

Abstract
Preclinical studies have shown synergistic effects when combining PARP1/2 inhibitors and platinum drugs in BRCA1/2 mutated cancer cell models. After a formulation change of olaparib from capsules to tablets, we initiated a dose finding study of olaparib tablets bidaily (BID) continuously with carboplatin to prepare comparative studies in this patient group. Patients were included in a 3 + 3 dose‐escalation schedule: olaparib 25 mg BID and carboplatin area under the curve (AUC) 3 mg*min/mL d1/d22, olaparib 25 mg BID and carboplatin AUC 4 mg*min/mL d1/d22, followed by increasing dose‐levels of olaparib from 50 mg BID, 75 mg BID, to 100 mg BID with carboplatin at AUC 4 mg*min/mL d1/d22. After two cycles, patients continued olaparib 300 mg BID as monotherapy. Primary objective was to assess the maximum tolerable dose (MTD). Twenty‐four patients with a confirmed diagnosis of advanced cancer were included. Most common adverse events were nausea (46%), fatigue (33%) and platelet count decrease (33%). Dose‐level 3 (olaparib 75 mg BID and carboplatin AUC 4 mg*min/mL; n = 6) was defined as MTD. Fourteen out of 24 patients (56%) had a partial response as best response (RECIST 1.1). Systemic exposure of the olaparib tablet formulation appeared comparable to the previous capsule formulation with olaparib tablet AUC0‐12 of 16.3 μg/mL*h at MTD. Polymers of ADP‐ribose levels in peripheral blood mononuclear cells were reduced by 98.7% ± 0.14% at Day 8 compared to Day 1 for dose‐level 3. Olaparib tablets 75 mg BID and carboplatin AUC 4 mg*min/mL for two cycles preceding olaparib monotherapy 300 mg is a feasible and tolerable treatment schedule for patients with advanced cancer.
Keywords: BRCA mutation, carboplatin, olaparib, phase I
Short abstract
What's new?
Preclinical studies have shown synergistic effects when combining PARP1/2‐inhibitors and platinum drugs in BRCA1/2 mutated cancer cell models. This phase I trial of olaparib tablets combined with carboplatin in advanced cancer patients showed that the combination has an acceptable side‐effect profile. The maximum tolerable dose was olaparib tablets 75 mg BID and carboplatin AUC 4 mg*min/ml. The observed preliminary anti‐tumor activity was encouraging, with 58% of patients having a decrease in tumor volume of more than 30%. This study shows that the tablet formulation of olaparib can be administered safely in combination with carboplatin, compared to the previous capsule formulation.
Abbreviations
- AE
adverse event
- AUC
area under the concentration‐time curve
- AUC0‐t
AUC from 0 to time t (12 hours for olaparib and 48 hours for carboplatin)
- BID
bidaily
- BRCA1
BReast CAncer 1
- BRCA2
BReast CAncer 2
- Cmax
maximum concentration
- DLT
dose‐limiting toxicity
- DSB
DNA double strand breaks
- ECOG‐PS
Eastern Cooperative Oncology Group performance status
- Gy
gray
- HPLC‐MS
high performance liquid chromatography‐mass spectrometry
- ICPMS
inductively coupled plasma mass spectometry
- MTD
maximum tolerable dose
- NCI
National Cancer Institute
- NER
nucleotide excision repair
- OS
overall survival
- PAR
polymers of ADP‐ribose
- PARP
poly(ADP‐ribose) polymerase
- PBMCs
peripheral blood mononuclear cells
- PD
progressive disease
- PFS
progression free survival
- PK
pharmacokinetic
- PR
partial response
- RECIST
response evaluation criteria in solid tumors
- REP‐assay
radiation‐enhanced‐PAR pharmacodynamic assay
- SSB
DNA single strand breaks
- WHO
World Health Organization
1. INTRODUCTION
BReast CAncer 1 (BRCA1) and BReast CAncer 2 (BRCA2) are the most important breast cancer susceptibility genes. The lifetime risk of breast cancer in BRCA1‐ and BRCA2‐mutation carriers is 45% to 80%. 1 , 2 BRCA1 and BRCA2 play important roles in the process of homologous recombination and the repair of DNA double strand breaks (DSB). 3 BRCA‐mutated tumors are often highly sensitive to drugs that induce DSB, such as alkylating agents. 4 Poly(ADP‐ribose) polymerase (PARP) plays an important role in the repair of DNA single strand breaks (SSB). Trapping of PARP on the DNA results in persistence of SSB leading to DSB. 5 The genetic interaction between PARP and BRCA can be described as synthetic lethality, which occurs where individual loss of either gene is compatible with cell survival, but simultaneous loss of both genes results in cell death. 6 Several preclinical studies have demonstrated that BRCA deficient cells are sensitive to PARP1/2 inhibition. 7 , 8 , 9 Furthermore, in clinical studies several selective PARP1/2‐inhibitors (talazoparib, niraparib, veliparib) and more broad PARP1,2,3,4,12,15,16‐inhibitors (rucaparib) have proven to be effective in patients with BRCA mutations. These studies have demonstrated efficacy of PARP1/2‐inhibitors in breast and ovarian cancers, and showed a tolerable safety profile. 10 , 11 , 12 , 13 , 14 Most clinical studies have been performed with the PARP1/2‐inhibitor olaparib. Proof of concept regarding olaparib treatment for advanced BRCA‐mutated breast cancer was shown by Tutt et al. 15 In a Phase I trial with olaparib in an oral capsule formulation, pharmacokinetic (PK) measurements showed a rapid absorption followed by a biphasic decline in plasma concentration. However, the area under the concentration‐time curve (AUC) relationship showed nonlinear absorption PKs. 16 Olaparib has also been investigated in combination with cytotoxic agents like paclitaxel, carboplatin and doxorubicin for solid tumors like ovarian and breast cancers. The benefit in efficacy of combining PARP1/2 inhibitors with cytotoxic chemotherapy has been shown, but more severe toxicity could be the result of combining these agents. 17 , 18 , 19 Olaparib combined with carboplatin showed more bone marrow toxicity compared to carboplatin alone. 17 , 19 Olaparib has been approved as maintenance treatment of patients with platinum sensitive high‐grade ovarian cancer. 20 Recently, there was a change in olaparib formulation from capsules to tablets. The approved capsule formulation of 400 mg bidaily (BID) required intake of eight 50 mg capsules twice daily. A tablet formulation has been designed and registered to overcome these disadvantages. The oral bioavailability of the tablet formulation is higher compared to the previous capsule formulation. 21 The AUC of the tablet formulation (300 mg) is 13% higher than the capsule formulation (400 mg). 22 As a result of the OlympiAD trial, olaparib tablets recently have been approved by the Food and Drug Administration as monotherapy for advanced BRCA‐mutated breast cancer. 23 Previous studies have determined maximum tolerable dose (MTD) of the olaparib capsule formulation when administered in combination with carboplatin. The MTD was found to be olaparib capsules 400 mg BID Day 1 to 7 and carboplatin target AUC 5 mg*min/mL once per 21‐day cycle. 24 Another Phase I study in which olaparib tablets were combined with carboplatin and paclitaxel showed increased myelosuppression requiring frequent dose modifications, including interruptions, delays and reductions. This toxicity appeared to be more frequent and severe with increasing doses of olaparib (ranging from 50 to 400 mg BID). 25 , 26 This supports lower dose olaparib in combination with carboplatin. The aim of our study was to investigate the MTD of the combination of olaparib in tablet formulation administered in combination with carboplatin for two cycles, followed by olaparib monotherapy.
2. PATIENTS AND METHODS
2.1. Patient selection
Patients were eligible if they were at least 18 years old and had a confirmed histological or cytological diagnosis of advanced cancer. A maximum of one prior line systemic chemotherapy and any number of prior lines of endocrine therapy for advanced disease was allowed. Patients were only included if benefit from the combination of olaparib and carboplatin could be expected. All patients had an Eastern Cooperative Oncology Group performance status (ECOG‐PS) of ≤2, adequate organ function and evaluable disease according to response evaluation criteria in solid tumor (RECIST) version 1.1. 27
2.2. Study design and drug treatment
The study design has been previously described elsewhere. 28 In brief, this was an investigator initiated 3 + 3 traditional Phase I dose‐escalation trial with predefined dose‐levels, conducted at the Netherlands Cancer Institute in Amsterdam, the Netherlands. Patients received two cycles of carboplatin intravenously with olaparib tablets, followed by olaparib monotherapy. Patients received carboplatin in 30 minute infusions on Day 1 of the first two cycles at a dose resulting in a target platinum AUC of 3 mg*min/mL (dose‐level −1) or AUC 4 mg*min/mL (all other dose‐levels). Olaparib was administered from Day 0 onwards at a dose ranging from 25 mg BID (dose‐level −1 and dose‐level 1) to 100 mg BID (dose‐level 4) continuously for a 21‐days cycle. After the first two cycles, patients continued with olaparib monotherapy at a dose of 300 mg BID (Supplemental Figure 1 shows an overview of the study design). Study treatment was continued until disease progression (≥20% increase in the sum of diameters of target lesions), unacceptable toxicity despite dose modifications or patient withdrawal. The Calvert formula, in which glomerular filtration rate was estimated using the Cockcroft‐Gault equation, was used to determine the carboplatin dose. 29
2.3. Objectives
The primary objective was to determine the MTD of two cycles carboplatin with olaparib tablets followed by olaparib monotherapy.
Secondary objectives were to investigate the systemic exposure of the olaparib tablet formulation, the pharmacodynamics and the preliminary response rate of this combination.
2.4. Dose‐escalation and dose‐limiting toxicities
A traditional 3 + 3 dose‐escalation scheme was used. The starting dose was olaparib 25 mg BID with carboplatin AUC 3 mg*min/mL followed by olaparib monotherapy 300 mg BID. Patients were enrolled per protocol in sequential cohorts of three patients based on the occurrence of dose‐limiting toxicity (DLT) within the first 21 days (one cycle) and only after study committee approval. If one of three patients experienced a DLT in the first cycle, the cohort was expanded to six patients. If a DLT was found in at least two out of six patients, the dose‐level was considered to be unsafe. The MTD was the highest dose‐level in which not more than one patient experienced a DLT. A DLT was defined as any of the following drug‐related adverse events (AEs) occurring in the first cycle of treatment (Day 1‐21): development of ˃ grade 2 toxicity during the DLT assessment period, toxicity that resulted in missing more than five doses of olaparib or that delayed the administration of carboplatin more than 7 days, a dose delay of 7 days or more of the second cycle of olaparib‐carboplatin (Supplemental Table 1). For some toxicities, ˃ grade 2 toxicity was accepted as supportive treatment was available. For those the DLT was defined when patients experienced: hematological toxicity: grade 4 anemia, grade 4 neutropenia ≥7 consecutive days, grade 3 or 4 febrile neutropenia, grade 4 thrombocytopenia or grade 3 thrombocytopenia with bleeding events. Nonhematological toxicities: ≥ grade 3 diarrhea, vomiting and nausea despite adequate supportive treatment, increased liver biochemistry (aspartate aminotransferase, alanine aminotransferase, alkaline phosphatase, gamma glutamyltransferase, lactate dehydrogenase) ≥ grade 3 lasting ˃3 days. In case of a DLT, dosing was interrupted until the toxicity was recovered to less than grade 2. Dose modifications according to protocol were allowed in the best interest of the patient.
2.5. Olaparib and platinum measurements
An high‐performance liquid chromatography‐mass spectrometry (HPLC‐MS/MS) method was used to determine olaparib in human plasma using Olaparib‐d8 (deuterated) as internal standard. The compounds were extracted from the plasma by liquid‐liquid extraction with tert‐butyl methyl ether. Chromatographic separation was performed on a Phenomenex HPLC Gemini C18 column using gradient elution. For detection, an AB Sciex API4000 tandem mass spectrometer equipped with an electrospray ionization interface was used operating in the positive ion mode. Further details have been described before. 30
An inductively coupled plasma mass spectometry (ICPMS) method was used to determine platinum from carboplatin in human plasma and plasma ultrafiltrate using iridium as internal standard. For detection, a Varian 810‐MS ICPMS is used. 31
2.6. Safety and assessments
During screening, information was gathered about medical history and demographics. At baseline and throughout treatment physical examination, vital signs, ECOG‐PS, concomitant medication and laboratory (hematology, chemistry, urine analysis) assessments were performed. Tumor response was evaluated using computer tomography scans at baseline and every two cycles according to RECIST version 1.1. AEs were graded according to the National Cancer Institute (NCI) Common Terminology Criteria version 4.03. 32
2.7. Statistical analysis
No formal sample size calculation was performed prior to our study because of the traditional 3 + 3 design. Upfront, we expected to enroll 15 to 20 patients in our study. Data were summarized with descriptive statistics and graphs. Disease progression was summarized with Kaplan‐Meier method. All analyses were performed using R software version 3.3.3.
2.8. PK assessments
To determine the PK parameters of olaparib from tablets in combination with carboplatin, an intensive blood sampling scheme was used. For olaparib 21 blood samples (21 × 4 mL) were collected: nine on Day 0: pre‐dose and 0.5, 1, 2, 4, 6, 8, 10 and 12 hours after administration. Twelve on Day 1 pre‐dose and 0.5, 1, 2, 2.25, 2.5, 3, 4, 6, 8, 10, 12 hours after olaparib administration. For carboplatin 10 blood samples (10 × 4 mL) were collected: 0.25, 0.5, 1, 2, 4, 6, 8, 10, 24 and 48 hours after infusion of carboplatin. For carboplatin PKs, concentrations of platinum were measured in both plasma and plasma ultrafiltrates. 31 Calculation of PK parameters included maximum concentration (C max), time to reach maximum concentration, AUC from 0 to time t (AUC0‐t; 12 hours for olaparib and 48 hours for carboplatin), AUC from time 0 to time of last measurable concentration and half‐life.
2.9. Pharmacodynamic assessments
Poly(ADP) ribose levels were determined in peripheral blood mononuclear cells (PBMCs) using the radiation‐enhanced‐polymers of ADP‐ribose (PAR) pharmacodynamic assay (REP assay). 33 In brief, 16 mL venous blood was collected in mononuclear cell preparation citrate tubes pre‐dose at Day 0, and at Day 8. PAR levels were assessed in three independent samples, containing 2 × 106 PBMCs, which were irradiated ex vivo with 8 gray (Gy) on ice and incubated for 1 hour on ice. Cellular PAR levels were measured by using the HT‐PARP in vivo pharmacodynamics Assay II, following the NCI protocol 34 using a Tecan‐Infinite‐200‐Pro. PAR levels on Day 8 of cycle one treatment were compared to the PAR levels before start of treatment to determine the balance of PARP and poly (ADP‐ribose) glycohydrolase (PARG) activity and the resulting relative reduction in PAR levels after 8 days of treatment.
The relative reduction in PAR levels was defined as ([PAR levels Day 0 − PAR levels Day 8]/PAR levels day 0) × 100%.
3. RESULTS
3.1. Patients
Between July 2015 and October 2017, we enrolled 24 eligible patients with advanced malignancies: 18 patients had breast cancer, 3 had ovarian cancer, 1 had eye melanoma, 1 colorectal cancer and 1 esophageal cancer. One patient had received more than one line of treatment in the advanced setting and was therefore not considered to be evaluable for safety and efficacy. Baseline characteristics of the 24 patients are presented in Table 1. The median patient age was 49 years (range, 27‐70). Most patients had a World Health Organization (WHO) performance status of zero (19/24; 79%). Nineteen patients (19/24; 79%) had a germline BRCA mutation (BRCA1 or BRCA2).
TABLE 1.
Table showing the baseline characteristics for all 24 evaluable patients included in our study
| N | % | |
|---|---|---|
| Gender | ||
| Female | 22 | 92 |
| Male | 2 | 8 |
| Age (median) (range) years | 49 (27‐70) | — |
| Tumor type primary disease | ||
| Breast | 18 | 75 |
| Ovarian | 3 | 13 |
| Colorectal | 1 | 4 |
| Esophageal | 1 | 4 |
| Eye melanoma | 1 | 4 |
| Ethnicity | ||
| Caucasian | 24 | 100 |
| WHO performance status | ||
| WHO 0 | 19 | 79 |
| WHO 1 | 4 | 17 |
| WHO 2 | 1 | 4 |
| BRCA‐status | ||
| BRCA‐1 mutated | 8 | 33 |
| BRCA‐2 mutated | 11 | 47 |
| BRCA‐2 like | 2 | 8 |
| Non‐carrier | 1 | 4 |
| Unknown | 2 | 8 |
| Previous platinum treatment | ||
| Yes | 6 | 25 |
| No | 18 | 75 |
| Lines of chemotherapy in adjuvant setting | ||
| 0 | 7 | 29 |
| 1 | 17 | 71 |
| Lines of chemotherapy for M1 disease | ||
| 0 | 18 | 25 |
| 1 | 6 | 75 |
| Previous hormonal therapy | ||
| Yes | 12 | 50 |
| No | 12 | 50 |
3.2. Treatment
Patients were enrolled in predefined dose cohorts (Supplemental Table 2); the lowest dose‐level started with 25 mg olaparib BID and carboplatin AUC 3 mg*min/mL, the highest dose‐level explored olaparib 100 mg BID and carboplatin AUC 4 mg*min/mL. Three patients were treated in the lowest dose‐level, six patients were treated at each dose‐levels with olaparib 50 mg BID, olaparib 75 mg BID and olaparib 100 mg BID, respectively. Since there were two dose‐limiting toxicities at the highest dose‐level of 100 mg BID, the MTD was determined to be olaparib 75 mg BID and carboplatin AUC 4 mg*min/mL. Dose‐reductions were applied in five patients. Two patients received olaparib maintenance at 250 mg BID instead of 300 mg BID because of hematological toxicity. Two patients received olaparib 200 mg BID instead of 300 mg because of malaise. One patient was allocated to the olaparib 100 mg BID and carboplatin AUC 4 mg*min/mL cohort and was switched to olaparib 75 mg BID in the second cycle because of hematologic toxicity in the first cycle. Supplemental Figure 2 shows an overview of the dose‐escalation scheme.
3.3. Safety
Three DLT events were observed in this trial. In dose‐level 2 (olaparib 50 mg BID and carboplatin AUC 4 mg*min/mL), one patient developed a grade 3 liver biochemistry increase lasting for more than 3 days. In dose‐level 4 (olaparib 100 mg BID and carboplatin AUC 4 mg*min/mL), two patients experienced a DLT consisting of ≥7 days dose delay of cycle 2 or missing ≥5 doses of olaparib due to hematologic toxicity. Therefore, the preceding dose‐level (olaparib 75 mg BID and carboplatin AUC 4 mg*min/mL) was expanded to six patients. No DLTs were observed at this dose‐level (Supplemental Table 2). Hence, the MTD was determined to be olaparib 75 mg BID combined with carboplatin AUC 4 mg*min/mL. The most common grade 1/2 AEs observed in our study were nausea (11/24; 46%), fatigue (8/24; 33%) and platelet count decrease (8/24; 33%). The majority of AEs (20/24; 83%) were grade 1/2 in severity (Supplemental Table 3). Most common grade 3/4 AE were hematological events: anemia (4/24; 17%), neutrophil count decrease (2/24; 8%) and platelet count decrease (2/24; 8%). Table 2 provides an overview of AEs that were possibly related to the treatment administration.
TABLE 2.
Adverse events at least possibly related to the treatment administration and occurring in >10% of patients in case of grade 1/2 events and all events in case of grade 3/4 events. All events were graded according to the Common Terminology for Adverse Events version 4.03
| Grade 1/2 | Grade 3/4 | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Dose‐level | −1 | 1 | 2 | 3 | 4 | All (%) | −1 | 1 | 2 | 3 | 4 | All (%) |
| Adverse event | ||||||||||||
| Alanine aminotransferase increased | 1 | 1 | 1 | 3 (13) | 1 | 1 (4) | ||||||
| Anemia | 1 | 1 (4) | 1 | 2 | 1 | 4 (17) | ||||||
| Anorexia | 1 | 2 | 1 | 4 (17) | ||||||||
| Aspartate aminotransferase increased | 1 | 1 | 2 (8) | 1 | 1 (4) | |||||||
| Dysgeusia | 2 | 1 | 3 (13) | |||||||||
| Fatigue | 2 | 2 | 1 | 3 | 8 (33) | |||||||
| Nausea | 6 | 3 | 2 | 11 (46) | ||||||||
| Neutrophil count decreased | 1 | 1 | 2 | 1 | 4 (17) | 2 | 2 (8) | |||||
| Platelet count decreased | 4 | 2 | 2 | 8 (33) | 1 | 1 | 2 (8) | |||||
| Vomiting | 1 | 1 | 2 | 4 (17) | ||||||||
3.4. Pharmacokinetics
PK parameters for all patients included in this Phase I study are presented in Supplemental Table 4. The mean C max of olaparib in the different dose‐levels show an increase with increasing olaparib dose. At MTD the median AUC0‐12 on day 0 was 15.5 μg/mL*h and on Day 1 16.3 μg/mL*h indicating minimal accumulation. Figure 1 shows summarized PK profiles of olaparib tablet formulation by dose‐level after receipt of a single olaparib dose. Mean AUC0‐48 for carboplatin target AUC 4 mg*min/mL, determined in plasma ultrafiltrates was 5.10 mg/mL*min. Supplemental Figure 3 shows the PK profile of carboplatin measured in plasma‐ultrafiltrate after a single dose.
FIGURE 1.
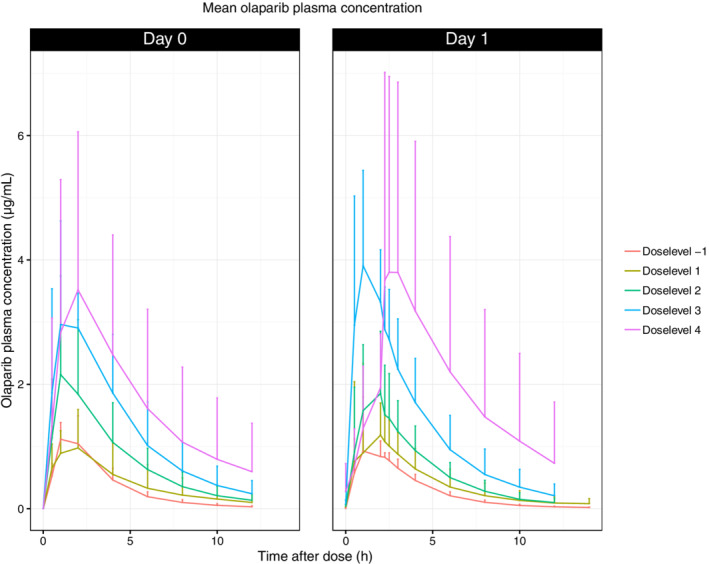
Mean olaparib plasma concentration. X‐axis: time after dose (h), Y‐axis: olaparib plasma concentration (ug/mL)
3.5. Pharmacodynamics
Supplemental Figure 4 shows the relative remaining PAR levels as a net result of the balance of PARP and PARG activity in PBMCs at Day 8 of cycle 1. The mean relative reduction in PAR levels at Day 8 was 97.5% ± 0.1%. The relative reduction in PAR levels increased with an increase of the olaparib dose and reached 99.1% ± 0.1% at dose‐level 4.
3.6. Efficacy
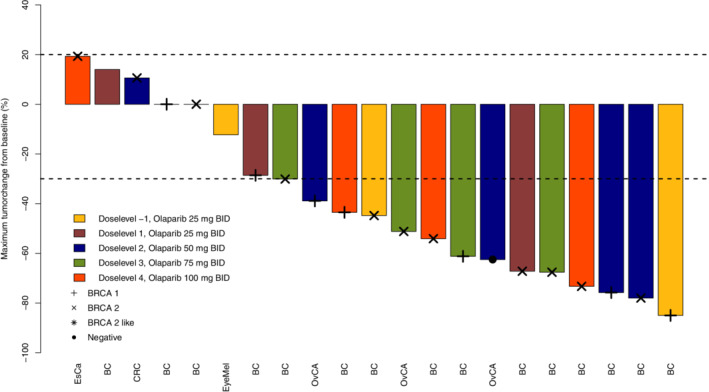
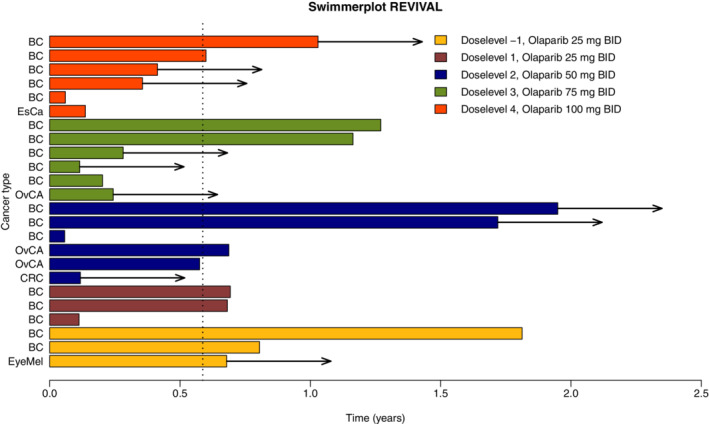
A total of 24 patients were evaluable for response. In one patient, physical examination was used for tumor evaluation, because of extensive skin metastases. Fourteen patients (14/24; 58%) had a partial response (PR) as best response. According to RE CIST 1.1, this was confirmed after at least 4 weeks. The group with PRs was represented by patients treated at all dose‐levels explored. Almost all patients achieved PR within the first 12 weeks of treatment (13/14; 93%). In seven patients (7/24; 29%) stable disease was the best response observed (Supplemental Table 5). The waterfall plot in Figure 2 shows the maximum change in target lesion diameter compared to baseline. Six patients (6/24; 25%) had a prolonged response of >12 months (Figure 3). The median progression free survival (PFS) was 7 months (Supplemental Figure 5).
FIGURE 2.
Swimmerplot. X‐axis: time (years), Y‐axis: individual patients. BC: breast cancer, EsCa: esophagus carcinoma, OvCa: ovarian cancer, CRC: colorectal cancer. EyeMel: eye melanoma. BID: bidaily
FIGURE 3.
Waterfall plot. X‐axis: individual patients, Y‐axis: maximum tumorchange from baseline (%). BC: breast cancer, EsCa: esophagus carcinoma, OvCa: ovarian cancer, CRC: colorectal cancer. EyeMel: eye melanoma, BID: bidaily
4. DISCUSSION
This Phase I trial of olaparib tablets, an oral PARP1/2 inhibitor, combined with carboplatin, showed that the combination is safe and has an acceptable side‐effect profile. The MTD was found to be olaparib tablets 75 mg BID and carboplatin AUC 4 mg*min/mL. Although the target AUC of carboplatin was AUC 4 mg*min/mL in 21 patients (dose‐level 1‐4), the mean AUC0‐48 measured in carboplatin ultrafiltrates of these 21 patients was 5.1 mg*min/mL. Therefore, the exposure of carboplatin was higher than the targeted AUC 4 mg*min/mL. Our study shows that the tablet formulation of olaparib can be administered safely in combination with carboplatin, compared to the previous capsule formulation. 16 Comparing the tablet PKs with the previous capsule formulation shows comparable exposure at the MTD, although there are slight differences in timeframe. 16 In our study, the AUC0‐12 is 16.3 μg/mL*h at 75 mg olaparib tablet BID, which is comparable to the AUC0‐12 of 13.2 μg/mL*h at 80 mg olaparib capsule BID. 16 However, in previous PK studies of olaparib tablet and capsule formulations, it was found that the exposure of the olaparib tablets was higher than the capsule formulation. 22 In our study, this was not confirmed. This could be explained by the low dosing of olaparib in our study compared to the bioequivalence testing dose of 400 mg before. At lower dose, nonlinear PKs may be less prominent which might explain this discrepancy. Pharmacodynamic analyses showed a high relative reduction in PAR levels, as read‐out for the balance of PARP and PARG activity in PBMCs, of 97.5% ± 0.1% at the lowest dose‐level indicating that already at the level of 25 mg olaparib BID there is almost complete PAR downregulation. Pharmacodynamic analyses showed a slight further relative reduction in PAR levels with an increase of the olaparib dose. Since olaparib treatment response may be dose dependent, we choose to abide to the registered monotherapy tablet dosage of 300 mg BID for maintenance therapy. 15 Future studies are necessary in order to explore whether pharmacodynamically guided reduction of maintenance dosing could lead to fewer AEs without compromising treatment response.
Before, only one validated enzyme‐linked immunosorbent assay for quantifying basal PAR levels was available for pharmacodynamic assessment of the effect of PARP1/2 inhibitors on the balance of PARP and PARG activity. Notably, the overall level of PAR measured is a reflection of both synthesis and degradation. 35 In that study, PAR levels were measured in PBMCs of patients who were administered veliparib in combination with topotecan, which resulted in a greater than 50% reduction in PAR levels in 19 out of 23 patients with measurable PAR levels. 36 A limitation of this previous progressive disease (PD) assay was that it is only applicable to patients with sufficient high levels of PAR. In our study, the radiation‐enhanced‐PAR pharmacodynamic assay (REP‐assay) was used. The REP‐assay uses 8 Gy of ex vivo radiation to strongly enhance the basal PARP1/2 activity in PBMCs, which allowed the sensitive determination of the lowest PAR levels present in PBMCs even from patients treated at the highest dose‐level. 33 This higher sensitivity of the REP‐assay probably explains the impressive relative reduction in PAR levels observed at all dose‐levels compared to previous studies and provides a better representation of the true biological inhibitory effect of PARP1/2 inhibitors on the balance of PARP and PARG activity.
Furthermore, since carboplatin induces DSB, it could therefore lead to higher PARP1/2 activity and higher PAR levels. So far, this has only been confirmed in nucleotide excision repair (NER) deficient tumor cell lines, but not in NER proficient tumor cell lines nor PBMCs. 37 Furthermore, the REP assay used in our study increased baseline PARP activity in PBMCs on average 121‐fold, which probably far outweighs any possible additional effect of carboplatin on PARP1/2 activity.
The most common AEs were mild and self‐limiting. However, in three patients, dose reductions were applied because of hematological toxicities. Anemia was the most common grade 3/4 AE (4/24; 17%). The level of hematological toxicities observed was comparable to the one reported in trials with olaparib monotherapy. 38 Serious toxicities that previously have been described for olaparib, such as pneumonitis or the development of secondary malignancies were not observed 39 ; this could be related to the relatively short follow‐up. Regarding the tumor response, the patient with the most pronounced decrease in tumor volume was treated with the lowest dose of the olaparib‐carboplatin combination explored in our study. However, there was only slight difference in olaparib dose between the different dose‐levels. Furthermore, an almost complete relative reduction in PAR levels was observed at the lowest dose‐levels. This raises the question whether treatment at a higher dose‐level would have an additional therapeutic effect. The increase in dose could lead to a more durable response but our study was not aimed at nor powered for in‐depth analyses of PFS or overall survival (OS), so conclusions on differences in response duration are not possible.
Recently, the results of the OlympIAD trial were published in which olaparib monotherapy was administered to patients with advanced BRCA‐mutated breast cancer. 40 The PFS in the olaparib group was significantly longer compared to the reference group (nonplatinum containing therapy) (7 vs 4.2 months). The main question is what the addition of carboplatin to olaparib would do on the end points of PFS and OS. Looking at the mechanism of action, the addition of carboplatin is a rational choice. However, the addition of carboplatin to olaparib could result in more and more severe (hematological) toxicities, 17 although not observed in our study even with the higher exposure to carboplatin of 5 mg*min/mL instead of the targeted AUC 4 mg*min/mL. Carboplatin monotherapy is also a promising therapy in patients with advanced BRCA‐mutated triple negative breast cancer. 41 Comparing in a 3‐arm study, olaparib monotherapy with the combination olaparib‐carboplatin and with carboplatin monotherapy might give useful information. Although our study provides valuable information on safety and anti‐tumor effect, some questions remain. First, it would have been interesting to have tumor tissue available from the time of progression in order to study the resistance mechanisms involved. Second, we did not measure PAR levels at the end of treatment. It would be interesting to see whether there remains sufficient reduction in PAR levels at the time of PD. Third, we did not perform any pretreatment genotyping of the tumor. Although most patients harbored a BRCA1 or BRCA2 mutation, treatment responses varied considerably. This may have been due to multiple factors, 42 , 43 , 44 including the molecular make‐up of the tumor, tumor heterogeneity or differences in the tumor microenvironment. Future studies addressing all these factors are highly desirable in order to select the most appropriate treatment for a certain patient. Finally, the small number of patients (n = 24) in this trial makes it difficult to draw firm conclusions on anti‐tumor activity and a prospective trial comparing olaparib with or without carboplatin would be needed.
Overall, our study provided the MTD of olaparib tablets in combination with two cycles of carboplatin. Furthermore, our study showed that this combination can be applied safely and is reasonably well tolerated. The observed preliminary anti‐tumor activity is encouraging with 58% of the patients having a decrease in tumor volume of more than 30%.
CONFLICT OF INTEREST
Dr. Linn reports grants and nonfinancial support from AstraZeneca, during the conduct of the study; grants from Agendia, grants and nonfinancial support from AstraZeneca, grants from Eurocept‐pharmaceuticals, grants and nonfinancial support from Genetech/Roche, grants and nonfinancial support from Novartis, grants from Pfizer, grants and nonfinancial support from Tesaro (now owned by GSK), grants and nonfinancial support from Immunomedics, other from Cergentis, other from IBM, other from Bayer, other from Daiichi‐Sankyo, outside the submitted work; J.H.M. Schellens and J.H. Beijnen are employees of Modra Pharmaceuticals, they have shares Modra Pharmaceuticals and are co‐patent holders on oral pharmaceutical formulations of taxanes. Dr. Schouten reports that a direct family member is employed at Astra Zeneca. Dr. Sonke reports institutional research support from AstraZeneca, Merck, Novartis and Roche.
ETHICS STATEMENT
This study (ClinicalTrials.gov identifier: NCT02418624) was approved by the institute's medical ethics committee and was conducted in accordance with the Declaration of Helsinki and guidelines for Good Clinical Practice. All patients gave written informed consent prior to study inclusion.
Supporting information
Appendix S1: Supporting Information
ACKNOWLEDGMENTS
The authors would like to thank the patients for participating in this study. In addition, the authors thank the NKI‐AvL trial office for collecting patient information. Also thanks to the Clinical Research Unit (CRU), the oncology nurses and the nursing specialists for their excellent care of the patients and their help to carry out the study investigations. This study was supported by an unrestricted research grant from AstraZeneca and by olaparib supply free of charge by AstraZeneca.
Geenen JJJ, Dackus GMHE, Schouten PC, et al. A Phase I dose‐escalation study of two cycles carboplatin‐olaparib followed by olaparib monotherapy in patients with advanced cancer. Int. J. Cancer. 2021;148:3041–3050. 10.1002/ijc.33498
Jill J. J. Geenen and Gwen M. H. E. Dackus contributed equally to this study.
Funding information AstraZeneca
Contributor Information
Jan H. M. Schellens, Email: j.schellens@gmail.com.
Sabine C. Linn, Email: s.linn@nki.nl.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the authors with permission of AstraZeneca. Restrictions apply to the availability of these data which were used under license for this study.
REFERENCES
- 1. King MC, Marks JH, Mandell JB. Breast and ovarian cancer risks due to inherited mutations in BRCA1 and BRCA2. Science (New York, NY). 2003;302(5645):643‐646. [DOI] [PubMed] [Google Scholar]
- 2. Antoniou A, Pharoah PD, Narod S, et al. Average risks of breast and ovarian cancer associated with BRCA1 or BRCA2 mutations detected in case series unselected for family history: a combined analysis of 22 studies. Am J Hum Genet. 2003;72(5):1117‐1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Evers B, Helleday T, Jonkers J. Targeting homologous recombination repair defects in cancer. Trends Pharmacol Sci. 2010;31(8):372‐380. [DOI] [PubMed] [Google Scholar]
- 4. Andreopoulou E, Schweber SJ, Sparano JA, McDaid HM. Therapies for triple negative breast cancer. Expert Opin Pharmacother. 2015;16(7):983‐998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Murai J, Huang SY, Das BB, et al. Trapping of PARP1 and PARP2 by clinical PARP inhibitors. Cancer Res. 2012;72(21):5588‐5599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hartwell LH, Szankasi P, Roberts CJ, Murray AW, Friend SH. Integrating genetic approaches into the discovery of anticancer drugs. Science (New York, NY). 1997;278(5340):1064‐1068. [DOI] [PubMed] [Google Scholar]
- 7. Farmer H, McCabe N, Lord CJ, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434(7035):917‐921. [DOI] [PubMed] [Google Scholar]
- 8. Bryant HE, Schultz N, Thomas HD, et al. Specific killing of BRCA2‐deficient tumours with inhibitors of poly(ADP‐ribose) polymerase. Nature. 2005;434(7035):913‐917. [DOI] [PubMed] [Google Scholar]
- 9. Rottenberg S, Jaspers JE, Kersbergen A, et al. High sensitivity of BRCA1‐deficient mammary tumors to the PARP inhibitor AZD2281 alone and in combination with platinum drugs. Proc Natl Acad Sci USA. 2008;105(44):17079‐17084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Mirza MR, Monk BJ, Herrstedt J, et al. Niraparib maintenance therapy in platinum‐sensitive, recurrent ovarian cancer. N Engl J Med. 2016;375(22):2154‐2164. [DOI] [PubMed] [Google Scholar]
- 11. Litton JK, Rugo HS, Ettl J, et al. Talazoparib in patients with advanced breast cancer and a germline BRCA mutation. N Engl J Med. 2018;379(8):753‐763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Somlo G, Frankel PH, Arun BK, et al. Efficacy of the PARP inhibitor Veliparib with carboplatin or as a single agent in patients with germline BRCA1‐ or BRCA2‐associated metastatic breast cancer: California cancer consortium trial NCT01149083. Clin Cancer Res. 2017;23(15):4066‐4076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. de Bono J, Ramanathan RK, Mina L, et al. Phase I, dose‐escalation, two‐part trial of the PARP inhibitor talazoparib in patients with advanced germline BRCA1/2 mutations and selected sporadic cancers. Cancer Discov. 2017;7(6):620‐629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Oza AM, Tinker AV, Oaknin A, et al. Antitumor activity and safety of the PARP inhibitor rucaparib in patients with high‐grade ovarian carcinoma and a germline or somatic BRCA1 or BRCA2 mutation: integrated analysis of data from study 10 and ARIEL2. Gynecologic Oncology. 2017;147(2):267‐275. [DOI] [PubMed] [Google Scholar]
- 15. Tutt A, Robson M, Garber JE, et al. Oral poly(ADP‐ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and advanced breast cancer: a proof‐of‐concept trial. Lancet (London, England). 2010;376(9737):235‐244. [DOI] [PubMed] [Google Scholar]
- 16. Fong PC, Boss DS, Yap TA, et al. Inhibition of poly(ADP‐ribose) polymerase in tumors from BRCA mutation carriers. N Engl J Med. 2009;361(2):123‐134. [DOI] [PubMed] [Google Scholar]
- 17. Oza AM, Cibula D, Benzaquen AO, et al. Olaparib combined with chemotherapy for recurrent platinum‐sensitive ovarian cancer: a randomised phase 2 trial. Lancet Oncol. 2015;16(1):87‐97. [DOI] [PubMed] [Google Scholar]
- 18. Del Conte G, Sessa C, von Moos R, et al. Phase I study of olaparib in combination with liposomal doxorubicin in patients with advanced solid tumours. Br J Cancer. 2014;111(4):651‐659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. van der Noll R, Marchetti S, Steeghs N, et al. Long‐term safety and anti‐tumour activity of olaparib monotherapy after combination with carboplatin and paclitaxel in patients with advanced breast, ovarian or fallopian tube cancer. Br J Cancer. 2015;113(3):396‐402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ledermann JA, Harter P, Gourley C, et al. Overall survival in patients with platinum‐sensitive recurrent serous ovarian cancer receiving olaparib maintenance monotherapy: an updated analysis from a randomised, placebo‐controlled, double‐blind, phase 2 trial. Lancet Oncol. 2016;17(11):1579‐1589. [DOI] [PubMed] [Google Scholar]
- 21. Mateo J, Moreno V, Gupta A, et al. An adaptive study to determine the optimal dose of the tablet formulation of the PARP inhibitor olaparib. Target Oncol. 2016;11(3):401‐415. [DOI] [PubMed] [Google Scholar]
- 22. Zhou D, Li J, Bui K, et al. Bridging Oolaparib capsule and tablet formulations using population pharmacokinetic meta‐analysis in oncology patients. Clin Pharmacokinet. 2019;58(5):615‐625. [DOI] [PubMed] [Google Scholar]
- 23. Robson M, Im SA, Senkus E, et al. Olaparib for metastatic breast cancer in patients with a germline BRCA mutation. N Engl J Med. 2017;377(6):523‐533. [DOI] [PubMed] [Google Scholar]
- 24. Lee JM, Hays JL, Annunziata CM, et al. Phase I/Ib study of olaparib and carboplatin in BRCA1 or BRCA2 mutation‐associated breast or ovarian cancer with biomarker analyses. J Natl Cancer Inst. 2014;106(6):dju089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Van der Noll RAJ, Jager A, Marchetti S, et al. Phase I study of olaparib in combination with carboplatin and/or paclitaxel in patients with advanced solid tumours. J Clin Oncol. 2013a;31:2579. [Google Scholar]
- 26. Van der Noll R, de Grève J, Jager A, et al. Safety results from a phase I study with a new tablet formulation of olaparib (O) in combination with carboplatin (C) and paclitaxel (Pa). Eur J Cancer. 2013;49:S174‐S175. [Google Scholar]
- 27. Eisenhauer EA, Therasse P, Bogaerts J, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer (Oxford, England: 1990). 2009;45(2):228‐247. [DOI] [PubMed] [Google Scholar]
- 28. Schouten PC, Dackus GM, Marchetti S, et al. A phase I followed by a randomized phase II trial of two cycles carboplatin‐olaparib followed by olaparib monotherapy versus capecitabine in BRCA1‐ or BRCA2‐mutated HER2‐negative advanced breast cancer as first line treatment (REVIVAL): study protocol for a randomized controlled trial. Trials. 2016;17(1):293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Calvert AH, Newell DR, Gumbrell LA, et al. Carboplatin dosage: prospective evaluation of a simple formula based on renal function. J Clin Oncol. 1989;7(11):1748‐1756. [DOI] [PubMed] [Google Scholar]
- 30. Nijenhuis CM, Lucas L, Rosing H, Schellens JH, Beijnen JH. Development and validation of a high‐performance liquid chromatography‐tandem mass spectrometry assay quantifying olaparib in human plasma. J Chromatogr B. 2013;940:121‐125. [DOI] [PubMed] [Google Scholar]
- 31. Brouwers EE, Tibben MM, Rosing H, et al. Sensitive inductively coupled plasma mass spectrometry assay for the determination of platinum originating from cisplatin, carboplatin, and oxaliplatin in human plasma ultrafiltrate. J Mass Spectrom. 2006;41(9):1186‐1194. [DOI] [PubMed] [Google Scholar]
- 32. Common Terminology Criteria for Adverse Events (CTCAE) version 4.03; 2010.
- 33. de Haan R, Pluim D, van Triest B, et al. Improved pharmacodynamic (PD) assessment of low dose PARP inhibitor PD activity for radiotherapy and chemotherapy combination trials. Radiother Oncol. 2018;126(3):443‐449. [DOI] [PubMed] [Google Scholar]
- 34. NCI DoCTaD [cited 2015 20‐10‐2015]; Available from: http://dctd.cancer.gov/ResearchResources/biomarkers/PolyAdenosylRibose.htm>.
- 35. Alvarez‐Gonzalez R, Althaus FR. Poly(ADP‐ribose) catabolism in mammalian cells exposed to DNA‐damaging agents. Mutat Res. 1989;218(2):67‐74. [DOI] [PubMed] [Google Scholar]
- 36. Kummar S, Chen A, Ji J, et al. Phase I study of PARP inhibitor ABT‐888 in combination with topotecan in adults with refractory solid tumors and lymphomas. Cancer Res. 2011;71(17):5626‐5634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Cheng H, Zhang Z, Borczuk A, et al. PARP inhibition selectively increases sensitivity to cisplatin in ERCC1‐low non‐small cell lung cancer cells. Carcinogenesis. 2013;34(4):739‐749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kaufman B, Shapira‐Frommer R, Schmutzler RK, et al. Olaparib monotherapy in patients with advanced cancer and a germline BRCA1/2 mutation. J Clin Oncol. 2015;33(3):244‐250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bendell J, O'Reilly EM, Middleton MR, et al. Phase I study of olaparib plus gemcitabine in patients with advanced solid tumours and comparison with gemcitabine alone in patients with locally advanced/metastatic pancreatic cancer. Ann Oncol. 2015;26(4):804‐811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Robson M, Im SA, Senkus E, et al. Olaparib for metastatic breast cancer in patients with a germline BRCA mutation. N Engl J Med. 2017;377:523‐533. [DOI] [PubMed] [Google Scholar]
- 41. Tutt A, Tovey H, Cheang MCU, et al. Carboplatin in BRCA1/2‐mutated and triple‐negative breast cancer BRCAness subgroups: the TNT trial. Nat Med. 2018;24(5):628‐637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Coffelt SB, de Visser KE. Immune‐mediated mechanisms influencing the efficacy of anticancer therapies. Trends Immunol. 2015;36(4):198‐216. [DOI] [PubMed] [Google Scholar]
- 43. Swanton C. Intratumor heterogeneity: evolution through space and time. Cancer Res. 2012;72(19):4875‐4882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Tannock IF, Hickman JA. Limits to personalized cancer medicine. N Engl J Med. 2016;375(13):1289‐1294. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1: Supporting Information
Data Availability Statement
The data that support the findings of this study are available from the authors with permission of AstraZeneca. Restrictions apply to the availability of these data which were used under license for this study.