

Abstract
Hypomagnesemia, seizures, and intellectual disability (HSMR) syndrome is a rare disorder caused by mutations in the cyclin M2 (CNNM2) gene. Due to the limited number of cases, extensive phenotype analyses of these patients have not been performed, hindering early recognition of patients. In this study, we established the largest cohort of HSMR to date, aiming to improve recognition and diagnosis of this complex disorder. Eleven novel variants in CNNM2 were identified in nine single sporadic cases and in two families with suspected HSMR syndrome. 25Mg2+ uptake assays demonstrated loss‐of‐function in seven out of nine variants in CNNM2. Interestingly, the pathogenic mutations resulted in decreased plasma membrane expression. The phenotype of those affected by pathogenic CNNM2 mutations was compared with five previously reported cases of HSMR. All patients suffered from hypomagnesemia (0.44–0.72 mmol/L), which could not be fully corrected by Mg2+ supplementation. The majority of patients (77%) experienced generalized seizures and exhibited mild to moderate intellectual disability and speech delay. Moreover, severe obesity was present in most patients (89%). Our data establish hypomagnesemia, seizures, intellectual disability, and obesity as hallmarks of HSMR syndrome. The assessment of these major features offers a straightforward tool for the clinical diagnosis of HSMR.
Keywords: CNNM2, HSMR, hypomagnesemia, intellectual disability, obesity
Eleven novel variants in CNNM2 were identified in nine single sporadic cases and in two families with hypomagnesemia. Our data establish hypomagnesemia, seizures, intellectual disability, and obesity as hallmarks of HSMR syndrome, caused by CNNM2 mutations
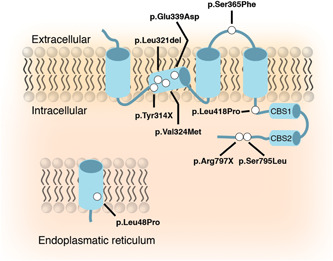
1. INTRODUCTION
Hypomagnesemia, seizures, and intellectual disability syndrome (HSMR; MIM# 616418) is a rare hereditary disorder, comprising hypomagnesemia due to renal magnesium (Mg2+) wasting, epileptic seizures, and intellectual disability (ID). In single HSMR syndrome patients, additional phenotypic features such as severe obesity, impaired motor skills, or autism spectrum disorder (ASD) have been reported (Arjona et al., 2014). However, the incidence of these features and their penetrance in HSMR syndrome have remained unknown.
In 2011, loss‐of‐function mutations in cyclin M2 (CNNM2) were identified to cause HSMR. Although the initial two families demonstrated autosomal‐dominant inheritance (Stuiver et al., 2011), patients in a follow‐up study carried de novo mutations (Accogli et al., 2018; Arjona et al., 2014; de Baaij et al., 2012). In addition, an autosomal‐recessive inheritance pattern was described in two families in which patients suffered from a severe neurological phenotype comprising brain malformations, enlargement of outer cerebrospinal liquor spaces, myelinization defects, and epileptic encephalopathy (Accogli et al., 2018; Arjona et al., 2014).
Within the kidney, CNNM2 is exclusively expressed at the basolateral membrane of cells in the distal convoluted tubule (DCT) and cortical thick ascending limb of Henle's loop (de Baaij et al. 2012; Stuiver et al., 2011). Although the exact function of CNNM2 is still under debate, mutations in CNNM2 are associated with defective Mg2+ reabsorption resulting in renal Mg2+ wasting (Arjona & de Baaij, 2018; Funato et al., 2018).
Interestingly, genome‐wide association studies (GWAS) linked the CNNM2 locus to cardiovascular traits such as blood pressure (Li et al., 2017)], coronary artery disease (Dichgans et al., 2014), and myocardial infarction (Matsuoka et al., 2015), and also to glycemic traits and body mass index (Hruby et al., 2013; Lv et al., 2017). Moreover, common variants within the CNNM2 locus have been associated with neuropsychiatric disorders including schizophrenia and bipolar disorder (Grigoroiu‐Serbanescu et al., 2015; Lotan et al., 2014) as well as brain morphologic parameters such as gray matter volume (Ohi et al., 2013; Rose et al., 2014). Despite these data, the role of CNNM2 in brain development and function remains unknown.
In this study, phenotypical and molecular analyses were performed in the largest novel cohort of HSMR patients to date. By combining data from all published HSMR cases, we aim to provide clinical guidance for the diagnosis of this complex disorder.
2. PATIENTS AND METHODS
2.1. Patient data collection
This study reports on 11 novel patients with CNNM2 variants that were genetically tested from 2015 to 2018 under the suspicion of HSMR or when hypomagnesemia or intellectual disability was observed without a known cause. Informed consent for genetic studies was obtained from each proband or from their parents (for minors). Clinical characteristics were analyzed and reported (Tables 1, S1, and S2). For the cohort analysis, five additional patients were included that have been described before (Arjona et al., 2014; Stuiver et al., 2011). All genetic studies were approved by the local ethics committee. Patients or their parents provided written informed consent in accordance with the Declaration of Helsinki.
Table 1.
Clinical manifestations of patients with novel identified CNNM2 mutations
| Proband | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | |
|---|---|---|---|---|---|---|---|---|---|---|
| Genetic findings | ||||||||||
| DNA | NC_000010.10:g.(?_104678237)_(104816721_?)del | NC_000010.10:g.104814162_104814164del | NM_017649.4:c.143T>C | NM_017649.4:c.942C>G | NM_017649.4:c.961_963del | NM_017649.4:c.970G>A | NM_017649.4:c.1253T>C | NM_017649.4:c.2384C>T | NM_017649:c.2389C>T | |
| Protein | N/A | N/A | p.Leu48Pro | p.Tyr314* | p.Leu321del | p.Val324Met | p.Leu418Pro | p.Ser795Leu | p.Arg797* | |
| Genetic diagnosis | Next‐generation sequencing panel for hypomagnesemia | Exome sequencing | Sanger sequencing | Sanger sequencing | Exome sequencing | Sanger sequencing | Exome sequencing | Exome sequencing | Next‐generation sequencing panel for ID | |
| General parameters | ||||||||||
| Gender | F | M | M | M | M | M | F | F | F | |
| Inherited | De novo | Ongoing | Dominant | Dominant | De novo | De novo | De novo | De novo | De novo | |
| Ethnicity | Caucasian | Sub‐Saharan African | Caucasian | Caucasian | Sub‐Saharan African | Caucasian | Caucasian | Caucasian | Caucasian | |
| Age at manifestation | 6 years | 3 months | 2 years | 3 years | 8 months | 13 months | 16 years | 8 months | 1–1.5 year | |
| Body mass index (percentile) | 24.6 (>P97) | 22.6 (P50–P85) | N/A | 25 (>P97) | 27 (>P97) | 27.4 (>P97) | N/A | 20.5 (P50‐P85) | 18.5 (>P97) | |
| eGFR (ml/min) | 112 | 84 | N/A | 93 | 120 | N/A | N/A | 56 | N/A | |
| Neurological manifestations | ||||||||||
| Seizures | Y | Y | Y | N | Y | Y | Y | N | Y | |
| Brain malformations | N | N | N | N | N | N | N | N | Y | |
| Intellectual disability | Y | Y | N | Y | Y | Y | Y | Y | Y | |
| Speech/Communication | Limited | Y | N | Y | Delayed | Delayed | N/A | Delayed | Limited | |
| ASD | N | N | N | N | N | N | N | N | Y | |
| Motor skill defects | Y | Y | Y | N | Y | Y | N | Y | N | |
| Electrolyte levels | ||||||||||
| Serum Na+ (mmol/L) | 139 | 138 | N/A | 141 | 139 | 141 | 143 | 140 | 138 | |
| Serum K+ (mmol/L) | 4.2 | 4.4 | N/A | 4.1 | 3.6 | 4.2 | 4.3 | 4.2 | 4.1 | |
| Serum Mg2+ (mmol/L) | 0.63 | 0.57 | 0.45 | 0.48 | 0.5 | 0.54 | 0.49 | 0.72 | 0.57 | |
| Serum Ca2+ (mmol/L) | 2.4 | 2.49 | normal | 1.32 (ionized) | 2.2 | 2.5 | 1.87 | 2,5 | 2.5 | |
| Urinary Mg2+ (%FE) | 1.14 mmol/L | N/A | 15.82 mmol/L | N/A | 2.9 | 5.8 mmol/L | 6.7 | 3.5 | Normal | |
| Treatment | ||||||||||
| Mg2+ supplementation | Oral | N/A | Intravenous | N/A | Oral | Oral | Oral | Oral | Oral | |
| Serum Mg2+ after supplementation (mmol/L) | 0.65 | N/A | 0.53‐0.66 | N/A | 0.51 | 0.52 | 0.58 | 0.7 | 0.69 | |
Abbreviations: ASD, autism spectrum disorder; CNNM2, cyclin M2; eGFR, estimated glomerular filtration rate; FE, fractional excretion; ID, intellectual disorder; N, no, N/A, unknown; Y, yes.
2.2. Genetic analysis
Genomic DNA was extracted from whole blood using standard methods. In four single patients, whole‐exome sequencing was used and panels containing tubulopathy, hypomagnesemia, or intellectual disability‐specific genes were used for screening. Identified CNNM2 variants were verified using Sanger sequencing. In one single case (P2), the Illumina OmniExpres microarray (Illumina Inc.) was used. In six individuals, CNNM2 was directly subjected to Sanger sequencing due to suspicion of HSMR syndrome. In silico prediction was done with Combined Annotation Dependent Depletion (CADD) GRCh37 v1.6 (https://cadd.gs.washington.edu/) including annotations which provided scores and predication consequences on the protein level. The novel identified variants have been deposited in ClinVar (accessions: CV001478379 ‐ SCV001478389).
2.3. Structure modeling
The structural impact of point mutations CNNM2‐p.Ser795Leu and p.Arg797* on CNNM2 was analyzed with RosettaBackrub (Lauck et al., 2010) using the crystal structure of the CNBH domain of CNNM2 (Chen et al., 2018) both in its monomeric and in its dimeric species, as a three‐dimensional template (PDB code: 6DJ3). The local changes observed in CNNM2 were compared with those resulting from an equivalent analysis done with the crystal structure of CNNM4 (PDB code 6G52), which yielded similar results. The comparison of native and mutant protein structure and the figures were done with UCSF Chimera (https://www.cgl.ucsf.edu/chimera/).
2.4. DNA constructs
A mouse wild‐type Cnnm2 complementary DNA construct was cloned into the pCINeo HA IRES GFP vector as described earlier (Stuiver et al., 2011). Mutant Cnnm2 constructs were made using the QuikChange site‐directed mutagenesis kit (Stratagene) according to the manufacturer's instructions by using primers listed in Tables S3 and S4. All constructs were verified by Sanger sequencing.
2.5. Cell culture
HEK293 cells were cultured in Dulbecco's modified eagle's medium (Lonza) containing 10% (v/v) fetal calf serum (VWR International), 2 mM l‐glutamine (Sigma‐Aldrich), and 10 μg/ml nonessential amino acids (Lonza) at 37°C and 5% (v/v) CO2. The cells were transfected with wild‐type or mutant CNNM2 using Lipofectamine 2000 (Invitrogen) at 1:2 DNA:Lipofectamine ratio for 48 h.
2.6. 25Mg2+ isotope assay
HEK293 cells were seeded as described previously (Arjona et al., 2014). Briefly, HEK293 cells transfected with WT or mutant CNNM2 were seeded in poly‐l‐lysine‐coated 12‐well plates. Cells were washed with 1× phosphate‐buffered saline (PBS) and incubated in a culture medium deprived of Mg2+ for 2.5 h, followed by washing with basic uptake buffer deprived of Mg2+ (Table S5) and incubated in a basic uptake buffer containing 1 mM 25Mg2+ (CortecNet). Subsequently, cells were washed with PBS, lysed in HNO3, and subjected to inductively coupled plasma mass spectrometry analysis.
2.7. Immunocytochemistry
HEK293 cells were washed 48 h posttransfection and fixated for 10 min with 4% (w/v) methanol‐free formaldehyde (Thermo Fisher Scientific) followed by 10 min incubation of 0.1% (v/v) Triton‐X100 and 0.3% (w/v) bovine serum albumin solution. After incubation for 10 min in a 50 mM NH4Cl solution, samples were washed and subsequently blocked in 16% (v/v) normal goat serum (Merck Millipore) for 30 min. Cells were then incubated with human influenza hemagglutinin (HA; amino acid sequence: YPYDVPDYA) (Cell Signaling) or FLAG (amino acid sequence: DYKDDDDK) (Sigma‐Aldrich) antibodies. Cells were mounted using a mounting medium supplemented with 4′,6‐diamidino‐2‐phenylindole (Southern Biotech). Fluorescence microscopy was performed on the FluoView 1000 inverted confocal microscope (Olympus Corporation).
2.8. Cell surface biotinylation
HEK293 cells were subjected to a cell surface biotinylation assay as described previously (Arjona et al., 2014). Briefly, the cell surface was biotinylated at 4°C in 0.5 mg/ml sulfo‐NHS‐LC‐LC‐biotin (Thermo Fisher Scientific). Cells were subsequently lysed in lysis buffer (Table S6) for 1 h and the lysate was incubated O/N with NeutrAvidin agarose beads. The beads were then resuspended in lysis buffer, Laemmli buffer, and 100‐mM dithiothreitol followed by denaturation at 37°C for 30 min. Samples were then subjected to sodium dodecyl sulfate polyacrylamide gel electrophoresis followed by immunoblotting.
2.9. Statistical analysis
Data are presented as mean ± standard error of the mean and the statistical significance was determined using a one‐way analysis of variance, followed by a Bonferroni posthoc test for all data. p < .05 was considered statistically significant.
3. RESULTS
3.1. Genetic analyses in novel patients suspected of suffering from HSMR syndrome
In eleven individuals suffering from suspected HSMR syndrome, variants in the CNNM2 gene were identified using next‐generation sequencing or targeted Sanger sequencing (Figures 1a,b). Eleven different variants were detected including six missense, two nonsense mutations, two large deletions, and one in‐frame single amino acid deletion. The deletion of exon 3–8 in CNNM2 observed in patient 2 also affected the neighboring gene NT5C2, encoding the 5′‐nucleotidase, cytosolic II, involved in purine and pyrimidine nucleotide metabolism. Loss of function mutations has been implicated in corticospinal motor neuron disease (SPG45; MIM# 613162) (Dursun et al., 2009; Novarino et al., 2014). For patient 1, it was not possible to determine the extent of the deletion. Genetic analyses of the parents, as well as available family members, demonstrated that eight patients carried de‐novo variants in CNNM2. In contrast, CNNM2 variants in families 3 and 4 were inherited in a dominant fashion (Figure S1). None of the variants was reported in the ExAC Exome or gnomAD Genome Aggregation Database. All missense variants were predicted to have deleterious effects on the protein using Polyphen‐2, SIFT, and CADD, except for the mutation CNNM2‐p.Leu48Pro, localized in the signal peptide domain, which was predicted to have no to mild effects on protein function (Table S7).
Figure 1.
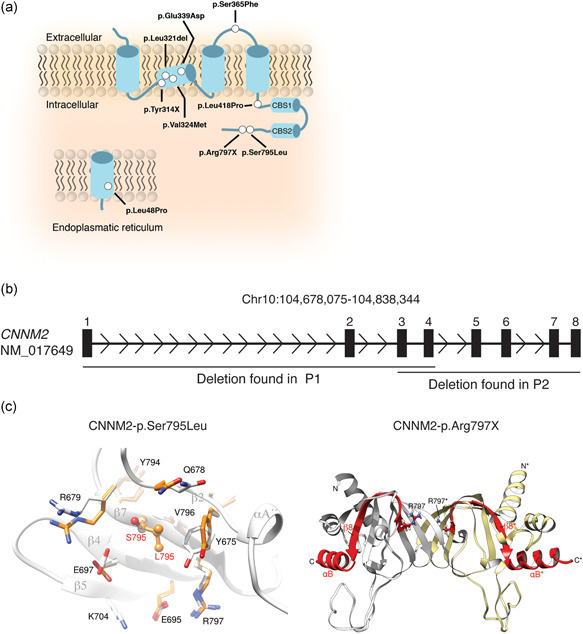
Identification of novel cyclin M2 (CNNM2) variants and their structural effects on the protein level. (a) Schematic overview of the localization of the mutations in the CNNM2 protein. Mutations are depicted as white dots. (b) Schematic overview of the genomic deletions found in the CNNM2 gene (h19; NM_017649) patients 1 and 2 (Table 1). Numbered boxes depict the exons within CNNM2. (c) The effect of mutation p.Ser795Leu was modeled using the crystal structure of CNNM2 (PDB code: 6DJ3) as a template. Left: Super composition of amino acid residues in the p.Ser795Leu environment in the native (light gray) and in the modeled mutation protein (orange). Substitution of serine to leucine (orange) at location 795 causes displacement of p.Glu697, Glu678, Arg679, and Tyr675, without affecting secondary elements (labeled in dark gray) significantly. Right: Crystal structure of the cNMP domain of CNNM2 (PDB code 6DJ3); the cNMP domain presents an α/β structure and self‐associates to form a tight elongated dimer (complementary subunits are colored in light gray and pale yellow, respectively), The β‐sheets occupy the central region and are sandwiched by α‐helices at both sides. The premature stop codon p.Arg797* disrupts the structure of strand β7 and causes the disappearance of the subsequent elements, strand β8 and helix αB (depicted in red) from the protein sequence. Arginine at position 797 is highlighted in balls and sticks. The long unstructured loop located in the bottom is visible only partially in one of the subunits in the crystals and has been modeled in the figure by applying the two‐fold symmetry that relates the two complementary subunits
Two variants located in the C‐terminus of the protein, p.Ser795Leu and p.Arg797*, were modeled to assess their effects on a structural level (Figure 1c). Due to limited structural information of the signal peptide and transmembrane domains of CNNM2, only the effects of the aforementioned mutations were modeled. The substitution of a serine to a leucine at position 795 was predicted to have local effects by exerting a hydrophobic hindrance that promotes displacement of residues Glu697, Gln678. Arg679, and Tyr675 and may destabilize the cNMP domain. The replacement of the serine with a leucine promoted the salt link formation of Arg679 and Glu697 instead of Ser795 and Arg679. The premature stop codon on position 797 disrupts the integrity of strand β7, thereby destabilizing the central B‐sheet, and consequently, the formation of the entire cNMP domain.
3.2. Molecular characterization of novel variants in CNNM2
To investigate the pathogenicity of newly identified CNNM2 variants, HEK293 cells overexpressing the mutant CNNM2 proteins were analyzed by 25Mg2+ uptake assays. For this, we used nonpolarized HEK293 cells and overexpressed the different CNNM2 variants to determine the CNNM2‐dependent Mg2+ influx. Seven variants showed a significant reduction in uptake of 25Mg2+ compared to wild‐type CNNM2, reflecting the impaired function of the protein (Figure 2a). The large deletions (F1 and F2) were not functionally tested. Cell surface biotinylation analysis showed that the plasma membrane expression was reduced in all mutants, except the p.Leu48Pro variant (Figure 3a,b). On the basis of the functional data, patients carrying the p.Gly339Asp and p.Ser365Phe variants could not be confirmed as pathogenic and were excluded from further analysis (clinical data of these two patients are summarized in Table S1).
Figure 2.
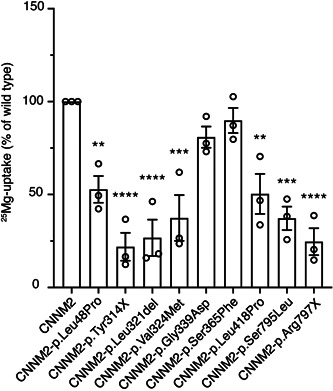
Mutations in cyclin M2 (CNNM2) affect Mg2+uptake in HEK293 cells. Column bars for 25Mg2+ uptake after 5 min. 25Mg2+ uptake was normalized to CNNM2 wild‐type (100%). *p < .05, ** p < .01, ***p < .001, and ****p < .0001, significant compared to wild‐type CNNM2 using a one‐way analysis of variance followed by a Bonferroni posthoc test
Figure 3.
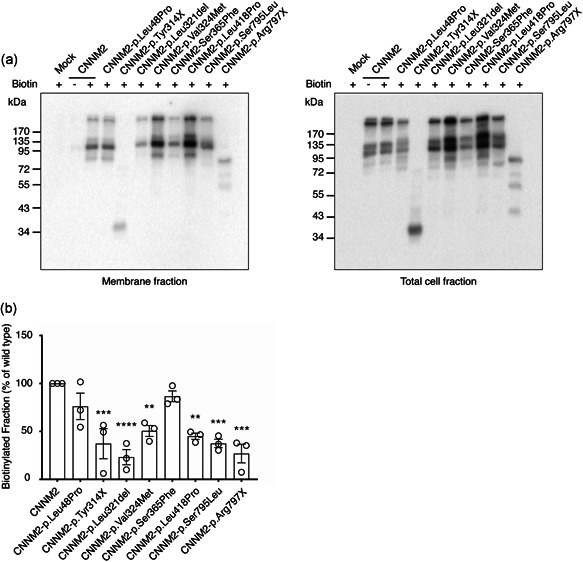
Mutations in cyclin M2 (CNNM2) disturb trafficking to the plasma membrane. (a) Representative immunoblots showing that mutations in CNNM2 reduce cell surface expression (left blot) compared to total CNNM2 expression (right blot). (b) Quantification of cell surface expression of CNNM2 normalized to total fraction and wild‐type CNNM2. Results are mean of duplicates ± standard error of the mean of three independent experiments in which within experiments data were normalized to wild‐type. *Statistical difference to wild‐type CNNM2‐transfected HEK293 cells (p < .05)
To assess the effects of the missense mutation in the signal peptide (p.Leu48Pro), confocal microscopy was employed to investigate the effects on the localization of the protein (Figure 4). The C‐terminal HA‐epitope showed the presence of wild‐type CNNM2 at the plasma membrane. Expression of the FLAG‐tag, before the signal peptide sequence, was absent in wild‐type, in line with normal cleavage and degradation. Interestingly, a prominent expression of the FLAG‐tag in the CNNM2‐p.Leu48Pro variant could be observed, indicating that the signal peptide was not correctly processed. In addition, localization of the HA‐epitope was detected with a more perinuclear pattern, suggestive of partial retention of the protein in the endoplasmic reticulum.
Figure 4.
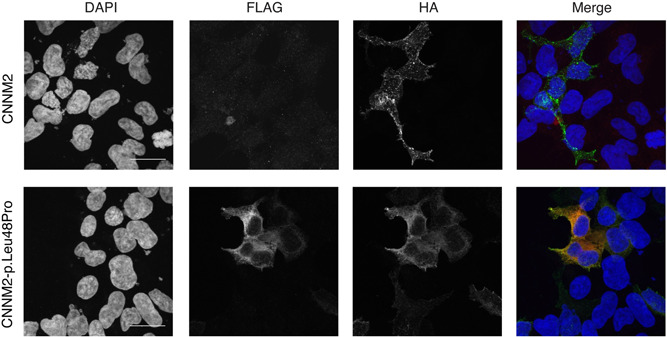
Mutations in cyclin M2 (CNNM2)‐p.Leu48Pro cause retention in the endoplasmic reticulum due to failed signal peptide. Maximum intensity projection of immunofluorescence images of HEK293 cells transiently transfected with wild‐type or mutant CNNM2‐p.Leu48Pro with a FLAG‐tag at the N‐terminus and an HA‐tag at the C‐terminus of the protein. Bars indicate 20 μm. Nuclei are stained with DAPI (4′,6‐diamidino‐2‐phenylindole) shown in blue, the FLAG‐epitope is shown in red, and the HA‐epitope in green
3.3. Phenotypical assessment of patients with CNNM2 mutations
Subsequently, the phenotype of the nine patients carrying pathogenic CNNM2 mutations was examined in detail (Tables 1). In all index patients, symptoms manifested in early life, ranging from birth to young adolescence with a median of 1.54 years (range, birth–16 years). Most mutations were of de novo origin. However, the p.Leu48Pro and p.Tyr314* variants were inherited in a dominant fashion. Clinical data of affected members carrying the p.Leu48Pro are described in Table S2.
The majority of patients suffered from recurrent generalized seizures at a young age (75%), which represented the predominant clinical phenotype. Interestingly, seizures ceased over time allowing termination of antiepileptic treatment. During follow‐up, a certain degree of developmental delay and ID was diagnosed in the majority of patients. Remarkably, a considerable impairment of speech development was noted which varied from a delayed initiation of speech to poor articulation and dysarthria resulting in limited communication skills in six patients. In approximately two‐thirds (63%) of the patients, motor skills were impaired and patients were reported to have hyperkinesia. Magnetic resonance imaging of the brain was available for eight patients showing no structural abnormalities, except in patient 8 (CNNM2‐p.Arg797*) slightly hyperintense white matter located in the corona radiata and centrum semiovale was reported (Figure S2).
Extraneurological symptoms particularly included severe obesity in five out of eight patients (body mass index [BMI] ≥ 97th percentile after correction for age, based on WHO guidelines; World Health Organization, 2019). Patients had hyponormomagnesemia ranging between 0.45 and 0.72 mmol/L (normal: 0.70–1.05 mmol/L) at initial manifestation and normal serum Na+, K+, and Ca2+ levels accompanied with an inappropriate fractional excretion of Mg2+ in the normal range despite a low serum Mg2+ level. Patient 8 (p.Ser795Leu) had a serum Mg2+ concentration of 0.72 mmol/L, yet was already taking Mg2+ supplementation before the initial assessment. Although supplementation with Mg2+ did not fully normalize serum Mg2+ levels concentrations in the hypomagnesemic patients, a general increase was observed.
3.4. Clinical presentation of HSMR syndrome patients carrying CNNM2 mutations
To determine the phenotypic spectrum of HSMR patients, the clinical information of the eight novel patients was combined with five previously described cases with heterozygous, pathogenic CNNM2 mutations (Table 2). The clinical and biochemical hallmarks of these fourteen patients comprised hypomagnesemia, ID, and obesity, which were together present in more than 80% of the patients. The mean serum Mg2+ value in the entire cohort was 0.57 mmol/L and could usually not be fully corrected by Mg2+ supplementation.
Table 2.
Clinical manifestations of HSMR syndrome patients with known deleterious heterozygous CNNM2 mutations
| CNNM2 variants previously reported | CNNM2 variants in novel cohort | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Mutation (ref) | p.Ile40SerfsX15 (Stuiver et al., 2011) | p.Ser269Trp (Arjona et al., 2014) | p.Gly357Lys F1 (Arjona et al., 2014) | p.Gly357Lys F2 (Arjona et al., 2014) | p.Thr568Ile (Stuiver et al., 2011) | Del ex1‐4 (F1) | Del ex3‐8 (F2) | p.Leu48Pro (F3) | p.Tyr314X (F4) | p.Leu321del (F5) | p.Val324Met (F6) | p.Leu418Pro (F7) | p.Ser795Leu (F8) | p.Arg797X (F9) | Average |
| Function (% of WT) | 0 | 17 | 1 | 1 | 0 | 0 | 0 | 49 | 12 | 20 | 16 | 46 | 48 | 29 | |
| Serum Mg2+(mmol/L) | 0.51 | 0.44 | 0.56 | 0.5 | 0.6 | 0.63 | 0.57 | 0.45 | 0.48 | 0.5 | 0.54 | 0.49 | 0.72 | 0.57 | 0.54 ± 0.08 |
| Age at onset (year) | 1 | 1 | 0.58 | 0.08 | 16 | 6 | 0.25 | 2 | 3 | 0.67 | 1.08 | 16 | 0.67 | 1 | 3.52 ± 5.50 |
| Obesity (BMI >97th percentile) | N/A | Y | Y | Y | N/A | Y | N | N/A | Y | Y | Y | N/A | N | Y | 80% (8/10) |
| Intellectual disability | N/A | Y | Y | Y | N/A | Y | Y | N | Y | Y | Y | Y | Y | Y | 92% (11/12) |
| Seizures | Y | Y | Y | Y | N | Y | Y | Y | N | Y | Y | Y | N | Y | 79% (11/14) |
| Speech/Communication | N/A | Y | Y | Y | N/A | Y | Y | N | Y | Y | Y | N/A | Y | Y | 91% (10/11) |
| Motor skills defects | N | Y | Y | Y | N | Y | Y | Y | N | Y | Y | N | Y | N | 64% (9/14) |
| Correction by Mg2+ supplementation | N/A | N | N | N | N | N/A | N/A | N | N/A | N | N | N | N | N | 0% (0/10) |
Note: Full correction by Mg2+ supplementation was defined as increasing serum Mg2+ levels to the normal range (0.70–0.05 mmol/L). The average for categorical parameters was determined by (affected probands/total) × 100% of those with available data. Average for continuous parameters are presented as mean ± SD.
Abbreviations: BMI, body mass index; F, family number in Table 1.; N, no; N/A, unknown; WT, wild‐type; Y, yes.
In particular, in early life, the clinical picture of HSMR is dominated by generalized seizure episodes, which subsided during follow‐up in the majority (11/14) of patients. Therefore, the permanent antiepileptic medication could be stopped in several patients without reoccurrence of seizures. However, a mild to moderate degree of neurodevelopmental delay represented a uniform feature of HSMR syndrome comprising intellectual deficits as well as impaired functional skills. Formal intelligence testing performed in two patients using the Snijders Oomen nonverbal intelligence test (SON‐R 5½–17) revealed a mild to moderate degree of ID (IG 55–59) (Arjona et al., 2014). Notably, a disturbed speech development with dysarthria and an impaired expressive language represent prominent clinical features in HSMR patients (90% of patients). In addition, two‐thirds of the patients exhibited deficits in motor functioning with difficulties in both fine and gross motor skills and hyperkinetic movements. It remains unclear whether this is related to the mild ID (Vuijk et al., 2010) or represents an independent feature of the disease itself. The majority of patients attend schools for special education and require further support such as physiotherapy and ergotherapy. Of note, there was no indication of an increased frequency of signs of autism spectrum disorder. Apart from neurologic manifestations, a large proportion of patients suffered from severe obesity (88%, BMI > 97) irrespective of gender.
4. DISCUSSION
To date, the spectrum of clinical features of patients with HSMR remained insufficiently defined, rendering the clinical diagnosis of the disease a challenge. A limited number of patients has been reported only by a small number of groups suggesting a large proportion of unrecognized and undiagnosed cases. In this study, the largest cohort of patients to date has been characterized to develop improved diagnostic criteria for HSMR syndrome. Our phenotypic data show that hypomagnesemia, cerebral seizures, obesity, and ID are essential features of CNNM2‐mediated disease. Additional characteristic neurodevelopmental findings comprise impairments of speech development and vocal communication as well as disturbed motor skills.
Generally, children and adolescents with ID are at greater risk of developing overweight and obesity (Maïano, 2011). However, the frequency, as well as the extent of obesity observed in HSMR patients by far, exceeds this increased risk observed in ID cohorts. Our findings rather suggest that obesity represents a primary feature of HSMR syndrome. Inline, a genetic association between the CNNM2 locus and obesity has been identified (Lv et al., 2017). Given that obesity is not a prominent finding of other hereditary hypomagnesemia syndromes, CNNM2 may have an Mg2+‐independent role in metabolism.
In contrast, features of ASD as observed in one HSMR patient described previously were not present in additional patients of this larger cohort (Arjona et al., 2014). However, similar features have been reported in single patients with hypomagnesemia due to ATP1A1 defects and global developmental delay (Schlingmann et al., 2018). The developmental delay and specifically the speech impairment present in CNNM2 patients might be easily confused with ASD in some HSMR patients without standardized ASD diagnostic tools.
Our genetic analyses demonstrate that CNNM2 variants primarily affect one allele and, in the majority of patients, arise de novo explaining the large proportion of sporadic cases. In contrast, CNNM2 mutations in two novel families were inherited in a dominant fashion as in the two originally reported families (Stuiver et al., 2011). Hypomagnesemia was present in all genetically affected family members of family 3 (CNNM2‐p.Leu48Pro). However, neurological symptoms in these individuals were largely absent or only mild (Table S2). Unfortunately, detailed phenotypic data of affected family members of family 3 (CNNM2‐p.Tyr314*) were not available. Of note, a neurological phenotype was also absent in the two initially reported families with dominantly inherited CNNM2 mutations (Meij et al., 2003; Stuiver et al., 2011).
Contrasting these moderate neurological findings in patients with heterozygous mutations, a severe brain phenotype has been described in patients with homozygous CNNM2 mutations (Accogli et al., 2018; Arjona et al., 2014). The disease manifestation in the neonatal period, the extent of seizures, the severe degree of intellectual disability, and the presence of structural brain abnormalities clearly render recessive CNNM2 disease a distinct clinical entity.
The nonsense mutations leading to a preterminal stop of translation as well as the deletion of multiple exons of the CNNM2 gene identified in two patients clearly argue for a loss of CNNM2 function as the general underlying disease mechanism of HSMR and for functional haploinsufficiency in monoallelic cases. Patients suffering from recessive CNNM2 mutations especially display severe structural brain defects, whereas patients with monoallelic CNNM2 mutations usually lack structural brain abnormalities suggesting that at least 50% expression of wild‐type CNNM2 appear to be sufficient to support physiologic brain development.
HSMR syndrome belongs to the clinically and genetically heterogeneous group of renal Mg2+ wasting disorders (Accogli et al., 2018; Arjona et al., 2014; Stuiver et al., 2011). Epileptic seizures and ID have also been described in patients with TRPM6 (HSH; MIM# 602014) and ATP1A1 (HOMGSMR2; MIM# 618314) mutations (Schlingmann et al., 2002, 2005, 2018). Although HSH patients usually efficiently respond to oral Mg2+ supplementation that typically leads to a cessation of seizures and an undisturbed psychomotor development, serum Mg2+ concentration in HSMR patients usually remains low despite Mg2+ supplementation. At the same time, there is an amelioration of seizure activity or seizures might cease completely allowing for discontinuation of antiepileptic medication during childhood or adolescence. Patients with ATP1A1 mutations experience generalized seizures in early infancy and severely low serum Mg2+ levels (approximately 0.35 mmol/L) at manifestation. Particularly in early life, the clinical presentation of children with ATP1A1 defects might be highly similar to children with recessive CNNM2 mutations.
The different modes of inheritance of HSMR syndrome as well as the diverse clinical presentations of affected patients and family members underpin the importance of experimental verification of rare CNNM2 variants to determine pathogenicity. In our functional assays, we overexpress CNNM2 and determine the functionality by changes of intracellular 25Mg2+ levels in the cells, which is a measure of Mg2+ influx. Although the function of CNNM2 is yet to be completely elucidated, it is postulated to transport Mg2+ itself, but to regulate Mg2+ import or export depending on the expression system (Arjona et al., 2014).
In the present study, two CNNM2 variants, p.Gly339Asp and p.Ser365Phe, displayed preserved functionality despite results of in silico prediction tools. In these cases, we were not able to confirm pathogenicity using our functional assay. Therefore, the respective patients were excluded from the further characterization of the HSMR phenotype. Indeed, the patient suffering the p.Ser365Phe variant did not experience hypomagnesemia and suffered other symptoms, such as microcephaly and cardiovascular disease, which was absent in all other reported cases. This led to the conclusion that this patient's phenotype may not be caused by the observed CNNM2 variant and that the genetic cause for the phenotype had yet to be identified.
Recently, several novel CNNM2 variants have been identified in patients suffering from electrolyte disorders, such as Bartter syndrome and clinical familial hypocalciuric, and hypercalcemia (García‐Castaño et al., 2020). Interestingly, one patient also had a premature stop codon at CNNM2‐p.Ser795, which is exactly the same amino acid that has been affected in one of our patients (p.Ser795Leu). However, as these novel variants are classified as variants of unknown significance, causality has not been definitely established. This emphasizes that functional testing is required for clinical confirmation of HSMR syndrome.
To date, only individual cases of HSMR syndrome had been described, which did not allow for detailed phenotypic analysis of patients. The identification of nine new patients demonstrates that CNNM2 mutations explain a substantial group of patients with hereditary hypomagnesemia and are more common than other forms of isolated hypomagnesemia, such as EGF, KCNA1, and FXYD2 (de Baaij et al., 2015; Glaudemans et al., 2009; Groenestege et al., 2007; Meij et al., 2000). In this study, we were able to combine reported cases with our cohort, resulting in a detailed description of prominent features of the disorder. A limitation of our study is that a significant portion of patients was already suspected of HSMR syndrome, potentially causing a selection bias. A more extensive genetic screening of patients with isolated hypomagnesemia or intellectual disability may, therefore, further expand the spectrum of CNNM2‐mediated disease and improve the characterization of the HSMR phenotype.
In conclusion, we characterized a novel patient cohort carrying heterozygous CNNM2 mutations on the clinical, genetic, and molecular levels. Combined with all known HSMR syndrome cases caused by defective CNNM2, this approach allowed us to distinguish common features between patients enabling the clinical diagnosis of HSMR. Physicians should include CNNM2 into a genetic screen upon receiving patients with lowered serum Mg2+ concentrations (±0.5–0.7 mmol/L), ID, transient seizures, obesity, and speech/language defects. To confirm HSMR syndrome, functional verification studies are recommended.
WEB RESOURCES
For this study, the following web resources have been consulted: https://cadd.gs.washington.edu for the in silico prediction; https://www.who.int/toolkits/child-growth-standards/standards/weight-for-age to determine the BMI percentile of the patients; https://gnomad.broadinstitute.org and http://exac.broadinstitute.org to validate the novelty of our observed variants.
CONFLICT OF INTERESTS
The authors declare that there are no conflict of interests.
Supporting information
Supplementary information.
ACKNOWLEDGMENTS
We thank Daan Viering and Dorien Lugtenberg for their help regarding the genetic analyses. This work was financially supported by ZonMW under the frame of EJPRD, the European Joint Programme on Rare Diseases (EJPRD2019‐40). In addition, this project has received funding from the European Union's Horizon 2020 research and innovation programme under the EJP RD COFUND‐EJP No. 825575. This contribution of Jeroen H. F. de Baaij and Joost G. J. Hoenderop was financially supported by a grant from the Netherlands Organization for Scientific Research: NWO Veni 016.186.012 and VICI 016.130.668, respectively. The contribution of coauthor F. Claverie‐Martin was supported by grant PI14/00760 cofinanced by the ISCIII‐Subdirección General de Evaluación y Fomento de la Investigación and the European Regional Development Fund “Another way to build Europe.” The funders had no role in study design, data collection, and analysis, decision to publish, or preparation of the manuscript.
Franken, G. A. C. , Müller, D. , Mignot, C. , Keren, B. , Lévy, J. , Tabet, A.‐C. , Germanaud, D. , Tejada, M‐I. , Kroes, H. Y. , Nievelstein, R. A. J. , Brimble, E. , Ruzhnikov, M. , Claverie‐Martin, F. , Szczepańska, M. , Ćuk, M. , Latta, F. , Konrad, M. , Martínez‐Cruz, L. A. , Bindels, R. J. , … de Baaij, J. H. F. (2021). The phenotypic and genetic spectrum of patients with heterozygous mutations in Cyclin M2 (CNNM2). Human Mutation, 42, 473–486. 10.1002/humu.24182
Karl‐Peter Schlingmann and Jeroen H. F. de Baaij contributed equally to this study.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- Accogli, A. , Scala, M. , Calcagno, A. , Napoli, F. , Di Iorgi, N. , Arrigo, S. , Mancardi, M. M. , Prato, G. , Pisciotta, L. , Nagel, M. , Severino, M. , & Capra, V. (2018). CNNM2 homozygous mutations cause severe refractory hypomagnesemia, epileptic encephalopathy and brain malformations. European Journal of Medical Genetics, 62, 198–203. 10.1016/j.ejmg.2018.07.014 [DOI] [PubMed] [Google Scholar]
- Arjona, F. J. , & de Baaij, J. H. (2018). CNNM proteins are not Na+/Mg2+ exchangers, but Mg2+ transport regulators playing a central role in transepithelial Mg2+ (re)absorption. Journal of Physiology, 596, 747–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arjona, F. J. , de Baaij, J. H. F. , Schlingmann, K. P. , Lameris, A. L. L. , van Wijk, E. , Flik, G. , Regele, S. , Korenke, G. C. , Neophytou, B. , Rust, S. , Reintjes, N. , Konrad, M. , Bindels, R. J. M. , & Hoenderop, J. G. J. (2014). CNNM2 mutations cause impaired brain development and seizures in patients with hypomagnesemia. PLOS Genetics, 10(4), e1004267. 10.1371/journal.pgen.1004267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, Y. S. , Kozlov, G. , Fakih, R. , Funato, Y. , Miki, H. , & Gehring, K. (2018). The cyclic nucleotide‐binding homology domain of the integral membrane protein CNNM mediates dimerization and is required for Mg2+ efflux activity. Journal of Biological Chemistry, 293, 19998–20007. 10.1074/jbc.RA118.005672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Baaij, J. H. F. , Dorresteijn, E. M. , Hennekam, E. A. M. , Kamsteeg, E. J. , Meijer, R. , Dahan, K. , Muller, M. , van den Dorpel, M. A. , Bindels, R. J. M. , Hoenderop, J. G. J. , Devuyst, O. , & Knoers, N. V. A. M. (2015). Recurrent FXYD2 p.Gly41Arg mutation in patients with isolated dominant hypomagnesaemia. Nephrology, Dialysis, Transplantation, 30(6), 952–957. 10.1093/ndt/gfv014 [DOI] [PubMed] [Google Scholar]
- de Baaij, J. H. F. , Stuiver, M. , Meij, I. C. , Lainez, S. , Kopplin, K. , Venselaar, H. , Müller, D. , Bindels, R. J. M. , & Hoenderop, J. G. J. (2012). Membrane topology and intracellular processing of cyclin M2 (CNNM2). Journal of Biological Chemistry, 287(17), 13644–13655. 10.1074/jbc.M112.342204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dichgans, M. , Malik, R. , König, I. R. , Rosand, J. , Clarke, R. , Gretarsdottir, S. , Thorleifsson, G. , Mitchell, B. D. , Assimes, T. L. , Levi, C. , O'Donnell, C. J. , Fornage, M. , Thorsteinsdottir, U. , Psaty, B. M. , Hengstenberg, C. , Seshadri, S. , Erdmann, J. , Bis, J. C. , Peters, A. , … Kooner, J. S. (2014). Shared genetic susceptibility to ischemic stroke and coronary artery disease: A genome‐wide analysis of common variants. Stroke, 45(1), 24–36. 10.1161/strokeaha.113.002707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dursun, U. , Koroglu, C. , Kocasoy Orhan, E. , Ugur, S. A. , & Tolun, A. (2009). Autosomal recessive spastic paraplegia (SPG45) with mental retardation maps to 10q24.3‐q25.1. Neurogenetics, 10(4), 325–331. 10.1007/s10048-009-0191-3 [DOI] [PubMed] [Google Scholar]
- Funato, Y. , Furutani, K. , Kurachi, Y. , & Miki, H. (2018). CNNM proteins are Na+/Mg2+ exchangers playing a central role in transepithelial Mg2+ (re)absorption. Journal of Physiology, 596, 743–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García‐Castaño, A. , Madariaga, L. , Antón‐Gamero, M. , Mejia, N. , Ponce, J. , Gómez‐Conde, S. , Pérez de Nanclares, G. , de la Hoz, A. B. , Martínez, R. , Saso, L. , Martínez de LaPiscina, I. , Urrutia, I. , Velasco, O. , Aguayo, A. , Castaño, L. , & Gaztambide, S. (2020). Novel variant in the CNNM2 gene associated with dominant hypomagnesemia. PLOS One, 15(9), e0239965. 10.1371/journal.pone.0239965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glaudemans, B. , van der Wijst, J. , Scola, R. H. , Lorenzoni, P. J. , Heister, A. , van der Kemp, A. W. , Knoers, N. V. , Hoenderop, J. G. , & Bindels, R. J. (2009). A missense mutation in the Kv1.1 voltage‐gated potassium channel‐encoding gene KCNA1 is linked to human autosomal dominant hypomagnesemia. Journal of Clinical Investigation, 119(4), 936–942. 10.1172/jci36948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grigoroiu‐Serbanescu, M. , Diaconu, C. C. , Heilmann‐Heimbach, S. , Neagu, A. I. , & Becker, T. (2015). Association of age‐of‐onset groups with GWAS significant schizophrenia and bipolar disorder loci in Romanian bipolar I patients. Psychiatry Research, 230(3), 964–967. 10.1016/j.psychres.2015.11.008 [DOI] [PubMed] [Google Scholar]
- Groenestege, W. M. T. , Thébault, S. , van der Wijst, J. , van den Berg, D. , Janssen, R. , Tejpar, S. , van den Heuvel, L. P. , van Cutsem, E. , Hoenderop, J. G. , Knoers, N. V. , & Bindels, R. J. (2007). Impaired basolateral sorting of pro‐EGF causes isolated recessive renal hypomagnesemia. Journal of Clinical Investigation, 117(8), 2260–2267. 10.1172/jci31680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hruby, A. , Ngwa, J. S. , Renström, F. , Wojczynski, M. K. , Ganna, A. , Hallmans, G. , Houston, D. K. , Jacques, P. F. , Kanoni, S. , Lehtimäki, T. , Lemaitre, R. N. , Manichaikul, A. , North, K. E. , Ntalla, I. , Sonestedt, E. , Tanaka, T. , van Rooij, F. J. A. , Bandinelli, S. , Djoussé, L. , … Nettleton, J. A. (2013). Higher magnesium intake is associated with lower fasting glucose and insulin, with no evidence of interaction with select genetic loci, in a meta‐analysis of 15 CHARGE Consortium Studies. Journal of Nutrition, 143(3), 345–353. 10.3945/jn.112.172049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauck, F. , Smith, C. A. , Friedland, G. F. , Humphris, E. L. , & Kortemme, T. (2010). RosettaBackrub—A web server for flexible backbone protein structure modeling and design. Nucleic Acids Research, 38(Web Server issue), W569–W575. 10.1093/nar/gkq369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, C. , Kim, Y. K. , Dorajoo, R. , Li, H. , Lee, I. T. , Cheng, C. Y. , He, M. , Sheu, W. H. , Guo, X. , Ganesh, S. K. , He, J. , Lee, J. , Liu, J. , Hu, Y. , Rao, D. C. , Tsai, F. J. , Koh, J. Y. , Hu, H. , Liang, K. W. , … Kelly, T. N. (2017). Genome‐wide association study meta‐analysis of long‐term average blood pressure in East Asians. Circulation: Cardiovascular Genetics, 10(2), e001527. 10.1161/circgenetics.116.001527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lotan, A. , Fenckova, M. , Bralten, J. , Alttoa, A. , Dixson, L. , Williams, R. W. , & van der Voet, M. (2014). Neuroinformatic analyses of common and distinct genetic components associated with major neuropsychiatric disorders. Frontiers in Neuroscience, 8, 331. 10.3389/fnins.2014.00331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lv, W.‐Q. , Zhang, X. , Zhang, Q. , He, J.‐Y. , Liu, H.‐M. , Xia, X. , Fan, K. , Zhao, Q. , Shi, X. Z. , Zhang, W. D. , Sun, C. Q. , & Deng, H. W. (2017). Novel common variants associated with body mass index and coronary artery disease detected using a pleiotropic cFDR method. Journal of Molecular and Cellular Cardiology, 112, 1–7. 10.1016/j.yjmcc.2017.08.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuoka, R. , Abe, S. , Tokoro, F. , Arai, M. , Noda, T. , Watanabe, S. , Horibe, H. , Fujimaki, T. , Oguri, M. , Kato, K. , Minatoguchi, S. , & Yamada, Y. (2015). Association of six genetic variants with myocardial infarction. International Journal of Molecular Medicine, 35(5), 1451–1459. 10.3892/ijmm.2015.2115 [DOI] [PubMed] [Google Scholar]
- Maïano, C. (2011). Prevalence and risk factors of overweight and obesity among children and adolescents with intellectual disabilities. Obesity Reviews, 12(3), 189–197. 10.1111/j.1467-789X.2010.00744.x [DOI] [PubMed] [Google Scholar]
- Meij, I. C. , van den Heuvel, L. P. , Hemmes, S. , van der Vliet, W. A. , Willems, J. L. , Monnens, L. A. , & Knoers, N. V. (2003). Exclusion of mutations in FXYD2, CLDN16 and SLC12A3 in two families with primary renal Mg2+ loss. Nephrology, Dialysis, Transplantation, 18(3), 512–516. 10.1093/ndt/18.3.512 [DOI] [PubMed] [Google Scholar]
- Meij, I. C. , Koenderink, J. B. , van Bokhoven, H. , Assink, K. F. H. , Tiel Groenestege, W. , de Pont, J. J. H. H. M. , Bindels, R. J. M. , Monnens, L. A. H. , van den Heuvel, L. P. W. J. , & Knoers, N. V. A. M. (2000). Dominant isolated renal magnesium loss is caused by misrouting of the Na(+),K(+)‐ATPase gamma‐subunit. Nature Genetics, 26(3), 265–266. 10.1038/81543 [DOI] [PubMed] [Google Scholar]
- Novarino, G. , Fenstermaker, A. G. , Zaki, M. S. , Hofree, M. , Silhavy, J. L. , Heiberg, A. D. , Abdellateef, M. , Rosti, B. , Scott, E. , Mansour, L. , Masri, A. , Kayserili, H. , Al‐Aama, J. Y. , Abdel‐Salam, G. M. H. , Karminejad, A. , Kara, M. , Kara, B. , Bozorgmehri, B. , Ben‐Omran, T. , … Gleeson, J. G. (2014). Exome sequencing links corticospinal motor neuron disease to common neurodegenerative disorders. Science, 343(6170), 506–511. 10.1126/science.1247363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohi, K. , Hashimoto, R. , Yamamori, H. , Yasuda, Y. , Fujimoto, M. , Umeda‐Yano, S. , Fukunaga, M. , Watanabe, Y. , Iwase, M. , Kazui, H. , & Takeda, M. (2013). The impact of the genome‐wide supported variant in the cyclin M2 gene on gray matter morphology in schizophrenia. Behavioral and Brain Functions, 9, 40. 10.1186/1744-9081-9-40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose, E. J. , Hargreaves, A. , Morris, D. , Fahey, C. , Tropea, D. , Cummings, E. , Caltagirone, C. , Bossú, P. , Chiapponi, C. , Piras, F. , Spalletta, G. , Gill, M. , Corvin, A. , & Donohoe, G. (2014). Effects of a novel schizophrenia risk variant rs7914558 at CNNM2 on brain structure and attributional style. British Journal of Psychiatry, 204(2), 115–121. 10.1192/bjp.bp.113.131359 [DOI] [PubMed] [Google Scholar]
- Schlingmann, K. P. , Bandulik, S. , Mammen, C. , Tarailo‐Graovac, M. , Holm, R. , Baumann, M. , König, J. , Lee, J. J. Y. , Drögemöller, B. , Imminger, K. , Beck, B. B. , Altmüller, J. , Thiele, H. , Waldegger, S. , van't Hoff, W. , Kleta, R. , Warth, R. , van Karnebeek, C. D. M. , Vilsen, B. , … Konrad, M. (2018). Germline de novo mutations in ATP1A1 cause renal hypomagnesemia, refractory seizures, and intellectual disability. The American Journal of Human Genetics, 103(5), 808–816. 10.1016/j.ajhg.2018.10.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlingmann, K. P. , Sassen, M. C. , Weber, S. , Pechmann, U. , Kusch, K. , Pelken, L. , Lotan, D. , Syrrou, M. , Prebble, J. J. , Cole, D. E. C. , Metzger, D. L. , Rahman, S. , Tajima, T. , Shu, S. G. , Waldegger, S. , Seyberth, H. W. , & Konrad, M. (2005). Novel TRPM6 mutations in 21 families with primary hypomagnesemia and secondary hypocalcemia. Journal of the American Society of Nephrology, 16(10), 3061–3069. 10.1681/asn.2004110989 [DOI] [PubMed] [Google Scholar]
- Schlingmann, K. P. , Weber, S. , Peters, M. , Niemann Nejsum, L. , Vitzthum, H. , Klingel, K. , Kratz, M. , Haddad, E. , Ristoff, E. , Dinour, D. , Syrrou, M. , Nielsen, S. , Sassen, M. , Waldegger, S. , Seyberth, H. W. , & Konrad, M. (2002). Hypomagnesemia with secondary hypocalcemia is caused by mutations in TRPM6, a new member of the TRPM gene family. Nature Genetics, 31, 166–170. 10.1038/ng889 [DOI] [PubMed] [Google Scholar]
- Stuiver, M. , Lainez, S. , Will, C. , Terryn, S. , Günzel, D. , Debaix, H. , Sommer, K. , Kopplin, K. , Thumfart, J. , Kampik, N. B. , Querfeld, U. , Willnow, T. E. , Němec, V. , Wagner, C. A. , Hoenderop, J. G. , Devuyst, O. , Knoers, N. V. A. M. , Bindels, R. J. , Meij, I. C. , & Müller, D. (2011). CNNM2, encoding a basolateral protein required for renal Mg2+ handling, is mutated in dominant hypomagnesemia. American Journal of Human Genetics, 88(3), 333–343. 10.1016/j.ajhg.2011.02.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vuijk, P. J. , Hartman, E. , Scherder, E. , & Visscher, C. (2010). Motor performance of children with mild intellectual disability and borderline intellectual functioning. Journal of Intellectual Disability Research, 54(11), 955–965. 10.1111/j.1365-2788.2010.01318.x [DOI] [PubMed] [Google Scholar]
- World Health Organization . (2019). Child growth standards. Weight‐for‐age. https://www.who.int/childgrowth/standards/weight_for_age/en/
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary information.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.