

Abstract
Objective
Progressive multifocal leukoencephalopathy (PML) is still burdened by high mortality in a subset of patients, such as those affected by hematological malignancies. The aim of this study was to analyze the safety and carry out preliminary evaluation of the efficacy of polyomavirus JC (JCPyV)‐specific T cell therapy in a cohort of hematological patients with PML.
Methods
Between 2014 and 2019, 9 patients with a diagnosis of “definite PML” according to the 2013 consensus who were showing progressive clinical deterioration received JCPyV‐specific T cells. Cell lines were expanded from autologous or allogenic peripheral blood mononuclear cells by stimulation with JCPyV antigen‐derived peptides.
Results
None of the patients experienced treatment‐related adverse events. In the evaluable patients, an increase in the frequency of circulating JCPyV‐specific lymphocytes was observed, with a decrease or clearance of JCPyV viral load in cerebrospinal fluid. In responsive patients, transient appearance of punctate areas of contrast enhancement within, or close to, PML lesions was observed, which was interpreted as a sign of immune control and which regressed spontaneously without the need for steroid treatment. Six of 9 patients achieved PML control, with 5 alive and in good clinical condition at their last follow‐up.
Interpretation
Among other novel treatments, T cell therapy is emerging as a viable treatment option in patients with PML, particularly for those not amenable to restoration of specific immunity. Neurologists should be encouraged to refer PML patients to specialized centers to allow access to this treatment strategy. ANN NEUROL 2021;89:769–779
Progressive multifocal leukoencephalopathy (PML) is a severe brain infection caused by the JC polyomavirus (JCPyV) following prolonged immune suppression, 1 , 2 as observed in association with human immunodeficiency virus (HIV) infection, 3 hematological malignancies, 4 , 5 congenital immune deficiencies, 6 , 7 , 8 solid organ/hematopoietic stem cell transplantation, 9 or after treatment with monoclonal antibodies. 10 To date, effective antiviral agents to treat JCPyV infections are not available, 11 and infection control relies on restoration of the host immune competence, which cannot always be attained. This explains why PML‐associated mortality is heavily dependent on whether immune suppression is reversible or not, ranging from 20% in patients with immune‐mediated disorders who develop PML during treatment with monoclonal antibodies 12 up to 90% in patients with hematological malignancies. 4 , 5 , 9 The need to find effective treatment options is even more urgent considering that the incidence of PML is increasing dramatically in the population of HIV‐negative patients, as a consequence of more widespread use of monoclonal antibodies and immune modulators for the management of hematological malignancies and autoimmune disorders. 13 Moreover, the indications for hematopoietic stem cell transplantation (HSCT) are increasing, as is the survival of transplant recipients, resulting in a continual rise in the number of patients at risk.
Enhancing immunity to JCPyV currently seems the most promising approach to counteract PML. Treatment with immune checkpoint inhibitors 14 , 15 , 16 , 17 , 18 , 19 , 20 and vaccination 21 , 22 have been attempted in isolated cases or small case series. However, these approaches have been associated with inconsistent results, because patients require a functional immune system to mount an effective cellular response towards the JCPyV. Passive immunization through the adoptive transfer of JCPyV‐specific T cells has the potential to be effective in patients with profound cellular immune suppression, who have little chance of responding to treatments that stimulate the endogenous immune system. This approach has been used for more than 2 decades in the treatment of refractory herpesvirus infections, 23 , 24 but has only recently been used in patients with PML. Since the first case was reported by our group in 2011, 25 only 4 additional patients have been treated with T cell therapy. 26 , 27 Here, we describe the clinical and paraclinical features in a series of 9 HIV‐negative patients treated with T cell therapy at our Institution, with the aim of reporting preliminary data on the efficacy and safety of this approach in view of launching a clinical trial in a larger cohort.
Patients and Methods
Patients
Patients were referred from different Italian centers to the Good Manufacturing Practice (GMP) Laboratory of the Fondazione IRCCS Policlinico San Matteo (Pavia, Italy). Criteria for treatment included a diagnosis of “definite PML” according to the consensus of the American Academy of Neurology of 2013 28 and ongoing neurological deterioration. Approval from the local ethics committee and from the national authority were obtained. All patients gave their written informed consent for study participation. The patient's previous medical history and clinical/paraclinical features at the time of PML diagnosis, including magnetic resonance imaging (MRI) scans, were gathered from the referring centers and reviewed centrally. From January 2014 to April 2019, a total of 9 HIV‐negative patients were treated with JCPyV‐specific lymphocytes.
Production and Testing of JCPyV‐Specific T Lymphocytes
Cell lines were produced within a GMP facility, according to a previously described method. 25 Briefly, peripheral blood mononuclear cells (PBMCs), obtained from the patient (autologous treatment) or from an allogeneic donor (HLA‐haploidentical HSCT donor, HLA‐haploidentical third‐party family member, or HLA‐partly matched third‐party donor), were pulsed with 15‐mer peptide pools, spanning the entire JCPyV viral capsid 1 protein (VP1) and large T (LT) proteins (Miltenyi Biotech, Bergisch Gladbach, Germany). On day +12, cultured cells were recovered, counted, and replated in the presence of autologous irradiated stimulator PBMCs, pulsed with JCPyV peptide mixes, and cultured for a further 12–14 days in the presence of IL‐2 (20U/ml). T cell lines were characterized by immunophenotype and tested for sterility, alloreactivity, and potency by JCPyV‐specific interferon gamma (IFNγ) enzyme‐linked immunospot (ELISPOT) assay 29 and cytotoxicity by standard 51Cr‐release assay. 29 , 30
Biological Characterization of JCPyV‐Targeted T Cell Lines
JCPyV‐specific T cells (JCPyV‐LTCs) were generated successfully ex vivo. Flow‐cytometry analyses showed that JCPyV‐LTCs included 71% CD4+, 13% CD8+, and 5% CD56+/CD3− natural killer cells. However, LTCs expanded from the patients included higher numbers of CD8+ cells (43 vs 8% in allogeneic LTCs). T cells were specific for JCPyV, because they produced IFNγ in response to VP1 and LT peptides (median: 73 spot‐forming units [SFU]/105 cells, range 27–900SFU/105 cells) and exerted virus‐specific cytotoxicity, showing specific lysis toward JCPyV antigen‐pulsed targets (median lysis at the effector‐to‐target [E:T] ratio of 2.5:1 after subtracting control lysis: 31%, range 3–43%). The median lysis of patients' or third‐party allogeneic phytohemagglutinin blasts pulsed with control antigen at the same E:T ratio was 4% (range 0–14%), indicating that allogeneic LTCs were devoid of alloreactivity.
Treatment Schedule and Assessment of Response
The JCPyV‐LTC starting dose was 1 × 105/kg, followed by a second dose of 2 × 105/kg, 15 days apart. Adverse events were recorded after each infusion and classified according to the Common Terminology Criteria for Adverse Events, v.4.0. Patients were re‐evaluated clinically 15 days after the second infusion. Ongoing neurological deterioration after the first 2 JCPyV‐LTC infusions was considered a reason for treatment discontinuation because of inefficacy. Improvement and stabilization were considered encouraging features, and T cell therapy was continued monthly at the dose of 2 × 105/kg as long as considered potentially beneficial, using the JCPyV viral load in the cerebrospinal fluid (CSF) and radiological evolution as complementary assessments reflecting disease activity. The JCPyV‐specific immune response was evaluated, when feasible, in PBMCs obtained from peripheral blood sampled at different time points after LTC administration, by 24 h IFNγ ELISPOT assay in the presence of 0.1μg/ml JCPyV VP1 and LT‐derived peptides. 29 Patients were evaluated regularly during follow‐up by neurological examination and contrast brain MRI. The size of PML lesions was calculated as the sum of the products of maximum perpendicular diameters of major lesions on axial Fluid Attenuated Inversion Recovery (FLAIR) sequences. Percentage variations in the size of PML lesions were calculated by comparing the size of PML lesions at the discontinuation of T cell therapy with the size of PML lesions before starting T cell therapy. After discontinuation of T cell therapy, patients were followed by their referring neurologist, who provided us with information on their neurological status at last follow‐up.
Results
Patient Demographic Features and Previous Medical History
Previous medical histories and the clinical–paraclinical features at the time of PML in the 9 patients are reported in Table 1 and Supplementary Table S1. Details on treatment and outcome are reported in Table 2.
TABLE 1.
Clinical–Paraclinical Features at the Time of PML Diagnosis in the 9 Patients Treated with T Cell Therapy
| Pt | Age, yr/Sex | Delay from Symptom Onset to PML Diagnosis (mo) | Neurological Presentation | Brain MRI Features | JC Virus DNA in the CSF, Copies/ml |
|---|---|---|---|---|---|
| 1 | 59/F | 4 | Right arm motor deficit | Single lesion in frontal prerolandic region | Negative a |
| 2 | 55/M | 4.8 | Cognitive impairment, left arm segmental ataxia | Multiple bilateral lesions in the frontal lobes, centrum semiovale, and in the posterior limb of the internal capsule | 3,950 |
| 3 | 70/F | 1.4 | Aphasia, agraphia, alexia | Single lesion involving the temporoparietal lobe | 2,440 |
| 4 | 50/M | 0.8 | Left LHH | Multiple lesions involving bilaterally the temporo‐occipital lobes and the corpus callosum | 9,000 |
| 5 | 68/M | 4 | Cognitive impairment, left LHH | Multiple lesions involving unilaterally the right parietal and occipital lobes | 2,277 |
| 6 | 54/M | 1 | Left arm motor deficit | Multiple lesions involving unilaterally the right frontal and parietal lobes | 1,024 |
| 7 | 66/M | 1 | Left hemiparesis | Multiple lesions located bilaterally in the frontal lobes | 1,300 |
| 8 | 54/F | 3.7 | Left arm segmental ataxia, nausea, gait ataxia | Multiple lesions located in the brainstem and the cerebellum | 300 |
| 9 | 17/M | 0.3 | Cognitive impairment, left hemiparesis | Multiple lesions in right parietal region, left temporoparietal regions, frontomesial region, corpus callosum, and basal ganglia | 384 |
This patient had 109 copies of JC virus DNA/100,000 cells on brain biopsy tissue.
CSF = cerebrospinal fluid; F = female; LHH = lateral homonymous hemianopia; M = male; MRI = magnetic resonance imaging; PML = progressive multifocal leukoencephalopathy; Pt = patient.
TABLE 2.
Treatment and Outcome in the 9 Patients Treated with T Cell Therapy for PML
| Pt | T Cell Therapy | Last FU | |||||
|---|---|---|---|---|---|---|---|
| Delay from PML Diagnosis to T Cell Therapy, mo | Neurological Status at the Time of T Cell Therapy Initiation | Number of Infusions | JC Virus DNA in the CSF After T Cell Therapy, Copies/ml | FU Duration from PML Diagnosis, mo | Patient Status at Last FU | Neurological Status at Last FU, Residual Deficits | |
| 1 | 4.7 | Severe right hemiparesis, aphasia, behavioral changes (mRS 4) | 4 | — | 69.0 | Alive | Severe hemiparesis, aphasia (mRS 4) |
| 2 | 9.9 | Cognitive impairment, left arm segmental sensory ataxia, epilepsy (mRS 2) | 3 | 290 | 27.7 | Alive | No residual deficit (mRS 1) |
| 3 | 3 | Global aphasia, agraphia, alexia, right hemiparesis (mRS 3) | 1 | — | 3.6 | Deceased (PML) | — |
| 4 | 2.6 | Cortical blindness, left sensory deficit (mRS 2) | 3 | — | 4.4 | Deceased (VZV encephalitis) | — |
| 5 | 5 | Mild cognitive impairment, LHH, seizures (mRS 2) | 6 | 1,860 | 19.3 | Alive | Mild cognitive impairment, LHH (mRS 2) |
| 6 | 2 | Vigilance impairment, left LHH, severe left hemiparesis, hypoesthesia and hemineglect (mRS 5) | 2 | — | 5.0 | Deceased (PML) | — |
| 7 | 2 | Dysarthria, cognitive impairment, severe left hemiparesis (mRS 4) | 4 | — | 6.0 | Deceased (PML) | — |
| 8 | 1.3 | Dysarthria, dysphagia, left arm segmental ataxia, severe gait ataxia (mRS 4) | 6 | Negative | 50.3 | Alive | Mild dysarthria and gait ataxia (mRS 2) |
| 9 | 1.1 | Cognitive impairment, aphasia, dysphagia, agraphia, seizures, vigilance impairment, severe left hemiparesis (mRS 4) | 5 | Negative | 12 | Alive | Mild ataxia and tremor (mRS 2) |
CSF = cerebrospinal fluid; FU = follow‐up; LHH = lateral homonymous hemianopia; mRS = modified Rankin Score; PML = progressive multifocal leukoencephalopathy; Pt = patient; VZV = varicella‐zoster virus; — = not applicable.
Six patients were male and 3 female. The median age at the time of PML diagnosis was 55 years (range 17–70 years). The underlying condition for immune suppression was represented by idiopathic CD4+ lymphopenia (n = 1; Patient 8), Wiskott–Aldrich syndrome (n = 1; Patient 9), or hematological malignancies (n = 7) [non‐Hodgkin lymphoma (n = 4), Hodgkin lymphoma (n = 1), multiple myeloma (n = 1), and chronic lymphocytic leukemia (n = 1); Patients 1–7). All 7 patients with hematological malignancies had received chemotherapy as part of their anti‐neoplastic treatment, and 6 of the 7 received B cell‐depleting monoclonal antibodies (rituximab, obinutuzumab, or ofatumumab). A total of 5 patients had undergone autologous (n = 3) or allogenic (n = 2) HSCT (median delay from HSCT to the development of PML: 19 months, range 2–36 months).
Clinical and Paraclinical Features at the Time of PML Diagnosis
The median CD4+ cell count at the time of PML diagnosis was 82 cells/mm3 (range 40–309 cells/mm3). Neurological presentation consisted of subacute progressive symptoms, including cognitive impairment, lateral homonymous hemianopia, aphasia, motor deficits, and segmental ataxia. Brain MRI showed findings typical of PML, with 1 or multiple T2 hyperintense white matter lesions involving U fibers, appearing hypointense on T1 images, located unilaterally (4 patients) or bilaterally (4 patients) in the supratentorial white matter (eg, Fig 1A, F) or confined to the infratentorial region (1 patient). Diffusion‐weighted imaging disclosed a rim of diffusion restriction along the margins of PML lesions, corresponding to the active demyelination front, in 8 of 9 patients (see Fig 1H, I). An intralesional T2‐gradient linear rim of hypointensity at the juxtacortical level was evident in 6 patients (Patients 1, 2, 5–7, and 9; see Fig 1B). In 2 patients (Patients 5 and 8), small areas of punctate contrast enhancement were present in the perilesional white matter, suggestive of an accompanying inflammatory response. Magnetic resonance spectroscopy displayed nonspecific alterations, including mild choline elevation, N‐acetylaspartate reduction and lactate elevation in all 3 cases in which it was performed. CSF analysis showed normal protein levels and cell counts except for a single patient (Patient 8) who had a mild lymphocytic pleocytosis (8 cells/m3). JCPyV DNA was detected in the CSF of 8 patients (viral load ranging from 300 to 9000 copies/ml), whereas the remaining patient (Patient 1) had undetectable JCPyV DNA in the CSF despite repeated lumbar punctures. In this patient, PML diagnosis was based on typical histopathological findings on brain biopsy (classic histopathologic triad plus positive tissue reverse transcriptase–polymerase chain reaction for JCPyV). The median delay from onset of neurological symptoms to diagnosis of PML was 1.4 months (range 0.3–4.8 months).
FIGURE 1.
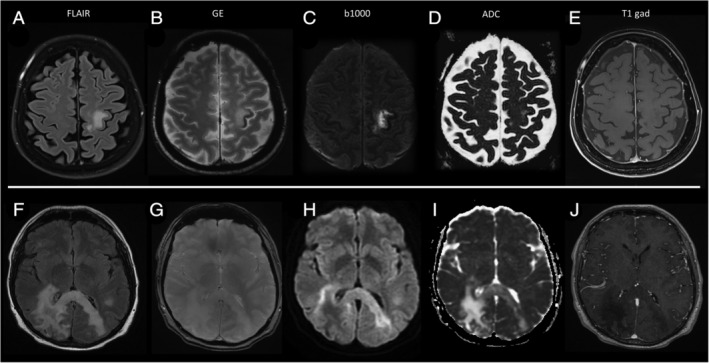
Magnetic resonance imaging findings at the time of progressive multifocal leukoencephalopathy (PML) diagnosis in Patient 1, in A–E. and Patient 4, in F–L. (A) Patient 1 had a single progressive multifocal leukoencephalopathy (PML) lesion located in the left rolandic white matter, hyperintense on axial FLAIR images. (B) The adjacent cortex shows a characteristic rim of hypointensity on gradient echo (GE) images. (C, D) No diffusion restriction is evident on b1000 sequences, in C, and ADC sequences, in D, and (E) no contrast enhancement was evident on T1 sequences after gadolinium injection. (F) Patient 4 had a large PML lesion in the right parietal white matter that involved the corpus callosum splenium and reached the contralateral hemisphere, evident as hyperintense on FLAIR images. (G) No cortical rim of hypointensity was evident on GE images. (H, I) Linear rims of diffusion restriction were present along the lateral margins of the PML lesion, corresponding to the front of active demyelination. (J) The PML lesion showed marked hypointensity on T1 sequences after gadolinium injection, but no contrast enhancement. FLAIR, Fluid Attenuated Inversion Recovery.
Treatment and Outcome
Two patients had previously received pharmacological agents, such as cidofovir (Patient 4) or mefloquine and mirtazapine (Patient 2), based on anecdotal efficacy results reported in the literature, 11 but none of these agents proved effective in the control of PML progression. As a result, all of the patients were in a state of clinical deterioration and were moderately to severely disabled (modified Rankin Score [mRS] 2–5) when they were referred to our center for T cell therapy.
The first JCPyV‐LTC infusion was administered at a median of 2.6 months after PML diagnosis (range 1.1–9.9 months). LTCs were generated from autologous PBMCs in 4 cases and from HLA‐partly matched allogeneic donors in the remaining 5 cases. JCPyV‐LTC characteristics are reported in Table 3. The median number of infusions per patient was 4 (range 1–6).
TABLE 3.
Characteristics of JCPyV‐Specific T Cell Products Used in the Cohort
| Pt | PBMC Source | IFNγ Production, Spots/105 Cells | Cytotoxic activity, % Lysis at 2.5:1 E:T Ratio | Phenotype, % Positive Cells | |||||
|---|---|---|---|---|---|---|---|---|---|
| JCPyV VP1 + LT Peptides | Allogeneic PBMCs | JCPyV VP1 + LT Peptides | Allogeneic PHA Blasts | CD3+ | CD4+ | CD8+ | CD3−/CD56+ | ||
| 1 | Allogeneic (related third party) | 560 | 14 | 18 | 0 | 95 | 89 | 7 | 1 |
| 2 | Autologous | 900 | — | 41 | — | 93 | 56 | 35 | 6 |
| 3 | Autologous | 27 | — | 3 | — | 38 | 24 | 12 | 57 |
| 4 | Allogeneic (unrelated third party) | 43 | 13 | 35 | 8 | 97 | 84 | 13 | 2 |
| 5 | Autologous | 35 | — | 31 | — | 90 | 39 | 52 | 5 |
| 6 | Allogeneic (related third party) | 202 | nd | 43 | nd | 99 | 73 | 21 | 1 |
| 7 | Allogeneic (unrelated third party) | 123 | 8 | 37 | 14 | 82 | 71 | 8 | 15 |
| 8 | Autologous | 73 | — | 29 | — | 88 | 2 | 80 | 19 |
| 9 | Allogeneic (HSCT donor) | 63 | 3 | 10 | 0 | 95 | 85 | 8 | 2 |
E:T ratio = effector‐to‐target ratio; HSCT = hematopoietic stem cell transplantation; JCPyV = polyomavirus JC; IFNγ = interferon gamma; LT = large T protein; nd = not done; PBMCs = peripheral blood mononuclear cells; PHA = phytohemagglutinin; VP1 = viral capsid 1 protein.
T cell therapy was well tolerated, and none of the patients experienced infusion‐related adverse events.
Three patients (Patients 3, 6, and 7) died owing to progression of PML, and 1 (Patient 4) died owing to an intervening varicella‐zoster virus reactivation related to his state of immune suppression. The remaining 5 patients achieved long‐term survival (median follow‐up duration: 39 months after PML diagnosis, range 19–69 months). One patient achieved neurological stabilization with a moderate level of disability (mRS 4), and 4 patients experienced substantial neurological improvement, with only mild or minimal residual disability at the last follow‐up (mRS 1–2).
PML lesions progressively reduced in size and evolved to cerebral atrophy in all surviving patients (Fig 2A, D, G). Intermediate control MRI performed during or shortly after administration of T cell therapy revealed the appearance of small areas of punctate contrast enhancement within, or close to, PML lesions in 4 patients (Patients 1, 2, 7, and 9; see Fig 2E). In cases in which punctate enhancement was already present before initiation of therapy (Patients 5 and 8), it remained unchanged at the control MRI performed during T cell therapy. Punctate enhancing MRI abnormalities were not associated with new or worsening symptoms and resolved spontaneously within 1 to 3 months (see Fig 2H), without the need for steroid treatment.
FIGURE 2.
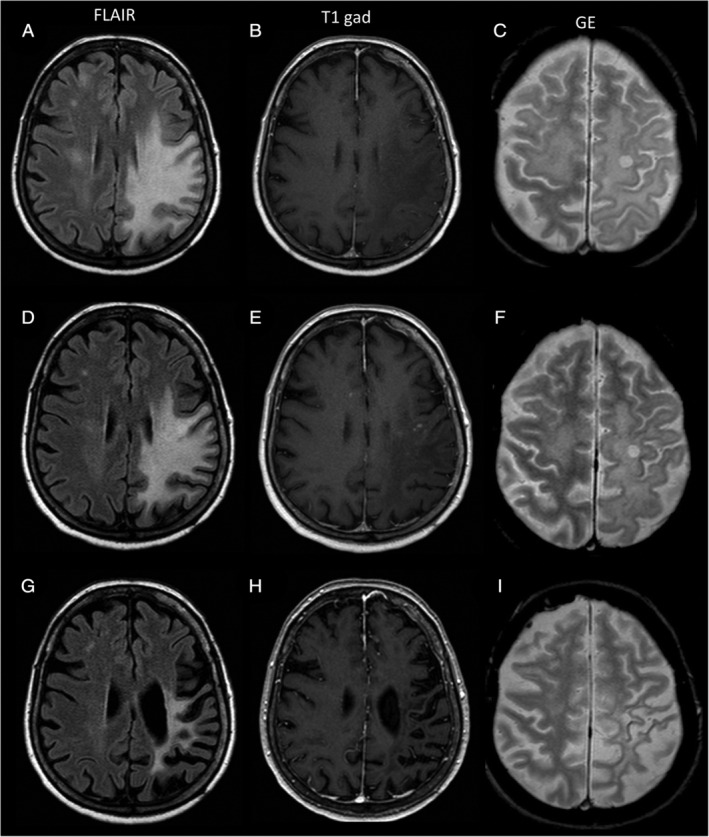
Magnetic resonance imaging (MRI) findings in Patient 1. (A–C) Before starting T cell therapy. (D–F) 1 month after T cell therapy discontinuation. (G–I) 2.5 years after T cell therapy discontinuation. (A, D, G) Axial FLAIR images. (B, E, H) T1‐weighted images after gadolinium injection. (C, F, I) Gradient echo (GE) images. (A, B) MRI at initiation of T cell therapy showed a large frontoparietal progressive multifocal leukoencephalopathy (PML) lesion that was hyperintense on FLAIR sequences, shown in A, and hypointense on T1 sequences, with no contrast enhancement after gadolinium injection, shown in B. (D) MRI acquired 1 month after the last T cell therapy infusion showed an initial reduction in the size of the PML lesion on FLAIR images, together with a sharp reduction of its mass effect. (E) On T1 sequences after gadolinium injection, small punctate areas of contrast enhancement had appeared within (or close to) the PML lesion. (G, H) MRI acquired at the last follow‐up showed further reduction of the FLAIR hypersignal, in G, and a major cortical–subcortical atrophy across the left frontoparietal lobes, in G and H. FLAIR, Fluid Attenuated Inversion Recovery.
Control CSF analysis was available for 4 patients who were stable or improved under treatment, showing a reduced (Patients 2 and 5) or undetectable (Patients 8 and 9) JCPyV load, consistent with clinical and radiological evolution. A repeat lumbar puncture was not performed in Patient 1, because CSF tested negative for JCPyV DNA at diagnosis, whereas Patients 3, 4, 6, and 7 did not have lumbar punctures performed because they deteriorated rapidly or died from other causes before control CSF analysis could be performed.
The JCPyV‐specific cellular immune response before and after cell therapy could be analyzed in 5 patients. The median frequency of circulating JCPyV‐specific IFNγ‐secreting cells before T cell administration was 0SFU/105 PBMCs (range 0–16SFU/105 PBMCs), with a median frequency of cells potentially able to respond (response to the lymphocyte mitogen phytohemagglutinin) of 96SFU/105 PBMCs (range 2–395SFU/105 PBMCs), which is well below the normal range for healthy subjects. After T cell therapy, the number of circulating JCPyV‐specific lymphocytes increased in 4 of the 5 patients, with a median frequency of 47SFU/105 PBMCs (range 6–106SFU/105 PBMCs).
Table 4 compares the clinical, biological, and radiological characteristics of patients surviving PML with those of patients who succumbed to PML or other intervening infections. Patients surviving PML had disability scores at the time of initiation of T cell therapy comparable to patients who died from PML progression (4 vs 3.5 on the modified Rankin Scale), although they had longer intervals from PML diagnosis to T cell therapy (median: 4.7 vs 2.3 months) and lower copy numbers of JCPyV DNA in their CSF (median: 384 vs 1870 copies/ml). All 5 surviving patients had evidence of punctate contrast enhancement, which was already present before (n = 2) or developed during T cell therapy (n = 3), compared with a single patient who developed contrast enhancement but ultimately died from PML progression (5 of 5, 100%, vs 1 of 4, 25%). Notably, 2 of the patients treated with autologous T cells resumed treatment for the underlying hematologic malignancy after receiving T cell therapy and continued in good clinical condition without developing new lesions.
TABLE 4.
Clinical, Biological, and Radiological Features in Patients Surviving PML (n = 5) and in Patients Dying Because of PML or Another Infection Related to Immune Suppression (n = 4)
| Characteristic | Survivors (n = 5) | Deceased (n = 4) |
|---|---|---|
| Age, yr, median (range) | 55 | 60 |
| Male gender, n (%) | 3 of 5 (60) | 3 of 4 (75) |
| Median delay from diagnosis to T cell therapy, mo | 4.7 | 2.3 |
| Median modified Rankin Score at T cell therapy | 4 | 3.5 |
| Median CD4+ cell count at PML diagnosis, cells/mm3 | 94 | 82 |
| Median JC viral load in the CSF at diagnosis, copies/ml | 384 | 1,870 |
| Median percentage variation in the size of PML lesions at last available MRI | −25 | +258 |
| Subcortical rim of T2* hypointensity before or during treatment, n (%) | 4 of 5 (80) | 2 of 4 (50) |
| Development of punctate contrast enhancement on MRI before and/or during T cell therapy, n (%) | 5 of 5 (100) | 1 of 4 (25) |
CSF = cerebrospinal fluid; MRI = magnetic resonance imaging; PML = progressive multifocal leukoencephalopathy.
Discussion
This study reports a series of 9 HIV‐negative patients who developed PML in the context of hematological malignancies or congenital immune deficiencies and who were treated with infusions of JCPyV‐specific T lymphocytes. None of the patients experienced treatment‐related adverse events, strengthening the evidence that T cell therapy is a safe and feasible treatment strategy in this fragile population. 25 , 26 , 27
Patients in our series had multiple risk factors for the development of PML, including a history of HSCT and treatment with monoclonal antibodies, and none of them was amenable to immune suppressant drug modulation. They were referred to our center to receive T cell therapy based on clinical grounds, because they were deteriorating rapidly despite conventional measures. Cell lines were generated ad hoc for 7 patients, from autologous or allogenic PBMCs, whereas in 2 cases third‐party banked JCPyV‐specific T cells were used. Whenever possible, the use of an HSCT donor or an HLA‐haploidentical third‐party family donor is preferred in patients with a severe impairment of cellular immunity. 27 However, the significant amount of time required to find an HLA‐compatible family donor, including HLA typing and donor work‐up for leukapheresis, and the lack of availability of suitably matched third‐party T cells prompted us to explore the possibility of expanding JCPyV‐specific T cells from the patient, after performing a small‐scale in vitro virus‐specific immunity test. In 4 patients, in vitro JCPyV immunity testing suggested the feasibility of establishing a functional T cell line, and autologous LTCs were expanded and used therapeutically. Using this approach, we even succeeded in expanding a functional, mainly CD8+, T cell line from 1 patient with idiopathic CD4+ T lymphopenia, which proved efficient in achieving control of PML.
The expansion and testing of virus‐specific T cells with the method reported here required 3–4 weeks. The delay to starting treatment when using autologous or family‐derived T cells probably represents the main limitation of our approach. Patients received the first T cell infusion after a median of 2.6 months from PML diagnosis, and this delay might be fatal in patients with rapidly progressive disease. The use of third‐party banked virus‐specific T cells, 26 , 31 as we did in 2 patients, or a reduction of the time needed to expand JCPyV‐LTCs 32 might represent viable solutions to speed up administration of treatment. In patients with rapid PML progression, we did not attempt IFNγ selection, 33 because the frequency of circulating JCPyV‐specific memory T cells was expected to be relatively low, and the supposedly small number of recovered and infused cells would have required time for in vivo expansion.
Only 3 patients (33%) in our series ultimately died because of PML progression. This observation is encouraging, considering that mortality rates in patients with hematological malignancies 4 , 5 , 13 or congenital immune deficiencies 6 generally approach 90%. Long‐term survival was achieved in 5 patients (56%), whose neurological improvement was paralleled by a decrease in JCPyV viral load in the CSF and a shrinkage of PML lesions. The long follow‐up available for these patients further strengthens the clinical and paraclinical evidence suggesting that PML was controlled permanently. Although promising, these results should be viewed with caution in light of the cohort characteristics and possible confounding factors. Some of the patients in our series showed baseline features that have been associated with favorable outcome in the population of HIV‐positive patients, such as the presence of contrast enhancement 34 and low JCPyV viral loads in the CSF. 35 , 36 Indeed, a few patients in our cohort showed a relatively indolent clinical course that allowed them to wait for JCPyV‐specific T cell lines to be generated and administered, suggesting that our approach might have selected involuntarily for patients with a favorable prognosis. However, it should be noted that all patients showed a steady clinical deterioration until T cell therapy was administered and that neurological improvement/stabilization strictly coincided with administration of JCPyV‐LTCs. Moreover, at the time of T cell preparation, JCPyV‐specific T cell responses were not detectable by standard analysis in the patients, and only in 4 patients were we able to force a response by repeated in vitro stimulation. Although we cannot exclude the possibility that favorable baseline features 34 , 35 , 36 or the natural course 37 might have contributed to the successful outcomes observed in our series, the close temporal relationship between the start of T cell therapy and neurological improvement suggests that T cell therapy played a role in this fortunate process.
Contrast enhancement, observed before or during treatment in all long‐term survivors, seemed to be the strongest predictor of a favorable outcome in our series. The punctate enhancing alterations observed within (or close to) PML lesions in our study and other reports 26 are likely to reflect an early inflammatory response related to the homing of infused JCPyV‐specific lymphocytes or, at least, the support of JCPyV‐LTCs to endogenous T cells present in the lesions, as suggested by other studies. 26 We did not consider these patients to have an immune reconstitution inflammatory syndrome (IRIS), because none developed other defining features of this condition, such as accompanying clinical deterioration, paradoxical enlargement of PML lesions on T2/FLAIR sequences, edema, or mass effect. IRIS is the result of a broad and exaggerated immune activation following an abrupt restoration of immune competence, whereas virus‐specific cytotoxic T cell therapy induces rapid lysis of infected cells with minimal inflammatory infiltrates in perivascular spaces, without the massive bystander activation of effector cells and the associated tissue damage that occurs in clinical IRIS. 38 In this regard, the high targeted cytotoxic activity of our products might have prevented development of IRIS. Our T cells were expanded by stimulation with peptides derived from JCPyV antigens, unlike the study by Muftuoglu and colleagues,26 who used third‐party T cells specific for BKPyV. Although it has been shown that there is a high degree of homology between the 2 viruses, and a certain cross‐reactivity in T cell responses might be expected, we believe that the higher specificity of JCPyV‐LTCs might have been beneficial in reducing the risk of IRIS, because 66% of the patients from the MD Anderson case series developed IRIS versus none in our cohort.
Another promising radiological predictor of improved outcome is the evidence of subcortical areas of low T2* signal adjacent to PML lesions (seen better on susceptibility imaging but also visible on traditional gradient echo T2‐weighted sequences), which seems to reflect the presence of activated glial cells and microglia/macrophages with high intracytoplasmic levels of iron and ferromagnetic pigments. 39 The presence of this radiological sign has also been associated with a better outcome, 39 , 40 and, like contrast enhancement, probably reflects some degree of immune response towards JCPyV.
We did not observe any difference in response rates between patients receiving JCPyV‐LTCs generated from autologous or allogenic PBMCs. The 3 patients who succumbed to progression of PML received autologous, related third‐party, and unrelated third‐party T cells, respectively, and, in general, patients treated with unrelated third‐party T cells were those with the worst clinical conditions, which did not allow the time needed for a dedicated cell therapy product. Thus, it is impossible to draw any conclusion on this particular aspect.
This study has, undoubtedly, the intrinsic limitations of an uncontrolled case series. Control MRI and CSF analyses were not performed at the same time intervals in all patients or by using standardized protocols. No central nervous system cell trafficking analysis was performed, and the main consideration supporting a role for lymphocyte infusions in clinical improvement is that all patients were in a state of steady neurological progression until T cell therapy was administered. Despite these limitations and the exiguity of the cohort, our results are extremely promising with regard to both mortality and neurological outcome. The individual patient‐dedicated approach and the requirement for regulatory agency‐certified production facilities represent the main obstacles to a tailored T cell therapy. Generation of cell lines is time consuming, especially when lines are generated ad hoc for each patient, and this might result in a crucial delay in administration of treatment. The establishment of third‐party T cell banks will possibly make this approach more feasible and allow more widespread access to cellular therapies for PML treatment. Neurologists should try to reach a PML diagnosis as promptly as possible and refer patients to specialized centers to enable them to access these novel treatments.
Author Contributions
P.C. and E.M. contributed to the conception and design of the study. G.B., S.B., L.S., A.P., A.P., M.P., F.L., S.G., E.V., L.D., P.B., M.G., S.D., C.P., E.S., M.F., M.L., F.B., M.Z., E.M., and P.C. contributed to the acquisition, analysis, and interpretation of the data. G.B., E.M., and P.C. contributed to drafting the text. A.P., M.P., F.L., A.R., and S.G. contributed to preparation of the figures.
Potential Conflicts of Interest
Nothing to report.
Supporting information
Table S1. Supporting information
Acknowledgment
S.B. was supported by Ministero della Salute, Ricerca Corrente grant RCR 08071519, and P.C. by Ministero della Salute, Ricerca Corrente grant RCR 08069113, and Ministero della Salute grant RF‐2019‐12371492.
References
- 1. Tan CS, Koralnik IJ. Progressive multifocal leukoencephalopathy and other disorders caused by JC virus: clinical features and pathogenesis. Lancet Neurol 2010;9:425–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Pavlovic D, Patel MA, Patera AC, et al. T cell deficiencies as a common risk factor for drug associated progressive multifocal leukoencephalopathy. Immunobiology 2018;223:508–517. [DOI] [PubMed] [Google Scholar]
- 3. Engsig FN, Hansen A‐BE, Omland LH, et al. Incidence, clinical presentation, and outcome of progressive multifocal leukoencephalopathy in HIV‐infected patients during the highly active antiretroviral therapy era: a nationwide cohort study. J Infect Dis 2009;199:77–83. [DOI] [PubMed] [Google Scholar]
- 4. García‐Suárez J, de Miguel D, Krsnik I, et al. Changes in the natural history of progressive multifocal leukoencephalopathy in HIV‐negative lymphoproliferative disorders: impact of novel therapies. Am J Hematol 2005;80:271–281. [DOI] [PubMed] [Google Scholar]
- 5. Neil EC, DeAngelis LM. Progressive multifocal leukoencephalopathy and hematologic malignancies: a single cancer center retrospective review. Blood Adv 2017;1:2041–2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zerbe CS, Marciano BE, Katial RK, et al. Progressive multifocal leukoencephalopathy in primary immune deficiencies: Stat1 gain of function and review of the literature. Clin Infect Dis 2016;62:986–994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gheuens S, Pierone G, Peeters P, Koralnik IJ. Progressive multifocal leukoencephalopathy in individuals with minimal or occult immunosuppression. J Neurol Neurosurg Psychiatry 2010;81:247–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hadjadj J, Guffroy A, Delavaud C, et al. Progressive multifocal leukoencephalopathy in primary immunodeficiencies. J Clin Immunol 2019;39:55–64. [DOI] [PubMed] [Google Scholar]
- 9. Mateen FJ, Muralidharan R, Carone M, et al. Progressive multifocal leukoencephalopathy in transplant recipients. Ann Neurol 2011;70:305–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Molloy ES, Calabrese CM, Calabrese LH. The risk of progressive multifocal leukoencephalopathy in the biologic era: prevention and management. Rheum Dis Clin N Am 2017;43:95–109. [DOI] [PubMed] [Google Scholar]
- 11. Pavlovic D, Patera AC, Nyberg F, et al. Progressive multifocal leukoencephalopathy: current treatment options and future perspectives. Ther Adv Neurol Disord 2015;8:255–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Williamson EML, Berger JR. Diagnosis and treatment of progressive multifocal leukoencephalopathy associated with multiple sclerosis therapies. Neurotherapeutics 2017;14:961–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Iacobaeus E, Burkill S, Bahmanyar S, et al. The national incidence of PML in Sweden, 1988‐2013. Neurology 2018;90:e498–e506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Cortese I, Muranski P, Enose‐Akahata Y, et al. Pembrolizumab treatment for progressive multifocal leukoencephalopathy. N Engl J Med 2019;380:1597–1605. [DOI] [PubMed] [Google Scholar]
- 15. Rauer S, Marks R, Urbach H, et al. Treatment of progressive multifocal leukoencephalopathy with pembrolizumab. N Engl J Med 2019;380:1676–1677. [DOI] [PubMed] [Google Scholar]
- 16. Walter O, Treiner E, Bonneville F, et al. Treatment of progressive multifocal leukoencephalopathy with nivolumab. N Engl J Med 2019;380:1674–1676. [DOI] [PubMed] [Google Scholar]
- 17. Hoang E, Bartlett NL, Goyal MS, et al. Progressive multifocal leukoencephalopathy treated with nivolumab. J Neurovirol 2019;25:284–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Pawlitzki M, Schneider‐Hohendorf T, Rolfes L, et al. Ineffective treatment of PML with pembrolizumab: exhausted memory T‐cell subsets as a clue? Neurol Neuroimmunol Neuroinflamm 2019;6:e627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Medrano C, Vergez F, Mengelle C, et al. Effectiveness of immune checkpoint inhibitors in transplant recipients with progressive multifocal leukoencephalopathy. Emerg Infect Dis 2019;25:2145–2147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Küpper C, Heinrich J, Kamm K, et al. Pembrolizumab for progressive multifocal leukoencephalopathy due to primary immunodeficiency. Neurol Neuroimmunol Neuroinflamm 2019;6:e628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sospedra M, Schippling S, Yousef S, et al. Treating progressive multifocal leukoencephalopathy with interleukin 7 and vaccination with JC virus capsid protein VP1. Clin Infect Dis 2014;59:1588–1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ray U, Cinque P, Gerevini S, et al. JC polyomavirus mutants escape antibody‐mediated neutralization. Sci Transl Med 2015;7:306ra151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Riddell SR, Watanabe KS, Goodrich JM, et al. Restoration of viral immunity in immunodeficient humans by the adoptive transfer of T cell clones. Science 1992;257:238–241. [DOI] [PubMed] [Google Scholar]
- 24. Rooney CM, Smith CA, Ng CY, et al. Use of gene‐modified virus‐specific T lymphocytes to control Epstein‐Barr‐virus‐related lymphoproliferation. Lancet 1995;345:9–13. [DOI] [PubMed] [Google Scholar]
- 25. Balduzzi A, Lucchini G, Hirsch HH, et al. Polyomavirus JC‐targeted T‐cell therapy for progressive multiple leukoencephalopathy in a hematopoietic cell transplantation recipient. Bone Marrow Transplant 2011;46:987–992. [DOI] [PubMed] [Google Scholar]
- 26. Muftuoglu M, Olson A, Marin D, et al. Allogeneic BK virus‐specific T cells for progressive multifocal leukoencephalopathy. N Engl J Med 2018;379:1443–1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Steinhardt MJ, Wiercinska E, Pham M, et al. Progressive multifocal leukoencephalopathy in a patient post Allo‐HCT successfully treated with JC virus specific donor lymphocytes. J Transl Med 2020;18:177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Berger JR, Aksamit AJ, Clifford DB, et al. PML diagnostic criteria: consensus statement from the AAN neuroinfectious disease section. Neurology 2013;80:1430–1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ginevri F, Azzi A, Hirsch HH, et al. Prospective monitoring of polyomavirus BK replication and impact of pre‐emptive intervention in pediatric kidney recipients. Am J Transplant 2007;7:2727–2735. [DOI] [PubMed] [Google Scholar]
- 30. Comoli P, Basso S, Azzi A, et al. Dendritic cells pulsed with polyomavirus BK antigen induce ex vivo polyoma BK virus‐specific cytotoxic T‐cell lines in seropositive healthy individuals and renal transplant recipients. J Am Soc Nephrol 2003;14:3197–3204. [DOI] [PubMed] [Google Scholar]
- 31. Tzannou I, Papadopoulou A, Naik S, et al. Off‐the‐shelf virus‐specific T cells to treat BK virus, human herpesvirus 6, cytomegalovirus, Epstein‐Barr virus, and adenovirus infections after allogeneic hematopoietic stem‐cell transplantation. J Clin Oncol 2017;35:3547–3557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Vera JF, Brenner LJ, Gerdemann U, et al. Accelerated production of antigen‐specific T cells for preclinical and clinical applications using gas‐permeable rapid expansion cultureware (G‐Rex). J Immunother 2010;33:305–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Feucht J, Opherk K, Lang P, et al. Adoptive T‐cell therapy with hexon‐specific Th1 cells as a treatment of refractory adenovirus infection after HSCT. Blood 2015;125:1986–1994. [DOI] [PubMed] [Google Scholar]
- 34. Berger JR, Levy RM, Flomenhoft D, Dobbs M. Predictive factors for prolonged survival in acquired immunodeficiency syndrome‐associated progressive multifocal leukoencephalopathy. Ann Neurol 1998;44:341–349. [DOI] [PubMed] [Google Scholar]
- 35. García De Viedma D, Díaz Infantes M, Miralles P, et al. JC virus load in progressive multifocal leukoencephalopathy: analysis of the correlation between the viral burden in cerebrospinal fluid, patient survival, and the volume of neurological lesions. Clin Infect Dis 2002;34:1568–1575. [DOI] [PubMed] [Google Scholar]
- 36. Taoufik Y, Gasnault J, Karaterki A, et al. Prognostic value of JC virus load in cerebrospinal fluid of patients with progressive multifocal leukoencephalopathy. J Infect Dis 1998;178:1816–1820. [DOI] [PubMed] [Google Scholar]
- 37. Sanjo N, Nose Y, Shishido‐Hara Y, et al. A controlled inflammation and a regulatory immune system are associated with more favorable prognosis of progressive multifocal leukoencephalopathy. J Neurol 2019;266:369–377. [DOI] [PubMed] [Google Scholar]
- 38. Bauer J, Gold R, Adams O, Lassmann H. Progressive multifocal leukoencephalopathy and immune reconstitution inflammatory syndrome (IRIS). Acta Neuropathol 2015;130:751–764. [DOI] [PubMed] [Google Scholar]
- 39. Thurnher MM, Boban J, Rieger A, Gelpi E. Susceptibility‐weighted MR imaging hypointense rim in progressive multifocal leukoencephalopathy: the end point of neuroinflammation and a potential outcome predictor. AJNR Am J Neuroradiol 2019;40:994–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hodel J, Outteryck O, Verclytte S, et al. Brain magnetic susceptibility changes in patients with natalizumab‐associated progressive multifocal leukoencephalopathy. AJNR Am J Neuroradiol 2015;36:2296–2302. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Supporting information