD (Yi et al., 2016). Opening of the Cx43 HCs can be triggered by the presence of Aβ (Aβ‐induced) inflammation, or an increased concentration of intracellular calcium ([Ca2+]i) in astrocytes, inducing the release of gliotransmitters such as ATP and glutamate from reactive astrocytes (Yi et al., 2016). ATP activates P2X7 or P2Y1 receptors on microglia and astrocytes, leading to increased calcium transients and calcium waves in astrocytes, as has been shown in another AD mouse model: the APPPS1 model driven by the Thy1 promoter (Delekate et al., 2014). HC‐dependent glutamate and ATP release amplify a vicious circle in astrocytes by inducing high [Ca2+]i levels that trigger Cx43 HC opening contributing to a maintenance of elevated [Ca2+]i. The effect of the resulting chronic release of glutamate and ATP mediated by activated HCs can be detrimental and/or protective for neurons (Yi et al., 2016). This is shown by cell culture experiments that indicate that glutamate release can contribute to synaptic loss by activation of extrasynaptic NMDA receptors (Talantova, Sanz‐Blasco, & Zhang, 2013), or mediate neuronal survival by activation of the astroglial mGluR3 (Durand et al., 2014). And, excessive ATP release can mediate a neurotoxic effect by stimulation of IL‐1β secretion by microglia (Pelegrin & Surprenant, 2009) or a neuroprotective effect by increasing the release of IL‐6 (Fujita, Tozaki‐Saitoh, & Inoue, 2009).
Astrocyte‐specific Cx43 deficiency was obtained by crossing a GFAP‐cre mouse line with a floxed‐Cx43 mouse line (Ren et al., 2018; Yi et al., 2016). Astroglial Cx43 deficiency in 9‐month‐old APPswePS1dE9 mice resulted in a reduced release of the gliotransmitters glutamate and ATP. The release of GABA was not affected in these mice (Yi et al., 2016). This latter finding is in line with the study by Jo et al. (2014), which shows that the release of GABA is mediated by Best1 channels in astrocytes (Jo et al., 2014), and thus not by HCs.
Besides the reduced gliotransmitter release, there was a significant decrease in GFAP levels in the cortical and hippocampal astrocytes of 12‐month‐old Cx43‐deficient APPswePS1dE9 mice, as compared to APPswePS1dE9 astrocytes. The decrease in GFAP was not caused by a decrease in Aβ plaque load, as this was similar in both mouse strains (Ren et al., 2018). The Cx43 deficiency also led to a reduction in neuronal damage, as shown by a decrease in oxidative stress and neuritic dystrophy in the hippocampus (Yi et al., 2016), and an increase in dendritic spine density and postsynaptic density 95 (PSD95) protein level (Ren et al., 2018). The Cx43 deficient APPswePS1dE9 mice had a better spatial and nonspatial memory compared to APPswePS1dE9 mice. Re‐expression of Cx43 by a viral vector in 7‐month‐old mice resulted in reactive astrocytes and aggravated synaptic function assessed 2 months later (Ren et al., 2018). Thus, both studies support that astroglial connexins, that is, Cx43 in particular, are potential therapeutic molecular targets to prevent reactive astrocytes and synaptic degeneration in AD (Ren et al., 2018; Yi et al., 2016).
4.1.2. Inducible expression of human Apolipoprotein E isoforms in astrocytes
The lipid/cholesterol carrier apolipoprotein E (ApoE) is coded by the polymorphic ApoE gene. The gene has three major alleles, ε2, ε3, and ε4. Genetically, the ApoE gene is a well‐established AD risk locus. The presence of 1 or 2 ε4 alleles strongly increases the risk to develop late‐onset AD (Esquerda‐Canals et al., 2017). ApoE is mainly expressed in glia, with the highest expression levels in astrocytes and microglia, followed by oligodendrocyte precursors. The expression in neurons is low (Zhang et al., 2014).
To determine at which age ApoE has the most impact on Aβ pathology, Liu et al. (2017) developed APPswePS1dE9 mice with inducible expression of human ApoE3 or ApoE4 in astrocytes. They generated a mouse line with cre‐dependent and doxycycline regulated ApoE3 or ApoE4 expression. Crossing with a GFAP‐cre mouse line resulted in astrocyte‐specific, inducible ApoE expression. ApoE3 expression in APPswePS1dE9 mice at any stage of Aβ pathology had no effect on insoluble Aβ levels and the Aβ plaque development, but led to decreased levels of cortical and hippocampal GFAP, implying that ApoE3 reduces Aβ‐induced reactive astrocytes. APPswePS1dE9‐ApoE3 mice also had elevated levels of PSD95, suggesting that ApoE3 can prevent synaptic impairment. In contrast, ApoE4 expressed at 0–6 months revealed a critical role of ApoE4 before and during the onset of Aβ pathology. During this stage, astroglial expression of ApoE4 significantly increased insoluble Aβ levels and Aβ plaque burden, impaired hippocampal Aβ clearance, and induced reactive gliosis in the cortex and hippocampus (i.e., increased GFAP and microglial Iba1 levels). Notably, inducing ApoE4 expression later in life, when Aβ plaques are present (6–9 months), barely affected amyloidosis (Liu et al., 2017). Surprisingly, crossing the original version of the APPswePS1dE9 mice, resulting from the crossing of the APPswe line C3‐3 and the PSdE9 line S‐9, with a mouse line expressing human ApoE4 driven by the mouse prion protein promoter did not enhance Aβ pathology at 6 months (Lesuisse et al., 2001).
These findings show that the astrocyte‐specific expression of ApoE3 or ApoE4 impact Aβ pathology and astrogliosis differently and indicate that the expression of ApoE isoforms in astrocytes is a potential therapeutic target to modulate reactive astrogliosis and synaptic degeneration in AD.
4.1.3. Increasing reactive astrocytes by oxidative stress
Oxidative stress plays an important role in the pathogenesis of AD (Bhatia & Sharma, 2021). Chun et al. (2020) studied whether the severity of reactive astrocytes could affect other hallmarks of AD. In this study, the diphtheria toxin receptor (DTR) was expressed in astrocytes by injecting AAVs with DTR driven by the GFAP104 promotor in the CA1 region of the hippocampus of 10‐ to 12‐month‐old APPswePS1dE9 mice. Administration of diphtheria toxin (DT) in these mice induced astroglial hydrogen peroxide (H2O2) production and resulted in an increase of reactive astrocytes, based on GFAP expression. Furthermore, it reduced the expression of NeuN, and increased expression of cleaved caspase‐3, suggesting the induction of neurodegeneration. Moreover, the severe reactive astrocytes in astrocyte‐specific DTR‐expressing APPswePS1dE9 mice impaired spike probability compared to APPswePS1dE9 mice (Chun et al., 2020). Whether this was due to synaptic degeneration and/or increased tonic inhibition by the severe reactive astrocytes, as indicated by a previous study (Jo et al., 2014; Section 4.2), was not determined. The DTR‐expressing APPswePS1dE9 mice showed impaired spatial memory assessed by the novel place recognition test, known as a hippocampal memory task. The effect on Aβ burden was not assessed in this study (Chun et al., 2020).
Taken together, the administration of DT induced severe reactive astrocytes, triggered neurodegeneration, and impaired spatial memory in DTR‐expressing APPswePS1de9 mice. This study indicates that reducing oxidative stress could reduce reactive astrocytes, presenting a therapeutic strategy for AD.
4.1.4. Reducing reactive astrocytes by overexpression of clusterin
Clusterin (CLU), also known as apolipoprotein J (APOJ), is a multifunctional glycoprotein, involved in heterogeneous processes, including lipid homeostasis, complement inhibition, and apoptosis (Foster, Dangla‐Valls, Lovestone, Ribe, & Buckley, 2019). Large‐scale genome‐wide association studies have identified an association of common polymorphisms within the CLU gene with the risk of developing AD (Lambert et al., 2013). CLU could form complexes with Aβ, thereby influencing its solubility and preventing amyloid fibril formation.
Astrocytes are a major source of CLU expression in the brain (Zhang et al., 2016). Wojtas et al., 2020 investigated the impact of overexpression of astroglial CLU on AD pathology. An AAV vector expressing CLU under the control of the GFAP promoter was used to induce a ~30% overexpression of CLU in astrocytes in 8‐month‐old APPswePS1dE9 mice. This CLU overexpression ameliorates amyloid pathology and reduced the number of dystrophic neurites in the cortex and hippocampus of APPswePS1dE9 mice. Also, gliosis, assessed by GFAP and Iba1 immunostaining, was reduced in 8‐month‐old AAV‐CLU‐APPswePS1dE9 mice compared to APPswePS1dE9 mice injected with control AAV‐GFP vectors. These results indicate that the overexpression of CLU in astrocytes impacts amyloid burden and associated pathologies and offers a novel therapeutic molecular target to reduce reactive astrocytes and synaptic degeneration in AD (Wojtas et al., 2020).
4.1.5. Reducing reactive astrocytes by disrupting or inhibiting the JAK2/STAT3 pathway
Reactive astrocytes can be directly induced by activating the Janus kinase/signal transducer and activator of transcription 3 (JAK/STAT3) pathway (Ceyzériat et al., 2018). The JAK/STAT3 pathway is activated in astrocytes of adult APPswePS1dE9 mice (8 months) (Haim et al., 2015). Binding of several cytokines and growth factors to their receptors activates JAK2, results in the phosphorylation and nuclear translocation of STAT3. Nuclear STAT3 induces the transcription of multiple target genes, including GFAP (Ceyzériat et al., 2018), the classical marker for reactive astrocytes.
Inhibition of the JAK2/STAT3 pathway via adeno‐associated virus‐based vectors (AAV)‐induced overexpression of Suppressor of Cytokine Signaling 3 (SOCS3) in the hippocampus of 3‐month‐old APPswePS1dE9 mice prevented the upregulation of GFAP and Aβ plaque load assessed 6 months later (Ceyzériat et al., 2018). In this experiment, SOCS3 was driven by the gfaABC1D promoter, which makes the expression astrocyte‐specific. The SOCS3‐mediated inhibition of JAK2/STAT3 normalized the expression of multiple reactive astrocyte markers, including the expression of cytokines and complement factors in 9‐month‐old APPswePS1dE9 mice. Moreover, inhibition of STAT3 improved spatial learning in APPswePS1dE9 mice, as assessed by the Morris water maze test. Vice versa, stimulation of the JAK2/STAT3 pathway induced reactive astrocytes in wild‐type mice and enhanced reactive astrogliosis in APPswePS1dE9 mice. More Aβ plaques were present in APPswePS1dE9 mice with an enhanced astrocyte reactivity (Ceyzériat et al., 2018). However, this study is in contrast with previous studies in which astrocyte reactivity was reduced by GFAP and vimentin deficiency (see next section).
Astrocyte‐specific STAT3 deficiency can be obtained by crossing a tamoxifen‐inducible Cx43‐Cre mouse line with a floxed‐STAT3 mouse line. Deletion of STAT3 in astrocytes from an early age, that is, 6 weeks, resulted in a reduced Aβ burden at the age of 8 months and an increase in internalization of Aβ in microglia (Reichenbach et al., 2019). While Ceyzériat et al., 2018 observed reduced GFAP immunoreactivity, this conditional STAT3 deficiency in astrocytes did not change the overall coverage of astrocytes and microglia (Reichenbach et al., 2019). Around plaques, even an increase in astrocyte volume was found, which is a characteristic of reactive astrocytes. The morphology of microglia near plaques was affected, displaying a higher number of branches and longer processes. It was suggested that STAT3 deletion triggers a switch in astrocytes from a neurotoxic to a protective astrocyte phenotype, based on the expression of specific markers (including Amigo2, C3, and Tm4sf1). Moreover, STAT3 deletion in astrocytes reduced the astrocyte calcium hyperactivity, and induced an improvement in spatial learning and memory. The application of a preclinical systemic STAT3 inhibitor for 6 weeks also protected 8‐month‐old APPswePS1dE9 mice from cognitive decline (Reichenbach et al., 2019). These results show that reactive astrocytes contribute to AD pathogenesis and that blocking the JAK/STAT3 pathway in astrocytes, therefore, could offer a novel therapeutic strategy for AD (Ceyzériat et al., 2018; Reichenbach et al., 2019).
4.1.6. Reducing reactive astrocytes by deleting intermediate filament proteins
Upregulation of the IF proteins GFAP and vimentin is a well‐known characteristic of reactive astrogliosis, the functional alterations associated with it, however, remain up to now largely unknown. The response of astrocytes to Aβ seems dependent on the presence of GFAP, as astrocytes of GFAP‐ deficient (−/−) mice failed to form a barrier‐like structure around an Aβ plaque in the hippocampus, as shown in slice cultures (Xu, Malouf, Messing, & Silver, 1999). By comparing the transcriptome of cortical astrocytes of 15‐ to 18‐month‐old APPswePS1dE9‐GFAP−/−, APPswePS1dE9‐GFAP−/−vimentin−/−, and control APPswePS1dE9 mice, Kamphuis et al. (2015) discovered that the transcriptional response of reactive astrocytes was different in astrocytes with and without GFAP and vimentin. Astrocytes of GFAP‐ and GFAP‐vimentin‐deficient APPswePS1dE9 mice displayed an aggravated inflammatory response and the decrease of neuronal support genes, as shown earlier (Orre, Kamphuis, Osborn, Jansen, et al., 2014), was absent. Whether this indicates a deteriorated or improved inflammatory status is still unclear, as the changed genes include pro‐inflammatory and anti‐inflammatory genes. No differences in Aβ plaque load were found between both IF‐deficient and APPswePS1dE9 mice with IFs in mice of 6, 9, and 15 months of age, indicating that the IFs themselves do not play a prominent role in amyloidosis (Kamphuis et al., 2015). These results are in contrast with earlier findings of Kraft et al., 2013, which showed an increase in plaque load and insoluble Aβ in 8‐ and 12‐month‐old APPswePS1dE9‐GFAP−/−vimentin−/− mice, without an effect on the expression or processing of APP (Kraft et al., 2013). They found that the number of astrocytes was unaffected, but the overlap between astrocyte processes and amyloid plaques was reduced after deletion of IF proteins (which is also seen in the study of Kamphuis et al.), whereas microglia number and overlap with plaques were increased. These results suggest that reactive astrocytes are needed to limit plaque growth (Kraft et al., 2013). Discrepancies between the studies can be caused by the different age of animals used in the studies, the mixed backgrounds of the IF‐deficient animals, or different staining techniques used to visualize Aβ plaques. Kamphuis et al., 2015 kept the animals at a genetic background of predominantly C57BL/6 with a variable contribution of 129Sv and 129Ola while Kraft et al., 2013 kept them on a mixed B6C3, C57BL/6, 129Sv, and 129Ola background (Kamphuis et al., 2015; Kraft et al., 2013). Discrepancies with the study by Ceyzériat et al., 2018, can be explained by the different interventions used. In contrast to the inhibition of the JAK2/STAT3 pathway to prevent the upregulation of GFAP (Ceyzériat et al., 2018), the crossings by Kraft et al., 2013 and Kamphuis et al., 2015 resulted in the complete removal of GFAP and vimentin. Additional factors that might contribute to the differences are the genetic background of the mouse lines and the timing of the intervention, as disruption of the JAK/STAT3 pathway was only induced from the age of 3–4 months (Ceyzériat et al., 2018), in contrast to a complete deficiency of GFAP (Kamphuis et al., 2015; Kraft et al., 2013).
In summary, although IF‐deficiencies affect the transcriptional response of reactive astrocytes (Kamphuis et al., 2015), contradicting findings have been reported on the effect on the Aβ burden (Kamphuis et al., 2015; Kraft et al., 2013). How background strain and pathology stage exactly affect this outcome is still unknown. Moreover, the effect of IF‐deficiency on cognitive decline in the APPswePS1dE9 still needs to be established.
4.2. Interventions indirectly affecting reactive astrocytes
Reactive astrocytes were also indirectly affected by various intervention studies, which we categorized on the main target of their intervention, Aβ, inflammation, transmitter release, neuroprotection, or environmental risk factors. In the following sections, we give a short overview of the findings of these studies, which are listed in Table 2.
4.2.1. Amyloid‐beta
To reduce the Aβ load or prevent the formation of Aβ in APPswePS1dE9 mice, many different strategies have been used over the years, such as Aβ immunization (Qu et al., 2007; Wang et al., 2016), stimulation of Aβ clearance (Bernstein et al., 2014; Gao et al., 2020; Koronyo‐Hamaoui et al., 2020; Luo et al., 2020), prevention of Aβ oligomerization (Tao et al., 2020), and modulation of APP cleavage enzymes (Mori et al., 2012, 2020; Prikhodko et al., 2020). Besides reducing the Aβ load, almost all of these interventions also reduced reactive astrogliosis based on GFAP expression and, when studied, showed improved memory of the APPswePS1dE9 mice (Table 2). In one study, however, the intervention to stimulate Aβ clearance resulted in an upregulation of GFAP around Aβ plaques. Also, LC3, an autophagy‐related protein, was increased in these astrocytes. These findings suggest that the intervention recruited astrocytes to plaques and enhanced Aβ clearance via autophagy by astrocytes (Luo et al., 2020).
4.2.2. Inflammation
Expectedly, many interventions that also affected reactive astrocytes were aimed at the modulation of inflammation, for example through inhibiting NF‐κB (Malm et al., 2007; Zhang et al., 2009), interfering with chemokine binding (Sui et al., 2019; Zhu et al., 2017), or crossbreeding the APPswePS1dE9 mice with mice that were deficient in genes that led to an altered inflammatory response (Lee et al., 2016; Manocha et al., 2017). In the majority of studies, reducing inflammation also decreased the expression of GFAP, and in some studies also led to a decrease in the Aβ load. In the studies in which also the cognitive function was studied, an improvement of memory was found in the studies that report on reduced reactive astrogliosis.
In three studies, however, the inflammation‐targeted interventions led to an upregulation of GFAP (Button et al., 2019; Jin et al., 2008; Lee et al., 2016). For two of these studies, this was expected considering the genotypes of the mice that were generated by cross‐breeding with APPswePS1dE9 mice, the LPSd/d‐APPswePS1dE9 mice (Jin et al., 2008) and the ApoA‐I deficient APPswePS1dE9 mice (Button et al., 2019). The LPSd/d genotype is known to enhance AD pathology in APPswePS1dE9 mice (see 3.2, Tahara et al., 2006; Song et al., 2011) and ApoA‐I, a primary component of high‐density lipoprotein (HDL) complex, is proposed to have a protective role in AD pathogenesis (Button et al., 2019). In the third study, the transient receptor potential cation channel, subfamily A1 (TRPA1)‐deficient APPswePS1dE9 mice were investigated. In TRPA‐1 deficient APPswePS1dE9 mice a reduction of Aβ load, inflammatory cytokine secretion, and improved cognition were found. Even though GFAP was increased in TRPA1‐deficient mice, a reduced expression of IL‐1β, IL‐6, and IL‐4 in astrocytes was detected. These findings indicate that despite an increased GFAP expression, astroglial inflammation was reduced. However, as TRPA1 is expressed in endothelial cells, neurons, and astrocytes, it remains unclear whether it was the TRPA1 deficiency in astrocytes in APPswePS1dE9 mice that contributed to the reduced inflammatory response and improvement in learning and memory.
Astrocytes are known to closely interact with microglia; both cells are involved in reactive gliosis in AD. The studies by Zhu et al. (2017) and Sui et al. (2019) highlight the role of microglia in AD pathogenesis. They used a C–C chemokine receptor type 3 (CCR3) antagonist and CCR3‐deficient APPswePS1dE9 mice to target microgliosis, which led to a reduced GFAP expression and Aβ load and an improved memory (Sui et al., 2019; Zhu et al., 2017).
4.2.3. Transmitter release
The study by Yi et al. (2017) emphasizes the important role of hemichannel Cx43 function (Section 4.1; Yi et al., 2016; Ren et al., 2018). They used Boldine, which has been shown in in vitro experiments to block HCs and not gap junctions. Boldine administration to 6‐month‐old APPswePS1dE9 mice for 3 months, via drinking water, reduced the release of ATP and glutamate, without affecting the release of d‐serine or GABA. Although not specifically mentioned, these transmitters can either be released from astrocytes (Araque et al., 2014; Volterra & Meldolesi, 2005) or neurons. Aβ load was unaffected and the number of dystrophic neurites was reduced (Yi et al., 2017). The studies by Jo et al. (2014) and Park et al. (2019) also focus on the role of transmitter release by astrocytes, specifically GABA. Both studies used MAO‐B inhibitors and showed a reduction in GABA production and release by astrocytes. Neuronal plasticity was increased and the inhibitors prevented memory impairment in APPswePS1dE9 mice (Jo et al., 2014; Park et al., 2019). It is important to note, that the drugs used in these three studies are not astrocyte‐specific and thus a contribution of neurons or microglia has to be considered (Jo et al., 2014; Park et al., 2019; Yi et al., 2017).
4.2.4. Neuroprotection
Because AD is a progressive neurodegenerative disease, various interventions have been targeted at neuroprotection in APPswePS1dE9 mice. These interventions included choline supplementation (Mellott et al., 2017), drugs targeting peroxisomes (Abbott, Toledo, Aranguiz, Inestrosa, & Varela‐Nallar, 2013), and crossing APPswePS1dE9 mice with mice that are lacking a neuroprotective protein (Wang et al., 2015) or late‐onset AD risk factor Pyk2 (Salazar et al., 2019). All studies categorized in the neuroprotective group, except one, showed a reduction of GFAP expression and Aβ burden, and some also showed an improvement in memory function (when examined), in response to the neuroprotective intervention.
Crossbreeding of Endophilin‐B1 (Endo‐B1) deficient mice with APPswePS1dE9 mice resulted in the upregulation of GFAP, an increase in Aβ burden, aggravation of synaptic degeneration, and behavioral impairment compared to APPswePS1dE9 mice (Wang et al., 2015). Endo‐B1 (also known as Bax‐interacting factor 1) has multiple functions, including the regulation of apoptosis, autophagy, and mitochondrial function. Endo‐B1 is neuroprotective, as overexpression of Endo‐B1 in neuronal cultures protected against Aβ‐ induced apoptosis and mitochondrial dysfunction (Wang et al., 2014). In cortical tissue of AD patients and 10‐month‐old APPswePS1dE9 mice, Endo‐B1 protein levels are decreased compared to controls (Wang et al., 2015).
Other neuroprotective interventions were aimed at the cholinergic network, modulated oxidative stress, or targeted the AD risk factor Pyk2. The basal forebrain cholinergic system is vulnerable to degeneration in AD patients (Hampel et al., 2018; Whitehouse et al., 1982). Even though the number of cholinergic neurons in the forebrain remained unchanged, dystrophic cholinergic neurites were found in the cortex, hippocampus, and striatum of APPswePS1dE9 mice (Perez, Dar, Ikonomovic, DeKosky, & Mufson, 2007). The use of perinatal choline supplementation prevented the reduction of the cholinergic marker choline acetyltransferase in 9‐ and 12‐month‐old APPswePS1dE9 mice. It also reduced amyloidosis and the GFAP levels specifically in 9‐month‐old female APPswePS1dE9 mice (Mellott et al., 2017).
One of the mechanisms by which Aβ induces neurotoxicity is oxidative stress (Inestrosa et al., 2013). Reducing oxidative stress by applying a semi‐synthetic derivative of the St. John's Wort (Cerpa et al., 2010) or a drug that enhances peroxisomal proliferation (Inestrosa et al., 2013) both resulted in an increased synaptic density and improved cognitive function in APPswePS1dE9 mice. These interventions also reduced the Aβ burden and GFAP levels (Cerpa et al., 2010; Inestrosa et al., 2013). Vice versa, increasing reactive astrocytes by oxidative stress in astrocyte‐specific DTR expressing APPswePS1dE9 mice resulted in neurodegeneration and impaired spatial memory compared to APPswePS1dE9 mice (Chun et al., 2020).
The kinase Pyk2 (PTK2B) has been implicated as a late‐onset AD risk factor, and is primarily expressed by neurons. Pyk2‐deficiency in APPswePS1dE9 prevented synapse loss and improved cognitive functioning compared to 12‐month‐old APPswePS1dE9 mice. Also, the Aβ burden and GFAP levels were reduced in Pyk2‐deficient mice (Salazar et al., 2019).
4.2.5. Environmental risk factors
Environmental factors can either be protective, such as exercise and social interaction, or a risk factor for developing AD, such as smoking, stress, and social isolation. Voluntary wheel running reduced GFAP and Aβ levels (Tapia‐Rojas et al., 2016), whereas exposure to cigarette smoke or restraint stress increased GFAP expression in the APPswePS1dE9 mice (Moreno‐Gonzalez et al., 2013; Perez‐Nievas et al., 2011). Liang et al., 2019 performed a social housing experiment with APPswePS1dE9 mice, which showed that enhancing social interaction with one or multiple cage mates, improved cognitive function, but also increased the expression of GFAP without affecting Aβ burden (Liang et al., 2019).
To conclude, the majority of interventions reduced both Aβ burden and GFAP expression, although some studies showed that these could be independently affected. When the studies also included cognitive testing, an improvement in cognitive function was detected in most studies. As most interventions summarized in Table 2 were aimed to reduce Aβ load, inflammation, or neuronal damage, the expression of GFAP could be affected in various ways. It is important to note that these studies impact reactive astrocytes indirectly and they were not designed to determine causality between reactive astrogliosis and AD pathogenesis. Therefore, conclusions should be made with caution.
Although GFAP expression is a valuable, but limited readout for reactive astrocytes, in almost all intervention studies (listed in Table 2) except five (Abbink et al., 2020; Jo et al., 2014; Malm et al., 2007; Park et al., 2019; Yi et al., 2017), GFAP expression was used to determine the effect on reactive astrocytes. Jo et al. (2014) and Park et al. (2019) showed a reduced GABA release as a readout to show the effect of their intervention on reactive astrocytes, and Yi et al. (2017) showed a reduction in the release of the gliotransmitters glutamate and ATP. Malm et al. (2007) found an increase in the level of astroglial GLT‐1 after inhibiting NF‐κB, although GFAP levels did not change. Abbink et al., 2020 found an increase in Fasn expression, without changes in other astrocyte‐related genes after early life stress exposure in APPswePS1dE9 mice (Abbink et al., 2020). Even though studies using alternative markers for reactive astrocytes are limited, this indicates that (functional) changes can be induced in reactive astrocytes by interventions, in some cases without affecting the expression of GFAP (Abbink et al., 2020; Malm et al., 2007; Yi et al., 2017). The causality and underlying mechanisms that mediate a positive effect on Aβ burden, reactive astrocytes, and cognitive performance require further investigation. An astrocyte‐specific approach to unravel the contribution of astrocytes in the interventions listed in Table 2 is needed to further explore astrocytes as a therapeutic target to treat AD patients.
5. DISCUSSION
In this review, we have listed direct and indirect interventions that attenuate the occurrence of reactive astrocytes in the APPswePS1dE9 AD mouse model. Considering their crucial role in proper brain functioning, the functional changes associated with the reactive state of astrocytes can have a direct impact on synapse function and neuronal circuit activity, potentially contributing to cognitive problems in AD (Chung et al., 2015; Kuchibhotla et al., 2008; Ortinski et al., 2010; Osborn et al., 2016). Classically, most studies investigating the changes in reactive astrocytes focus on GFAP expression, rather than on functional changes.
An increased GFAP expression and changed morphology are the most often used markers for reactive astrocytes (Escartin et al., 2019; Liddelow et al., 2017), also evident from the studies listed in Tables 1 and 2. However, it remains unclear whether an increase in GFAP expression is an appropriate marker for all reactive astrocytes. Moreover, this focus on GFAP expression and morphological changes in astrocytes have so far not been sufficient to reveal what happens in the early stages of AD pathology. While an increase in soluble Aβ42 is detected in 3‐month‐old APPswePS1dE9 mice (Van Tijn et al., 2012) and plaques in 4‐month‐old animals (Garcia‐Alloza et al., 2006; Ruan et al., 2009), reactive GFAP positive astrocytes around amyloid deposits are first detected at 6 months of age in APPswePS1dE9 mice (Ruan et al., 2009; Van Tijn et al., 2012; Végh et al., 2014). Whether the early increase in Aβ oligomers is sufficient to induce reactive astrocytes, and whether GFAP upregulation is a sensitive marker for the early and subtle functional changes in astrocytes is unknown.
To determine the contribution of reactive astrocytes to AD pathogenesis, it is necessary to unravel the effect of their functional changes to synaptic dysfunction and/or loss and eventually dementia. The consequences of disturbances in astroglial function, including but not limited to the release of gliotransmitters, uptake of neurotransmitters, ion buffering, and BBB maintenance (Osborn et al., 2016; Verkhratsky & Nedergaard, 2018) are largely unstudied in AD. There is an emerging interest in astrocyte function and more tools are becoming available to study this using in vivo, ex vivo, and in vitro systems (Almad & Maragakis, 2018). Astrocyte‐specific techniques are being developed to manipulate astrocyte function and to study neuron–glia interactions. Optogenetics and chemogenetics enable the manipulation of astrocytes (Agulhon et al., 2013; Mederos et al., 2018; Xie, Yang, Song, Quan, & Qing, 2019). Genetically encoded calcium indicators and sensors for transmitters allow for the functional measurement in astrocytes (Almad & Maragakis, 2018). The field is relatively young, and in recent years, an increase in studies focusing on astrocyte heterogeneity, function, and astrocyte–neuron interactions (Escartin et al., 2019) is observed. This will undoubtedly lead to more insight in astrocyte function in health and disease, which is essential to understand the full spectrum of AD pathogenesis.
To develop astrocyte‐specific and well‐timed interventions, changes in astroglial function at different stages of the disease need to be determined. This information will enable astrocyte‐directed therapies to be effective in reversing the astrocyte‐induced damage or allow for repair and/or recovery. When the damage is too severe, it might be irreversible. As more therapeutics will be targeted towards the earlier stages of AD, identifying pathological changes in astrocyte function in the early stages of pathology is important. This should coincide with the development of accurate biomarkers for specific disease stages to make the treatment of AD patients possible.
Transcriptomics and proteomics are important tools to find changes in astrocytes at the gene or protein level (Pestana, Edwards‐Faret, Belgard, Martirosyan, & Holt, 2020; Westergard & Rothstein, 2020). The A1 and A2 profiles identifying neuroinflammation and ischemia‐induced subtypes of reactive astrocytes, respectively (Liddelow et al., 2017), are a good start of this endeavor. However, limitations of this classification have also been revealed, as aged astrocytes take on a A1‐like neuroinflammatory phenotype (Clarke et al., 2018) and reactive astrocytes co‐expressing A1 and A2 genes are observed in APPswePS1dE9 mice, the MCAO model, and the aging mouse brain (Clarke et al., 2018; Orre, Kamphuis, Osborn, Jansen, et al., 2014). These findings indicate that it may be difficult to define classes of reactive astrocytes with only a few genes. Recent single‐cell RNAseq approaches are likely to reveal more and more disease and cell‐type‐specific profiles. A recent single‐cell RNA sequencing study identified five transcriptomically distinct types of astrocytes in the hippocampus and cortex of adult mice (Batiuk et al., 2020). Still little is known about the effect of the molecular and functional heterogeneous subtypes of astrocytes and reactive astrocytes and their contribution to AD pathogenesis. Using hippocampal astrocytes of 7‐month‐old 5xFAD AD mice, Habib et al., 2020 identified a disease‐associated astrocyte (DAA) population (Habib et al., 2020). Not all genes in the DAA profile are unique for AD‐associated astrocytes as the profile shared a significant number of upregulated genes compared with the Gfap‐high population, a population also present in wild‐type mice. It remains to be determined whether these markers for reactive astrocytes are universal for different AD mouse models, pathology stages, and brain regions studied.
Overall, state‐of‐the‐art sequencing approaches will discover novel disease‐ and subtype‐specific profiles, which also need to be validated in AD patient material. These profiles will help to identify specific pathways that can be targeted with the use of cell‐specific tools and enable the development of astrocyte‐specific therapies. Connecting gene and protein changes to functional changes will be the first step in the development of astrocyte‐ and pathway‐specific AD therapies.
6. CONCLUSION
Reactive astrocytes are present in the APPswePS1dE9 mice when plaques become apparent, coinciding with cognitive decline (Ruan et al., 2009; Van Tijn et al., 2012; Végh et al., 2014). Still little is known about the effect of the functional changes associated with their reactive state and their contribution to AD pathogenesis. Specific treatment windows and astrocyte‐specific interventions need to be further developed. The few astrocyte‐specific interventions highlighted in this review produced encouraging results (Ceyzériat et al., 2018; Reichenbach et al., 2019; Ren et al., 2018; Yi et al., 2016), indicating that targeting reactive astrocytes could be a promising therapeutic area for AD.
ACKNOWLEDGEMENTS
JM was supported by ZonMw (733050504), EMH and TS by ZonMw (733050505) and EMH and JM by ZonMw (733050816)—The ZonMw, dementia Research and Innovation Program “Memorabel”. EMH and JM were supported by Alzheimer Nederland (WE.March 4, 2017). ZonMw and Alzheimer Nederland projects are part of the cooperative “Deltaplan for Dementia”, the Dutch national platform to address and manage the growing problem of dementia. DRB was supported by the National Institute on Aging (P50AG047266) and by the Santa Fe HealthCare Alzheimer's Disease Research Center.
Smit T, Deshayes NAC, Borchelt DR, Kamphuis W, Middeldorp J, Hol EM. Reactive astrocytes as treatment targets in Alzheimer's disease—Systematic review of studies using the APPswePS1dE9 mouse model. Glia. 2021;69:1852–1881. 10.1002/glia.23981
Jinte Middeldorp and Elly M. Hol have equally shared the joint senior authorship.
Funding information Alzheimer Nederland, Grant/Award Number: WE.03‐2017‐04; National Institute on Aging, Grant/Award Number: P50AG047266; Santa Fe HealthCare Alzheimer's Disease Research Center; ZonMw, Grant/Award Numbers: 733050504, 733050505, 733050816
DATA AVAILABILITY STATEMENT
Data sharing is not applicable to this article as no new data were created or analyzed in this study.
REFERENCES
- Abbink, M. R. , Kotah, J. M. , Hoeijmakers, L. , Mak, A. , Yvon‐Durocher, G. , van der Gaag, B. , … Korosi, A. (2020). Characterization of astrocytes throughout life in wildtype and APP / PS1 mice after early‐life stress exposure. Journal of Neuroinflammation, 17(91), 1–16. 10.1186/s12974-020-01762-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abbott, A. C. , Toledo, C. C. , Aranguiz, F. C. , Inestrosa, N. C. , & Varela‐Nallar, L. (2013). Tetrahydrohyperforin increases adult hippocampal neurogenesis in wild‐type and APPswe/PS1DeltaE9 mice. Journal of Alzheimer's Disease, 34(4), 873–885. 10.3233/JAD-121714 [DOI] [PubMed] [Google Scholar]
- Agulhon, C. , Boyt, K. M. , Xie, A. X. , Friocourt, F. , Roth, B. L. , & Mccarthy, K. D. (2013). Modulation of the autonomic nervous system and behaviour by acute glial cell Gq protein‐coupled receptor activation in vivo. Journal of Physiology, 591(22), 5599–5609. 10.1113/jphysiol.2013.261289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allaman, I. , Bélanger, M. , & Magistretti, P. J. (2011). Astrocyte‐neuron metabolic relationships: For better and for worse. Trends in Neurosciences, 34(2), 76–87. 10.1016/j.tins.2010.12.001 [DOI] [PubMed] [Google Scholar]
- Almad, A. , & Maragakis, N. J. (2018). A stocked toolbox for understanding the role of astrocytes in disease. Nature Reviews Neurology, 14(6), 351–362. 10.1038/s41582-018-0010-2 [DOI] [PubMed] [Google Scholar]
- Araque, A. , Carmignoto, G. , Haydon, P. G. , Oliet, S. H. R. , Robitaille, R. , Volterra, A. , & Oliet, H. R. (2014). Gliotransmitters travel in time and space. Neuron, 81(4), 728–739. 10.1016/j.neuron.2014.02.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araque, A. , Parpura, V. , Sanzgiri, R. P. , & Haydon, P. G. (1998). Glutamate‐dependent astrocyte modulation of synaptic transmission between cultured hippocampal neurons. European Journal of Neuroscience, 10, 2129–2142. 10.1046/j.1460-9568.1998.00221.x [DOI] [PubMed] [Google Scholar]
- Araque, A. , Parpura, V. , Sanzgiri, R. P. , & Haydon, P. G. (1999). Tripartite synapses: Glia, the unacknowledged partner. Trends in Neurosciences, 22(5), 208–215. 10.1016/S0166-2236(98)01349-6 [DOI] [PubMed] [Google Scholar]
- Barber, S. A. , Perera, P.‐Y. , & Vogel, S. N. (1995). Defective ceramide response in C3H/HeJ (Lpsd) macrophages. Journal of Immunology, 155(5), 2303–2305. [PubMed] [Google Scholar]
- Batiuk, M. Y. , Martirosyan, A. , Wahis, J. , de Vin, F. , Marneffe, C. , Kusserow, C. , … Holt, M. G. (2020). Identification of region‐specific astrocyte subtypes at single cell resolution. Nature Communications, 11(1), 1–15. 10.1038/s41467-019-14198-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bazzigaluppi, P. , Weisspapir, I. , Stefanovic, B. , Leybaert, L. , & Carlen, P. L. (2017). Astrocytic gap junction blockade markedly increases extracellular potassium without causing seizures in the mouse neocortex. Neurobiology of Disease, 101, 1–7. 10.1016/j.nbd.2016.12.017 [DOI] [PubMed] [Google Scholar]
- Belyaev, N. D. , Kellett, K. A. B. , Beckett, C. , Makova, N. Z. , Revett, T. J. , Nalivaeva, N. N. , … Turner, A. J. (2010). The transcriptionally active amyloid precursor protein (APP) intracellular domain is preferentially produced from the 695 isoform of APP in Aβ‐secretase‐dependent pathway. Journal of Biological Chemistry, 285(53), 41443–41454. 10.1074/jbc.M110.141390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein, K. E. , Koronyo, Y. , Salumbides, B. C. , Sheyn, J. , Pelissier, L. , Lopes, D. H. J. , … Koronyo‐Hamaoui, M. (2014). Angiotensin‐converting enzyme overexpression in myelomonocytes prevents Alzheimer's‐like cognitive decline. Journal of Clinical Investigation, 124(3), 1000–1012. 10.1172/JCI66541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatia, V. , & Sharma, S. (2021). Role of mitochondrial dysfunction, oxidative stress and autophagy in progression of Alzheimer's disease. Journal of the Neurological Sciences, 421, 117253. 10.1016/j.jns.2020.117253 [DOI] [PubMed] [Google Scholar]
- Borchelt, D. R. , Davis, J. , Fischer, M. , Lee, M. K. , Slunt, H. H. , Ratovitsky, T. , … Price, D. L. (1996). A vector for expressing foreign genes in the brains and hearts of transgenic mice. Genetic Analysis ‐ Biomolecular Engineering, 13(6), 159–163. 10.1016/S1050-3862(96)00167-2 [DOI] [PubMed] [Google Scholar]
- Borchelt, D. R. , Ratovitski, T. , Lare van, J. , Lee, M. K. , Gonzales, V. , Jenkins, N. A. , … Sisodia, S. S. (1997). Accelerated amyloid deposition in the brains of transgenic mice Coexpressing mutant Presenilin 1 and amyloid precursor proteins. Neuron, 19, 939–945. 10.1016/s0896-6273(00)80974-5 [DOI] [PubMed] [Google Scholar]
- Borchelt, D. R. , Thinakaran, G. , Eckman, C. B. , Lee, M. K. , Davenport, F. , Ratovitsky, T. , … Sisodia, S. S. (1996). Familial Alzheimer's disease‐linked presenilin I variants elevate Aβ1‐ 42/1‐40 ratio in vitro and in vivo. Neuron, 17(5), 1005–1013. 10.1016/S0896-6273(00)80230-5 [DOI] [PubMed] [Google Scholar]
- Braak, H. , & Braak, E. (1991). Neuropathological stageing of Alzheimer‐related changes. Acta Neuropathologica, 82(4), 239–259. 10.1109/ICINIS.2015.10 [DOI] [PubMed] [Google Scholar]
- Buffo, A. , Rolando, C. , & Ceruti, S. (2010). Astrocytes in the damaged brain: Molecular and cellular insights into their reactive response and healing potential. Biochemical Pharmacology, 79(2), 77–89. 10.1016/j.bcp.2009.09.014 [DOI] [PubMed] [Google Scholar]
- Bukhari, H. , Glotzbach, A. , Kolbe, K. , Leonhardt, G. , Loosse, C. , & Müller, T. (2017). Small things matter: Implications of APP intracellular domain AICD nuclear signaling in the progression and pathogenesis of Alzheimer's disease. Progress in Neurobiology, 156, 189–213. 10.1016/j.pneurobio.2017.05.005 [DOI] [PubMed] [Google Scholar]
- Bushong, E. A. , Martone, M. E. , Jones, Y. Z. , & Ellisman, M. H. (2002). Protoplasmic astrocytes in CA1 stratum radiatum occupy separate anatomical domains. Journal of Neuroscience, 22(1), 183–192. 10.1523/jneurosci.22-01-00183.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Button, E. B. , Boyce, G. K. , Wilkinson, A. , Stukas, S. , Hayat, A. , Fan, J. , … Wellington, C. L. (2019). ApoA‐I deficiency increases cortical amyloid deposition, cerebral amyloid angiopathy, cortical and hippocampal astrogliosis, and amyloid‐associated astrocyte reactivity in APP/PS1 mice. Alzheimer's Research & Therapy, 11(1), 1–18. 10.1186/s13195-019-0497-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calió, M. L. , Mosini, A. C. , Marinho, D. S. , Salles, G. N. , Massinhani, F. H. , Ko, G. M. , & Porcionatto, M. A. (2021). Leptin enhances adult neurogenesis and reduces pathological features in a transgenic mouse model of Alzheimer's disease. Neurobiology of Disease, 148, 1–22. [DOI] [PubMed] [Google Scholar]
- Cao, X. , & Südhof, T. C. (2004). Dissection of amyloid‐β precursor protein‐dependent transcriptional transactivation. Journal of Biological Chemistry, 279(23), 24601–24611. 10.1074/jbc.M402248200 [DOI] [PubMed] [Google Scholar]
- Cerpa, W. , Hancke, J. L. , Morazzoni, P. , Bombardelli, E. , Riva, A. , Marin, P. , & Inestrosa, N. C. (2010). The Hyperforin derivative IDN5706 occludes spatial memory impairments and Neuropathological changes in a double transgenic Alzheimers mouse model. Current Alzheimer Research, 7(2), 126–133. 10.2174/1567210199392302050 [DOI] [PubMed] [Google Scholar]
- Ceyzériat, K. , Haim, L. B. , Denizot, A. , Pommier, D. , Matos, M. , Guillemaud, O. , … Escartin, C. (2018). Modulation of astrocyte reactivity improves functional deficits in mouse models of Alzheimer's disease. Acta Neuropathologica Communications, 6, 1–23. 10.1186/s40478-018-0606-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chartier‐Harlin, M. C. , Crawford, F. , Houlden, H. , Warren, A. , Hughes, D. , Fidani, L. , … Mullan, M. (1991). Early‐onset Alzheimer's disease caused by mutations at codon 717 of the β‐amyloid precursor protein gene. Nature, 353(6347), 844–846. 10.1038/353844a0 [DOI] [PubMed] [Google Scholar]
- Chun, H. , Im, H. , Kang, Y. J. , Kim, Y. , Shin, J. H. , Won, W. , … Lee, C. J. (2020). Severe reactive astrocytes precipitate pathological hallmarks of Alzheimer's disease via H2O2− production. Nature Neuroscience, 23(12), 1555–1566. 10.1038/s41593-020-00735-y [DOI] [PubMed] [Google Scholar]
- Chung, W.‐S. S. , Welsh, C. A. , Barres, B. A. , & Stevens, B. (2015). Do glia drive synaptic and cognitive impairment in disease? Nature Neuroscience, 18(11), 1539–1545. 10.1038/nn.4142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Citron, M. , Oltersdorf, T. , Haass, C. , McConlogue, L. , Hung, A. Y. , Seubert, P. , … Selkoe, D. J. (1992). Mutation of the beta‐amyloid precursor protein in familial Alzheimer's disease increases beta‐protein production. Nature, 360, 672–674. 10.1038/360672a0 [DOI] [PubMed] [Google Scholar]
- Clarke, L. E. , Liddelow, S. A. , Chakraborty, C. , Münch, A. E. , Heiman, M. , & Barres, B. A. (2018). Normal aging induces A1‐like astrocyte reactivity. Proceedings of the National Academy of Sciences of the United States of America, 115(8), E1896–E1905. 10.1073/pnas.1800165115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crook, R. , Verkkoniemi, A. , Perez‐Tur, J. , Mehta, N. , Baker, M. , Houlden, H. , … Haltia, M. (1998). A variant of Alzheimer's disease with spastic paraparesis and unusual plaques due to deletion of exon 9 of presenilin 1. Nature Medicine, 4(4), 452–455. 10.1038/nm0498-452 [DOI] [PubMed] [Google Scholar]
- De Bock, M. , Leybaert, L. , & Giaume, C. (2017). Connexin channels at the Glio‐vascular Interface: Gatekeepers of the brain. Neurochemical Research, 42(9), 2519–2536. 10.1007/s11064-017-2313-x [DOI] [PubMed] [Google Scholar]
- De Strooper, B. (2003). Aph‐1, Pen‐2, and Nicastrin with Presenilin generate an active γ‐Secretase complex. Neuron, 38(1), 9–12. 10.1016/S0896-6273(03)00205-8 [DOI] [PubMed] [Google Scholar]
- De Strooper, B. , & Karran, E. (2016). The cellular phase of Alzheimer's disease. Cell, 164(4), 603–615. 10.1016/j.cell.2015.12.056 [DOI] [PubMed] [Google Scholar]
- De Strooper, B. , Saftig, P. , Craessaerts, K. , Vanderstichele, H. , Guhde, G. , Annaert, W. , … Van Leuven, F. (1998). Deficiency of presenilin‐1 inhibits the normal cleavage of amyloid precursor protein. Nature, 391, 387–390. [DOI] [PubMed] [Google Scholar]
- Delekate, A. , Füchtemeier, M. , Schumacher, T. , Ulbrich, C. , Foddis, M. , & Petzold, G. C. (2014). Metabotropic P2Y1 receptor signalling mediates astrocytic hyperactivity in vivo in an Alzheimer's disease mouse model. Nature Communications, 5, 5422. 10.1038/ncomms6422 [DOI] [PubMed] [Google Scholar]
- Ding, Y. , Qiao, A. , Wang, Z. , Goodwin, J. S. , Lee, E. S. , Block, M. L. , … Fan, G. H. (2008). Retinoic acid attenuates beta‐amyloid deposition and rescues memory deficits in an Alzheimer's disease transgenic mouse model. The Journal of Neuroscience, 28, 11622–11634. 10.1523/JNEUROSCI.3153-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dossi, E. , Vasile, F. , & Rouach, N. (2018). Human astrocytes in the diseased brain. Brain Research Bulletin, 136(136), 139–156. 10.1016/j.brainresbull.2017.02.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doustar, J. , Rentsendorj, A. , Torbati, T. , Regis, G. C. , Fuchs, D. T. , Sheyn, J. , … Koronyo‐Hamaoui, M. (2020). Parallels between retinal and brain pathology and response to immunotherapy in old, late‐stage Alzheimer's disease mouse models. Aging Cell, 19(11), 1–25. 10.1111/acel.13246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duggan, S. P. , & McCarthy, J. V. (2016). Beyond γ‐secretase activity: The multifunctional nature of presenilins in cell signalling pathways. Cellular Signalling, 28(1), 1–11. 10.1016/j.cellsig.2015.10.006 [DOI] [PubMed] [Google Scholar]
- Durand, D. , Carniglia, L. , Beauquis, J. , Caruso, C. , Saravia, F. , & Lasaga, M. (2014). Astroglial mGlu3 receptors promote alpha‐secretase‐mediated amyloid precursor protein cleavage. Neuropharmacology, 79, 180–189. 10.1016/j.neuropharm.2013.11.015 [DOI] [PubMed] [Google Scholar]
- Duyckaerts, C. , Potier, M. C. , & Delatour, B. (2008). Alzheimer disease models and human neuropathology: Similarities and differences. Acta Neuropathologica, 115(1), 5–38. 10.1007/s00401-007-0312-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elder, G. A. , Gama Sosa, M. A. , & De Gasperi, R. (2010). Transgenic mouse models of Alzheimer's disease. Mount Sinai Journal of Medicine, 77, 69–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escartin, C. , Galea, E. , Lakatos, A. , O’Callaghan, J. P. , Petzold, G. C. , Serrano‐Pozo, A. , … Verkhratsky, A. (2021). Reactive astrocyte nomenclature, definitions, and future directions. Nature Neuroscience, 10.1038/s41593-020-00783-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escartin, C. , Guillemaud, O. , & Carrillo‐de Sauvage, M. A. (2019). Questions and (some) answers on reactive astrocytes. Glia, 67(12), 2221–2247. 10.1002/glia.23687 [DOI] [PubMed] [Google Scholar]
- Esquerda‐Canals, G. , Montoliu‐Gaya, L. , Güell‐Bosch, J. , & Villegas, S. (2017). Mouse models of Alzheimer's disease. Journal of Alzheimer's Disease, 57(4), 1171–1183. 10.3233/JAD-170045 [DOI] [PubMed] [Google Scholar]
- Foster, E. M. , Dangla‐Valls, A. , Lovestone, S. , Ribe, E. M. , & Buckley, N. J. (2019). Clusterin in Alzheimer's disease: Mechanisms, genetics, and lessons from other pathologies. Frontiers in Neuroscience, 13, 1–27. 10.3389/fnins.2019.00164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francis, R. , McGrath, G. , Zhang, J. , Ruddy, D. A. , Sym, M. , Apfeld, J. , … Curtis, D. (2002). Aph‐1 and pen‐2 are required for notch pathway signaling, γ‐secretase cleavage of βAPP, and presenilin protein accumulation. Developmental Cell, 3(1), 85–97. 10.1016/S1534-5807(02)00189-2 [DOI] [PubMed] [Google Scholar]
- Fujita, T. , Tozaki‐Saitoh, H. , & Inoue, K. (2009). P2Y1 receptor signaling enhances neuroprotection by astrocytes against oxidative stress via IL‐6 release in hippocampal cultures. Glia, 57(3), 244–257. 10.1002/glia.20749 [DOI] [PubMed] [Google Scholar]
- Gao, J. , Littman, R. , Diamante, G. , Xiao, X. , Ahn, S. , Yang, X. , … Tontonoz, P. (2020). Therapeutic IDOL reduction ameliorates amyloidosis and improves cognitive function in APP/PS1 mice. Molecular and Cellular Biology, 40, 1–19. 10.1128/MCB.00518-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia‐Alloza, M. , Robbins, E. M. , Zhang‐Nunes, S. X. , Purcell, S. M. , Betensky, R. A. , Raju, S. , … Frosch, M. P. (2006). Characterization of amyloid deposition in the APPswe/PS1dE9 mouse model of Alzheimer disease. Neurobiology of Disease, 24, 516–524. 10.1016/j.nbd.2006.08.017 [DOI] [PubMed] [Google Scholar]
- Giaume, C. , Koulakoff, A. , Roux, L. , Holcman, D. , & Rouach, N. (2010). Astroglial networks: A step further in neuroglial and gliovascular interactions. Nature Reviews Neuroscience, 11(2), 87–99. 10.1038/nrn2757 [DOI] [PubMed] [Google Scholar]
- Goate, A. , Chartier‐Harlin, M.‐C. , Mullan, M. , Brown, J. , Crawford, F. , Fidani, L. , … Hardy, J. (1991). Segregation of a missense mutation in the amyloid β‐protein precursor gene with familial Alzheimer's disease. Nature, 349, 704–706. 10.1038/349704a0 [DOI] [PubMed] [Google Scholar]
- Gong, Z. , Huang, J. , Xu, B. , Ou, Z. , Zhang, L. , Lin, X. , … Xuan, A. (2019). Urolithin a attenuates memory impairment and neuroinflammation in APP/PS1 mice. Journal of Neuroinflammation, 16(62), 1–13. 10.1186/s12974-019-1450-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goutte, C. , Tsunozaki, M. , Hale, V. A. , & Priess, J. R. (2002). APH‐1 is a multipass membrane protein essential for the notch signaling pathway in Caenorhabditis elegans embryos. Proceedings of the National Academy of Sciences of the United States of America, 99(2), 775–779. 10.1210/endo-63-5-707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo, Q. , Li, H. , Cole, A. L. , Hur, J. Y. , Li, Y. , & Zheng, H. (2013). Modeling Alzheimer's disease in mouse without mutant protein overexpression: Cooperative and independent effects of Aβ and tau. PLoS One, 8(11), 1–14. 10.1371/journal.pone.0080706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haass, C. , Lemere, C. A. , Capell, A. , Citron, M. , Seubert, P. , Lannfelt, L. , & Selkoe, D. J. (1995). The Swedish mutation causes early‐onset Alzheimer's disease by bèta‐secretase cleavage within the secretory pathway. Nature Medicine, 1(12), 1291–1296. 10.1038/nm1295-1291 [DOI] [PubMed] [Google Scholar]
- Habib, N. , Mccabe, C. , Medina, S. , Varshavsky, M. , Kitsberg, D. , Dvir‐Szternfeld, R. , … Schwartz, M. (2020). Disease‐associated astrocytes in Alzheimer's disease and aging. Nature Neuroscience, 23, 701–706. 10.1038/s41593-020-0624-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haim, L. B. , Ceyzeriat, K. , Carrillo‐de Sauvage, M. A. , Aubry, F. , Auregan, G. , Guillermier, M. , … Escartin, C. (2015). The JAK/STAT3 pathway is a common inducer of astrocyte reactivity in Alzheimer's and Huntington's diseases. Journal of Neuroscience, 35(6), 2817–2829. 10.1523/JNEUROSCI.3516-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall, A. M. , & Roberson, E. D. (2012). Mouse models of Alzheimer's disease. Brain Research Bulletin, 88(4), 3–12. 10.1016/j.brainresbull.2011.11.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hampel, H. , Mesulam, M. M. , Cuello, A. C. , Farlow, M. R. , Giacobini, E. , Grossberg, G. T. , … Khachaturian, Z. S. (2018). The cholinergic system in the pathophysiology and treatment of Alzheimer's disease. Brain, 141(7), 1917–1933. 10.1093/brain/awy132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy, J. (1997). Amyloid, the presenilins and Alzheimer's disease. Trends in Neurosciences, 20(4), 154–159. 10.1016/S0166-2236(96)01030-2 [DOI] [PubMed] [Google Scholar]
- Hardy, J. , & Allsop, D. (1991). Amyloid deposition as the central event in the aetiology of Alzheimer's disease. Trends in Pharmacological Sciences, 12, 383–388. 10.1016/0165-6147(91)90609-V [DOI] [PubMed] [Google Scholar]
- Hardy, J. , & Selkoe, D. J. (2002). The amyloid hypothesis of Alzheimer's disease: Progress and problems on the road to therapeutics. Science, 297, 353–356. 10.1126/science.1072994 [DOI] [PubMed] [Google Scholar]
- Hébert, S. S. , Serneels, L. , Tolia, A. , Craessaerts, K. , Derks, C. , Filippov, M. A. , … De Strooper, B. (2006). Regulated intramembrane proteolysis of amyloid precursor protein and regulation of expression of putative target genes. EMBO Reports, 7(7), 739–745. 10.1038/sj.embor.7400704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiltunen, M. , Helisalmi, S. , Mannermaa, A. , Alafuzoff, I. , Koivisto, A. M. , Lehtovirta, M. , … Soininen, H. (2000). Identification of a novel 4.6‐kb genomic deletion in presenilin‐1 gene which results in exclusion of exon 9 in a Finnish early onset Alzheimer's disease family: An Alu core sequence‐stimulated recombination? European Journal of Human Genetics, 8(4), 259–266. 10.1038/sj.ejhg.5200423 [DOI] [PubMed] [Google Scholar]
- Hol, E. M. , & Pekny, M. (2015). Glial fibrillary acidic protein (GFAP) and the astrocyte intermediate filament system in diseases of the central nervous system. Current Opinion in Cell Biology, 32, 121–130. 10.1016/j.ceb.2015.02.004 [DOI] [PubMed] [Google Scholar]
- Holubová, M. , Hrubá, L. , Popelová, A. , Bencze, M. , Pražienková, V. , Gengler, S. , … Maletínská, L. (2019). Liraglutide and a lipidized analog of prolactin‐releasing peptide show neuroprotective effects in a mouse model of β‐amyloid pathology. Neuropharmacology, 144, 377–387. 10.1016/j.neuropharm.2018.11.002 [DOI] [PubMed] [Google Scholar]
- Hong, S. , Beja‐Glasser, V. F. , Nfonoyim, B. M. , Frouin, A. , Li, S. , Ramakrishnan, S. , … Stevens, B. (2016). Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science, 352(6286), 712–716. 10.1126/science.aad8373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes, E. G. , Maguire, J. L. , McMinn, M. T. , Scholz, R. E. , & Sutherland, M. L. (2004). Loss of glial fibrillary acidic protein results in decreased glutamate transport and inhibition of PKA‐induced EAAT2 cell surface trafficking. Molecular Brain Research, 124(2), 114–123. 10.1016/j.molbrainres.2004.02.021 [DOI] [PubMed] [Google Scholar]
- Hyman, B. , & Tanzi, R. E. (2019). Effects of species‐specific genetics on Alzheimer's mouse models. Neuron, 101(3), 351–352. 10.1016/j.neuron.2019.01.021 [DOI] [PubMed] [Google Scholar]
- Inestrosa, N. C. , Carvajal, F. J. , Zolezzi, J. M. , Tapia‐Rojas, C. , Serrano, F. , Karmelic, D. , … Santos, M. J. (2013). Peroxisome proliferators reduce spatial memory impairment, synaptic failure, and neurodegeneration in brains of a double transgenic mice model of Alzheimer's disease. Journal of Alzheimer's Disease, 33(4), 941–959. 10.3233/JAD-2012-120397 [DOI] [PubMed] [Google Scholar]
- Ishii, K. , Ii, K. , Hasegawa, T. , Shoji, S. , Doi, A. , & Mori, H. (1997). Increased Aβ 42(43)‐plaque deposition in early‐onset familial Alzheimer's disease brains with the deletion of exon 9 and the missense point mutation (H163R) in the PS‐1 gene. Neuroscience Letters, 228(1), 17–20. 10.1016/S0304-3940(97)00347-9 [DOI] [PubMed] [Google Scholar]
- Itagaki, S. , Mcgeer, P. L. , & Akiyama, S. (1989). Relationship of microglia and astrocytes to amyloid deposits of Alzheimer disease. Journal of Neuroimmunology, 24, 173–182. 10.1017/CBO9781107415324.004 [DOI] [PubMed] [Google Scholar]
- Jackson, H. M. , Onos, K. D. , Pepper, K. W. , Graham, L. C. , Akeson, E. C. , Byers, C. , … Howell, G. R. (2015). DBA/2J genetic background exacerbates spontaneous lethal seizures but lessens amyloid deposition in a mouse model of Alzheimer's disease. PLoS One, 10(5), 1–18. 10.1371/journal.pone.0125897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jankowsky, J. L. , Fadale, D. J. , Anderson, J. , Xu, G. M. , Gonzales, V. , Jenkins, N. A. , … Borchelt, D. R. (2004). Mutant presenilins specifically elevate the levels of the 42 residue beta‐amyloid peptide in vivo: Evidence for augmentation of a 42‐specific γ‐secretase. Human Molecular Genetics, 13(2), 159–170. 10.1093/hmg/ddh019 [DOI] [PubMed] [Google Scholar]
- Jankowsky, J. L. , Slunt, H. H. , Ratovitski, T. , Jenkins, N. A. , Copeland, N. G. , & Borchelt, D. R. (2001). Co‐expression of multiple transgenes in mouse CNS: A comparison of strategies. Biomolecular Engineering, 17(6), 157–165. 10.1016/S1389-0344(01)00067-3 [DOI] [PubMed] [Google Scholar]
- Jankowsky, J. L. , Younkin, L. H. , Gonzales, V. , Fadale, D. J. , Slunt, H. H. , Lester, H. A. , … Borchelt, D. R. (2007). Rodent Aβ modulates the solubility and distribution of amyloid deposits in transgenic mice. The Journal of Biological Chemistry, 282(31), 22707–22720. 10.1074/jbc.M611050200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jankowsky, J. L. , & Zheng, H. (2017). Practical considerations for choosing a mouse model of Alzheimer's disease. Molecular Neurodegeneration, 12(1), 1–22. 10.1186/s13024-017-0231-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiao, S.‐S. , Bu, X.‐L. , Liu, Y.‐H. , Zhu, C. , Wang, Q.‐H. , Shen, L.‐L. , … Wang, Y.‐J. (2016). Sex dimorphism profile of Alzheimer's disease‐type pathologies in an APP/PS1 mouse model. Neurotoxicity Research, 29(2), 256–266. 10.1007/s12640-015-9589-x [DOI] [PubMed] [Google Scholar]
- Jin, J. J. , Kim, H. D. , Maxwell, J. A. , Li, L. , & Fukuchi, K. I. (2008). Toll‐like receptor 4‐dependent upregulation of cytokines in a transgenic mouse model of Alzheimer's disease. Journal of Neuroinflammation, 5, 1–10. 10.1186/1742-2094-5-23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo, S. , Yarishkin, O. , Hwang, Y. J. , Chun, Y. E. , Park, M. , Woo, D. H. , … Lee, C. J. (2014). GABA from reactive astrocytes impairs memory in mouse models of Alzheimer's disease. Nature Medicine, 20(8), 886–896. 10.1038/nm.3639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston, J. , O'Neill, C. , Lannfelt, L. , Winblad, B. , & Cowburn, R. F. (1994). The significance of the Swedish APP670/671 mutation for the development of Alzheimer's disease amyloidosis. Neurochemistry International, 25(1), 73–80. 10.1016/0197-0186(94)90056-6 [DOI] [PubMed] [Google Scholar]
- Joo, Y. , Ha, S. , Hong, B. H. , Kim, J. A. , Chang, K. A. , Liew, H. , … Kim, H. S. (2010). Amyloid precursor protein binding protein‐1 modulates cell cycle progression in fetal neural stem cells. PLoS One, 5(12), 1–12. 10.1371/journal.pone.0014203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakuda, N. , Funamoto, S. , Yagishita, S. , Takami, M. , Osawa, S. , Dohmae, N. , & Ihara, Y. (2006). Equimolar production of amyloid β‐protein and amyloid precursor protein intracellular domain from β‐carboxyl‐terminal fragment by γ‐secretase. Journal of Biological Chemistry, 281(21), 14776–14786. 10.1074/jbc.M513453200 [DOI] [PubMed] [Google Scholar]
- Kamphuis, W. , Kooijman, L. , Orre, M. , Stassen, O. , Pekny, M. , & Hol, E. M. (2015). GFAP and Vimentin deficiency alters gene expression in astrocytes and microglia in wild‐type mice and changes the transcriptional response of reactive glia in mouse model for Alzheimer's disease. Glia, 63, 1036–1056. 10.1002/glia.22800 [DOI] [PubMed] [Google Scholar]
- Kamphuis, W. , Middeldorp, J. , Kooijman, L. , Sluijs, J. a. , Kooi, E.‐J. , Moeton, M. , … Hol, E. M. (2014). Glial fibrillary acidic protein isoform expression in plaque related astrogliosis in Alzheimer's disease. Neurobiology of Aging, 35(3), 492–510. 10.1016/j.neurobiolaging.2013.09.035 [DOI] [PubMed] [Google Scholar]
- Kamphuis, W. , Orre, M. , Kooijman, L. , Dahmen, M. , & Hol, E. M. (2012). Differential cell proliferation in the cortex of the APPswePS1dE9 Alzheimer's disease mouse model. Glia, 60, 615–629. 10.1002/glia.22295 [DOI] [PubMed] [Google Scholar]
- Kato, S. , Gondo, T. , Hoshii, Y. , Takahashi, M. , Yamada, M. , & Ishihara, T. (1998). Confocal observation of senile plaques in Alzheimer's disease: Senile plaque morphology and relationship between senile plaques and astrocytes. Pathology International, 48(5), 332–340. 10.1111/j.1440-1827.1998.tb03915.x [DOI] [PubMed] [Google Scholar]
- Kettenmann, H. , & Verkhratsky, A. (2008). Neuroglia: The 150 years after. Trends in Neurosciences, 31(12), 653–659. 10.1016/j.tins.2008.09.003 [DOI] [PubMed] [Google Scholar]
- Kim, J. , Kleizen, B. , Choy, R. , Thinakaran, G. , Sisodia, S. S. , & Schekman, R. W. (2007). Biogenesis of γ‐secretase early in the secretory pathway. Journal of Cell Biology, 179(5), 951–963. 10.1083/jcb.200709012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirischuk, S. , Parpura, V. , & Verkhratsky, A. (2012). Sodium dynamics: Another key to astroglial excitability? Trends in Neurosciences, 35(8), 497–506. 10.1016/j.tins.2012.04.003 [DOI] [PubMed] [Google Scholar]
- Koronyo, Y. , Salumbides, B. C. , Sheyn, J. , Pelissier, L. , Li, S. , Ljubimov, V. , … Koronyo‐hamaoui, M. (2015). Therapeutic effects of glatiramer acetate and grafted CD115 + monocytes in a mouse model of Alzheimer's disease. Brain, 138, 2399–2422. 10.1093/brain/awv150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koronyo‐Hamaoui, M. , Sheyn, J. , Hayden, E. Y. , Li, S. , Fuchs, D. T. , Regis, G. C. , … Rentsendorj, A. (2020). Peripherally derived angiotensin converting enzyme‐enhanced macrophages alleviate Alzheimer‐related disease. Brain, 143(1), 336–358. 10.1093/brain/awz364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraft, A. W. , Hu, X. , Yoon, H. , Yan, P. , Xiao, Q. , Wang, Y. , … Lee, J. M. (2013). Attenuating astrocyte activation accelerates plaque pathogenesis in APP/PS1 mice. FASEB Journal, 27(1), 187–198. 10.1096/fj.12-208660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuchibhotla, K. V. , Goldman, S. T. , Lattarulo, C. R. , Wu, H.‐Y. Y. , Hyman, B. T. , & Bacskai, B. J. (2008). Aβ plaques lead to aberrant regulation of calcium homeostasis in vivo resulting in structural and functional disruption of neuronal networks. Neuron, 59(2), 214–225. 10.1016/j.neuron.2008.06.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuchibhotla, K. V. , Lattarulo, C. R. , Hyman, B. T. , & Bacskai, B. J. (2009). Synchronous hyperactivity and intercellular calcium waves in astrocytes in Alzheimer mice. Science, 323, 1211–1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert, J. C. , Ibrahim‐Verbaas, C. A. , Harold, D. , Naj, A. C. , Sims, R. , Bellenguez, C. , … Seshadri, S. (2013). Meta‐analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer's disease. Nature Genetics, 45(12), 1452–1458. 10.1038/ng.2802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson, J. , Lynch, G. , Games, D. , & Seubert, P. (1999). Alterations in synaptic transmission and long‐term potentiation in hippocampal slices from young and aged PDAPP mice. Brain Research, 840, 23–35. 10.1016/S0006-8993(99)01698-4 [DOI] [PubMed] [Google Scholar]
- Lee, K. , Lee, H. , Lin, H. , Tsay, H. , Tsai, F. , Shyue, S. , & Lee, T. (2016). Role of transient receptor potential ankyrin 1 channels in Alzheimer's disease. Journal of Neuroinflammation, 13(92), 1–16. 10.1186/s12974-016-0557-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, M. K. , Borchelt, D. R. , Kim, G. , Thinakaran, G. , Slunt, H. H. , Ratovitski, T. , & Martin, L. J. (1997). Hyperaccumulation of FAD‐linked presenilin 1 variants in vivo. Nature Medicine, 3(7), 756–760. 10.1038/nm0797-756 [DOI] [PubMed] [Google Scholar]
- Lesuisse, C. , Xu, G. , Anderson, J. , Wong, M. , Jankowsky, J. , Holtz, G. , … Borchelt, D. R. (2001). Hyper‐expression of human apolipoprotein E4 in astroglia and neurons does not enhance amyloid deposition in transgenic mice. Human Molecular Genetics, 10(22), 2525–2537. [DOI] [PubMed] [Google Scholar]
- Levy‐lahad, E. , Wasco, W. , Poorkaj, P. , Romano, D. M. , Pettingell, W. H. , Yu, C. , … Tanzi, R. E. (1995). Candidate gene for the chromosome 1 familial Alzheimer's disease locus. Science, 269, 973–977. 10.1126/science.7638622 [DOI] [PubMed] [Google Scholar]
- Li, L. , Lundkvist, A. , Andersson, D. , Wilhelmsson, U. , Nagai, N. , Pardo, A. C. , … Pekny, M. (2008). Protective role of reactive astrocytes in brain ischemia. Journal of Cerebral Blood Flow and Metabolism, 28(3), 468–481. 10.1038/sj.jcbfm.9600546 [DOI] [PubMed] [Google Scholar]
- Liang, F. , Yang, S. , Zhang, Y. , & Hao, T. (2019). Social housing promotes cognitive function through enhancing synaptic plasticity in APP/PS1 mice. Behavioural Brain Research, 368, 1–11. 10.1016/j.bbr.2019.111910 [DOI] [PubMed] [Google Scholar]
- Liddelow, S. A. , Guttenplan, K. A. , Clarke, L. E. , Bennett, F. C. , Bohlen, C. J. , Schirmer, L. , … Barres, B. A. (2017). Neurotoxic reactive astrocytes are induced by activated microglia. Nature, 541, 481–487. 10.1038/nature21029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liedtke, W. , Edelmann, W. , Bieri, P. L. , Chiu, F. C. , Cowan, N. J. , Kucherlapati, R. , & Raine, C. S. (1996). GFAP is necessary for the integrity of CNS white matter architecture and long‐term maintenance of myelination. Neuron, 17(4), 607–615. 10.1016/S0896-6273(00)80194-4 [DOI] [PubMed] [Google Scholar]
- Liedtke, W. , Edelmann, W. , Chiu, F. C. , Kucherlapati, R. , & Raine, C. S. (1998). Experimental autoimmune encephalomyelitis in mice lacking glial fibrillary acidic protein is characterized by a more severe clinical course and an infiltrative central nervous system lesion. American Journal of Pathology, 152(1), 251–259. [PMC free article] [PubMed] [Google Scholar]
- Liu, C. C. , Zhao, N. , Fu, Y. , Wang, N. , Linares, C. , Tsai, C. W. , & Bu, G. (2017). ApoE4 accelerates early seeding of amyloid pathology. Neuron, 96(5), 1024–1032. 10.1016/j.neuron.2017.11.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, Z. , Li, Y. , Cui, Y. , Roberts, C. , Lu, M. , Wilhelmsson, U. , … Chopp, M. (2014). Beneficial effects of gfap/vimentin reactive astrocytes for axonal remodeling and motor behavioral recovery in mice after stroke. Glia, 62(12), 2022–2033. 10.1002/glia.22723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo, R. , Su, L. , Li, G. , Yang, J. , Liu, Q. , Yang, L. , & Zhang, D. (2020). Activation of PPARA‐mediated autophagy reduces Alzheimer disease‐like pathology and cognitive decline in a murine model. Autophagy, 16(1), 52–69. 10.1080/15548627.2019.1596488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malm, T. , Koistinaho, J. , & Kanninen, K. (2011). Utilization of APPswe/PS1dE9 transgenic mice in research of Alzheimer's disease: Focus on gene therapy and cell‐based therapy applications. International Journal of Alzheimer's Disease, 2011, 1–8. 10.4061/2011/517160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malm, T. M. , Iivonen, H. , Goldsteins, G. , Keksa‐Goldsteine, V. , Ahtoniemi, T. , Kanninen, K. , … Koistinaho, J. (2007). Pyrrolidine dithiocarbamate activates Akt and improves spatial learning in APP/PS1 mice without affecting β‐amyloid burden. The Journal of Neuroscience, 27(14), 3712–3721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann, D. M. A. , Takeuchi, A. , Sato, S. , Cairns, N. J. , Lantos, P. L. , Rossor, M. N. , … Iwatsubo, T. (2001). Cases of Alzheimer's disease due to deletion of exon 9 of the presenilin‐1 gene show an unusual but characteristic β‐amyloid pathology known as “cotton wool” plaques. Neuropathology and Applied Neurobiology, 27(3), 189–196. 10.1046/j.1365-2990.2001.00316.x [DOI] [PubMed] [Google Scholar]
- Manocha, G. , Ghatak, A. , Puig, K. , & Combs, C. (2018). Anti‐α4β1 integrin antibodies attenuated brain inflammatory changes in a mouse model of Alzheimer's disease. Current Alzheimer Research, 15(12), 1123–1135. 10.2174/1567205015666180801111033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manocha, G. D. , Ghatak, A. , Puig, K. L. , Kraner, S. D. , Norris, C. M. , & Combs, C. K. (2017). NFATc2 modulates microglial activation in the AβPP/PS1 mouse model of Alzheimer's disease. Journal of Alzheimer's Disease, 58(3), 775–787. 10.3233/JAD-151203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mederos, S. , Hernández‐Vivanco, A. , Ramírez‐Franco, J. , Martín‐Fernández, M. , Navarrete, M. , Yang, A. , … Perea, G. (2018). Melanopsin for precise optogenetic activation of astrocyte‐neuron networks. Glia, 67(5), 915–934. 10.1002/glia.23580 [DOI] [PubMed] [Google Scholar]
- Mellott, T. J. , Huleatt, O. M. , Shade, B. N. , Pender, S. M. , Liu, Y. B. , Slack, B. E. , & Blusztajn, J. K. (2017). Perinatal choline supplementation reduces amyloidosis and increases choline acetyltransferase expression in the hippocampus of the APPswePS1dE9 Alzheimer's disease model mice. PLoS One, 12(1), 1–22. 10.1371/journal.pone.0170450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendes, F. R. , Leclerc, J. L. , Liu, L. , Kamat, P. K. , Naziripour, A. , Hernandez, D. , … Doré, S. (2020). Effect of experimental ischemic stroke and PGE2 EP1 selective antagonism in Alzheimer's disease mouse models. Journal of Alzheimer's Disease, 74, 173–187. 10.3233/jad-191069 [DOI] [PubMed] [Google Scholar]
- Minkeviciene, R. , Rheims, S. , Dobszay, M. B. , Zilberter, M. , Hartikainen, J. , Fulop, L. , … Tanila, H. (2009). Amyloid beta‐induced neuronal hyperexcitability triggers progressive epilepsy. Journal of Neuroscience, 29, 3453–3462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minter, M. R. , Hinterleitner, R. , Meisel, M. , Zhang, C. , Leone, V. , Zhang, X. , … Sisodia, S. S. (2017). Antibiotic‐induced perturbations in microbial diversity during post‐natal development alters amyloid pathology in an aged APPSWE/PS1ΔE9murine model of Alzheimer's disease. Scientific Reports, 7(1), 1–18. 10.1038/s41598-017-11047-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moechars, D. , Dewachter, I. , Lorent, K. , Reversé, D. , Baekelandt, V. , Naidu, A. , … Van Leuven, F. (1999). Early phenotypic changes in transgenic mice that overexpress different mutants of amyloid precursor protein in brain. Journal of Biological Chemistry, 274(10), 6483–6492. 10.1074/jbc.274.10.6483 [DOI] [PubMed] [Google Scholar]
- Moreno‐Gonzalez, I. , Estrada, L. D. , Sanchez‐Mejias, E. , & Soto, C. (2013). Smoking exacerbates amyloid pathology in a mouse model of Alzheimer's disease. Nature Communications, 4, 1495. 10.1038/ncomms2494 [DOI] [PubMed] [Google Scholar]
- Mori, T. , Koyama, N. , Yokoo, T. , Segawa, T. , Maeda, M. , Sawmiller, D. , … Town, T. (2020). Gallic acid is a dual α/β‐secretase modulator that reverses cognitive impairment and remediates pathology in Alzheimer's mice. Journal of Biological Chemistry, 295(48), 16251–16266. 10.1074/jbc.ra119.012330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori, T. , Rezai‐Zadeh, K. , Koyama, N. , Arendash, G. W. , Yamaguchi, H. , Kakuda, N. , … Town, T. (2012). Tannic acid is a natural β‐Secretase inhibitor that prevents cognitive impairment and mitigates Alzheimer‐like pathology in transgenic mice. Journal of Biological Chemistry, 287(9), 6912–6927. 10.1074/jbc.M111.294025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullan, M. , Crawford, F. , Axelman, K. , Houlden, H. , Lilius, L. , Winblad, B. , & Lannfelt, L. (1992). A pathogenic mutation for probable Alzheimer's disease in the APP gene at the N‐terminus of β‐amyloid. Nature Genetics, 1(5), 345–347. 10.1038/ng0892-345 [DOI] [PubMed] [Google Scholar]
- Mungenast, A. E. , Siegert, S. , & Tsai, L. H. (2016). Modeling Alzheimer's disease with human induced pluripotent stem (iPS) cells. Molecular and Cellular Neuroscience, 73, 13–31. 10.1016/j.mcn.2015.11.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nalivaeva, N. N. , & Turner, A. J. (2013). The amyloid precursor protein: A biochemical enigma in brain development, function and disease. FEBS Letters, 587, 2046–2054. 10.1016/j.febslet.2013.05.010 [DOI] [PubMed] [Google Scholar]
- Nelson, P. T. , Alafuzoff, I. , Bigio, E. H. , Bouras, C. , Braak, H. , Cairns, N. J. , … Beach, T. G. (2012). Correlation of Alzheimer disease neuropathologic changes with cognitive status: A review of the literature. Journal of Neuropathology and Experimental Neurology, 71(5), 362–381. 10.1097/NEN.0b013e31825018f7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neuner, S. M. , Heuer, S. E. , Huentelman, M. J. , O'Connell, K. M. S. , & Kaczorowski, C. C. (2019). Harnessing genetic complexity to enhance translatability of Alzheimer's disease mouse models: A path toward precision medicine. Neuron, 101(3), 399–411.e5. 10.1016/j.neuron.2018.11.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikolaev, A. , McLaughlin, T. , O'Leary, D. D. M. , & Tessier‐Lavigne, M. (2009). APP binds DR6 to trigger axon pruning and neuron death via distinct caspases. Nature, 457(7232), 981–989. 10.1038/nature07767 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Oberheim, N. A. , Takano, T. , Han, X. , He, W. , Lin, J. H. C. , Wang, F. , … Nedergaard, M. (2009). Uniquely hominid features of adult human astrocytes. Journal of Neuroscience, 29(10), 3276–3287. 10.1523/JNEUROSCI.4707-08.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Octave, J. N. , Pierrot, N. , Ferrao Santos, S. , Nalivaeva, N. N. , & Turner, A. J. (2013). From synaptic spines to nuclear signaling: Nuclear and synaptic actions of the amyloid precursor protein. Journal of Neurochemistry, 126(2), 183–190. 10.1111/jnc.12239 [DOI] [PubMed] [Google Scholar]
- Orre, M. , Kamphuis, W. , Dooves, S. , Kooijman, L. , Chan, E. T. , Kirk, C. J. , … Hol, E. M. (2013). Reactive glia show increased immunoproteasome activity in Alzheimer's disease. Brain, 136, 1415–1431. 10.1093/brain/awt083 [DOI] [PubMed] [Google Scholar]
- Orre, M. , Kamphuis, W. , Osborn, L. M. , Jansen, A. H. P. P. , Kooijman, L. , Bossers, K. , & Hol, E. M. (2014). Isolation of glia from Alzheimer's mice reveals inflammation and dysfunction. Neurobiology of Aging, 35(12), 2746–2760. 10.1016/j.neurobiolaging.2014.06.004 [DOI] [PubMed] [Google Scholar]
- Orre, M. , Kamphuis, W. , Osborn, L. M. , Melief, J. , Kooijman, L. , Huitinga, I. , … Hol, E. M. (2014). Acute isolation and transcriptome characterization of cortical astrocytes and microglia from young and aged mice. Neurobiology of Aging, 35(1), 1–14. 10.1016/j.neurobiolaging.2013.07.008 [DOI] [PubMed] [Google Scholar]
- Ortinski, P. I. , Dong, J. , Mungenast, A. , Yue, C. , Takano, H. , Watson, D. J. , … Coulter, D. A. (2010). Selective induction of astrocytic gliosis generates deficits in neuronal inhibition. Nature Neuroscience, 13(5), 584–591. 10.1038/nn.2535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osborn, L. M. , Kamphuis, W. , Wadman, W. J. , & Hol, E. M. (2016). Astrogliosis: An integral player in the pathogenesis of Alzheimer's disease. Progress in Neurobiology, 144, 121–141. 10.1016/j.pneurobio.2016.01.001 [DOI] [PubMed] [Google Scholar]
- Park, J. H. , Ju, Y. H. , Choi, J. W. , Song, H. J. , Jang, B. K. , Woo, J. , … Park, K. D. (2019). Newly developed reversible MAO‐B inhibitor circumvents the shortcomings of irreversible inhibitors in Alzheimer's disease. Science Advances, 5(3), 1–12. 10.1126/sciadv.aav0316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park, J. H. , Widi, G. A. , Gimbel, D. A. , Harel, N. Y. , Lee, D. H. S. , & Strittmatter, S. M. (2006). Subcutaneous Nogo receptor removes brain amyloid‐ and improves spatial memory in Alzheimer's transgenic mice. Journal of Neuroscience, 26(51), 13279–13286. 10.1523/JNEUROSCI.4504-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park, T.‐S. , Ryu, Y.‐K. , Park, H.‐Y. , Kim, J. Y. , Go, J. , Noh, J.‐R. , … Kim, K.‐S. (2017). Humulus japonicus inhibits the progression of Alzheimer's disease in a APP/PS1 transgenic mouse model. International Journal of Molecular Medicine, 39, 21–30. 10.3892/ijmm.2016.2804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pekny, M. , Pekna, M. , Messing, A. , Steinhäuser, C. , Lee, J. M. , Parpura, V. , … Verkhratsky, A. (2016). Astrocytes: A central element in neurological diseases. Acta Neuropathologica, 131(3), 323–345. 10.1007/s00401-015-1513-1 [DOI] [PubMed] [Google Scholar]
- Pelegrin, P. , & Surprenant, A. (2009). The P2X7 receptor—Pannexin connection to dye uptake and IL‐1β release. Purinergic Signalling, 5(2), 129–137. 10.1007/s11302-009-9141-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez, S. E. , Dar, S. , Ikonomovic, M. D. , DeKosky, S. T. , & Mufson, E. J. (2007). Cholinergic forebrain degeneration in the APPswe/PS1ΔE9 transgenic mouse. Neurobiology of Disease, 28(1), 3–15. 10.1016/j.nbd.2007.06.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez‐Nievas, B. G. , Hammerschmidt, T. , Kummer, M. P. , Terwel, D. , Leza, J. C. , & Heneka, M. T. (2011). Restraint stress increases neuroinflammation independently of amyloid β levels in amyloid precursor protein/PS1 transgenic mice. Journal of Neurochemistry, 116(1), 43–52. 10.1111/j.1471-4159.2010.07083.x [DOI] [PubMed] [Google Scholar]
- Perez‐Tur, J. , Froelich, S. , Prihar, G. , Crook, R. , Baker, M. , Duff, K. , … Hutton, M. (1995). A mutation in Alzheimer's disease destroying a splice acceptor site in the presenili‐1 gene. Neuroreport, 7, 297–301. [PubMed] [Google Scholar]
- Pestana, F. , Edwards‐Faret, G. , Belgard, T. G. , Martirosyan, A. , & Holt, M. G. (2020). No longer underappreciated: The emerging concept of astrocyte heterogeneity in neuroscience. Brain Sciences, 10(168), 1–21. 10.3390/brainsci10030168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters, O. , Schipke, C. G. , Philipps, A. , Haas, B. , Pannasch, U. , Wang, L. P. , … Kettenmann, H. (2009). Astrocyte function is modified by Alzheimer's disease‐like pathology in aged mice. Journal of Alzheimer's Disease, 18(1), 177–189. 10.3233/JAD-2009-1140 [DOI] [PubMed] [Google Scholar]
- Poltorak, A. , He, X. , Smirnova, I. , Liu, M. Y. , Van Huffel, C. , Du, X. , … Beutler, B. (1998). Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: Mutations in Tlr4 gene. Science, 282(11), 2085–2088. 10.1126/science.282.5396.2085 [DOI] [PubMed] [Google Scholar]
- Prikhodko, O. , Rynearson, K. D. , Sekhon, T. , Mante, M. M. , Nguyen, P. D. , Rissman, R. A. , … Wagner, S. L. (2020). The GSM BPN‐15606 as a potential candidate for preventative therapy in Alzheimer's disease. Journal of Alzheimer's Disease, 73(4), 1541–1554. 10.3233/JAD-190442 [DOI] [PubMed] [Google Scholar]
- Qu, B.‐X. , Rosenberg, R. N. , Johnston, S. A. , Hynan, L. S. , Li, L. , & Xiang, Q. (2007). Aβ42 gene vaccine prevents Aβ42 deposition in brain of double transgenic mice. Journal of the Neurological Sciences, 260, 204–213. 10.1016/j.jns.2007.05.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajendran, L. , Honsho, M. , Zahn, T. R. , Keller, P. , Geiger, K. D. , Verkade, P. , & Simons, K. (2006). Alzheimer's disease β‐amyloid peptides are released in association with exosomes. Proceedings of the National Academy of Sciences of the United States of America, 103(30), 11172–11177. 10.1073/pnas.0603838103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reichenbach, N. , Delekate, A. , Plescher, M. , Schmitt, F. , Krauss, S. , Blank, N. , … Petzold, G. C. (2019). Inhibition of Stat3‐mediated astrogliosis ameliorates pathology in an Alzheimer's disease model. EMBO Molecular Medicine, 11(2), 1–16. 10.15252/emmm.201809665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren, R. , Zhang, L. , & Wang, M. (2018). Specific deletion connexin43 in astrocyte ameliorates cognitive dysfunction in APP/PS1 mice. Life Sciences, 208, 175–191. 10.1016/j.lfs.2018.07.033 [DOI] [PubMed] [Google Scholar]
- Rogaev, E. I. , Sherrington, R. , Rogaeva, E. A. , Levesque, G. , Ikeda, M. , Liang, Y. , … St George‐Hyslop, P. H. (1995). Familial Alzheimer's disease in hindreds with missense mutations in a gene on chromosome 1 related to the Alzheimer's disease type 3 gene. Nature, 376, 775–778. 10.1038/376775a0 [DOI] [PubMed] [Google Scholar]
- Rouach, N. , Koulakoff, A. , Abudara, V. , Willecke, K. , & Giaume, C. (2008). Astroglial metabolic networks sustain hippocampal synaptic transmission. Science, 322, 1551–1555. 10.1126/science.1164022 [DOI] [PubMed] [Google Scholar]
- Ruan, L. , Kang, Z. , Pei, G. , & Le, Y. (2009). Amyloid deposition and inflammation in Appswe/Ps1de9 mouse model of Alzheimer's disease. Current Alzheimer Research, 6, 531–540. [DOI] [PubMed] [Google Scholar]
- Saito, T. , Matsuba, Y. , Mihira, N. , Takano, J. , Nilsson, P. , Itohara, S. , … Saido, T. C. (2014). Single app knock‐in mouse models of Alzheimer's disease. Nature Neuroscience, 17(5), 661–663. 10.1038/nn.3697 [DOI] [PubMed] [Google Scholar]
- Salazar, S. V. , Cox, T. O. , Lee, S. , Brody, A. H. , Chyung, A. S. , Haas, L. T. , & Strittmatter, S. M. (2019). Alzheimer's disease risk factor Pyk2 mediates amyloid‐β‐induced synaptic dysfunction and loss. The Journal of Neuroscience, 39(4), 758–772. 10.1523/jneurosci.1873-18.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salles, G. N. , Calió, M. L. , Hölscher, C. , Pacheco‐Soares, C. , Porcionatto, M. , & Lobo, A. O. (2020). Neuroprotective and restorative properties of the GLP‐1/GIP dual agonist DA‐JC1 compared with a GLP‐1 single agonist in Alzheimer's disease. Neuropharmacology, 162, 107813. 10.1016/j.neuropharm.2019.107813 [DOI] [PubMed] [Google Scholar]
- Salminen, A. , Kaarniranta, K. , Kauppinen, A. , Ojala, J. , Haapasalo, A. , Soininen, H. , & Hiltunen, M. (2013). Impaired autophagy and APP processing in Alzheimer's disease: The potential role of Beclin 1 interactome. Progress in Neurobiology, 106(107), 33–54. 10.1016/j.pneurobio.2013.06.002 [DOI] [PubMed] [Google Scholar]
- Sannerud, R. , & Annaert, W. (2009). Trafficking, a key player in regulated intramembrane proteolysis. Seminars in Cell and Developmental Biology, 20(2), 183–190. 10.1016/j.semcdb.2008.11.004 [DOI] [PubMed] [Google Scholar]
- Sannerud, R. , Declerck, I. , Peric, A. , Raemaekers, T. , Menendez, G. , Zhou, L. , … Annaert, W. (2011). ADP ribosylation factor 6 (ARF6) controls amyloid precursor protein (APP) processing by mediating the endosomal sorting of BACE1. Proceedings of the National Academy of Sciences of the United States of America, 108(34), E559–E568. 10.1073/pnas.1100745108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sannerud, R. , Esselens, C. , Ejsmont, P. , Mattera, R. , Rochin, L. , Tharkeshwar, A. K. , … Annaert, W. (2016). Restricted location of PSEN2/γ‐Secretase determines substrate specificity and generates an intracellular Aβ Pool. Cell, 166(1), 193–208. 10.1016/j.cell.2016.05.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaguri, H. , Nilsson, P. , Hashimoto, S. , Nagata, K. , Saito, T. , De Strooper, B. , … Saido, T. C. (2017). APP mouse models for Alzheimer's disease preclinical studies. The EMBO Journal, 36(17), 2473–2487. 10.15252/embj.201797397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selkoe, D. J. (1991). The molecular pathology of Alzheimer's disease. Neuron, 6, 487–498. 10.1016/0896-6273(91)90052-2 [DOI] [PubMed] [Google Scholar]
- Selkoe, D. J. , & Hardy, J. (2016). The amyloid hypothesis of Alzheimer's disease at 25 years. EMBO Molecular Medicine, 8(6), 595–608. 10.15252/emmm.201606210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selkoe, D. J. , Yamazaki, T. , Citron, M. , Podlisny, M. B. , Koo, E. H. , Teplow, D. B. , & Haass, C. (1996). The role of APP processing and trafficking pathways in the formation of amyloid β‐protein. Annals of the New York Academy of Sciences, 777(617), 57–64. 10.1111/j.1749-6632.1996.tb34401.x [DOI] [PubMed] [Google Scholar]
- Serrano, M. P. , Herrero‐Labrador, R. , Futch, H. S. , Serrano, J. , Romero, A. , Fernandez, A. P. , … Martínez‐Murillo, R. (2017). The proof‐of‐concept of ASS234: Peripherally administered ASS234 enters the central nervous system and reduces pathology in a male mouse model of Alzheimer's disease. Journal of Psychiatry and Neuroscience, 42(1), 59–69. 10.1503/jpn.150209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shemer, I. , Holmgren, C. , Min, R. , Fülöp, L. , Zilberter, M. , Sousa, K. M. , … Harkany, T. (2006). Non‐fibrillar β‐amyloid abates spike‐timing‐dependent synaptic potentiation at excitatory synapses in layer 2/3 of the neocortex by targeting postsynaptic AMPA receptors. European Journal of Neuroscience, 23(8), 2035–2047. 10.1111/j.1460-9568.2006.04733.x [DOI] [PubMed] [Google Scholar]
- Shen, J. (2014). Function and dysfunction of Presenilin. Neurodegeneration Diseases, 13(0), 61–63. 10.1159/000354971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherrington, R. , Rogaev, E. I. , Liang, Y. , Rogaeva, E. A. , Levesque, G. , Ikeda, M. , … St George‐Hyslop, P. H. (1995). Cloning of a gene bearing missense mutations in early‐onset familial Alzheimer's disease. Nature, 375(6534), 754–760. 10.1038/375754a0 [DOI] [PubMed] [Google Scholar]
- Shrivastava, A. N. , Kowalewski, J. M. , Renner, M. , Bousset, L. , Koulakoff, A. , Melki, R. , … Triller, A. (2013). β‐Amyloid and ATP‐induced diffusional trapping of astrocyte and neuronal metabotropic glutamate Type‐5 receptors. Glia, 61(10), 1673–1686. 10.1002/glia.22548 [DOI] [PubMed] [Google Scholar]
- Simpson, J. E. , Ince, P. G. , Lace, G. , Forster, G. , Shaw, P. J. , Matthews, F. , … Wharton, S. B. (2010). Astrocyte phenotype in relation to Alzheimer‐type pathology in the ageing brain. Neurobiology of Aging, 31(4), 578–590. 10.1016/j.neurobiolaging.2008.05.015 [DOI] [PubMed] [Google Scholar]
- Smith, M. J. , Kwok, J. B. , McLean, C. A. , Kril, J. J. , Broe, G. A. , Nicholson, G. A. , … Brooks, W. S. (2001). Variable phenotype of Alzheimer's disease with spastic paraparesis. Annals of Neurology, 49(1), 104–106. [DOI] [PubMed] [Google Scholar]
- Sofroniew, M. V. (2015). Astrocyte barriers to neurotoxic inflammation. Nature Reviews Neuroscience, 16(5), 249–263. 10.1038/nrn3898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song, M. , Jin, J. , Lim, J. E. , Kou, J. , Pattanayak, A. , Rehman, J. A. , … Fukuchi, K. (2011). TLR4 mutation reduces microglial activation, increases Abeta deposits and exacerbates cognitive deficits in a mouse model of Alzheimer's disease. Journal of Neuroinflammation, 8(92), 1–14. 10.1186/1742-2094-8-92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steiner, H. , Romig, H. , Grim, M. G. , Philipp, U. , Pesini, P. , Citron, M. , … Haass, C. (1999). The biological and pathological function of the Presenilin‐1 ΔExon 9 mutation is independent of its defect to undergo Proteolytic processing. Journal of Biological Chemistry, 274(12), 7615–7618. 10.1074/jbc.274.12.7615 [DOI] [PubMed] [Google Scholar]
- Steinhäuser, C. , Seifert, G. , & Bedner, P. (2012). Astrocyte dysfunction in temporal lobe epilepsy: K + channels and gap junction coupling. Glia, 60(8), 1192–1202. 10.1002/glia.22313 [DOI] [PubMed] [Google Scholar]
- Sui, Y. , Zhang, Y. , Dong, C. , Xu, B. , & Sun, X. (2019). The small molecular CCR3 antagonist YM344031 attenuates neurodegenerative pathologies and improves learning and memory performance in a mouse model of Alzheimer's disease. Brain Research, 1719, 1–10. 10.1016/j.brainres.2019.05.022 [DOI] [PubMed] [Google Scholar]
- Sullivan, S. E. , & Young‐Pearse, T. L. (2017). Induced pluripotent stem cells as a discovery tool for Alzheimer's disease. Brain Research, 1656, 98–106. 10.1016/j.brainres.2015.10.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szaruga, M. , Munteanu, B. , Lismont, S. , Veugelen, S. , Horré, K. , Mercken, M. , … Chávez‐Gutiérrez, L. (2017). Alzheimer's‐causing mutations shift Aβ length by destabilizing γ‐Secretase‐Aβn interactions. Cell, 170(3), 443–456. 10.1016/j.cell.2017.07.004 [DOI] [PubMed] [Google Scholar]
- Tahara, K. , Kim, H. D. , Jin, J. J. , Maxwell, J. A. , Li, L. , & Fukuchi, K. (2006). Role of toll‐like receptor signalling in Aβ uptake and clearance. Brain, 129, 3006–3019. 10.1093/brain/awl249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takami, M. , Nagashima, Y. , Sano, Y. , Ishihara, S. , Morishima‐Kawashima, M. , Funamoto, S. , & Ihara, Y. (2009). γ‐Secretase: Successive tripeptide and tetrapeptide release from the transmembrane domain of β‐carboxyl‐terminal fragment. The Journal of Neuroscience, 29(41), 13042–13052. 10.1523/JNEUROSCI.2362-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talantova, M. , Sanz‐Blasco, S. , & Zhang, X. (2013). Aβ induces astrocytic glutamate release, extrasynaptic NMDA receptor activation, and synaptic loss. Proceedings of the National Academy of Sciences of the United States of America, 110(33), 13690–13691. 10.1073/pnas.1313266110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao, C. , Cheng, K. , Chen, Y. M. W. H. Y. , Fuh, J. , Lee, W. , & Lee, C. C. E. H. Y. (2020). Galectin‐3 promotes Aβ oligomerization and Aβ toxicity in a mouse model of Alzheimer's disease. Cell Death and Differentiation, 27, 192–209. 10.1038/s41418-019-0348-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tapia‐Rojas, C. , Aranguiz, F. , Varela‐Nallar, L. , & Inestrosa, N. C. (2016). Voluntary running attenuates memory loss, decreases neuropathological changes and induces neurogenesis in a mouse model of Alzheimer's disease. Brain Pathology, 26, 62–74. 10.1111/bpa.12255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tasic, B. , Yao, Z. , Graybuck, L. T. , Smith, K. A. , Nguyen, T. N. , Bertagnolli, D. , … Zeng, H. (2018). Shared and distinct transcriptomic cell types across neocortical areas. Nature, 563(7729), 72–78. 10.1038/s41586-018-0654-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turdi, S. , Guo, R. , Huff, A. F. , Wolf, E. M. , Culver, B. , & Ren, J. (2009). Cardiomyocyte contractile dysfunction in the APPswe/PS1dE9 mouse model of Alzheimer's disease. PLoS One, 4(6), 1–12. 10.1371/journal.pone.0006033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Tijn, P. , Dennissen, F. J. A. A. , Gentier, R. J. G. G. , Hobo, B. , Hermes, D. , Steinbusch, H. W. M. , … Fischer, D. F. (2012). Mutant ubiquitin decreases amyloid β plaque formation in a transgenic mouse model of Alzheimer's disease. Neurochemistry International, 61(5), 739–748. 10.1016/j.neuint.2012.07.007 [DOI] [PubMed] [Google Scholar]
- Végh, M. J. , Heldring, C. M. , Kamphuis, W. , Hijazi, S. , Timmerman, A. J. , Li, K. W. , … van Kesteren, R. E. (2014). Reducing hippocampal extracellular matrix reverses early memory deficits in a mouse model of Alzheimer's disease. Acta Neuropathologica Communications, 2(76), 1–11. 10.1186/s40478-014-0076-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verkhratsky, A. , & Nedergaard, M. (2018). Physiology of astroglia. Physiological Reviews, 98(1), 239–389. 10.1152/physrev.00042.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volianskis, A. , Kostner, R. , Molgaard, M. , Hass, S. , Jensen, M. S. , Køstner, R. , … Jensen, M. S. (2010). Episodic memory deficits are not related to altered glutamatergic synaptic transmission and plasticity in the CA1 hippocampus of the APPswe/PS1∆E9‐deleted transgenic mice model of β‐amyloidosis. Neurobiology of Aging, 31, 1173–1187. 10.1016/j.neurobiologyaging.2008.08.005 [DOI] [PubMed] [Google Scholar]
- Volterra, A. , & Meldolesi, J. (2005). Astrocytes, from brain glue to communication elements: The revolution continues. Nature, 6, 626–640. 10.1038/nrn1722 [DOI] [PubMed] [Google Scholar]
- Wakabayashi, T. , & De Strooper, B. (2008). Presenilins: Members of the γ‐secretase quartets, but part‐time soloists too. Physiology, 23(4), 194–204. 10.1152/physiol.00009.2008 [DOI] [PubMed] [Google Scholar]
- Wallraff, A. , Köhling, R. , Heinemann, U. , Theis, M. , Willecke, K. , & Steinhäuser, C. (2006). The impact of astrocytic gap junctional coupling on potassium buffering in the hippocampus. The Journal of Neuroscience, 26(20), 5438–5447. 10.1523/JNEUROSCI.0037-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh, D. M. , Klyubin, I. , Fadeeva, J. V. , Cullen, W. K. , Anwyl, R. , Wolfe, M. S. , … Selkoe, D. J. (2002). Naturally secreted oligomers of amyloid β protein potently inhibit hippocampal long‐term potentiation in vivo. Nature, 416(6880), 535–539. 10.1038/416535a [DOI] [PubMed] [Google Scholar]
- Walter, S. , Letiembre, M. , Liu, Y. , Heine, H. , Hao, W. , Bode, B. , … Faßbender, K. (2007). Role of the toll‐like receptor 4 in neuroinflammation in Alzheimer's disease. Cellular Physiology and Biochemistry, 20, 947–956. [DOI] [PubMed] [Google Scholar]
- Wang, D. B. , Kinoshita, Y. , Kinoshita, C. , Uo, T. , Sopher, B. L. , Cudaback, E. , … Morrison, R. S. (2015). Loss of endophilin‐B1 exacerbates Alzheimer's disease pathology. Brain, 138(7), 2005–2019. 10.1093/brain/awv128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, D. B. , Uo, T. , Kinoshita, C. , Sopher, B. L. , Lee, R. J. , Murphy, S. P. , … Morrison, R. S. (2014). Bax interacting factor‐1 promotes survival and mitochondrial elongation in neurons. Journal of Neuroscience, 34(7), 2674–2683. 10.1523/JNEUROSCI.4074-13.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, T. , Xie, X. , Ji, M. , Wang, S. , Zha, J. , Zhou, W.‐w. , … Liu, R. (2016). Naturally occurring autoantibodies against Aβ oligomers exhibited more beneficial effects in the treatment of mouse model of Alzheimer's disease than intravenous immunoglobulin. Neuropharmacology, 105, 561–576. 10.1016/j.neuropharm.2016.02.015 [DOI] [PubMed] [Google Scholar]
- Wang, Z. , Cao, M. , Xiang, H. , Wang, W. , Feng, X. , & Yang, X. (2019). WBQ5187, a multitarget, directed agent, ameliorates cognitive impairment in a transgenic mouse model of Alzheimer's disease and modulates cerebral β‐amyloid, gliosis, cAMP levels and Neurodegeneration. ACS Chemical Neuroscience, 10, 4787–4799. 10.1021/acschemneuro.9b00409 [DOI] [PubMed] [Google Scholar]
- West, M. J. , Bach, G. , Søderman, A. , & Jensen, J. L. (2009). Synaptic contact number and size in stratum radiatum CA1 of APP/PS1ΔE9 transgenic mice. Neurobiology of Aging, 30(11), 1756–1776. 10.1016/j.neurobiolaging.2008.01.009 [DOI] [PubMed] [Google Scholar]
- Westergard, T. , & Rothstein, J. D. (2020). Astrocyte diversity: Current insights and future directions. Neurochemical Research, 45, 1–8. 10.1007/s11064-020-02959-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weyer, S. W. , Klevanski, M. , Delekate, A. , Voikar, V. , Aydin, D. , Hick, M. , … Müller, U. C. (2011). APP and APLP2 are essential at PNS and CNS synapses for transmission, spatial learning and LTP. EMBO Journal, 30(11), 2266–2280. 10.1038/emboj.2011.119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitehouse, P. J. , Price, D. L. , Struble, R. G. , Clark, A. W. , Coyle, J. T. , & DeLong, M. R. (1982). Alzheimer's disease and senile dementia: Loss of neurons in the basal forebrain. Science, 215(4537), 1237–1239. 10.1126/science.7058341 [DOI] [PubMed] [Google Scholar]
- Wirz, K. T. S. , Bossers, K. , Stargardt, A. , Kamphuis, W. , Swaab, D. F. , Hol, E. M. , & Verhaagen, J. (2013). Cortical beta amyloid protein triggers an immune response, but no synaptic changes in the APPswe/PS1dE9 Alzheimer's disease mouse model. Neurobiology of Aging, 34(5), 1328–1342. 10.1016/j.neurobiolaging.2012.11.008 [DOI] [PubMed] [Google Scholar]
- Wojtas, A. M. , Sens, J. P. , Kang, S. S. , Baker, K. E. , Berry, T. J. , Kurti, A. , … Fryer, J. D. (2020). Astrocyte‐derived clusterin suppresses amyloid formation in vivo. Molecular Neurodegeneration, 15(1), 1–14. 10.1186/s13024-020-00416-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfe, M. S. , Xia, W. , Ostaszewski, B. L. , Diehl, T. S. , Kimberly, W. T. , & Selkoe, D. J. (1999). Two transmembrane aspartates in presenilin‐1 required for presenilin endoproteolysis and γ‐secretase activity. Nature, 398, 513–517. 10.1038/19077 [DOI] [PubMed] [Google Scholar]
- Woo, J. A. , Zhao, X. , Khan, H. , Penn, C. , Wang, X. , Joly‐Amado, A. , … Kang, D. E. (2015). Slingshot‐Cofilin activation mediates mitochondrial and synaptic dysfunction via Aβ ligation to β1‐integrin conformers. Cell Death and Differentiation, 22(6), 921–934. 10.1038/cdd.2015.5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, Z. , Guo, Z. , Gearing, M. , & Chen, G. (2012). Tonic inhibition in dentate gyrus impairs long‐term potentiation and memory in an Alzheimer's disease model. Nature Communications, 29, 997–1003. 10.1016/j.biotechadv.2011.08.021.Secreted [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia, W. (2000). Role of presenilin in γ‐secretase cleavage of amyloid precursor protein. Experimental Gerontology, 35, 453–460. 10.1016/S0531-5565(00)00111-X [DOI] [PubMed] [Google Scholar]
- Xie, Z. , Yang, Q. , Song, D. , Quan, Z. , & Qing, H. (2019). Optogenetic manipulation of astrocytes from synapses to neuronal networks: A potential therapeutic strategy for neurodegenerative diseases. Glia, 68(2), 215–226. 10.1002/glia.23693 [DOI] [PubMed] [Google Scholar]
- Xing, L. , Yang, T. , Cui, S. , & Chen, G. (2019). Connexin Hemichannels in astrocytes: Role in CNS disorders. Frontiers in Molecular Neuroscience, 12(23), 1–10. 10.3389/fnmol.2019.00023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong, H. , Callaghan, D. , Wodzinska, J. , Xu, J. , Premyslova, M. , Liu, Q.‐Y. , … Zhang, W. (2011). Biochemical and behavioral characterization of the double transgenic mouse model (APPswe/PS1dE9) of Alzheimer's disease. Neuroscience Bulletin, 27(4), 221–232. 10.1007/s12264-011-1015-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, K. , Malouf, A. T. , Messing, A. , & Silver, J. (1999). Glial fibrillary acidic protein is necessary for mature astrocytes to react to β‐amyloid. Glia, 25(4), 390–403. [DOI] [PubMed] [Google Scholar]
- Xuan, A. , Pan, X. , Wei, P. , Ji, W. , Zhang, W. , Liu, J. , … Long, D. (2015). Valproic acid alleviates memory deficits and attenuates amyloid‐β deposition in transgenic mouse model of Alzheimer's disease. Molecular Neurobiology, 51(1), 300–312. 10.1007/s12035-014-8751-4 [DOI] [PubMed] [Google Scholar]
- Yi, C. , Ezan, P. , Fernández, P. , Schmitt, J. , Sáez, J. C. , Giaume, C. , & Koulakoff, A. (2017). Inhibition of glial hemichannels by boldine treatment reduces neuronal suffering in a murine model of Alzheimer's disease. Glia, 65(10), 1607–1625. 10.1002/glia.23182 [DOI] [PubMed] [Google Scholar]
- Yi, C. , Mei, X. , Ezan, P. , Mato, S. , Matias, I. , Giaume, C. , & Koulakoff, A. (2016). Astroglial connexin43 contributes to neuronal suffering in a mouse model of Alzheimer's disease. Cell Death and Differentiation, 23(10), 1691–1701. 10.1038/cdd.2016.63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin, Z. , Raj, D. , Saiepour, N. , Van Dam, D. , Brouwer, N. , Holtman, I. R. , … Boddeke, E. (2017). Immune hyperreactivity of Aβ plaque‐associated microglia in Alzheimer's disease. Neurobiology of Aging, 55, 115–122. 10.1016/j.neurobiolaging.2017.03.021 [DOI] [PubMed] [Google Scholar]
- Yu, G. , Nishimura, M. , Arawaka, S. , Levitan, D. , Zhang, L. , Tandon, A. , … George‐Hyslop, P. S. (2000). Nicastrin modulates presenilin‐mediated notch/glp‐1 signal transduction and βAPP processing. Nature, 407, 48–54. 10.1038/35024009 [DOI] [PubMed] [Google Scholar]
- Zhang, L. , Ma, Q. , Yang, W. , Qi, X. , Yao, Z. , Liu, Y. , … Qin, C. (2013). Recombinant DNA vaccine against neurite outgrowth inhibitors attenuates behavioral deficits and decreases Abeta in an Alzheimer's disease mouse model. Neuropharmacology, 70, 200–210. 10.1016/j.neuropharm.2012.10.023 [DOI] [PubMed] [Google Scholar]
- Zhang, X. , Luhrs, K. J. , Ryff, K. A. , Malik, W. T. , Driscoll, M. J. , & Culver, B. (2009). Suppression of NF‐kappa B ameliorates astrogliosis but not amyloid burden in APPswe/PS1dE9 mice. Neuroscience, 161, 53–58. 10.1016/j.neuroscience.2009.03.010 [DOI] [PubMed] [Google Scholar]
- Zhang, Y. , Chen, K. , Sloan, S. A. , Bennett, M. L. , Scholze, A. R. , O'Keeffe, S. , … Wu, J. Q. (2014). An RNA‐sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. Journal of Neuroscience, 34(36), 11929–11947. 10.1523/jneurosci.1860-14.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Y. , Sloan, S. A. , Clarke, L. E. , Caneda, C. , Plaza, C. A. , Blumenthal, P. D. , … Barres, B. A. (2016). Purification and characterization of progenitor and mature human astrocytes reveals transcriptional and functional differences with mouse. Neuron, 89(1), 37–53. 10.1016/j.neuron.2015.11.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, Y. , Song, W. M. , Andhey, P. S. , Swain, A. , Levy, T. , Miller, K. R. , … Colonna, M. (2020). Human and mouse single‐nucleus transcriptomics reveal TREM2‐dependent and TREM2‐independent cellular responses in Alzheimer's disease. Nature Medicine, 26(1), 131–142. 10.1038/s41591-019-0695-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu, C. , Xu, B. , Sun, X. , Zhu, Q. , & Sui, Y. (2017). Targeting CCR3 to reduce amyloid‐β production, tau hyperphosphorylation, and synaptic loss in a mouse model of Alzheimer's disease. Molecular Neurobiology, 54, 7964–7978. 10.1007/s12035-016-0269-5 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing is not applicable to this article as no new data were created or analyzed in this study.