

Abstract
Icaritin, a small molecule currently being investigated in phase III clinical trials in China (NCT03236636 and NCT03236649) for treatment of advanced hepatocellular carcinoma (HCC), is a prenylflavonoid derivative obtained from the Epimedium genus. Previously, it was found that Icaritin decreased the expression of PD‐L1, but its direct molecular targets and the underlying mechanisms have not been identified. In this study, we report the identification of IKK‐α as the protein target of Icaritin by biotin‐based affinity binding assay. The further mutagenesis assay has provided evidence that C46 and C178 in IKK‐α were essential amino acids for Icaritin binding to IKK‐α, revealing the binding sites of Icaritin to IKK‐α for the first time. Functionally, Icaritin inhibited the NF‐κB signalling pathway by blocking IKK complex formation, which led to decreased nuclear translocation of NF‐κB p65, and subsequent downregulation of PD‐L1 expression in a dose–dependent manner. More importantly, PD‐L1‐positive patients exhibited longer overall survival upon Icaritin therapy. Finally, Icaritin in combination with checkpoints antibodies, such as α‐PD‐1, has demonstrated much better efficacy than any single therapy in animal models. This is the first report that anticancer effects of Icaritin are mediated, at least in part, by impairing functions of IKK‐α.
Keywords: HCC, Icaritin, NF‐κB, PD‐L1, Immunotherapy
In this study, IKK‐α was identified as the protein target of Icaritin, by blocking the complex formation of IKK‐α/‐β, Icaritin inhibits NF‐κB p65 translocation and then downregulates PD‐L1 expression in tumor cells and immunosuppressive cells, which enhance the effect of immunotherapy.
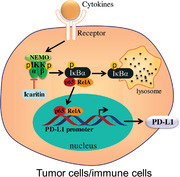
Introduction
Icaritin is a prenylflavonoid derivative obtained from the Epimedium genus that has long been used in Chinese traditional medicine. Icaritin has shown antitumor activities by inhibiting endometrial [1] and breast [2] cancer cell proliferation and prostate cancer cell growth [3]. Icaritin can also induce apoptosis of prostate cancer cells [4], AML cells [5], hepatocellular carcinima cells (HCCs) [6], and glioblastoma cells [7]. In its phase I/II clinic trials, Icaritin had demonstrated favourable safety, tolerability, and more durable overall survival rate compared with sorafenib in advanced HCC patients [8]. Currently Icaritin is in its late stage of phase III trials treating patients with advanced HCC as a single agent.
The programmed cell death‐ligand 1 (PD‐L1)/programmed cell death protein 1 (PD‐1) pathway has been identified as the most critical immune checkpoint in immunotherapy [9]. PD‐L1 is expressed on immune cells and multiple cancer cells including HCC [10]. PD‐L1 binds to its receptor PD‐1, which is expressed on CTLs, and thus, inhibits the antitumor function of CTLs and leads to tumor evasion [11]. Studies have reported that PD‐L1 was regulated, at least partially, by the NF‐κB signaling pathway [12], therefore, drugs that inhibit NF‐κB leading to PD‐L1 expression suppression and eventually restoration of CTL function are promising for cancer immunotherapy. Our previous study found that Icaritin treatment significantly decreased PD‐L1 expression in HL‐60 cells but the underlying molecular mechanisms have not been elucidated since no molecular targets was identified or proposed in that article [13]. Though several proteins have been summarized in a recent review as the binding targets of Icartin [14], the underlying molecular mechanisms of Icaritin inhibiting PD‐L1 expression are still not elucidated with these targets. Herein, we present physical evidence that Icaritin directly interacts with IKK‐α, indicating that Icaritin interferes with the NF‐κB pathways. The present study may provide some explanation of the molecular mechanisms of Icaritin, especially regarding its antitumor effect, and implicates other clinical applications of this compound.
Results
Icaritin inhibits tumor growth in vivo
We previously reported that Icaritin treatment dose‐dependently decreased tumor burden of murine B16F10 melanoma and MC38 colorectal tumors in a T cell‐dependent manner. The treatment effects are associated with increased CD8 T‐cell infiltration and increased effector memory T‐cell frequency [13]. To further examine antitumor activities of Icaritin in vivo, hepatocellular carcinoma Hepa1‐6, colorectal cancer MC‐38, or melanoma B16F10 cells were employed to establish a syngeneic mouse model and explore Icaritin antitumor activity through immune modulation. The syngeneic model has been proved to be the best model to study immunocheckpoint PD‐1 in‐vivo efficacy as single agent or in combinational therapy. Mice were treated with 70 mg/kg Icaritin as described in previous report [13] (at its maximum dose without apparent toxicity and its clinical dosage), while tumor volume and tumor weight were measured. Both tumor volume and tumor weight were significantly decreased in the Icaritin group compared with those of vehicle group in Hepa1‐6 and B16F10 models, and the tumor volume and tumor weight were also slightly decreased in MC‐38 model (Fig. 1, Supporting information Fig. S1). The results clearly demonstrated that Icaritin inhibited tumor growth in these mouse models.
Figure 1.
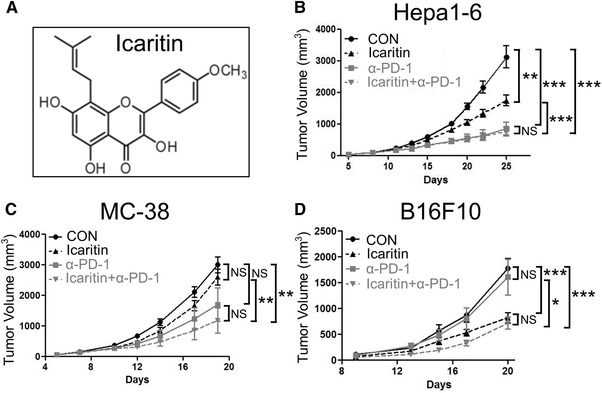
Icaritin suppress tumor growth cumulatively with PD‐1 mAb. (A) The chemical structure of Icaritin. C57BL/6 mice (n = 8) inoculated with 2 × 105 Hepa1‐6 (B), MC‐38 (C), or B16F10 (D) tumor cells were treated with 70 mg/kg Icaritin (p.o. bid, dissolved in corn oil) or 5 mg/kg anti‐PD‐1 mAb (i.v. biw) or both. Tumor growth of tumor‐bearing mice measured by tumor volume. Data are from a single experiment representative of three independent experiments with eight mice in each group per experiment. Values are mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001, NS: no significance (by Student's t test).
Exploratory biomarker studies in phase II Icaritin clinical trial have demonstrated that patients with positive PD‐L1 expression exhibited longer overall survival than patients with negative PD‐L1 expression (Supporting information Fig. S2). PD‐L1 positive patients might be more sensitive to Icaritin, and Icaritin might increase the efficacy of PD(L)‐1 antibody. To test this hypothesis, Hepa1‐6, MC‐38, and B16F10 mice were treated with Icaritin and in combination with anti‐PD‐1 antibody. Although Icaritin or anti‐PD‐1 mAb individually showed modest tumor growth inhibition, Icaritin and anti‐PD‐1 mAb together inhibited tumor growth cumulatively, leading to significantly greater tumor growth inhibition in all three models (Fig. 1/Supporting information Fig. 1). Furthermore, even though PD‐1 mAb in MC‐38 and B16F10 models showed no obviously efficacy, significant tumor growth inhibition was observed when it was combined with Icartin (Fig. 1C and D), which strongly indicated that animals resistant to anti‐PD‐1 therapy could benefit from the combined treatment of Icaritin and PD‐1 mAb. Those results demonstrated and confirmed that Icaritin inhibited tumor growth in these mouse models in combination with PD‐1 as described in previous report that Icaritin demonstrated the combinational in‐vivo efficacy with PD‐1 and CTLA‐4 as triple drug therapy [13].
Icaritin regulates the expression of checkpoints
To confirm whether Icaritin regulate PD‐L1 expression in monocytes and cancer cells, we treated HCC cell line SMMC‐7721 and monocytes THP‐1 with Icaritin and analysed the expression level of PD‐L1. As shown in Fig. 2, Icaritin pretreatment significantly decreased TNF‐α‐induced PD‐L1 at both the protein (Fig. 2A) and mRNA (Fig. 2B) levels in SMMC‐7721 and THP‐1 cells. The expression level of PD‐L1 on THP‐1 cells surface was also measured use flow cytometry. Figure 3C and D shows that Icaritin pretreatment also obviously decreased PD‐L1 expression on THP‐1 cell surface. These results indicated that Icaritin is able to inhibit PD‐L1 expression in both immune cells and cancer cells, leading to its combinational in‐vivo efficacy with anti‐PD1 antibody in inhibition of malignant tumor growth, because Icaritin functions through immune modulation, particularly immune cells with myeloid lineage, so that THP‐1 cell line was employed along with SMMC‐7221 cell line.
Figure 2.
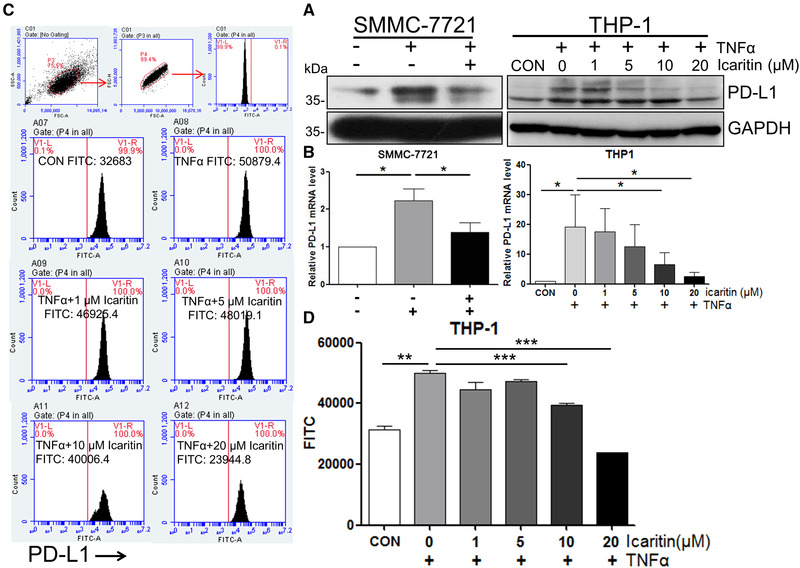
Icaritin inhibits PD‐L1 expression. Cells were treated with 10 μM (SMMC‐7721) or indicated concentrations (THP‐1) of Icaritin for 1 h, followed by 50 ng/mL TNF‐α for 24 h (mRNA level) or 48 h (protein level). The level of PD‐L1 was determined by western blotting, GAPDH was used as the loading control. Blots are representative of three independent experiments (A) or real‐time RT‐qPCR (normalized to GAPDH) (B). (C and D) THP‐1 cells were pretreated with the indicated concentrations of Icaritin for 1 h, followed by 50 ng/mL TNF‐α treatment for 48 h. The level of PD‐L1 was determined by flow cytometry. Gating strategy used to identify THP‐1 cells‐expressing PD‐L1 on their plasma membrane. A first gate was set in living cells, then on physical parameters (FSC‐A vs FSC‐H) to eliminate doublets, and then on PD‐L1+ cells. Data are from one single experiment representative of three independent experiments, three samples are included in each experiment. Values are mean ± SEM (n = 3), *p < 0.05, **p < 0.01, ***p < 0.001 (by Student's t‐test).
Figure 3.
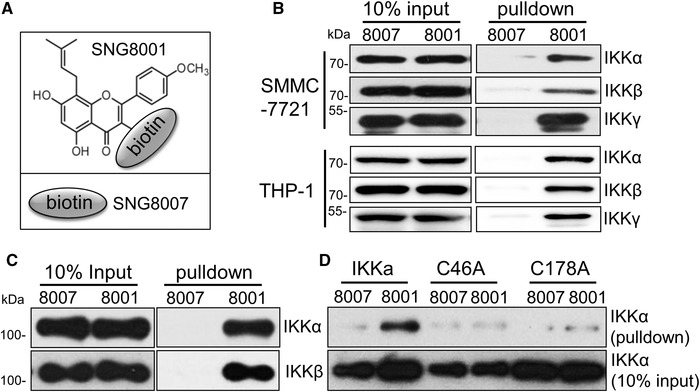
Cysteines 46 and 178 are essential for Icaritin binding to IKK‐α. (A) SNG8001: biotin+linker was linked to Icaritin 3’‐OH, SNG8007: biotin+linker, serve as negative control. (B) A total of 500 μg aliquot of cell lysate from SMMC‐7721 (upper) or THP‐1 (lower) cells, or 200 ng of purified protein (C and D) was incubated with biotin labelled Icaritin overnight, followed by the addition of 20 μL of streptavidin magnetic beads and incubation for 2 h. The pulled‐down samples were analyzed by western blotting, 50 μg cell lysate or 20 ng purified protein serve as 10% input loading control. Data are representative of three independent experiments.
Icaritin binding to the IKK complex
To elucidate the molecular mechanisms of Icaritin in regulating the expression of checkpoint proteins, a Surface Plasmon Resonance assay (SPRi) was performed by fixing Icaritin onto a photocrosslinker chip (Supporting information Fig. S3A). Cell lysate from SMMC‐7721 was used for SPRi affinity binding assay as described in Materials and methods. After two rounds of screening, the eluted samples were subject to LC‐MS profiling. Hundreds of proteins were identified, which included immune‐responsive proteins IKKs, MyD88, NFKB1, IKBKB, TRAF2/6, TRADD, and many others (data not shown).
The specific binding targets of Icaritin were further defined by biotin‐labelled Icaritin (Fig. 3A) pull‐down assays. SNG8007 and SNG8001 were separately incubated with cell lysate from SMMC‐7721 or THP‐1 cells, then the pulldown samples were analysed by Western Blot. As shown in Fig. 3B, SNG8001 (but not SNG8007) pulled down a significant amount of IKK‐α/‐β/‐γ, which proved that Icaritin can specifically and selectively interact with IKK complex. In a binding assay using purified IKK‐α and IKK‐β, it was discovered that purified IKK‐α and IKK‐β signals could both be detected after incubation with SNG8001 but not with SNG8007 (Fig. 3C). Additionally, the binding of SNG8001 to IKK‐α was dose dependent (Supporting information Fig. S3B). These results clearly proved that Icaritin can directly interact with the IKK complex, especially IKK‐α.
Previous studies by other researchers have shown that cysteine 46 and cysteine 179 in IKKβ are novel drug‐binding sites [15, 16]. To determine whether these two cysteines in IKK‐α are also the binding sites of Icaritin, the two cysteines were mutated to alanines, and purified IKKα‐C46A and IKKα‐C178A were generated (in IKK‐α is cysteine 178). Interestingly, SNG8001 could no longer pull‐down IKK‐α after the introduction of the mutations (Fig. 3D), which supported that cysteine 46 and cysteine 178 are critical for Icaritin binding to IKK‐α.
Icaritin blocks IKK complex formation
As Icaritin can selectively bind to IKK‐α, Icaritin was further examined to inhibit IKK‐α kinase activity. The purified IKK‐α was treated by Icaritin and staurosporine (a nonselective inhibitor of protein kinases as positive control), the kinase activities were quantitated by fluorescence resonance energy transfer (FRET) assay. Surprisingly, the activity of IKK‐α was not inhibited by Icaritin, while nonselective kinase inhibitor staurosporine did inhibit IKK‐α significantly with an IC50 at around 10 micromolar (Fig. 4A). Those results revealed that Icaritin had no effect on IKK‐α kinase activity even though it can selectively and specifically bind to IKK‐α. IKK‐α forms a complex with IKK‐β to play key roles in its downstream NF‐κB signal transduction regulation, we next examined whether Icaritin could block the IKK complex formation after binding to IKK‐α. Indeed, unlabeled Icaritin treatment could decrease the signal of IKK‐β pulled‐down by IKK‐α (Fig. 4B), which confirmed that Icaritin had its inhibitory roles by interfering with IKK complex formation.
Figure 4.
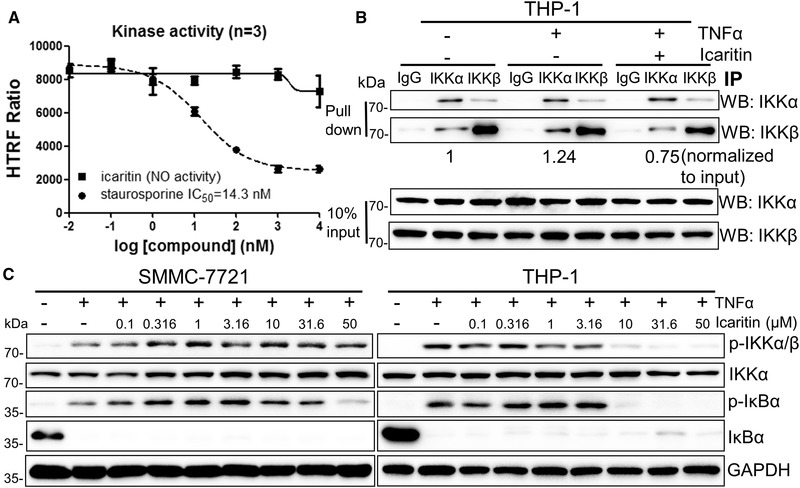
Icaritin inhibits IKK complex formation. (A) Biochemical assay of IKK‐α in the presence of Icaritin or staurosporine. Values are mean ± SEM. (B) THP‐1 cells were pre‐treated with 20 μM Icaritin overnight, followed by 50 ng/mL TNFα treatment for 0.5 hr. After 300 ug protein pull‐down by the indicated antibody, the levels of IKK‐α and IKK‐β were examined by western blotting, 10% input serves as loading control. (C) THP‐1 or SMMC‐7721 cells were pretreated with indicated concentration of Icaritin overnight, followed by 50 ng/mL TNF‐α treatment for 0.5 h. The levels of p‐IKKα/β, IKK‐α, p‐IκBα, and IκB‐α were examined by western blotting, GAPDH was used as the loading control. Data are from a single experiment representative of three independent experiments.
Since Icaritin can block IKK complex formation, we next studied whether Icaritin would interfere with function of the NF‐κB signaling pathway in such a manner. As shown in Fig. 4C, the levels of p‐IKK‐α/‐β and p‐IκBα in SMMC‐7721 and THP‐1 cells all decreased after Icaritin treatment (Fig. 4C), while monocyte THP‐1 cells exhibited much more sensitivity to Icaritin than SMMC‐7721 HCC cells did, suggesting that Icaritin with a relative high activity in reducing NF‐κB signaling pathway through direct inhibition of IKK complex formation in monocytes.
Icaritin decrease p65 onccupancy on PD‐L1 promoter
The NF‐κB family consists of highly conserved transcription factors that regulate key important cellular functions, particularly the inflammatory and immune responses [17]. IκBα phosphorylated by the IKK complex can be modified by ubiquitination and degraded by the proteasome. Subsequently, the NF‐κB complex is freed to enter the nucleus, where it binds to the promoters of downstream genes and regulates the expression of these genes [18, 19]. As Icaritin binds directly to IKK‐α and inhibits NF‐κB signalling pathway, it may inhibit NF‐κB p65 translocation to the nucleus. To determine whether this is true or not, the level of p65 in SMMC‐7721 and Hela cells nucleus was detected using western blot and immunofluorescence assay. As shown in Fig. 5A and Supporting information Fig. S4, the level of p65 in the nucleus was substantially increased after TNF‐α treatment, whereas it was significantly decreased after Icaritin pretreatment was performed. The results supported that Icaritin can inhibit p65 recruitment to the nuclear compartment. Since p65 drives downstream genes by activating their promoters, Icaritin may also inhibit PD‐L1 promoter activity. The PD‐L1 promoter was cloned into luciferase reporter plasmids and transfected into SMMC‐7721 cells, then luciferase activities were measured at 48 h after transfection. As shown in Fig. 5B, PD‐L1 promoter activity was markedly increased by approximately fivefold after TNF‐α induction. However, it was significantly decreased in the Icaritin pretreatment group. To further evaluate the binding efficiency of p65 to the PD‐L1 promoter, a qPCR‐based chromatin immunoprecipitation (ChIP) assay was performed in SMMC‐7721 cells. As shown in Fig. 5C, p65 binding to the PD‐L1 promoter showed a fivefold increase upon TNF‐α induction, while it was significantly decreased to the baseline level under Icaritin pretreatment. In summary, these results further supported the critical roles of Icaritin in transcriptionally regulating PD‐L1 activity.
Figure 5.
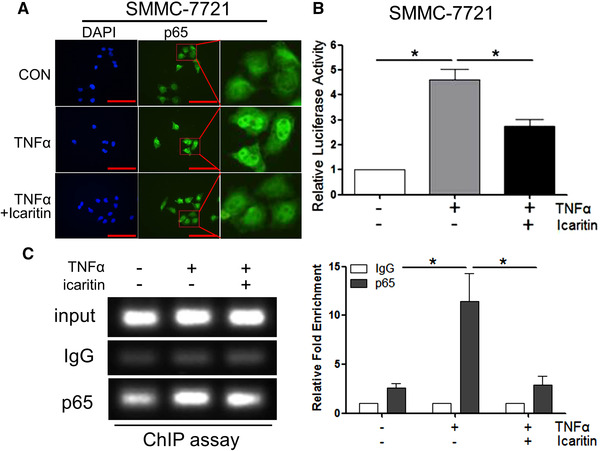
Icaritin inhibits NF‐κB p65 translocation and decrease p65 occupancy on PD‐L1 promoter. (A) Confocal microscopy image showing p65 localization after pretreatment with 20 μM Icaritin for 4 h, followed by treatment with 15 ng/mL TNF‐α for 0.5 h in SMMC‐7721 cells. One representative image from three independent experiments is shown. Scale bar represents 100 μm. (B) Luciferase activity was measured and normalized according to Renilla luciferase activity in SMMC‐7721 cells transiently transfected with the PD‐L1 luciferase promoter (PD‐L1‐Luc). Cells were pretreated with the indicated concentration of Icaritin for 1 h, followed by treatment with TNF‐α for 24 h. (C) Soluble chromatin from SMMC‐7721 cells was precipitated with anti‐NF‐κB p65 or control IgG. The final DNA samples from input or pull‐down group were amplified via qPCR with primers for the PD‐L1 promoter p65‐binding region, data were normalized to input. Each group of cells with three replications were pretreated with 20 μM Icaritin for 12 h, followed by treatment with 50 ng/mL TNFα for 0.5 h. Data are from three independent experiments. Values are mean ± SEM (n = 3). *p < 0.05, **p < 0.01, ***p < 0.001 (by Student's t‐test).
Discussion
Icaritin is a small molecule of prenylflavonoid derivative purified from the Epimedium genus. Similar to many other Traditional Chinese Medicines and natural compounds, Icaritin and its structural analogues have long been used to treat various disorders. Icaritin has been proven to possess antitumor activity through extensive mechanisms in in‐vitro cancer cell growth inhibition and in‐vivo animal tumor models, which originated from both solid tumor and hematological cancer. The common cytotoxicity of Icaritin in different malignancies includes inhibiting proliferation, inducing apoptosis, and blocking cell cycle. In its phase I/II clinic trials, Icaritin has demonstrated favourable safety, tolerability, and more durable overall survival rate compared to sorafenib in advanced HCC patients [8]. Currently, Icaritin is in its late stage of phase III trials in treatment of the advanced HCC patient as a single reagent. Exploratory biomarker studies in phase II Icaritin clinical trial have demonstrated that patients with positive PD‐L1 expression exhibited longer overall survival than patients with negative PD‐L1 expression. PD‐L1 positive patients might be more sensitive to Icaritin, and Icaritin might increase the efficacy of PD‐1 or PD‐L1 antibody. In this report, we demonstrated that Icaritin and anti‐PD‐1 mAb together inhibited tumor growth cumulatively, leading to significant tumor growth inhibition. Furthermore, animals resistant to anti‐PD‐1 therapy could benefit from the combined treatment of Icaritin and PD‐1 mAb.
It remains unknown that combinational enhancement between Icaritin and PD‐1 has been demonstrated on the cancer cell growth inhibition in multiple tumor models. Immune cells and cancer cells can facilitate immune suppression via the elevated expression of PD‐L1 [20]. Recently, it was discovered that Icaritin can downregulate PD‐L1 protein expression in neutrophils, which implied that Icaritin may mechanistically exerted its anticancer effects by regulating immune‐checkpoint protein expression at cellular level [13]. DC and macrophages are APCs that are crucial for the induction of antitumor T‐cell responses [21, 22], monocytes can differentiate into inflammatory DCs or macrophages during inflammation [23]. Through extensive analysis of Icaritin regulation on PD‐L1 expression in THP‐1 monocytes and HCC cells, we have found out that Icaritin can significantly decreased TNF‐α‐induced PD‐L1 at both the protein and mRNA levels in both immune cells and cancer cells leading to its combinational in‐vivo efficacy with anti‐PD1 antibody in inhibition of malignant tumor growth.
Similar to many other Traditional Chinese Medicines and natural compounds, Icaritin and its structural analogues have long been used to treat various disorders. But the lack of mechanisms explaining their pleiotropic and pharmacological functions often leads to confusions and sometimes disbelief. Even now, the underlying mechanisms of Icaritin's anticancer activity by regulating immune response are still not fully elucidated. The protein binding affinities of flavonoids are usually low, but their specificities are broad. This is the result of the various chemical properties of flavonoids. The antitumor activities of flavonoids are moderate, but they exhibit very low cellular toxicities. In fact, Havsteen indicated that “the margin of safety for the therapeutic use of flavonoids in humans, therefore, is very large and probably not surpassed by any other drug in current use” [24]. The low protein affinities of flavonoids may explain their low toxicity because high toxicity often results from high affinity, which is required for high potency and efficacy. However, it would be difficult to understand why such low affinities of flavonoids can exert significant biological and therapeutic activities. Several proteins have been reported directly interacting with apigenin or Icaritin such as hnRNPA2 [25], RPS9 [26], SphK1 and ERα36 [14, 27]. Molecular docking studies have identified that Icaritin can function also as Janus kinase 3 (JAK3) and fatty acid synthase (FASN) inhibitors, binding selectively to its binding pocket within the enzymes [28, 29]. Those finding have partially consolidated the mechanistic knowledge of Icaritin function. In this report, we have addressed the molecular and cellular mechanisms of Icaritin in inhibition of tumor growth by SPRi and biotin‐based affinity pull‐down studies. Icaritin was demonstrated selectively binding to IKK‐α and inhibiting IKK complex formation. The cysteine 46 and cysteine 178 were critical for Icaritin binding to IKK, which has led to downregulation of NF‐κB signalling pathway in monocytes. This is the first time that protein targets have been clearly identified for Icaritin, which provides better understanding of the pleiotropism of Icaritin and other flavonoid compounds in their anticancer and anti‐inflammatory efficacy.
Mechanistically, we provide strong evidence that Claritin can downregulate PD‐L1 gene expression at mRNA expression and protein levels directly and indirectly by interfering with TNF‐α mediated NF‐κB signal transduction pathway. Our findings provide insight into how apparent cumulative in‐vivo inhibitions of cancer cell growth were achieved in multiple mouse models when Icaritin combined with checkpoint antibodies (Fig. 1, Supporting information Fig. S1). It has been reported that upon PD‐1 on T‐cell surface binding to its ligand PD‐L1 on APCs, a pair of tyrosines within the cytoplasmic tail of PD‐1 becomes phosphorylated and recruits the protein tyrosine phosphatases SHP2 and SHP1, which dephosphorylate both the TCR and costimulatory signaling components [30, 31, 32, 33]. These biochemical events ultimately lead to the attenuation of T‐cell proliferation, cytokine production, and cytolytic activities [34]. PD‐L1 are expressed on both tumor cells and host immune cells and both contribute to tumor immune evasion. However, PD‐L1 plays distinct roles on immune cells and tumor cells. For an example, on expressing PD‐1, tumor cell PD‐L1 binding suppresses effector T‐cell TCR or costimulatory signaling, resulting in T‐cell apoptosis, anergy, and exhaustion. On the other hand, since DCs and macrophages can activate effector T cells by presenting specific antigens to TCR and activate T cells, PD‐1 binding of PD‐L1 from macrophages and DCs inhibits T‐cell activation and expansion. Indeed, blockade of PD‐L1 on immune cells or tumor cells contribute differently in different tumor immune environment settings, as reviewed by Tang and Zheng [35]. In present study, we found that monocyte cell line THP‐1 cells were more sensitive to Icaritin than tumor cell lines such as SMMC‐7221 (Fig. 4C). We speculate that Icaritin may exert its effect mainly on host immune cells, particularly those with monocyte lineage, such as macrophages and myeloid DCs, which activate T cells by antigen presenting. After PD‐L1 expression level inhibited by Icaritin, the activity of APCs to stimulate T cells will increase, leading to increased T‐cell activation and expansion. While PD‐L1 antibody achieve its effect by blocking individual PD‐L1 molecules, Icaritin achieves its effect by decrease PD‐L1 expression level intracellularly, which appears to be a more efficient approach. PD‐1 antibody would further increase the simulating activity of APCs by blocking PD‐1 on T cells, so Icaritin and PD‐1 antibody cumulatively enhance the activation of T cells (Fig. 6 upper). On the other hand, PD‐1 antibody also prevents the engagement of PD1/PD‐L1 between T cells and cancer cells, thus, reactivating effector T cell initially activated by antigen presenting cells (Fig. 6 lower).
Figure 6.
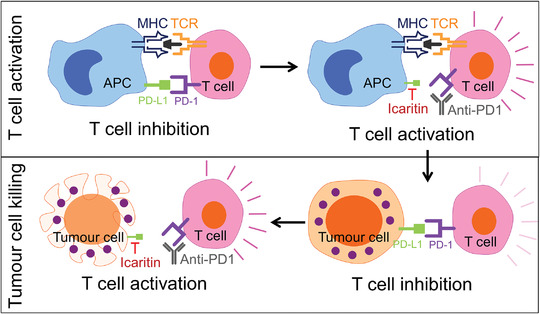
Schematic illustration of the mechanism of action of Icaritin‐induced immune modulation. At the T cell activation stage, upon PD‐1 on T cells binding to the ligand PD‐L1 on APCs, the cell proliferation, cytokine production and cytolytic activities of T cells will be attenuated. While T cell suppression will be reversed following disruption of the interaction between PD‐1 and PD‐L1 after Icaritin down‐regulate PD‐L1 expression as well as PD‐1 antibody block PD‐1 on T cells. On the other hand, at the tumor killing stage, T cell activity will further be inhibited after PD‐1 binding to PD‐L1 on tumor cell, which can also be restored by Icartin and PD‐1 antibody.
In addition to PD‐L1 expression inhibition, Icaritin may execute its antitumor effect through other approaches yet again via NF‐κB signaling pathway. NF‐κB pathway is probably the most important intracellular signal transduction pathway. As a master regulator, NF‐κB regulates chronic inflammation, cell survival and apoptosis, cell proliferation, metastasis, angiogenesis, metabolism, and finally immune evasion. The roles of NF‐κB in cancer development have been well documented. For instance, IL‐8 expression is regulated by NF‐κB and IL‐8 plays important role in tumorigenesis, and the combination therapy between IL‐8 antibodies and PD‐(L)1 antibodies have been evaluated in clinical trials [36, 37]. As a matter of fact, our clinical data showed that patients whose serum IL‐6 and IL‐8 were reduced were more likely to benefit from treatment in terms of overall survival.
In conclusion, IKK‐α has been experimentally identified as direct targets of Icaritin, which was verified at the molecular, cellular, and in‐vivo levels. Additionally, Icaritin showed good efficacy when combined with checkpoint antibodies. Based on the proposed mechanisms, the pleiotropic biological and therapeutic activities of Icaritin are well explained. Further experimental and clinical studies are proposed to verify the application of Icaritin on cancer therapy.
Materials and methods
Chemicals and antibodies
Icaritin (structure shown in Fig. 1A) was an in‐house purified hydrolytic product from traditional Chinese herbal medicine Epimedium genus with a purity of up to 99.5%; It was formulated in DMSO. TNF‐α was purchased from Peprotech (AF300‐01A), streptavidin magnetic beads were obtained from New England Biolab (NEB, #S1420S), staurosporine was from Selleck (1421), and IKK‐α protein was from Carna (05‐112). The antibodies used for western blot analysis, immunofluorescence and immunoprecipitation are listed in Supporting information Table 1.
Mice and cell line
C57BL/6 mice (6 to 8 weeks old, male and female) were purchased from the Laboratory Animal Center (Army Medical University, China). Mice were maintained in a specific pathogen‐free facility in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Mice were used under protocols approved by the Army Medical University Animal Care and Use Committee. The Hepa1‐6, MC‐38, B16F10, SMMC‐7721, Jurkat, HEK293T, and THP‐1 cell lines were obtained from American Type Culture Collection (ATCC) and cultured according to ATCC protocol. The cell lines were authenticated by LGC Standards (UK) Cell Line Authentication service in December 2018. Cells passaged less than 20 times were used in this work.
Construction of plasmids, point mutation, and protein purification
Human IKK‐α was cloned into a pcDNA3.1 vector containing an N‐terminal polyhistidine (6× His) tag and an HA‐tag sequence at the C‐terminal region. Site‐directed mutagenesis was performed with a Fast Mutagenesis System (TransGen, FM111‐01). These proteins were expressed in HEK293 T cells and subsequently purified. HEK293 T cells were transfected with the indicated plasmids using the Lipofectamine®3000 transfection reagent (Invitrogen, L3000‐015) in Opti‐MEM (Gibco, 11058021) for 48 h, after which the cells were collected, and the total lysates were prepared. The recombinant proteins were purified by using BabyBio Ni‐NTA.
SPRi measurements
The SPRi measurements were performed in a temperature‐controlled HORIBA Scientific SPRi‐PLEX II apparatus. Briefly, Icaritin was added onto the photo‐crosslingker chip and was irradiated with UV photocrosslinker chamber at 365 nm wavelength for 15 min. After washed with enthanol and water, protein lysate from 1 × 107 SMMC‐7721 cells was loaded onto the chip and washed with PBS to remove unbound proteins. Then the bound proteins were eluted with 1% SDS and reloaded onto the chip for a second round screening. After two rounds screening, the eluted samples were subject to LC‐MS profiling.
Nuclear protein extraction
SMMC‐7721 cells were pretreated with 0, 1, 5, or 10 μM of Icaritin for 1 h, followed by TNF‐α (50 ng/mL) treatment for 4 h. Then nuclear protein were extracted with Nuclear and Cytoplasmic Protein Extraction Kit (Byeotime, #P0027). Briefly, cells were washed with PBS and resuspended with 200 μL of cytoplasmic protein extraction buffer A, incubated on ice for 15min, then 10 μL of cytoplasmic protein extraction buffer B was added to the sample. After vortex for 5 s and incubated on ice for 1 min, the sample was centrifuged at 16 000 g for 5 min. The supernatant was removed as much as possible, and the pellet was resuspended with 50 μL of nuclear protein extraction buffer. After incubated on ice for 30 min, sample was centrifuged at 16 000 g for 5 min and the supernatant was analyzed using western bolt assay.
PD‐L1 reporter gene activation assay
A 605‐bp fragment of the PD‐L1 gene promoter region (‐478 to +127) containing the NF‐κB‐binding sequence used for the luciferase reporter assay was amplified by PCR using the following primer pair: forward: 5’‐GCGGTACCtatgggtctgctgctgact‐3; reverse: 5’‐CCGCTCGAGgagctagccagagatact‐3’. The PCR fragment was then cloned into the KpnI/XhoI sites of the pGL4.10‐Basic vector (Promega, E6651) and verified by sequencing. Cells were transiently cotransfected with the PD‐L1_luc reporter plasmid and Renilla plasmid (Promega, E6921) at a 2:1 ratio using the Lipofectamine®3000 transfection reagent for 48 h and then pretreated with Icaritin (20 μM) for 1 h, followed by 50 ng/mL TNF‐α treatment for 24 h. The cells were subsequently lysed and analysed for luciferase activity using the Dual Luciferase Reporter Gene Assay System (Promega, E2920) in a fluorescence spectrophotometer.
Kinase assay
Active IKK‐α kinase was purchased from Carna (05‐112‐5 μg). IC50 measurements were made using a fluorescence resonance energy transfer (FRET)‐based assay (cisbio, # 62ST0PEB). All measurements were performed at 25℃ and run in the presence of 1 ng/μL IKK‐α, 2 μM substrate, and 3 μM ATP. IKK‐α were preincubated with indicated concentration of compounds for 80 min, then reactions were quenched by adding detection agents. The signal was monitored and curves were fit using GraphPad Prism software.
Chromatin immunoprecipitation (ChIP) assay
The ChIP assay was performed with the SimpleChIP® Enzymatic Chromatin IP Kit (CST, 9003). Briefly, cells were incubated with 1% formaldehyde in cell culture medium at room temperature for 10 min, followed by incubation in 1× glycine at room temperature for 5 min to halt cross‐linking. Thereafter, the cross‐linked cells were washed twice with ice‐cold PBS and lysed. The nuclear extracts were digested with micrococcal nuclease and sonicated to fragment the DNA to lengths of approximately 150–900 bp. The chromatin was then diluted with ChIP buffer, and the sheared DNA mixture was subjected to immunoprecipitation with 1 μg of a p65 antibody (CST, 8242) or an equivalent amount of normal rabbit IgG (CST, 2729) at 4°C overnight with rotation. The protein–DNA complexes were then precipitated, reverse cross‐linked, washed, and eluted for PCR analysis. The following primers were used for DNA quantification: PD‐L1 promoter forward: 5’‐TGGACTGACATGTTTCACTTTCT‐3’ and reverse: 5’‐CAAGGCAGCAAATCCAGTTT‐3’.
Quantitative reverse transcription (qRT) PCR assays
Cells were washed twice with cold PBS and immediately lysed in NucleoZol (MN, 740404.6). The lysed samples were subjected to total RNA extraction using the RNeasy Mini Kit (Qiagen, 74106). To measure the expression of mRNA, we synthesized cDNA from 2 μg purified total RNA using the RevertAid First Strand cDNA Synthesis Kit (Thermo, K1622) following manufacturer's instructions. qPCR was performed in a real‐time PCR machine using the following primers (Supporting information Table 2), and all data analysis was performed using the comparative CT method. The results were normalized to the internal control GAPDH mRNA.
Western blot analysis and binding assay
The western blot procedure was conducted as follows:In brief, proteins were separated on 10% SDS‑PAGE by electrophoresis. The separated proteins were transferred onto nitrocellulose membranes, and the membranes were blocked with 2.5% nonfat milk in TBST (150 mM Nacl, 10 mM Tris, 01.% Tween‐20) at room temperature for 1 h. Subsequently, the membranes were probed with primary antibody overnight at 4˚C. Following three rapid washes in TBST, the membranes were incubated with secondary antibody at room temperature for 1 h. The immune complexes were detected using an enhanced chemiluminescence kit (Engreen, #29050). For Icaritin binding assay, the cells were lysed in reaction buffer (10 mM Tris•HCl, pH 7.6, 50 mM KCl, 1 mM EDTA) and centrifuged at 16 000 × g for 30 min to remove debris. A total of 500 μg cleared lysates or 200 ng purified protein were subjected to immunoprecipitation with Icaritin beads in 500 μL reaction buffer. After incubation overnight with rotation, beads were washed five times with 1 mL reaction buffer and the binding proteins were analysed using western blot.
Statistics
All data are presented as the mean ± SEM. p values were calculated using a one‐tailed Student's t test. All statistical analyses were performed with GraphPad Prism 5 (GraphPad Software, CA). Significance was indicated as p < 0.05 (*), p < 0.01 (**), p < 0.001 (***).
Study approval
Mice were maintained in a specific pathogen‐free facility in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Mice were used under protocols approved by the Army Medical University Animal Care and Use Committee. The clinical study was performed in accordance with good clinical practice (GCP) and Declaration of Helsinki guidelines. The study protocol was approved by an institutional review board (IRB), and written informed consent was obtained from all participating patients for enrolment. Approval from the appropriate ethics committees and institutional review boards was obtained and documented before the study.
Peer review
The peer review history for this article is available at https://publons.com/publon/10.1002/eji.202048905.
Conflict of Interest
The authors declare no commercial or financial conflicts of interest.
Abbreviations
- ChIP
chromatin immunoprecipitation
- HCCs
hepatocellular carcinima cells
- PD‐L
programmed cell death‐ligand 1
- SPRi
surface plasmon resonance assay
Supporting information
Supporting Information
Acknowledgments
We thank Limin Zheng from Sun Yat‐sen University and Bin Ye from Beijing Shenogen Pharma Group. Ltd for helpful comments and Guilin Qian from Beijing Shenogen Pharma Group. Ltd for language polishing, respectively. This work was supported by the grants from National Science and Technology Major Project of China (grant nos. 2017ZX09309023).
Data availability statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- 1. Tong, J. S. , Zhang, Q. H. , Huang, X. , Fu, X. Q. , Qi, S. T. , Wang, Y. P. , Hou, Y. et al. Icaritin causes sustained ERK1/2 activation and induces apoptosis in human endometrial cancer cells. PLoS One 2011. 6: e16781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Tiong, C. T. , Chen, C. , Zhang, S. J. , Li, J. , Soshilov, A. , Denison, M. S. , Lee, L. S. et al. A novel prenylflavone restricts breast cancer cell growth through AhR‐mediated destabilization of ERalpha protein. Carcinogenesis 2012. 33: 1089–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sun, F. , Indran, I. R. , Zhang, Z. W. , Tan, M. H. , Li, Y. , Lim, Z. L. , Hua, R. et al. A novel prostate cancer therapeutic strategy using icaritin‐activated arylhydrocarbon‐receptor to co‐target androgen receptor and its splice variants. Carcinogenesis 2015. 36: 757–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sun, F. , Zhang, Z. W. , Tan, E. M. , Lim, Z. L. R. , Li, Y. , Wang, X. C. , Chua, S. E. et al. Icaritin suppresses development of neuroendocrine differentiation of prostate cancer through inhibition of IL‐6/STAT3 and Aurora kinase A pathways in TRAMP mice. Carcinogenesis 2016. 37: 701–711. [DOI] [PubMed] [Google Scholar]
- 5. Li, Q. , Huai, L. , Zhang, C. , Wang, C. , Jia, Y. , Chen, Y. , Yu, P. et al. Icaritin induces AML cell apoptosis via the MAPK/ERK and PI3K/AKT signal pathways. Int J Hematol 2013. 97: 617–623. [DOI] [PubMed] [Google Scholar]
- 6. He, J. , Wang, Y. , Duan, F. , Jiang, H. , Chen, M. F. and Tang, S. Y. , Icaritin induces apoptosis of HepG2 cells via the JNK1 signaling pathway independent of the estrogen receptor. Planta Med 2010. 76: 1834–1839. [DOI] [PubMed] [Google Scholar]
- 7. Li, Z. P. , Meng, X. W. and Jin, L. , Icaritin induces apoptotic and autophagic cell death in human glioblastoma cells. Am J Transl Res 2016.8: 4628–4643. [PMC free article] [PubMed] [Google Scholar]
- 8. Fan, Y. , Li, S. , Ding, X. , Yue, J. , Jiang, J. , Zhao, H. , Hao, R. et al. First‐in‐class immune‐modulating small molecule Icaritin in advanced hepatocellular carcinoma: preliminary results of safety, durable survival and immune biomarkers. BMC Cancer 2019. 19: 279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Constantinidou, A. , Alifieris, C. and Trafalis, D. T. , Targeting Programmed Cell Death ‐1 (PD‐1) and ligand (PD‐L1): A new era in cancer active immunotherapy. Pharmacol Ther 2019. 194: 84–106. [DOI] [PubMed] [Google Scholar]
- 10. Calderaro, J. , Rousseau, B. , Amaddeo, G. , Mercey, M. , Charpy, C. , Costentin, C. , Luciani, A. et al. Programmed death ligand 1 expression in hepatocellular carcinoma: Relationship with clinical and pathological features. Hepatology 2016. 64: 2038–2046. [DOI] [PubMed] [Google Scholar]
- 11. Zou, W. P. , Wolchok, J. D. and Chen, L. , PD‐L1 (B7‐H1) and PD‐1 pathway blockade for cancer therapy mechanisms, response biomarkers, and combinations. Sci. Transl. Med. 2016. 8: 328rv4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Maeda, T. , Hiraki, M. , Jin, C. , Rajabi, H. , Tagde, A. , Alam, M. , Bouillez, A. et al. MUC1‐C induces PD‐L1 and immune evasion in triple‐negative breast cancer. Cancer Res 2018. 78: 205–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hao, H. , Zhang, Q. , Zhu, H. , Wen, Y. , Qiu, D. , Xiong, J. , Fu, X. et al. Icaritin promotes tumor T‐cell infiltration and induces antitumor immunity in mice. Eur J Immunol 2019. 49: 2235–2244. [DOI] [PubMed] [Google Scholar]
- 14. Bailly, C. , Molecular and cellular basis of the anticancer activity of the prenylated flavonoid icaritin in hepatocellular carcinoma. Chem Biol Interact 2020. 325: 109124. [DOI] [PubMed] [Google Scholar]
- 15. Li, T. , Wong, V. K. , Jiang, Z. H. , Jiang, S. P. , Liu, Y. , Wang, T. Y. , Yao, X. J. et al. Mutation of cysteine 46 in IKK‐beta increases inflammatory responses. Oncotarget 2015. 6: 31805–31819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kapahi, P. , Takahashi, T. , Natoli, G. , Adams, S. R. , Chen, Y. , Tsien, R. Y. and Karin, M. , Inhibition of NF‐kappa B activation by arsenite through reaction with a critical cysteine in the activation loop of Ikappa B kinase. J Biol Chem 2000. 275: 36062–36066. [DOI] [PubMed] [Google Scholar]
- 17. Barkett, M . and Gilmore, T. D. , Control of apoptosis by Rel/NF‐kappaB transcription factors. Oncogene 1999. 18: 6910–6924. [DOI] [PubMed] [Google Scholar]
- 18. Senftleben, U. , Cao, Y. , Xiao, G. , Greten, F. R. , Krähn, G. , Bonizzi, G. , Chen, Y. et al. Activation by IKKalpha of a second, evolutionary conserved, NF‐kappa B signaling pathway. Science 2001. 293: 1495–1499. [DOI] [PubMed] [Google Scholar]
- 19. Nelson, D. E. , Ihekwaba, A. E. , Elliott, M. , Johnson, J. R. , Gibney, C. A. , Foreman, B. E. , Nelson, G. et al. Oscillations in NF‐kappaB signaling control the dynamics of gene expression. Science 2004. 306: 704–708. [DOI] [PubMed] [Google Scholar]
- 20. Dong, H. , Strome, S. E. , Salomao, D. R. , Tamura, H. , Hirano, F. , Flies, D. B. , Roche, P. C. et al. Tumor‐associated B7‐H1 promotes T‐cell apoptosis: A potential mechanism of immune evasion. Nat Med 2002. 8: 793–800. [DOI] [PubMed] [Google Scholar]
- 21. Nierkens, S. and Janssen, E. M. , Harnessing dendritic cells for tumor antigen presentation. Cancers (Basel) 2011. 3: 2195–2213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Unanue, E. R. , Antigen‐presenting Function of the Macrophage. Annu Rev Immunol 1984. 2: 395–428. [DOI] [PubMed] [Google Scholar]
- 23. Geissmann, F. , Manz, M. G. , Jung, S. , Sieweke, M. H. , Merad, M. and Ley, K. , Development of monocytes, macrophages, and dendritic cells. Science 2010. 327: 656–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Havsteen, B. H. , The biochemistry and medical significance of the flavonoids. Pharmacol Ther 2002. 96: 67–202. [DOI] [PubMed] [Google Scholar]
- 25. Arango, D. , Morohashi, K. , Yilmaz, A. , Kuramochi, K. , Parihar, A. , Brahimaj, B. , Grotewold, E. and Doseff, A. I. , Molecular basis for the action of a dietary flavonoid revealed by the comprehensive identification of apigenin human targets. Proc Natl Acad Sci U S A 2013. 110: E2153–2162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Iizumi, Y. , Oishi, M. , Taniguchi, T. , Goi, W. , Sowa, Y. and Sakai, T. The flavonoid apigenin downregulates CDK1 by directly targeting ribosomal protein S9. PLoS One 2013. 8: e73219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lu, P. H. , Chen, M. B. , Liu, Y. Y. , Wu, M. H. , Li, W. T. , Wei, M. X. , Liu, C. Y. et al. Identification of sphingosine kinase 1 (SphK1) as a primary target of icaritin in hepatocellular carcinoma cells. Oncotarget 2017. 8: 22800–22810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Su, D. , Gao, Y. Q. , Deng, Y. J. , Zhang, H. H. , Wu, Y. R. , Hu, Y. and Mei, Q. X. Identification of Chinese herbal compounds with potential as JAK3 inhibitors. Evid Based Complement Alternat Med 2019. 4982062: eCollection 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wu, J. F. , Du, J. , Fu, X. Q. , Liu, B. , Cao, H. H. , Li, T. , Su, T. et al. Icaritin, a novel FASN inhibitor, exerts anti‐melanoma activities through IGF‐1R/STAT3 signaling. Oncotarget 2016. 7: 51251–51269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hui, E. , Cheung, J. , Zhu, J. , Su, X. , Taylor, M. J. , Wallweber, H. A. , Sasmal, D. K. et al. T cell costimulatory receptor CD28 is a primary target for PD‐1‐mediated inhibition. Science 2017. 355: 1428–1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Parry, R. V. , Chemnitz, J. M. , Frauwirth, K. A. , Lanfranco, A. R. , Braunstein, I. , Kobayashi, S. V. , Linsley, P. S. et al. CTLA‐4 and PD‐1 receptors inhibit T‐cell activation by distinct mechanisms. Mol Cell Biol 2005. 25: 9543–9553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sheppard, K. A. , Fitz, L. J. , Lee, J. M. , Benander, C. , George, J. A. , Wooters, J. , Qiu, Y. et al. PD‐1 inhibits T‐cell receptor induced phosphorylation of the ZAP70/CD3zeta signalosome and downstream signaling to PKCtheta. FEBS Lett 2004. 574: 37–41. [DOI] [PubMed] [Google Scholar]
- 33. Yokosuka, T. , Takamatsu, M. , Kobayashi‐Imanishi, W. , Hashimoto‐Tane, A. , Azuma, M. and Saito, T. Programmed cell death 1 forms negative costimulatory microclusters that directly inhibit T cell receptor signaling by recruiting phosphatase SHP2. J Exp Med 2012. 209: 1201–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Keir, M. E. , Butte, M. J. , Freeman, G. J. and Sharpe, A. H. PD‐1 and its ligands in tolerance and immunity. Annu Rev Immunol 2008. 26: 677–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Tang, F. and Zheng, P. Tumor cells versus host immune cells: whose PD‐L1 contributes to PD‐1/PD‐L1 blockade mediated cancer immunotherapy? Cell Biosci 2018. 8: 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Yuen, K. C. , Liu, L. F. , Gupta, V. , Madireddi, S. , Keerthivasan, S. , Li, C. , Rishipathak, D. et al. High systemic and tumor‐associated IL‐8 correlates with reduced clinical benefit of PD‐L1 blockade. Nat Med 2020. 26: 693–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Schalper, K. A. , Carleton, M. , Zhou, M. , Chen, T. , Feng, Y. , Huang, S. P. , Walsh, A. M. et al. Elevated serum interleukin‐8 is associated with enhanced intratumor neutrophils and reduced clinical benefit of immune‐checkpoint inhibitors. Nat Med 2020. 26: 688–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.