

Abstract
Fluorescence microscopy imaging enables receptor proteins to be investigated within their biological context. A key challenge is to site‐specifically incorporate reporter moieties into proteins without interfering with biological functions or cellular networks. Small peptide tags offer the opportunity to combine inducible labeling with small tag sizes that avoid receptor perturbation. Herein, we review the current state of live‐cell labeling of peptide‐tagged cell‐surface proteins. Considering their importance as targets in medicinal chemistry, we focus on membrane receptors such as G protein‐coupled receptors (GPCRs) and receptor tyrosine kinases (RTKs). We discuss peptide tags that i) are subject to enzyme‐mediated modification reactions, ii) guide the complementation of reporter proteins, iii) form coiled‐coil complexes, and iv) interact with metal complexes. Given our own contributions in the field, we place emphasis on peptide‐templated labeling chemistry.
Keywords: bioorganic chemistry, fluorescence microscopy, membrane proteins, protein modification, signal transduction
Fluorescence microscopy imaging provides information about the localization and trafficking of G protein‐coupled receptors and receptor tyrosine kinases. To visualize these proteins, reporter groups must be introduced on the surface of live cells. This review discusses live‐cell protein labelling by small, genetically encoded peptide tags serving as enzyme substrates or recognition sites for interactions with proteins, coiled‐coil peptides and metal reagents.
1. Introduction
In order to survive, eukaryotic cells need to adapt to environmental changes. For this purpose, receptor proteins in the cell membrane convert extracellular stimuli into intracellular signal responses. Three important families of membrane receptors encoded in the human genome are G protein‐coupled receptors (GPCR), receptor tyrosine kinases (RTK) and ligand‐controlled ion channels. Herein, we will focus on the former two families. The GPCRs are the largest family of membrane receptors which are characterized by a common architecture with a transmembrane helix bundle, an extracellular N terminus and an intracellular C terminus. [1] GPCR ligands are diverse ranging from ions and small molecules to peptides and proteins. [2] Ligand binding to GPCRs induces the stabilization of distinct conformations, leading to the activation of G proteins and the generation of intracellular second messengers like Ca2+ or cyclic adenosine monophosphate (cAMP).[ 3 , 4 ] To terminate GPCR signaling, receptors are phosphorylated and translocated into cytoplasmic compartments.[ 5 , 6 ] Internalized GPCRs are subsequently transported back to the cell membrane or degraded in lysosomes. [7] RTKs comprise a single‐pass transmembrane domain with an extracellular N terminus, containing the ligand interaction motifs, and an intracellular C terminus, necessary for activation by phosphorylation. [8] Upon ligand binding to RTKs, the C‐terminal phosphorylation motifs are exposed and cross‐phosphorylated in RTK dimers/oligomers by an intramolecular kinase domain.[ 9 , 10 ] RTK signaling is propagated by multiple intracellular steps, for example, phosphorylation‐dependent activation of Ras proteins or transcription factors like signal transducer and activator of transcription (STAT) proteins.[ 11 , 12 , 13 , 14 , 15 ] RTK signaling can be terminated upon translocation from the cell membrane into cytoplasmic vesicles. [16] As phosphorylation of the RTK is essential for activation, phosphatases control RTK signaling by removal of the phosphate groups. [17] Similar to GPCRs, RTKs can recycle back to the cell membrane or are degraded with their ligand in lysosomal compartments.[ 18 , 19 ]
Mechanistic studies of receptor function call for methods that provide information about their localization, trafficking and interactions within the native environment. Live cell fluorescence microscopy has contributed significantly to our current knowledge. This method requires fluorescent reporter groups that do not perturb receptor function. Immunochemistry allows attachment of reporter groups by means of antibody binding. However, only a small number of selective antibodies against membrane‐embedded GPCRs and RTKs are commercially available.[ 20 , 21 ] To extend the scope of immunostaining, also to nanobodies, exogenous peptide antigens such as the FLAG‐tag, [22] HA‐tag, [23] V5‐tag, [24] ALFA‐tag, [25] or BC2‐tag [26] are fused to the protein of interest (POI) N terminus. Common labeling protocols containing different blocking steps, long staining periods, and multiple washing steps exclude immunostaining from being applied to the analysis of short‐lived changes in receptor signal transduction in live cells.[ 27 , 28 ] Furthermore, the large size of antibodies is problematic for the live cell analysis of proximity‐dependent processes. Labeling with nanobodies can provide a solution to the size problem.[ 25 , 26 ] It has to be considered that cell fixation and permeabilization can cause artefacts. [29]
An established approach to visualize cellular proteins relies on the genetically encoded fusion of the POI with an auto‐fluorescent protein. Since their discovery, fluorescent proteins (FP) such as the green FP (GFP) have been widely used to elucidate membrane expression and intracellular trafficking routes. Although easily implemented, the size of the FPs can perturb protein function and localization.[ 30 , 31 , 32 , 33 , 34 ] In rare cases, overexpression of FPs can induce apoptosis. [35] Furthermore, the maturation of the chromophore creates equivalent amounts of hydrogen peroxide as by‐product.[ 36 , 37 ] The analysis of membrane receptors can be plagued by background from intracellularly retained protein subpopulations due to aggregation of the FP.[ 38 , 39 ] Methods that allow the introduction of reporter groups at specified time points can limit label‐induced perturbations. The use of labeling agents that act on the cell surface also provides a solution to background issues. For example, self‐modifying enzyme tags such as the Halo‐ and SNAP‐tag have been used to validate membrane localization and identify novel receptor ligands.[ 40 , 41 , 42 , 43 , 44 , 45 , 46 ] Enzyme‐based tags with peroxidase activity such as APEX [47] or APEX2 [48] use H2O2 to generate fluorescent signals. Enzymatic catalysis provides for high sensitivity. Of note, APEX‐type tags convert diaminobenzidine to a polymeric material that can be stained with osmium substrates. This enables imaging by electron microscopy. Enzyme tags have a molecular weight comparable to FPs (GFP: 27 kDa; Halo‐tag: 33 kDa; SNAP‐tag: 20 kDa; APEX2: 28 kDa). Often, the label approaches the size of the POI, potentially hindering proximity‐dependent interactions and limiting the imaging resolution.
In recent years, powerful methods have been developed that allow live cell labeling with genetically encoded peptide tags smaller than 50 amino acids (aa). Such short, encodable tags display various advantages in comparison to auto‐fluorescent reporters and enzyme tags. Owing to their small size it is less likely that localization and proximity‐dependent protein interactions are perturbed. Furthermore, a smaller cargo offers the prospect of a more fine‐grained imaging than possible with enzyme‐sized labels. Common to all methods based on fusion proteins is that the respective POI is provided by eukaryotic expression vectors under the control of a promoter sequence. By choosing a suitable promoter sequence, the translation strength is tunable and allows to control protein expression levels. [49] Furthermore, different promoters have been developed to enable inducible expression in mammalian cells. [50] As encoded peptide tags do not contain a reporter in themselves, they provide for unrestricted experimental application as the reporter can be individually chosen for the desired assay readout. Additionally, inducible labeling facilitates high spatial and temporal resolution of protein behavior in response to extracellular stimuli. Complementing and updating reviews on protein fluorescence imaging,[ 51 , 52 , 53 , 54 , 55 , 56 ] we focus in this review on techniques used for live cell analysis of GPCRs and RTKs based on genetically encoded, small peptide tags.
2. Visualization of Receptor Proteins on the Surfaces of Live Cells
For successful labeling of membrane proteins, accessibility on the cell surface is required. The protein needs to be correctly embedded in the cell membrane without impairing the tightly regulated intracellular transport.[ 57 , 58 , 59 ] Thus, confirming cell‐surface expression is a vital part in characterizing tagged membrane proteins. As the membrane of mammalian cells poses a natural barrier for compound uptake into the cell, intracellular labeling is complicated. However, this is advantageous for excluding intracellularly localized receptors from labeling and facilitates the analysis/tracking of membrane‐embedded receptors.
2.1. Overview of genetically encoded peptide tags for receptor visualization on live‐cell surfaces
Short peptide tags (<50 aa) fused to the N terminus of GPCRs or RTKs are readily accessible for labeling reagents without interfering with ligand/receptor interactions. Different techniques have been developed over the years, which visualize GPCRs and RTKs in live cell experiments. A short summary of commonly applied N‐terminal tags discussed in this review is provided in Table 1.
Table 1.
Commonly used methodologies and peptide tag sequences for labeling of membrane‐embedded proteins like G protein‐coupled receptors and receptor tyrosine kinases in live mammalian cell systems.
|
Labeling approach |
Tag sequence |
Labeling conditions for live cell application |
|---|---|---|
|
Biotin ligase [60] |
GLNDIFEAQKIEWHE (AP‐tag) |
Biotinylation: 0.5 μM BirA, 10 μM biotin and 2 mM ATP in PBS (reaction time: 20 min at room temperature). [61] Biotin detection requires reporter‐equipped streptavidin. Ketone probe: 0.2 μM BirA, 1 mM ketone probe, and 1 mM ATP in DPBS (reaction time: 10–60 min at 32 °C). Subsequent ketone conversion requires 1 mM hydrazine probe (e. g., incubation for 10–60 min at 16 °C). [62] |
|
DEVLVEIETDKAVLEVPGGEEE (LAP‐tag) GFEIDKVWYDLDA (LAP2‐tag) |
High concentrations of enzyme (10 μM LplA) and lipoic acid derivative (350 μM azide substrate) and additional co‐factors (1 mM ATP) required for labeling within 1 h. [63] Probe detection depends on installed reactive handle. |
|
|
DSLEFIASKLA (ybbR‐tag) GDSLSWLLRLLN (S6‐tag) GDSLDMLEWSLM (A1‐tag) |
Labeling is performed within 30 min, using 5 μM CoA substrate, 1 μM enzyme, and 10 μM Mg2+).[ 65 , 68 ] |
|
|
PNPQLPF (Q1‐tag) PKPQQFM (Q2‐tag) GQQQLG (Q3‐tag) |
Micromolar concentrations of the enzyme (∼1 μM) and the labeling agent (04–0.5 mM) in the presence of 12 mM CaCl2 (reaction time: ∼30 min at 4 or 37 °C). [72] |
|
|
Transpeptidation by sortase A[ 73 , 74 , 75 , 76 , 77 , 78 ] |
LPETGGn (n depending on the POI) |
For protein labeling in live cells: 30 μM SrtA and 10 μM labeling agent (reaction time up to 1 h at 37 °C). [75] |
|
Tubulin tyrosine ligase [79] |
VDSVEGEGEEEGEE (Tub‐tag) |
For protein modification, 1 μM tubulin tyrosine ligase, 1 mM tyrosine derivative, and 2.5 mM ATP (reaction time 1–3 h). [79] Subsequent reporter transfer onto the reactive handle depends on the bio‐orthogonal chemistry. |
|
splitNluc complementation [80] |
VSGWRLFKKIS (HiBiT‐tag) |
Luminophore reassembly: addition of 10 nM LgBiT (15 min at 37 °C) prior to luminescence readout after furimazine addition. [81] |
|
splitGFP reassembly [82] |
NHYLSTQTVLSKDPNEKRDHMVLHEYVNAAGIT (GFP10‐11; shown without flanking Gly4) |
Reporter reassembly: 2 μM of GFP 1–9 (reaction time: 20 min at 37 °C in PBS). [82] |
|
SpyTag/SpyCatcher [83] |
AHIVMVDAYKPTK |
5 μM SpyCatcher‐Alexa Fluor 555 in PBS, supplemented with 5 mM Mg2+ (reaction time: 15 min at 25 °C). [83] |
|
Heterodimerization of coiled‐coil peptides[ 84 , 85 , 86 , 87 , 88 , 89 , 90 , 91 ] |
see Table 2 |
|
|
CCXXCC (Tetracysteine motif) |
Extracellular labeling: incubation with 5 mM MES and 0.5 mM TCEP (for 20–30 min) prior to addition of FlAsH reactant (2–5 μM). [92] |
|
|
His6 or His10 |
15 nM of dye‐conjugated, Ni2+‐loaded NTA for noncovalent labeling in <15 min. [96] Covalent modification: pre‐incubation with 0.5 mM TCEP prior to addition of the labeling probe (0.5 μM) at room temperature. [95] |
|
|
Split intein [97] |
Gp41‐1 system: N‐intein, 88 aa; C‐intein, 37 aa |
For live cell labeling: 0.5 μM of N‐intein, equipped with biotin, and 20 mM reduced glutathione (reaction time: 8 h). [98] Biotin detection requires reporter‐equipped streptavidin. |
2.2. Recognition peptides for chemoenzymatic labeling on cell membranes
Various enzymes recognize specific peptide sequences and install posttranslational modifications. Some classes of enzymes have been successfully applied in labeling of human membrane proteins. Biotin ligase (BirA), derived from Escherichia coli, allows the site‐specific transfer of biotin to the side chain of a lysine residue within the recognition sequence of the acceptor peptide, the AP‐tag (GLNDIFEAQKIEWHE; Figure 1B). [60] A two‐step protocol is required for fluorescent protein labeling: firstly, on‐surface biotinylation of the POI by BirA, followed by biotin recognition by streptavidin (carrying the desired reporter). As shown for the Na‐K‐2Cl cotransporter, protein biotinylation is efficiently performed within 20 min at room temperature, but requires micromolar concentrations of enzyme and substrate (e. g., 0.5 μM biotin ligase and 10 μM biotin) as well as wash steps to remove excess biotinylation agent. [61] Beyond imaging, biotin ligase methods have proven invaluable for proximity ligation assays. Fusion of the biotinylation target sequence, the AP‐tag, and BirA, the biotinylating enzyme, to different POI enables proximity induced biotinylation for investigation of protein homo‐ and heterodimerization. This approach was used to study the dimerization behavior of the C‐X‐C motif chemokine receptor 4 (CXCR4), a rhodopsin‐like GPCR. For proximity induced biotinylation, the AP‐CXCR4 and BirA‐CXCR4 were co‐expressed. Only upon close vicinity of the two GPCR species in a homodimer, biotinylation of AP‐CXCR4 occurs. Since BirA displays a high affinity towards its substrates (e. g., K D(biotin) ∼45 nM), [99] a shortened AP’‐tag was exploited to exclude the BirA/AP‐interaction as driving force for GPCR dimerization.[ 100 , 101 ] The removal of only three residues in the AP sequence lead to a K m increase from 5 μM to ∼345 mM. [101] Investigations with this system suggested that the GPCRs exist as a homodimer in the membrane of live cells. In addition, CXCR4 heterodimerization with the chemokine receptors 2 and 5 (CCR2/5) was revealed by this approach. [100] Proximity‐based biotinylation of the GPCR was performed efficiently in 15 min at 100 μM biotin and 1 mM ATP. Amongst the most prominent BirA mutants used for proximity assays are BioID (R118G mutation), [102] TurboID [103] (15 mutations), and miniTurbo (N‐terminal truncation and 13 mutations) [103] . Not only the enzyme and its target sequence have been optimized for BirA labeling. To reduce the cargo added upon visualization of biotinylated proteins with streptavidin (monomeric streptavidin: ∼17 kDa), Chen, et al. reported on a ketone analog of biotin, compatible with BirA labeling, which allows selective modification of the ketone by hydrazide‐ and hydroxylamine‐equipped reporter as shown for the epidermal growth factor receptor (EGFR). [62] To increase rates, the reactions were performed at slightly acidic pH 6.2.
Figure 1.
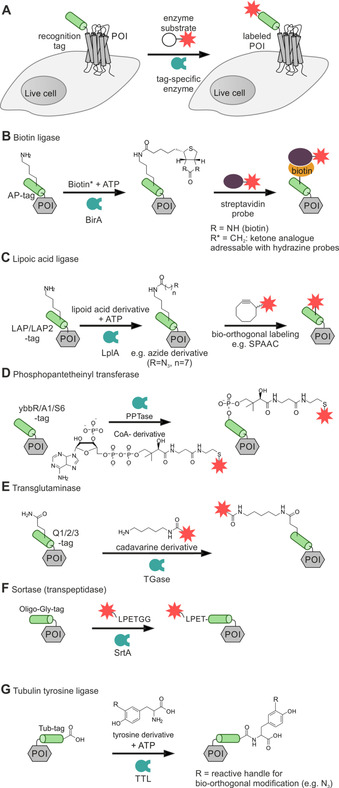
Covalent enzymatic labeling of recognition tags at the N terminus of live cells. A protein of interest (POI, gray box) tagged with a peptide recognition tag (green) at the N terminus is selectively labeled with a cargo (red star) by employing an enzymatically (blue) catalyzed reaction (A). Enzymatic labeling methodologies: Biotin ligase (BirA, B), lipoic acid ligase (LplA, C), phosphopantetheinyl transferase (PPTase, D), transglutaminase (TGase, E), transpeptidase sortase A (SrtA, F), and tubulin tyrosine ligase (TTL, G).
Lipoic acid ligase (LplA) site‐specifically ligates lipoic acid to a lysine residue of the extracellularly exposed acceptor peptide LAP (DEVLVEIETDKAVLEVPGGEEE) or its optimized shorter variant LAP2 (GFEIDKVWYDLDA; Figure 1C).[ 63 , 64 ] This enzyme recognizes various lipoic acid derivatives as substrate for the modification of membrane POI. Azide handles allowed subsequent fluorescent labeling by azide‐alkyne cycloaddition. Other chemical handles have been introduced to allow second step labeling via inverse‐electron demand Diels‐Alder reaction or tetrazine ligation.[ 104 , 105 ] Engineered LplA variants allowed labeling of proteins with coumarin [106] or red‐emitting resorufin. [107] Similar to BirA reaction conditions, LplA requires high concentrations of both enzyme and substrate (e. g., 10 μM LplA, 350 μM azide substrate) and addition of a co‐factor (1 mM ATP) for efficient labeling within 1 h incubation time. [63] Due to the similarity in reaction conditions of LplA and BirA, and the orthogonality of the peptide tags, the two enzymatic labeling platforms can be simultaneously combined for dual labeling of different protein species as demonstrated for AP‐tagged EGFR and LAP‐carrying low‐density lipoprotein receptor (LAP‐LDLR) in live HEK293 (human embryonic kidney) cells. [63]
Covalent labeling at N‐terminal tags can also be achieved by transferases like the phosphopantetheinyl transferase (PPTase; Figure 1D). This enzyme transfers 4‘‐phosphopantetheine from coenzyme A (CoA) onto a serine residue of the recognition sequence. [65] The use of PPTases from different species, for example, the Sfp enzyme from Bacillus subtilis and the AcpS enzyme from E. coli allows orthogonal dual labeling.[ 66 , 67 ] Sfp recognizes the ybbR‐tag (DSLEFIASKLA) and S6‐tag (GDSLSWLLRLLN), whereas AcpS targets the A1‐tag (GDSLDMLEWSLM). Transfer of the reporter moiety from CoA to the POI has been achieved within 30 min by applying 5 μM CoA substrate and 1 μM enzyme in the presence of high amounts of cations (e. g., 10 μM Mg2+).[ 65 , 68 ] PPTase‐induced live cell labeling has been applied to the imaging of the human epidermal growth factor 2 (HER2), EGFR, and the neurokinin‐1 receptor (NK1R).[ 68 , 69 , 108 ] A recent study suggests that CoA‐dye conjugates can enter the cell interior and induce modification of endogenous proteins. [68]
Transglutaminase (TGase) catalyzes the condensation between the side chains of lysine and glutamine residues. The genetically encodable tags Q1 (PNPQLPF), Q2 (PKPQQFM), and Q3 (GQQQLG) present glutamine for conjugation with labeling reagents based on cadaverine (1,5‐diaminopentan) or spermine, a polyamine, equipped with the desired reporter moiety (e. g., reporter dye; Figure 1E).[ 70 , 71 , 72 ] TGase has been applied for in vitro modification of proteins and was successfully used for EGFR visualization on live HEK293 cells. Typically, micromolar concentrations of the enzyme (∼1 μM) and the labeling agent (∼0.5 mM) in the presence of high salt concentrations (e. g., 12 mM CaCl2) are required for efficient transglutamination. [72]
Sortase A (SrtA) is an enzyme that catalyzes transpeptidation reactions. To label extracellularly exposed protein N‐termini, the POI is equipped with the LPETG motif, which can be converted into the poly‐glycine‐tag by SrtA‐mediated transpepdidation using a triglycine probe. [75] In a subsequent second reaction, the poly‐glycine tag is converted by SrtA and addition of a reporter‐LPETG probe. This reaction reconstitutes the initial LPETG motif at the POI with the addition of the N‐terminal reporter group (e. g., a fluorescent dye). The labeling reaction involves cleavage of the labeling agent (cargo‐LPETGG) between threonine and glycine and transfer onto the N‐terminal poly‐glycine sequence of the POI (Figure 1F).[ 73 , 74 , 75 ] In a typical “sortagging” experiment, 30 μM SrtA and 10 μM labeling agent was used for labeling of a membrane‐anchored cyan FP (CFP). [75] With only a few recognition features needed, sortase is an extremely versatile enzyme since labeling can be performed N‐ and C‐terminally. The reversibility of the reaction and the need for rather high enzyme loads can be obstacles. However, an impressive amount of reports provides testimony to the user‐friendliness of sortase methods. [109]
The tubulin tyrosine ligase (TTL) can be applied for protein labeling at the C terminus, fused to the 14 aa Tub‐tag. [79] The TTL enzyme tolerates various modification of its substrate tyrosine, which can be equipped with different reactive handles (e. g., azides) for subsequent two‐step labeling as demonstrated for the GFP‐specific nanobody GBP4. [79] Tyrosine transfer by TTL onto the Tub‐tag is performed in 1–3 h, using 1 mM tyrosine substrate, but requires the supplementation with 2.5 mM ATP for 1 μM TTL. The reaction time of the subsequent transfer of the second reporter moiety onto the reactive handle strongly depends on the applied orthogonal chemistry. [79] However, labeling of membrane‐embedded proteins on live cells by this methodology is yet to be demonstrated.
2.3. Complementation of reporter units on cell membrane receptors guided by short peptide tags
Split fluorescent proteins provide the opportunity to express proteins with a small sized tag and restore fluorescence at specific time points. For example, the non‐fluorescent strands 10 and 11 of GFP act as receptor tag, which reassemble the native fluorophore upon addition of GFP strands 1–9. [82] In a typical experiment, fluorescence was induced upon a 20 min incubation of the N‐terminally GFP 10‐11‐tagged GPCR with 2 μM of GFP 1–9. [82] The split GFP system allows to move the recognition sequence from the very N terminus into the extracellular loops (ECL) of GPCRs, increasing the intrinsic assay flexibility.[ 110 , 111 ] Recently, the splitGFP technique was applied to other GFP derivatives such as the red fluorescent mCherry. [112] The tagged GPCR needs to be carefully characterized prior to protein labeling since the introduction of protein moieties within the ECL can abolish receptor‐ligand interactions.[ 113 , 114 ]
Split luciferases offer an alternative to split FPs. A particularly noteworthy example involves the nanoLuciferase (Nluc), which provides brighter chemiluminescence than Renilla luciferase. [115] The luminophore was split into an 11 aa peptide‐tag (∼1.3 kDa, HiBiT) and a cell‐impermeable protein fragment (∼18 kDa, LgBiT; Figure 2). Since HiBiT and LgBiT display high affinity towards each other (K D=0.7 nM), the luminophore is readily reassembled at low concentration (10 nM) of the LgBiT agent.[ 80 , 116 ] In a typical assay, LgBiT and furimazine are added to cells expressing the HiBiT‐tagged POI without washing. Luminescence imaging is performed after 20 min incubation. [81] Applied to the monitoring of internalization of the adenosine A1 receptor (A1AR) and β2 adrenergic receptor (β2AR) in live cells, a decrease of the luminescent signal is observed when internalized receptors are excluded from interactions with the cell‐impermeable LgBiT.[ 81 , 117 ] Recently, surface‐specific labeling by the HiBiT‐LgBiT system was used to study the potential of the relaxin family receptor‐1 (RXFP1) to undergo ligand‐induced homodimerization. [118] The assay was based on the distance‐dependent bioluminescence resonance energy transfer (BRET) between a luciferase‐ and mCitrine‐tagged RXFP1. Pairs comprised of the constitutively active Nluc and mCitrine reporters showed BRET responses indicative of ligand‐independent formation of RXFP1 dimers. This assay collects signals from intracellular receptors and surface receptors. By contrast, the combination of HiBiT‐LgBiT and mCitrine reporters provides a surface‐specific read‐out and showed that the RXFP1‐RXFP1 proximity on the surface is not due to stable interactions but most likely due to overexpression.
Figure 2.
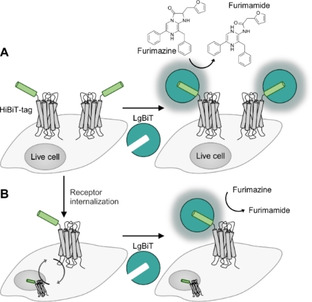
Reassembly of reporter proteins on GPCR based on small peptide tags. A) The luciferase Nluc can be reconstituted by an N‐terminal peptide tag (HiBiT, light green) and addition of LgBiT (jade green). The reassembled enzyme catalyzes the oxidative decarboxylation of furimazine. Formation of furimamide is accompanied by luminescence. Substrate consumption, correlates to the number of proteins (gray) on the cell surface. B) To quantify receptor internalization, control cells and cells treated with agonist are incubated with LgBiT. The bioluminescence signal is proportional to the number of reassembled luciferase.
Besides the reconstitution of fluorescent or luminescent enzymes on membrane‐embedded proteins, complementation platforms have the potential to introduce different reporters on the POI. The fibronectin‐binding protein FbaB from Streptococcus pyogenes, contains a reactive domain, which is capable of spontaneously forming an isopeptide bond between Lys and Asp. [83] Howarth and co‐worker split the FbaB into two fragments and developed a self‐assembling protein labeling methodology. The SpyTag, a short (13 aa), Asp‐containing peptide, is thereby fused to the POI, reacts with the Lys‐containing SpyCatcher (∼15 kDa), forming an amide bond between the two reactive side chains, and covalently attaching the SpyCatcher onto the POI. [83] SpyTag‐carrying ICAM1 (intercellular adhesion molecule 1) were fluorescently labeled on the surface of HeLa cells with SpyCatcher, equipped with Alexa Fluor 555 by incubation with 5 μM SpyCatcher‐Alexa Fluor 555 (in PBS, supplemented with 5 mM Mg2+) for 15 min. [83]
2.4. Coiled‐coil heterodimers enabling detailed analysis of membrane protein interactions on live cells
Inducible labeling facilitates real‐time monitoring of GPCR or RTK subpopulations that localize to or pass through the cell membrane. High rates of labeling are desirable to enable analyses of fast processes. Peptide‐peptide interactions based on coiled‐coil motifs combine high speed of labeling with small tag size. Coiled‐coil motifs are based on heptad repeats, displaying defined patterns of ionic and hydrophobic residues on the interaction interface.[ 119 , 120 , 121 , 122 ] For receptor labeling, one of the peptide sequences is fused to the N terminus of the POI. Addition of a suitable “complementary” peptide triggers the formation of a superhelix comprised of two peptides, which wrap around each other.[ 123 , 124 ] Labeling the added peptide with a suitable reporter, for example, a fluorescent organic dye enables protein visualization upon heterodimerization. A variety of different coiled‐coil peptide pairs have been established for selective labeling of membrane proteins, ranging from metal‐dependent leucine zippers to artificially generated coiled‐coil motifs (see Table 2 for an overview). Because coiled‐coil sequences can be designed de novo, the heterodimers have the intrinsic potential to be tailored towards specific applications. [125] Nanomolar affinities are achieved with ≥21 aa, resulting in only minor molecular weights added to the POI (ca. 2–6 kDa). Compared to globular tags such as the reassembled Nluc and GFP, coiled‐coil heterodimers are less bulky, and, thus, less likely to impair vesicular transport and membrane insertion of the tagged receptor. Due to the small size, coiled‐coil tags are less likely to interfere with ligand binding to the POI and lateral protein/protein interactions.
Table 2.
Coiled‐coil interactions established as tag/probe systems for labeling membrane‐embedded proteins on live mammalian cells. Core interaction sequences are given for each tag and probe peptide combination (1‐letter code). Additional sequences needed for genetic fusion or bacterial expression are excluded. Underlined tag/probe sequences have been equipped with reactive moieties for covalent labeling of proteins by different chemistries.
|
Coiled‐coil system |
Labeling conditions |
|
|---|---|---|
|
|
||
|
E3‐tag |
EIAALEKEIAALEKEIAALEK |
Noncovalent labeling: incubation with 20–60 nM fluorescently labeled K3 or K4 peptide for 2 min. [127] See also Figure 4. Covalent labeling: incubation of Cys‐E3‐tagged POI with 100 nM probe peptide (equipped with cargo through thioester linkage) for 2–5 min.[ 87 , 136 , 137 ] See also Figure 7C. |
|
E4‐tag |
EIAALEKEIAALEKEIAALEKEIAALEK |
|
|
K3 probe |
KIAALKEKIAALKEKIAALKE |
|
|
K4 probe |
KIAALKEKIAALKEKIAALKEKIAALKE |
|
|
ZIP tag/probe [a][85] |
|
|
|
A2‐tag[a] |
ALKKELEAAKKELEALKKELA‐GGCGG‐ALEKELEALEKEAEALEKELA |
Labeling of the A2‐tag is performed by applying 1 μM of the A2 probe for 15 min. [85] See also Figure 5. |
|
A2 probe |
ALKKKLEALKKKXEALKKKLA |
|
|
|
|
|
|
CCE/CCK system [84] |
|
|
|
CCE3‐tag |
EVAALEKEVAALEKEVAALEK |
The labeling reaction is performed after a 10 min incubation with 0.5 mM TCEP (at room temperature) by administration of 0.2–1 μM peptide probe (reaction time: 20 min at room temperature). [84] See also Figure 7A. |
|
CCK3 probe |
KVAALKEKVAALKEKVAALKE |
|
|
ER3/RCL3 system [138] |
|
|
|
ER3‐tag |
EIAALEREIAALEREIAALERGKGSIEGR |
Covalent labeling is performed by applying 150 nM fluorescently labeled probe peptide (reaction time: 10–20 min at 25 °C). [136] See also Figure 7B. |
|
R3CL probe |
RIAALRERIAALRERIAALREGC |
|
|
VIP Y/Z [b][86] |
|
|
|
CoilY |
NTVKELKNYIQELEERNAELKNLKEHLKFAKAELEFELAAHKFE |
After a blocking step with BSA, optimal labeling was achieved when cells were treated with 300 nM probe for 30 min in buffered cell medium after a BSA blocking step. [86] |
|
CoilZ |
QKVAQLKNRVAYKLKENAKLENIVARLENDNANLEKDIANLEKDIANLERDVAR |
|
|
VIPER [b][90] |
|
|
|
CoilE |
LEIEAAFLERENTALETRVAELRQRVQRLRNRVSQYRTR |
After a blocking step with BSA, labeling was performed using 100–500 nM peptide probe (reaction time: 15–30 min at 4 °C). [90] See also Figure 6. |
|
CoilR |
LEIRAAFLRQRNTALRTEVAELEQEVQRLENEVSQYETR |
|
|
MiniVIPER [b][91] |
|
|
|
MiniE |
LEIEAAFLERENTALETRVAELRQRVQRLRNE |
After a blocking step with BSA, labeling was performed with 100 nM peptide probe (reaction time: 30 min at 4 °C). [91] |
|
MiniR |
LEIRVAFLRQRNTALRTEVAELEQEVQRLENR |
|
|
|
||
|
P1‐tag |
EIQALEEENAQLEQENAALEEEIAQLEY |
Covalent cargo transfer is carried out on in <5 min using 100 nM peptide probe at room temperature. [89] See also Figure 10. |
|
P2 probe |
KIAQLKEKNAALKEKNQQLKEKIQALKY |
|
[a] X=2,3‐diaminopropionic acid. [b] Core sequences enabling coiled‐coil interaction of the tag/probe system. For VIP/VIPER/MiniVIPER both sequences can be used as tag or probe respectively, but require additional tags for purification upon bacterial expression.
The first coiled‐coil system used for receptor imaging was based on the de novo designed “IAAL” E/K coil system. [126] The E peptide contains glutamic acid residues on positions “e” and “g” (Figure 3). In the K peptide, these positions are occupied by lysine residues. Both peptides have hydrophobic isoleucine at the “a” position, and leucine at the “d” position. Positions “b” and “c” are occupied by alanine. The interaction of a 21 aa E3 peptide (=3 heptad repeats) with a 21 aa K3 peptide results in the formation of a heterodimeric coiled‐coil with a stability in the range of K D ∼70 nM. [126] Extension to four heptad repeats in the E4 (28 aa)/K4 (28 aa) pair provides even higher stability (K D ∼6 nM).[ 126 , 127 ] In their pioneering work, Matsuzaki and co‐workers used the E/K coiled‐coils for imaging of a prostaglandin E2 receptor (EP3βR) and a β2‐adrenergic receptor (β2AR) expressed on living CHO (Chinese hamster ovary) cells. [127] Like in all other applications of the E3/K3 methodology, the E3‐tag was fused to the N terminus of the receptors. For labeling, cells were incubated with 20–60 nM fluorescence labeled K3 or K4 peptide for 2 min, which were prepared by solid phase peptide synthesis (SPPS). Using a mixture of two distinct K4 probes carrying either a tetramethylrhodamine (TAMRA) dye or a pH‐sensitive fluorescein dye enabled a rapid read‐out of agonist‐induced uptake of E3‐tagged β2AR into endosomes. [88] Fluorescein emission is quenched in acidic compartments leading to increases of the TAMRA/fluorescein emission ratio when receptors localize to acidic compartments. It should be noted, however, that E/K coils are not stable at acidic conditions. Furthermore, this technique has been applied in nonfluorescent applications. Nakase, et al. used K4‐coated exosomes to cluster E3‐tagged EGFR and induce internalization. [128] Charging K4‐coated exosomes with cell‐toxic amounts of saponin allowed selective killing of E3‐POI expressing cells.
Figure 3.
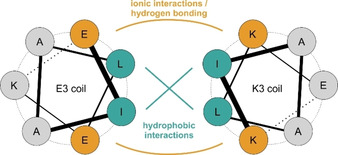
Helix orientation in the parallel E3/K3 heterodimer. Interaction interfaces of the E3 (sequence: (EIAALEK)3) and K3 (sequence: (KIAALKE)3) coils are depicted by the hydrophobic interface (jade green); adjacent ionic interactions (yellow) stabilize the hydrophobic interaction of the peptide helices.
An important aspect of the behavior of membrane‐embedded receptors are lateral receptor/receptor interactions within the lipid bilayer. For example, RTKs form dimers and various oligomerization modes have been reported for this protein family.[ 129 , 130 ] To clarify the oligomerization state of the EGFR in absence and presence of its ligand EGF, Yamashita, et al. labeled the E3‐tagged RTK with a mixture of Alexa568‐ and Alexa647‐K4 peptides added at defined ratios and determined the apparent efficiency of fluorescence resonance energy transfer (FRET) as a function of donor molar fraction [131] as previously described for the neurokinin‐1 receptor labeled by means of an ACP fusion. [132] Using this approach, it was shown that the RTK predominantly exists as monomer in the absence of ligand, whereas agonist addition lead to the formation of EGFR dimers (Figure 4A). Of note, the investigation revealed that the EGF concentration required to evoke half‐maximal dimerization is lower than that for half‐maximal auto‐phosphorylation (Figure 4B).
Figure 4.
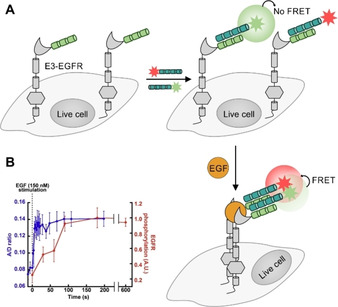
Application of N‐terminal protein labeling by coiled‐coil interaction to investigate EGFR dimerization on the surface of live cells. A) Heteromeric coiled‐coil interactions enable competitive labeling of E3‐tagged GPCR (light green) by using K4 donor probes (jade green) with two FRET‐compatible dyes (red/green stars). In the absence of ligand, EGFRs remain as monomers on the cell surface. B) Addition of EGF (yellow) induces E3‐EGFR dimerization, triggering the FRET between the two Using FRET as readout revealed that EGFR dimerization occurs prior to EGFR cross‐phosphorylation (left). FRET data derived from Yamashita, et al. [131]
In light of the controversial discussion about lateral interactions between GPCRs, Matsuzaki and co‐workers investigated the homodimerization of the CXCR4, the dopamine 2 receptor (D2R) and the prostaglandin E receptor type 1 (EP1R) by coiled‐coil labeling. [133] The dependence of FRET on donor molar fraction showed that, while no constitutive homodimerization of the tested GPCRs was detectable, CXCR4 and EP1R clustering was detectable upon ligand administration. However, the time course of FRET emergence was lacking behind the time course of the ligand‐induced Ca2+ influx. The authors hypothesized for certain GPCRs, that dimerization can lack behind signaling, suggesting that signaling occurs from receptor monomers followed by clustering upon ligand‐induced internalization through clathrin‐coated pits during the course of internalization. The notion of signaling in a monomeric state contradicts data obtained by bimolecular fluorescence complementation (BiFC) assays.[ 134 , 135 ] However, measurements of dimerization by BiFC is arguably less accurate because i) interaction between split FPs can induce proximity, ii) slow dissociation kinetics of complemented split FPs perturb equilibria and iii) variations of molar ratios of two artificially expressed proteins is difficult. In contrast, coiled‐coil labeling is reversible, excludes reporter reassembly as driving force of the POI interaction and allows variations of FRET donor/acceptor ratios which facilitates quantitative measurements.
Recently, the Jungmann and Seitz groups joined in order to develop a super resolution microscopy technique which is based on the reversible formation of E/K coiled‐coils. [140] In this method, transient interactions between a fluorescent molecule and the POI lead to localized blinking which is the basis for subdiffraction resolution imaging method known as PAINT (in points accumulation in nanoscale topography). A systematic study provided an E/K coiled‐coil that is in a dynamic equilibrium between bound an unbound states. It was shown that this optimum equilibrium of binding and dissociation was obtained with an E3 peptide (21 aa) interacting with 18 or 19 aa K peptides (K D≈1.7 μM or 81 nM, respectively). By conjugation of the modified E3‐tag to (secondary) anti‐α‐tubulin and anti‐vimentin antibodies using DBCO‐sulfo‐NHS ester chemistry, the intricate cellular cytoskeleton could be visualized by super‐resolution microscopy. Imaging speed and efficiency of PAINT enabled by the E/K coiled‐coil interaction outperformed the previously reported DNA PAINT method.
The Tamamura group developed a fluorogenic labeling method, which relies on the formation of trimeric coiled‐coil. Two α‐helical peptides derived from the GCN4 leucine zipper were linked through a 5 aa loop. The 47 aa construct (A2‐tag) was genetically fused to the N terminus of the GPCR CXCR4. A 23 aa probe peptide (A2‐peptide) was labeled with a 4‐nitrobenzenzo‐2‐oxa‐1,3‐diazole (NBD) dye, which is positioned within a hydrophobic environment upon formation of the 70 aa tag/probe complex (Figure 5). [85] A wavelength shift and enhancements of fluorescence enables a wash‐free imaging. In the pursuit of a heterodimeric pair combining high affinity with a balance of basic and acidic residues within each strand, Beatty and co‐workers introduced the VIP (versatile interacting peptide) tag. [86] This system consists of two genetically encoded peptide tags: CoilY and CoilZ, derived from synthetic heterodimeric coiled‐coils (SYNZIP). [141] Both sequences can be used as protein tag or peptide probe respectively (CoilY‐tag/CoilZ probe and CoilZ‐tag/CoilY probe). To establish labeling by the VIP system, the 44 aa tags were fused to membrane‐anchored fluorescent proteins GFP and mCherry. The 69 aa labeling probes were generated by bacterial expression and equipped with the fluorescent reporter or a biotin moiety through a cysteine residue. For protein labeling in live cells, a blocking step with high concentration of BSA (6 %, w/v) was applied prior to incubation of probes. Optimal labeling was achieved when cells were treated with 300 nM probe for 30 min in buffered cell medium. [86]
Figure 5.
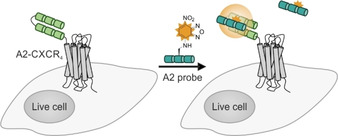
GPCR labeling on cell surfaces by using trimeric coiled‐coils. Two α‐helical peptides (A2‐tag, light green), linked by a short loop, are genetically fused to the N terminus of the CXC‐motif chemokine receptor 4(CXCR4, gray). The A2 peptide probe is equipped with 4‐nitrobenzenzo‐2‐oxa‐1,3‐diazole (NBD, yellow). Upon formation of a tag‐probe complex, NBD is positioned within a hydrophobic environment, inducing fluorescence enhancement (compared to unbound dye‐peptide probes).
The Beatty lab also introduced the VIPER system, which is comprised of the 43 aa CoilE tag and the 69 aa CoilR tag and involves the formation of a very stable coiled‐coil (K D=13 pM). [90] The system was applied (Figure 6) to investigate the internalization of the transferrin receptor 1 (TfR1) protein complex in transfected CHO cells by using quantum dots as reporter groups for electron microscopy. Labeling succeeded within 15 min. The method enables correlated light‐electron microscopy (CLEM) imaging. Of further notice is the application of VIPER to intracellular targets. [142] The CoilE‐acceptor peptide was genetically fused to the histone protein H2B, whereas the CoilR probe was delivered to the POI by using nanoshells equipped with cell‐penetrating peptides. Very recently, Beatty and co‐workers reduced the size of their coiled‐coil sequences and introduced the 32 aa MiniE and MiniR tags, which are recognized by 64 aa MiniR and MiniE probes. [91]
Figure 6.
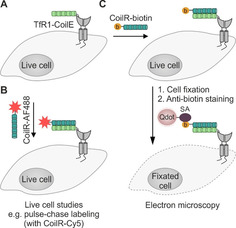
Labeling of membrane proteins by using the VIPER system. A) The transferrin receptor 1 (TfR1, gray) is fused to the CoilE‐tag. B) Administration of the CoilR probe, equipped with an AF488 fluorophore (red star) enables visualization of the POI and live cell experiments. C) The CoilR can be equipped with a biotin group (yellow) to label the protein. Upon cell fixation, the biotin label is addressed by a streptavidin probe (SA, purple) quarrying a quantum dot (Qdot, light pink), allowing electron microscopy.
Labeling by formation of coiled‐coils is reversible. Large coiled‐coils provide very high stability. The size of ultrastable coiled‐coil tags such as the VIPER‐tag (≈13 kDa) still compares favorably with that of enzyme‐based tags such as the SNAP‐tag (20 kDa). Smaller tags such as the E3/K3 tag add less molecular weight and SPPS provides facile access to various functionalized labeling probes given the peptides are short. However, with short coiled‐coils dissociation may occur at dilution conditions after prolonged times and under acidic conditions within endosomal compartments.
Coiled‐coil guided conjugation reactions offer the prospect of combining high stability of labeling with small tag size. Xia and co‐workers developed a method for the proximity‐induced covalent labeling upon formation of an E3/K3 coiled‐coil derivative (CCE3/CCK3 system) featuring valine instead of isoleucine. [84] The reaction involved a cysteine residue at an internal position of the CCE3‐tag and a chloroacetyl group that was connected to the “side chain” of 2,3‐diaminopropanoic acid of a CCK3 peptide derived labeling probe (Figure 7A). For labeling, CHO cells expressing the CCE3‐tagged POI were incubated for >20 min with a bidentate, 53 aa reactive CCK3 peptide in 0.2–1 μM concentration. Yano, et al. reported on a covalent cargo transfer method based on the free N‐terminal amino function of the E3‐tagged POI. [138] The R3CL3 peptide probe was C‐terminally armed with a sulfosuccinimidyl ester. For this purpose, a cysteine side chain was modified in a reaction with a maleimide‐based bifunctional reagent. Formation of the coiled‐coil brings the reactive moiety into close proximity of a lysine side chain near the C terminus of the ER3‐tag. This covalently crosslinks the R3CL probe with the ER3‐tag, yielding a 5.5–6 kDa label (Figure 7B). For efficient labeling, CHO cells expressing ER3‐tagged β2AR were incubated with 150 nM probe for 2×10 min. In contrast to experiments with unreactive coiled‐coils, the label remained after 5 washes with PBS, indicating the stability provided by covalent bonds. Within the context of work funded by the Deutsche Forschungsgemeinschaft (DFG) Priority Program SPP1623 (BE1264‐15) we aimed for a labeling method that provided for very high reaction rates at small tags. To achieve this aim, we repurposed the E3/K3 system for covalent labeling by a proximity‐triggered acyl transfer reaction.[ 87 , 137 ] The E3‐tag was elongated by an additional cysteine residue at its N terminus (Cys‐E3‐tag). This cysteine served as an acceptor of the reporter group. To enable fast labeling by native chemical ligation (NCL)‐type reactions,[ 143 , 144 ] the reporter group was linked as a thioester to a mercaptoaryl group mounted at the N terminus of the K3 peptide probe (Figure 7C). The end‐of‐helix arrangement brings the reactive groups into very close proximity, and coiled‐coil‐induced label transfer proceeded with t 1/2=1–2 min. Of note, the reaction does not ligate the E3‐tag with the K3 probe and the overall label size remains small (≈3 kDa). A typical labeling reaction involves 100 nM K3 conjugate and is performed for 5 min only. Using this peptide‐templated NCL, several pharmacologically interesting GPCRs like the human neuropeptide Y receptor type 1, 2, 4, and 5 (hY1/2/4/5R), the neuropeptide FF receptors 1 and 2 (NPFF1/2R), and the dopamine receptor 1 (D1R) were efficiently visualized in live cell experiments, circumventing the need for C‐terminal auto‐fluorescent proteins (Figure 8). [87] We showed that efficient labeling with the fluorophore TAMRA, attached to the K3 peptide, was only observable for HEK293 cells expressing the respective receptor. No off‐target signals were detected, highlighting the selectivity of the peptide‐templated NCL‐type reaction. As expected, the label withstands mild acidic (pH 5) and mild basic (pH 9) conditions, which would not be possible with labels installed by noncovalent coiled‐coils. As a result, the label persists the passage through endosomal compartments when tagged receptors internalize upon stimulation with agonist and recycle to the membrane. The high speed of labeling by the peptide‐templated acyl transfer is advantageous for investigations of short‐lived cellular processes. Furthermore, the stability provided by covalently attached labels facilitates long‐term pulse‐chase experiments and circumvents the need for auto‐fluorescent proteins fused to the POI. In an exemplary study, we examined intracellular sorting of internalized hY2R by two color pulse‐chase labeling in live mammalian cells (Figure 9). [136] Prior to agonist‐mediated internalization, the Cys‐E3‐hY2R was pulse labeled by means of a 3 min incubation with 150 nM TAMRA‐armed K3 conjugate. Chase labeling of membrane‐retained Y2R subpopulations was performed using a green Atto488‐carrying donor probe. This enabled the intracellular tracking of internalized hY2R subpopulations by multi‐color visualization with high spatial and temporal resolution due to the short labeling protocol. It was shown that hY2R populations, which have been internalized at different time points, are not trafficked separately but instead are pooled within minutes in early or sorting endosomes. Recently, Gavins, et al. reported on a novel coiled‐coil labeling system which enables the erasable imaging of GPCRs and RTKs (Figure 10A). [89] The method relies on the 28 aa P1 and P2 coiled‐coil peptides introduced by Jerala. [139] An N‐terminal Cys‐P1‐tag was fused to the human EGFR, and the endothelin B receptor (ETBR), a rhodopsin‐like GPCR and labeled upon incubation with a “complementary” peptide probe P2. The labeling reaction installed a peptide nucleic acid (PNA) strand which was transferred from the thioester linked PNA‐P2 conjugate to Cys‐P1‐POI. Like the E3/K3 system, the covalent transfer of the cargo relies on a proximity‐triggered NCL and can be carried out on different mammalian cell lines (HEK293 and CHO) in less than 5 min using low probe concentrations (e. g., 100 nM PNA‐MPAA‐P2). Once the POI is equipped with the PNA cargo, the PNA label subsequently serves as barcode for the hybridization with fluorescence labeled complementary DNA, which is complete upon <5 min incubation with 200 nM DNA probe. Labeling of human EGFR in live cells with organic dyes like TAMRA, Cy3, Cy7, and Atto565 demonstrated the high versatility offered by the modularity of the approach and the commercial availability of dye‐labeled oligonucleotides. The method provides a convenient way to increase the brightness of labels. For this purpose, the PNA tag is hybridized with a sticky end DNA complex prepared by alignment of multiple fluorescent DNA strands along a DNA adaptor strand. An interesting aspect of the PNA barcoding approach is the removal of hybridized fluorescent DNA by toehold‐mediated strand displacement, allowing erasure of the fluorescent signals from the cell membrane (Figure 10B). This is especially interesting for GPCRs and RTKs with only minor internalization rates. In this case, internalized subpopulations can be tracked without noise from membrane‐residing POI after incomplete receptor internalization.
Figure 7.
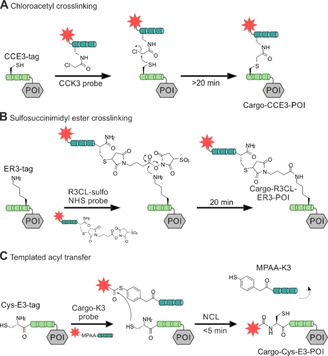
Covalent protein labeling at the protein mediated by coiled‐coil peptide heterodimers. A protein of interest (POI, grey box) tagged with a peptide recognition tag (green) at the N terminus is selectively labeled with a cargo (red star) by means of a proximity‐triggered reaction. Covalent protein labeling is achieved by chloroacetyl crosslinking (A), sulfosuccinimidyl ester crosslinking (B) or templated acyl transfer (C).
Figure 8.
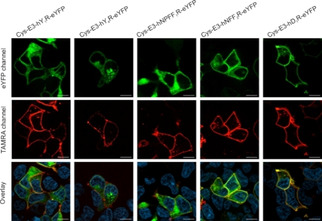
Peptide‐templated labeling of N‐terminally Cys‐E3‐tagged G protein‐coupled receptors (GPCR) and its application to live cell imaging. Labeling was performed on GPCRs carrying a C‐terminal eYFP (enhanced yellow fluorescent protein) in transient transfected HEK293 cells. N‐terminal fusion of the Cys‐E3‐tag was performed to the human receptors neuropeptide Y receptor 1 and 4 (hY1/4R), the neuropeptide FF receptor 1 and 2 (hNPFF1/2R), and the dopamine receptor 1 (hD1R). Microscopy data adapted from Reinhardt, et al. [87]
Figure 9.
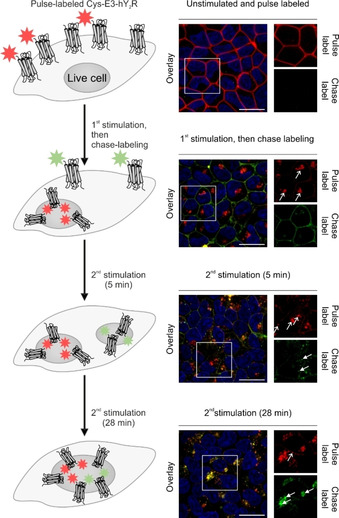
Investigation of distinct human neuropeptide Y receptor 2 (hY2R) subpopulations by pulse‐chase labeling. Cys‐E3‐tagged hY2R were pulse labeled (red) by peptide‐templated NCL prior to agonist‐induced internalization. After the first stimulation, the remaining Cys‐E3‐hY2R, residing in the cell membrane, were chase labeled (light green) by peptide‐templated NCL. Chase‐labeled receptors were internalized upon agonist addition and co‐localized with chase‐labeled hY2R populations after >10 min (fused vesicles indicated by white arrows). Fusion of pulse‐ and chase‐labeled vesicles shown by (merged) yellow structures in the overlay pictures. Data adapted from Lotze, et al. [136]
Figure 10.
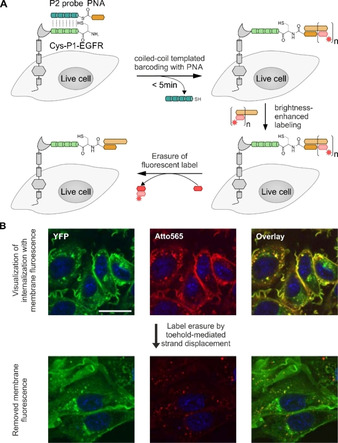
Labeling of membrane proteins by coiled‐coil‐templated barcoding with peptide nucleic acid (PNA). A) The POI (gray) was N‐terminally fused to the peptide tag Cys‐P1‐tag (light green). Addition of peptide probe P2 (jade green) triggers a coiled‐coil‐induced labeling reaction that installs a PNA tag. The PNA‐Cys‐P1‐POI is fluorescently labeled by hybridization with a complementary oligonucleotide (light brown bar) equipped with a fluorescent dye (red star). Brightness‐enhanced labeling of PNA‐Cys‐P1‐POI is achieved by hybridization with an elongated DNA strand, allowing multiple hybridization steps with smaller fluorescently labeled oligonucleotides (light red bar). Using toehold‐mediated strand displacement, the fluorescence is erased from the PNA by displacing the small fluorescent DNA fragments with complementary DNA sequences (dark red bar). B) EGFR was labeled with Atto565 by coiled‐coil‐induced PNA barcoding and DNA hybridization, and subsequently treated with EGF. It is difficult to detect internalized EGFR behind the background of EGFRs on the membrane (upper row). Internalized receptors are visualized with higher signal‐to‐noise ratios upon toehold‐mediated strand displacement (lower row). Microscopy data adapted from Gavins, et al. [89]
Recently, Jerala introduced a set of six orthogonal coiled‐coil pairs. In a noteworthy application, three coiled‐coil pairs were used to equip fluorescent proteins with localizers based on nuclear localization sequences, nuclear export signals or a membrane localization protein. [145] Beatty combined two coiled‐coils, the MiniVIPER and the VIPER systems, for orthogonal noncovalent labeling of transferrin receptor TfR1 and histon protein H2B as examples for two distinct protein targets. [91]
2.5. Recognition of small amino acid tags by (semi)‐metals
A very small size is the characteristic feature of the self‐labeling tetracysteine motif (CCXXCC; X=A, G, or P). Covalent labeling of this motif is achieved by fluorescein arsenical helix binder (FlAsH), containing two arsonous acid thioester centers (Figure 11A). [146] The typically used FlAsH derivatives provide fluorescence enhancement upon binding to the tetracysteine motif. For extracellular labeling, cells expressing the tagged protein need to be pre‐incubated with reducing agents (e. g., 5 mM 2‐(N‐morpholino)ethanesulfonic acid (MES) and 0.5 mM TCEP in HBSS for 20–30 min) to ensure that free thiols are accessible for on surface labeling prior to addition of the labeling reactant. [92] Subsequent labeling involves 2–5 μM concentrations of the arsenic FlAsH for efficient tag conversion. [92] Despite reports about background signals and off‐target labeling[ 147 , 148 , 149 ] FlAsH tagging has been widely applied to study GPCR interactions e. g. by determination of pure rotational diffusion of the β2aAR on live cells. [93] For an alternative to FlAsH, the cysteine residues were substituted by serine residues (tetraserine motif; SSXXSS; X=A, G, or P), which enable recognition by less toxic bisboronic acid probes. [150] However, the abundancy of polyserine‐containing proteins causes a high background.
Figure 11.
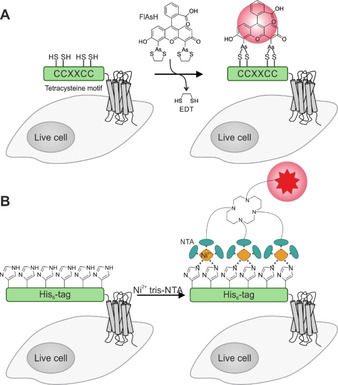
Labeling of membrane proteins such as GPCRs by metal ions recognizing small amino acid tags on live cells. A) A tetracysteine fragment (CCXXCC; X=A, G, or P; green) is genetically fused to the N terminus of a GPCR (gray). Addition of FlAsH (fluorescein arsenical hairpin binder) transfers the bisarsenic fluorescein onto the free thiols of the cysteine stretch, thus enhancing the fluorescence of the dye. B) The His6‐tag (green), fused to the N terminus of a membrane protein like a GPCR (gray), can be labeled with Ni2+‐loaded tris‐NTA (N‐nitrilotriacetic acid). The metal ion is selectively chelated by clustered imidazole side chains of the oligo‐histidine sequence. To visualize the POI, the tris‐NTA is equipped with a respective reporter moiety like an organic dye.
Selective ion chelation is commonly used in protein purification, for example, for immobilizing oligo‐His‐tagged proteins on substrates containing N‐nitrilotriacetic acid (NTA) complexes of Co2+, Ni2+, or Zn2+.[ 151 , 152 ] This approach has been repurposed to live cellfluorescence imaging (Figure 11B). [153] NTA was equipped with an organic fluorophore and used to label N‐terminally oligo‐His‐tagged platelet derived growth factor (PDGF) receptor. [96] Visualization can be performed in <15 min, using only 15 nM of dye‐conjugated, Ni2+‐loaded NTA. The affinity of the His‐stretch for the metal ion correlates with the length of the oligo‐His sequence. His10 displays a sixfold higher Ni2+affinity than the shorter His6‐tag (K D∼166 nM and 1.05 μM, respectively). [94] The binding of NTA‐chelated Ni2+ to histidine involves two distinct coordination sites, able to recognize separate imidazole moieties with a binding affinity of approximately ∼10 μM.[ 154 , 155 ] The remaining four coordination sites of the cation are occupied by the chelating NTA. [155] Simultaneous binding of multiple Ni‐NTA units to multiple imidazole groups along a His6‐tag boosts the binding affinity as shown for different NTAs binding to oligo‐His6‐tags: mono‐NTA (K D∼13 μM)<bis‐NTA (KD∼68 nM)<tris‐NTA (K D∼2.1 nM). [154] The graded affinity of tris‐NTA probes for His6‐and His10‐tags enabled imaging of intracellular His10‐tagged POIs by complexation with His6‐tagged cell penetrating peptides.[ 156 , 157 ]
For covalent POI modification, a chloroalkane NTA entity was developed. The method allowed imaging of the GPCR B2R on live mammalian cells.[ 95 , 158 ] To facilitate crosslinking of the NTA with the POI a cysteine residue was added at the N terminus of the His‐tag (Cys‐His6‐tag). As shown for the bradykinin N2 receptor (B2R), a pre‐incubation with 0.5 mM TCEP was required prior to addition of 0.5 μM labeling probe. Efficient GPCR labeling was performed in less than 30 min. In addition to histidine‐mediated ion chelation, aspartate repeats can be applied for protein labeling as well. Several oligo‐aspartate‐tags are described (D3, D4, D4x2) to fine tune the affinity of Zn2+‐loaded probes, which have been successfully applied for N‐terminal labeling of the B2R and muscarinic acetylcholine receptor M1 using protocols, comparable to oligo‐His‐tag labeling.[ 159 , 160 , 161 ] Similar to the oligo‐His‐tags, oligo‐Asp‐mediated labeling is reversible due to the transient interaction with the metal ion, but can be converted into covalent POI modification by introducing a chloroalkane moiety into the metal loaded NTA and adding an N‐terminal cysteine residue into the oligo‐Asp‐tag. [158]
2.6. Special case: Traceless labeling by split‐intein‐mediated splicing
Despite small tag sizes, all of the aforementioned labeling platforms induce modifications of the endogenous protein sequence to generate reaction specificity. Ideally, protein labeling technologies do not alter or add to the POI sequence. This can be achieved by split intein‐mediated protein trans‐splicing (PTS) since the intein is removed from the POI during the ligation of the flanking exteins. [162] PTS is commonly applied to protein semi‐synthesis and modification.[ 163 , 164 , 165 , 166 ] For split intein‐based techniques, one fragment is synthetically generated and equipped with the reporter, whereas the complementary fragment is fused to the POI (Figure 12). Different split‐intein platforms have been developed based on natural occurring systems. The split α‐subunit of the DNA polymerase III (DnaE), derived from Nostoc punctiforme (Npu) [167] and Anabaena variabilis (Ava), [168] are amongst the most common split‐intein PTS platforms applied to protein labeling in cellular contexts.[ 169 , 170 , 171 ] High‐affinity inteins have been developed as demonstrated for the engineering of the split DnaB M86 mini‐intein system (Ssp DnaB M86), which displays a 10 nM fragment affinity. [172] For modification of proteins embedded in cell membranes, C‐terminal labeling was demonstrated for the TfR1 in CHO cells. [173] The C terminus of TfR1 was fused to the 150 aa N‐intein of the noncanonical split‐intein Ssp GyrB S11. The synthetic 6 aa short C‐intein was equipped with fluorescein for covalent labeling. PTS was efficient after 18 h incubation with micromolar C‐intein concentrations (1.25–12.5 μM). N‐terminal protein labeling on live cells was demonstrated for the red fluorescent protein (mRFP), anchored in the membrane of CHO cells. [98] The DnaBC(12–154) fragment (C‐intein) was fused to mRFP and labeled with 0.5 μM of the respective N‐intein, equipped with biotin, requiring 20 mM reduced glutathione and a reaction time of 8 h. Subsequent POI visualization was performed by anti‐biotin staining using fluorescent streptavidin. Recently, Mootz and co‐workers reported on the Gp41‐1 split intein and in cellulo semisynthesis of intracellular proteins in live mammalian cells using relatively short peptide fragments (N‐intein: 88 aa, C‐intein: 37 aa). [97] This split intein displays rapid PTS (t1/2∼5 s at 37 °C) with high yields.[ 174 , 175 ] However, split intein approaches are still limited to rather big POI modifications for efficient labeling, when targeting the N terminus of proteins like GPCRs and RTKs.[ 176 , 177 ] Despite the big POI modification, the reaction occurs traceless and removes the recognition site at the POI. [178]
Figure 12.
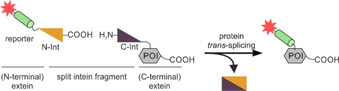
Reaction scheme of split‐intein‐mediated trans‐splicing (PTS) of protein N termini. For split‐intein‐based techniques applied to the protein N terminus, the C‐terminal fragment (C‐Int, purple) is fused to the target protein (POI, gray), while the N‐terminal fragment (N‐Int, yellow) is artificially generated to carry the required reporter unit (green with red star). Split‐intein‐mediated PTS is a traceless labeling approach as the intein fragments are removed from the POI during the ligation of the exteins.
3. Summary and Outlook
Our overview of the methods available for labeling of cell surface receptors points to a remarkable development. Early research had to rely on fusion of the POI with a fluorescent protein. More than two decades ago, Tsien, et al. introduced the FlAsH method which allowed labeling by short peptide tags. [146] After their seminal contribution, the field took off in the 2000s with a series of papers from the Johnsson, Ting and Ploegh groups on exploiting enzymes for protein labeling.[ 73 , 101 , 179 ] Using phosphopantetheinyl transferases (AcpS, Sfp), biotin ligase (BirA) and lipoic acid ligase (LplA) to transfer reporter groups from a donor substrate to specific peptide sequences was also based on genetically encodable tags. However, even after labeling the peptide tags are much smaller than fluorescent or bioluminescent fusion proteins. Furthermore, the reagents can be tailored to exclude intracellular protein populations from labeling. Enzyme‐based techniques facilitate the analysis of cell surface expression and internalization of receptors, and allow orthogonal labeling. However, substrates are often added in high concentration, co‐factors may be required, and the enzyme's specificity frequently limits the choice of the reporter group. In this case, bioorthogonal chemistries provide remedy by allowing secondary labeling of modified conjugates in a two‐step protocol. In the pursuit of highly efficient labeling methods, the self‐modifying SNAP‐ and Halo‐tags were developed. Surface‐specific labeling requires cell‐impermeable labeling agents.[ 180 , 181 ] In general, the size of the enzyme tags is a concern in investigating proximity‐dependent protein interactions. Complementation methods, in which short peptide tags guide the assembly of a fluorescent or luminescent protein also provide for robust labeling. The cargo added to the POI remains small prior to labeling, however, the size issue resurfaces after labeling.
Aiming for small tag sizes similar to peptide tags used for enzymatic labeling, but enabling labeling at much higher speed, Matsuzaki introduced the coiled‐coil labeling system. [127] With this method labeling succeeds on small tags (2–3 kDa for each tag and labeling probe) at nanomolar concentration within less than 5 min without extensive washing protocols. The method has proven valuable for the analysis of GPCR homodimerization by FRET measurements. [133] Contributions of the Tamamura and Beatty labs on fluorogenic labeling and stability‐improved coiled‐coils, showcased the versatility of the coiled‐coil method.[ 85 , 86 , 91 , 142 ] The adjustability of coiled‐coil interactions allowed the development of peptide PAINT for subdiffraction resolution imaging. [140] Seeking to combine both stability and speed of labeling, coiled‐coil interactions have been used to template proximity‐triggered chemical reactions. For example, acyl reporter groups can be transferred from thioester‐linked peptide probes onto cysteinyl peptide tags within 2–5 minutes.[ 87 , 136 ] An extension of the method enabled tagging of receptor proteins with PNA, which can serve as a universal landing hub for functional units through oligonucleotide hybridization. We have shown that erasable fluorescence labeling by toehold‐mediated displacement of oligonucleotide‐dye conjugates facilitated the analysis of EGFR internalization. [89]
Focusing on the application of known fusion proteins, metal ion recognition tags based on the interaction between oligo‐histidine tags and NiII⋅trisNTA complexes are noteworthy contributions due to their tunable affinity based on the oligo‐histidine length. [154]
The intein technology approaches an ideal labeling methodology, because the tag is excised upon labeling. For C‐terminal labeling, the short C‐terminal intein fragment carries the fluorophore and a bigger N‐terminal fragments is fused to the POI.N‐terminal labelling calls for fluorescence labelled N‐intein agents, which are – given their size – more difficult to prepare.
The breadth of methods allowing a cell surface specific labeling of receptor proteins is extremely useful for GPCR and RTK research. These receptors are amongst the most frequently addressed targets in medicinal chemistry, pharmacology and cell biology. Typical research questions refer to localization, internalization, trafficking, interaction partners and oligomerization of the receptors, preferably within their native environment. Fluorescence microscopy and the methods described in this review can provide answers to these questions.
In future research, multiplexed labeling will likely be important to study interactions/trafficking of a set of receptors. In this regard, a combination of orthogonal labeling methods will be needed as demonstrated by Jerala [145] and Beatty [91] labs using coiled‐coil heterodimers. Protein‐protein interactions need to be studied under native conditions – ideally, in tissues or in vivo models. Using gene editing tools like CRISPR/Cas9, it was already demonstrated that short tags can be used for POI labeling under native expression levels in cell culture models. [116] Additionally, efficient in vivo labeling requires cargos like quantum dots,[ 182 , 183 ] to reduce tissue‐derived noise. A major obstacle is the delivery of the labeling probe to the desired tissue, which is limiting to labeling platforms requiring huge probes with impaired tissue penetration properties. [184] Therefore, we should focus on methods that enable a free choice of labels to allow the introduction of superbright fluorophores with extreme selectivity. We believe that coiled‐coil platforms provide a suitable mix of properties. We also foresee an increasing importance for inducible labeling systems enabling the monitoring or even manipulation of receptor properties. For example, methods allowing an inducible homo‐ or heterodimerization by small tags are expected to shine light on the significance of receptor oligomerization and cross activation. This calls for modular methods that allow facile variations of the transferred label. It seems that such methods are available by now and the authors of this review article are confident that new imaging modalities and applications will emerge soon.
Conflict of interest
The authors declare no conflict of interest.
Biographical Information
Philipp Wolf earned his bachelor‘s and master's degrees in biochemistry from Leipzig University (Germany), broadening the scope of his studies with internships at the CSIC‐IQAC (Spain) and NSTDA‐BIOTEC (Thailand). He is now pursuing his PhD in Professor Annette G. Beck‐Sickinger's group at Leipzig University, focusing on investigating G protein‐coupled receptors by orthogonal labeling on the surface of live cells.

Biographical Information
Georgina Gavins completed her MChem in Medicinal and Biological Chemistry at The University of Edinburgh (UK), including an industry year at F. Hoffman‐la Roche (Basel, Switzerland). She is now working toward her PhD in chemistry in the working group of Professor Oliver Seitz at the Humboldt Universität zu Berlin (Germany) where she explores selective acyl transfer reactions on the membrane of living cells.

Biographical Information
Annette G. Beck‐Sickinger is a professor of biochemistry and bioorganic chemistry at Leipzig University (Germany). Her major research fields include structure‐activity relationships of peptide/protein hormones and G protein‐coupled receptors. She has been awarded many prizes including the Leonidas Zervas Award of the European Peptide Society, the gold medal of the Max‐Bergmann‐Kreis (2009), the Albrecht Kossel Award of Biochemistry of the GDCh (2018) and the Du Vigneaud Award of the American Peptide Society (2019). She is a member of the National Academy of Science Leopoldina in Germany and was awarded with the Saxonian Order of Merit in 2017.

Biographical Information
Oliver Seitz obtained his PhD from the University of Mainz (Germany) in 1995. After postdoctoral research at the Scripps Research Institute in La Jolla (USA) he moved to the University of Karlsruhe (Germany). In 2000, he became group leader at the Max‐Planck‐Institute of Molecular Physiology in Dortmund (Germany), and in 2003 he was appointed Full Professor at the Humboldt‐Universität zu Berlin. Oliver Seitz has a keen interest in developing chemistry that allows the interrogation and perturbation of cellular processes. Recently, he has focused on protein synthesis, peptide‐ and nucleic acid‐templated chemistry and RNA and protein imaging.

Acknowledgements
The financial support by the Deutsche Forschungsgemeinschaft (SPP1623, BE1264‐15) is kindly acknowledged. P.W. acknowledges the support of the FCI and the graduate school “Leipzig School of Natural Sciences – Building with Molecules and Nano‐objects” (BuildMoNa). Open access funding enabled and organized by Projekt DEAL.
P. Wolf, G. Gavins, A. G. Beck-Sickinger, O. Seitz, ChemBioChem 2021, 22, 1717.
Dedicated to Horst Kunz on the occasion of his 80th birthday.
Contributor Information
Prof. Annette G. Beck‐Sickinger, Email: abeck-sickinger@uni-leipzig.de.
Prof. Oliver Seitz, Email: oliver.seitz@chemie.hu-berlin.de.
References
- 1. Fredriksson R., Lagerström M. C., Lundin L.-G., Schiöth H. B., Mol. Pharmacol. 2003, 63, 1256–1272. [DOI] [PubMed] [Google Scholar]
- 2. Wingler L. M., Lefkowitz R. J., Trends Cell Biol. 2020, 30, 736–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gilman A. G., Annu. Rev. Biochem. 1987, 56, 615–649. [DOI] [PubMed] [Google Scholar]
- 4. Oldham W. M., Hamm H. E., Nat. Rev. Mol. Cell Biol. 2008, 9, 60–71. [DOI] [PubMed] [Google Scholar]
- 5. Zidar D. A., Violin J. D., Whalen E. J., Lefkowitz R. J., Proc. Natl. Acad. Sci. USA 2009, 106, 9649–9654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chaturvedi M., Maharana J., Shukla A. K., Cell 2020, 180, 1041–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Tian X., Kang D. S., Benovic J. L., Handb. Exp. Pharmacol. 2014, 219, 173–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Fantl W. J., Johnson D. E., Williams L. T., Annu. Rev. Biochem. 1993, 62, 453–481. [DOI] [PubMed] [Google Scholar]
- 9. Schlessinger J., Science 2004, 306, 1506–1507. [DOI] [PubMed] [Google Scholar]
- 10. Zhang X., Gureasko J., Shen K., Cole P. A., Kuriyan J., Cell 2006, 125, 1137–1149. [DOI] [PubMed] [Google Scholar]
- 11. Beadling C., Guschin D., Witthuhn B. A., Ziemiecki A., Ihle J. N., Kerr I. M., Cantrell D. A., EMBO J. 1994, 13, 5605–5615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Tian S. S., Lamb P., Seidel H. M., Stein R. B., Rosen J., Blood 1994, 84, 1760–1764. [PubMed] [Google Scholar]
- 13. Pessin J. E., Okada S., Endocr. J. 1999, 46, S11–6. [DOI] [PubMed] [Google Scholar]
- 14. Katz M., Amit I., Yarden Y., Biochim. Biophys. Acta 2007, 1773, 1161–1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Guo X.-X., An S., Yang Y., Liu Y., Hao Q., Xu T.-R., Cell. Physiol. Biochem. 2016, 39, 137–156. [DOI] [PubMed] [Google Scholar]
- 16. Lemmon M. A., Schlessinger J., Cell 2010, 141, 1117–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Östman A., Trends Cell Biol. 2001, 11, 258–266. [DOI] [PubMed] [Google Scholar]
- 18. Sigismund S., Argenzio E., Tosoni D., Cavallaro E., Polo S., Di Fiore P. P., Dev. Cell 2008, 15, 209–219. [DOI] [PubMed] [Google Scholar]
- 19. Sorkin A., Goh L. K., Exp. Cell Res. 2009, 315, 683–696. [DOI] [PubMed] [Google Scholar]
- 20. Jensen B. C., Swigart P. M., Simpson P. C., Naunyn-Schmiedeberg's Arch. Pharmacol. 2009, 379, 409–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Michel M. C., Wieland T., Tsujimoto G., Naunyn-Schmiedeberg's Arch. Pharmacol. 2009, 379, 385–388. [DOI] [PubMed] [Google Scholar]
- 22. Knappik A., Plückthun A., BioTechniques 1994, 17, 754–761. [PubMed] [Google Scholar]
- 23. Field J., Nikawa J., Broek D., MacDonald B., Rodgers L., Wilson I. A., Lerner R. A., Wigler M., Mol. Cell. Biol. 1988, 8, 2159–2165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hanke T., Young D. F., Doyle C., Jones I., Randall R. E., J. Virol. Methods 1995, 53, 149–156. [DOI] [PubMed] [Google Scholar]
- 25. Götzke H., Kilisch M., Martínez-Carranza M., Sograte-Idrissi S., Rajavel A., Schlichthaerle T., Engels N., Jungmann R., Stenmark P., Opazo F., et al., Nat. Commun. 2019, 10, 4403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Virant D., Traenkle B., Maier J., Kaiser P. D., Bodenhöfer M., Schmees C., Vojnovic I., Pisak-Lukáts B., Endesfelder U., Rothbauer U., Nat. Commun. 2018, 9, 930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Brown L. S., Ladizhansky V., Protein Sci. 2015, 24, 1333–1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Krug U., Gloge A., Schmidt P., Becker-Baldus J., Bernhard F., Kaiser A., Montag C., Gauglitz M., Vishnivetskiy S. A., Gurevich V. V., Beck-Sickinger A. G., Glaubitz C., Huster D., Angew. Chem. Int. Ed. 2020, 59, 23854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Schnell U., Dijk F., Sjollema K. A., Giepmans B. N. G., Nat. Methods 2012, 9, 152–158. [DOI] [PubMed] [Google Scholar]
- 30. Bertens P., Heijne W., van der Wel N., Wellink J., van Kammen A., Arch. Virol. 2003, 148, 265–279. [DOI] [PubMed] [Google Scholar]
- 31. Margolin W., J. Bacteriol. 2012, 194, 6369–6371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Stadler C., Rexhepaj E., Singan V. R., Murphy R. F., Pepperkok R., Uhlén M., Simpson J. C., Lundberg E., Nat. Methods 2013, 10, 315–323. [DOI] [PubMed] [Google Scholar]
- 33. Heo M., Nord A. L., Chamousset D., van Rijn E., Beaumont H. J. E., Pedaci F., Sci. Rep. 2017, 7, 12583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Küey C., Larocque G., Clarke N. I., Royle S. J., J. Cell Sci. 2019, 132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Liu H. S., Jan M. S., Chou C. K., Chen P. H., Ke N. J., Biochem. Biophys. Res. Commun. 1999, 260, 712–717. [DOI] [PubMed] [Google Scholar]
- 36. Lu C., Albano C. R., Bentley W. E., Rao G., Biotechnol. Bioeng. 2005, 89, 574–587. [DOI] [PubMed] [Google Scholar]
- 37. Zhang L., Patel H. N., Lappe J. W., Wachter R. M., J. Am. Chem. Soc. 2006, 128, 4766–4772. [DOI] [PubMed] [Google Scholar]
- 38. Jain R. K., Joyce P. B., Molinete M., Halban P. A., Gorr S. U., Biochem. J. 2001, 360, 645–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Yanushevich Y. G., Staroverov D. B., Savitsky A. P., Fradkov A. F., Gurskaya N. G., Bulina M. E., Lukyanov K. A., Lukyanov S. A., FEBS Lett. 2002, 511, 11–14. [DOI] [PubMed] [Google Scholar]
- 40. Keppler A., Gendreizig S., Gronemeyer T., Pick H., Vogel H., Johnsson K., Nat. Biotechnol. 2003, 21, 86–89. [DOI] [PubMed] [Google Scholar]
- 41. Janezic E. M., Lauer S. M.-L., Williams R. G., Chungyoun M., Lee K.-S., Navaluna E., Lau H.-T., Ong S.-E., Hague C., Sci. Rep. 2020, 10, 7209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kountz T. S., Lee K.-S., Aggarwal-Howarth S., Curran E., Park J.-M., Harris D.-A., Stewart A., Hendrickson J., Camp N. D., Wolf-Yadlin A., et al., J. Biol. Chem. 2016, 291, 18210–18221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Levoye A., Zwier J. M., Jaracz-Ros A., Klipfel L., Cottet M., Maurel D., Bdioui S., Balabanian K., Prézeau L., Trinquet E., et al., Front. Endocrinol. (Lausanne) 2015, 6, 167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Los G. V., Encell L. P., McDougall M. G., Hartzell D. D., Karassina N., Zimprich C., Wood M. G., Learish R., Ohana R. F., Urh M., et al., ACS Chem. Biol. 2008, 3, 373–382. [DOI] [PubMed] [Google Scholar]
- 45. So M.-k., Yao H., Rao J., Biochem. Biophys. Res. Commun. 2008, 374, 419–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lesiak L., Zhou X., Fang Y., Zhao J., Beck J. R., Stains C. I., Org. Biomol. Chem. 2020, 18, 2459–2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Martell J. D., Deerinck T. J., Sancak Y., Poulos T. L., Mootha V. K., Sosinsky G. E., Ellisman M. H., Ting A. Y., Nat. Biotechnol. 2012, 30, 1143–1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Lam S. S., Martell J. D., Kamer K. J., Deerinck T. J., Ellisman M. H., Mootha V. K., Ting A. Y., Nat. Methods 2015, 12, 51–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Qin J. Y., Zhang L., Clift K. L., Hulur I., Xiang A. P., Ren B.-Z., Lahn B. T., PLoS One 2010, 5, e10611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Forster K., Helbl V., Lederer T., Urlinger S., Wittenburg N., Hillen W., Nucleic Acids Res. 1999, 27, 708–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Giepmans B. N. G., Adams S. R., Ellisman M. H., Tsien R. Y., Science 2006, 312, 217–224. [DOI] [PubMed] [Google Scholar]
- 52. Jing C., Cornish V. W., Acc. Chem. Res. 2011, 44, 784–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Xue L., Karpenko I. A., Hiblot J., Johnsson K., Nat. Chem. Biol. 2015, 11, 917–923. [DOI] [PubMed] [Google Scholar]
- 54. Chen X., Wu Y.-W., Org. Biomol. Chem. 2016, 14, 5417–5439. [DOI] [PubMed] [Google Scholar]
- 55. Lotze J., Reinhardt U., Seitz O., Beck-Sickinger A. G., Mol. BioSyst. 2016, 12, 1731–1745. [DOI] [PubMed] [Google Scholar]
- 56. Schneider A. F. L., Hackenberger C. P. R., Curr. Opin. Biotechnol. 2017, 48, 61–68. [DOI] [PubMed] [Google Scholar]
- 57. Rodrigues A. R., Sousa D., Almeida H., Gouveia A. M., Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 1217–1226. [DOI] [PubMed] [Google Scholar]
- 58. Ulloa-Aguirre A., Zariñán T., Dias J. A., Conn P. M., Mol. Cell. Endocrinol. 2014, 382, 411–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Margeta-Mitrovic M., Jan Y. N., Jan L. Y., Neuron 2000, 27, 97–106. [DOI] [PubMed] [Google Scholar]
- 60. Beckett D., Kovaleva E., Schatz P. J., Protein Sci. 1999, 8, 921–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Jaykumar A. B., Caceres P. S., Sablaban I., Tannous B. A., Ortiz P. A., Am. J. Physiol. 2016, 310, F183–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Chen I., Howarth M., Lin W., Ting A. Y., Nat. Methods 2005, 2, 99–104. [DOI] [PubMed] [Google Scholar]
- 63. Fernández-Suárez M., Baruah H., Martínez-Hernández L., Xie K. T., Baskin J. M., Bertozzi C. R., Ting A. Y., Nat. Biotechnol. 2007, 25, 1483–1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Puthenveetil S., Liu D. S., White K. A., Thompson S., Ting A. Y., J. Am. Chem. Soc. 2009, 131, 16430–16438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. George N., Pick H., Vogel H., Johnsson N., Johnsson K., J. Am. Chem. Soc. 2004, 126, 8896–8897. [DOI] [PubMed] [Google Scholar]
- 66. Lambalot R. H., Gehring A. M., Flugel R. S., Zuber P., LaCelle M., Marahiel M. A., Reid R., Khosla C., Walsh C. T., Chem. Biol. 1996, 3, 923–936. [DOI] [PubMed] [Google Scholar]
- 67. Yin J., Lin A. J., Golan D. E., Walsh C. T., Nat. Protoc. 2006, 1, 280–285. [DOI] [PubMed] [Google Scholar]
- 68. Stüber J. C., Plückthun A., PLoS One 2019, 14, e0226579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Zhou Z., Cironi P., Lin A. J., Xu Y., Hrvatin S., Golan D. E., Silver P. A., Walsh C. T., Yin J., ACS Chem. Biol. 2007, 2, 337–346. [DOI] [PubMed] [Google Scholar]
- 70. Sato H., Ikeda M., Suzuki K., Hirayama K., Biochemistry 1996, 35, 13072–13080. [DOI] [PubMed] [Google Scholar]
- 71. Hu B.-H., Messersmith P. B., J. Am. Chem. Soc. 2003, 125, 14298–14299. [DOI] [PubMed] [Google Scholar]
- 72. Lin C.-W., Ting A. Y., J. Am. Chem. Soc. 2006, 128, 4542–4543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Popp M. W., Antos J. M., Grotenbreg G. M., Spooner E., Ploegh H. L., Nat. Chem. Biol. 2007, 3, 707–708. [DOI] [PubMed] [Google Scholar]
- 74. Tanaka T., Yamamoto T., Tsukiji S., Nagamune T., ChemBioChem 2008, 9, 802–807. [DOI] [PubMed] [Google Scholar]
- 75. Yamamoto T., Nagamune T., Chem. Commun. (Camb.) 2009, 1022–1024. [DOI] [PubMed] [Google Scholar]
- 76. Huang Y.-H., Su Y.-S., Chang C.-J., Sun W.-H., J. Recept. Signal Transduction Res. 2016, 36, 633–644. [DOI] [PubMed] [Google Scholar]
- 77. Tan M., Yamaguchi S., Yamahira S., Nakamura M., Nagamune T., Lab Chip 2017, 17, 1933–1938. [DOI] [PubMed] [Google Scholar]
- 78. Tan M., Yamaguchi S., Nakamura M., Nagamune T., J. Biosci. Bioeng. 2018, 126, 363–370. [DOI] [PubMed] [Google Scholar]
- 79. Schumacher D., Helma J., Mann F. A., Pichler G., Natale F., Krause E., Cardoso M. C., Hackenberger C. P. R., Leonhardt H., Angew. Chem. Int. Ed. 2015, 54, 13787–13791; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 13992–13996. [Google Scholar]
- 80. Dixon A. S., Schwinn M. K., Hall M. P., Zimmerman K., Otto P., Lubben T. H., Butler B. L., Binkowski B. F., Machleidt T., Kirkland T. A., et al., ACS Chem. Biol. 2016, 11, 400–408. [DOI] [PubMed] [Google Scholar]
- 81. Soave M., Kellam B., Woolard J., Briddon S. J., Hill S. J., SLAS Discovery 2020, 25, 186–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Jiang W.-X., Dong X., Jiang J., Yang Y.-H., Yang J., Lu Y.-B., Fang S.-H., Wei E.-Q., Tang C., Zhang W.-P., Sci. Rep. 2016, 6, 20568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Zakeri B., Fierer J. O., Celik E., Chittock E. C., Schwarz-Linek U., Moy V. T., Howarth M., Proc. Natl. Acad. Sci. USA 2012, 109, E690–E697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Wang J., Yu Y., Xia J., Bioconjugate Chem. 2014, 25, 178–187. [DOI] [PubMed] [Google Scholar]
- 85. Tsutsumi H., Nomura W., Abe S., Mino T., Masuda A., Ohashi N., Tanaka T., Ohba K., Yamamoto N., Akiyoshi K., et al., Angew. Chem. Int. Ed. 2009, 48, 9164–9166; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 9328–9330. [Google Scholar]
- 86. Zane H. K., Doh J. K., Enns C. A., Beatty K. E., ChemBioChem 2017, 18, 470–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Reinhardt U., Lotze J., Mörl K., Beck-Sickinger A. G., Seitz O., Bioconjugate Chem. 2015, 26, 2106–2117. [DOI] [PubMed] [Google Scholar]
- 88. Takeda Y., Yano Y., Matsuzaki K., Anal. Chem. 2012, 84, 1754–1759. [DOI] [PubMed] [Google Scholar]
- 89. Gavins G., Groeger K., Bartoscheck M. D., Wolf P., Beck-Sickinger A. G., Bultmann S., Seitz O., Nat. Chem. 2021, 13, 15–23. [DOI] [PubMed] [Google Scholar]
- 90. Doh J. K., White J. D., Zane H. K., Chang Y. H., López C. S., Enns C. A., Beatty K. E., Proc. Natl. Acad. Sci. USA 2018, 115, 12961–12966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Doh J. K., Tobin S. J., Beatty K. E., Biochemistry 2020, 59, 3051–3059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Adams S. R., Campbell R. E., Gross L. A., Martin B. R., Walkup G. K., Yao Y., Llopis J., Tsien R. Y., J. Am. Chem. Soc. 2002, 124, 6063–6076. [DOI] [PubMed] [Google Scholar]
- 93. Spille J.-H., Zürn A., Hoffmann C., Lohse M. J., Harms G. S., Biophys. J. 2011, 100, 1139–1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Guignet E. G., Hovius R., Vogel H., Nat. Biotechnol. 2004, 22, 440–444. [DOI] [PubMed] [Google Scholar]
- 95. Takahira I., Fuchida H., Tabata S., Shindo N., Uchinomiya S., Hamachi I., Ojida A., Bioorg. Med. Chem. Lett. 2014, 24, 2855–2858. [DOI] [PubMed] [Google Scholar]
- 96. Giannone G., Hosy E., Levet F., Constals A., Schulze K., Sobolevsky A. I., Rosconi M. P., Gouaux E., Tampé R., Choquet D., et al., Biophys. J. 2010, 99, 1303–1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Bhagawati M., Hoffmann S., Höffgen K. S., Piehler J., Busch K. B., Mootz H. D., Angew. Chem. Int. Ed. 2020, 59, 21007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Ando T., Tsukiji S., Tanaka T., Nagamune T., Chem. Commun. (Camb.) 2007, 4995–4997. [DOI] [PubMed] [Google Scholar]
- 99. Kwon K., Beckett D., Protein Sci. 2000, 9, 1530–1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Steel E., Murray V. L., Liu A. P., PLoS One 2014, 9, e93646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Fernández-Suárez M., Chen T. S., Ting A. Y., J. Am. Chem. Soc. 2008, 130, 9251–9253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Roux K. J., Kim D. I., Raida M., Burke B., J. Cell Biol. 2012, 196, 801–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Branon T. C., Bosch J. A., Sanchez A. D., Udeshi N. D., Svinkina T., Carr S. A., Feldman J. L., Perrimon N., Ting A. Y., Nat. Biotechnol. 2018, 36, 880–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Best M., Degen A., Baalmann M., Schmidt T. T., Wombacher R., ChemBioChem 2015, 16, 1158–1162. [DOI] [PubMed] [Google Scholar]
- 105. Baalmann M., Ziegler M. J., Werther P., Wilhelm J., Wombacher R., Bioconjugate Chem. 2019, 30, 1405–1414. [DOI] [PubMed] [Google Scholar]
- 106. Uttamapinant C., White K. A., Baruah H., Thompson S., Fernández-Suárez M., Puthenveetil S., Ting A. Y., Proc. Natl. Acad. Sci. USA 2010, 107, 10914–10919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Liu D. S., Nivón L. G., Richter F., Goldman P. J., Deerinck T. J., Yao J. Z., Richardson D., Phipps W. S., Ye A. Z., Ellisman M. H., et al., Proc. Natl. Acad. Sci. USA 2014, 111, E4551–E4559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Meyer B. H., Martinez K. L., Segura J.-M., Pascoal P., Hovius R., George N., Johnsson K., Vogel H., FEBS Lett. 2006, 580, 1654–1658. [DOI] [PubMed] [Google Scholar]
- 109. Dai X., Böker A., Glebe U., RSC Adv. 2019, 9, 4700–4721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Ghosh I., Hamilton A. D., Regan L., J. Am. Chem. Soc. 2000, 122, 5658–5659. [Google Scholar]
- 111. Pédelacq J.-D., Cabantous S., Tran T., Terwilliger T. C., Waldo G. S., Nat. Biotechnol. 2006, 24, 79–88. [DOI] [PubMed] [Google Scholar]
- 112. Feng S., Varshney A., Coto Villa D., Modavi C., Kohler J., Farah F., Zhou S., Ali N., Müller J. D., van Hoven M. K., et al., Commun. Biol. 2019, 2, 344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Shihoya W., Izume T., Inoue A., Yamashita K., Kadji F. M. N., Hirata K., Aoki J., Nishizawa T., Nureki O., Nat. Commun. 2018, 9, 4711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Yang Z., Han S., Keller M., Kaiser A., Bender B. J., Bosse M., Burkert K., Kögler L. M., Wifling D., Bernhardt G., et al., Nature 2018, 556, 520–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Hall M. P., Unch J., Binkowski B. F., Valley M. P., Butler B. L., Wood M. G., Otto P., Zimmerman K., Vidugiris G., Machleidt T., et al., ACS Chem. Biol. 2012, 7, 1848–1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Schwinn M. K., Machleidt T., Zimmerman K., Eggers C. T., Dixon A. S., Hurst R., Hall M. P., Encell L. P., Binkowski B. F., Wood K. V., ACS Chem. Biol. 2018, 13, 467–474. [DOI] [PubMed] [Google Scholar]
- 117. Boursier M. E., Levin S., Zimmerman K., Machleidt T., Hurst R., Butler B. L., Eggers C. T., Kirkland T. A., Wood K. V., Friedman Ohana R., J. Biol. Chem. 2020, 295, 5124–5135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Hoare B. L., Kocan M., Bruell S., Scott D. J., Bathgate R. A. D., Pharmacol. Res. Perspect. 2019, 7, e00513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. McLachlan A. D., Stewart M., J. Mol. Biol. 1976, 103, 271–298. [DOI] [PubMed] [Google Scholar]
- 120. McLachlan A. D., J. Mol. Biol. 1978, 121, 493–506. [DOI] [PubMed] [Google Scholar]
- 121. Conway J. F., Parry D. A., Int. J. Biol. Macromol. 1990, 12, 328–334. [DOI] [PubMed] [Google Scholar]
- 122. Conway J. F., Parry D. A., Int. J. Biol. Macromol. 1991, 13, 14–16. [DOI] [PubMed] [Google Scholar]
- 123. Crick F. H. C., Acta Crystallogr. 1953, 6, 689–697. [Google Scholar]
- 124. Pauling L., Corey R. B., Nature 1953, 171, 59–61. [DOI] [PubMed] [Google Scholar]
- 125. Lupas A., van Dyke M., Stock J., Science 1991, 252, 1162–1164. [DOI] [PubMed] [Google Scholar]
- 126. Litowski J. R., Hodges R. S., J. Biol. Chem. 2002, 277, 37272–37279. [DOI] [PubMed] [Google Scholar]
- 127. Yano Y., Yano A., Oishi S., Sugimoto Y., Tsujimoto G., Fujii N., Matsuzaki K., ACS Chem. Biol. 2008, 3, 341–345. [DOI] [PubMed] [Google Scholar]
- 128. Nakase I., Ueno N., Katayama M., Noguchi K., Takatani-Nakase T., Kobayashi N. B., Yoshida T., Fujii I., Futaki S., Chem. Commun. (Camb.) 2017, 53, 317–320. [DOI] [PubMed] [Google Scholar]
- 129. Liang S. I., van Lengerich B., Eichel K., Cha M., Patterson D. M., Yoon T.-Y., von Zastrow M., Jura N., Gartner Z. J., Cell Rep. 2018, 22, 2593–2600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Westerfield J. M., Barrera F. N., J. Biol. Chem. 2020, 295, 1792–1814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Yamashita H., Yano Y., Kawano K., Matsuzaki K., Biochim. Biophys. Acta 2015, 1848, 1359–1366. [DOI] [PubMed] [Google Scholar]
- 132. Meyer B. H., Segura J.-M., Martinez K. L., Hovius R., George N., Johnsson K., Vogel H., Proc. Natl. Acad. Sci. USA 2006, 103, 2138–2143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Kawano K., Yagi T., Fukada N., Yano Y., Matsuzaki K., J. Pept. Sci. 2017, 23, 650–658. [DOI] [PubMed] [Google Scholar]
- 134. Hamatake M., Aoki T., Futahashi Y., Urano E., Yamamoto N., Komano J., Cancer Sci. 2009, 100, 95–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Guo W., Urizar E., Kralikova M., Mobarec J. C., Shi L., Filizola M., Javitch J. A., EMBO J., 2008, 27, 2293–2304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Lotze J., Wolf P., Reinhardt U., Seitz O., Mörl K., Beck-Sickinger A. G., ACS Chem. Biol. 2018, 13, 618–627. [DOI] [PubMed] [Google Scholar]
- 137. Reinhardt U., Lotze J., Zernia S., Mörl K., Beck-Sickinger A. G., Seitz O., Angew. Chem. Int. Ed. 2014, 53, 10237–10241; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 10402–10406. [Google Scholar]
- 138. Yano Y., Furukawa N., Ono S., Takeda Y., Matsuzaki K., Biopolymers 2016, 106, 484–490. [DOI] [PubMed] [Google Scholar]
- 139. Gradišar H., Jerala R., J. Pept. Sci. 2011, 17, 100–106. [DOI] [PubMed] [Google Scholar]
- 140. Eklund A. S., Ganji M., Gavins G., Seitz O., Jungmann R., Nano Lett. 2020, 20, 6732–6737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Thompson K. E., Bashor C. J., Lim W. A., Keating A. E., ACS Synth. Biol. 2012, 1, 118–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Morgan E., Doh J., Beatty K., Reich N., ACS Appl. Mater. Interfaces 2019, 11, 36383–36390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Grossmann T. N., Seitz O., J. Am. Chem. Soc. 2006, 128, 15596–15597. [DOI] [PubMed] [Google Scholar]
- 144. Dawson P. E., Muir T. W., Clark-Lewis I., Kent S. B., Science 1994, 266, 776–779. [DOI] [PubMed] [Google Scholar]
- 145. Lebar T., Lainšček D., Merljak E., Aupič J., Jerala R., Nat. Chem. Biol. 2020, 16, 513–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Griffin B. A., Adams S. R., Tsien R. Y., Science 1998, 281, 269–272. [DOI] [PubMed] [Google Scholar]
- 147. Hearps A. C., Pryor M. J., Kuusisto H. V., Rawlinson S. M., Piller S. C., Jans D. A., J. Fluoresc. 2007, 17, 593–597. [DOI] [PubMed] [Google Scholar]
- 148. Langhorst M. F., Genisyuerek S., Stuermer C. A. O., Histochem. Cell Biol. 2006, 125, 743–747. [DOI] [PubMed] [Google Scholar]
- 149. Stroffekova K., Proenza C., Beam K. G., Pfluegers Arch. 2001, 442, 859–866. [DOI] [PubMed] [Google Scholar]
- 150. Halo T. L., Appelbaum J., Hobert E. M., Balkin D. M., Schepartz A., J. Am. Chem. Soc. 2009, 131, 438–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Morris P. J., Bruce Martin R., J. Inorg. Nucl. Chem. 1970, 32, 2891–2897. [Google Scholar]
- 152. Crowe J., Masone B. S., Ribbe J., Methods Mol. Biol. 1996, 58, 491–510. [DOI] [PubMed] [Google Scholar]
- 153. Wieneke R., Tampé R., Angew. Chem. Int. Ed. 2019, 58, 8278–8290. [DOI] [PubMed] [Google Scholar]
- 154. Lata S., Reichel A., Brock R., Tampé R., Piehler J., J. Am. Chem. Soc. 2005, 127, 10205–10215. [DOI] [PubMed] [Google Scholar]
- 155. Hochuli E., Döbeli H., Schacher A., J. Chromatogr. A 1987, 411, 177–184. [DOI] [PubMed] [Google Scholar]
- 156. Kollmannsperger A., Sharei A., Raulf A., Heilemann M., Langer R., Jensen K. F., Wieneke R., Tampé R., Nat. Commun. 2016, 7, 10372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Wieneke R., Labòria N., Rajan M., Kollmannsperger A., Natale F., Cardoso M. C., Tampé R., J. Am. Chem. Soc. 2014, 136, 13975–13978. [DOI] [PubMed] [Google Scholar]
- 158. Nonaka H., Fujishima S.-h., Uchinomiya S.-h., Ojida A., Hamachi I., J. Am. Chem. Soc. 2010, 132, 9301–9309. [DOI] [PubMed] [Google Scholar]
- 159. Fujishima S.-h., Nonaka H., Uchinomiya S.-h., Kawase Y. A., Ojida A., Hamachi I., Chem. Commun. (Camb.) 2012, 48, 594–596. [DOI] [PubMed] [Google Scholar]
- 160. Ojida A., Fujishima S.-h., Honda K., Nonaka H., Uchinomiya S.-h., Hamachi I., Chem. Asian J. 2010, 5, 877–886. [DOI] [PubMed] [Google Scholar]
- 161. Ojida A., Honda K., Shinmi D., Kiyonaka S., Mori Y., Hamachi I., J. Am. Chem. Soc. 2006, 128, 10452–10459. [DOI] [PubMed] [Google Scholar]
- 162. Wood D. W., Camarero J. A., J. Biol. Chem. 2014, 289, 14512–14519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Lue R. Y. P., Chen G. Y. J., Hu Y., Zhu Q., Yao S. Q., J. Am. Chem. Soc. 2004, 126, 1055–1062. [DOI] [PubMed] [Google Scholar]
- 164. Kalia J., Raines R. T., ChemBioChem 2006, 7, 1375–1383. [DOI] [PubMed] [Google Scholar]
- 165. Muir T. W., Annu. Rev. Biochem. 2003, 72, 249–289. [DOI] [PubMed] [Google Scholar]
- 166. Wood R. J., Pascoe D. D., Brown Z. K., Medlicott E. M., Kriek M., Neylon C., Roach P. L., Bioconjugate Chem. 2004, 15, 366–372. [DOI] [PubMed] [Google Scholar]
- 167. Zettler J., Schütz V., Mootz H. D., FEBS Lett. 2009, 583, 909–914. [DOI] [PubMed] [Google Scholar]
- 168. Shah N. H., Dann G. P., Vila-Perelló M., Liu Z., Muir T. W., J. Am. Chem. Soc. 2012, 134, 11338–11341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Jung D., Sato K., Min K., Shigenaga A., Jung J., Otaka A., Kwon Y., Chem. Commun. (Camb.) 2015, 51, 9670–9673. [DOI] [PubMed] [Google Scholar]
- 170. David Y., Vila-Perelló M., Verma S., Muir T. W., Nat. Chem. 2015, 7, 394–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Borra R., Dong D., Elnagar A. Y., Woldemariam G. A., Camarero J. A., J. Am. Chem. Soc. 2012, 134, 6344–6353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Braner M., Kollmannsperger A., Wieneke R., Tampé R., Chem. Sci. 2016, 7, 2646–2652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Volkmann G., Liu X.-Q., PLoS One 2009, 4, e8381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Böcker J. K., Dörner W., Mootz H. D., Chem. Commun. (Camb.) 2019, 55, 1287–1290. [DOI] [PubMed] [Google Scholar]
- 175. Carvajal-Vallejos P., Pallissé R., Mootz H. D., Schmidt S. R., J. Biol. Chem. 2012, 287, 28686–28696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Bhagawati M., Terhorst T. M. E., Füsser F., Hoffmann S., Pasch T., Pietrokovski S., Mootz H. D., Proc. Natl. Acad. Sci. USA 2019, 116, 22164–22172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Lee E., Min K., Chang Y.-T., Kwon Y., Protein Sci. 2018, 27, 1568–1574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Mootz H. D., ChemBioChem 2009, 10, 2579–2589. [DOI] [PubMed] [Google Scholar]
- 179. Gronemeyer T., Chidley C., Juillerat A., Heinis C., Johnsson K., Protein Eng. Des. Sel. 2006, 19, 309–316. [DOI] [PubMed] [Google Scholar]
- 180. Poc P., Gutzeit V. A., Ast J., Lee J., Jones B. J., D′Este E., Mathes B., Lehmann M., Hodson D. J., Levitz J., et al., Chem. Sci. 2020, 11, 7871–7883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Iwatate R. J., Yoshinari A., Yagi N., Grzybowski M., Ogasawara H., Kamiya M., Komatsu T., Taki M., Yamaguchi S., Frommer W. B., et al., Plant Cell 2020, 32, 3081–3094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Walling M. A., Novak J. A., Shepard J. R. E., Int. J. Mol. Sci. 2009, 10, 441–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Slotkin J. R., Chakrabarti L., Dai H. N., Carney R. S. E., Hirata T., Bregman B. S., Gallicano G. I., Corbin J. G., Haydar T. F., Dev. Dyn. 2007, 236, 3393–3401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Li Z., Krippendorff B.-F., Sharma S., Walz A. C., Lavé T., Shah D. K., mAbs 2016, 8, 113–119. [DOI] [PMC free article] [PubMed] [Google Scholar]