

Summary
Demand for cannabidiol (CBD), the predominant cannabinoid in hemp (Cannabis sativa), has favored cultivars producing unprecedented quantities of CBD. We investigated the ancestry of a new cultivar and cannabinoid synthase genes in relation to cannabinoid inheritance.
A nanopore‐based assembly anchored to a high‐resolution linkage map provided a chromosome‐resolved genome for CBDRx, a potent CBD‐type cultivar. We measured cannabinoid synthase expression by cDNA sequencing and conducted a population genetic analysis of diverse Cannabis accessions. Quantitative trait locus mapping of cannabinoids in a hemp × marijuana segregating population was also performed.
Cannabinoid synthase paralogs are arranged in tandem arrays embedded in long terminal repeat retrotransposons on chromosome 7. Although CBDRx is predominantly of marijuana ancestry, the genome has cannabidiolic acid synthase (CBDAS) introgressed from hemp and lacks a complete sequence for tetrahydrocannabinolic acid synthase (THCAS). Three additional genomes, including one with complete THCAS, confirmed this genomic structure. Only cannabidiolic acid synthase (CBDAS) was expressed in CBD‐type Cannabis, while both CBDAS and THCAS were expressed in a cultivar with an intermediate tetrahydrocannabinol (THC) : CBD ratio.
Although variation among cannabinoid synthase loci might affect the THC : CBD ratio, variability among cultivars in overall cannabinoid content (potency) was also associated with other chromosomes.
Keywords: cannabidiol (CBD), cannabinoids, domestication, hemp, marijuana, tetrahydrocannabinol (THC)
Introduction
Cannabis has been cultivated for millennia in the form of hemp for fiber and grain, or marijuana for psychoactive tetrahydrocannabinol (THC) (ElSohly et al., 2000; Merlin & Clark, 2013). Delta‐9‐tetrahydrocannabinolic acid (THCA) and cannabidiolic acid (CBDA) are chemicals uniquely produced by Cannabis plants. When decarboxylated, these molecules bind to endocannabinoid receptors in the nervous systems of vertebrates and elicit a broad range of neurological effects in humans (Russo, 2016). Archeological and forensic evidence suggests that the psychoactivity of THC played a role in the domestication of marijuana (Small, 2016; Zhao et al., 2019) and in selective breeding to increase THCA (hereafter THC) content during the late 20th century (ElSohly et al., 2000). On the contrary, hemp cultivars produce predominantly CBDA (hereafter CBD). Recent demand for cannabidiol (CBD) has favored the breeding of high‐potency CBD‐dominant (high‐CBD) plants (Small, 2016). Recently developed, high‐CBD cultivars commonly referred to as ‘hemp’ can exceed THC < 0.3% DW and fail to meet statutory definitions of industrial hemp when grown to maturity. Here, we restrict our use of the term ‘hemp’ to fiber and grain cultivars for the sake of clarity (Toth et al., 2020).
The enzymes THCAS and CBDAS compete for a common precursor (cannabigerolic acid or CBGA) and are implicated in alternative explanations for the THC : CBD ratio. Some investigators suggest that sequence variation among THCAS gene copies influences the ratio (van Bakel et al., 2011; Onofri et al., 2015), while others propose that a nonfunctional CBDAS allele in the homozygous state alters the ratio in favor of THC (Weiblen et al., 2015). Current explanations for differences among cultivars in the THC : CBD ratio focus on cannabinoid synthase gene loci (Sirikantaramas et al., 2004; van Bakel et al., 2011; Onofri et al., 2015; Weiblen et al., 2015). In spite of recent advances in genome sequencing, precisely how cannabinoid synthase genes influence the THC : CBD ratio and the overall abundance of cannabinoids (potency) is poorly understood (van Bakel et al., 2011). Among the obstacles to understanding the relationship between cannabinoid synthase gene diversity and phenotypes is genomic complexity that has frustrated attempts to assemble complete chromosomes until recently (Laverty et al., 2019; Kovalchuk et al., 2020).
We generated a chromosome‐resolved reference genome for a new, high‐CBD cultivar (CBDRx) with a nanopore‐based assembly anchored by a hemp × marijuana mapping population (Weiblen et al., 2015). The ancestry of the CBDRx genome was investigated through population genetic analysis of several hundred cultivars. We then compared the highly inbred CBDRx genome with other assemblies including three new, nanopore‐based genome assemblies. We examined the genomic structure of cannabinoid synthase gene and pseudogene copies across the assemblies in relation to cannabinoid phenotypes. Full‐length cDNA sequencing was used to associate patterns of gene expression to phenotype. Lastly, quantitative trait locus (QTL) mapping investigated the association of the THC : CBD ratio and overall cannabinoid content (potency).
Materials and Methods
Plant material
High‐CBD cultivars, CBDRx and First Light (FL), are related to Cherry cultivars originating from Colorado, USA (e.g. Cannatonic, Charlotte’s Web, ACDC). FL plants, FL48 and FL49, were full siblings and a third, FL18, descended from a different family. The F2 mapping population was described in Weiblen et al. (2015). In brief, parental high‐THC Cannabis (Skunk#1) and fiber hemp (Carmen) cultivars were sibling‐crossed for five generations to increase homozygosity. A single fifth‐generation Skunk#1 female was fertilized with pollen from a single fifth‐generation Carmen male. A single female F1 plant was isolated and propagated asexually. Development of staminate flowers in female clones was induced using silver nitrate, and clones were self‐pollinated to produce the F2 generation. Plant growth conditions are described in Supporting Information Methods S1.
Cannabinoid analysis
We refer to the decarboxylated forms (THC and CBD) as cannabinoid phenotypes for the sake of simplicity. CBDRx cannabinoid analysis was performed by Functional Remedies using high‐performance liquid chromatography (HPLC) and consistently produced a CBD : THC ratio of 18 : 1 (15% CBD and 0.8% THC). FL plants were analyzed for cannabinoid content multiple times, with a typical test result as shown in Table S1. While some testing laboratories reported different absolute values, the cannabinoid ratios were consistent. Cannabinoid contents of Skunk#1, Carmen, their F1 hybrid and 96 female F2 plants were measured by GC as described in Weiblen et al. (2015) (Tables S1, S2).
Illumina sequencing
Procedures for DNA isolation and library preparation from the mapping population, CBDRx, and FL are detailed in the Methods S1. Libraries were sequenced on an Illumina HiSeq 2500 SBS V4 (Illumina, San Diego, CA, USA) in 2 × 125 bp read high‐output mode to a coverage depth of 2X at the University of Minnesota Genomics Center for the F2. By contrast, 2 × 150 bp was generated for CBDRx and FL. The bioinformatic workflow from raw sequence data processing to genome assembly and analysis is summarized in Fig. S1. For all mapping population sequence data, we gently trimmed Illumina reads of low‐quality bases and adapter sequence with trimmomatic (Bolger et al., 2014), aligned them to the reference assembly with bwa (Li & Durbin, 2009), sorted and compressed the alignments with samtools (Li et al., 2009), and marked duplicates with picard tools (Broad Institute, 2016).
Nanopore genome assembly
Procedures for purification of high‐molecular‐weight genomic DNA samples of CBDRx and FL for Oxford Nanopore (ONT) sequencing are described in Methods S1. Nanopore sequence was generated on the MinION ONT platform, resulting in varying genome coverages (Table S3). The resulting raw reads in fastq format were aligned (overlap) with minimap2 (Li, 2018) and an assembly graph (layout) was generated with miniasm (Li, 2016). The resulting graph was inspected using bandage (Wick et al., 2015). A consensus sequence was generated by mapping reads to the assembly with minimap2 (Fig. S1), followed by mapping three times with racon (Vaser et al., 2017). Lastly, the assembly was polished with pilon (Walker et al., 2014) three times using the CBDRx Illumina paired‐end 2 × 150 bp sequence; the Illumina reads were mapped to the consensus assembly using bwa (Li & Durbin, 2009). All assembly steps (Michael et al., 2018) were carried out on a machine with 231 Gb RAM and 56 CPU. A Hi‐C library was prepared and sequenced by Phase Genomics on a CBDRx full‐sibling using a restriction enzyme cut site GATC and sequenced with 2 × 80 bp Illumina PE.
Genetic linkage map
We constructed a genetic linkage map from the segregating F2 population involving Carmen hemp crossed with Skunk#1 marijuana (Weiblen et al., 2015). A pseudo‐F1 dataset was constructed by concatenating all F2 Illumina reads followed by random subsampling to a target genomic coverage of ×100. The pseudo‐F1 and parental reads were independently error‐corrected using k‐mer histograms (with k = 25) with allpathslg (Gnerre et al., 2011). We then constructed a de Bruijn graph from the pseudo‐F1 dataset to identify alleles segregating in the F2 population. The graph was based on error‐corrected pseudo‐F1 reads using mccortex assembler at k = 19 (Iqbal et al., 2012). This program is unique in that genome assembly and variant discovery are performed simultaneously. As reads are assembled, paths through regions of the de Bruijn graph that diverge and rejoin (‘bubbles’) were retained as variants. The bubble‐read coverage distribution is used to classify bubbles as repeats, homologous alleles or errors. Sites at which the Carmen and Skunk#1 parents were fixed for alternate alleles were genotyped in the F2 population by comparing reads from individual plants with the population graph. Genotypes were imputed using a sliding‐window hidden Markov model as implemented in lb‐impute (Fragoso et al., 2016) that leverages physical linkage and coverage information within a window width of 10 variants.
Segregating genotype bins containing no missing data across the population that appeared at least 10 times were selected for use as map markers using a strategy adapted from Hahn et al. (2014). Markers exhibiting segregation distortion by a χ2 test were low in number and are retained in the map (c. 10% of markers). Linkage groups and marker order were inferred using the ant colony optimization in antmap (Iwata & Ninomiya, 2006) solution to the traveling salesman path. Recombinations were counted directly and divided by the number of gametes in the population (192) to infer genetic distance between adjacent markers and summed consecutively in linear order to give map position on a linkage group.
Pseudomolecule generation
Before aligning genetic map markers with CBDRx contigs, the CBDRx assembly was evaluated for library contaminants using blobtools (Laetsch & Blaxter, 2017) and the NCBI nonredundant database to exclude contigs derived from outside Viridiplantae. Map markers were aligned to the CBDRx contigs with bwa (Li & Durbin, 2009). Contigs mapped to different linkage groups or more than 10 cM away from each other were deemed chimeric and broken at the longest repeat between the different regions.
An initial set of rough pseudomolecules were constructed by assigning contigs to linkage groups, ordering contigs by mean cM, and orienting by cM position on either end (Badouin et al., 2017). The F2 population was genotyped again via alignment to the rough pseudomolecules followed by lb‐impute. Population marker bins from this second round of genotyping were used to further saturate the genetic map if they increased map density without increasing the map length (Dijkstra, 1959). Contigs were partitioned by linkage group and scaffolded with the Hi‐C library using three iterations of salsa (Ghurye et al., 2017). allmaps (Tang et al., 2015) was used to generate the contig order and orientation with the template genetic map positions, second‐round genetic map positions, and salsa contig positions as input. This scaffolding step ordered and oriented contigs but left gaps between them. Pseudomolecules were further polished with an additional 10 iterations of racon followed by 10 iterations of pilon. The racon consensus procedure facilitated alignment of additional reads bridging gaps represented by uncalled bases (Ns) in the scaffolds, and these were replaced by a consensus sequence of the bridging reads. After scaffolding and gap filling, 841 contiguous sequences spanning 714 498 588 bp were anchored to nuclear pseudomolecules.
Scaffolding with Hi‐C data alone did not completely resolve the CBDRx assembly and so the genetic map derived from the F2 segregating population was leveraged a third time to order and orient the remaining 841 contigs. This strategy involved genotyping the F2 population once more against CBDRx as a reference and visually inspecting marker bins for disorder. Most of the recombination breakpoints in the genetic map could be referenced to CBDRx contigs in the same order as on the genetic map. This strategy allowed us to validate the ordering and orientation of loci and to estimate the extent of CBDRx rearrangement relative to the independent F2 population. We found most contigs to be largely collinear in genetic and physical space but a few that had no corresponding recombination break points could not be ordered or oriented. This was the case for two of the three synthase‐bearing (those containing the functional copies of CBDAS and CBCAS) contigs on chromosome 7. For these, we manually reordered the synthase‐bearing contigs to be adjacent, as this was most parsimonious in the absence of evidence for an alternative arrangement. After we first assembled CBDRx (Grassa et al., 2018), McKernan et al. (2020) released a public Cannabis genome assembly that also located the synthases in a single contig, affirming their adjacency. We measured the completeness of the final chromosome‐resolved assembly of CBDRx with Benchmarking Universal Single Copy Orthologs (busco) using database version viridiplantae_odb10 with a protein set drawn from 57 species (Simao et al., 2015). The bioinformatic analysis workflow (Fig. S1) used for construction of the CBDRx chromosome‐resolved assembly is detailed further in Methods S1.
Pseudomolecules for the FL, Carmen and Skunk#1 assemblies were created using reveal (Linthorst et al., 2015) with the CBDRx pseudomolecules as reference. In order to close gaps, the FL pseudomolecules were polished again with three rounds of racon followed by four rounds of pilon.
Genome size estimation
The genome sizes of CBDRx, FL18, FL48 and FL49 were estimated using kmer frequency analysis. Kmer frequency was determined with Jellyfish (v.2.2.4) using an Illumina paired‐end sequence that was generated to polish the assemblies. All kmers were counted (no ‐U) to capture the high‐copy‐number kmers (kmer > 10 000). Genome size was estimated from the kmer (‐m 31) frequency using a custom script and Genomescope (http://qb.cshl.edu/genomescope/).
Copy number estimation using short‐read coverage analysis
Often in genome assemblies repeat sequences such as ribosomal, centromere, transposable elements and tandemly repeated genes are collapsed as a result of the sequencing technology or assembly method used. One way to estimate the copy number of these repeated sequences in a genome is through coverage analysis, which leverages the differential mapping of a short‐read dataset to single‐copy and repeat gene elements. Coverage analysis was performed by mapping Illumina reads from CBDRx, Purple Kush (SRR352150) and Finola (SRR7285294) (van Bakel et al., 2011) with minimap2 to the genomic versions of a single‐copy gene GIGANTEA (GI), one complete ribosomal cassette (rDNA:18S‐5S‐26S) and the three synthase arrays. Copy number was estimated by dividing the coverage over the repeat element (rDNA, synthase array) by the coverage over the single‐copy gene (see Methods S1). This analysis estimated 13 synthase copies in CBDRx and suggested 500–600 rDNA arrays, which is consistent with other genomes of this size. Seventeen and 25 synthase copies were estimated for Finola and Purple Kush, respectively.
Repeat and gene prediction and annotation
An overview of procedures for sequence prediction and annotation is given in Fig. S1. Full‐length long terminal repeat retrotransposons (LTR‐RTs) were predicted using LTR_FINDER using the standard settings and one mismatch (Xu & Wang, 2007). The resulting full‐length LTR‐RTs were used to mask the genome using repeatmasker (Chen, 2004). Four full‐length cDNA nanopore read libraries were aligned to the reference with minimap2 (Li, 2018) before and after error correction by canu (Koren et al., 2017) of co‐located batches (i.e. all reads aligning to the same locus in the genome). RNAseq libraries found on the Sequence Read Archive (Table S4) were aligned to the reference with gsnap (Wu et al., 2016) and assembled into transcripts with stringtie (Pertea et al., 2015). Four high‐coverage RNAseq libraries (Table S4) were assembled using trinity (Haas et al., 2008) in both de novo and reference‐guided modes. Contaminate sequence was removed using seqclean. The full‐length cDNAs, stringtie assembly and trinity transcripts were assembled into gene models with the Program to Assemble Spliced Alignments (pasa) (Haas et al., 2013). Additional transcriptome assemblies (Table S4) from Humulus lupulus (Hill et al., 2017) and Cannabis were aligned to the reference with gmap (Wu et al., 2016). Genes were predicted ab initio using augustus (Hoff & Stanke, 2013). augustus was trained for C. sativa using the CanSat3 genome and representative transcriptome (van Bakel et al., 2011) from the Augustus web portal. Nonredundant RefSeq proteins (Pruitt et al., 2007) for Viridiplantae were clustered at 90% identity with CD‐HIT (Fu et al., 2012). Representative sequences for each cluster were aligned to the reference genome using diamond (Buchfink et al., 2014). Pairwise hits were locally realigned with AAT (Huang et al., 1997) and Exonerate protein2genome. Repetitive sequence was identified using the set union, taken with bedtools merge, of three programs: repeatmasker, tephra and red using default parameters (Girgis, 2015). evidencemodeler was used to integrate all evidence for and against protein‐coding genes. Finally, the output of evidencemodeler was used to update the PASA database to refine exon and UTR boundaries of predicted transcripts.
QTL analysis
Quantative trait locus (QTL) analysis was based on a composite linkage map incorporating 1175 segregating marker bins derived from Illumina sequencing as described earlier with 48 amplified fragment length polymorphisms (AFLPs), 11 microsatellite markers and one Sanger‐sequenced marker from Weiblen et al. (2015). The map was constructed using Joinmap 4.1 (Wageningen, the Netherlands) with parents (Skunk#1 and Carmen) and 96 F2 plants. Linkage groups were assembled from independent log‐of‐odds scores (LOD) based on G‐tests for independence of two‐way contingency tables. Linkage groups with LOD > 3.0 and containing four or more markers were included in a map based on the Kosambi (1944) function.
Cannabinoid profiles of the same 96 F2 individuals (Table S2) were analyzed with respect to the composite linkage map using Windows QTL Cartographer v.2.5_011 (winqtlcart) (Wang et al., 2006). Composite interval mapping was used to estimate LOD over a walk speed of 1.0 cM with 10 cofactors and a 2.5 cM exclusion window. Significant associations between traits and linkage groups were identified using an experiment‐wise (P = 0.05) LOD threshold was estimated in winqtlcart using 1000 permutations. Results were plotted with Mapchart 2.32 (Wageningen, the Netherlands).
CBDRx ancestry and selection
We obtained previously published whole‐genome sequenced libraries (Sawler et al., 2015; Lynch et al., 2016; Soorni et al., 2017) representing diverse Cannabis populations for the purpose of investigating CBDRx ancestry. WGS sequences from 367 individuals were genotyped using bcftools and CBDRx as the reference genome. Plink and Plink2 were used to filter the genotype matrix and minimize structure originating from familial relatedness, potentially artefactual patterns of allele frequency, selection and genetic linkage as follows. We selected a single representative from closely groups related based on a KING‐robust kinship coefficient > 0.016. We retained biallelic sites called in at least 80% of individuals, with a minor allele frequency > 1% and observed heterozygosity < 0.60. We removed sites failing an exact test for Hardy–Weinberg with a mid‐p adjustment (Graffelman & Moreno, 2013). We eliminated individuals genotyped at < 90% of sites. We thinned sites for linkage disequilibrium in sliding windows with a width of 50 single nucleotide polymorphisms (SNPs), a slide of five SNPs, and a variance inflation factor threshold of 2. A principal components analysis was conducted on the filtered genotype matrix.
The filtered genotype matrix was used to estimate genome‐wide ancestry proportions at k = 3 using admixture (Alexander et al., 2009). Individuals identified as having > 99% ancestry were assigned to respective drug Cannabis and hemp populations. A subset of segregating sites was selected for assigning ancestry blocks along chromosomes using a method intended to maximize information and minimize linkage disequilibrium. Sites were ranked by Wright’s F ST (Wright, 1950). Genetic positions for all segregating sites were interpolated along a B‐spline function fitted to the empirically observed positions in the mapping population with penalized coefficients to maintain monotonicity (Pya & Wood, 2015). For each chromosome, the site with the highest F ST value and lowest genetic position was the first selected. Additional sites were selected in order of decreasing F ST when separated by at least 0.03 cM from previously selected sites. Ancestry blocks were assigned by ancestryhmm (Corbett‐Detig & Nielsen, 2017) assuming a single pulse from hemp to drug Cannabis eight generations in the past, based on discussions with the breeders. As described in Methods S1, we used the population branch statistic (PBS) (Yi et al., 2010), an F ST‐based three‐population test, to scan the genome for regions exhibiting different molecular evolutionary rates among populations that can be interpreted as evidence of selection.
Results
CBDRx ancestry
Single nucleotide polymorphisms segregating in a diverse sample of Cannabis genotypes indicate that marijuana and hemp cultivars are associated with a major axis of population genetic differentiation (F ST = 0.229) (Fig. 1a). CBDRx was grouped with marijuana cultivars despite having a THC : CBD ratio more similar to hemp. The genome‐wide ancestry of CBDRx was estimated to be 89% marijuana and 11% hemp (Figs 1b, S2). The PBS showed evidence of selection in the vicinity of cannabinoid synthases on chromosome 7 (Figs 2, S3).
Fig. 1.
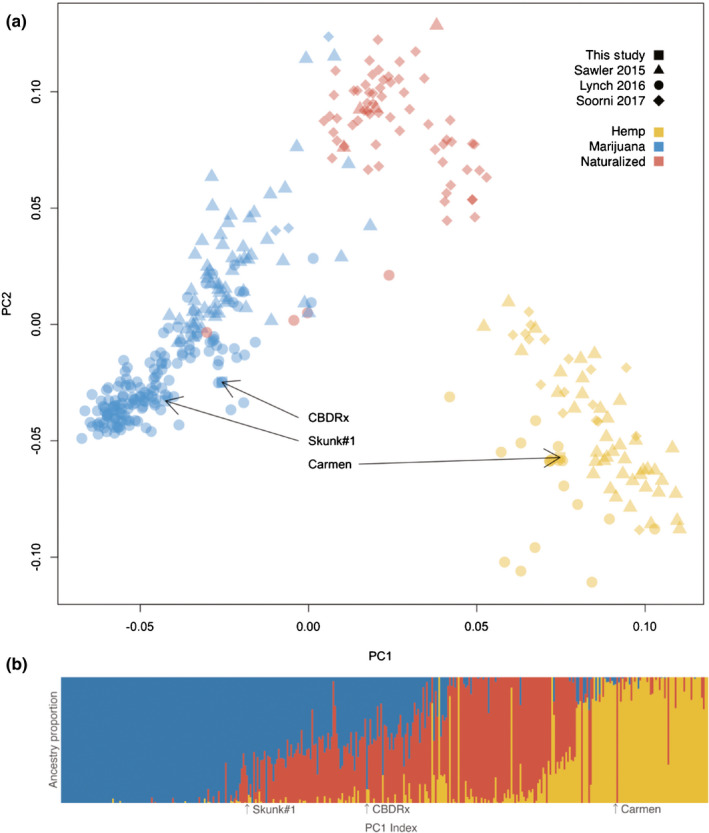
Population genetic structure inferred from 2051 single nucleotide polymorphisms and 367 accessions of hemp, marijuana and naturalized (established wild population) Cannabis sativa. We limit our definition of hemp to grain and fiber cultivars. (a) Principal components analysis (PCA) of the genotype matrix with circles, triangles and diamonds indicating data sources (Sawler et al., 2015; Lynch et al., 2016; Soorni et al., 2017). Arrows point to the intoxicating marijuana cultivar (Skunk#1), an industrial fiber hemp cultivar (Carmen), and a high‐cannabidiol (CBD) cultivar (CBDRx). Clusters were assigned from k‐means as cultivated hemp (yellow), marijuana cultivars (blue) or naturalized Cannabis (red). Naturalized individuals are defined as any plants derived from noncultivated populations. Wild populations referred to in the literature as landraces (Merlin & Clark, 2013) are here termed ‘naturalized’ to acknowledge ambiguity about nativity, recent escape from cultivation, orancient feral populations. The first component (PC1) divides cultivated hemp and marijuana, while the second (PC2) represents a continuum between naturalized and domesticated Cannabis. (b) Bayesian admixture plot indicting ancestry of accessions given k = 3 idealized donor populations. Accessions are ordered left to right according to their position along PC1 with estimated ancestry proportional to the color of the vertical segment. The Skunk#1 genome ancestry was estimated to be 78% marijuana and 22% naturalized. The Carmen genome ancestry was estimated to be 94% hemp and 6% marijuana. The CBDRx ancestry was estimated to be 89% marijuana and 11% hemp.
Fig. 2.
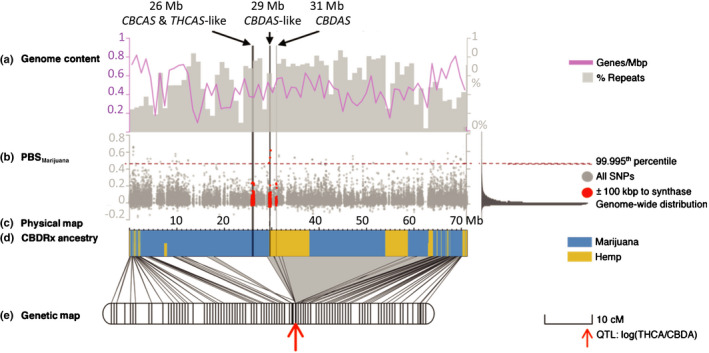
The location of cannabinoid synthase gene sequences on chromosome 7 in high‐cannabidiol (CBD) cultivar (CBDRx) Cannabis sativa is associated with evidence of recent selection for hemp ancestry introgressed into a marijuana background and maps to a quantitative trait locus (QTL) for the ratio of tetrahydrocannabinol (THC) : CBD. Vertical lines transecting the upper four panels indicate the locations of three cannabinoid synthase gene arrays. (a) Gene content (pink lines) and percentage repeat content (gray bars) of a 1 Mb sliding window across chromosome 7. (b) Manhattan plot of the population branch statistic (PBS), an F ST‐based, three‐population test with extreme values suggesting lineage‐specific evolutionary processes. Values for the marijuana branch are displayed in gray dots with a histogram of the genome‐wide distribution on the right. A dashed red line marks the 99.995th percentile of the distribution. Points within 100 kbp of cannabinoid synthase array are also colored red. (c) Physical length of chromosome 7. (d) Ancestry estimates for genomic segments derived from marijuana (blue) and hemp (yellow). (e) Genetic map of an F2 mapping population of Skunk#1 × Carmenanchored to the physical map using 1175 Illumina‐based whole genome sequencing (WGS) marker bins dervied from 22 280 segregating markers (211 106 for all chromosomes genome‐wide). Lines connecting the maps indicate the positions of markers in physical and genetic space. Shaded in gray are consecutive physical markers associated with genomic regions of low recombination. The red arrow marks the position of the only QTL associated with the THC : CBD ratio.
CBDRx genome
The CBDRx genome assembly was 97.4% complete with a 1.9% duplication percentage that is consistent with residual heterozygosity. The quality of scaffolding that supported the assembly is evident in a contact map of the Hi‐C library (Fig. S4). After a first round of CBDRx assembly, 1190 contigs representing 602 278 047 bp were anchored using the F2 genetic linkage map alone. Most contigs mapped to individual linkage groups, but 61 contigs mapped to multiple linkage groups. Discordance between the genetic and physical maps could be attributed to either assembly error or chromosomal rearrangement. In either case, chromosomal rearrangement in CBDRx relative to the mapping population would seem to be rather low given the high percentage of contigs that were assigned to a single linkage group. We opted to break ambiguously mapped contigs and assign their pieces to different linkage groups.
After splitting ambiguously mapped contigs, 685 of 1306 contigs were anchored with two or more adjacent and unique genetic map positions that facilitated the granular ordering and orientation of 65% of the genome. Another 78 contigs representing 3% of the genome mapped to a unique genetic position but could not be oriented unambiguously. The remaining 543 contigs (32% of the genome) did not map to a unique genetic position within a linkage group and could not be ordered or oriented. Nearly all of the contigs mapped to the presumed centromeric regions with a few mapped to the nucleolus‐organizing region or to telomeric regions.
Genes in CBDRx were predicted using a combination of ab initio and empirical data including full‐length cDNA sequenced using ONT long‐read sequencing. After masking 63% of the genome for repeats consisting of 17 536 full‐length LTR‐RTs, we predicted the presence of 42 052 protein‐coding genes in the assembly. The busco analysis identified 97% out of 425 buscos as complete, including 62% single‐copy and 35% duplicated buscos. Only 10 buscos were missing and two were fragmented. We also identified a 345–355 bp subtelomeric repeat similar to that observed in H. lupulus (Divashuk et al., 2014) and a 224 bp centromeric repeat (Melters et al., 2013). The percentage of all reads mapping to each repeat is a proxy for their overall abundance in the genome. Consistent with the expectation of extreme abundance of these repeats, 14% of the reads mapped to the subtelomeric repeat and 17% mapped to the centromeric repeat.
CBDRx cannabinoid synthase genes
We conducted a coverage analysis to compare the quality of the CBDRx assembly to other published Cannabis assemblies (e.g. Laverty et al., 2019) with special regard for the genomic position of cannabinoid synthase genes. Coverage analysis confirmed that we identified all cannabinoid synthase gene copies present in the CBDRx assembly (Tables S5, S6). By contrast, genomic data sets for Purple Kush and Finola (Laverty et al., 2019) included unanchored, synthase‐bearing contigs (Tables S7, S8). Only 52% (13/25) of synthase homologs were assembled in Purple Kush, compared with 94% (16/17) in Finola.
In CBDRx, 13 cannabinoid synthase gene sequences and pseudogenes were grouped on chromosome 7 at locations near 26, 29 and 31 Mb (Fig. 2; Tables S5, S9). Cannabinoid synthase homologs positioned in this highly repetitive, pericentromeric region of suppressed recombination were linked in physical and genetic space (Fig. 2). Aside from a solitary CBDAS copy at 31 Mb, the other 12 homologs were located in tandemly repeated arrays consisting of seven and five copies each at 26 and 29 Mb, respectively. Each homolog within a tandem array shared at least 1000 bp of identical upstream sequence. All but two homologs were pseudogenes consisting either of incomplete coding sequence or containing stop codons in the reading frame. A complete copy of cannabichromenic acid synthase (CBCAS), five CBCAS‐like pseudogenes, and a THCAS‐like pseudogene were present in the 26 Mb array (Fig. 3). A complete coding sequence for THCAS was not detected in CBDRx. Among the five homologs in the 29 Mb array, only one had a full‐length open reading frame. This sequence shared only 93% similarity with CBDAS and was nearly identical to what Taura et al. (2007), labeled ‘CBDAS2’. This sequence and its truncated homologs in the 29 Mb array are hereafter referred to as CBDAS‐like.
Fig. 3.
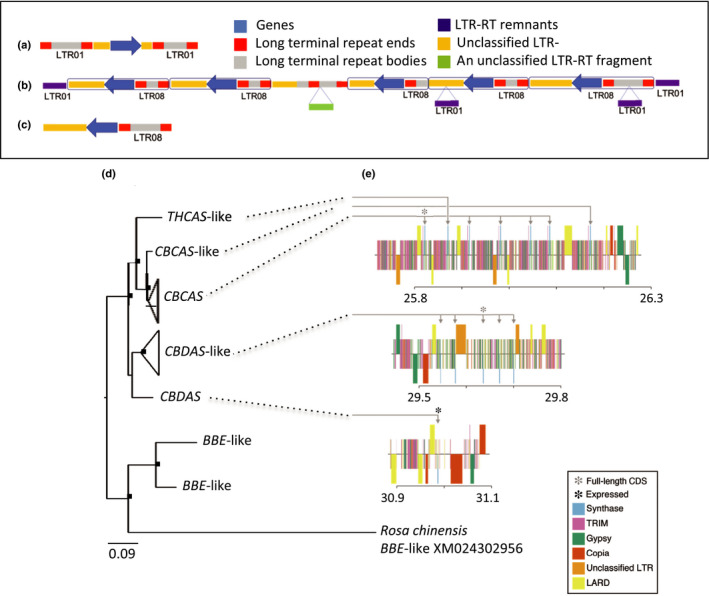
High‐cannabidiol (CBD) cultivar (CBDRx) Cannabis sativa cannabinoid synthase genes located among long terminal repeat retrotransposons (LTR‐RTs). Shown are LTR‐RT ends, LTR‐RT bodies, unclassified LTR‐RTs, LTR‐RT remnants and an unclassified LTR‐RT fragment. Synthase copies and LTR‐RTs occurred in tandemly repeated cassettes at 26 and 29 Mb. (a) Each of seven cassettes in the 26 Mb array consisted of a synthase paralog flanked by a pair of LTR01s. (b) The array at 29 Mb was flanked by a pair of LTR01s and included five cassettes, each consisting of a synthase sequence with a downstream LRT08. (c) The solitary synthase sequence at 31 Mb was similarly associated with a downstream LTR08. (d) CBDRx cannabinoid synthase gene tree rooted with closely related berberine bridge enzyme (BBE‐like) sequences from CBDRx and rose (Rosa). Sequences > 97% similar are collapsed at the tips of the tree. Nodes bearing black squares indicate Bayesian posterior probabilities > 0.90. The classification of paralogs is based on a broader phylogenetic analysis of published cannabinoid synthase sequences (Weiblen et al., 2015). Tandem synthase copies located at 26 Mb comprised a clade including a partial THCAS‐like sequence, a partial CBCAS‐like sequence and five sequences matching CBCAS (Laverty et al., 2019). Four of the five CBCAS sequences in this array were truncated but otherwise identical to the complete gene. Synthase copies at 29 Mb were more closely similar to CBDAS than to THCAS or CBCAS. (e) Functionally annotated maps of the cannabinoid synthase arrays in CBDRx. Synthase arrays are depicted in blue, terminal repeat retrotransposons in miniature (TRIM) in pink, gypsy‐like transposable elements in green, copia‐like transposons in dark orange, unclassified long terminal repeats in light orange, and large retrotransposon derivative elements (LARD) in yellow. Color bands are arbitrarily offset above and below the center line to facilitate visual location of synthase arrays within regions of highly repetitive DNA. Four of the five CBCAS sequences at 26 Mb were incomplete and no expression of the single full‐length CBCAS sequence was detected. At 29 Mb, only one of the CBDAS‐like copies was full‐length and again no expression was detected. Only the solitary CBDAS sequence at 31 Mb was highly expressed in CBDRx.
Each of the three synthase locations consisted of cassettes comprising 31–45 kb tandem repeats nested between LTR‐RTs in regions otherwise riddled with abundant transposable elements (Fig. 3). An LTR‐RT associated with the 26 Mb array (LTR01; gypsy‐related) occurred in great abundance over the entire genome, such that almost every long read had at least some LTR01 sequence. By contrast, a rare LTR‐RT (LTR08; gypsy‐related) was associated with the CBDAS and CBDAS‐like arrays, bearing similarity only to small sequence fragments elsewhere in the genome.
First light genome assemblies
We sequenced three closely related individuals of FL with ONT long reads to examine whether tandemly repeated synthase arrays are present in other Cannabis genomes. The FL plants included two high‐CBD phenotypes and an intermediate phenotype expressing both CBD and THC (Table S1). Contig‐level assemblies were on par or slightly more contiguous than CBDRx, and busco completeness scores confirmed that assembly quality was similar to CBDRx (Table S3). The high‐CBD lines (FL18 and FL49) displayed the same synthase tandem array structure as CBDRx with a CBCAS and THCAS‐like array at 26 Mb, a CBDAS‐like array at 29 Mb and a single CBDAS at 31 Mb (Fig. 4). The co‐location of all three synthase‐containing regions on the same contig in these two assemblies validated our methodological assumption that separate, contig‐bearing arrays are contiguous in CBDRx. However, the assembly of FL48, the intermediate cultivar producing both THC and CBD, unlike the three high‐CBD assemblies, failed to gather CBCAS, CBCAS‐like, THCAS‐like and THCAS sequences into a single contig. Based on the structure of the assembly graph, we attribute this to heterozygosity in this region of FL48 (Fig. 4), and mapping the separate contigs back to CBDRx confirmed that the presumably heterozygous arrays were located in the correct chromosomal locations.
Fig. 4.
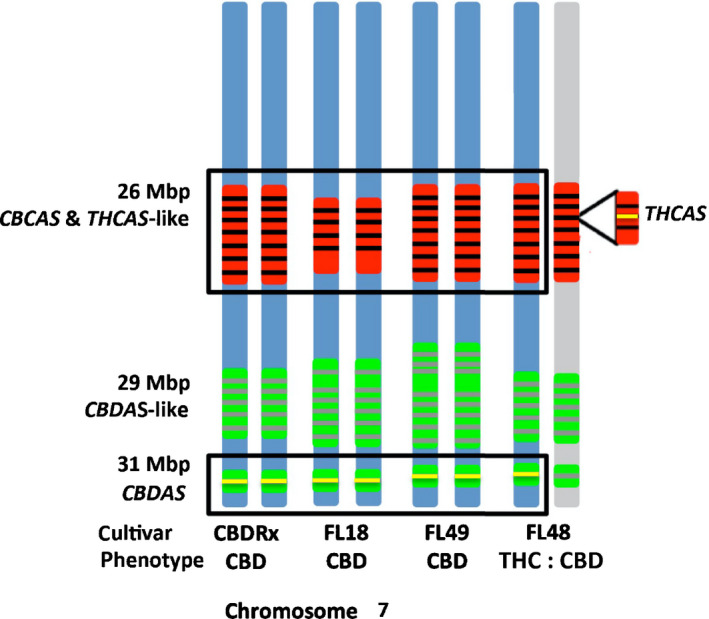
Haplotype diagram of cannabinoid synthase arrays on chromosome 7 in the high‐cannabidiol (CBD) cultivar (CBDRx) and First Light Cannabis sativa genomes. High‐CBD cultivars (CBDRx, FL18, FL49) lacked a functional THCAS at 26 Mb and carried a single functional CBDAS per homologous chromosome at 31Mb. The intermediate cultivar FL48 produced both THC and CBD, and carried both functional CBDAS and THCAS, which are, respectively, identical to those carried by the high‐CBD cultivars and Skunk#1, a high‐THC cultivar (Weiblen et al., 2015). FL48 is heterozygous at the THCAS locus based on coverage analysis, whereas the CBDAS‐like array at 29 Mb assembled on the same contig and is homozygous.
Heterozygosity estimates based on kmer frequency for FL48 (0.58%), CBDRx (0.38%), FL18 (0.50%) and FL49 (0.54%) are consistent with the interpretation of Fig. 4 (Table S3). Despite the highly homozygous condition of each FL genome and CBDRx, FL48 had the greatest heterozygosity. Genotype‐based estimates of heterozygosity from Illumina reads mapped to the assemblies are likewise consistent (Table S3). Genome size estimation plots serve to further illustrate this interpretation (Fig. S5). Unimodal kmer distributions with slight shoulders at lower coverage in CBDRx, FL18 and FL49 suggest the presence of residual heterozygosity in highly inbred cultivars (Fig. S5), whereas a bimodal distribution in FL48 points to greater genome‐wide heterozygosity than in the other three genomes. FL48 could be heterozygous in the vicinity of 26 Mb and hemizygous with a single THCAS‐bearing allele (Fig. 4). The difference between the predicted genome size of CBDRx based on kmer frequency (639 Mb; Table S3) from the physical length of the CBDRx assembly (737 Mb; Table S3) could also be attributed to residual heterozygosity. Although kmer frequency estimates and busco duplication percentages are not interchangeable, they are at least concordant in suggesting low degrees of residual heterozygosity that are also consistent with genotype‐based estimates. Regardless, the completeness of the CBDRx and FL assemblies according to busco was comparable to other published genomes (Table S3).
THCAS and CBDAS gene expression
We confirmed the predicted association between cannabinoid synthase gene expression and cannabinoid phenotypes by examining THC : CBD ratios and full‐length cDNA transcripts from CBDRx and FL cultivars. The close similarity of different synthases and the absence of introns complicates their discrimination by short‐read sequencing technology so we used ONT sequencing to obtain complete transcripts from pistillate flowers of each cultivar, as described in Methods S1. High‐CBD cultivars expressed only the single CBDAS synthase gene located at 31 Mb, whereas the heterozygous intermediate (FL48) expressed both the THCAS gene located at 26 Mb and the 31 Mb CBDAS gene. Copy DNAs for THCAS and CBDAS were inspected to confirm full length, but the open reading frames for CBCAS and CBDAS‐like genes showed no evidence of expression (Fig. 3e).
Genetic map and cannabinoid QTL
A segregating mapping population derived from Skunk#1 marijuana × Carmen hemp was used to associate THC : CBD ratios with the genomic positions of THCAS and CBDAS in CBDRx and FL. The high‐density composite linkage map comprised 10 linkage groups, 1235 total segregating markers, a map distance of 818.6 cM and a mean intermarker distance of 0.66 cM (Fig. 5; Table S10). Each linkage group corresponded to a separate CBDRx chromosome. A single QTL accounting for > 90% of variance in the THC : CBD ratio (r 2 = 0.92) was mapped to the location of the synthase arrays on chromosome 7 (Figs 2e, 5; Table S11). We also identified significant QTLs for the fraction of inflorescence DW composed of CBD and THC, cannabigerol (CBG) and cannabichromene (CBC). The strongest QTLs for CBD (partial r 2 = 0.40) and THC (partial r 2 = 0.52) content were located near the THC : CBD ratio QTL. A single QTL for CBG (r 2 = 0.15) was detected on chromosome 3 and comparably weak QTLs for CBC were located on chromosomes 3, 7 and the X chromosome. We also investigated QTLs for potency defined as total cannabinoid content. Six significant QTLs located on five different chromosomes together accounted for more than half of variance in potency (0.53 = ∑ partial r 2). Although several of the potency QTLs were co‐located with QTL for individual cannabinoids, we did not find experiment‐wise significant (LOD ≥ 4.0) potency association (max LR = 13.9; LOD = 3.02) with the THCAS and CBDAS arrays of chromosome 7.
Fig. 5.
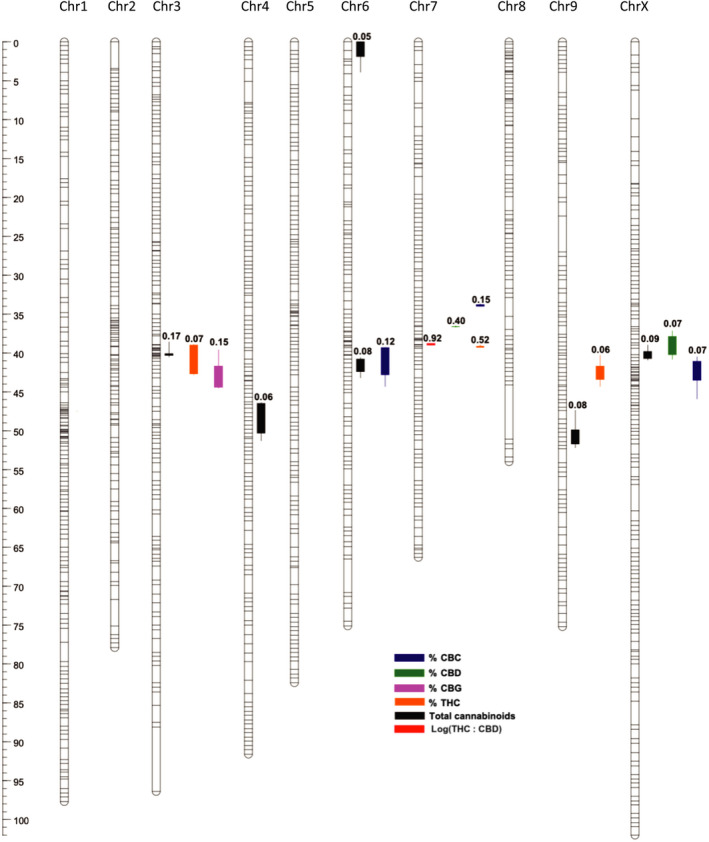
Composite genetic linkage and quantitative trait locus (QTL) map derived from a high‐tetrahydrocannabinol (THC) (Skunk#1) × hemp (Carmen) Cannabis sativa experimental cross. The map consists of 10 linkage groups based on 1175 segregating marker bins drawn from Illumina‐based whole‐genome sequencing (WGS), 48 amplified fragment length polymorphisms, 11 microsatellite markers and one Sanger‐sequenced marker. Ninety‐six F2 female plants were scored for each of 1175 WGS marker bins, while a subset of 62 plants was scored for the other 60 markers (Weiblen et al., 2015). Horizontal lines represent segregating markers along the length of each linkage group. QTLs for 10 phenotypes detected by composite interval mapping (P < 0.05; 1000 permutations) are indicated by vertical bar‐and‐whisker plots (1‐log‐of‐odds (1‐LOD) and 2‐LOD intervals, respectively) to the right of corresponding linkage groups. Partial r 2 for additive and dominance effects are indicated above QTL plots. Bar (at left), genetic distance in cM.
Discussion
CBDRx genome assembly
The genomic positions of CBDAS and THCAS in CBDRx and FL cultivars as assembled from long‐read sequences mapped to the location of a QTL for the THC : CBD ratio in a segregating population derived from Skunk#1 marijuana × Carmen hemp. The high‐density genetic map derived from this segregating population (Weiblen et al., 2015) was further leveraged in assembling long‐read sequences from CBDRx to resolve nine autosomes and the X chromosome. The high quality of the CBDRx assembly is supported by a busco completeness score of 97.2% and a genome size (736 Mb) matching predictions from kmer frequency (Table S3). The number of CBDAS‐like and THCAS‐like paralogs that we identified also matched estimates based on coverage analysis (Tables S5, S6). The CBDRx assembly is highly collinear with published chromosomal assemblies for Finola hemp and Purple Kush marijuana (Fig. S6) but with more genes anchored, an order of magnitude fewer contigs, and higher contig N50 values (Tables S3, S5, S8). However, CBDRx and other available genome assemblies were derived from female (pistillate) plants such that an assembly derived from a male (staminate) plant is yet needed to shed light on the Y chromosome (Kovalchuk et al., 2020).
Genomic structure of cannabinoid synthases in CBDRx
The association of cannabinoid synthases with highly repetitive elements as detected by long‐read sequencing (Tables S6, S7, S8) provides some insight into why these complex gene regions did not assemble previously (van Bakel et al., 2011). Fig. 3(e) illustrates where the synthases are located in CBDRx relative to a variety of repetitive DNA including terminal‐repeat retrotransposons in miniature (TRIM), gypsy‐like transposable elements, copia‐like transposons, large retrotransposon derivative elements (LARD), and numerous unclassified long terminal repeats. Cannabinoid synthase arrays on chromosome 7 of CBDRx consist of multiple paralogs with LTR‐RTs in proximity. That each of the seven paralogs in the 26 Mb array is flanked by pairs of LRT01 remnants (Fig. 3a) suggests a potential mechanism for the independent movement of multiple synthase ‘cassettes’ (Chuong et al., 2017). A pair of LTR01 remnants also flanks the entire 29 Mb array, but each of the five synthase paralogs within it each has only a downstream LTR08 remnant (Fig. 3b). This configuration suggests that a single transposition event might have landed an entire array in the vicinity of 29 Mb.
The solitary CBDAS at 31 Mb has a downstream LTR08 remnant similar to that of the CBDAS‐like paralogs in the 29 Mb array. The presence of LTR08 in CBDAS and CBDAS‐like cassettes could be interpreted as evidence of additional transposition events affecting synthase copy numbers. That each cannabinoid synthase homolog within a tandem array shares at least 1000 bp of identical upstream sequence further suggests that variation in copy number might have arisen by illegitimate recombination and/or the activation and movement of LTR‐RT associated with the synthases (Chuong et al., 2017).
Neither the CBDRx genome nor high‐CBD cultivars FL18 and FL49 possessed a complete coding sequence for THCAS but rather each had a single THCAS‐like pseudogene in the 26 Mb array (Fig. 4). That only the intermediate cultviar FL48 possessed a complete THCAS sequence could be explained by the presence of a single THCAS allele in the hemizygous condition. Along with this interpretation we would expect hemp ancestry in the vicinity of 26 Mb but this was not the case in CBDRx (Fig. 2d). How do we account for the location of the THCAS‐like tandem array 26 Mb in a region of drug‐type ancestry? There could be error associated with the inference of chromosomal ancestry blocks such that our sliding window analysis failed to detect a narrow band of hemp‐type introgression at 26 Mb. The potential for systematic error in the assignment of ancestry blocks has yet to be investigated (Corbett‐Detig & Nielsen, 2017). Alternatively, the absence of THCAS in a region of true marijuana ancestry could be the result of an as yet unknown mechanism or genomic complexity absent from our model (Fig. 4). A recent review suggested that the amount of discord among available C. sativa genome assemblies is evidence of such complexity (Kovalchuk et al., 2020). Although this may be the case, at least the assignment of THCAS and CBDAS to the same chromosome in the Purple Kush and Finola assemblies as updated after the original publication by Laverty et al. (2019) is consistent with the chromosomal placement of the synthases in CBDRx, Skunk#1, Carmen and FL cultivars. Variation in the location and number of cannabinoid synthase arrays on chromosome 7 requires further study.
Cannabinoid synthase activity and the THC : CBD ratio
The assembly of CBDAS and THCAS‐like synthases in three contigs located relatively near each other on CBDRx chromosome 7 (Fig. 2; Table S9) supports a genomic architecture that is consistent with the segregation of THC : CBD ratio phenotypes and putative loci (de Meijer et al., 2003; Weiblen et al., 2015). We sequenced and assembled three additional genomes to evaluate the generality of the model. FL genomes included two CBD‐type cultivars and an intermediate‐type cultivar producing an approximately even ratio of THC : CBD (Table S1). The contig‐level assemblies for FL plants were on a par with or slightly more contiguous than CBDRx (Table S3), had busco completeness scores > 90%, and included cannabinoid synthase arrays with the same genomic architecture as CBDRx. Synthases assembled into one or three contigs in the high‐CBD cultivars, but the cultivar with an intermediate THC : CBD ratio did not assemble well. Detailed annotation of synthase copies indicated that the high‐CBD cultivars carry only one complete CBDAS and lack a complete THCAS sequence (Fig. 4). Consistent with this observation were full‐length cDNA libraries showing expression of but a single full‐length CBDAS and no THCAS expression in each of the three high‐CBD cultivars. By contrast, the intermediate cultivar (FL48) expressed both full‐length CBDAS and THCAS. Difficulty assembling the FL48 genome in the vicinity of 26 Mb is consistent with a potentially hemizygous state where but a single allele of THCAS is present with no alternative allele in the THCAS‐like array. Regardless, genome structure and transcript expression together suggest that, although CBDAS and THCAS are not allelic, the three main cannabinoid profiles can be conferred by the expression of either a single full‐length CBDAS or THCAS, or both in the case of intermediate cultivars.
The close physical linkage of CBDAS and THCAS loci provides a mechanistic explanation for the appearance of single‐locus inheritance observed previously where predominantly CBD, intermediate and predominantly THC profiles segregate 1 : 2 : 1 in the F2 generation of hemp × marijuana experimental crosses (de Meijer et al., 2003). An F2 genetic linkage map derived from the cross of Carmen hemp with Skunk#1 marijuana (Weiblen et al., 2015) is highly collinear with genetics maps for Finola and Purple Kush (Fig. S6d,e). Comparing the F2 map with the CBDRx assembly (Fig. 2c,e), the position of CBDAS and the CBDAS‐like array in CBDRx corresponds to a major QTL for the THC : CBD ratio that accounts for 92% of the trait variance in the F2 population (r 2 = 0.92; Fig. 5). This association and the inferred ancestry of chromosome 7 (Fig. 2d) lend additional support to the interpretation that the genomic segment responsible for the predominance of CBD in CBDRx was introgressed from hemp‐type Cannabis into a marijuana genetic background.
Cannabinoid potency
It is known from other systems that increases in copy number of biosynthetic gene clusters can elevate secondary metabolite production (Manderscheid et al., 2016) and it has been suggested that cannabinoid synthase gene copy numbers might play a role in determining overall cannabinoid content (Vergara et al., 2019). Although multiple copies are present in tandemly repeated arrays in CBDRx, only a single copy of CBDAS was expressed. Also, contrary to the prediction of the copy number hypothesis, none of the six significant QTLs for total cannabinoid content (potency) in a segregating population was associated with the cannabinoid synthase arrays on chromosome 7 (Fig. 5; Table S11). However, a potential marker association near the cannabinoid ratio QTL, at 38.89 cM, did not reach the experiment‐wise threshold for statistical significance (max LR = 13.9, LOD = 3.02, at 31.65 cM). Repeating the potency analysis with additional marker cofactors on chromosome 7 increased the likelihood ratio to 17.95 (LOD = 3.9) but still fell just short of significance.
We might expect genes expressing other metabolic enzymes upstream of THCAS and CBDAS in the cannabinoid pathway to be associated with potency. The hexanoate pathway, the methylerythritol phosphate (MEP) pathway, and the geranyl diphosphate pathway produce essential substrates for cannabinoid synthesis. Previous experimental work identified each of the enzymes involved in these pathways (Laverty et al., 2019). We located these genes in our assemblies and verified their expression with full‐length cDNA libraries (Table S12). We then compared their physical map positions with our genetic map and found two candidate genes proximal to potency QTLs. The gene coding Acyl‐activating enzyme 1 (AAE1), the last enzyme of the hexanoate pathway (Gagne et al., 2012), is located at 39.7 cM on chromosome 3 and a QTL (LOD peak 40.2 cM) associated with 17% of the variance in potency. The gene coding for 4‐hydroxy‐3‐methylbut‐2‐enyl diphosphate reductase (HDR), the last enzyme in the MEP pathway, is located 1.61 cM from a QTL (LOD peak at 40.59 cM) on the X chromosome that is associated with 9% of potency variance. These associations point to potential directions for future studies of mechanisms enhancing cannabinoid expression and selection of this economically important trait.
CBDRx ancestry and the origin of CBD‐type Cannabis
Weiblen et al. (2015) argued that marijuana breeding would favor plants lacking CBDAS. Without the enzyme that competes with THCAS for the same precursor, CBG, the cannabinoid ratio is skewed in favor of intoxicating THC (Onofri et al., 2015). Evidence from dN : dS ratios suggests strong, positive selection for nonfunctional variants of CBDAS in marijuana (Weiblen et al., 2015). Marijuana breeders also possibly selected other independently inherited traits affecting cannabinoid content (potency) such as inflorescence architecture or trichome size (Small & Naraine, 2016). Once highly potent marijuana cultivars were developed (ElSohly et al., 2000) they could be crossed with hemp‐type Cannabis, decoupling the THC : CBD ratio from overall cannabinoid content so that highly potent CBD‐type Cannabis could be selected by introgressing functional CBDAS into marijuana.
Multiple lines of evidence from CBDRx support this scenario. The CBDRx genome is predominantly of marijuana ancestry (Figs 1, S2) and much of the hemp‐derived ancestry in the CBDRx genome is found on chromosome 7 where CBDAS is located (Fig. 2). The lone QTL for the cannabinoid ratio maps to the 31 Mb position of CBDAS on this chromosome (Fig. 2d) and there is population genetic evidence of recent, positive selection in the vicinity (PBS; Figs 2b, S3). These observations are consistent with the interpretation that a CBD‐type cannabinoid profile is the result of introgression of hemp‐like alleles into a drug‐type genetic background to elevate CBD production at the expense of THC. Further evaluation of this hypothesis will require additional genome assemblies from THC‐type C. sativa.
It appears that introgression followed by artificial selection has yielded new types of Cannabis like CBDRx with unprecedented combinations of phenotypic traits, as has been observed in other domesticated plants, including sunflower (Rieseberg et al., 2003). Marijuana and hemp cultivars have a history of independent breeding and reduced gene flow between domesticated populations selected for divergent traits. We suggest that breeders have responded to recent interest in CBD with targeted introgression to produce marijuana cultivars with exceptionally high concentrations of CBD.
Conclusion
We trace the origin of a Cannabis cultivar with elevated CBD content to chromosome 7 where the introgression of CBDAS from hemp into a marijuana background has shifted the predominant cannabinoid from THC to CBD while maintaining an overall quantity of cannabinoids that is typical of drug‐type Cannabis. Cannabinoid synthase gene clusters could be further manipulated, but QTL analysis suggests that other genetic regions on different chromosomes can be targeted to either enhance or reduce potency. It is highly abnormal for a plant to allocate 20–30% of its flowering biomass to one or two specialized metabolites, as do modern Cannabis cultivars. Dissecting this trait will require much additional study. We speculate that integrating cell biology and developmental genetics with existing knowledge of the relevant metabolic pathways will be necessary. Although controlled substance regulations have hindered Cannabis science for decades, economic trends, recent changes in law and the genomic results described here have the potential to accelerate the study of a plant that has coevolved with human culture since the origins of agriculture.
Author contributions
GDW, CJG, and CJS designed the study. GDW, JPW and CD developed the mapping population and prepared materials for genomic analysis. TPM, SGP and STM sequenced the CBDRx genome, FL plants and full‐length cDNA. CJG and TPM assembled, annotated and analyzed the genomes. CJG integrated the maps and analyzed the populations. JPW and CD measured phenotypes and performed QTL analysis. CJG, GDW, CD, JPW, TPM and CJS wrote the manuscript.
Supporting information
Fig. S1 Bioinformatic analysis workflow.
Fig. S2 Genome‐wide ancestry of CBDRx.
Fig. S3 Population branch statistic.
Fig. S4 Hi‐C to CBDRx contact map.
Fig. S5 Kmer genome size estimates for Cannabis lines.
Fig. S6 Chromosome scale alignment of Cannabis genomes, pairwise comparisons of genetic maps, and CBDRx cannabinoid synthase alignments.
Methods S1 Detailed description of plant growth, DNA isolation, genome sequencing, cDNA sequencing and bioinformatic, population branch statistic and comparative genomic analyses.
Table S1 Cannabinoid profiles (% DW) for six Cannabis genomes reported in this study.
Table S2 Mean (SD) cannabinoid content in mature pistillate inflorescences from 96 drug‐type, hemp‐type and intermediate‐type F2 plants as a percentage of total DW.
Table S3 Cannabis genome statistics at the level of sequencing reads, contigs, pseudomolecules, genome size and busco scores.
Table S4 cDNA libraries referenced for annotation.
Table S5 Coverage analysis using sequence reads and the assembled CBDAS and THCAS cassettes.
Table S6 Sequenced Cannabis genomes, data sources, numbers of contigs, depth of coverage, numbers of cannabinoid synthase copies and sequencing methods.
Table S7 Purple Kush (PK) cannabinoid synthase Blast matches (> 82%) for THCAS mRNA (AB057805).
Table S8 Finola (FN) cannabinoid synthase blast matches (>82%) for THCAS mRNA (AB057805).
Table S9 CBDRx cannabinoid synthase blast matches (>82%) for THCAS mRNA (AB057805).
Table S10 Marker density and description of the 10 pseudomolecules and correspondence with the Purple Kush and Finola chromosomes.
Table S11 QTL composite interval mapping results of phenotypic traits.
Table S12 Protein‐coding genes involved in the cannabinoid synthase and precursor pathways.
Please note: Wiley Blackwell are not responsible for the content or functionality of any Supporting Information supplied by the authors. Any queries (other than missing material) should be directed to the New Phytologist Central Office.
Acknowledgements
This work was supported by a David and Lucile Packard Fellowship (GDW), J. Craig Venter Institute (TPM), Sunrise Genetics Inc. and the Minnesota Department of Agriculture. Veronica Tonnell contributed DNA isolation for the mapping population. We thank Matthew Gibbs and Jason Schwartz for support from Sunrise Genetics Inc. and access to the FL germplasm. Timothy Gordon of Functional Remedies provided the high‐CBDA material (CBDRx). We are grateful to the anonymous referees for questions and insights that prompted us to strengthen the manuscript.
Contributor Information
George D. Weiblen, Email: gweiblen@umn.edu.
Todd P. Michael, Email: tmichael@salk.edu.
C. J. Schwartz, Email: gweiblen@umn.edu, Email: tmichael@salk.edu, Email: cj@ihempgene.com.
Data availability
The Cannabis CBDRx chromosome assembly, annotation and genome raw data have been deposited in ENA under project PRJEB29284, GenBank assembly accession GCA_900626175.2, and are also available for download from http://cannabisgenome.org. NCBI has also chosen the CBDRx (cs10) genome as the reference genome for Cannabis and produced a high‐quality gene prediction that is available for users to search, download and visualize at NCBI (https://www.ncbi.nlm.nih.gov/genome/annotation_euk/Cannabis_sativa/100/). Chromosome assemblies of three First Light cultivars have been deposited at CoGe https://genomevolution.org/coge/ under Genome ID FL18 60292, FL48 60293 and FL49 60294. A collection of Bash and Perl scripts documenting the assembly of the CBDRx genome are available via GitHub (https://github.com/grassa/CBDRx).
References
- Alexander DH, Novembre J, Lange K. 2009. Fast model‐based estimation of ancestry in unrelated individuals. Genome Research 19: 1655–1664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badouin H, Gouzy J, Grassa CJ, Murat F, Staton SE, Cottret L, Lelandais‐Briere C, Owens GL, Carrere S, Mayjonade B et al. 2017. The sunflower genome provides insights into oil metabolism, flowering and Asterid evolution. Nature 546: 148–152. [DOI] [PubMed] [Google Scholar]
- van Bakel H, Stout JM, Cote AG, Tallon CM, Sharpe AG, Hughes TR, Page JE. 2011. The draft genome and transcriptome of Cannabis sativa . Genome Biology 12: R102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, Usadel B. 2014. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30: 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broad Institute . 2016. Picard tools. Broad Institute, Github repository. [WWW document] URL: https://github.com/broadinstitute/picard. Accessed November 1, 2018.
- Buchfink B, Xie C, Huson D. 2014. Fast and sensitive protein alignment using DIAMOND. Nature Methods 12: 59–60. [DOI] [PubMed] [Google Scholar]
- Chen N. 2004. Using RepeatMasker to identify repetitive elements in genomic sequences. Current Protocols in Bioinformatics 5: 4.10.1–4.10.14. [DOI] [PubMed] [Google Scholar]
- Chuong EB, Elde NC, Feschotte C. 2017. Regulatory activities of transposable elements: from conflicts to benefits. Nature Reviews Genetics 18: 71–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corbett‐Detig R, Nielsen R. 2017. A hidden Markov model approach for simultaneously estimating local ancestry and admixture time using next generation sequence data in samples of arbitrary ploidy. PLoS Genetics 13: e1006529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dijkstra E. 1959. A note on two problems in connexion with graphs. Numerische Mathematik 1: 269–271. [Google Scholar]
- Divashuk MG, Alexandrov OS, Razumova OV, Kirov IV, Karlov GI. 2014. Molecular cytogenetic characterization of the dioecious Cannabis sativa with an XY chromosome sex determination system. PLoS ONE 9: e85118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ElSohly MA, Ross SA, Mehmedic Z, Arafat R, Yi B, Banahan BF. 2000. Potency trends of delta9‐THC and other cannabinoids in confiscated marijuana from 1980–1997. Journal of Forensic Sciences 45: 24–30. [PubMed] [Google Scholar]
- Fragoso CA, Heffelfinger C, Zhao H, Dellaporta SL. 2016. Imputing genotypes in biallelic populations from low‐coverage sequence data. Genetics 202: 487–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu L, Niu B, Zhu Z, Wu S, Li W. 2012. CD‐HIT: accelerated for clustering the next‐generation sequencing data. Bioinformatics 28: 3150–3152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gagne SJ, Stout JM, Liu E, Boubakir Z, Clark SM, Page JE. 2012. Identification of olivetolic acid cyclase from Cannabis sativa reveals a unique catalytic route to plant polyketides. Proceedings of the National Academy of Sciences, USA 109: 12811–12816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghurye J, Pop M, Koren S, Bickhart D, Chin C. 2017. Scaffolding of long read assemblies using long range contact information. BMC Genomics 18: 527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girgis HZ. 2015. Red: an intelligent, rapid, accurate tool for detecting repeats de‐novo on the genomic scale. BMC Bioinformatics 16: 227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gnerre S, Maccallum I, Przybylski D, Ribeiro FJ, Burton JN, Walker BJ, Sharpe T, Hall G, Shea TP, Sykes S et al. 2011. High‐quality draft assemblies of mammalian genomes from massively parallel sequence data. Proceedings of the National Academy of Sciences, USA 108: 1513–1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graffelman J, Moreno V. 2013. The mid p‐value in exact tests for Hardy‐Weinberg equilibrium. Statistical Applications in Genetics and Molecular Biology 12: 433–448. [DOI] [PubMed] [Google Scholar]
- Grassa CJ, Wenger JP, Dabney C, Poplawski SG, Motley T, Michael TP, Schwartz CJ, Weiblen GD. 2018. A complete Cannabis chromosome assembly and adaptive admixture for elevated cannabidiol (CBD) content. bioRxiv: 458083.
- Haas BJ, Papanicolaou A, Yassour M, Grabherr M, Blood PD, Bowden J, Couger MB, Eccles D, Li B, Lieber M et al. 2013. De novo transcript sequence reconstruction from RNA‐seq using the Trinity platform for reference generation and analysis. Nature Protocols 8: 1494–1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas BJ, Salzberg SL, Zhu W, Pertea M, Allen JE, Orvis J, White O, Buell CR, Wortman JR. 2008. Automated eukaryotic gene structure annotation using EVidenceModeler and the program to assemble spliced alignments. Genome Biology 9: R7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn MW, Zhang SV, Moyle LC. 2014. Sequencing, assembling, and correcting draft genomes using recombinant populations. G3: Genes, Genomes, Genetics 4: 669–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill ST, Sudarsanam R, Henning J, Hendrix D. 2017. HopBase: a unified resource for Humulus genomics. Database 2017: bax009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoff KJ, Stanke M. 2013. WebAUGUSTUS‐a web service for training AUGUSTUS and predicting genes in eukaryotes. Nucleic Acids Research 41: W123–W128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X, Adams MD, Zhou H, Kerlavage AR. 1997. A tool for analyzing and annotating genomic sequences. Genomics 46: 37–45. [DOI] [PubMed] [Google Scholar]
- Iqbal Z, Caccamo M, Turner I, Flicek P, McVean G. 2012. De novo assembly and genotyping of variants using colored de Bruijn graphs. Nature Genetics 44: 226–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwata H, Ninomiya S. 2006. AntMap: constructing genetic linkage maps using an ant colony optimization algorithm. Breeding Science 56: 371–377. [Google Scholar]
- Koren S, Walenz BP, Berlin K, Miller JR, Bergman NH, Phillippy AM. 2017. Canu: scalable and accurate long‐read assembly via adaptive k‐mer weighting and repeat separation. Genome Research 27: 722–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosambi DD. 1944. The estimation of map distances from recombination values. Annals of Eugenics 12: 172–175. [Google Scholar]
- Kovalchuk I, Pellino M, Rigault P, van Velzen R, Ebersbach J, Ashnest JR, Mau M, Schranz ME, Alcorn J, Laprairie RB et al. 2020. The genomics of Cannabis and its close relatives. Annual Review of Plant Biology 71: 713–739. [DOI] [PubMed] [Google Scholar]
- Laetsch DR, Blaxter ML. 2017. BlobTools: Interrogation of genome assemblies [version 1; peer review: 2 approved with reservations]. F1000Research 6: 1287. [Google Scholar]
- Laverty KU, Stout JM, Sullivan MJ, Shah H, Gill N, Holbrook L, Deikus G, Sebra R, Hughes TR, Page JE et al. 2019. A physical and genetic map of Cannabis sativa identifies extensive rearrangements at the THC:CBD acid synthase loci. Genome Research 29: 146–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. 2016. Minimap and miniasm: fast mapping and de novo assembly for noisy long sequences. Bioinformatics 32: 2103–2110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. 2018. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34: 3094–3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R. 2009. Fast and accurate short read alignment with Burrows‐Wheeler transform. Bioinformatics 25: 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, Proc GPD. 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25: 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linthorst J, Hulsman M, Holstege H, Reinders M. 2015. Scalable multi whole‐genome alignment using recursive exact matching. bioRxiv: 022715.
- Lynch RC, Vergara D, Tittes S, White K, Schwartz CJ, Gibbs MJ, Ruthenburg TC, deCesare K, Land DP, Kane NC. 2016. Genomic and chemical diversity in Cannabis . Critical Reviews in Plant Sciences 35: 349–363. [Google Scholar]
- Manderscheid N, Bilyk B, Busche T, Kalinowski J, Paululat T, Bechthold A, Petzke L, Luzhetskyy A. 2016. An influence of the copy number of biosynthetic gene clusters on the production level of antibiotics in a heterologous host. Journal of Biotechnology 232: 110–117. [DOI] [PubMed] [Google Scholar]
- McKernan KJ, Helbert Y, Kane LT, Ebling H, Zhang L, Liu B, Eaton Z, McLaughlin S, Kingan S, Baybayan P et al. 2020. Sequence and annotation of 42 cannabis genomes reveals extensive copy number variation in cannabinoid synthesis and pathogen resistance genes. bioRxiv: 894428.
- de Meijer EPM, Bagatta M, Carboni A, Crucitti P, Moliterni VMC, Ranalli P, Mandolino G. 2003. The inheritance of chemical phenotype in Cannabis sativa L. Genetics 163: 335–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melters DP, Bradnam KR, Young HA, Telis N, May MR, Ruby JG, Sebra R, Peluso P, Eid J, Rank D et al. 2013. Comparative analysis of tandem repeats from hundreds of species reveals unique insights into centromere evolution. Genome Biology 14: R10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merlin MD, Clark RC. 2013. Cannabis: evolution and ethnobotany. Oakland, CA, USA: University of California Press. [Google Scholar]
- Michael TP, Jupe F, Bemm F, Motley ST, Sandoval JP, Lanz C, Loudet O, Weigel D, Ecker JR. 2018. High contiguity Arabidopsis thaliana genome assembly with a single nanopore flow cell. Nature Communications 9: 541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onofri C, de Meijer EPM, Mandolino G. 2015. Sequence heterogeneity of cannabidiolic‐ and tetrahydrocannabinolic acid‐synthase in Cannabis sativa L. and its relationship with chemical phenotype. Phytochemistry 116: 57–68. [DOI] [PubMed] [Google Scholar]
- Pertea M, Pertea GM, Antonescu CM, Chang TC, Mendell JT, Salzberg SL. 2015. StringTie enables improved reconstruction of a transcriptome from RNA‐seq reads. Nature Biotechnology 33: 290–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pruitt KD, Tatusova T, Maglott DR. 2007. NCBI reference sequences (RefSeq): a curated non‐redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Research 35: D61–D65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pya N, Wood SN. 2015. Shape constrained additive models. Statistics and Computing 25: 543–559. [Google Scholar]
- Rieseberg LH, Raymond O, Rosenthal DM, Lai Z, Livingstone K, Nakazato T, Durphy JL, Schwarzbach AE, Donovan LA, Lexer C. 2003. Major ecological transitions in wild sunflowers facilitated by hybridization. Science 301: 1211–1216. [DOI] [PubMed] [Google Scholar]
- Russo EB. 2016. Beyond Cannabis: plants and the endocannabinoid system. Trends in Pharmacological Sciences 37: 594–605. [DOI] [PubMed] [Google Scholar]
- Sawler J, Stout JM, Gardner KM, Hudson D, Vidmar J, Butler L, Page JE, Myles S. 2015. The genetic structure of marijuana and hemp. PLoS ONE 10: e0133292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simao FA, Waterhouse RM, Ioannidis P, Kriventseva EV, Zdobnov EM. 2015. BUSCO: assessing genome assembly and annotation completeness with single‐copy orthologs. Bioinformatics 31: 3210–3212. [DOI] [PubMed] [Google Scholar]
- Sirikantaramas S, Morimoto S, Shoyama Y, Ishikawa Y, Wada Y, Shoyama Y, Taura F. 2004. The gene controlling marijuana psychoactivity: molecular cloning and heterologous expression of Delta1‐tetrahydrocannabinolic acid synthase from Cannabis sativa L. Journal of Biological Chemistry 279: 39767–39774. [DOI] [PubMed] [Google Scholar]
- Small E. 2016. Cannabis: a complete guide. Boca Raton, FL, USA: CRC Press. [Google Scholar]
- Small E, Naraine SGU. 2016. Size matters: evolution of large drug‐secreting resin glands in elite pharmaceutical strains of Cannabis sativa (marijuana). Genetic Resources and Crop Evolution 63: 349–359. [Google Scholar]
- Soorni A, Fatahi R, Haak DC, Salami SA, Bombarely A. 2017. Assessment of genetic diversity and population structure in Iranian Cannabis germplasm. Scientific Reports 7: 15668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang H, Zhang X, Miao C, Zhang J, Ming R, Schnable JC, Schnable PS, Lyons E, Lu J. 2015. ALLMAPS: robust scaffold ordering based on multiple maps. Genome Biology 16: 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taura F, Sirikantaramas S, Shoyama Y, Yoshikai K, Shoyama Y, Morimoto S. 2007. Cannabidiolic‐acid synthase, the chemotype‐determining enzyme in the fiber‐type Cannabis sativa . FEBS Letters 581: 2929–2934. [DOI] [PubMed] [Google Scholar]
- Toth JA, Stack GM, Cala AR, Carlson CH, Wilk RL, Crawford JL, Viands DR, Philippe G, Smart CD, Rose JKC et al. 2020. Development and validation of genetic markers for sex and cannabinoid chemotype in Cannabis sativa L. Global Change Biology Bioenergy 12: 213–222. [Google Scholar]
- Vaser R, Sovic I, Nagarajan N, Sikic M. 2017. Fast and accurate de novo genome assembly from long uncorrected reads. Genome Research 27: 737–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vergara D, Huscher EL, Keepers KG, Givens RM, Cizek CG, Torres A, Gaudino R, Kane NC. 2019. Gene copy number is associated with phytochemistry in Cannabis sativa . AoB PLANTS 11: plz074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker BJ, Abeel T, Shea T, Priest M, Abouelliel A, Sakthikumar S, Cuomo CA, Zeng Q, Wortman J, Young SK et al. 2014. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE 9: e112963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Basten CJ, Zeng ZB. 2006. Windows QTL Cartographer 2.5: North Carolina State University. Raleigh, NC, USA: Department of Statistics. [Google Scholar]
- Weiblen GD, Wenger JP, Craft KJ, ElSohly MA, Mehmedic Z, Treiber EL, Marks MD. 2015. Gene duplication and divergence affecting drug content in Cannabis sativa . New Phytologist 208: 1241–1250. [DOI] [PubMed] [Google Scholar]
- Wick RR, Schultz MB, Zobel J, Holt KE. 2015. Bandage: interactive visualization of de novo genome assemblies. Bioinformatics 31: 3350–3352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright S. 1950. Genetical structure of populations. Nature 166: 247–249. [DOI] [PubMed] [Google Scholar]
- Wu TD, Reeder J, Lawrence M, Becker G, Brauer MJ. 2016. GMAP and GSNAP for genomic sequence alignment: enhancements to speed, accuracy, and functionality. In: Mathé E, Davis S, eds. Statistical genomics: methods and protocols. New York, NY, USA: Springer, New York, 283–334. [DOI] [PubMed] [Google Scholar]
- Xu Z, Wang H. 2007. LTR_FINDER: an efficient tool for the prediction of full‐length LTR retrotransposons. Nucleic Acids Research 35(Web Server issue): W265–W268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi X, Liang Y, Huerta‐Sanchez E, Jin X, Cuo ZX, Pool JE, Xu X, Jiang H, Vinckenbosch N, Korneliussen TS et al. 2010. Sequencing of 50 human exomes reveals adaptation to high altitude. Science 329: 75–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao MY, Jiang HG, Grassa CJ. 2019. Archaeobotanical studies of the Yanghai cemetery in Turpan, Xinjiang, China. Archaeological and Anthropological Sciences 11: 1143–1153. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1 Bioinformatic analysis workflow.
Fig. S2 Genome‐wide ancestry of CBDRx.
Fig. S3 Population branch statistic.
Fig. S4 Hi‐C to CBDRx contact map.
Fig. S5 Kmer genome size estimates for Cannabis lines.
Fig. S6 Chromosome scale alignment of Cannabis genomes, pairwise comparisons of genetic maps, and CBDRx cannabinoid synthase alignments.
Methods S1 Detailed description of plant growth, DNA isolation, genome sequencing, cDNA sequencing and bioinformatic, population branch statistic and comparative genomic analyses.
Table S1 Cannabinoid profiles (% DW) for six Cannabis genomes reported in this study.
Table S2 Mean (SD) cannabinoid content in mature pistillate inflorescences from 96 drug‐type, hemp‐type and intermediate‐type F2 plants as a percentage of total DW.
Table S3 Cannabis genome statistics at the level of sequencing reads, contigs, pseudomolecules, genome size and busco scores.
Table S4 cDNA libraries referenced for annotation.
Table S5 Coverage analysis using sequence reads and the assembled CBDAS and THCAS cassettes.
Table S6 Sequenced Cannabis genomes, data sources, numbers of contigs, depth of coverage, numbers of cannabinoid synthase copies and sequencing methods.
Table S7 Purple Kush (PK) cannabinoid synthase Blast matches (> 82%) for THCAS mRNA (AB057805).
Table S8 Finola (FN) cannabinoid synthase blast matches (>82%) for THCAS mRNA (AB057805).
Table S9 CBDRx cannabinoid synthase blast matches (>82%) for THCAS mRNA (AB057805).
Table S10 Marker density and description of the 10 pseudomolecules and correspondence with the Purple Kush and Finola chromosomes.
Table S11 QTL composite interval mapping results of phenotypic traits.
Table S12 Protein‐coding genes involved in the cannabinoid synthase and precursor pathways.
Please note: Wiley Blackwell are not responsible for the content or functionality of any Supporting Information supplied by the authors. Any queries (other than missing material) should be directed to the New Phytologist Central Office.
Data Availability Statement
The Cannabis CBDRx chromosome assembly, annotation and genome raw data have been deposited in ENA under project PRJEB29284, GenBank assembly accession GCA_900626175.2, and are also available for download from http://cannabisgenome.org. NCBI has also chosen the CBDRx (cs10) genome as the reference genome for Cannabis and produced a high‐quality gene prediction that is available for users to search, download and visualize at NCBI (https://www.ncbi.nlm.nih.gov/genome/annotation_euk/Cannabis_sativa/100/). Chromosome assemblies of three First Light cultivars have been deposited at CoGe https://genomevolution.org/coge/ under Genome ID FL18 60292, FL48 60293 and FL49 60294. A collection of Bash and Perl scripts documenting the assembly of the CBDRx genome are available via GitHub (https://github.com/grassa/CBDRx).