

Abstract
SMYD3 is a multifunctional epigenetic enzyme with lysine methyltransferase activity and various interaction partners. It is implicated in the pathophysiology of cancers but with an unclear mechanism. To discover tool compounds for clarifying its biochemistry and potential as a therapeutic target, a set of drug‐like compounds was screened in a biosensor‐based competition assay. Diperodon was identified as an allosteric ligand; its R and S enantiomers were isolated, and their affinities to SMYD3 were determined (K D=42 and 84 μM, respectively). Co‐crystallization revealed that both enantiomers bind to a previously unidentified allosteric site in the C‐terminal protein binding domain, consistent with its weak inhibitory effect. No competition between diperodon and HSP90 (a known SMYD3 interaction partner) was observed although SMYD3–HSP90 binding was confirmed (K D=13 μM). Diperodon clearly represents a novel starting point for the design of tool compounds interacting with a druggable allosteric site, suitable for the exploration of noncatalytic SMYD3 functions and therapeutics with new mechanisms of action.
Keywords: biophysical methods, epigenetic enzymes, ligand discovery, SMYD3 biology
From a distance: SMYD3 lysine methyltransferase is an epigenetic enzyme with multiple cellular functions and the ability to recognize broad range of substrates from histones to cytosolic proteins. An SPR biosensors‐based biophysical strategy for screening SMYD3 ligands was developed and revealed an allosteric binding site. Subsequent crystallographic studies provided a structural description of the novel binding site of SMYD3.
Introduction
SET‐ and MYND‐domain‐containing protein 3 (SMYD3) was originally discovered as a lysine methyltransferase (KTMase), playing an important role in transcriptional regulation and potentially in colorectal and hepatocellular carcinomas. [1] SMYD3 catalyzes S‐adenosyl‐l‐methionine (SAM)‐dependent mono‐, di‐ and trimethylation of lysine residues in proteins. The enzyme was initially identified to have histone H3‐lysine 4 (H3‐K4)‐specific methyltransferase activity, [1] but it has later been found to act also on other protein substrates, including histone H4,[ 2 , 3 ] protein kinase MAP3K2[ 4 , 5 ] and growth factor receptor VEGFR1. [6]
SMYD3 has been associated with additional functions besides catalysis. For example, the protein is a part of the RNA polymerase II transcription complex, [7] and it interacts in a sequence‐specific manner with DNA. [1] Moreover, SMYD3 physically associates with the molecular chaperone HSP90, which increases its catalytic activity in vitro.[ 1 , 8 ] The MYND domain and the tetratricopeptide repeat (TPR) motif in the C‐terminal domain of SMYD3 have been reported as recognition sites for the HSP90 interactions. [8] The presence of multiple interaction interfaces suggests that the function and in vivo activity of SMYD3 is complex, and that the enzyme can be regulated through a variety of indirect mechanisms.
Many of the various functions and features of SMYD3 have implications for cancer progression. Via the induction of tumor cell proliferation and metastasis, its overexpression is associated with multiple oncogenic activities and a poor disease outcome, reviewed by Giakountis et al. [9] Moreover, it has been shown that its pharmacological inhibition with a small molecule compound reduces proliferation of various cancer cell types by arresting cell cycle at the S/G2 boundary. [10] Consequently, there is an interest in developing strategies that reduce SMYD3 expression, methylation activity, regulatory roles and/or otherwise interfere directly or indirectly with its functions. It is therefore encouraging that deletion of SMYD3 gene in animal models does not translate into significant pathologies, developmental disorders or infertility (Mouse Resource Portal, Wellcome Sanger Institute).
However, the use of SMYD3 as a therapeutic target has been questioned since inhibition of the catalytic activity of SMYD3, as well as knockout of SMYD3 genes by CRISPR/Cas9 in cancer cell lines show no proliferative effect demonstrating the elusive nature of SMYD3 functions. [11] Still, the assessment of the relevance of a potential target has to consider its tissue‐specific and subcellular localization and interacting partners, as has been emphasized by Giakountis et al. [9] Also, the complexity of the role of SMYD3 in cancer progression has been highlighted in a recent review by Bottino et al. [12] In summary, despite the challenges and somewhat conflicting results, the search for modulators of SMYD3 functions is still of importance for clarifying its biochemistry and potential as a therapeutic target.
To discover therapeutics targeting SMYD3, research has so far focused on compounds inhibiting its methylation activity. These have been designed to interact with one of two canonical binding sites, the co‐factor binding site (SAM site) or the protein substrate binding site. Although SAM analogues selective for SMYD3 can in principle be developed, [13] there is a risk that they may elicit toxic effects due to a generic metabolic role of the co‐factor. An alternative is to design inhibitors that interact with key amino acid residues in the protein substrate binding site, that is, the site that binds the peptide motif containing the lysine side chain that is methylated. This could be a challenging task, as active sites of enzymes acting on protein substrates (e. g., epigenetic enzymes, proteases, protein kinases) are typically relatively large and open, lacking suitable determinants for an efficient recognition of small molecules. The design of inhibitors towards this type of targets therefore often involves using macrocycles to afford sufficient selectivity and affinity, or reactive groups that bind covalently and inactivate the protein irreversibly.[ 14 , 15 ] Still, several studies have reported the successful discovery and development of potent compounds acting as active site binding inhibitors of SMYD3 activity, both in enzymatic and cellular assays.[ 16 , 17 , 18 , 19 ] They confirm that effective inhibitors of SMYD3 activity can be developed using orthosteric targeting.
Another option for discovery of function modulators is to identify allosteric ligands. These can affect catalysis through long‐range conformational effects, but can also interfere with noncatalytic functions, such as protein‐protein interactions (PPIs). Because allosteric sites are typically not as well conserved as active sites, this avoids specificity problems originating from interactions with SAM or substrate recognition sites in similar proteins, like other SAM‐dependent KTMases. Importantly, a very different structural repertoire can be explored as allosteric sites often have features very different to those from enzyme active sites.
In this study, we exploited a previously developed surface plasmon resonance (SPR) biosensor assay [20] to screen a small library of compounds. The assay was adapted for the explicit identification of allosteric ligands by using multiple sensor surfaces, including a target surface where a specific tight‐binding inhibitor of SMYD3 (EPZ031686 [17] ) blocked the active site of the enzyme. An advantage of SPR‐biosensor assays is that they can detect the interaction as such and do not rely on a functional readout assuming a certain mechanism, for example, an inhibitory effect, or interference with other functions. This is particularly advantageous when screening for SMYD3 inhibitors, as methyltransferase activity assays are typically either impractical, relying on radioactivity[ 11 , 17 ] or mass‐spectrometry‐based readouts,[ 16 , 20 ] or have a coupled design that requires multiple control experiments. [21] SPR‐biosensor and cell‐based assays for exploring the previously reported SMYD3‐HSP90 interaction [8] were also developed. In parallel to experimental studies, in silico methods were used to assess the druggability of possible binding sites.
Results
Selection of compounds for screening
A screening library of 61 compounds was assembled from an internal library of known drugs from the Prestwick Chemical Library, in‐house synthetic compounds and previously purchased drug‐like compounds. The selection resulted in a diversity set of chemical scaffolds spanning compounds from 183–712 g mol−1 and various chemotypes. No consideration was taken to their potential suitability for a certain binding site or mechanism of action (e. g., they were not substrate‐based or peptidomimetics). To avoid problems due to low solubility of the compounds in the screening buffer at the concentration required for screening (200 μM), their solubility was evaluated by UV‐visible spectroscopy by monitoring the scattering of light in the 300–600 nm region. Compounds with a scattering higher than 0.05 absorbance units (AU) were discarded from further experiments (21 compounds), leaving 40 compounds for the SPR screen. Structures of the screened compounds and their molecular weights are provided in the Supporting Information.
Development of competitive SPR biosensor screening assay
A competitive SPR biosensor assay was specifically designed for screening of the selected set of compounds. It was devised to probe and discriminate interactions with the canonical SAM and protein substrate binding sites of SMYD3, and potential sites distant from the active site. Experiments were performed at 15 °C, with SMYD3 immobilization levels of 10–13 kRU, employing an experimental procedure for immobilization and data collection described by Fabini et al. [20]
The strategy exploited a four‐surface sensor chip (Figure 1A). Two analytical surfaces with SMYD3 immobilized in a native form were used, one with SMYD3 alone and the other with SMYD3 in complex with the inhibitor EPZ031686 (surface: bSMYD3, Figure S1). EPZ031686 binds deep into the protein substrate‐recognizing gorge of the enzyme and forms a complex with a relatively long residence time (k off≪8×10−5 s−1, as shown by means of SPR biosensors for the given system [20] ); this ensures the blocking of the binding pocket for the time required for the screening of the whole compounds set. Two surfaces were used as references: one with denatured protein (dSMYD3) to correct for nonspecific interactions with the unfolded protein, and another with an unmodified surface (Mock) to monitor possible interactions between the compounds and the sensor chip matrix.
Figure 1.
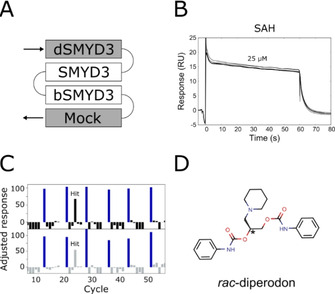
Competitive SPR biosensor screen for SMYD3 and identified allosteric hit. A) Layout of the four surfaces on the sensor chip: Native SMYD3 alone (SMYD3) and in complex with active site‐specific tight‐binding inhibitor EPZ031686 (bSMYD3) were used as analytical surfaces, denatured protein (dSMYD3) and an empty intact surface (Mock) were used for referencing. B) Overlay of unreferenced sensorgrams for 25 μM SAH injected repeatedly over SMYD3 over the course of 10 h. C) Data for 40 compounds screened at 200 μM. Molecular weight‐adjusted responses from SMYD3 (top) and bSMYD3 (bottom) surfaces. Recurring injections of 25 μM SAH (positive control, blue bars). D) Chemical structure of the identified screening hit.
S‐Adenosyl‐l‐homocysteine (SAH), a second product of the reaction catalyzed by SMYD3, served as a control of sensor surface functionality. Repeated injections showed that the kinetics or signal levels for SAH interactions did not change over the course of 10 h (Figure 1B). Moreover, SAH was an ideal internal control compound as it has a moderate affinity, rapid rate of dissociation and did not interact with the denatured protein, Figures S2 and S3. [20]
Identification of diperodon as allosteric screening hit
The output from the screening of the 40 compounds is shown in Figure 1C. A majority of the tested compounds accumulated on the surface with the denatured enzyme, reflecting their promiscuous behavior at high concentrations. However, the drug diperodon (CAS 537‐12‐2, Figure 1D), was found as an assay hit. There was a minimal accumulation of diperodon on the reference surfaces and the interaction kinetics and signal responses were comparable to those for SAH, confirming that it interacted specifically with native SMYD3 and had a well‐defined interaction mechanism. Interestingly, diperodon showed a similar response for the native SMYD3 sensor surface and the surface functionalized with the SMYD3‐EPZ031686 complex, Figures 1C, S4 and S5. In addition, the diperodon interaction with SMYD3 was not affected by the absence or presence of SAM at saturating concentrations of 1 μM in the assay buffer. These results suggest that diperodon did not interact with either of the two known canonical binding sites of the protein, that is, the substrate or co‐factor binding sites.
Isolation of diperodon enantiomers
Since diperodon was present in the library as a racemic mixture (rac‐diperodon), the two enantiomers were isolated by preparative enantioselective high‐performance liquid chromatography (eHPLC). The procedure resulted in the recovery of two enantiomerically pure fractions from the racemic mixture (Figure 2A–C). The two enantiomers were not seen to interconvert in the conditions used for the eHPLC separation, at least over the time (hours) used for experiments.
Figure 2.
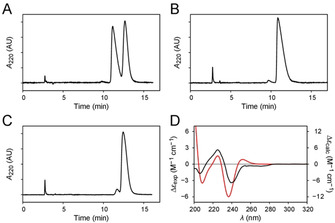
Separation and stereochemical characterization of diperodon enantiomers. A) Enantioresolution of rac‐diperodon by enantioselective HPLC on analytical Lux Cellulose‐2 column. B) Chromatogram of the least‐retained enantiomer (ee: 99.9 %). C) Chromatogram of the most‐retained enantiomer (ee: 90.4 %). D) CD spectrum of the least‐retained enantiomer of diperodon (black) and the theoretical CD spectrum of (R)‐diperodon (red).
A combination of circular dichroism (CD) spectroscopy and time‐dependent density functional theory (TD‐DFT) calculations[ 22 , 23 ] were subsequently used to assess the absolute configurations of the isolated enantiomers. Molecular mechanics (MM) and DFT conformational searches found 65 different conformations with a non‐negligible population at equilibrium (Table S1, Figure S6), indicating that diperodon is endowed with a high degree of structural flexibility. Thus, the TD‐DFT determination of the electronic properties of the hit compound (Table S2, Figure S7) was a rather challenging task, particularly prone to inaccuracies in the calculation of the optimized structures and energies of the conformers. Nevertheless, the computational protocol yielded an acceptable degree of correlation (r=0.445) between the theoretical CD spectrum of the R enantiomer and the experimental spectrum of the least retained enantiomer (Figure 2D), thus allowing the stereochemical characterization of the two enantiomers.
Interaction and inhibition analysis of diperodon enantiomers with SMYD3
SPR biosensor experiments were conducted with the two isolated enantiomers of diperodon to establish which, if any, was the more active and if both could interact with immobilized SMYD3. The results showed that both enantiomers interacted with SMYD3 with only minor differences in their apparent affinities: the K D values were determined to be (42±8) μM for (S)‐diperodon and (84±12) μM for (R)‐diperodon (Figure 3) using steady state analysis and concentration series with 100 μM as the highest concentration. Analytical experiments with higher concentrations of diperodon were not performed due to the limited solubility of the compound, a consequence of its relatively high lipophilicity (Crippen log P=4.2). The racemic mixture had an affinity intermediate to that for the pure enantiomers (Figure S4). As previously described for the racemic mixture interaction with EPZ031686‐blocked enzyme (Figure S5), the isolated enantiomers interacted equally well with the SMYD3 and the EPZ031686‐SMYD3 surfaces. Additionally, and also in line with what was described for rac‐diperodon, both enantiomers showed nearly identical binding profiles in the presence and in the absence of saturating concentration of SAM in the running buffer.
Figure 3.
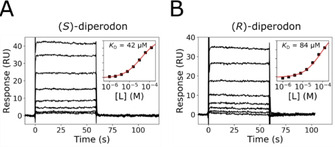
Interaction kinetic analysis of A) (S)‐diperodon and B) (R)‐diperodon with SMYD3 using an SPR biosensor‐based assay. The compounds were injected in a concentration series over immobilized SMYD3. Insets: Steady‐state analysis used to estimate K D values using a 1 : 1 interaction model (red line).
The inhibition of SMYD3 by rac‐diperodon and its isolated enantiomers was evaluated at 100 μM by monitoring the degree of MAP3K2249–274 peptide methylation [20] (Figure S8 and Table S3). EPZ031686, a known SMYD3 inhibitor, was used as refence. The percentages of inhibition are listed in Tables 1 and S3. The compounds acted as very weak inhibitors with no significant difference for the two enantiomers. Because of the weak effect, no detailed or mechanistic inhibition analysis was meaningful and the compounds were not considered to be effective inhibitor of the catalytic activity of SMYD3.
Table 1.
Inhibition of SMYD3 methylation activity by diperodon in the form of racemic mixture, isolated enantiomers and reconstituted racemic mixture. The reference inhibitor EPZ031686 was used at a concentration close to its IC50 value (0.7 μM [20] ). Values are presented as averages with a corresponding standard deviation.
|
Compound |
Concentration [μM] |
Inhibition [%] |
|---|---|---|
|
rac‐diperodon |
100 |
44.5±2.0 |
|
(S)‐diperodon |
100 |
45.8±1.5 |
|
(R)‐diperodon |
100 |
40.4±1.1 |
|
reconstituted rac‐diperodon |
100 |
39.7±1.6 |
|
EPZ031686 |
0.63 |
52.8±1.7 |
Localization of allosteric site and comparison of binding modes of diperodon enantiomers
A set of crystallization trials was performed to confirm the presence and location of allosteric binding site(s) for the two diperodon enantiomers on SMYD3. SAM‐saturated SMYD3 was co‐crystallized with the isolated enantiomers of diperodon and with the reconstituted racemic mixture. The crystals belonged to space group P212121 and diffracted to a nominal resolution of 1.6 Å and 1.9 Å for the SMYD3 complexes with the S and R enantiomers, respectively. Data reduction and model refinement statistics are given in Table S4.
The two enantiomers of diperodon were found to occupy the same allosteric binding site, located at a SMYD3 surface adjacent to that containing the active site (Figure 4A). This location is consistent with the absence of competition between both enantiomers and SAM or the active site binding inhibitor EPZ031686. The overall data quality was high enough to independently confirm the assignment of the absolute stereochemistry, despite the very weak difference density peaks for the piperidine moiety of the ligand (Figure 4B). This saturated heterocycle was found to be solvent‐exposed and did not participate in a direct contact with the protein.
Figure 4.
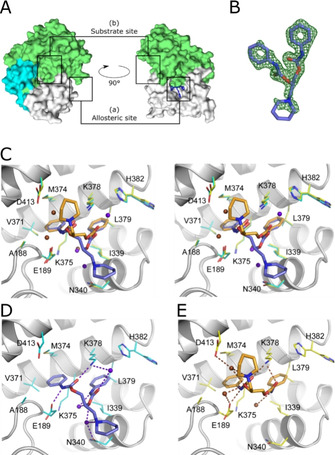
Crystal structures of SMYD3 in complex with diperodon. A) SMYD3 with diperodon (blue sticks) bound to the allosteric site (a), distinctly separated from the substrate binding site (b). The surface is colored to visualize the three main domains of SMYD3: SET and MYND domains (green), post‐SET domain (cyan), C‐terminal domain (white). B) (F 0−F c) difference density for (S)‐diperodon (green mesh, contoured at 3 σ). C) Visualization of the binding modes of (S)‐ and (R)‐diperodon in the allosteric site (stereoview, PDB IDs: 6Z2R and 6YUH). The ligands are shown as thick sticks colored by atom, with the carbon atoms of the S enantiomer in steel‐blue and those of the R enantiomer in orange. Amino acid residues within a 4 Å radius of either ligand are shown as thin sticks with carbon atoms in cyan and yellow for complexes with the S and R enantiomers, respectively. Water molecules interacting with the S and R enantiomers are colored brown and blue/purple, respectively. Details of the hydrogen‐bonding interactions of D) (S)‐diperodon (ligand in steel blue) and E) (R)‐diperodon (ligand in orange). Ligands and interacting SMYD3 residues are depicted as in (B), hydrogen bonds are shown as dashed lines.
The phenyl rings of diperodon are both bound in well‐defined hydrophobic pockets on the protein surface (Figure 4C). For the S enantiomer, the phenyl ring of the extended N‐phenylcarbamate arm is placed in the pocket lined by M374, V371, A188, and K375, whereas the pocket for the other phenyl ring is formed by H382, I339, and K378 (Figure 4D). Direct hydrogen bonds connect the amine nitrogen of the extended N‐phenylcarbamate arm with the side chain of E189, and the carbonyl oxygen of the same arm with the side chain of K387. Water‐mediated hydrogen bonds between K378 and the carbonyl oxygen, and between N340 and the alkoxy atom of the shorter N‐phenylcarbamate arm further stabilize the binding of (S)‐diperodon to the allosteric site. The nitrogen atoms of the latter moiety and of the piperidine ring are both hydrogen‐bonded to N340, via two and one water molecule(s), respectively.
The placement of the phenyl groups of the R enantiomer are essentially identical to those for the S enantiomer (Figure 4E), but with a swapped position. As a consequence, the carbonyl oxygens of both N‐phenylcarbamate arms are moved closer to K378 and directly hydrogen bond to its amino group (Figure 4D). From the remaining hydrogen bonds of the S enantiomer, only the interactions of the carbamoyl nitrogen atoms are conserved. The amide group of the piperidine ring and the alkoxy atom of the shorter N‐phenylcarbamate arm of (S)‐diperodon both form water‐mediated hydrogen bonds with the carboxyl group of D413, and a further water‐mediated hydrogen bond connects the alkoxy atom with E189. The piperidine ring points in the opposite direction as compared to (S)‐diperodon, placing it near D413.
The electron density map obtained for SMYD3 co‐crystallized with the reconstituted racemic mixture indicates that both enantiomers bind, but with a larger proportion of the allosteric sites being occupied by the S enantiomer (not shown). No additional binding sites were observed.
Computational exploration of SMYD3 surface and diperodon interactions
Structural in silico studies were performed to substantiate the hypothesis that diperodon interacted with an allosteric site on SMYD3 and evaluate the druggability of the protein surface outside the active site. The analysis was carried out on the basis of the interaction data, before the co‐crystal structures of SMYD3 and diperodon were determined (above), and the sites annotated and re‐analyzed once experimental data were available.
To identify conceivable allosteric sites on the surface of SMYD3, fPocket[ 24 , 25 ] calculations were carried out using a previously published crystal structure of SMYD3 in complex with SAM and EPZ030456 (PDB ID: 5CCM [17] ). The analysis identified 16 potential ligand‐binding pockets, of which only five had a druggability score (based on static surface properties) above 0.1 (Table 2, Figure 5). The remaining pockets (all having a score below 0.03) were predicted to be undruggable, and therefore not considered. Pocket 1, with a score of 0.5, represented the SAM binding site (Figure 5A). Pockets 4, 8, 9 and 15 (Figure 5B) with scores 0.26, 0.12, 0.65 and 0.12, respectively, were further investigated.
Table 2.
Data for F‐pocket calculations. The computationally predicted binding sites are visualized in Figure 5.
|
Site |
|
|
Druggability score[a] |
|---|---|---|---|
|
active site |
1 |
|
0.5 |
|
diperodon site |
4 |
|
0.26 |
|
|
8 |
|
0.12 |
|
others |
9 |
|
0.65 |
|
|
15 |
|
0.12 |
[a] A “druggability score” >0.5 indicates that binding is likely.
Figure 5.
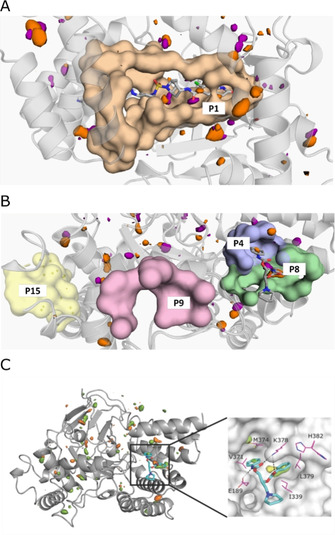
Identification of potential allosteric sites by in silico pocket detection and solvent mapping. Potentially druggable cavities identified by fPocket calculations. The surfaces show contours of residues lining the pockets; hydrophobic and polar binding hotspots identified by solvent mapping are shown as orange and purple contours, respectively. Druggability scores are presented in Table 2. SAM and (S)‐diperodon (sticks) are displayed for reference but were not included in the calculation. A) SAM binding pocket (P1). B) Diperodon binding site (split into two pockets: P4 and P8) and other pockets with high fPocket druggability score. C) Left: Structure of SMYD3 highlighting hotspots for ligand binding identified through mixed‐solvent MD simulations using MDMix. All binding pockets indicated by fPocket were probed, and high‐ and low‐energy areas identified. The low‐energy areas probed by ethanol (orange) help to identify donor or acceptor features that may be exploited for ligand binding. Hydrophobic sites (orange) were also probed. Right: Close up of the interaction hotspots within the allosteric diperodon site, highlighted using ethanol–water (yellow) and acetamide–water (green) descriptors. The two phenyl substituents of (S)‐diperodon occupy two distinct hydrophobic pockets, whereas the carbamates form polar contacts with the protein.
In silico solvent mapping was performed with the MDMix approach to explore their potential to interact with small ligands (Figure 5C).[ 26 , 27 ] The system was probed with a set of molecules containing polar and non‐polar groups, which recapitulate the most common moieties of drug‐like ligands. The simulations identified multiple interaction hotspots over the surface, including in the SAM site, and pockets 4 and 8 (Figure 5). As no hotspots were detected in pockets 9 or 15, the combination of pockets 4 and 8 were deemed the most probable allosteric site. The predicted allosteric site, later identified to bind diperodon, contains two hydrophobic hotspots and one polar binding feature. These features overlap well with the hydrophobic interactions diperodon makes with M374, V371, A188, K375 (pocket 4) and H382, I339, and K378 (pocket 8).
This analysis indicates that the diperodon binding pocket is indeed druggable and could be exploited by other ligands with completely different chemical scaffolds. Future design should take into account that the piperidine moiety is not essential for binding and could be easily replaced, or even removed. This is because it is facing the solvent, disordered (lack of electron density) and rather distant from the protein surface. This is also supported by the absence of binding hotspots in the MDMix analysis. Further, the same type of analysis reveals more prominent hydrophobic hotspots in pocket 8 than pocket 4 (Figure 5), suggesting that the former is more stable and less flexible than the later. The other major interaction point that future ligands should satisfy is a binding hot spot for an acceptor moiety, to form a hydrogen bond with Lys378 (which diperodon is also making).
Biosensor‐based analysis of SMYD3 and HSP90 interactions and effect of diperodon
The newly identified allosteric binding site is located in the C‐terminal domain of SMYD3. This region features a TPR‐like domain,[ 8 , 28 , 29 ] an α‐helix tandem repeat module frequently recurring in many proteins, described to mediate PPIs. In SMYD3, the C‐terminal domain has been reported to interact with HSP90, thereby enhancing its catalytic activity and affecting its nuclear localization and association with chromatin. [8] One of the proposed HSP90 recognition hotspots includes amino acid residues I339, N340, K375, K378 and H382, all part of or adjacent to the discovered allosteric site (Figure 4). To evaluate if diperodon could interfere with the proposed SMYD3‐HSP90 interaction, a set of biophysical and cellular experiments was performed.
The direct interaction between SMYD3 and the C‐terminal domain of HSP90 (HSP90626–732) was explored using SPR biosensor experiments specifically designed for the purpose. Two experimental designs were used (Figure 6). In Figure 6A, HSP90626–732 was injected as analyte over a low density SMYD3 surface. However, the responses were very low, and much lower than expected from responses observed with the reference compound SAH. Since HSP90626–732 has a low isoelectric point (pI ca. 4.2), the poor response could be due to non‐specific repulsion from the negatively charged dextran matrix, or steric hindrance after the immobilization, considering that many lysine amino acid residues are located in the potential SMYD3‐HSP90 interface. To resolve this problem, the assay was reversed. However, the low pI of HSP90626–732 did not allow sufficient pre‐concentration of the protein on the dextran matrix for efficient immobilization, thus excluding the possibility to develop useful surfaces with the HSP90626–732 domain alone. Instead, a GST‐HSP90626–732 fusion‐protein was successfully used to create an HSP90‐functionalized surface for experiments where SMYD3 was injected as analyte (Figure 6B).
Figure 6.
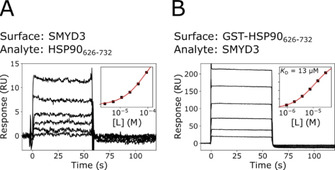
Analysis of interactions between SMYD3 and the C‐terminal domain of HSP90. Two sensor surface orientations were used. A) SMYD3 surface and HSP90626–732 as analyte. B) GST‐HSP90626–732 surface and SMYD3 as analyte. Insets show a steady‐state analysis with a 1 : 1 Langmuir interaction model, used to estimate K D.
Both experimental setups confirmed the proposed interaction between SMYD3 and HSP90626–732. They showed that the interactions were rapid and of low affinity, irrespective of which protein was immobilized or used as an analyte (Figure 6). However, only the GST‐HSP90626–732‐functionalized surface allowed a sufficiently wide concentration range of to be injected for a quantitative analysis. The K D was estimated to (13±1) μM at 25 °C (n=3), which is similar to what has been reported previously with an orthogonal technique. [8]
Several experimental procedures were used to detect competition between HSP90626–732 and rac‐diperodon, or the isolated enantiomers, for binding to SMYD3. These included injection of single concentrations or titration series of SMYD3 together with constant concentrations (100–200 μM) of the ligand over a GST‐HSP90626–732 surface, and the same set up for the inverted assay where HSP90626–732 and the ligand were injected over a SMYD3 surface. However, no significant competition or indication whether diperodon has any specific effect on SMYD3‐HSP90 interaction was observed in any of these experiments, as exemplified for (S)‐diperodon in Figure S9.
Analysis of diperodon on SMYD3 function in colon cancer cells
The potential biological effect of diperodon on SMYD3 was analyzed in colon cancer cells. Since HSP90 was previously described as a SMYD3 interactor able to modulate its localization within the nucleus, [8] the effect of diperodon on this interaction was evaluated in a HCT116 cell line. Co‐immunoprecipitation experiments confirmed the interaction between SMYD3 and HSP90 in cellulo (Figure 7A, left) and revealed that, whereas the isolated enantiomers had no observable effect on the interaction (Figure 7A, middle), the SMYD3‐HSP90 interaction was disrupted by rac‐diperodon as well as by the reconstituted racemic mixture (Figure 7A, right).
Figure 7.
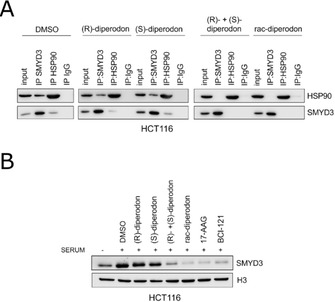
Effects of diperodon on SMYD3 in HCT116 colon cancer cells. A) Co‐immunoprecipitates stained with anti‐SMYD3 and anti‐HSP90 after treatment with 50 μM of diperodon in the form of a racemic mixture, isolated enantiomers or reconstituted racemic mixture. IgG was used as negative control. B) Chromatin fractions stained with anti‐SMYD3 and anti‐H3. HCT116 cells were cultured in serum deprivation and treated for 24 h with 50 μM of diperodon in the form of a racemic mixture, isolated enantiomers or reconstituted racemic mixture, 1 μM of 17‐AAG (Sigma), 100 μM of BCI‐121. SMYD3 recruitment on chromatin was enhanced by serum stimulation for 4 h with regular 10 % FBS cell medium. Uncropped blots are shown in Figure S10.
The influence of diperodon on the sub‐nuclear localization of SMYD3 was subsequently analyzed in chromatin fractions (Figure 7B). It showed that the chromatin association of SMYD3 was significantly reduced in the presence of rac‐diperodon or the reconstituted racemic mixture, with an effect similar to the pharmacological disruption of HSP90 or SMYD3 with 17‐AAG [30] and BCI‐121 compounds. [10] No effect was observed for either of the isolated enantiomers (Figure 7B). The results showed that rac‐diperodon causes a loss of SMYD3‐HSP90 interaction in colon cancer cells and leads to an altered SMYD3 localization within the nucleus, thereby preventing its chromatin association.
Discussion
In this study we adapted an SPR‐based biosensor assay for analysis of interactions with SMYD3 [20] for the identification of compounds that can help resolve the complex biochemistry of SMYD3 and potentially act as modulators of the complex noncatalytic functions of SMYD3. The assay was designed to identify SMYD3 ligands without assuming interactions with a certain binding site or having a certain mode of action. This was achieved by strategically using all four sequential surfaces in the same flow cell. To discriminate ligands that bound to the active site from those that bound elsewhere, screening was done against native SMYD3 surfaces and surfaces where SMYD3 was blocked with an active‐site inhibitor. In addition, to identify promiscuous binders, screening was also done against an empty reference surface and a denatured protein‐modified surface and employing a dual referencing strategy in the data analysis. SAH was used as an internal reference to monitor that the immobilized target protein was not blocked or lost binding activity during the screening experiment.
A small, structurally diverse, library consisting of 40 known drugs and other synthetic compounds was screened using the new assay. Although compounds with poor solubility at the screening concentration and in the buffers used for screening, had been removed before the screening, a majority of the tested compounds accumulated on the surface with the denatured enzyme, reflecting their promiscuous behavior. This is expected when screening libraries of drug‐like compounds at relatively high concentrations, here 200 μM.
The screening resulted in the identification of one hit, namely the drug diperodon (3‐piperidinopropane‐1,2‐diol dicarbanilate). Diperodon was discovered and used as a local topical anesthetic in the 1930s. More recently, it has been proposed as a potential inhibitor of HL60 cell proliferation. [31] However, no further studies appear to have been published relating diperodon to any target‐specific interaction. Its interaction with SMYD3 is therefore coincidental and with no direct relevance beyond the possibility of using diperodon as a starting point for design of allosteric ligands and modulators of noncatalytic SMYD3 functions or therapeutics with novel mechanisms of action.
The experimental setup and analysis of the screening results revealed that diperodon interacted equally well with SMYD3 alone and SMYD3 in complex with an active site inhibitor, thus providing direct evidence that it binds to an allosteric site already when it was identified as a hit. A complicating factor was the fact that diperodon was included in the library as a racemic mixture. The enantiomers of diperodon therefore had to be isolated, characterized and studied separately to identify the active enantiomer. X‐ray crystallography was used to localize the allosteric diperodon binding site and the mode of binding. Interestingly, both enantiomers interacted with the same site, but in flipped orientations. This is a consequence of the symmetry in the molecule and shows that there is room for optimization of the interaction.
Computational modeling (MDMix) predicted the diperodon binding site to be the most druggable, after the SAM site. It was found to overlap with a previously reported module for PPIs located in the C‐terminal domain of SMYD3. In SMYD3, this module has been reported to interact with HSP90, and it might be responsible for the recruitment also of other proteins to form multiprotein complexes. [8] This is potentially important since the current understanding of SMYD3 biology suggests that its pro‐oncogenic role is not only dependent on its methyltransferase activity, but that also SMYD3 expression levels and subcellular localization are relevant for cancer insurgence and progression. [12] It is therefore notable that SMYD3‐HSP90 complex formation has been proposed to be the basis for SMYD3 chromatin association and subcellular localization. [8] Additionally, SMYD3 interactions with key promoter regions have also been reported as a basis for its function as an enhancer of gene transcription. [32] Thus, a potential therapeutic strategy involving the targeting of SMYD3 interactions with HSP90 and other proteins can be envisaged.
Although the current study confirmed an interaction between SMYD3 and HSP90 using the biosensor assay, and could estimate the affinity to be in the low‐micromolar range (13 μM), no direct competition between diperodon and HSP90 for their interaction with SMYD3 was observed. This can most likely be attributed to the lower affinity of diperodon for SMYD3 (K D=42–84 μM), and its small size relative the interface for the SMYD3‐HSP90 interaction. In silico studies revealed probable binding hotspots in the diperodon binding site that could aid in the design of ligands with higher affinity and that extend further out in the HSP90 interaction interface.
The results from experiments in cells were more elusive, as effects were only observed with racemic mixtures but not with pure enantiomers of diperodon. However, the experiments were carried out using very high concentrations of the compounds (50 μM), deliberately chosen because of the low affinity of diperodon to SMYD3 observed in biophysical experiments. This makes the cellular data prone to artefacts and rather preliminary. Nevertheless, since the final effect on the SMYD3 and HSP90 interaction and the nuclear localization of SMYD3 was observed with the racemic mixture of diperodon, it is consistent with a real synergy between the enantiomers. Further studies are required to understand whether other players in cells are targeted by the enantiomers of diperodon, since no data were obtained to support a particular mechanism of action. Still, it can be speculated that a loss of SMYD3‐HSP90 interaction in colon cancer cells can lead to an altered SMYD3 localization within the nucleus, thereby preventing its chromatin association. A mis‐localization of SMYD3 may disrupt its methyltransferase activity, thus resulting in an impairment of SMYD3 modulation of cancer‐related pathways and therefore of its oncogenic activity by affecting its nuclear signaling. Targeting this specific interaction might consequently be a therapeutic option and, assuming a good match between the new allosteric small‐molecule binding site reported here and the localization of the SMYD3‐HSP90 interaction interface proposed by Brown et al., [8] this study provides methodological and structural basis for the development of novel SMYD3 probes with an unorthodox mechanism of action.
The obscure results from the analysis of SMYD3‐HSP90 interactions are in line with the opinion that PPIs are difficult to exploit as drug targets. [33] They are often seen as interacting via shallow binding interfaces with large areas for the intermolecular contacts, lacking contiguous epitopes. However, these challenges can be overcome, for example using macrocycles.[ 34 , 35 ] Of relevance for the current study is the realization that cellular signaling and protein regulation may occur via weak interactions between peptide binding domains in proteins and short linear motifs. [36]
Conclusion
This study showed that large screening libraries are clearly not a prerequisite for ligand discovery, providing that the compounds are well selected and the methods used have a very high sensitivity and suitable selection criteria. The competitive biosensor‐based assay used had the required sensitivity to detect allosteric ligands interacting with relatively weak affinity. A combined computational and experimental approach revealed the binding site and provided insights into its druggability. The novel allosteric site located on the hypothesized SMYD3‐HSP90 interaction interface, and the two specific tool compounds identified, can be of interest for elucidating the biology of SMYD3, especially noncatalytic functions, and exploring the potential of SMYD3 as a therapeutic target. The new experimental strategy and in silico tools employed for this study are expected to be useful for exploration also of other allosteric sites in SMYD3 and evolution of ligands.
Experimental Section
Protein expression and purification
SMYD3. The details for purification of SMYD3 are given elsewhere. [20] Briefly, a pET15b‐SMYD3 plasmid, encoding a hexa‐histidine tag followed by a thrombin cleavage site and full‐length SMYD3, was transformed in Esherichia coli Rosetta 2 cells. Expression was induced at 22 °C for 12 h with 0.4 mM IPTG, and the growth medium was supplemented with ZnSO4 to 50 μM. Cells were lysed in a buffer composed of 50 mM Tris, 300 mM NaCl, 2 mM MgCl2, 0.5 mM CaCl2, 10 μg mL−1 DNAse I, 10 μg mL−1 RNAse I, 5 mM phenylmethylsulfonyl fluoride, 5 mM 2‐mercaptoethanol (2ME), pH 8.0. The lysate was clarified. A crude SMYD3‐containing fraction was obtained by Ni2+‐immobilized chromatography using a mobile phase consisting of 50 mM Tris, 300 mM NaCl, 5 mM 2ME, 10 to 300 mM imidazole, pH 8.0. The IMAC fraction was desalted into TBS buffer (50 mM Tris, 150 mM NaCl, 2 mM dithiothreitol, pH 8.0) and subjected to proteolysis with human thrombin (Merck KgAA) overnight. The next day, the mixture was passed through the IMAC column, the flow‐through was desalted into a buffer consisting of 50 mM Tris, 50 mM NaCl, 5 mM 2ME, pH 7.6, and applied on Sepharose Q (GE Healthcare). The protein of interest was eluted at 200 mM NaCl, desalted into TBS and concentrated to >10 mg mL−1.
GST‐HSP90626 –732. The expression plasmid encoding GST‐HSP90626–732 (Addgene plasmid No. 22483; pGEX4T3 backbone) was a gift from William Sessa. E. coli Rosetta 2 cells bearing the expression plasmid were grown in Lysogeny Broth, supplemented with ampicillin and chloramphenicol to 100 and 35 μg mL−1, respectively, to OD600 nm=0.8 and induced with 0.5 mM IPTG for 4 h at 37 °C. Cells were harvested and lysed in cold TBS buffer. The lysate was clarified and incubated under gentle agitation with glutathione agarose CL4‐B (in‐house preparation) on ice for 3 h. The beads were washed with the same buffer, and the fused protein was eluted with TBS supplemented with reduced neutralized glutathione to 30 mM. The eluate was desalted into TBS and concentrated to >10 mg mL−1.
HSP90626 –732. The cDNA encoding the C‐terminal domain of HSP90 was amplified from the GST‐fused construct (above) using AACTGACATATGGACCAACCGATGGAGG and AATAGCCTCGAGTTCAGCCTCATCATCGCTTAC as primers. The PCR product was dual digested and ligated into pET15b as recipient plasmid (NdeI/XhoI restriction endonucleases, T4 DNA ligase). The final construct had N‐terminal hexa‐histidine tag, followed by a thrombin site and HSP90626–732.
The conditions for culturing cells and induction of expression were identical to those for the GST‐HSP90626–732 fusion protein. Also, procedures for IMAC purification, proteolytic digestion using thrombin and reverse IMAC were identical to those for purification of SMYD3. The flow through was desalted into a citric acid buffer (50 mM citric acid, 50 mM NaCl, pH 6.0) and loaded on Sepharose Q. The column was washed with 240 mM NaCl and the protein of interest was eluted with 335 mM NaCl. The HSP90626–732‐containing fraction was desalted into TBS and concentrated to >10 mg mL−1.
Compounds
SAM and SAH were purchased from Sigma‐Aldrich. Stock solutions were prepared to 20 mM in 50 mM HCl for SAH, and 10 mM in DMSO for SAM dihydrochloride. The screening library was assembled as diversity set, taking into account different factors such as chemotype diversity, diversity of molecular weight, synthetic complexity and presence of in the library of known drugs with the general aim to search novel scaffolds as binders of SMYD3 without any preconception about the mechanism of action. Compounds were solubilized in DMSO to 10 mM. Buffer solubility was assessed by UV/Vis spectroscopy via monitoring light scattering in the 300–600 nm range at the same concentrations used in kinetic experiments.
Interaction kinetic analysis
Interaction kinetic analysis was carried out on Biacore 2000, 3000 and T200 SPR‐based flow biosensors and CM5‐type sensor chips (GE Healthcare). Sensor surfaces with SMYD3 immobilization levels of 10–13 kRU were prepared at 25 °C using a modified amine coupling protocol: [20] SMYD3 (200 μg mL−1 in 10 mM Bis–Tris, pH 7.0) was injected over both test and reference surfaces using 10 mM HEPES, 150 mM NaCl, pH 7.5, as a running buffer. The surface was not deactivated. Instead, immediately after immobilization, the running buffer was changed to TBS, supplemented with 0.05 % (v/v) Tween‐20 and 2 % (v/v) DMSO (TBS‐TD) and the system temperature was reduced to 15 °C. The system was let to equilibrate for at least 8 h.
Prior to the kinetic experiments, the protein on the reference surface was denatured with two 10 min injection of 6 M guanidine hydrochloride. In addition, for screening, the protein on one of the analytical surfaces was saturated with a tight‐binding inhibitor EPZ031686, [17] by injecting 1 μM of the compound for 5 min.
Compounds were screened at a single concentration of 200 μM and a flow rate of 50 μL min−1. Injections of buffer and 25 μM SAH served as negative and positive controls, respectively. Association and dissociation phases were monitored for 30 s. At the end of each cycle, the sample line was washed with TBS supplemented to 25 % (v/v) DMSO. Acquired sensorgrams were referenced against either denatured SMYD3 or mock surfaces, and solvent corrected. In the screening experiments, responses were molecular weight adjusted using Biacore T200 Evaluation Software (GE Healthcare). Hit characterization was performed using a similar protocol as for the screening experiments, extending the association and dissociation phases to 60 s.
Protein‐protein interaction experiments were performed at 25 °C in TBS−T buffer. Two types of surfaces were used: a low density SMYD3 surface (ca. 4000 RU surface density) was prepared as described above, while a GST‐HSP90626–732 surface was prepared following a standard amine coupling protocol, immobilizing the protein at 50 μg mL−1 in 10 mM sodium acetate buffer, pH 5.5, to an immobilization level of 2000–3000 RU.
SMYD3 methyltransferase activity inhibition assay
SMYD3 stock solution (267 μM) was stored at −80 °C before use. MAP3K2249–274 peptide (DYDNPIFEKFGK260GGTYPRRYHVSYHH) and stock solutions of rac‐diperodon (Sigma‐Aldrich), isolated enantiomers, reconstituted racemic mixture and EPZ031686, used as a reference inhibitor, were prepared at 10 mM in DMSO. A stock solution of SAM at 37.7 mM was prepared in water. All further dilutions were performed in assay buffer (Tris 20 mM pH 8.0 containing, MgCl2 4 mM, Tween‐20 0.2 % (w/w) and DTT 2 mM). MAP3K2249–274 peptide dilutions were performed with the assay buffer containing 10 % DMSO (v/v). SMYD3 (2 μM) was incubated in the absence and in the presence of diperodon, single enantiomers and the reconstituted mixture at 200 μM at 23 °C (Thermomixer Eppendorf Comfort) for 1 h. DMSO was 2 % (v/v). 15 μL of these solutions were finally incubated with 5 μL of SAM 300 μM, 5 μL of MAP3K2249–274 peptide 75 μM and 5 μL of assay buffer. In the final conditions, SMYD3 was 1 μM, rac‐, (S)‐, (R)‐diperodon or reconstituted racemic mixture were 100 μM, MAP3K2249–274 peptide was 12.5 μM, SAM was 50 μM and the final percentage of DMSO was 2 % (v/v). The reference inhibitor was assayed at the final concentration of 0.63 μM, a concentration close to its IC50 value [20] ). After 1 h of incubation at 30 °C, the methyltransferase activity of SMYD3 was stopped adding 30 μL of stop solution consisting of H2O/AcCN/FA (50 : 50:0.1, v/v/v) and 10 μL were analyzed by LC‐ESI‐MS. The entire experiment was performed in duplicate.
LC‐MS analyses were carried out on an Agilent 1200 HPLC instrument equipped with a thermostated autosampler and a C4 reversed‐phase Jupiter 300 column (150×2 mm i.d., 5 μM particle size, 300 Å pore size; Phenomenex, USA) kept at 60 °C, coupled to a Q‐ToF mass‐spectrometer equipped with a Z‐Spray ion source (Micromass). Mobile phases A (H2O/AcCN/FA, 99 : 1:0.1, v/v/v) and B (AcCN/H2O/FA, 99 : 1:0.1, v/v/v) were used to develop a solvent gradient set as follows: 10–60 % B over 2 min and 60 % B for 3 min. MS detection was performed with the following settings: source temperature 120 °C, desolvation temperature 300 °C, capillary voltage 3.0 kV, cone voltage 35 V. Chromatograms were recorded in total ion current (TIC), in the m/z range 500–1700 and the scan time was 1 s. MAP3K2249–274 peptide baseline‐subtracted spectrum (m/z 700–1700) was deconvoluted onto a true mass scale using the maximum entropy (MaxEnt1)‐based software supplied with MassLynx software. Output parameters were: mass range 3000–3300 Da and resolution 2 Da/channel. The uniform Gaussian model was used, with 0.7 Da width at half height. The degree of substrate methylation was calculated by dividing the intensity of the methylated MAP3K2249–274 peptide by the sum of the intensities of the methylated and non‐methylated forms (total amount of petide), and multiplying the result by 100. Percentage of inhibition was calculated by comparing the amount of methylated MAP3K2249–274 in the presence and in the absence of tested compound.
Enantioselective HPLC
The enantioresolution of rac‐diperodon was performed using an HPLC system composed as follow: Waters 600 pump, Waters 600 control unit and Waters 2487 dual absorbance detector. Waters empower software was used for the data analysis. As stationary phase, Phenomenex Lux Cellulose‐2 preparative column (250×10 mm i.d., 5 μm particle size) was used. Mobile phase was composed by n‐hexane/propan‐2‐ol/diethylamine (80 : 20:0.2, v/v) mixture and the flow rate was set to 5 mL min−1. The first‐eluting fraction was collected from early peak onset to peak maximum and the second‐eluting fraction was collected from peak maximum to peak disappearance to improve the enantiomeric excess of enantiomers. The collected fractions were vacuum‐dried to eliminate all residual solvent and stored as dry powder for further analysis.
The enantiomeric excess (ee) of isolated enantiomers was determined by analytical HPLC. The HPLC system consisted of a Jasco PU‐980 pump, a LG‐2080‐02 ternary gradient unit, a DG‐2080‐53 degasser, MD‐910 photodiode array, a Rheodyne (Cotati) 7725i syringe loading injector and a 20 μL sample loop. Purified fractions of (R)‐ and (S)‐diperodon were injected on a Lux Cellulose‐2 column (250×4.6 mm i.d., 3 μm particle size), purchased using a n‐hexane/propan‐2‐ol/diethylamine (80 : 20:0.2, v/v) mobile phase at a flow rate of 1 mL min−1. HPLC‐grade solvents were purchased from Sigma‐Aldrich. Samples were dissolved in the mobile phase and injected at the concentration of 0.5 mg mL−1.
Stereochemical characterization
The stereochemical characterization of the enantiomers of diperodon was performed according to a standard protocol.[ 22 , 23 ] The experimental circular dichroism (CD) spectrum of the least retained enantiomeric fraction of diperodon (250 μM) was measured in the 350–195 nm spectral range using (HCl 0.1 N)/propan‐2‐ol (91 : 9, v/v) as solvent. Measurements were carried out on a Jasco J‐810 spectropolarimeter using Hellma QS quartz cells with optical path lengths of 1 cm (350–250 nm) and 0.1 cm (260–195 nm), a spectral bandwidth of 2 nm, a scanning speed of 50 nm min−1, a data integration time of 2 s, a data interval of 0.2 nm and an accumulation cycle of 3 scans per spectrum. CD data were corrected from the contribution of the solvent and converted to molar units (▵ϵ, in M−1 cm−1).
Quantum chemical (QC) calculations based on time‐dependent density functional theory (TD‐DFT) were then performed on (R)‐diperodon in conjugate acid form using the Gaussian 09 Rev. D.01 software (Gaussian Inc.). A preliminary conformational search by molecular mechanics (MM) was performed with the Spartan'02 (Wavefunction Inc.) using the MMFF94 s force field [37] and Monte‐Carlo sampling. The geometries of the MM conformers found within a 15 kcal mol−1 energy window were optimized at the DFT level, using the B97D [38] functional in combination with the def2‐TZVP basis set,[ 39 , 40 ] the density fitting approximation[ 41 , 42 ] and the IEFPCM solvation model for water. [43] Optimized conformers with a root‐mean‐square deviation (RMSD) below 0.01 Å for heavy atoms were clustered, while conformers displaying imaginary frequencies or relative electronic energies (▵E) above 2.5 kcal mol−1 were discarded. The results of the MM and DFT conformational search on (R)‐diperodon are reported in Table S1, while a graphical representation of the lowest‐energy conformers is given in Figure S1.
TD‐DFT calculations were carried out on the resulting low‐energy DFT conformers using the PBE0 functional[ 44 , 45 ] in combination with the def2‐TZVP basis set and the IEFPCM solvation model for water. The theoretical UV and CD spectra of each conformer were calculated by approximation of oscillator strengths (f j) and rotational strengths (R j, dipole length form) to Gaussian bands (▵σ=0.2 eV) [46] and sum over the 50 excited states with the lowest excitation energies. The overall theoretical spectra of (R)‐diperodon were then derived by averaging the contribution of all conformers according to their population at equilibrium, as predicted by Boltzmann statistics based on electronic energies (298.15 K, 1 atm), and finally compared to the experimental UV and CD spectra of the least retained enantiomeric fraction of diperodon using the Pearson correlation coefficient (r). The results of the TD‐DFT calculations on (R)‐diperodon are reported in Table S2, while the comparison between the experimental and theoretical UV spectra is given in Figure S2.
Computational studies
Initial computational studies used the PDB structure 5CCM [17] since the SMYD3‐(S)‐diperodon co‐crystal structure was not yet available. Pocket calculations followed standard protocols.[ 24 , 25 ] To generate a suitable starting structure for solvent mapping, it was stripped of all ligands and a short molecular dynamics simulation (MD) was ran to see that the system was equilibrated. The resulting structure was very similar to the crystal structure of 5CCM, showing that the structures were stable in silico. The solvent mapping was subsequently done by MD using two solvent mixtures: ethanol‐water and acetamide‐water. The system was probed with a set of molecules containing polar and nonpolar groups, which recapitulate the most common moieties of drug‐like ligands.
The MDMix simulation used the co‐crystal structure of SMYD3 in complex with (S)‐diperodon. Bound ligands and all crystallographic waters were removed. The protein was protonated and all termini were capped using MOE 2016. Using MDMix, 3 replicas of 50 ns were simulated without restrains. All other settings were kept at default. MDMix settings for solvent maps were used from previous studies.[ 26 , 27 ] The system was solvated using the following mixtures: 20 % ethanol‐water (ETA identified), 20 % acetamide‐water (MAM) and 100 % water (WAT).
Protein crystallization, data collection and model building
SMYD3 was co‐crystallized with (S)‐diperodon under the following conditions: protein at 10 mg mL−1 (ca. 200 μM) was mixed with 1 mM of the synthetic compound in TBS buffer, supplemented with 10 % (v/v) DMSO, and combined at a 1 : 1 (v/v) ratio with the reservoir solution (100 mM Tris, 50 mM magnesium acetate, 11 % PEG3350, pH 8.25). Co‐crystallization of SMYD3 with (R)‐diperodon was done with 7 mg mL−1 (ca. 140 μM) of protein incubated with 2.5 mM of synthetic compound in TBS buffer and 10 % (v/v) DMSO, combined at a 1 : 1 (v/v) ratio with reservoir solution (100 mM Tris, 100 mM magnesium acetate, 16 % PEG3350, pH 8.25).
Crystallization experiments were performed in a hanging drop manner at 22 °C, with a total drop volume of 2 μL. Needle‐like crystals nucleated within 12 h and grew to maximal dimensions within 2 d; prior to cryo‐cooling, crystals were cryo‐protected in reservoir solution supplemented with 10 % (v/v) glycerol for ca. 5 s.
Diffraction data was collected at the ESRF (Grenoble, France) beamline ID‐24 and the MAX IV (Lund, Sweden) BioMAX beamline. Data was indexed, autoprocessed, scaled and merged on‐site using the implemented data processing routines and software. Phases were obtained through molecular replacement with PhaserMR [47] employing the ligand‐free structure with PDB ID: 5CCM [17] as a search model. Ligand dictionaries were created using eLBOW, [48] model building was performed using Coot, [49] and structure refinement using phenix.refine [50] and REFMAC5. [51] Model quality was evaluated using Rampage, [52] figures were prepared with PyMol. The coordinates and structure factors of the co‐crystal structures were deposited in the Protein Data Bank with the IDs 6YUH (SMYD3‐(R)‐diperodon complex) and 6Z2R (SMYD3‐(S)‐diperodon complex), respectively.
Cellular experiments
HCT116 colon cancer cells (ATCC) were cultured in DMEM with 10 % FBS (Gibco) and 100 IU/mL penicillin‐streptomycin (Gibco) at 37 °C and in 5 % CO2. Co‐Immunoprecipitation and immunoblot analysis were performed after lysis of cells in IP lysis buffer (150 mM NaCl, 50 mM Tris pH 7.5, 0.3 % CHAPS, 1 mM EDTA, 1 % NP40, 10 % glycerol, 1 % Triton X‐100) supplemented with protease inhibitors (Roche). 1 μg of anti‐SMYD3 (Cell Signaling Technology), anti‐HSP90 (Sigma) was coupled to Dynabeads Protein A (10002D, Thermo Fisher Scientific) or G (10004D, Thermo Fisher Scientific) in 100 μL of 0.01 % Tween‐20‐1X PBS for 45 min at room temperature on a rocking platform. 10 % of the cell lysate was incubated with antibody‐Dynabeads complexes for 1 h at room temperature on a rocking platform. Immunocomplexes were washed three times with the lysis buffer, boiled in 4x Laemmli sample buffer (BioRad) and subjected to sodium dodecyl sulphate‐polyacrylamide gel (SDS‐PAGE) for immunoblot analysis with anti‐SMYD3 (Cell Signaling Technologies) and anti‐HSP90 (Sigma). HRPO‐conjugated antibodies (GE Healthcare) were used as secondary antibodies and revealed using the ECL‐plus chemiluminescence reagent, following manufacturer's instructions (GE Healthcare).
Cells were collected and chromatin fractions were isolated as described by Mendez and Stillman. [53] 20 μg of protein extracts from each sample were denatured in 4x Laemmli sample buffer and separated by SDS‐PAGE for immunoblot analysis. Immunoblots were performed with anti‐SMYD3 (Cell Signaling Technologies) and anti‐Histone H3 (Abcam).
Conflict of interest
The authors declare no conflict of interests.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
The authors are grateful to the Biophysical Screening and Characterisation Facility (SciLifeLab, Uppsala University, Sweden) for providing access to Biacore T200 instrument, Prof. A. Mazzanti (Dept. of Industrial Chemistry, University of Bologna) for assistance with preparative HPLC, Prof. R. Zanasi (Dept. of Chemistry and Biology, University of Salerno, Italy) for providing access to the computing cluster used for QC calculations, and the staff of beamlines ID24 (ESRF, Grenoble, France) and BioMAX (MAX IV, Lund, Sweden) for assistance with the diffraction data collection. This work was supported by AIRC IG N. 19172 and IG N. 23794, POR FSE 2014/2020 project ONCOPENTA, Italian MoH Ricerca Corrente 2018–2020, 2019–2021 and SG‐2019‐12371540, and PRIN grant N.2017WNKSLRLS4, EU Framework Programme for Research and Innovation Horizon 2020 (2014‐2020) under the Marie‐Skoldowska‐Curie grant agreement number ID 675899 (FRAGNET). U.H.D. acknowledges the Swedish Research Council for support (grant no. D0571301).
V. O. Talibov, E. Fabini, E. A. FitzGerald, D. Tedesco, D. Cederfeldt, M. J. Talu, M. M. Rachman, F. Mihalic, E. Manoni, M. Naldi, P. Sanese, G. Forte, M. Lepore Signorile, X. Barril, C. Simone, M. Bartolini, D. Dobritzsch, A. Del Rio, U. H. Danielson, ChemBioChem 2021, 22, 1597.
Contributor Information
Dr. Alberto Del Rio, Email: alberto.delrio@isof.cnr.it.
Prof. U. Helena Danielson, Email: helena.danielson@kemi.uu.se.
References
- 1. Hamamoto R., Furukawa Y., Morita M., Iimura Y., Silva F. P., Li M., Yagyu R., Nakamura Y., Nat. Cell Biol. 2004, 6, 731–740. [DOI] [PubMed] [Google Scholar]
- 2. Foreman K. W., Brown M., Park F., Emtage S., Harriss J., Das C., Zhu L., Crew A., Arnold L., Shaaban S., Tucker P., PLoS One 2011, 6, e22290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. van Aller G. S., Reynoird N., Barbash O., Huddleston M., Liu S., Zmoos A. F., McDevitt P., Sinnamon R., Le B. C., Mas G., Annan R., Sage J., Garcia B. A., Tummino P. J., Gozani O., Kruger R. G., Epigenetics 2012, 7, 340–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Mazur P. K., Reynoird N., Khatri P., Jansen P. W. T. C., Wilkinson A. W., Liu S., Barbash O., Van Aller G. S., Huddleston M., Dhanak D., Tummino P. J., Kruger R. G., Garcia B. A., Butte A. J., Vermeulen M., Sage J., Gozani O., Nature 2014, 510, 283–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Fu W., Liu N., Qiao Q., Wang M., Min J., Zhu B., Xu R. M., Yang N., J. Biol. Chem. 2016, 291, 9173–9180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kunizaki M., Hamamoto R., Silva F. P., Yamaguchi K., Nagayasu T., Shibuya M., Nakamura Y., Furukawa Y., Cancer Res. 2007, 67, 10759–10765. [DOI] [PubMed] [Google Scholar]
- 7. Sarris M. E., Moulos P., Haroniti A., Giakountis A., Talianidis I., Cancer Cell 2016, 29, 354–366. [DOI] [PubMed] [Google Scholar]
- 8. Brown M. A., Foreman K., Harriss J., Das C., Zhu L., Edwards M., Shaaban S., Tucker H., Oncotarget 2015, 6, 4005–4019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Giakountis A., Moulos P., Sarris M. E., Hatzis P., Talianidis I., Semin. Cancer Biol. 2017, 42, 70–80. [DOI] [PubMed] [Google Scholar]
- 10. Peserico A., Germani A., Sanese P., Barbosa A. J., Di Virgilio V., Fittipaldi R., Fabini E., Bertucci C., Varchi G., Moyer M. P., Caretti G., Del Rio A., Simone C., J. Cell. Physiol. 2015, 230, 2447–2460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Thomenius M. J., Totman J., Harvey D., Mitchell L. H., Riera T. V., Cosmopoulos K., Grassian A. R., Klaus C., Foley M., Admirand E. A., Jahic H., Majer C., Wigle T., Jacques S. L., Gureasko J., Brach D., Lingaraj T., West K., Smith S., Rioux N., Waters N. J., Tang C., Raimondi A., Munchhof M., Mills J. E., Ribich S., Porter Scott M., Kuntz K. W., Janzen W. P., Moyer M., Smith J. J., Chesworth R., Copeland R. A., Boriack-Sjodin P. A., PLoS One 2018, 13, e0197372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bottino C., Peserico A., Simone C., Caretti G., Cancers (Basel). 2020, 12, 142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Campagna-Slater V., Mok M. W., Nguyen K. T., Feher M., Najmanovich R., Schapira M., J. Chem. Inf. Model. 2011, 51, 612–623. [DOI] [PubMed] [Google Scholar]
- 14. Shunmugam L., Ramharack P., Soliman M. E. S., Protein J. 2017, 36, 397–406. [DOI] [PubMed] [Google Scholar]
- 15. Pillaiyar T., Namasivayam V., Manickam M., Curr. Med. Chem. 2016, 23, 3404–3447. [DOI] [PubMed] [Google Scholar]
- 16. Van Aller G. S., Graves A. P., Elkins P. A., Bonnette W. G., McDevitt P. J., Zappacosta F., Annan R. S., Dean T. W., Su D. S., Carpenter C. L., Mohammad H. P., Kruger R. G., Structure 2016, 24, 774–781. [DOI] [PubMed] [Google Scholar]
- 17. Mitchell L. H., Boriack-Sjodin P. A., Smith S., Thomenius M., Rioux N., Munchhof M., Mills J. E., Klaus C., Totman J., Riera T. V., Raimondi A., Jacques S. L., West K., Foley M., Waters N. J., Kuntz K. W., Wigle T. J., Scott M. P., Copeland R. A., Smith J. J., Chesworth R., ACS Med. Chem. Lett. 2016, 7, 134–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Huang C., Liew S. S., Lin G. R., Poulsen A., Ang M. J. Y., Chia B. C. S., Chew S. Y., Kwek Z. P., Wee J. L. K., Ong E. H., Retna P., Baburajendran N., Li R., Yu W., Koh-Stenta X., Ngo A., Manesh S., Fulwood J., Ke Z., Chung H. H., Sepramaniam S., Chew X. H., Dinie N., Lee M. A., Chew Y. S., Low C. B., Pendharkar V., Manoharan V., Vuddagiri S., Sangthongpitag K., Joy J., Matter A., Hill J., Keller T. H., Foo K., ACS Med. Chem. Lett. 2019, 10, 978–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Su D., Qu J., Schulz M., Blackledge C. W., Yu H., Zeng J., Burgess J., Reif A., Stern M., Nagarajan R., Pappalardi M. B., Wong K., Graves A. P., Bonnette W., Wang L., Elkins P., Knapp-reed B., Carson D., Mchugh C., Mohammad H., Kruger R., Luengo J., Heerding D. A., Creasy C. L., ACS Med. Chem. Lett. 2020, 11, 133–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Fabini E., Talibov V. O., Mihalic F., Naldi M., Bartolini M., Bertucci C., Del Rio A., Danielson U. H., Biochemistry 2019, 58, 3634–3645. [DOI] [PubMed] [Google Scholar]
- 21. Acker M. G., Auld D. S., Perspect. Sci. 2014, 1, 56–73. [Google Scholar]
- 22. Bertucci C., Tedesco D., J. Chromatogr. A 2012, 1269, 69–81. [DOI] [PubMed] [Google Scholar]
- 23. Sardella R., Carotti A., Manfroni G., Tedesco D., Martelli A., Bertucci C., Cecchetti V., Natalini B., J. Chromatogr. A 2014, 1363, 162–168. [DOI] [PubMed] [Google Scholar]
- 24. Le Guilloux V., Schmidtke P., Tuffery P., BMC Bioinf. 2009, 10, 168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Schmidtke P., Barril X., J. Med. Chem. 2010, 53, 5858–5867. [DOI] [PubMed] [Google Scholar]
- 26. Seco J., Luque F. J., Barril X., J. Med. Chem. 2009, 52, 2363–2371. [DOI] [PubMed] [Google Scholar]
- 27. Alvarez-Garcia D., Barril X., J. Med. Chem. 2014, 57, 8530–8539. [DOI] [PubMed] [Google Scholar]
- 28. Xu S., Wu J., Sun B., Zhong C., Ding J., Nucleic Acids Res. 2011, 39, 4438–4449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Blatch G. L., Lässle M., BioEssays 1999, 21, 932–939. [DOI] [PubMed] [Google Scholar]
- 30. Schulte T. W., Neckers L. M., Cancer Chemother. Pharmacol. 1998, 42, 273–279. [DOI] [PubMed] [Google Scholar]
- 31. Manzotti G., Parenti S., Ferrari-Amorotti G., Soliera A. R., Cattelani S., Montanari M., Cavalli D., Ertel A., Grande A., Calabretta B., Cell Cycle 2015, 14, 2578–2589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Giakountis A., Moulos P., Sarris M. E., Hatzis P., Talianidis I., Semin. Cancer Biol. 2017, 42, 70–80. [DOI] [PubMed] [Google Scholar]
- 33. Wells J. A., McClendon C. L., Nature 2007, 450, 1001–1009. [DOI] [PubMed] [Google Scholar]
- 34. Dougherty P. G., Qian Z., Pei D., Biochem. J. 2017, 474, 1109–1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Tyagi M., Begnini F., Poongavanam V., Doak B. C., Kihlberg J., Chem. Eur. J. 2020, 26, 49–88. [DOI] [PubMed] [Google Scholar]
- 36. Davey N. E., Seo M., Yadav V. K., Jeon J., Nim S., Krystkowiak I., Blikstad C., Dong D., Markova N., Kim P. M., Ivarsson Y., FEBS J. 2017, 284, 485–498. [DOI] [PubMed] [Google Scholar]
- 37. Halgren T. A., J. Comput. Chem. 1999, 20, 720–729. [DOI] [PubMed] [Google Scholar]
- 38. Grimme S., J. Comput. Chem. 2006, 27, 1787–1799. [DOI] [PubMed] [Google Scholar]
- 39. Weigend F., Ahlrichs R., Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [DOI] [PubMed] [Google Scholar]
- 40. Weigend F., Phys. Chem. Chem. Phys. 2006, 8, 1057–1065. [DOI] [PubMed] [Google Scholar]
- 41. Dunlap B. I., J. Chem. Phys. 1983, 78, 3140–3142. [Google Scholar]
- 42. Dunlap B. I., J. Mol. Struct. 2000, 529, 37–40. [Google Scholar]
- 43. Tomasi J., Mennucci B., Cancès E., J. Mol. Struct. 1999, 464, 211–226. [Google Scholar]
- 44. Perdew J. P., Burke K., Ernzerhof M., Phys. Rev. Lett. 1996, 77, 3865–3868. [DOI] [PubMed] [Google Scholar]
- 45. Adamo C., Barone V., J. Chem. Phys. 1999, 110, 6158–6170. [Google Scholar]
- 46. Stephens P. J., Harada N., Chirality 2010, 22, 229–233. [DOI] [PubMed] [Google Scholar]
- 47. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., Read R. J., J. Appl. Crystallogr. 2007, 40, 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Moriarty N. W., Grosse-Kunstleve R. W., Adams P. D., Acta Crystallogr. Sect. D Biol. Crystallogr. 2009, 65, 1074–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Emsley P., Lohkamp B., Scott W. G., Cowtan K., Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., Zwart P. H., Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Murshudov G. N., Skubák P., Lebedev A. A., Pannu N. S., Steiner R. A., Nicholls R. A., Winn M. D., Long F., Vagin A. A., Acta Crystallogr. D. Biol. Crystallogr. 2011, 67, 355–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Lovell S. C., Davis I. W., W. B. Arendall III , De Bakker P. I. W., Word J. M., Prisant M. G., Richardson J. S., Richardson D. C., Proteins Struct. Funct. Genet. 2003, 450, 437–450. [DOI] [PubMed] [Google Scholar]
- 53. Méndez J., Stillman B., Mol. Cell. Biol. 2000, 20, 8602–8612. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary