

Summary
Kelps are dominant primary producers in temperate coastal ecosystems. Large amounts of kelp biomass can be exported to the seafloor during the algal growth cycle or following storms, creating new ecological niches for the associated microbiota. Here, we investigated the bacterial community associated with the kelp Laminaria hyperborea during its accumulation and degradation on the seafloor. Kelp tissue, seawater and sediment were sampled during a 6‐month in situ experiment simulating kelp detritus accumulation. Evaluation of the epiphytic bacterial community abundance, structure, taxonomic composition and predicted functional profiles evidenced a biphasic succession. Initially, dominant genera (Hellea, Litorimonas, Granulosicoccus) showed a rapid and drastic decrease in sequence abundance, probably outcompeted by algal polysaccharide‐degraders such as Bacteroidia members which responded within 4 weeks. Acidimicrobiia, especially members of the Sva0996 marine group, colonized the degrading kelp biomass after 11 weeks. These secondary colonizers could act as opportunistic scavenger bacteria assimilating substrates exposed by early degraders. In parallel, kelp accumulation modified bacterial communities in the underlying sediment, notably favouring anaerobic taxa potentially involved in the sulfur and nitrogen cycles. Overall, this study provides insights into the bacterial degradation of algal biomass in situ, an important link in coastal trophic chains.
Introduction
Brown macroalgae of the order Laminariales, collectively known as kelps, are crucial foundation species that thrive in coastal regions of temperate and Arctic seas worldwide. Kelps feature some of the highest growth rates and primary productivity on the planet (Mann, 1973). Kelp biomass thus represents an abundant standing stock of organic matter, primarily composed of polysaccharides and proteins that account for ca. 50% and 3%–15% of the dry weight, respectively (Kloareg and Quatrano, 1988; Fleurence, 1999). Up to 82% of the kelp primary production can be exported as dissolved or particulate organic matter to the shores or neighbouring intertidal, subtidal and benthic zones (Krumhansl and Scheibling, 2012). This may be due to the drift of naturally detached old blades as well as the fragmentation or dislodging of kelps caused by hydrodynamic forces such as strong storm events (Krumhansl and Scheibling, 2012; de Bettignies et al., 2015; Krause‐Jensen and Duarte, 2016). This massive input of organic matter can strongly influence the stranding ecosystems, creating new niches for meiofauna and microorganisms (Duarte and Cebrián, 1996). This is particularly relevant in benthic subtidal habitats where sediments are otherwise devoid of kelps. The turnover of algal biomass largely depends on bacterial degradation processes (Mann, 1982; Newell et al., 1982) and is influenced by several physico‐chemical parameters. For example, small fragments are degraded faster, and degradation is favoured when temperature and hydrodynamics increase (Krumhansl and Scheibling, 2012).
Live macroalgae host diverse and abundant bacterial communities (Burke et al., 2011), with about 106 to 109 bacterial cells.cm−2 on algal surfaces (Martin et al., 2014). Bacteria play an important role in algal health and physiology: they can provide vitamins to the host, defend the host against pathogens via antibiotic production, or degrade algal tissues (Egan et al., 2013, 2014; Minich et al., 2018). Epiphytic bacterial communities have been thoroughly characterized for an increasing number of kelp species and generally comprise large proportions of Alphaproteobacteria, Gammaproteobacteria, Bacteroidetes, Verrucomicrobia and Planctomycetes (Bengtsson and Øvreås, 2010; Lemay et al., 2018). Studies further revealed that kelp epiphytic communities are stable over large spatial scales (Marzinelli et al., 2015), can be species‐specific (Lemay et al., 2018), depend on the age of algal tissues (Weigel and Pfister, 2019; Ramirez‐Puebla et al., 2020) and follow reproducible seasonal successions that may be explained by water temperature (Bengtsson et al., 2010; Minich et al., 2018). In addition, physiological parameters varying at the algal surface may impact communities. Oxygen release by the alga can promote the growth of aerobic bacteria but also harm epiphytic bacteria by producing reactive oxygen species (ROS) (Goecke et al., 2010; Egan et al., 2013). Kelps may also control resident or pathogenic microorganisms by secreting secondary metabolites such as halogenated compounds (Egan et al., 2014). In contrast to epiphytic communities on intact macroalgae, the specific bacterial communities colonizing detached kelps and their succession are still understudied. Total microflora has been determined essentially from kelp detritus stranded on intertidal areas (Koop et al., 1982; Bouvy et al., 1986; Delille and Perret, 1991). Even less information is available on the taxonomy, function and succession of bacterial communities colonizing large submersed accumulations of kelp fragments on benthic subtidal habitats. Furthermore, kelp accumulation and degradation processes are also expected to influence sediment communities, which are known to harbour contrasted and more diverse microbial communities compared to the overlying water column (Gobet et al., 2012).
We previously reported an experimental approach to mimic dislodgement and accumulation of the kelp Laminaria hyperborea on a subtidal sandy bottom ecosystem of the north‐western coast of Brittany, France (de Bettignies et al., 2020). We showed that algal biomass decomposition started after 2 weeks and reached a critical step after 11 weeks, with an increase in respiration rate and phlorotannin content. After 24 weeks, 85% of the algal biomass was lost. Using samples collected during the same experiment, we investigated here the changes in bacterial community abundance and composition and predicted functions associated with the in situ degradation of the kelp biomass over 6 months, in four compartments: L. hyperborea's tissues, sediments directly underneath the experimental cages (hereafter ‘underlying sediment’), control sediments 2 m away from the cages and devoid of kelp tissues (hereafter ‘external sediment’), and the overlying seawater 1 m above the cages. We tested the following hypotheses: (i) the bacterial diversity and community structure are specific to a given compartment; (ii) there is a succession of different bacterial groups associated with the degradation of L. hyperborea's tissues; and (iii) bacterial communities in the underlying sediment are affected by the degrading kelp tissues and different from the external sediment.
Results
Fluctuations and diversity patterns of the bacterial community during kelp degradation
The characteristics of the 92 samples analysed in this study are listed in the Supporting Information Table S1. Total bacterial abundance was estimated by quantitative PCR in each compartment using 16S rRNA gene copy number as a proxy. The average number of 16S rRNA gene copies on kelp surfaces was initially 2.27 × 108 copies.cm−2. Considering an average of four copies of 16S rRNA gene per bacterial cell (Větrovský and Baldrian, 2013), it corresponds to ca. 6 × 107 cells.cm−2. It decreased significantly to 5.65 × 107 copies.cm−2 (ca. 1.4 × 107 cells.cm−2) during the first 2 weeks and remained stable afterwards [analysis of variance (ANOVA), F 7,16 = 7.37, P < 0.001; Fig. 1A]. Over the 6 months, 16S rRNA gene copy number significantly increased in the surrounding seawater (ANOVA, F 7,16 = 4.02, P = 0.010), the underlying sediment (ANOVA F 7,16 = 3.83, P = 0.012) and to a lesser extent in the external sediment (ANOVA F 7,14 = 4.32, P = 0.010) (Fig. 1B and C). Notably, the number of 16S rRNA gene copies was similar between the two sediment compartments the first 2 weeks, but the increase tended to be stronger in the underlying sediment (4.6‐fold change between 0 and 24 weeks) than in the external sediment (1.7‐fold change).
Fig 1.
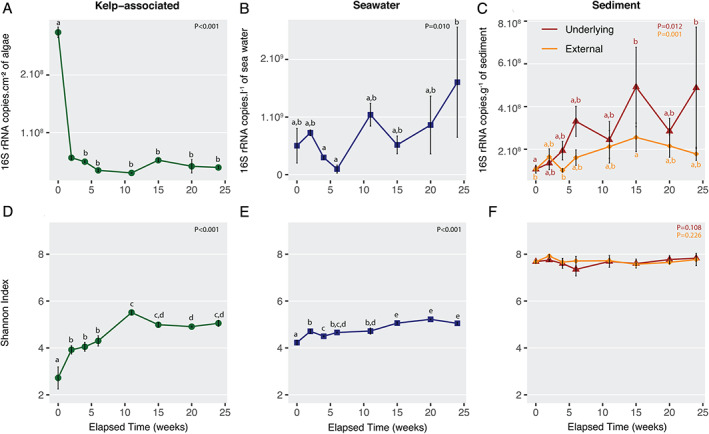
Abundance and diversity estimates of the bacterial community in the four compartments. Fluctuation of bacterial 16S rRNA gene copy number in the kelp‐associated (A), the water column (B), and the underlying and the external sediment (C) compartments, and of the Shannon index in the kelp‐associated (D), the water column (E), and the underlying and the external sediment (F) compartments during the degradation of L. hyperborea. ANOVA analysis was conducted to compare diversity indices over time. When significant ANOVA results were found (P < 0.05), a post hoc Tukey HSD test was calculated. Accordingly, different letters indicate significant differences between sampling dates. Values are mean ± standard deviation (n = 3, except n = 2 at 0 week in kelp‐associated and 11 weeks in external sediment for Shannon index and at 2 and 11 weeks in external sediment for 16S rRNA copy number). [Color figure can be viewed at wileyonlinelibrary.com]
Alpha‐diversity indices were two to three times higher in sediment samples compared to kelp‐associated and water column samples (Fig. 1D and F; Supporting Information Fig. S1). The OTU richness and the Shannon index from kelp‐associated samples increased significantly in the first 11 weeks (OTU richness from 351–493 to 2090–2312, Shannon index from 2.4–3.1 to 5.4–5.6; ANOVA, F 7,15 = 44.68, P < 0.001 and F 7,15 = 54.78, P < 0.001, respectively) before reaching a plateau. Bacterial diversity slightly increased in the water column throughout the experiment (ANOVA, F 7,16 = 48.73, P = 0.001 for OTU richness and F 7,16 = 62.50, P = 0.001 for Shannon index) while it stayed stable in the underlying and the external sediment samples (P > 0.05). Further, sample compartment showed a strong impact on the bacterial community structure (permutational analysis of variance PERMANOVA, F 3,84 = 35.77, P < 0.001, Fig. 2A). Pairwise comparisons showed significant dissimilarities between each compartment (P = 0.001), although less pronounced between external and underlying sediments (Supporting Information Table S2). A higher dispersion was visible on the principal coordinates analysis (PCoA) within the algal samples (mean dispersion: 0.60) compared to the other compartments (seawater 0.55, underlying sediment 0.56, external sediment 0.53; P < 0.001). Only 1 946 OTUs, i.e. 2.2% of the total number of OTUs were shared between the four compartments (Fig. 2B). The sequences contained in this core set of OTUs are present in different proportions of the complete dataset in each compartment, they represented 12% of the sequence abundance in kelp‐associated samples, 17% in seawater, 12% in underlying sediment and 7% in external sediment. In contrast, 58.2% (51 344 OTUs) of the total number of OTUs were specific to a given compartment, but they represented only 10% of the total number of sequences in the dataset.
Fig 2.
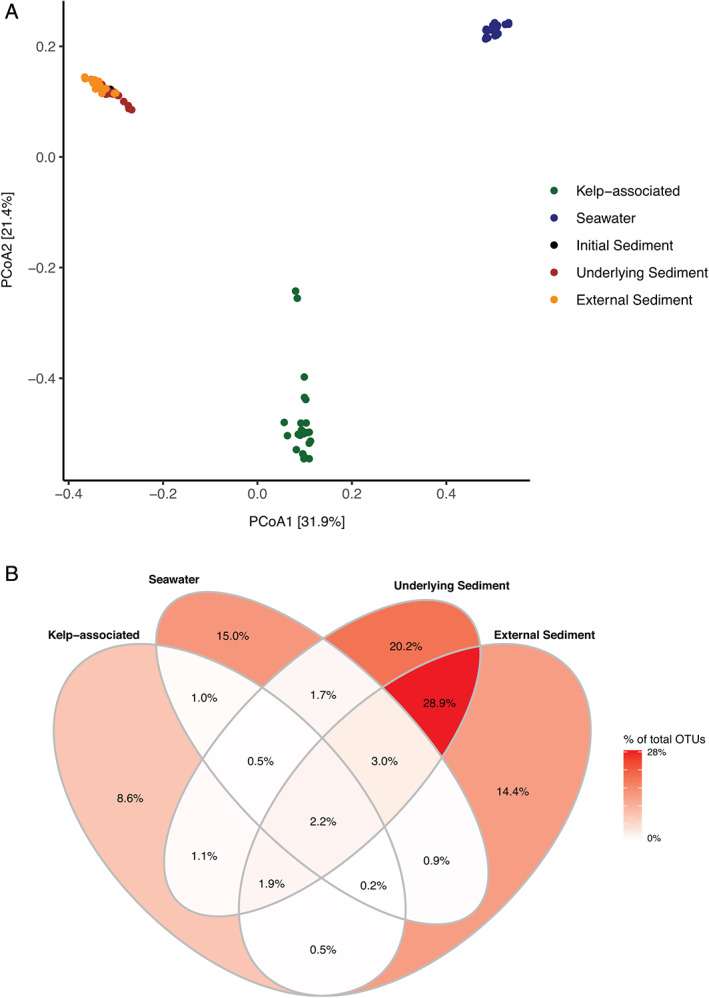
Bacterial community structure in the four compartments during the degradation of detached L. hyperborea. A. Two‐dimensional representation of a PCoA calculated from the Bray–Curtis dissimilarity index. The plot represents samples from all time points. B. Proportion of OTUs unique to one compartment or shared between compartments, calculated from the total number of 88 194 OTUs in the dataset. [Color figure can be viewed at wileyonlinelibrary.com]
Succession of epiphytic bacteria on kelp tissues
The structure of the kelp‐associated bacterial community was impacted by the elapsed degradation time (PERMANOVA, F 7,15 = 6.33, P = 0.001), showing distinct groups for freshly detached kelps (week 0) and samples collected between weeks 2–6 or 11–24 (Fig. 3A). Pairwise ANOVA comparisons confirmed that samples from weeks 2–6 significantly differed from samples from weeks 11–24 (F 1,19 = 13.46, P = 0.003). Although visible on the ordination plot, the difference between samples from week 0 and the other groups could not be reliably tested due to one missing sample. Constrained ordination using distance‐based redundancy analysis (db‐RDA) showed that four environmental parameters were significant explanatory variables, namely remaining biomass (F 1,15 = 11.09, P = 0.001), respiration (F 1,15 = 6.35, P = 0.001), temperature (F 1,15 = 3.80, P = 0.002) and light (F 1,15 = 2.39, P = 0.025).
Fig 3.
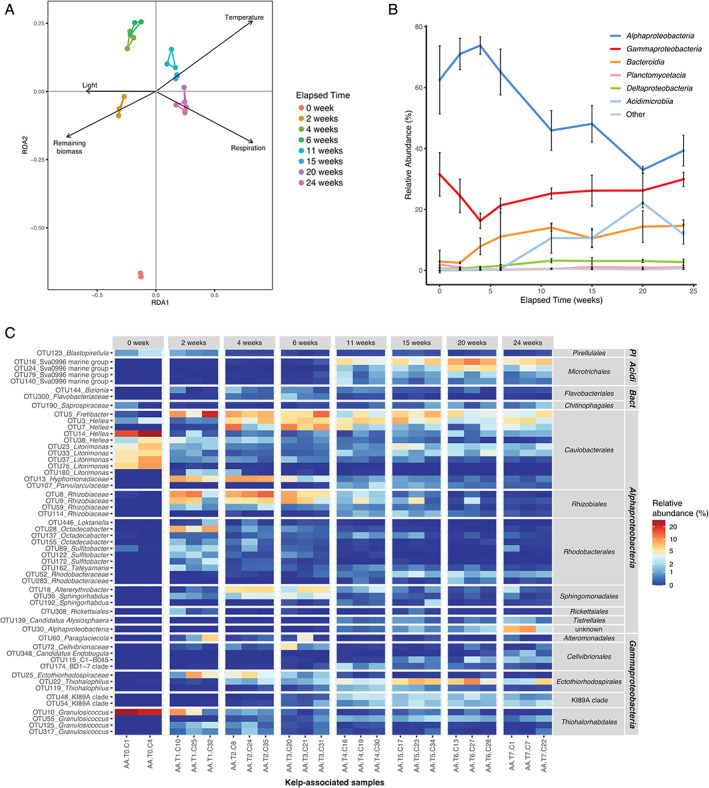
Fluctuations of kelp‐associated bacterial communities during the degradation of detached L. hyperborea. A. db‐RDA plot with vectors representing significant contextual predictors. Samples from the same sampling date are connected by coloured segments. B. Relative abundance of bacterial classes in kelp‐associated samples during the 6 month‐experiment. Sequence relative abundances of classes representing less than 0.5% of all sequences at each sampling time were summed into the ‘Other’ category. Values are mean ± standard deviation (n = 3, except for T0 n = 2). C. Relative abundance of differentially abundant kelp‐associated OTUs during the experiment. OTUs showing significant differential abundance (P < 0.05) and representing at least 1.5% of the sequences in at least one sample are shown. OTUs were annotated at the genus level. For unclassified genera, taxa were assigned to the lowest taxonomic level identified. OTUs were grouped by class and order. Pl: Planctomycetacia; Acidi: Acidimicrobiia; Bact: Bacteroidia. [Color figure can be viewed at wileyonlinelibrary.com]
To further understand these patterns of the community structure, we investigated fluctuations of bacterial taxonomic groups at the kelp surface. The epiphytic community of freshly detached kelps was dominated by Alphaproteobacteria and Gammaproteobacteria (ca. 62% and 32% of all sequences at week 0, respectively), while other classes represented less than 3% (Fig. 3B). In particular, 83% of the sequences retrieved from kelp‐associated samples at week 0 belonged to only 10 OTUs, with four of them assigned to Litorimonas and three to Hellea (Supporting Information Fig. S2). The in situ degradation of kelp biomass was associated with a succession pattern in the taxonomic composition of the epiphytic community (Fig. 3B; Supporting Information Fig. S2). Alphaproteobacteria remained the most abundant class throughout the experiment, although their contribution started decreasing after 4 weeks and reached 40% after 24 weeks. The proportion of Bacteroidia increased early on from 2.5% at week 2 to 14.6% after 24 weeks. Acidimicrobiia were rare (<0.5%) until week 6 but increased afterwards, peaking at 22% relative sequence abundance at week 20. The contribution of Deltaproteobacteria increased slightly from the initial sampling date (0.6%) to week 11 (3.2%) and stayed stable afterwards. This pattern differed from the one observed in the surrounding seawater (Supporting Information Fig. S3A), where a seasonal increase of Gammaproteobacteria during summer (weeks 11–15) coincided with a decrease of Alphaproteobacteria and Bacteroidia.
A differential abundance test detected 249 OTUs (i.e. 1.8% of the total OTUs) for which the relative sequence abundance significantly changed with kelp degradation time (P < 0.05) (Supporting Information Table S3). For clarity, we only display in Fig. 3C the 54 differential OTUs that represent ≥1.5% of the total community in at least one sample. Among these, seven OTUs affiliated to Granulosicoccus (Gammaproteobacteria) and Litorimonas and Hellea (Alphaproteobacteria) were abundant at the beginning of the experiment and decreased over time (Tukey; P < 0.001 between week 0 and 2). This decrease was particularly drastic for OTU10 (Granulosicoccus) and OTU14 (Hellea), which accounted for 25.4% and 22.5% at week 0 and only 0.03% and 1.1% after 24 weeks respectively (P < 0.001). The sequence abundance of 18 Alphaproteobacteria OTUs out of the 32 represented in Fig. 3C increased during the first 4 weeks of the experiment and then decreased. The sequence abundance of 8 out of 14 Gammaproteobacteria OTUs, including members of Cellvibrionales, Ectothiorhodospirales and the KI89A clade, increased after 11 weeks of degradation. In particular, one OTU related to Thiohalophilus (OTU22) peaked at 7% mean relative sequence abundance at week 20. The sequence abundance of four OTUs related to the Sva0996 marine group (Microtrichales, Acidimicrobiia) showed an important increase between 6 and 11 weeks of experiment (P = 0.003). The most abundant one (OTU16) represented 0.1% of the communities after 6 weeks vs. 5.6% after 6 months. Finally, 14% (35/249) of the differential OTUs belonged to Bacteroidia (Supporting Information Table S3) but are not represented in Fig. 3C since they did not pass the 1.5% abundance threshold we chose for clarity.
We built a correlation network of the kelp‐associated bacterial communities to search for potential associations (Fig. 4). The final network comprised 134 nodes and 572 edges. Sixty‐seven percent of the associations were positive, including those with a maximum delay of one sampling date (384 and 188 positive and negative associations, respectively). Among the 10 contextual variables included in the dataset, eight showed significant associations within the network. The variables with the most associations were ‘Temperature’ (23 significant edges), “C/N" (22 significant edges) and ‘Respiration’ (19 significant edges) (Supporting Information Fig. S4). Most positive associations of OTU relative sequence abundance to respiration and C/N ratio were delayed. Community respiration was negatively correlated with algal biomass, indicating that the bulk of oxygen consumption did not come from algal respiration. Instead, respiration was positively correlated with temperature, which is expected to increase bacterial activity. Eight clusters were detected in the network (Supporting Information Table S4), which might represent strong microbial associations, i.e. nodes that are highly connected to each other but weakly connected to nodes outside of their module. In particular, the two strongest clusters 1 and 2 comprised 66% of the nodes in the network (Fig. 4, inset) and were almost mutually exclusive, suggesting they denote two different communities during the bacterial succession. Most OTUs from cluster 2 showed a major change in their relative sequence abundance during the first 6 weeks (Supporting Information Fig. S5). This suggests that cluster 2 represents associations occurring early during the bacterial succession. Seventy‐five percent of these OTUs from cluster 2 belonged to Alphaproteobacteria (Supporting Information Table S5). None of these OTUs had significant associations to environmental parameters (Fig. 4). In contrast, OTUs from cluster 1 were more diverse (Supporting Information Table S5), significantly associated with a number of environmental parameters (Fig. 4) and showed a later response (Supporting Information Fig. S6). Only four OTUs were shared between clusters 1 and 2 (Fig. 4, inset, nodes in crimson colour), including OTU923 (Arenicella, Gammaproteobacteria), OTU190 (Saprospiraceae, Bacteroidia), OTU5587 (Hellea, Alphaproteobacteria) and OTU361 (Litorimonas, Alphaproteobacteria). These shared OTUs showed the highest closeness centrality (a measure indicating the distance of a node to all other nodes) of the entire network (CC > 0.4) and might therefore act as hubs connecting the two types of communities found in clusters 1 and 2.
Fig 4.
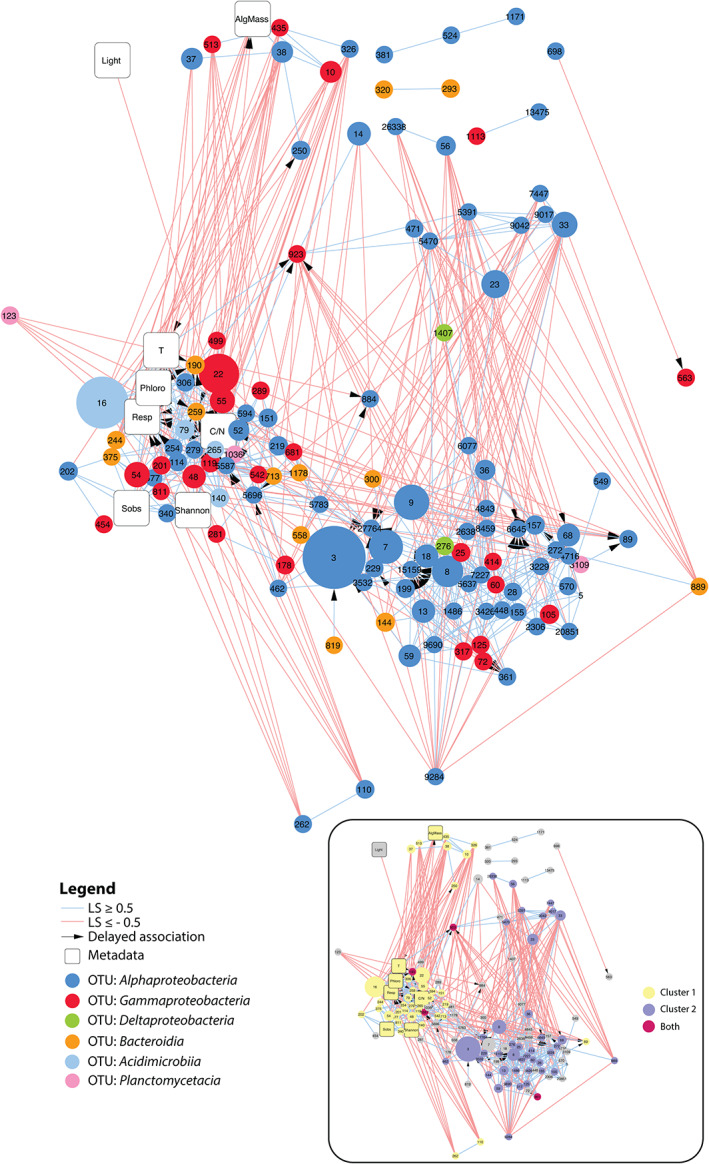
Extended local similarity analysis (eLSA) network obtained for kelp‐associated bacterial communities throughout the eight sampling time points. OTU nodes are colour‐coded based on taxonomy, and their size is proportional to the median relative abundance throughout the experiment. Positive and negative associations are depicted by light blue and light red lines, respectively. Arrows indicate delayed temporal associations (maximum delay = 1 sampling date). Network characteristics were as follows: clustering coefficient 0.358; characteristic path length 3.171; diameter 8; average number of neighbours 8.54; maximum number of neighbours 23; network density 0.064. Inset: the same network where nodes are coloured depending on their belonging to the main cluster 1 (yellow) and cluster 2 (purple) or both (crimson). AlgMass: remaining algal biomass; T: water temperature; Phloro: concentration of phlorotannins in kelp tissue; Resp: community respiration; Sobs: number of bacterial OTUs; C/N: carbon to nitrogen ratio of kelp tissue. [Color figure can be viewed at wileyonlinelibrary.com]
Putative functional profiles were inferred from 16S rRNA data using FAPROTAX for the kelp‐associated communities (Fig. 5), leading to the assignation of 3 753 OTUs to at least one functional group (31.4% of all OTUs). The predicted abundance of functional groups involved in aerobic chemoheterotrophy and the oxidation of sulfur compounds increased from 2 weeks on before a slight decrease at 11 weeks. Moreover, predicted groups involved in fermentation and nitrate reduction were virtually absent initially (< 18 and 28 estimated sequence read counts, respectively) and more represented during the degradation (55–997 and 177–899 estimated sequence read counts, respectively). Since sugars constitute a major part of algal biomass, we specifically assessed the predicted relative sequence abundance of glycoside hydrolases (GH) and polysaccharide lyases (PL) with PICRUSt2. The mean Nearest Sequenced Taxon Index (NSTI) was 0.21 ± 0.03. NSTI estimates the extent to which microorganisms in a given sample are related to sequenced genomes used to infer metagenomes and ranges from 0 (high similarity) to 1 (no similarity). The relatively high NSTI values obtained here denote that few genomes of kelp‐associated bacteria are available yet, and that predictions should be interpreted carefully. The predicted relative sequence abundance of endolytic enzymes such as cellulase and endo‐1,4‐β‐xylanase decreased over time (Supporting Information Fig. S7). In contrast, the predicted relative sequence abundance of several exolytic GHs increased significantly with elapsed time of degradation, either at an early stage, from 2 weeks (i.e. α‐glucosidase, α‐galactosidase and β‐mannosidase) or later, after 6 weeks (i.e. α‐amylase, 4‐α‐d‐{(1‐ > 4)‐α‐d‐Glc}trehalose trehalohydrolase, α‐mannosidase) .
Fig 5.
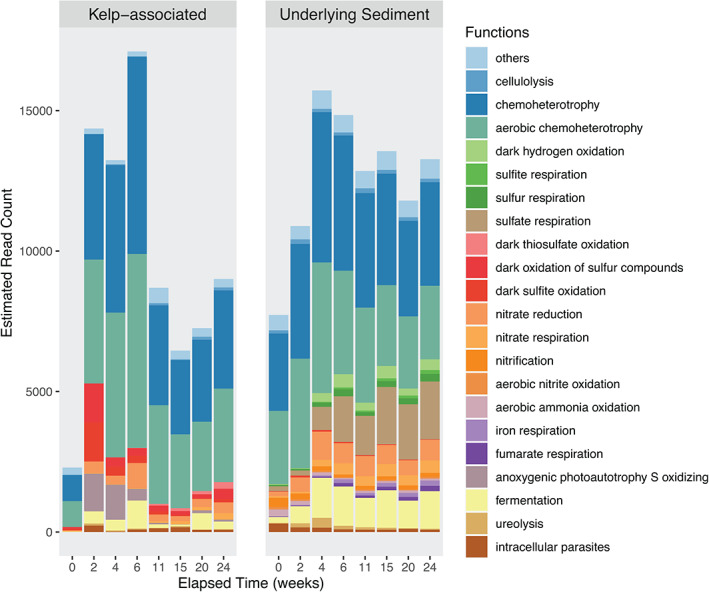
Putative functional groups of kelp‐associated and underlying sediment communities throughout kelp degradation. The OTUs from the rarefied dataset were parsed against the FAPROTAX database to assign putative functions to taxonomically defined OTUs. Only functions varying significantly across time based on the ALDEx2 analysis were represented (P < 0.05). [Color figure can be viewed at wileyonlinelibrary.com]
Effect of algal degradation on sediment bacterial communities
We further focused on the effect of kelp accumulation on sediment bacterial communities of the receiving ecosystem. Beta‐diversity analysis of the underlying sediment samples showed an effect of the elapsed degradation time on the bacterial community structure (Fig. 6A) (PERMANOVA, F 7,16 = 2.10, P < 0.001). Constrained ordination using db‐RDA showed that three environmental parameters were significant explanatory variables, namely remaining biomass (F 1,16 = 4.53, P = 0.001), respiration (F 1,16 = 1.97, P = 0.009) and temperature (F 1,16 = 1.95, P = 0.012). Sediment communities were initially dominated by Gammaproteobacteria and Bacteroidia (about 36% and 23% of all sequences at week 0, respectively) (Fig. 6B ). Only ca. 9.5% of OTUs present in the underlying or external sediments were shared with the algal epiphytic microbiota (Fig. 2B). The abundance of these OTUs in the sediments and in the kelp‐associated compartments represented 22.3% and 20.8% of the overall sequence abundance, respectively. Bacterial communities in external sediments were stable with a slight increase of Bacteroidia during kelp degradation (Supporting Information Fig. S3B), while changes were more pronounced in the underlying sediment (Fig. 6B). Furthermore, about 40% of the OTUs detected in the underlying sediments (containing 13% of the number of sequences in the underlying sediments) were not found in the external sediments (Fig. 2B). In underlying sediments, the sequence relative abundance of Gammaproteobacteria decreased until week 6, reaching 22% before an increase at 31% and stabilization around 27%. This was different from external sediments, where Gammaproteobacteria gradually decreased to 27% until week 15. The sequence relative abundance of Bacteroidia increased rapidly in underlying sediments, peaking at about 40% after 6 weeks before stabilization around 31%. This increase was less pronounced in external sediments. The relative sequence abundance of Alphaproteobacteria in underlying sediments slightly decreased from about 14% at week 0 to 10% at week 24, while it stayed stable in external sediments. Deltaproteobacteria were enriched along the time course, representing about 9% of the total sequence relative abundance at week 0 and reaching 15% and 11% at week 24 in underlying and external sediments, respectively. Differential abundance analysis detected 177 OTUs (i.e. 0.3% of the total number of OTUs) for which the relative sequence abundance significantly changed in the underlying sediment during kelp degradation (P < 0.05), while 27 were detected in external sediments (Supporting Information Table S6 and S7). Among these, only 11 OTUs affiliated with Bacteroidia, Deltaproteobacteria and Gammaproteobacteria were found to significantly vary with time in both underlying and external sediments (Supporting Information Fig. S8). For consistency, we applied the same filter to differential OTUs in underlying sediment than that used for kelp‐associated samples (>1.5% relative abundance in at least one sample), but only seven OTUs passed this criteria. Consequently, we lowered the cut‐off at 0.5% to show 35 differential OTUs (Fig. 6C). The sequence abundance of the 35 represented OTUs stayed generally stable during the first 2 weeks. The sequence abundance of 16 members of Bacteroidales and Flavobacteriales (Bacteroidia) as well as two Clostridiales (Clostridia) and two Rhodobacterales (Alphaproteobacteria) increased between 0 and 4 weeks (Tukey; P = 0.015, P = 0.005, P = 0.010, respectively) and decreased afterwards (P = 0.001, P = 0.044, P = 0.037 between 6 and 24 weeks, respectively). Five Desulfobacterales OTUs (Deltaproteobacteria) increased between 0 and 6 weeks (P = 0.015) and two Sulfurovum OTUs (Campylobacteria) increased between 0 and 15 weeks (P = 0.011).
Fig 6.
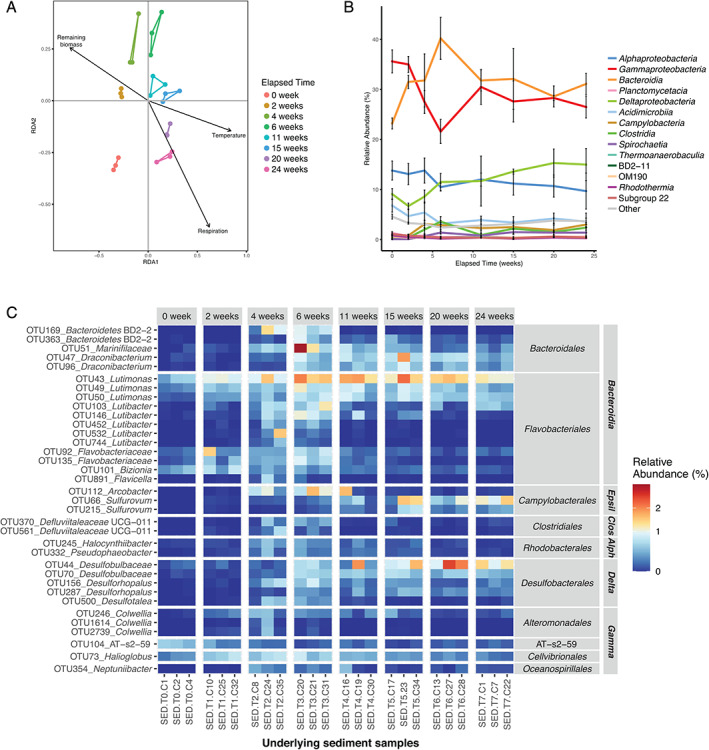
Fluctuations of bacterial community structure in the sediment underlying L. hyperborea during its degradation. A. db‐RDA plot with vectors representing significant contextual predictors. Samples from the same sampling date are connected by coloured segments. B. Relative abundance of bacterial classes in underlying sediment samples during the 6 month‐experiment. Sequence relative abundances of classes representing less than 0.5% of all sequences at each sampling time were summed into the ‘Other’ category. Values are mean ± standard deviation (n = 3). C. Relative abundance of differentially abundant OTUs in the underlying sediment during the experiment. OTUs showing significant differential abundance (P < 0.05) and representing at least 0.5% of the sequences in at least one sample are shown. OTUs were annotated at the genus level. For unclassified genera, taxa were assigned to the lowest taxonomic level identified. OTUs were grouped by class and order. Epsil: Epsilonproteobacteria; Clos: Clostridia; Alph: Alphaproteobacteria; Delta: Deltaproteobacteria; Gamma: Gammaproteobacteria. [Color figure can be viewed at wileyonlinelibrary.com]
Putative functional profiles were inferred for underlying sediment communities (Fig. 5). The 10 657 OTUs were assigned to at least one functional group (24.5%). The predicted abundance of functional groups involved in aerobic chemoheterotrophy, nitrogen cycle and fermentation increased during the first 4 weeks and remained stable afterwards. In contrast, sulfate‐reducing functional groups increased throughout the degradation process. The predicted sequence abundance of the sulfate respiration group increased 10 times over 24 weeks.
Discussion
In temperate coastal ecosystems, large amounts of kelp tissues are exported to the seafloor naturally or following seasonal storms. To investigate the degradation dynamics and the effect of kelp tissue accumulation on receiving ecosystems, a 6‐month experiment was deployed on a low subtidal sandy floor. Here, we investigated the bacterial community succession associated with the degradation of L. hyperborea tissue accumulated on the seafloor in four compartments: the degrading kelp, the seawater, the underlying and the external sediment.
Microbial communities are specific to a given compartment
Bacterial diversity patterns showed specific communities and succession in the four compartments. In particular, half of the kelp‐associated OTUs were not detected in any other compartment. This observation was expected as each compartment features contrasting biotic and abiotic conditions. The kelp surface is an interface between the alga and its surrounding environment where specific metabolic exchanges occur (e.g. defence, biofilm control). This is an organic‐rich environment, ideal for copiotrophs but with high competition between microorganisms for surface colonization, which might select for K‐strategists (Andrews and Harris, 1986; Fierer et al., 2007). In contrast, the water column is more substrate‐depleted, which may select for oligotrophs and K‐strategists. Finally, sediments concentrate decaying organic matter, available for r‐strategist copiotrophs, and oxygen availability as well as chemical conditions varying with depth offer multiple microniches. This likely explains the higher diversity with various microbial life strategy‐types in this compartment compared to kelp‐associated and seawater samples. Overall, bacterial diversity was not strongly impacted in the seawater and external sediments during the 6‐month period, pointing to a local effect of kelp accumulation. Indeed, constant water renewal due to tides and currents likely dilutes the compounds liberated upon degradation (Masson, 2002). In the following paragraphs, we focus on the major changes observed in kelp‐associated and underlying sediment communities.
Bacterial communities associated with healthy kelp tissue
Throughout the experiment, kelp‐associated bacterial communities were dominated by Alphaproteobacteria and Gammaproteobacteria. These bacterial classes are regularly found among the most abundant on brown algal tissue (Lachnit et al., 2011; Stratil et al., 2013; Martin et al., 2015; Ramirez‐Puebla et al., 2020). At the initial sampling date, in April 2017, the surface of fresh L. hyperborea hosted an abundant resident bacterial community, concurring with previous studies showing 106–109 cells.cm−2 on kelp surfaces (Corre and Prieur, 1990; Martin et al., 2014). The initial community had a low diversity and was dominated by a few OTUs affiliated to Granulosicoccus (Gammaproteobacteria), Hellea and Litorimonas (Alphaproteobacteria). These genera were always present in the epiphytic community of L. hyperborea and were previously detected on other brown algae such as Macrocystis pyrifera (Florez et al., 2019), Taonia atomaria (Paix et al., 2019), Fucus vesiculosus (Lachnit et al., 2011; Stratil et al., 2013), and L. digitata (Gobet, personal communication). We detected < 1% Planctomycetia and Verrucomicrobia in the initial communities. This contrasts with previous studies using fluorescent in situ hybridization and denaturing gel gradient electrophoresis where these taxa contributed up to 50% of epiphytic bacteria on L. hyperborea at the Norwegian coast (Bengtsson and Øvreås, 2010; Bengtsson et al., 2010). This discrepancy may be due to (i) ecological differences between Norway and Brittany coasts, (ii) seasonal variations and (iii) the technique used to survey the microbial community (Zinger et al., 2012). The primer used for our metabarcoding approach, designed to minimize amplification of plastid sequences, may partly miss Planctomycetia and Verrucomicrobia (Thomas et al., 2020).
Dynamics of bacterial communities at the surface of degrading kelp tissues
The kelp‐associated bacterial community shifted after 2 weeks. 16S rRNA gene copies decreased fourfold and stayed stable during the 6 months. This could reflect both a decrease in the total number of bacteria attached to the kelp surface and a shift towards taxa featuring fewer 16S rRNA gene copies per genome such as Acidobacteria (Větrovský and Baldrian, 2013). Analysis of kelp‐associated community composition further showed a biphasic succession, with changes during the first 11 weeks followed by stabilization of the bacterial community composition until 24 weeks. This corroborates the kinetics of algal biomass degradation (de Bettignies et al., 2020), which showed a rapid loss of algal biomass until week 11 that slowed down until week 24. During the 6‐month experiment, oxygen consumption increased in cages while algal biomass decreased. Although this could partially be due to colonization by fauna (de Bettignies et al., 2020), it likely indicates the important contribution of kelp‐degrading bacteria to the holobiont respiration (here, the kelp host and its associated fauna and microbiota) (de Bettignies et al., 2020). Overall, we suggest that the observed changes in kelp‐associated bacterial communities result from a succession of heterotrophic taxa with different substrate niches, as previously observed for the degradation of terrestrial plant biomass (Jiménez et al., 2017) and marine polysaccharide particles (Enke et al., 2019). During the first phase, kelp biomass was colonized by bacteria specialized in the degradation of complex algal polysaccharides and exudates. This corroborates results we previously obtained, showing an increase of mannitol‐ and alginate‐utilizing bacteria between 0 and 2 weeks (de Bettignies et al., 2020). The activity of these early degraders likely opened new substrate niches and increased the accessibility for opportunistic scavenger bacteria that responded during the second phase. Some of these changes might be partly due to seasonal variations that would also occur on intact, non‐detached algal tissue. This is suggested by the significant effect of temperature and light found with db‐RDA and the number of associations between individual OTUs and temperature in the network. Previous studies showed that seasonal parameters influence brown algal microbiomes, notably increasing temperature that can favour the growth of Flavobacteriales, Rhodobacterales and Rhizobiales (Stratil et al., 2013; Minich et al., 2018). In addition, the observed increase in OTU richness is stronger than what was previously measured on kelps due to seasons (Weigel and Pfister, 2019) tissue aging (Bengtsson et al., 2012) or temperature elevation (Minich et al., 2018), suggesting a combined effect of degradation and seasonal variations. Furthermore, kelp defence reactions in early time points (e.g. emission of reactive oxygen species and halogenated compounds) that occur in stress situations such as blade detachment, cutting, displacement and degradation (Cosse et al., 2007) may select for a resistant fraction of the resident bacterial community. The control exerted by living kelps on biofilm formation through surface metabolites (Bengtsson et al., 2012; Salaün et al., 2012) might also be relaxed during algal decay, possibly explaining some of the observed changes.
During the first phase of the succession (0–6 weeks), the diversity of the kelp‐associated community increased, due to a decrease in the abundance of initially dominant OTUs affiliated to Litorimonas, Hellea and Granulosicoccus. These genera were reported as primary surface colonizers (Dang and Lovell, 2016; Ramirez‐Puebla et al., 2020) but might be outcompeted by specialist algae‐degraders. In parallel, a number of OTUs that were initially absent or rare at the kelp surface showed a transient increase in abundance during the first 6 weeks of degradation, including Alphaproteobacteria (e.g. Fretibacter, Hellea, unclassified Rhizobiaceae, Octadecabacter, Sulfitobacter, Tateyamaria, Altererythrobacter) and Gammaproteobacteria (e.g. Paraglaciecola, unclassified Ectothiorhodospiraceae). The same changes were not detected in the surrounding seawater, suggesting that they result more from an enrichment of initially rare taxa at the kelp surface than from a colonization by free‐living bacteria. The rapid and transient enrichment of these bacteria on detached blades might be due to their capacity to assimilate low‐molecular weight algal compounds. For instance, a study on the Fucus spiralis microbiota showed that Sulfitobacter and Octadecabacter isolates were able to grow on mannitol (Dogs et al., 2017), a brown algal polyol whose exudation is enhanced upon stress such as blade damage. Members of the genus Tateyamaria, previously found on kelp (Bengtsson et al., 2012), can use the brown algal storage compound laminarin (Sass et al., 2010). The first phase of the succession was also characterized by a global increase in the relative sequence abundance of numerous Bacteroidia OTUs which individually represented a low proportion of the microbiota, and stayed stable afterwards. Bacteroidia are commonly known as particle‐associated taxa (Thiele et al., 2015) and positively respond to an input of marine polysaccharides (Wietz et al., 2015; Balmonte et al., 2018, 2019; Enke et al., 2019). In particular, members of the family Flavobacteriaceae are specialized in the degradation of algal biomass (Thomas et al., 2011; Teeling et al., 2012). Flavobacteriaceae are known to colonize algal surfaces and form biofilms (Mann et al., 2013) and to possess numerous polysaccharide utilization loci (PULs) (Grondin et al., 2017) that encode a great number and diversity of carbohydrate‐active enzymes for the breakdown of algal polysaccharides, together with regulators and transporters. Genomes of marine flavobacteria frequently feature PULs predicted to be involved in the degradation of brown algal polysaccharides such as alginate, laminarin and fucoidan (Barbeyron et al., 2016; Kappelmann et al., 2019). Notably, the PUL dedicated to alginate, the most abundant polysaccharide in kelps, was biochemically characterized and shown to account for the full substrate degradation into simple sugars (Thomas et al., 2012; Dudek et al., 2020). Therefore, the Bacteroidia detected here on detached kelps likely contributed to the decomposition of the algal biomass.
During the second phase of the succession (11–24 weeks), kelp‐associated bacterial community was the most diverse, suggesting that new niches were available for bacterial colonization. The most striking observation is the enrichment in Acidimicrobiia, mainly due to OTUs related to the Sva0996 marine group. This group may be ubiquitous as it was previously detected on the brown macroalgae Taonia atomaria (Paix et al., 2019) and L. digitata (Gobet, personal communication), as well as sponges and ascidians (Steinert et al., 2015; Verhoeven et al., 2017; Dat et al., 2018), in deep marine sediments (Chen et al., 2016) and in the water column of oligotrophic environments (Reintjes et al., 2019). Little is known about the Sva0996 marine group, except its ability to use phytoplankton‐derived dissolved proteins and potential importance in the dissolved organic nitrogen cycle (Orsi et al., 2016). Here, the enrichment in Sva0996‐affiliated bacteria during the second phase of kelp degradation could suggest a liberation of soluble material from the kelp tissue. The second phase of the succession was also characterized by an enrichment in several gammaproteobacterial OTUs. The increase in Cellvibrionaceae might reflect their ability to use complex polysaccharides as those found in kelp biomass, a widespread trait in the family (Spring et al., 2015). Also increasing after 11 weeks, OTUs affiliated to the clades KI89A and BD1‐7 belong to the oligotrophic marine Gammaproteobacteria (OMG) group (Cho and Giovannoni, 2004; Spring et al., 2015). Although more typical of oligotrophic environments, these clades were previously found on brown macroalgae (Dittami et al., 2016; Paix et al., 2019). Their increase in relative abundance towards the end of the 6‐month period might reflect a seasonal effect, as previously observed in coastal marine bacterioplankton whereby an OTU affiliated to the KI89A clade was more abundant during the summer (Alonso‐Sáez et al., 2015). Finally, the strong increase observed for an OTU related to Thiohalophilus, a sulfur‐oxidizing chemolithoautotrophic gammaproteobacteria, could result from a boosted sulfur cycle in the underlying sediment (see next section).
Effect of kelp degradation on sediment communities
Bacterial diversity in sediments was higher and stable throughout the experiment compared to algal surfaces and water samples. This likely reflects the complexity of the sediment environment, that receives organic deposits all year‐round and offers contrasted microscale niches (Boudreau et al., 2001; de Beer et al., 2005; Ettinger et al., 2017). Comparisons of underlying and external sediments indicate that algal degradation had mostly a local effect directly underneath the accumulated kelps. The low proportion of OTUs shared between kelp‐associated and underlying sediment communities suggests that this change in community composition is not due to a direct transfer of OTUs from algae, but to a change of environmental conditions linked to kelp accumulation and degradation. Indeed, in the underlying sediment, there was a shift in abundance between the two most abundant bacterial classes, with the Gammaproteobacteria decreasing in sequence relative abundance until week 6 and the Bacteroidia increasing and remaining the most abundant group until 24 weeks. As stated in the section above, bacteria from the class Bacteroidia such as the Flavobacteriaceae are involved in degradation of complex molecules such as algal polysaccharides. This taxonomic group is likely enriched in the underlying sediment through the experiment as fragments of degrading algal tissue are continuously deposited onto the seafloor. Both the limited oxygen diffusion due to kelp accumulation and the activity of aerobic degrading bacteria are expected to rapidly deplete O2 in underlying sediments. Accordingly, differential sequence abundance analyses and functional predictions indicated an increase of taxa involved in sulfur and nitrogen cycles, as well as fermentation. Although some of these changes might partially be due to seasonal variations (Gobet et al., 2012; Marchant et al., 2014), our data showed a significant effect of algal accumulation since early changes (2–4 weeks) were not seen in external sediments. Further, during tissue degradation, there was an increase in Deltaproteobacteria, a group regularly found in temperate coastal sediment and known for sulfate and nitrate reduction (Mußmann et al., 2005). Sulfate reduction is one of the most common anaerobic degradation pathways for organic matter in marine sediments (Wasmund et al., 2017). Subsequently, several OTUs related to Desulfobacterales and Clostridiales were enriched during the degradation. Members of these orders are well‐known sulfate reducers and are frequently detected in temperate marine sediments rich in organic matter (Muyzer and Stams, 2008; Gobet et al., 2012; Mahmoudi et al., 2015; Ettinger et al., 2017). Desulfobacterales feature metabolically versatile and larger genomes than other sulfate‐reducing bacteria (SRB), which might allow a faster response to organic matter inputs (Strittmatter et al., 2009). OTUs related to Arcobacter and Sulfurovum were also enriched in underlying sediments during the degradation. These genera can oxidize reduced sulfur compounds produced by SRB (e.g. H2S, thiosulfate) back to sulfate, therefore completing the sulfur cycle (Wasmund et al., 2017). Furthermore, FAPROTAX analysis showed a functional enrichment of nitrate reduction pathways. Nitrate reducers use either denitrification to N2 or dissimilatory nitrate reduction to ammonium (DNRA). Here, the massive input of organic matter and probable increase in sediment sulfide concentrations due to SRB enrichment would be expected to favour DNRA (Burgin and Hamilton, 2007; Hardison et al., 2015). Therefore, we can speculate that kelp degradation allows retaining nitrogen inputs as bioavailable ammonium instead of loss as N2. Similar to these results, a recent study (Aires et al., 2019) also showed that additions of red or green algal biomass (Gracilaria vermiculophylla or Ulva rigida, respectively) strongly favoured Desulfobacterales, Bacteroidales, Clostridiales and Campylobacterales. Therefore, at the order level changes in sediment bacterial communities due to seaweed accumulation do not appear to be specific to the type of algae.
Conclusion
In conclusion, following kelp detachment and accumulation on the seafloor, the structure and composition of the epiphytic bacterial community shifted, showing a biphasic succession of early degraders and secondary colonizers that might exploit different substrate niches. Besides this direct effect on kelp‐associated bacteria, we also evidenced changes in the underlying sediments, showing that kelp accumulation likely has a global effect on the microbial communities of receiving ecosystems. This work paves the way for future studies investigating the functional determinants, mechanisms and bacterial interactions of the epiphytic microbiota during the kelp breakdown process.
Experimental procedures
In situ experiment and sampling procedure
The in situ experiment was conducted from April 2017 to October 2017 in the bay of Morlaix (48°42'33.78'' N, 003°57'12.36'' W) on the north‐western coast of Brittany (France) and detailed elsewhere (de Bettignies et al., 2020). Briefly, cages were randomly filled with 1 kg of new blades from young healthy adult L. hyperborea sporophytes (140–200 cm). A total of 35 cages were set at 9 m depth on a sandy floor to simulate an accumulation of kelp fragments. Three cages were randomly selected at each of eight sampling times (0, 2, 4, 6, 11, 15, 20, 24 weeks) to sample algal surfaces, underlying and external sediments and overlying water as follows. Ten algal pieces of 1.3 cm diameter were subsampled from each cage using a sterilized stainless steel punch. The surface sediments (0–4 cm depth) directly below each cage (‘underlying sediment’) or 2 m away from the cages (‘external sediment’) were sampled in sterile plastic containers. ‘Initial sediment’ was collected immediately prior to the cages set‐up. Three litres of the overlying water (1 m above the cages) were taken using Niskin bottles. Triplicates of 1 l were prefiltered at 3 μm to remove large particles and then filtered through a 0.2 μm Sterivex filter (Merck, Darmstadt, Germany). All samples were stored at −80°C before DNA extraction. Measurements of contextual parameters related to physico‐chemical conditions (temperature, light, percent organic matter in sediment) and state of the kelp tissue (remaining biomass, oxygen consumption, photosynthetic capacity, carbon/nitrogen ratio, phlorotannin content) were detailed elsewhere (de Bettignies et al., 2020) and summarized in the Supporting Information text. The list of samples and contextual parameters is available in the Supporting Information Table S1.
DNA extraction
We adapted a protocol for DNA extraction from the algal surface. Algal pieces were immersed in a bacterial lysis buffer, without previous grinding of the tissues to prevent extraction of endophytic bacteria and algal plastids while efficiently lyzing epiphytic bacteria. After incubation, algal pieces were discarded and DNA was extracted only from the soluble lysate using a phenol:chloroform:isoamyl alcohol extraction combined with filtration steps of the NucleoSpin PlantII kit (Macherey Nagel, Hoerdt, France). For water samples, DNA was extracted from the 0.2 μm filters following a similar protocol as used for the algal samples. For sediment samples, DNA was extracted from 0.5 g of sediment using the DNeasy PowerLyzer PowerSoil Kit (Qiagen, Courtaboeuf, France). For further details, please see the Supporting Information text.
16S rRNA gene copies quantification
The total number of bacterial 16S rRNA gene copies in each sample was assessed by quantitative real‐time PCR as described previously (Bacchetti De Gregoris et al., 2011), using the universal primers 926F/1062R. This primer pair has a predicted coverage of 55% for chloroplast‐derived 16S rRNA genes (Silva Testprime on SSU r138.1). Therefore, we cannot exclude that a small proportion of quantified 16S rRNA gene copies originates from chloroplasts. Amplification and detection were performed in a LightCycler 480 Instrument II. For each sample, triplicate reactions were prepared as described in the Supporting Information text. Serial dilutions of purified bacterial genomic DNA ranging from 103 to 108 16S rRNA gene copies equivalent were used as a standard curve and were amplified in triplicate in the same run as the environmental samples. A non‐template control was included in the run. Results were analysed using the LightCycler 480 Software v1.5. One‐way ANOVA analyses followed by pairwise post‐hoc Tukey HSD were conducted with time as fixed factor, individually for each compartment. Details on qPCR assays are given in the Supporting Information Table S8, following the MIQE guidelines (Bustin et al., 2009).
Library preparation, sequencing and sequence processing
As a positive control, we constructed a mock community based on the genomic DNA of 32 pure marine bacterial isolates as reported previously (Thomas et al., 2020). Libraries were prepared for the 94 samples (24 algal samples, 24 water samples, 3 initial sediment samples, 21 underlying sediment samples, 20 external sediment samples, one mock community and one negative control (PCR molecular grade water)). Libraries were prepared using the primers S‐D‐Bact‐0341‐b‐S‐17 (5' CCTACGGGNGGCWGCAG 3′) and 799F_rc (5' CMGGGTATCTAATCCKGTT 3′) targeting the V3‐V4 region of the 16S rRNA gene and sequenced on a MiSeq paired‐end sequencing run (300 cycles × 2, Illumina, San Diego, CA, USA) as described previously (Thomas et al., 2020). 16S rRNA gene amplicon sequences are available at NCBI under BioProject ID PRJNA667331. Raw Illumina sequence reads were trimmed to remove low‐quality bases and Illumina adapters using Trimmomatic v0.38 (Bolger, Lohse and Usadel, 2014, for details see Supporting Information text). Overlapping paired‐end sequence reads were assembled using PANDAseq v2.11 and assembled sequences between 400 and 500 bp long were kept (Masella et al., 2012). All the following steps for sequence processing were performed using the sequence processing pipeline FROGS (Find Rapidly OTU with Galaxy Solution) developed for the Galaxy platform (http://galaxy3.sb-roscoff.fr, Escudié et al., 2018). Sequences were clustered into OTUs using SWARM v2 (Mahé et al., 2014). PCR chimera and singletons were filtered out and OTUs were taxonomically assigned using the RDP classifier (Wang et al., 2007) on the Silva 16S rRNA (v132) database.
Multivariate and statistical analyses
Prior to further analysis, one kelp‐associated sample from week 0 (AA‐T0‐C2 in the Supporting Information Table S1 ) was removed from the dataset due to its poor sequencing depth (3 302 sequences) compared to other samples (range 24 482–123 368 sequences). Samples from the bacterial community dataset were rarefied (100 permutations) to 24 482 sequences prior to OTU richness and Shannon diversity index calculation. Dissimilarities in community structure were calculated using the Bray–Curtis dissimilarity index (Bray and Curtis, 1957) before principal coordinate analysis (PCoA). Permutational analysis of variance (PERMANOVA, 999 permutations) was applied using adonis to discriminate groups of samples according to time or type of compartment (Anderson, 2001), followed by multivariate pairwise comparisons using pairwise.perm.manova. Mean dispersions within the compartments were calculated using betadisper (999 permutations). The effect of environmental factors on kelp‐associated and underlying sediment communities were assessed using distance‐based redundancy analysis (db‐RDA) (Legendre and Andersson, 1999) with capscale function followed by ANOVA. Environmental factors were log10‐transformed prior to this analysis. Only significant environmental factors were selected for db‐RDA visualization (ANOVA, P < 0.05). The above multivariate and statistical analyses were performed in R v3.5.0 with the packages phyloseq, vegan, ggplot and EcolUtils (Wickham, 2009; Mcmurdie and Holmes, 2013; Oksanen et al., 2013; R Core Team, 2018). Differential sequence abundance analysis through time was performed at the OTU level for kelp‐associated and underlying sediment communities using the GLM test with the ALDEx2 R package (Fernandes et al., 2014). The variation of selected differential OTUs through time was further tested using ANOVA followed by Tukey HSD post hoc test.
An association network was built for kelp‐associated bacterial communities. Extended Local Similarity Analysis (eLSA v1.0.4) (Xia et al., 2011) was performed on a dataset comprising 10 contextual parameters (eight environmental factors and two bacterial diversity measures) and the relative sequence abundance of OTUs found in at least 19 samples (135 OTUs). This prevalence filter was set according to recommendations in Röttjers and Faust (2018). A delay of one sampling date was allowed for temporal associations. P‐values were estimated using the permutation method, with 1 000 permutations. Q‐values were estimated to control for false positives. Default eLSA parameters were used otherwise. The network was visualized in Cytoscape v3.3.0 (Shannon et al., 2003). Only associations with |LS| ≥ 0.5, Q ≤ 0.05 and P ≤ 0.001 were represented with the ‘edge‐weighted, spring‐embedded layout’ (nodes are strongly repelled or attracted as a function of their LS value; spring strength = 50, spring rest length = 100, strength of disconnected spring = 0.05, rest length of disconnected spring = 500, and strength to avoid collisions = 500). Clusters were detected using the MCODE algorithm (Bader and Hogue, 2003) with ‘haircut’ and ‘fluff’ options and otherwise default parameters.
Functional profiles for kelp‐associated and underlying sediment communities were inferred using the FAPROTAX v1.2.2 database and script (Louca et al., 2016) against the rarefied OTU dataset with the assigned taxonomy. In addition, PICRUSt2 v2.1.4 (Douglas et al., 2020) was run with the default parameters (except min_reads set to 5) to get metagenome predictions for EC numbers. Differences in sequence abundance of functional groups and enzymes over time were assessed using the GLM test with ALDEx2.
Supporting information
Appendix S1: Supporting Information
Acknowledgements
The authors thank the marine operation staff of Roscoff Biological Station (Service Mer & Observation SBR), especially Laurent Levêque, Yann Fontana, Wilfried Thomas, Matthieu Camusat, Noël Guidal and François Le Ven for their help with scuba diving fieldwork and experimental set‐up. The authors thank Dr. Simon Dittami for critical reading of this manuscript, as well as Murielle Jam and Dr. Hetty Kleinjan for their help during sampling. The authors thank two anonymous reviewers for their useful comments that helped improved this manuscript. This work has benefited from the facilities of the Genomer platform and from the computational resources of the ABiMS bioinformatics platform (FR 2424, CNRS‐Sorbonne Université, Roscoff), which are part of the Biogenouest core facility network. This work has benefited from the support of the UMR 8227 and from the French Government via the National Research Agency programs IDEALG (ANR‐10‐BTBR‐04) and ALGAVOR (ANR‐18‐CE02‐0001‐01). A.G. acknowledges support by the Institut Français de Recherche pour l'Exploitation de la Mer (IFREMER).
Contributor Information
Angélique Gobet, Email: angelique.gobet@ifremer.fr.
François Thomas, Email: francois.thomas@sb-roscoff.fr.
References
- Aires, T. , Muyzer, G. , Serrão, E.A. , and Engelen, A.H. (2019) Seaweed loads cause stronger bacterial community shifts in coastal lagoon sediments than nutrient loads. Front Microbiol, 9: 3283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alonso‐Sáez, L. , Díaz‐Pérez, L. , and Morán, X.A.G. (2015) The hidden seasonality of the rare biosphere in coastal marine bacterioplankton. Environ Microbiol 17: 3766–3780. [DOI] [PubMed] [Google Scholar]
- Anderson, M.J. (2001) A new method for non‐parametric multivariate analysis of variance. Austral Ecol 26: 32–46. [Google Scholar]
- Andrews, J.H. , and Harris, R.F. (1986) R‐ and K‐selection and microbial ecology. In Advances in Microbial Ecology, Marshall, K.C. (ed). Boston, MA: Springer, pp. 99–147. [Google Scholar]
- Bacchetti De Gregoris, T. , Aldred, N. , Clare, A.S. , and Burgess, J.G. (2011) Improvement of phylum‐ and class‐specific primers for real‐time PCR quantification of bacterial taxa. J Microbiol Methods 86: 351–356. [DOI] [PubMed] [Google Scholar]
- Bader, G.D. , and Hogue, C.W.V. (2003) An automated method for finding molecular complexes in large protein interaction networks. BMC Bioinformatics 4: 1–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balmonte, J.P. , Buckley, A. , Hoarfrost, A. , Ghobrial, S. , Ziervogel, K. , Teske, A. , and Arnosti, C. (2019) Community structural differences shape microbial responses to high molecular weight organic matter. Environ Microbiol 21: 557–571. [DOI] [PubMed] [Google Scholar]
- Balmonte, J.P. , Teske, A. , and Arnosti, C. (2018) Structure and function of high Arctic pelagic, particle‐associated and benthic bacterial communities. Environ Microbiol 20: 2941–2954. [DOI] [PubMed] [Google Scholar]
- Barbeyron, T. , Thomas, F. , Barbe, V. , Teeling, H. , Schenowitz, C. , Dossat, C. , et al. (2016) Habitat and taxon as driving forces of carbohydrate catabolism in marine heterotrophic bacteria: example of the model algae‐associated bacterium Zobellia galactanivorans DsijT . Environ Microbiol 18: 4610–4627. [DOI] [PubMed] [Google Scholar]
- de Beer, D.D. , Wenzhöfer, F. , Ferdelman, T.G. , Boehme, S.E. , Huettel, M. , van Beusekom, J.E.E. , et al. (2005) Transport and mineralization rates in North Sea sandy intertidal sediments. Limnol Ocean 50: 113–127. [Google Scholar]
- Bengtsson, M. , Sjøtun, K. , and Øvreås, L. (2010) Seasonal dynamics of bacterial biofilms on the kelp Laminaria hyperborea . Aquat Microb Ecol 60: 71–83. [Google Scholar]
- Bengtsson, M.M. , and Øvreås, L. (2010) Planctomycetes dominate biofilms on surfaces of the kelp Laminaria hyperborea . BMC Microbiol 10: 261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bengtsson, M.M. , Sjøtun, K. , Lanzén, A. , and Øvreås, L. (2012) Bacterial diversity in relation to secondary production and succession on surfaces of the kelp Laminaria hyperborea . ISME J 6: 2188–2198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Bettignies, F. , Dauby, P. , Thomas, F. , Gobet, A. , Delage, L. , Bohner, O. , et al. (2020) Degradation dynamics and processes associated with the accumulation of Laminaria hyperborea (Phaeophyceae) kelp fragments: an in situ experimental approach. J Phycol 56: 1481–1492. [DOI] [PubMed] [Google Scholar]
- de Bettignies, T. , Wernberg, T. , Lavery, P.S. , Vanderklift, M.A. , Gunson, J.R. , Symonds, G. , and Collier, N. (2015) Phenological decoupling of mortality from wave forcing in kelp beds. Ecology 96: 850–861. [DOI] [PubMed] [Google Scholar]
- Bolger, A.M. , Lohse, M. , and Usadel, B. (2014). Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 30: 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boudreau, B.P. , Huettel, M. , Forster, S. , Jahnke, R.A. , McLachlan, A. , Middelburg, J.J. , et al. (2001) Permeable marine sediments: overturning an old paradigm. Eos (Washington DC) 82: 133–136. [Google Scholar]
- Bouvy, M. , Le Romancer, M. , and Delille, D. (1986) Significance of microheterotrophs in relation to the degradation process of subantarctic kelp beds (Macrocystis pyrifera). Polar Biol 5: 249–253. [Google Scholar]
- Bray, J.R. , and Curtis, J.T. (1957) An ordination of the upland forest communities of southern Wisconsin. Ecol Monogr 27: 325–349. [Google Scholar]
- Burgin, A.J. , and Hamilton, S.K. (2007) Have we overemphasized the role of denitrification in aquatic ecosystems? A review of nitrate removal pathways. Front Ecol Environ 5: 89–96. [Google Scholar]
- Burke, C. , Thomas, T. , Lewis, M. , Steinberg, P. , and Kjelleberg, S. (2011) Composition, uniqueness and variability of the epiphytic bacterial community of the green alga Ulva australis . ISME J 5: 590–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bustin, S.A. , Benes, V. , Garson, J.A. , Hellemans, J. , Huggett, J. , Kubista, M. , et al. (2009) The MIQE guidelines: minimum information for publication of quantitative real‐time PCR experiments. Clin Chem 55: 611–622. [DOI] [PubMed] [Google Scholar]
- Chen, P. , Zhang, L. , Guo, X. , Dai, X. , Liu, L. , Xi, L. , et al. (2016) Diversity, biogeography, and biodegradation potential of actinobacteria in the deep‐sea sediments along the southwest Indian ridge. Front Microbiol 7: 1340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho, J.C. , and Giovannoni, S.J. (2004) Cultivation and growth characteristics of a diverse Group of Oligotrophic Marine Gammaproteobacteria . Appl Environ Microbiol 70: 432–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corre, S. , and Prieur, D. (1990) Density and morphology of epiphytic bacteria on Laminaria digitata . Bot Mar 33: 515–523. [Google Scholar]
- Cosse, A. , Leblanc, C. , and Potin, P. (2007) Dynamic Defense of marine macroalgae against pathogens: from early activated to gene‐regulated responses. Adv Bot Res 46: 221–266. [Google Scholar]
- Dang, H. , and Lovell, C.R. (2016) Microbial surface colonization and biofilm development in marine environments. Microbiol Mol Biol Rev 80: 91–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dat, T.T.H. , Steinert, G. , Cuc, N.T.K. , Smidt, H. , and Sipkema, D. (2018) Archaeal and bacterial diversity and community composition from 18 phylogenetically divergent sponge species in Vietnam. PeerJ 6: e4970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delille, D. , and Perret, E. (1991) The influence of giant kelp Macrocystis pyrifera on the growth of subantarctic marine bacteria. J Exp Mar Bio Ecol 153: 227–239. [Google Scholar]
- Dittami, S.M. , Duboscq‐Bidot, L. , Perennou, M. , Gobet, A. , Corre, E. , Boyen, C. , and Tonon, T. (2016) Host‐microbe interactions as a driver of acclimation to salinity gradients in brown algal cultures. ISME J 10: 51–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dogs, M. , Wemheuer, B. , Wolter, L. , Bergen, N. , Daniel, R. , Simon, M. , and Brinkhoff, T. (2017) Rhodobacteraceae on the marine brown alga Fucus spiralis are abundant and show physiological adaptation to an epiphytic lifestyle. Syst Appl Microbiol 40: 370–382. [DOI] [PubMed] [Google Scholar]
- Douglas, G.M. , Maffei, V.J. , Zaneveld, J. , Yurgel, S.N. , Brown, J.R. , Taylor, C.M. , et al. (2020) PICRUSt2: an improved and extensible approach for metagenome inference. bioRxiv 672295. [Google Scholar]
- Duarte, C.M. , and Cebrián, J. (1996) The fate of marine autotrophic production. Limnol Oceanogr 41: 1758–1766. [Google Scholar]
- Dudek, M. , Dieudonné, A. , Jouanneau, D. , Rochat, T. , Michel, G. , Sarels, B. , and Thomas, F. (2020) Regulation of alginate catabolism involves a GntR family repressor in the marine flavobacterium Zobellia galactanivorans DsijT . Nucleic Acids Res, 48: 7786–7800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egan, S. , Fernandes, N.D. , Kumar, V. , Gardiner, M. , and Thomas, T. (2014) Bacterial pathogens, virulence mechanism and host defence in marine macroalgae. Environ Microbiol 16: 925–938. [DOI] [PubMed] [Google Scholar]
- Egan, S. , Harder, T. , Burke, C. , Steinberg, P. , Kjelleberg, S. , and Thomas, T. (2013) The seaweed holobiont: understanding seaweed‐bacteria interactions. FEMS Microbiol Rev 37: 462–476. [DOI] [PubMed] [Google Scholar]
- Enke, T.N. , Datta, M.S. , Schwartzman, J. , Cermak, N. , Schmitz, D. , Barrere, J. , et al. (2019) Modular assembly of polysaccharide‐degrading marine microbial communities. Curr Biol 29: 1528–1535.e6. [DOI] [PubMed] [Google Scholar]
- Escudié, F. , Auer, L. , Bernard, M. , Mariadassou, M. , Cauquil, L. , Vidal, K. , et al. (2018) FROGS: find, rapidly, OTUs with galaxy solution. Bioinformatics, 34: 1287–1294. [DOI] [PubMed] [Google Scholar]
- Ettinger, C.L. , Voerman, S.E. , Lang, J.M. , Stachowicz, J.J. , and Eiser, J.A. (2017) Microbial communities in sediment from Zostera marina patches, but not the Z. marina leaf or root microbiomes, vary in relation to distance from patch edge. PeerJ 5.e3246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandes, A.D. , Reid, J.N.S. , Macklaim, J.M. , McMurrough, T.A. , Edgell, D.R. , and Gloor, G.B. (2014) Unifying the analysis of high‐throughput sequencing datasets: characterizing RNA‐seq, 16S rRNA gene sequencing and selective growth experiments by compositional data analysis. Microbiome 2: 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fierer, N. , Bradford, M.A. , Jackson, R.B. (2007). Toward an ecological classification of soil bacteria. Ecology, 88(6), 1354–1364. [DOI] [PubMed] [Google Scholar]
- Fleurence, J. (1999) Seaweed proteins. Trends Food Sci Technol 10: 25–28. [Google Scholar]
- Florez, J.Z. , Camus, C. , Hengst, M.B. , Marchant, F. , and Buschmann, A.H. (2019) Structure of the epiphytic bacterial communities of Macrocystis pyrifera in localities with contrasting nitrogen concentrations and temperature. Algal Res 44: 101706. [Google Scholar]
- Gobet, A. , Böer, S.I. , Huse, S.M. , Van Beusekom, J.E.E. , Quince, C. , Sogin, M.L. , et al. (2012) Diversity and dynamics of rare and of resident bacterial populations in coastal sands. ISME J 6: 542–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goecke, F. , Labes, A. , Wiese, J. , and Imhoff, J.F. (2010) Chemical interactions between marine macroalgae and bacteria. Mar Ecol Prog Ser 409: 267–300. [Google Scholar]
- Grondin, J.M. , Tamura, K. , Déjean, G. , Abbott, D.W. , and Brumer, H. (2017) Polysaccharide utilization loci: fuelling microbial communities. J Bacteriol: JB.00860‐16.199(15), e00860‐16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardison, A.K. , Algar, C.K. , Giblin, A.E. , and Rich, J.J. (2015) Influence of organic carbon and nitrate loading on partitioning between dissimilatory nitrate reduction to ammonium (DNRA) and N2 production. Geochim Cosmochim Acta 164: 146–160. [Google Scholar]
- Jiménez, D.J. , Dini‐Andreote, F. , DeAngelis, K.M. , Singer, S.W. , Salles, J.F. , and van Elsas, J.D. (2017) Ecological insights into the dynamics of plant biomass‐degrading microbial consortia. Trends Microbiol 25: 788–796. [DOI] [PubMed] [Google Scholar]
- Kappelmann, L. , Krüger, K. , Hehemann, J.H. , Harder, J. , Markert, S. , Unfried, F. , et al. (2019) Polysaccharide utilization loci of North Sea Flavobacteriia as basis for using SusC/D‐protein expression for predicting major phytoplankton glycans. ISME J 13: 76–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kloareg, B. , and Quatrano, R.S. (1988) Structure of the cell walls of marine algae and ecophysiological functions of the matrix polysaccharides. Ocean Mar Biol Annu Rev 26: 259–315. [Google Scholar]
- Koop, K. , Newell, R. , and Lucas, M. (1982) Biodegradation and carbon flow based on kelp (Ecklonia maxima) debris in a Sandy Beach microcosm. Mar Ecol Prog Ser 7: 315–326. [Google Scholar]
- Krause‐Jensen, D. , and Duarte, C.M. (2016) Substantial role of macroalgae in marine carbon sequestration. Nat Geosci 9: 737–742. [Google Scholar]
- Krumhansl, K.A. , and Scheibling, R.E. (2012) Production and fate of kelp detritus. Mar Ecol Prog Ser 467: 281–302. [Google Scholar]
- Lachnit, T. , Meske, D. , Wahl, M. , Harder, T. , and Schmitz, R. (2011) Epibacterial community patterns on marine macroalgae are host‐specific but temporally variable. Environ Microbiol 13: 655–665. [DOI] [PubMed] [Google Scholar]
- Legendre, P. , and Andersson, M.J. (1999) Distance‐based redundancy analysis: testing multispecies responses in multifactorial ecological experiments. Ecol Monogr 69: 1–24. [Google Scholar]
- Lemay, M. , Martone, P. , Keeling, P. , Burt, J. , Krumhansl, K. , Sanders, R. , and Parfrey, L. (2018) Sympatric kelp species share a large portion of their surface bacterial communities. Environ Microbiol 20: 658–670. [DOI] [PubMed] [Google Scholar]
- Louca, S. , Parfrey, L.W. , and Doebeli, M. (2016) Decoupling function and taxonomy in the global ocean microbiome. Science 353: 1272–1277. [DOI] [PubMed] [Google Scholar]
- Mahé, F. , Rognes, T. , Quince, C. , de Vargas, C. , and Dunthorn, M. (2014) Swarm: robust and fast clustering method for amplicon‐based studies. PeerJ 2: e593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahmoudi, N. , Robeson, M.S. , Castro, H.F. , Fortney, J.L. , Techtmann, S.M. , Joyner, D.C. , et al. (2015) Microbial community composition and diversity in Caspian Sea sediments. FEMS Microbiol Ecol 91: 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann, A.J. , Hahnke, R.L. , Huang, S. , Werner, J. , Xing, P. , Barbeyron, T. , et al. (2013) The genome of the alga‐associated marine flavobacterium Formosa agariphila KMM 3901T reveals a broad potential for degradation of algal polysaccharides. Appl Environ Microbiol 79: 6813–6822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann, K.H. (1982) Ecology of Coastal Waters : A Systems Approach, Berkeley: University of California Press. [Google Scholar]
- Mann, K.H. (1973) Seaweeds: their productivity and strategy for growth. Science 182: 975–981. [DOI] [PubMed] [Google Scholar]
- Marchant, H.K. , Lavik, G. , Holtappels, M. , and Kuypers, M.M.M. (2014) The fate of nitrate in intertidal permeable sediments. PLoS One 9: e104517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin, M. , Barbeyron, T. , Martin, R. , Portetelle, D. , Michel, G. , and Vandenbol, M. (2015) The cultivable surface microbiota of the brown alga Ascophyllum nodosum is enriched in macroalgal‐polysaccharide‐degrading bacteria. Front Microbiol 6: 1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin, M. , Portetelle, D. , Michel, G. , and Vandenbol, M. (2014) Microorganisms living on macroalgae: diversity, interactions, and biotechnological applications. Appl Microbiol Biotechnol 98: 2917–2935. [DOI] [PubMed] [Google Scholar]
- Marzinelli, E.M. , Campbell, A.H. , Zozaya Valdes, E. , Vergés, A. , Nielsen, S. , Wernberg, T. , et al. (2015) Continental‐scale variation in seaweed host‐associated bacterial communities is a function of host condition, not geography. Environ Microbiol 17: 4078–4088. [DOI] [PubMed] [Google Scholar]
- Masella, A.P. , Bartram, A.K. , Truszkowski, J.M. , Brown, D.G. , and Neufeld, J.D. (2012) PANDAseq: paired‐end assembler for Illumina sequences. BMC Bioinformatics 13: 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masson, D. (2002) Deep water renewal in the strait of Georgia. Estuar Coast Shelf Sci 54: 115–126. [Google Scholar]
- Mcmurdie, P.J. , and Holmes, S. (2013) Phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS One 8: 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minich, J.J. , Morris, M.M. , Brown, M. , Doane, M. , Edwards, M.S. , Michael, T.P. , and Dinsdale, E.A. (2018) Elevated temperature drives kelp microbiome dysbiosis, while elevated carbon dioxide induces water microbiome disruption. PLoS One 13: 1–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mußmann, M. , Ishii, K. , Rabus, R. , and Amann, R. (2005) Diversity and vertical distribution of cultured and uncultured Deltaproteobacteria in an intertidal mud flat of the Wadden Sea. Environ Microbiol 7: 405–418. [DOI] [PubMed] [Google Scholar]
- Muyzer, G. , and Stams, A.J.M. (2008) The ecology and biotechnology of sulphate‐reducing bacteria. Nat Rev Microbiol 6: 441–454. [DOI] [PubMed] [Google Scholar]
- Newell, R. , Field, J. , and Griffiths, C. (1982) Energy balance and significance of microorganisms in a kelp bed community. Mar Ecol Prog Ser 8: 103–113. [Google Scholar]
- Oksanen, J. , Blanchet, F.G. , Kindt, R. , Legendre, P. , Minchin, P.R. , O'Hara, R.B. , et al. (2013) Vegan: community ecology package.
- Orsi, W.D. , Smith, J.M. , Liu, S. , Liu, Z. , Sakamoto, C.M. , Wilken, S. , et al. (2016) Diverse, uncultivated bacteria and archaea underlying the cycling of dissolved protein in the ocean. ISME J 10: 2158–2173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paix, B. , Othmani, A. , Debroas, D. , Culioli, G. , and Briand, J.F. (2019) Temporal covariation of epibacterial community and surface metabolome in the Mediterranean seaweed holobiont Taonia atomaria . Environ Microbiol 21: 3346–3363. [DOI] [PubMed] [Google Scholar]
- R Core Team (2018) R: a language and environment for statistical computing.
- Ramirez‐Puebla, T. , Weigel, B. , Jack, L. , Schlundta, C. , Pfister, C. , and Mark Welch, J. (2020) Spatial organization of the kelp microbiome at micron scales. bioRxiv 2020. 10.1101/2020.03.01.972083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reintjes, G. , Tegetmeyer, H.E. , Bürgisser, M. , Orlić, S. , Tews, I. , Zubkov, M. , et al. (2019) On‐site analysis of bacterial communities of the Ultraoligotrophic South Pacific gyre. Appl Environ Microbiol 85: 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Röttjers, L. , and Faust, K. (2018) From hairballs to hypotheses–biological insights from microbial networks. FEMS Microbiol Rev 42: 761–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salaün, S. , La Barre, S. , Dos Santos‐Goncalvez, M. , Potin, P. , Haras, D. , and Bazire, A. (2012) Influence of exudates of the kelp Laminaria digitata on biofilm formation of associated and exogenous bacterial epiphytes. Microb Ecol 64: 359–369. [DOI] [PubMed] [Google Scholar]
- Sass, H. , Köpke, B. , Rütters, H. , Feuerlein, T. , Dröge, S. , Cypionka, H. , and Engelen, B. (2010) Tateyamaria pelophila sp. nov., a facultatively anaerobic alphaproteobacterium isolated from tidal‐flat sediment, and emended descriptions of the genus Tateyamaria and of Tateyamaria omphalii . Int J Syst Evol Microbiol 60: 1770–1777. [DOI] [PubMed] [Google Scholar]
- Shannon, P. , Markiel, A. , Ozier, O. , Baliga, N.S. , Wang, J.T. , Ramage, D. , et al. (2003) Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 13: 2498–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spring, S. , Scheuner, C. , Göker, M. , and Klenk, H.P. (2015) A taxonomic framework for emerging groups of ecologically important marine gammaproteobacteria based on the reconstruction of evolutionary relationships using genome‐scale data. Front Microbiol 6: 281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinert, G. , Taylor, M.W. , and Schupp, P.J. (2015) Diversity of Actinobacteria associated with the marine ascidian Eudistoma toealensis . Marine Biotechnol 17: 377–385. [DOI] [PubMed] [Google Scholar]
- Stratil, S.B. , Neulinger, S.C. , Knecht, H. , Friedrichs, A.K. , and Wahl, M. (2013) Temperature‐driven shifts in the epibiotic bacterial community composition of the brown macroalga Fucus vesiculosus . Microbiology 2: 338–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strittmatter, A.W. , Liesegang, H. , Rabus, R. , Decker, I. , Amann, J. , Andres, S. , et al. (2009) Genome sequence of Desulfobacterium autotrophicum HRM2, a marine sulfate reducer oxidizing organic carbon completely to carbon dioxide. Environ Microbiol 11: 1038–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teeling, H. , Fuchs, B.M. , Becher, D. , Klockow, C. , Gardebrecht, A. , Bennke, C.M. , et al. (2012) Substrate‐controlled succession of marine bacterioplankton populations induced by a phytoplankton bloom. Science 336: 608–611. [DOI] [PubMed] [Google Scholar]
- Thiele, S. , Fuchs, B.M. , Amann, R. , and Iversen, M.H. (2015) Colonization in the photic zone and subsequent changes during sinking determine bacterial community composition in marine snow. Appl Environ Microbiol 81: 1463–1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas, F. , Barbeyron, T. , Tonon, T. , Génicot, S. , Czjzek, M. , and Michel, G. (2012) Characterization of the first alginolytic operons in a marine bacterium: from their emergence in marine Flavobacteriia to their independent transfers to marine Proteobacteria and human gut Bacteroides . Environ Microbiol 14: 2379–2394. [DOI] [PubMed] [Google Scholar]
- Thomas, F. , Dittami, S.M. , Brunet, M. , Le Duff, N. , Tanguy, G. , Leblanc, C. , and Gobet, A. (2020) Evaluation of a new primer combination to minimize plastid contamination in 16S rDNA metabarcoding analyses of alga‐associated bacterial communities. Environ Microbiol Rep 12: 30–37. [DOI] [PubMed] [Google Scholar]
- Thomas, F. , Hehemann, J.‐H. , Rebuffet, E. , Czjzek, M. , and Michel, G. (2011) Environmental and gut Bacteroidetes: the food connection. Front Microbiol 2: 93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhoeven Joost, T.P. , Kavanagh, A.N. , and Dufour, S.C. (2017). Microbiome analysis shows enrichment for specific bacteria in separate anatomical regions of the deep‐sea carnivorous sponge Chondrocladia grandis . FEMS Microbiology Ecology 93: fiw214. [DOI] [PubMed] [Google Scholar]
- Větrovský, T. , and Baldrian, P. (2013) The variability of the 16S rRNA gene in bacterial genomes and its consequences for bacterial community analyses. PLoS One 8: 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Q. , Garrity, G.M. , Tiedje, J.M. , and Cole, J.R. (2007) Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol 73: 5261–5267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wasmund, K. , Mußmann, M. , and Loy, A. (2017) The life sulfuric: microbial ecology of sulfur cycling in marine sediments. Environ Microbiol Rep 9: 323–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weigel, B.L. , and Pfister, C.A. (2019) Successional dynamics and seascape‐level patterns of microbial communities on the canopy‐forming kelps Nereocystis luetkeana and Macrocystis pyrifera . Front Microbiol 10: 346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickham, H. (2009) ggplot2. New York, NY: Springer‐Verlag. [Google Scholar]
- Wietz, M. , Wemheuer, B. , Simon, H. , Giebel, H.A. , Seibt, M.A. , Daniel, R. , et al. (2015) Bacterial community dynamics during polysaccharide degradation at contrasting sites in the southern and Atlantic oceans. Environ Microbiol 17: 3822–3831. [DOI] [PubMed] [Google Scholar]
- Xia, L.C. , Steele, J.A. , Cram, J.A. , Cardon, Z.G. , Simmons, S.L. , Vallino, J.J. , et al. (2011) Extended local similarity analysis (eLSA) of microbial community and other time series data with replicates. BMC Syst Biol 5: S15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zinger, L. , Gobet, A. , and Pommier, T. (2012) Two decades of describing the unseen majority of aquatic microbial diversity. Mol Ecol 21: 1878–1896. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1: Supporting Information