

Abstract
Background
Homozygous and compound heterozygous variants in glucocerebrosidase (GBA) can cause Gaucher disease (GD), whereas heterozygous variants increase the risk of developing Parkinson's disease (PD). GD patients display altered peripheral immune proteins. However, it is unknown if these are altered in GBA carriers with PD.
Objectives
To determine whether plasma cytokines and immune biomarkers associated with GD are also altered in GBA carriers with or without PD.
Methods
Inflammatory cytokines and established GD biomarkers, ferritin, CD162, CCL18, and chitotriosidase (28 biomarkers) were measured in GBA pathogenic variant carriers with (n = 135) and without (n = 83) PD, and non‐carriers with (n = 75) and without PD (n = 77).
Results
PD patients with biallelic pathogenic variants in GBA had elevated plasma levels of ferritin, CCL18, and MIP1α. These biomarkers were not elevated in heterozygous GBA carriers.
Conclusion
GD plasma biomarkers are not promising candidates for stratifying the risk for PD in carriers of heterozygous GBA pathogenic variants. © 2021 The Authors. Movement Disorders published by Wiley Periodicals LLC on behalf of International Parkinson and Movement Disorder Society
Keywords: Parkinson's disease, cytokine, monocyte, inflammation, Gaucher disease, glucocerebrosidase
Homozygous and compound heterozygous mutations in GBA, which encodes the enzyme β‐glucocerebrosidase (GCase), cause the lysosomal storage disorder Gaucher disease (GD). 1 GD patients, as well as heterozygous carriers of pathogenic variants, are at risk for Parkinson's disease (PD).2, 3, 4, 5 The penetrance of pathogenic GBA variants for PD is estimated at 10%–30%, indicating that the majority of mutation carriers will never develop PD.6, 7, 8, 9 Thus, biomarkers that can inform which GBA variant carriers are more likely to develop PD are required. Moreover, a number of strategies targeting the GCase pathway are currently being explored as potential therapeutics for PD,10, 11, 12 so there is much interest in finding biomarkers that can identify efficacious trial compounds.
The link between pathogenic GBA variants and PD highlights the potential role of the immune system in PD. Cells of the reticuloendothelial system (eg, macrophages), are particularly affected in GD. Accumulation of lipids (primarily glucocerebroside) in macrophages results in macrophage dysfunction and increased systemic inflammation in GD patients 13 and in preclinical models.14, 15 Indeed, a number of monocyte activation markers have been validated as biomarkers for monitoring patient responses to GCase enzyme replacement therapy or substrate reduction therapy, the standard treatments for GD. These include CD163, 16 chitotriosidase (CHIT1), 17 CCL18, 18 and IL‐1β. 19 Importantly, monocyte dysfunction and elevated peripheral inflammation have also been reported in PD patients.20, 21, 22
In the present study we used plasma from a cohort of GBA pathogenic variant carriers with and without PD, as well as PD patients without GBA mutations, and matched controls to determine whether cytokines and/or peripheral immune biomarkers associated with GD, were also elevated in carriers of GBA pathogenic variants associated with PD.
Materials and Methods
Samples were obtained from participants of the Spot study at Columbia University Irving Medical Center (CUIMC) and the Icahn School of Medicine at Mount Sinai (ISMMS). Details of patient recruitment and assessment are provided in the supplementary methods. All clinical study procedures were approved by the Columbia University IRB (and ISMMS IRB, if collected at Mount Sinai, NY), and all participants signed informed consent. All samples were shipped to Sydney, Australia on dry ice. All biomarker studies were approved by the University of Sydney Human Research Ethics Committee (2017/076 and 2017/857).
Multiplex Cytokine ELISAs
Bio‐Rad Bio‐Plex Pro Human Cytokine 27‐plex assays (#M500KCAF0Y, 1:4 plasma dilution) were used to measure cytokines and chemokines with details provided in the supplementary methods.
CCL18, CD163, and Ferritin ELISA Assays
Human PARC (CCL18, #EHCCL18, 1:500 plasma dilution), CD163 (#EHCD163, 1:50 plasma dilution), and ferritin (#EHFTL, 1:20 plasma dilution) were measured by enzyme‐linked immunosorbent assay (ELISA) (all Thermo Scientific) with details provided in the supplementary methods.
Chitotriosidase Activity Assay
CHIT1 activity was measured using a BioVision Chitotriosidase Activity Assay Kit (Fluorometric, #K512‐100, 1:4 dilution) with details provided in the supplementary methods.
Statistical Analysis
Statistical analyses were performed using SPSS v25 Statistics software 23 unless otherwise specified. The mean of each replicate sample was determined for each marker and those below the level of detection were given a notional value of 0. Principal component analysis was used to determine clustering of the measured plasma protein profiles. Multivariate analysis of covariance (MANCOVA) was performed on log10 +1 transformed variables and unless otherwise indicated included age, sex, and PD status as covariates. An overall significant effect for MANCOVA analysis was accepted at P < 0.05 using Wilks' Lambda test. Significant MANCOVA effects were followed with pairwise comparison post hoc tests using the estimated marginal means. A Bonferroni corrected P < 0.002 (0.05/28 variables) was applied where appropriate to correct for multiple testing. To detect differences between groups in clinical or demographic variables, one‐way ANOVA with least‐significant difference multiple comparison post hoc tests was used. Spearman's correlations were performed to identify any associations between plasma proteins and clinical data. Graphs were made with Prism (v8.00 GraphPad Software). More specific analysis details are provided in the supplementary methods.
Results
Demographic Comparisons
The demographics and phenotype of GBA carriers with PD (GBA+/PD+, n = 135), GBA carriers without PD (GBA+/PD−, n = 83), non‐carriers with PD (GBA−/PD+, n = 75), and non‐carriers without PD (GBA−/PD−, n = 77) are presented in Table 1. A breakdown of the mutation types is provided in Table S1. The groups were matched by age and sex, and the PD groups were also similar in age at onset, disease duration, levodopa equivalent daily dose, and Unified Parkinson's Disease Rating Scale Part III (UPDRS‐III) scores. The GBA+/PD+ group had a significantly lower Montreal Cognitive Assessment (MoCA) score than all other groups (P = 0.001), indicating greater cognitive dysfunction in this group. Principal component analysis was then used to determine any clustering of the measured plasma proteins across the entire cohort. This did not indicate any clear separation between groups; however, three high inflammatory individuals clearly separated from the cohort majority (Fig. 1A) and were removed from the analysis (see supplementary methods). The absolute values for the 28 proteins that could robustly be detected in plasma for the remaining participants are shown in Table S2.
TABLE 1.
Demographic and clinical characteristic data. Participants were grouped by the presence or absence of either Parkinson's disease or a GBA mutation
| Parameter | GBA−/PD− | GBA+/PD− | GBA−/PD+ | GBA+/PD+ |
|---|---|---|---|---|
| n | 77 | 83 | 75 | 135 |
| Age (yr) | 62.6 ± 1.2 | 62 ± 1.2 | 62.4 ± 1.2 | 64.4 ± 0.9 |
| Sex (M/F) | 40/37 | 32/51 | 37/38 | 84/51 |
| AAO | − | − | 57.5 ± 1.3 | 58.8 ± 0.9 |
| MoCA | 26.8 ± 0.3 | 26.5 ± 0.3 | 26.2 ± 0.4 | 25.1 ± 0.4 a b |
| Education (yr) | 16.7 ± 0.3 | 17.6 ± 0.4 | 17 ± 0.4 | 17 ± 0.3 |
| UPDRS‐III | 1 ± 0.2 | 1.5 ± 0.3 | 17.3 ± 1.2 a | 18.8 ± 1 a |
| LEDD | − | − | 413.8 ± 46.2 | 446.1 ± 33.4 |
Values are presented as mean ± SEM. Data were analyzed by one‐way ANOVA with a least‐significant difference post hoc test except sex, which was analyzed by Kruskal–Wallis followed by Mann–Whitney U.
P < 0.05 compared to the control group.
P < 0.05 compared to the PD group without a GBA mutation.
Abbreviations: PD, Parkinson's disease; M, male; F, female; AAO, age at clinical onset; MoCA, Montreal Cognitive Assessment; UPDRS‐III, Unified Parkinson's Disease Rating Scale Part III; LEDD, levodopa equivalent daily dosage.
FIG. 1.
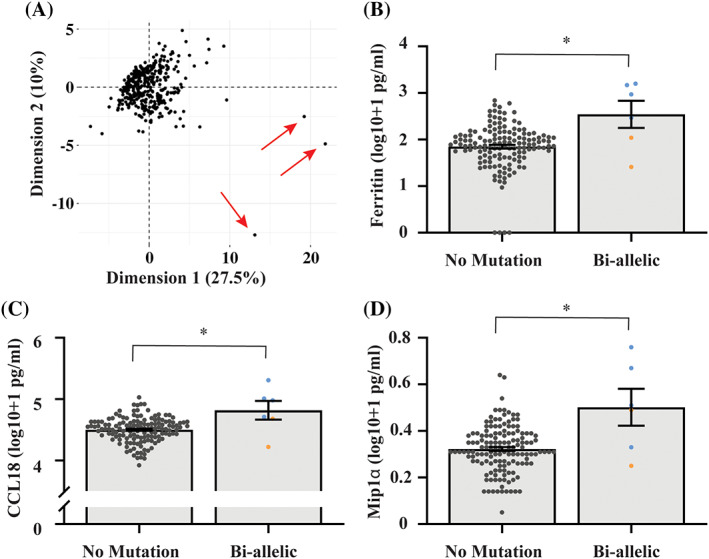
Increased plasma proteins in biallelic GBA mutation carriers. (A) Principal component analysis was performed to determine clustering of the measured 28 plasma proteins (n = 371). Three participants clearly separated from the cohort (indicated with red arrows) due to high expression of multiple inflammatory cytokines and these participants were excluded from downstream analysis. MANCOVA analysis covarying for age and sex was used to compare the 28 measured plasma proteins between biallelic GBA mutation carriers (n = 6) and subjects with no GBA mutation (n = 138). Ferritin (B), CCL18 (C), and MIP1α (D) were significantly higher in biallelic GBA mutation carriers compared to those with no mutation. Homozygous GBA N370S carriers are blue and compound heterozygous GBA carriers are orange. Graphs show individual values and also the mean ± SE. *P ≤ 0.002.
Increased Ferritin, MIP1α, and CCL18 in Biallelic GBA Pathogenic Variant Carriers
Six subjects in the study carried biallelic GBA pathogenic variants, four of which also had a diagnosis of GD, of whom two were receiving enzyme replacement therapy. As anticipated, multivariate analysis covarying for age and sex indicated that biallelic mutations had a significant overall effect on the plasma protein profile compared to no GBA mutation carriers (P = 0.002). Post hoc analysis indicated that ferritin (P = 0.002, Fig. 1B), CCL18 (P = 0.001, Fig. 1C), and MIP1α (P < 0.001, Fig. 1D) were significantly increased in the biallelic group. Chitotriosidase activity was also included in the multivariate analysis and was not increased in the biallelic group (Fig. S1).
No Change in Inflammatory Plasma Biomarkers in Heterozygous GBA Pathogenic Variants Carriers
To determine any differences in the levels of the 27 measured plasma proteins and chitotriosidase activity in heterozygous carriers of GBA mutations, two‐factor MANCOVA analysis using transformed data and covarying for age sex was performed. The results showed that neither PD (P = 0.097) nor GBA mutation status (P = 0.157) were significantly associated with the plasma levels of the measured proteins. The statistical analysis also revealed that there was no interactive or additive effect between GBA mutation status and PD status on the plasma proteins (P = 0.670). Removing the eight GBA mutation cases which also had a LRRK2 mutation from the analysis did not alter the results (all P > 0.05). The same analysis was performed using only heterozygous N370S mutation carriers, with the same result of no significant difference due to either PD (P = 0.108) nor N370S mutation status (P = 0.272).
Correlations Between Inflammatory Plasma Markers and Clinical Variables
Spearman's correlation analysis was performed to determine whether any of the measured plasma proteins correlated to the clinical scores for motor severity and cognitive dysfunction. The UPDRS III score significantly positively correlated to IL4, IL8, MCP1, TNFα, and MIP1α, while the MoCA score significantly negatively correlated to IL17RA, CXCL10, MIP1α, and CCL18, and significantly positively correlated to PDGF (Table S3).
Discussion
In the current study we measured 28 peripheral immune proteins across a cohort of 371 individuals. We found that biallelic GBA mutation carriers, which included four homozygous N370S carriers clinically diagnosed with both PD and GD, had significantly higher plasma levels of ferritin, CCL18, and MIP1α compared to those with no GBA mutation. Hyperferritinemia is commonly observed in GD patients and indicates immune dysregulation,24, 25, 26 and treatment of GD can be effective for decreasing or normalizing ferritin. 25 Likewise, CCL18 released from macrophages is markedly elevated in GD patient plasma, 27 and is useful for monitoring disease severity and response to treatment. 28 In contrast, these proteins were not increased in heterozygous GBA mutation carriers. That neither ferritin, MIP1α, nor CCL18 were increased in heterozygous N370S carriers suggests that the increased plasma levels of these proteins observed in biallelic carriers was due to the presence of GD rather than PD.
Elevated plasma levels of the inflammatory chemokine MIP1α (also known as CCL3) have also been documented in GD patients, 29 and MIP1α was another protein increased in biallelic GBA mutation carriers in the current study. Intriguingly, MIP1α was also strongly correlated with the UPDRS III PD severity scores. A recent study in a large PD cohort indicated that higher plasma levels of MIP1α were associated with faster PD disease progression, 30 and stimulation of peripheral immune cells from PD patients with the inflammatory agonist lipopolysaccharide (LPS) resulted in higher secretion of MIP1α, that again associated with disease severity. 31 Moreover, LPS stimulation of peripheral PD immune cells also results in higher secretion of MCP1, IL8, TNFα, CCL5, and IL‐1β, 31 with MCP1, IL8, and TNFα also correlating with disease severity in the current study. A number of reports have demonstrated an underlying inflammatory phenotype in PD patients; however, the extent of any inflammation certainly varies across studies. 32 Inflammatory medication use, recent illness, or the presence of other comorbid inflammatory diseases can contribute to variability in peripheral cytokine measures and were not recorded in the current study. Furthermore, it is possible that measurement of cytokines in other biofluids (eg, CSF) may provide additional information. Importantly, associations between inflammatory cytokines/chemokines and disease severity measures were independent of GBA pathogenic variant type or presence, and thus more likely a feature of PD in general.
The major strengths of this study are the relatively large number of GBA pathogenic variant carriers and the blinding of the laboratory that measured the cytokines. Our study did not include GD patients without PD however, and thus it could not be determined if the above biomarkers can distinguish between GD patients with and without PD. Also, among all genotypes, only the N370S group was sufficiently large enough to compare carriers with and without PD. Lastly, we did not genotype for CHIT1 mutations, which would affect chitotriosidase plasma levels. 33 Future studies exploring the potential role of plasma chitotriosidase as a PD biomarker should stratify analyses by CHIT1 genotype.
In summary, these results, based on a large number of GBA pathogenic variant carriers, indicate that plasma cytokines and biomarkers used to monitor the severity of GD, are not promising candidates for stratifying the risk of developing PD for carriers of heterozygous GBA pathogenic variants.
Disclosures
Jasmin Galper received scholarship funding from the University of Sydney and Australian Rotary Health. Manisha Balwani has the following declarations: Alnylam Pharma: clinical trial support, advisory board, honoraria, scientific advisory board Acute Hepatic Porphyria registry; Recordati Rare Diseases: honoraria for participation in advisory board; Genzyme/Sanofi: member of the ICGG North American advisory board, honoraria for participation, clinical trial support for enrollment in ICGG registry; Alexion: scientific advisory board member LALD registry, honoraria for participation; Mitsubishi Tanabe: clinical trial support; Takeda/Shire: honoraria for participation in advisory board; Freeline Therapeutics: honoraria for participation in advisory board; Prevail Therapeutics: honoraria for participation in advisory board. Stanley Fahn received consultation fees from Stoparkinson Healthcare Systems, LLC; research support from the Smart Family Foundation; lecture honoraria from the Movement Disorder Society; editor honoraria from Springer Publishers for serving as co‐editor of Current Neurology and Neuroscience Reports, and author royalties from Elsevier Publishers for co‐authorship of the book Principles and Practices of Movement Disorders. Cheryl Waters received research support from Biogen, Roche, and Sanofi; consulting fees from Kyowa, Alexza, and Sunovion; speaker's honoraria from Acadia, Acorda, Adamas, Amneal, Kyowa, Neurocrine, and US WorldMeds. Lynne Krohn has nothing to disclose. Ziv Gan‐Or received consulting fees from Lysosomal Therapeutics Inc. (LTI), Idorsia, Prevail Therapeutics, Inceptions Sciences (now Ventus), Ono Therapeutics, Neuron23, Handl Therapeutics, Denali, Lighthouse, Guidepoint, and Deerfield. Nicolas Dzamko received research support from grant funding from the Michael. J. Fox Foundation, Shake It Up Australia Foundation, and Inventia Life Sciences. Roy N. Alcalay is funded by the NIH, DoD, the Parkinson's Foundation, and the Michael. J. Fox Foundation. He received consultation fees from Sanofi and Janssen.
Author Roles
Study concept and design: N.D., R.N.A.
Subject recruitment, assessment, genotyping and blood collection: M.B., S.F., C.W., L.K., Z.G.‐O., R.N.A.
Biomarker assays: J.G., N.D.
Statistical analysis of data: J.G., N.D., R.N.A.
Initial drafting of manuscript: J.G., N.D., R.N.A.
Editing and approval of submitted manuscript: all authors.
Supporting information
Appendix S1. Supporting Information
Acknowledgments
We acknowledge the Australian Red Cross Blood service for the provision of materials.
Financial disclosure/Conflict of interest: The authors have no financial disclosures or conflicts of interest to report.
Funding Agencies: This project was funded by the Michael J. Fox Foundation and the Shake It Up Australia Foundation (grant# MJFF‐14764) awarded to N.D. and R.N.A. J.G. is funded by a scholarship from Australian Rotary Health and the David Henning Memorial Foundation. The Columbia University cohort (Spot) is supported by the Parkinson's Foundation and the National Institutes of Health (K02NS080915 and UL1 TR000040).
[The copyright line for this article was changed on 16 April 2021, after original online publication.]
Contributor Information
Nicolas Dzamko, Email: nicolas.dzamko@sydney.edu.au.
Roy N. Alcalay, Email: rna2104@cumc.columbia.edu.
References
- 1. Hruska KS, LaMarca ME, Scott CR, Sidransky E. Gaucher disease: mutation and polymorphism spectrum in the glucocerebrosidase gene (GBA). Hum Mutat 2008;29(5):567–583. [DOI] [PubMed] [Google Scholar]
- 2. Neudorfer O, Giladi N, Elstein D, et al. Occurrence of Parkinson's syndrome in type I Gaucher disease. QJM 1996;89(9):691–694. [DOI] [PubMed] [Google Scholar]
- 3. Tayebi N, Walker J, Stubblefield B, et al. Gaucher disease with parkinsonian manifestations: does glucocerebrosidase deficiency contribute to a vulnerability to parkinsonism? Mol Genet Metab 2003;79(2):104–109. [DOI] [PubMed] [Google Scholar]
- 4. Gan‐Or Z, Giladi N, Rozovski U, et al. Genotype‐phenotype correlations between GBA mutations and Parkinson disease risk and onset. Neurology 2008;70(24):2277–2283. [DOI] [PubMed] [Google Scholar]
- 5. Sidransky E, Nalls MA, Aasly JO, et al. Multicenter analysis of glucocerebrosidase mutations in Parkinson's disease. N Engl J Med 2009;361(17):1651–1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Anheim M, Elbaz A, Lesage S, et al. Penetrance of Parkinson disease in glucocerebrosidase gene mutation carriers. Neurology 2012;78(6):417–420. [DOI] [PubMed] [Google Scholar]
- 7. Alcalay RN, Dinur T, Quinn T, et al. Comparison of Parkinson risk in Ashkenazi Jewish patients with Gaucher disease and GBA heterozygotes. JAMA Neurol 2014;71(6):752–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Rana HQ, Balwani M, Bier L, Alcalay RN. Age‐specific Parkinson disease risk in GBA mutation carriers: information for genetic counseling. Genet Med 2013;15(2):146–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. McNeill A, Duran R, Hughes DA, Mehta A, Schapira AH. A clinical and family history study of Parkinson's disease in heterozygous glucocerebrosidase mutation carriers. J Neurol Neurosurg Psychiatry 2012;83(8):853–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sardi SP, Cedarbaum JM, Brundin P. Targeted therapies for Parkinson's disease: from genetics to the clinic. Mov Disord 2018;33(5):684–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Menozzi E, Schapira AHV. Enhancing the activity of glucocerebrosidase as a treatment for Parkinson disease. CNS Drugs 2020;34(9):915–923. [DOI] [PubMed] [Google Scholar]
- 12. Schneider SA, Alcalay RN. Precision medicine in Parkinson's disease: emerging treatments for genetic Parkinson's disease. J Neurol 2020;267(3):860–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mehta A. Epidemiology and natural history of Gaucher's disease. Eur J Intern Med 2006;17:S2–S5. [DOI] [PubMed] [Google Scholar]
- 14. Mizukami H, Mi Y, Wada R, et al. Systemic inflammation in glucocerebrosidase‐deficient mice with minimal glucosylceramide storage. J Clin Invest 2002;109(9):1215–1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Liu J, Halene S, Yang M, et al. Gaucher disease gene GBA functions in immune regulation. Proc Natl Acad Sci U S A 2012;109(25):10018–10023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Moller HJ, de Fost M, Aerts H, Hollak C, Moestrup SK. Plasma level of the macrophage‐derived soluble CD163 is increased and positively correlates with severity in Gaucher's disease. Eur J Haematol 2004;72(2):135–139. [DOI] [PubMed] [Google Scholar]
- 17. Hollak CE, van Weely S, van Oers MH, Aerts JM. Marked elevation of plasma chitotriosidase activity. A novel hallmark of Gaucher disease. J Clin Invest 1994;93(3):1288–1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Raskovalova T, Deegan PB, Yang R, et al. Plasma chitotriosidase activity versus CCL18 level for assessing type I Gaucher disease severity: protocol for a systematic review with meta‐analysis of individual participant data. Syst Rev 2017;6(1):87–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Barak V, Acker M, Nisman B, et al. Cytokines in Gaucher's disease. Eur Cytokine Netw 1999;10(2):205–210. [PubMed] [Google Scholar]
- 20. Grozdanov V, Bliederhaeuser C, Ruf WP, et al. Inflammatory dysregulation of blood monocytes in Parkinson's disease patients. Acta Neuropathol 2014;128(5):651–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ferrari CC, Tarelli R. Parkinson's disease and systemic inflammation. Parkinsons Dis 2011;2011:436813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Nissen SK, Shrivastava K, Schulte C, et al. Alterations in blood monocyte functions in Parkinson's disease. Mov Disord 2019;34(11):1711–1721. [DOI] [PubMed] [Google Scholar]
- 23. IBM Corp . IBM SPSS Statistics for Windows, Version 25.0. Armonk, NY: IBM Corp.; 2017. [Google Scholar]
- 24. Morgan MA, Hoffbrand AV, Laulicht M, Luck W, Knowles S. Serum ferritin concentration in Gaucher's disease. Br Med J (Clin Res Ed). 1983;286(6381):1864–1864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mekinian A, Stirnemann J, Belmatoug N, et al. Ferritinemia during type 1 Gaucher disease: mechanisms and progression under treatment. Blood Cells Mol Dis 2012;49(1):53–57. [DOI] [PubMed] [Google Scholar]
- 26. Regenboog M, van Dussen L, Verheij J, et al. Hepatocellular carcinoma in Gaucher disease: an international case series. J Inherit Metab Dis 2018;41(5):819–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Boot RG, Verhoek M, de Fost M, et al. Marked elevation of the chemokine CCL18/PARC in Gaucher disease: a novel surrogate marker for assessing therapeutic intervention. Blood 2004;103(1):33–39. [DOI] [PubMed] [Google Scholar]
- 28. Raskovalova T, Deegan PB, Mistry PK, et al. Accuracy of chitotriosidase activity and CCL18 concentration in assessing type I Gaucher disease severity. A systematic review with meta‐analysis of individual participant data. Haematologica 2021;106(2):437–445. 10.3324/haematol.2019.236083. Online ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. van Breemen MJ, de Fost M, Voerman JS, et al. Increased plasma macrophage inflammatory protein (MIP)‐1alpha and MIP‐1beta levels in type 1 Gaucher disease. Biochim Biophys Acta 2007;1772(7):788–796. [DOI] [PubMed] [Google Scholar]
- 30. Ahmadi Rastegar D, Ho N, Halliday GM, Dzamko N. Parkinson's progression prediction using machine learning and serum cytokines. NPJ Parkinsons Dis 2019;5:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Reale M, Iarlori C, Thomas A, et al. Peripheral cytokines profile in Parkinson's disease. Brain Behav Immun 2009;23(1):55–63. [DOI] [PubMed] [Google Scholar]
- 32. Dzamko N, Geczy CL, Halliday GM. Inflammation is genetically implicated in Parkinson's disease. Neuroscience 2015;302:89–102. [DOI] [PubMed] [Google Scholar]
- 33. Grace ME, Balwani M, Nazarenko I, Prakash‐Cheng A, Desnick RJ. Type 1 Gaucher disease: null and hypomorphic novel chitotriosidase mutations‐implications for diagnosis and therapeutic monitoring. Hum Mutat 2007;28(9):866–873. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1. Supporting Information