

Abstract
Half a century ago, Harry Angelman reported three patients with overlapping clinical features, now well known as Angelman syndrome. Angelman syndrome is caused by mutations affecting the maternally inherited UBE3A gene, which encodes an E3‐ubiquitin ligase that is critical for typical postnatal brain development. Emerging evidence indicates that UBE3A plays a particularly important role in the nucleus. However, the critical substrates that are controlled by UBE3A remain elusive, which hinders the search for effective treatments. Moreover, given the multitude of signalling mechanisms that are derailed, it is unlikely that targeting a single pathway is going to be very effective. Therefore, expectations are very high for approaches that aim to restore UBE3A protein levels. A particular promising strategy is an antisense oligonucleotide approach, which activates the silenced paternal UBE3A gene. When successful, such treatments potentially offer a disease‐modifying therapy for Angelman syndrome and several other neurodevelopmental disorders.
What this paper adds.
Loss of UBE3A affects multiple signalling pathways in the brain.
Emerging evidence suggests that UBE3A plays a critical role in the cell nucleus.
Trials using antisense oligonucleotides to restore UBE3A levels are continuing.
What this paper adds
Loss of UBE3A affects multiple signalling pathways in the brain.
Emerging evidence suggests that UBE3A plays a critical role in the cell nucleus.
Trials using antisense oligonucleotides to restore UBE3A levels are continuing.
Abbreviations
- ASO
Antisense oligonucleotide
- GABA
γ‐Aminobutyric acid
ANGELMAN SYNDROME: FROM THREE INDIVIDUALS TO A WELL‐CHARACTERIZED SYNDROME
In 1965, the English paediatrician Harry Angelman reported three patients with severe neurodevelopmental delay and overlapping features, now well known as Angelman syndrome. 1 The estimated birth incidence of Angelman syndrome is approximately 1 in 20 0002 and it is characterized by intellectual disability, impaired motor coordination, seizures, characteristic EEG abnormalities, sleep impairments, increased anxiety, lack of speech, and high comorbidity with autism spectrum disorder. 2 The first clinical manifestations usually present during the first year of life, when parents notice the lack of psychomotor activity and seizures. 3 , 4 , 5 Currently, only symptomatic treatments are available for Angelman syndrome, which aim to reduce seizures, improve sleep, or improve behavioural aspects. 6
Angelman syndrome is caused by the loss of UBE3A protein. 7 The UBE3A gene lies in the imprinted Angelman syndrome/Prader–Willi syndrome 15q11.2‐q13 locus, such that in neurons only the maternally inherited UBE3A gene is expressed. Four different genetic causes can lead to loss of functional UBE3A protein in neurons. 8 The most common cause (approximately 70%) represents a de novo deletion of maternal chromosomal region 15q11.2‐q13 encompassing the UBE3A gene. The second frequent cause is a de novo or inherited mutation in the maternal UBE3A gene itself, leading to a loss of functional UBE3A protein. A less frequent cause is paternal uniparental disomy of chromosome 15, which results in decreased UBE3A protein levels owing to two imprinted chromosomes. Similarly, imprinting defects, due to mutations affecting the Prader–Willi syndrome Imprinting Center, affect UBE3A protein expression through repression of the maternal UBE3A gene (Fig. 1).
Figure 1.
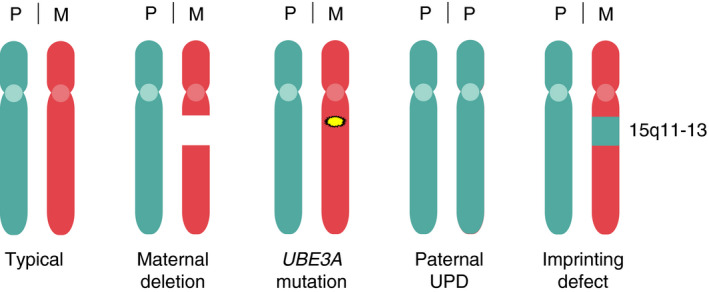
Genetic causes of Angelman syndrome. Representation of the paternal (P) and maternal (M) chromosomes in individuals with Angelman syndrome compared with neurotypical individuals. Note that in all cases the maternally inherited UBE3A gene is affected, either by a deletion of the maternal 15q11‐13 locus, an intragenic mutation in the UBE3A gene, the inheritance of two copies of the paternal chromosome (uniparental disomy [UPD]), or by imprinting defects that repress maternal UBE3A expression.
Some patients with Angelman syndrome‐like phenotypes have been reported who meet (most of) the clinical criteria for having Angelman syndrome but who do not fall into these aforementioned four genetic categories. These patients are typically affected by a mutation in a different gene, and hence represent a different syndrome. 8 , 9 For instance, patients with a HERC2 mutation share many clinical features with those who have Angelman syndrome. 10 Interestingly, it has been shown that HERC2 is a positive regulator of UBE3A activity. 11 It needs to be determined whether the genes that are identified in patients with Angelman syndrome‐like symptoms can also be directly linked to UBE3A function. Importantly, it should be noted that a deviation of the typical Angelman syndrome phenotype does not rule out a bona fide Angelman syndrome mutation. Recent reports have shown that certain UBE3A gene mutations lead to phenotypes that are milder than typical Angelman syndrome, and patients with these do not meet the clinical criteria for Angelman syndrome. 12 , 13 Given these recent findings, it would be useful within the field to discuss whether we should adopt genetic criteria rather than clinical criteria to define patients as having Angelman syndrome.
MOUSE MODELS PROVIDE INSIGHT INTO UBE3A FUNCTION
The identification of the UBE3A gene as the causal gene for Angelman syndrome 7 allowed for the generation of mouse models of Angelman syndrome to study its function. 14 The UBE3A gene encodes the E3 ubiquitin ligase UBE3A (previously identified as E6‐associated protein [E6‐AP] 15 ) which is an enzyme that is responsible for linking ubiquitin molecules to their target proteins. This can either result in the degradation of these target proteins (poly‐ubiquitination) or change the localization or function of target proteins (mono‐ubiquitination).
In humans, the UBE3A gene encodes for three isoforms that are generated by alternative splicing, such that they all display unique amino (N) termini. 16 , 17 The two most abundant human UBE3A isoforms, 1 and 3, make up more than 95% of human UBE3A protein and are conserved in nearly all placental animals. 17 The human UBE3A isoform 2 evolved during early primate evolution and is not found in other mammals. 17 Although the significance and function of the different UBE3A isoforms has yet to be revealed, it is clear that the unique N terminus of the isoforms dictates the localization of UBE3A. 17 , 18 The predominant expression of the nuclear isoforms in both mice and humans explains the highly enriched UBE3A staining observed in these neurons. 16 , 17 , 18 , 19 , 20 Notably, the mouse UBE3A isoform 2 (homologous to human UBE3A isoform 3) that is cytosolic in mice (and given the sequence conservation, probably in most other mammals as well) acquired a mutation during late primate evolution. Hence, in humans and Old World monkeys, this isoform is primarily targeted to the nucleus. 17 The finding that most UBE3A protein is nuclear could indicate that the critical targets of UBE3A might be nuclear as well. In support of that, loss of the cytosolic UBE3A isoform does not result in a discernible phenotype in mice. In contrast, loss of the major nuclear isoform results in typical Angelman syndrome phenotypes in mice and humans, although in humans this is milder than in typical cases of Angelman syndrome. 12 , 18 Although several nuclear targets have been identified 21 and UBE3A function has directly been linked to transcription regulation, 22 , 23 , 24 , 25 it is not clear which nuclear targets are really critical for Angelman syndrome pathophysiology. This greatly hinders the search for UBE3A‐target‐based treatments.
Apart from the nuclear targets that have been identified, several cytosolic targets have been reported. However, similar to the nuclear targets, it is unclear what their relevance is to Angelman syndrome pathophysiology. 14 , 21 , 26 It should be further noted that direct ubiquitination by UBE3A has not been demonstrated for most of the presumed UBE3A targets. Instead, increased levels of target protein in Ube3a mice are often used as an indication that the breakdown of a protein is UBE3A dependent. But if nuclear UBE3A plays a direct or indirect role in gene expression, increased protein levels could also be the result of transcriptional changes. This cautionary note is well illustrated by the observation that the most cited target of UBE3A, the synaptic protein ARC, 27 is no longer considered to be a direct substrate of UBE3A. 28 , 29 Instead, ARC is a target of the ubiquitin ligase TRIAD3A/RNF216 30 and UBE3A probably regulates ARC at a transcriptional level. 29
Several research laboratories have used mouse models of Angelman syndrome (or sometimes induced pluripotent stem cells) to identify novel therapeutics to treat Angelman syndrome. Such therapeutics can be classified into two major categories: (1) targeted treatments aimed at correcting pathophysiological deficits associated with Angelman syndrome; and (2) treatments aimed at restoring the loss of UBE3A expression in the brain.
TARGETED TREATMENTS FOR PATHOPHYSIOLOGICAL DEFICITS ASSOCIATED WITH ANGELMAN SYNDROME
It has been consistently shown that mouse models of Angelman syndrome show marked changes in synaptic plasticity and excitatory/inhibitory balance. 14 Therefore, correcting these changes could provide a great target for treatment. However, over recent years, the view has emerged that these neuronal changes cannot be attributed to just a single mechanism. Several mechanisms have been identified that could underly the changes in neuronal excitability: for instance, changes in the alpha1 subunit of Na/K ATPase, 31 in small‐conductance calcium‐activated potassium channels (SK channels) 32 in calcium and voltage‐dependent big potassium (BK) channels, 33 and in GABA (γ‐aminobutyric acid) transporter (GAT1) levels. 34 Moreover, multiple changes have been observed in signalling cascades that are indirectly critical to neuronal function, such as CAMK2, 35 , 36 Ephexin5‐EphB, 37 neuregulin‐ErbB4, 38 TrkB‐PSD‐95, 39 ERK, 40 mTORC, 41 and dopamine signalling pathways. 42 , 43 , 44 , 45 These widespread changes are consistent with the suggestion that UBE3A probably plays an important global regulatory role, such as transcriptional regulation and/or by regulating (nuclear) proteasome activity to which it is strongly attached. 14 , 18 , 22 , 23 , 24 , 25
In the face of these widespread changes, it is rather surprising that by specifically targeting a single mechanism, an electrophysiological or even a behavioural rescue could be observed in most of the aforementioned studies. 31 , 32 , 33 , 36 , 38 , 39 , 41 It is unclear how these highly specific treatments, which reversed the behavioural phenotypes in mouse models of Angelman syndrome, can be reconciled with the notion that so many mechanisms are affected. We believe that the observed rescue may in part be due to using underpowered studies in combination with behavioural readouts that are only weakly affected in mice with Angelman syndrome. The development of a standardized behavioural test battery of robust phenotypes, and the concomitant power analysis for each test, may allow better drug selection to then be moved to clinical trials. 46 For example, minocycline treatment was successful in a low‐powered study that focused on a limited number of readouts. 47 However, when tested in the standardized behavioural test battery with a sufficiently high number of mice with Angelman syndrome, there was no improvement with minocycline on any of the readouts, 46 nor in clinical trials of patients with Angelman syndrome. 48 Similarly, levodopa treatment did not show an improvement in the Angelman syndrome mouse behavioural test battery nor in a clinical trial. 46 , 49
Given that so many molecular mechanisms seem to be affected, a treatment that acts at a more global level may ultimately be more successful in a clinical trial. Several such treatments were successful in Ube3a mice. For some of these treatments, the precise mechanism targeted in Angelman syndrome is unclear, and it is conceivable that they act more generally as cognitive enhancers (e.g. treatment with reelin, 50 ampakines, 51 a neurogenesis stimulator, 52 or IGF‐2 treatment 53 ). Other drugs target the excitatory/inhibitory imbalance by the GABA modulators, such as ganaxolone and gabaxadol. 34 , 54 The latter drug was able to partly correct motor impairments and anxiety phenotypes in mice with Angelman syndrome. 34 But recent data of a randomized, double‐blind, placebo‐controlled, phase 3 study (Neptune trial, NCT04106557), which enrolled 97 patients treated with gabaxadol (OV101) or placebo, failed to show a change in the primary endpoint (overall score on the Clinical Global Impression‐Improvement‐Angelman syndrome scale). In addition, secondary outcome measures were not significantly changed. These disappointing results indicate that even identifying more broadly acting treatments is still very difficult.
As a cautionary note of the translational failures, it should be pointed out that all drug testing in mice to date has been performed on mouse models of Angelman syndrome that specifically lack Ube3a, whereas most trials have predominantly included patients with 15q11‐13Del Angelman syndrome lacking the entire (maternally inherited) 15q11‐13 gene cluster. The deletion of the additional genes in this locus may make it more difficult to obtain a clinical effect of the tested therapies. Hence, some of the translational failures might be caused by using an inappropriate mouse model. 14 Developing additional mouse models of Angelman syndrome that lack the equivalent genes of the 15q11‐13 gene cluster, or using 15q11‐13Del Angelman syndrome patient‐derived induced pluripotent stem cells, 33 , 55 might circumvent such issues.
TREATMENTS AIMED AT RESTORING UBE3A EXPRESSION IN THE BRAIN
Undoubtedly, the most promising therapeutic approach for treating Angelman syndrome aims at restoring UBE3A expression. Such a therapy would potentially be disease‐modifying and get as close to a cure as possible. There are two ways to achieve this: either by gene therapy that introduces UBE3A protein into the brain through viral vectors, or through activation of the imprinted (silenced) paternal UBE3A allele.
Viral‐mediated delivery of UBE3A in the brain was shown to be partly effective in mice with Angelman syndrome. 56 However, a major translational challenge will be to create safe viral vectors with a good biodistribution that allow UBE3A expression throughout the human brain. Moreover, getting the correct amount of UBE3A per cell is very important, as too much could potentially be detrimental and may result in autism. 57 Last but not least, such a viral vector would preferably express both dominant UBE3A isoforms (or possibly all three isoforms), to ensure that all UBE3A functions are restored. 18
To circumvent these issues, a very elegant approach to re‐express neuronal UBE3A is by activating the dormant UBE3A gene on the paternally inherited chromosome (commonly referred to as ‘unsilencing’). In both humans and mice, the paternally inherited UBE3A allele is silenced owing to the paternal expression of the long UBE3A‐ATS transcript (>460 kilobases in humans, approximately 1000 kilobases in mice) that runs antisense to the UBE3A locus. 58 Elegant experiments in mice have demonstrated that this interferes with the expression of the paternally inherited Ube3a allele 59 , 60 (Fig. 2). Interfering with the synthesis of the UBE3A‐ATS by topoisomerase inhibitors 61 or by Cas9‐mediated targeting 62 induces paternal UBE3A gene expression, without the risk of inducing too much UBE3A expression.
Figure 2.
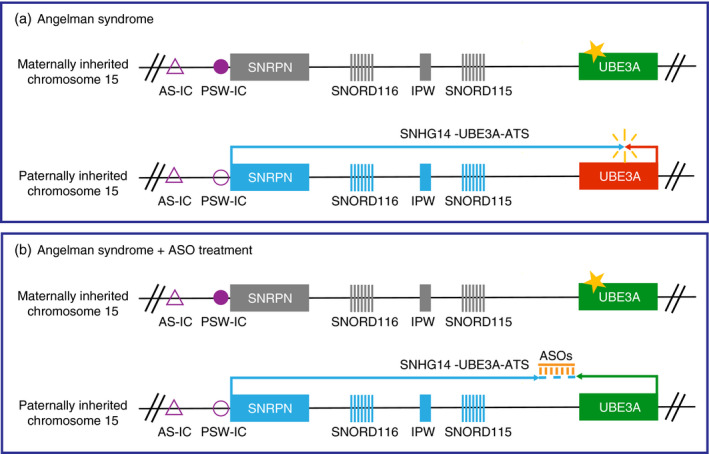
Mechanism of neuronal UBE3A imprinting and antisense oligonucleotide (ASO)‐mediated unsilencing of paternal UBE3A gene expression. (a) Overview of the UBE3A locus in neurons of an individual with Angelman syndrome with a mutation in the UBE3A gene (indicated with a star). Note that most patients with Angelman syndrome carry a maternally inherited deletion of the depicted region extending far beyond the Angelman syndrome Imprinting Center (AS‐IC) and the UBE3A gene. (b) Overview of paternal UBE3A expression in the Angelman syndrome condition upon ASO treatment (orange). Maternally imprinted genes are depicted in grey, the AS‐IC is indicated as an empty purple triangle. The lack of a methylated Prader–Willi syndrome Imprinting Center (PWS‐IC; indicated as an empty purple circle) allows for the transcription of the long non‐coding SNHG14 gene, also known as UBE3A‐ATS, which is responsible for suppressing paternal UBE3A transcription (red rectangle). The administration of ASOs leads to cleavage of the UBE3A‐ATS transcript, resulting in the unsilencing of the paternal UBE3A gene (depicted by a green rectangle), allowing restoration of synthesis of the UBE3A protein.
Alternatively, the UBE3A‐ATS can be targeted by antisense oligonucleotide (ASO) treatment. With this approach, binding of the ASO to the UBE3A‐ATS leads to RNase‐H‐mediated cleavage of the ASO‐RNA heteroduplex, thereby unsilencing the paternal UBE3A gene 63 (Fig. 2). ASO treatment would solve most of the limitations associated with viral injection, especially with respect to controlling the level and the type (isoform) of UBE3A expression as well as its biodistribution. ASOs are readily absorbed by neurons, and given the great success of ASO treatment for spinal muscular atrophy (a motor neuron disorder), such a therapy holds great promise for Angelman syndrome and many other (genetic) neurodevelopmental disorders.
Despite its great therapeutic promise, ASO treatment of mice with Angelman syndrome resulted in a rather disappointing lack of improvement of behavioural phenotypes, despite significant upregulation of UBE3A protein levels in most areas of the brain. 63 These disappointing results may have resulted from treating the mice when they were adult. Subsequent studies showed that the critical period for the full recovery of well‐established Angelman syndrome mouse phenotypes by Ube3a gene reinstatement lies around birth. 64 , 65 This suggests that UBE3A function is critical for typical brain development, which is further highlighted by the observation that deletion of the Ube3a gene in adult mice has little effect. 66 How this critical period in mice with Angelman syndrome relates precisely to the optimal treatment of patients with Angelman syndrome is unknown, but it is noteworthy that there is no critical period for restoring hippocampal plasticity. 64 Since hippocampal plasticity is often used as a cellular proxy for learning and memory formation, this would suggest that gene reinstatement will improve cognitive function, at least to some extent, at any treatment age. 65 , 66 Another important question is whether UBE3A reinstatement is equally beneficial in patients who carry the 15q11‐13 deletion as in those carrying the UBE3A mutation. Owing to the lack of a mouse model of 15q11‐13 deletion, 14 it is difficult to predict to what extent the effect of losing the additional genes in this locus is masked by the severe consequence of losing UBE3A. However, restoring UBE3A in these patients will probably still significantly improve symptoms.
Since the sequence of the UBE3A‐ATS is poorly conserved between mice and humans, the use of patient‐derived induced pluripotent stem cells 67 has been instrumental in identifying efficient ASOs that can be used in the clinic. Currently, two active clinical trials are taking advantage of the ASO‐mediated UBE3A gene reinstatement approach. One trial is sponsored by Hoffmann‐La Roche (molecule RO7248824; NCT04428281) and the other is sponsored by GeneTX Biotherapeutics (molecule GTX‐102; NCT04259281). A third trial (sponsored by Ionis/Biogen) is expected to start soon as well. Since ASOs do not cross the blood–brain barrier, they will need to be administered through intrathecal injections. It is important to note that all current studies are phase I trials, primarily aimed at investigating the safety and tolerability of ASOs, as well as looking into their pharmacokinetic and pharmacodynamic properties. The safety and tolerability aspects are particularly important as these ASOs cannot be tested in unaffected population norm volunteers, owing to the risk of inducing levels of UBE3A that are too high. Notably, all trials make use of different target sequences, as well as different phosphodiester linkages and sugar modifications that stabilize the ASO. 68 Hence, both in terms of efficacy (ASO stability and absorption, UBE3A‐ATS binding and breakdown) and (dose‐related) side effects, these trials may have different outcomes. Needless to say, the results of these ASO trials are eagerly awaited by the families and the medical and scientific communities alike. Success of these trials would not only transform the lives of patients with Angelman syndrome and their families, but could also offer great opportunities for treating many other genetic neurodevelopmental disorders.
Acknowledgements
We thank Ben Distel, Edwin Mientjes, Minetta Elgersma, and Kaitlin Dalby for reading the manuscript and fruitful discussions, Anouk Heuvelmans for creating Figure 1, and the parents and patient organizations Angelman Syndrome Foundation (ASF) and Angelman Syndrome Alliance (ASA) for their continuing support of our research. YE has acted as a paid consultant to Biogen and Hoffmann‐La Roche.
Data Availability Statement
Data sharing not applicable to this article as no datasets were generated or analysed during the current study.
REFERENCES
- 1. Angelman H. “Puppet” children. A report on three cases. Dev Med Child Neurol 1965; 7: 681–8. [DOI] [PubMed] [Google Scholar]
- 2. Mertz LGB, Christensen R, Vogel I, et al. Angelman syndrome in Denmark. Birth incidence, genetic findings, and age at diagnosis. Am J Med Genet A 2013; 161: 2197–203. [DOI] [PubMed] [Google Scholar]
- 3. Williams CA. The behavioral phenotype of the Angelman syndrome. Am J Med Genet C 2010; 154: 432–7. [DOI] [PubMed] [Google Scholar]
- 4. Heus KGCBB, Mous SE, Hooven‐Radstaake M, et al. An overview of health issues and development in a large clinical cohort of children with Angelman syndrome. Am J Med Genet A 2020; 182: 53–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Besten I, Jong RF, Geerts‐Haages A, et al. Clinical aspects of a large group of adults with Angelman syndrome. Am J Med Genet A 2021; 185: 168–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Tan W, Bird LM. Angelman syndrome: current and emerging therapies in 2016. Am J Med Genet C 2016; 172: 384–401. [DOI] [PubMed] [Google Scholar]
- 7. Kishino T, Lalande M, Wagstaff J. UBE3A/E6‐AP mutations cause Angelman syndrome. Nat Genet 1997; 15: 70–3. [DOI] [PubMed] [Google Scholar]
- 8. Buiting K, Williams C, Horsthemke B. Angelman syndrome — insights into a rare neurogenetic disorder. Nat Rev Neurol 2016; 12: 584–93. [DOI] [PubMed] [Google Scholar]
- 9. Tan W‐H, Bird LM, Thibert R, Williams CA. If not Angelman, what is it? A review of Angelman‐like syndromes. Am J Med Genet A 2014; 164: 975–92. [DOI] [PubMed] [Google Scholar]
- 10. Harlalka GV, Baple EL, Cross H, et al. Mutation of HERC2 causes developmental delay with Angelman‐like features. J Med Genet 2013; 50: 65. [DOI] [PubMed] [Google Scholar]
- 11. Kühnle S, Kogel U, Glockzin S, et al. Physical and functional interaction of the HECT ubiquitin‐protein ligases E6AP and HERC2. J Biol Chem 2011; 286: 19410–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Yang Y, Muzny DM, Xia F, et al. Molecular findings among patients referred for clinical whole‐exome sequencing. JAMA 2018; 312: 1870–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Geerts‐Haages A, Bossuyt SNV, Besten I, et al. A novel UBE3A sequence variant identified in eight related individuals with neurodevelopmental delay, results in a phenotype which does not match the clinical criteria of Angelman syndrome. Mol Genet Genom Med 2020; 8: e1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Rotaru DC, Mientjes EJ, Elgersma Y. Angelman syndrome: from mouse models to therapy. Neuroscience 2020; 445: 172–89. [DOI] [PubMed] [Google Scholar]
- 15. Scheffner M, Huibregtse JM, Vierstra RD, Howley PM. The HPV‐16 E6 and E6‐AP complex functions as a ubiquitin‐protein ligase in the ubiquitination of p53. Cell 1993; 75: 495–505. [DOI] [PubMed] [Google Scholar]
- 16. Sirois CL, Bloom JE, Fink JJ, et al. Abundance and localization of human UBE3A protein isoforms. Hum Mol Genet 2020; 29: 3021–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zampeta IF, Sonzogni M, Niggl E, et al. Conserved UBE3A subcellular distribution between human and mice is facilitated by non‐homologous isoforms. Hum Mol Genet 2020; 29: 3032–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Trezza RA, Sonzogni M, Bossuyt SNV, et al. Loss of nuclear UBE3A causes electrophysiological and behavioral deficits in mice and is associated with Angelman syndrome. Nat Neurosci 2019; 22: 1235–47. [DOI] [PubMed] [Google Scholar]
- 19. Burette AC, Judson MC, Burette S, Phend KD, Philpot BD, Weinberg RJ. Subcellular organization of UBE3A in neurons: UBE3A localization in the mouse brain. J Comp Neurol 2017; 525: 233–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Burette AC, Judson MC, Li AN, et al. Subcellular organization of UBE3A in human cerebral cortex. Mol Autism 2018; 9: 54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. LaSalle JM, Reiter LT, Chamberlain SJ. Epigenetic regulation of UBE3A and roles in human neurodevelopmental disorders. Epigenomics 2015; 7: 1213–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Low D, Chen K‐S. Genome‐wide gene expression profiling of the Angelman syndrome mice with Ube3a mutation. Eur J Hum Genet 2010; 18: 1228–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ramamoorthy S, Nawaz Z. E6‐associated protein (E6‐AP) is a dual function coactivator of steroid hormone receptors. Nucl Recept Signal 2008; 6: e006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hokayem JE, Nawaz Z. E6AP in the brain: one protein, dual function, multiple diseases. Mol Neurobiol 2013; 49: 827–39. [DOI] [PubMed] [Google Scholar]
- 25. Furumai R, Tamada K, Liu X, Takumi T. UBE3A regulates the transcription of IRF, an antiviral immunity. Hum Mol Genet 2019; 28: 1947–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sell GL, Margolis SS. From UBE3A to Angelman syndrome: a substrate perspective. Front Neurosci 2015; 9: 322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Greer PL, Hanayama R, Bloodgood BL, et al. The Angelman syndrome protein Ube3A regulates synapse development by ubiquitinating Arc. Cell 2010; 140: 704–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Mandel‐Brehm C, Salogiannis J, Dhamne SC, Rotenberg A, Greenberg ME. Seizure‐like activity in a juvenile Angelman syndrome mouse model is attenuated by reducing Arc expression. Proc Natl Acad Sci U S A 2015; 112: 5129–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kuhnle S, Mothes B, Matentzoglu K, Scheffner M. Role of the ubiquitin ligase E6AP/UBE3A in controlling levels of the synaptic protein Arc. Proc Natl Acad Sci U S A 2013; 110: 8888–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Mabb AM, Je HS, Wall MJ, et al. Triad3A regulates synaptic strength by ubiquitination of Arc. Neuron 2014; 82: 1299–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kaphzan H, Buffington SA, Ramaraj AB, et al. Genetic reduction of the α1 subunit of Na/K‐ATPase corrects multiple hippocampal phenotypes in Angelman syndrome. Cell Rep 2013; 4: 405–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sun J, Zhu G, Liu Y, et al. UBE3A regulates synaptic plasticity and learning and memory by controlling SK2 channel endocytosis. Cell Rep 2015; 12: 449–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sun AX, Yuan Q, Fukuda M, et al. Potassium channel dysfunction in human neuronal models of Angelman syndrome. Science 2019; 366: 1486–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Egawa K, Kitagawa K, Inoue K, et al. Decreased tonic inhibition in cerebellar granule cells causes motor dysfunction in a mouse model of Angelman syndrome. Sci Transl Med 2012; 4: 163ra157. [DOI] [PubMed] [Google Scholar]
- 35. Weeber EJ, Jiang Y, Elgersma Y, et al. Derangements of hippocampal calcium/calmodulin‐dependent protein kinase II in a mouse model for Angelman mental retardation syndrome. J Neurosci 2003; 23: 2634–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. van Woerden GM, Harris KD, et al. Rescue of neurological deficits in a mouse model for Angelman syndrome by reduction of αCaMKII inhibitory phosphorylation. Nat Neurosci 2007; 10: 280–2. [DOI] [PubMed] [Google Scholar]
- 37. Margolis SS, Salogiannis J, Lipton DM, et al. EphB‐mediated degradation of the RhoA GEF ephexin5 relieves a developmental brake on excitatory synapse formation. Cell 2010; 143: 442–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kaphzan H, Hernandez P, Jung JI, et al. Reversal of impaired hippocampal long‐term potentiation and contextual fear memory deficits in Angelman syndrome model mice by ErbB inhibitors. Biol Psychiatry 2012; 72: 182–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Cao C, Rioult‐Pedotti MS, Migani P, et al. Impairment of TrkB‐PSD‐95 signaling in Angelman syndrome. PLoS Biol 2013; 11: e1001478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Filonova I, Trotter JH, Banko JL, Weeber EJ. Activity‐dependent changes in MAPK activation in the Angelman syndrome mouse model. Learn Memory 2014; 21: 98–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sun J, Liu Y, Tran J, O’Neal P, Baudry M, Bi X. mTORC1–S6K1 inhibition or mTORC2 activation improves hippocampal synaptic plasticity and learning in Angelman syndrome mice. Cell Mol Life Sci 2016; 73: 4303–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Mulherkar SA, Jana NR. Loss of dopaminergic neurons and resulting behavioural deficits in mouse model of Angelman syndrome. Neurobiol Dis 2010; 40: 586–92. [DOI] [PubMed] [Google Scholar]
- 43. Farook MF, DeCuypere M, Hyland K, Takumi T, LeDoux MS, Reiter LT. Altered serotonin, dopamine and norepinepherine levels in 15q duplication and Angelman syndrome mouse models. PLoS ONE 2012; 7: e43030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Steinkellner T, Yang J‐WW, Montgomery TR, et al. Ca2+/calmodulin‐dependent protein kinase IIα (αCaMKII) controls the activity of the dopamine transporter: implications for Angelman syndrome. J Biological Chem 2012; 287: 29627–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Riday TT, Dankoski EC, Krouse MC, et al. Pathway‐specific dopaminergic deficits in a mouse model of Angelman syndrome. J Clin Invest 2012; 122: 4544–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Sonzogni M, Wallaard I, Santos SS, et al. A behavioral test battery for mouse models of Angelman syndrome: a powerful tool for testing drugs and novel Ube3a mutants. Mol Autism 2018; 9: 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Grieco JC, Ciarlone SL, Gieron‐Korthals M, et al. An open‐label pilot trial of minocycline in children as a treatment for Angelman syndrome. BMC Neurology. 2014; 14: 232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ruiz‐Antorán B, López AS, Cazorla R, et al. Randomized clinical trial, placebo compared to evaluate the efficacy and safety of minocycline in Angelman syndrome (A‐Manece Study). Clin Ther 2015; 37: e154–5. [Google Scholar]
- 49. Tan W, Bird LM, Sadhwani A, et al. A randomized controlled trial of levodopa in patients with Angelman syndrome. Am J Med Genet A 2017; 176: 1099–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Hethorn WR, Ciarlone SL, Filonova I, et al. Reelin supplementation recovers synaptic plasticity and cognitive deficits in a mouse model for Angelman syndrome. Eur J Neurosci 2015; 41: 1372–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Baudry M, Kramar E, Xu X, et al. Ampakines promote spine actin polymerization, long‐term potentiation, and learning in a mouse model of Angelman syndrome. Neurobiol Dis 2012; 47: 210–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Liu Y, Johe K, Sun J, et al. Enhancement of synaptic plasticity and reversal of impairments in motor and cognitive functions in a mouse model of Angelman syndrome by a small neurogenic molecule, NSI‐189. Neuropharmacology 2018; 144: 337–44. [DOI] [PubMed] [Google Scholar]
- 53. Cruz E, Descalzi G, Steinmetz A, Scharfman HE, Katzman A, Alberini CM. CIM6P/IGF‐2 receptor ligands reverse deficits in Angelman syndrome model mice. Autism Res 2020. 10.1002/aur.2418. Online ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Ciarlone SL, Wang X, Rogawski MA, Weeber EJ. Effects of the synthetic neurosteroid ganaxolone on seizure activity and behavioral deficits in an Angelman syndrome mouse model. Neuropharmacology 2016; 116: 142–50. [DOI] [PubMed] [Google Scholar]
- 55. Fink JJ, Robinson TM, Germain ND, et al. Disrupted neuronal maturation in Angelman syndrome‐derived induced pluripotent stem cells. Nat Commun 2017; 8: 15038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Daily JL, Nash K, Jinwal U, et al. Adeno‐associated virus‐mediated rescue of the cognitive defects in a mouse model for Angelman syndrome. PLoS ONE 2011; 6: e27221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Elgersma Y. A molecular tightrope. Nature 2015; 526: 50–1. [DOI] [PubMed] [Google Scholar]
- 58. Runte M, Kroisel PM, Gillessen‐Kaesbach G, et al. SNURF‐SNRPN and UBE3A transcript levels in patients with Angelman syndrome. Hum Genet 2004; 114: 553–61. [DOI] [PubMed] [Google Scholar]
- 59. Meng L, Person RE, Beaudet AL. Ube3a‐ATS is an atypical RNA polymerase II transcript that represses the paternal expression of Ube3a. Hum Mol Genet 2012; 21: 3001–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Meng L, Person RE, Huang W, Zhu PJ, Costa‐Mattioli M, Beaudet AL. Truncation of Ube3a‐ATS unsilences paternal Ube3a and ameliorates behavioral defects in the Angelman syndrome mouse model. PLoS Genet 2013; 9: e1004039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Huang H‐S. Topoisomerase inhibitors unsilence the dormant allele of Ube3a in neurons. Nature 2012; 481: 185–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Wolter JM, Mao H, Fragola G, et al. Cas9 gene therapy for Angelman syndrome traps Ube3a‐ATS long non‐coding RNA. Nature 2020; 587: 281–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Meng L, Ward AJ, Chun S, Bennett CF, Beaudet AL, Rigo F. Towards a therapy for Angelman syndrome by targeting a long non‐coding RNA. Nature 2015; 518: 409–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Silva‐Santos S, van Woerden GM, Bruinsma CF, et al. Ube3a reinstatement identifies distinct developmental windows in a murine Angelman syndrome model. J Clin Invest 2015; 125: 2069–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Sonzogni M, Zhai P, Mientjes EJ, van Woerden GM, Elgersma Y. Assessing the requirements of prenatal UBE3A expression for rescue of behavioral phenotypes in a mouse model for Angelman syndrome. Mol Autism 2020; 11: 70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Sonzogni M, Hakonen J, Kleijn MB, et al. Delayed loss of UBE3A reduces the expression of Angelman syndrome‐associated phenotypes. Mol Autism 2019; 10: 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Chamberlain SJ, Chen P‐F, Ng KY, et al. Induced pluripotent stem cell models of the genomic imprinting disorders Angelman and Prader‐Willi syndromes. Proc Natl Acad Sci U S A 2010; 107: 17668–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Rinaldi C, Wood MJA. Antisense oligonucleotides: the next frontier for treatment of neurological disorders. Nat Rev Neurol 2018; 14: 9–21. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing not applicable to this article as no datasets were generated or analysed during the current study.