

Abstract
Dysregulated angiogenesis of mesenchymal stem cells (MSCs) is closely related to inflammation and disrupted bone metabolism in patients with various autoimmune diseases. However, the role of MSCs in the development of abnormal angiogenesis in patients with ankylosing spondylitis (AS) remains unclear. In this study, we cultured human umbilical vein endothelial cells (HUVECs) with bone marrow‐derived MSCs from patients with AS (ASMSCs) or healthy donors (HDMSCs) in vitro. Then, the cocultured HUVECs were assayed using a cell counting kit‐8 (CCK‐8) to evaluate the cell proliferation. A wound healing assay was performed to investigate cell migration, and a tube formation assay was conducted to determine the angiogenesis efficiency. ASMSCs exhibited increased angiogenesis, and increased expression of SMAD‐specific E3 ubiquitin ligase 2 (Smurf2) in MSCs was the main cause of abnormal angiogenesis in patients with AS. Downregulation of Smurf2 in ASMSCs blocked angiogenesis, whereas overexpression of Smurf2 in HDMSCs promoted angiogenesis. The pro‐angiogenic effect of Smurf2 was confirmed by the results of a Matrigel plug assay in vivo. By functioning as an E3 ubiquitin ligase in MSCs, Smurf2 regulated the levels of pentraxin 3 (PTX3), which has been shown to suppress angiogenesis through the PTX3‐fibroblast growth factor 2 pathway. Moreover, Smurf2 transcription was regulated by activating transcription factor 4‐induced endoplasmic reticulum stress. In conclusion, these results identify novel roles of Smurf2 in negatively regulating PTX3 stability and promoting angiogenesis in ASMSCs.
Keywords: angiogenesis, arthritis, autoimmune disease, mesenchymal stem cells (MSCs), tube formation
SMAD‐specific E3 ubiquitin protein ligase 2 (Smurf2) transcription is regulated by activating transcription factor 4‐induced endoplasmic reticulum stress, and Smurf2 negatively regulates pentraxin 3 (PTX3) stability in ankylosing spondylitis mesenchymal stem cells and abnormally promotes angiogenesis through the PTX3‐fibroblast growth factor 2 axis.
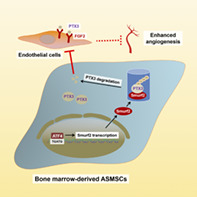
Significance statement.
Angiogenesis participates in the pathogenesis of ankylosing spondylitis (AS). Dysregulated angiogenesis of mesenchymal stem cells (MSCs) is closely related to inflammation and disturbance of bone metabolism in various diseases. This study uncovered the role of MSCs in patients with AS (ASMSCs) in promoting angiogenesis and identified SMAD‐specific E3 ubiquitin ligase 2 (Smurf2) as one of the key factors. Elevated Smurf2 levels in ASMSCs result in the polyubiquitination and degradation of pentraxin 3 (PTX3). And activating transcription factor 4 (ATF4) signaling may be an important mechanism leading to the upregulation of Smurf2. Above all, this study concluded that Smurf2 transcription is regulated by ATF4‐induced endoplasmic reticulum stress and that Smurf2 negatively regulates PTX3 stability and abnormally promotes angiogenesis in ASMSCs.
1. INTRODUCTION
Ankylosing spondylitis (AS) is a type of chronic, inflammatory arthritis that mainly affects the joints of the pelvic bones and spine. It causes symptoms such as pain and stiffness in the lower back and hips. Angiogenesis is an important pathological feature of AS. Data from histological studies suggest that the microvascular density, including the bone‐cartilage interface and subchondral bone marrow, is significantly increased in patients with AS. 1 Increased serum levels of angiogenesis factors have been documented in patients with AS and are closely correlated with the Bath Ankylosing Spondylitis Disease Activity Index (BASDAI), which is the most commonly used disease activity questionnaire for determining disease status. 2 , 3 , 4 Moreover, the progression of spine osteophytes is strongly predicted by the levels of pro‐angiogenic factors. 5 Vascularization of the synovium is more common in patients with spondyloarthritis than in patients with rheumatoid arthritis. 6 However, the molecular mechanism underlying abnormal angiogenesis in patients with AS is unclear, and further research is needed.
Mesenchymal stem cells (MSCs) support hematopoiesis, regulate immune functions, and differentiate into adipocytes, osteocytes, and chondrocytes as adult stem cells that are most abundant in bone marrow. 7 MSCs secrete a variety of angiogenic factors to promote endothelial cell proliferation, migration, and lumen formation under physiological conditions, 8 , 9 and these processes are abnormally enhanced under pathological conditions. 10 , 11 For instance, dysregulated angiogenesis of MSCs is closely related to inflammation and disrupted bone metabolism in patients with rheumatoid arthritis 12 and metastatic breast cancer. 13 However, the role of MSCs in patients with AS (ASMSCs) in the development of abnormal angiogenesis remains unclear.
The ubiquitin‐proteasome system (UPS) is a key component of the angiogenic switch in pathological angiogenesis. 14 A quantitative proteomic analysis revealed that the UPS also plays an important role in the pathogenesis of AS. 15 The UPS controls protein degradation through a series of steps, including substrate recognition, ubiquitin conjugation, and the degradation of ubiquitinated substrates by the proteasome. 16 SMAD‐specific E3 ubiquitin protein ligase 2 (Smurf2) is a novel E3 ubiquitin enzyme that was initially identified as a negative regulator of the bone morphogenetic protein and transforming growth factor beta signaling pathways. 17 Overexpression of Smurf2, which promotes metastasis by enhancing PI3K/AKT‐dependent cell proliferation and invasion, has been observed in various cancers, 18 , 19 whereas another series of studies showed that inactivation of Smurf2 might trigger the mesenchymal‐to‐epithelial transition (MET) and indicated that Smurf2 functions as a potent antitumor factor that prevents carcinogenesis in both mouse models and human cells. 20 , 21 These studies have described the importance and variability of Smurf2 in cell physiology and disease, but the question of whether Smurf2 participates in angiogenesis remains largely unexplored.
Pentraxin 3 (PTX3) is a soluble pattern‐recognition receptor with a high affinity for fibroblast growth factor 2 (FGF2). The PTX3‐FGF2 interaction exerts an inhibitory effect on angiogenesis. 22 , 23 , 24 , 25 Polymorphisms in the PTX3 gene have been shown to be positively correlated with AS—the CC genotype rs3816527 and CT genotype rs3845978 are present at markedly higher levels in patients with AS and increase the risk of AS. 26 In patients with AS presenting BASDAI scores >4, PTX3 has been used as a marker for vasculitis. However, in the general population of patients with AS, peripheral blood PTX3 levels are not significantly changed. 27 Researchers have not yet determined whether PTX3 directly participates in the pathogenesis of AS.
In this study, we illuminated the role of ASMSCs in abnormally promoting angiogenesis. We detected elevated levels of several E3 ubiquitination‐modifying enzymes in ASMSCs. Among them, Smurf2 levels were the most significantly increased and played the most important role. Downregulation of Smurf2 reduced the ability of ASMSCs to promote abnormal angiogenesis. Moreover, we identified PTX3, an angiogenesis repressor, as a substrate of Smurf2, which was controlled by Smurf2‐mediated ubiquitination. Elevated Smurf2 levels in ASMSCs resulted in degradation of PTX3 and promoted angiogenesis, revealing a novel mechanism of AS pathogenesis. This study identified Smurf2 as a key factor contributing to ASMSC‐mediated angiogenesis and revealed the broad cellular pathways by which Smurf2 regulated PTX3, particularly in angiogenesis.
2. MATERIALS AND METHODS
2.1. Cell culture
This study was approved by the Ethics Committee of The Eighth Affiliated Hospital, Sun Yat‐sen University, Shenzhen, China. Thirty healthy donors and 30 patients with AS who met the modified New York criteria were recruited. Written informed consent was obtained from all participants, and their characteristics are presented in our previous study. 28 In the present study, a biological replicate was defined as one cell line of MSCs obtained from a single human—either a healthy donor or a patient with AS. We used MSCs from healthy donors (HDMSCs) as a control for ASMSCs. MSCs were isolated, cultured, and identified using flow cytometry as described in a previous study. 29 Briefly, the MSCs were cultured in Dulbecco's Modified Eagle's Medium (DMEM, glucose 1000 mg/L; Gibco, Waltham, Massachusetts) containing 10% foetal bovine serum (FBS; Sijiqing, Hangzhou, China). Human umbilical vein endothelial cells (HUVECs) and the human embryonic kidney cell line (293T cells) were obtained from iCell Bioscience (Shanghai, China). HUVECs were cultured in endothelial growth medium 2 (EGM‐2; Lonza, Switzerland) containing 10% FBS and the supplied growth factors, and cells at the third to fourth passages were used in all experiments. 293T cells were cultured in high‐glucose DMEM (glucose 4500 mg/L; Gibco) supplemented with 10% FBS.
2.2. Immunohistochemical (IHC) staining
In many studies, tissue samples from patients undergoing surgery for other diseases have often been used as controls for AS tissue samples, such as osteoarthritis (OA) or rheumatoid arthritis. 1 , 6 , 30 In this study, we obtained the hip synovium of AS patients and OA patients during hip replacement surgeries and used the hip synovium of OA patients as a control for the hip synovium of AS patients.
As a marker of endothelial cell differentiation, IHC staining for CD31 was performed to identify local capillaries. IHC staining for Smurf2 was performed to analyze its expression levels in patients with AS. Sections were deparaffinized and hydrated, followed by antigen retrieval in citrate buffer. After quenching with 3% H2O2/H2O and blocking in goat serum, the sections were incubated with an anti‐CD31 antibody (3528; CST, Danvers, Massachusetts) or an anti‐Smurf2 antibody (393 848; Santa Cruz Bio, Santa Cruz, California) overnight at 4°C. Specific labeling was detected using Elivision (Maixin Biotech, Fuzhou, China) plus Polymer HRP Kits and DAB Plus Kits (Maixin Biotech).
A histomorphometric analysis was performed to evaluate the angiogenic capacity based on CD31 staining. Briefly, the IHC‐stained sections were scanned using a digital section scanner (Leica Aperio CS2, Germany) and scored with image analysis techniques (Leica Qwin Proimage analysis system, Germany), which recognize brown CD31 staining within the region of interest (ROI). Vessel‐like structures (VLS) were defined by the presence of CD31‐positive structures with lumens. The vessel density was used as a quantitative parameter, which was defined as the VLS number/ROI (mm2) and calculated using image analysis software.
2.3. Immunofluorescence (IF) staining
For IF staining, sections were deparaffinized and hydrated, followed by antigen retrieval in citrate buffer. After quenching with 3% H2O2/H2O and blocking in goat serum, the sections were incubated with the primary antibodies against CD105 (ab231774; Abcam, Cambridge, UK), CD31 (3528; CST), GFP (2956; CST), Smurf2 (ab94483; Abcam), and PTX3 (373951; Santa Cruz Bio) overnight at 4°C and then incubated with the respective secondary antibodies (4413, 4408, 4409, 4412; CST) at room temperature for 1 hour in the dark. Images of IF staining were acquired with a fluorescence microscope (Leica DM6B, Germany). The vessel density was quantified from at least three random visual fields per section using the Image‐Pro version 6.0 software.
Both HDMSCs and ASMSCs were fixed with 4% paraformaldehyde, blocked with 5% normal goat serum, incubated with primary antibodies against Smurf2 (ab94483; Abcam) and PTX3 (373951; Santa Cruz Bio) overnight at 4°C, and then incubated with the appropriate secondary antibodies and stained with DAPI to detect the colocalization of Smurf2 and PTX3. Images were obtained using an LSM 5 Exciter confocal imaging system (Carl Zeiss, Germany). Positively stained cells were quantified from at least three random visual fields per section using the Image‐Pro version 6.0 software.
2.4. Effects of MSCs on HUVECs in vitro
An indirect Transwell coculture system was employed to assess the different effects of HDMSCs and ASMSCs on HUVECs. HUVECs (2.5 × 105 cells/well) were seeded into the lower chambers, and MSCs (0.5 × 105 cells/well; HUVECs:MSCs = 5:1) were seeded into the upper chambers of 12‐well plates with Transwell inserts (0.4 μm pore size; Corning, New York). The cells were cocultured in complete EGM‐2 for 24 hours, and then the HUVECs were digested with 0.25% trypsin containing 0.53 mM EDTA for subsequent proliferation, migration, and tube formation assays. Neutralizing antibodies, including anti‐EGF (ab9695; Abcam; 0.1 μg/mL), anti‐FGF2 (AF‐233; R&D Systems, Minneapolis, Minnesota; 5 μg/mL) and their negative IgG controls (ab172730; Abcam and AB‐108‐C; R&D Systems), were added to the HUVECs and MSCs coculture system as appropriate.
For the proliferation assay, HUVECs (5 × 103 cells/well; five replicates per group) were seeded into 96‐well culture plates after coculturing with HDMSCs or ASMSCs for 24 hours. A well without cells served as the blank control. On days 1, 2, 3, 4, and 5, cell counting kit‐8 (CCK‐8) reagent (10 μL per well; 7 Sea Biotech, Shanghai, China) was added to the culture medium (100 μL per well). After incubation at 37°C for 2 hours, the absorbance of each well was measured at 450 nm according to the protocol, and the cell proliferation was calculated by subtracting the absorbance of the blank well from the mean absorbance of each individual well.
For the wound healing assay, a square space (0.8 cm2) devoid of cells was created by placing a biocompatible silicon self‐stick cellular stopper (Ibidi, Germany) in each well before seeding HUVECs (3.5 × 104 cells per chamber). After the cells reached confluence, the Ibidi inserts were removed, leaving a 500 μm cell‐free gap. HUVEC migration across the gap was monitored for 0, 12, and 24 hours within the serum‐free medium. Cell migration was determined by calculating the percentage of the area of migrated cells relative to the total area of the wound using the Image‐Pro version 6.0 software.
For the Transwell migration assay, after coculturing with HDMSCs or ASMSCs for 24 hours, HUVEC migration was assessed in 24‐well Transwell inserts with an 8 μm pore size (Corning). Briefly, 2 × 104 cells were plated in the upper chamber of each well in 100 μL of low‐serum (2% FBS) EGM‐2. The lower chamber was filled with 500 μL of complete medium (10% FBS). After 6 hours at 37°C, nonmigrating cells were removed from the filter by washing three times with PBS and gentle scraping. Migrated cells present on the lower surface of the filter were fixed with 10% formaldehyde for 30 minutes and stained with 0.1% crystal violet for 20 minutes. Five fields in each well were counted. Migrated cells were quantified using the Image‐Pro version 6.0 software.
The in vitro tube formation assay was performed using 15 wells of a μ‐slide angiogenesis plate (Ibidi) coated with Matrigel (10 mg/mL; BD, San Jose, California) according to the manufacturer's instructions. Briefly, the wells of the μ‐slide were coated with 10 μL of Matrigel and incubated at 37°C for 30 minutes to allow gelation. Then, 1.5 × 104 cells/well were suspended in EGM‐2 supplemented with a low level of serum (2% FBS) and plated onto a layer of Matrigel. The μ‐slide angiogenesis plate was then incubated for an additional 4 hours at 37°C, and capillary‐like tube formation was observed and photographed with a microscope. Quantitative image analysis of the cell‐covered area, loops, tube length, and the number of branch points was performed with the automated analysis tool WimTube using the Web platform (https://mywim.wimasis.com).
2.5. Matrigel plug assay in vivo
Animal procedures were performed in accordance with the guidelines for laboratory animal usage and used a protocol approved by Sun Yat‐sen University's Committee on the Use and Care of Animals. Eight‐week‐old female BALB/c‐nu mice (Laboratory Animal Center of Sun Yat‐sen University) were used in all experiments. We pretransfected HUVECs with an empty lentivirus vector expressing the GFP protein (sequence element: Ubi‐MCS‐SV40‐EGFP‐IRES‐puromycin), and the population of HUVECs expressing GFP was isolated using fluorescence‐activated cell sorting (FACS). All the lentiviruses used to transfect MSCs in the Matrigel plug assay were constructed by Obio Technology (Shanghai, China). In particular, lentiviruses encoding short hairpin RNA (shRNA) targeting Smurf2 (Sh‐Smurf2: 5′‐GGAAGCGAUUAAUGAUAAA‐3′) were constructed with the GL401 vector (sequence element: pCLenti‐U6‐shRNA‐CMV‐Puro‐WPRE). Lentiviruses for Smurf2 overexpression (OE‐Smurf2) were constructed with the GL119 vector (sequence element: pSLenti‐CMV‐MCS‐PGK‐Puro‐WPRE), and the negative control was named NC. Lentiviruses (109 TU/mL) and polybrene (5 μg/mL) were added to the medium at an multiplicity of infection (MOI) of 50 and incubated with the MSCs for 24 hours. Then, the cells were treated with puromycin (2.0 μg/mL; Solarbio, Beijing, China) for another 48 hours for selection. Each implant contained 3 × 106 total cells (MSCs only, HUVECs only, or cell mixtures at a 1:1 ratio) suspended in a total volume of 200 μL consisting of 100 μL of PBS and 100 μL of Matrigel (15 mg/mL; BD). The solution was subcutaneously injected into the dorsal flank of each mouse, with two implants per animal. Animals were maintained in a stationary position for 5 minutes to allow implant solidification and then placed in fresh cages for recovery. Three replicates (n = 3 different ASMSC lines per group) of each sample type were analyzed (ASMSC‐only, HUVEC‐only, HUVEC+ASMSCNC, HUVEC+ASMSCOE‐Smurf2, and HUVEC+ASMSCSh‐Smurf2 groups). Specimens were harvested 2 weeks after implantation. The implants were fixed with 4% paraformaldehyde, dehydrated, and subsequently embedded in paraffin. Sections (5 μm thickness) were stained with hematoxylin and eosin (H&E) and a CD31 (3528; CST) antibody. In addition, the anti‐CD31 antibody reacted only with the human protein, ensuring that the staining was specific for human cells.
2.6. Quantitative real‐time PCR (qRT‐PCR)
The total RNA was isolated from cells or tissue samples using TRIzol (Invitrogen, Carlsbad, California) and transcribed into cDNAs using the PrimeScript RT Reagent Kit (Takara Bio, Mountain View, California). QRT‐PCR was performed with a Light Cycler 480 Real‐Time PCR System (Roche, Switzerland) using SYBR Premix Ex Taq (Takara Bio.). Relative changes in the mRNA levels of genes were assessed using the 2(−ΔΔct) method and normalized to the housekeeping gene GAPDH. The specific primers are listed in Supplementary Table S1.
2.7. Western blotting
Cells were lysed in RIPA buffer supplemented with protease and phosphatase inhibitors, and the clarified lysates were resolved by SDS‐PAGE and transferred to PVDF membranes for Western blotting using ECL detection reagents (Beyotime, Shanghai, China). The immunoblots were processed according to standard procedures using primary antibodies against GAPDH (5174; CST), Smurf2 (12024; CST), PTX3 (ab125007; Abcam), Flag (8146; CST), Myc (2276; CST), HA (3724; CST), and activating transcription factor 4 (ATF4; 11815; CST) at 4°C overnight. Then, the membranes were incubated with the appropriate HRP‐conjugated secondary antibodies (Beyotime). We used VeriBlot for IP detection reagents (ab131366; Abcam), which recognizes only native (nonreduced) antibodies to avoid interference from the IgG heavy chain; therefore, detection of the heavy and light chains of IgG was sufficiently minimized.
2.8. Inhibition of lysosomal and proteasomal degradation pathways
MSCs or 293T cells were seeded into six‐well plates at a density of 3 × 105 cells/well. The cells were treated with the lysosomal inhibitor chloroquine (50 μM; 14774; CST) or the proteasomal inhibitor MG132 (10 μM; S2619; Selleck) or MG101 (10 μM; S7386; Selleck) for another 12 hours to block the autophagy‐lysosome pathway (ALP) or UPS. Protein lysates were subsequently harvested, and the level of the Myc‐PTX3 protein was evaluated using Western blotting.
2.9. Cycloheximide (CHX) chase assay
The half‐life of the PTX3 protein was evaluated using the CHX chase assay as described in a previous study. 31 Briefly, MSCs or 293T cells were seeded into six‐well plates at a density of 3 × 105 cells/well. Protein lysates were prepared at the indicated time points after the addition of CHX (10 μM). The levels of PTX3 or Myc‐PTX3 in the MSCs and 293T cells were determined using immunoblotting and quantified at the indicated time points.
2.10. Transfection
All of the following lentiviruses were constructed by GeneChem (Shanghai, China). In particular, lentiviruses encoding shRNAs targeting Smurf2 (Sh‐Smurf2: 5′‐GGAAGCGAUUAAUGAUAAA‐3′), PTX3 (Sh‐PTX3: 5′‐GGGAUAGUGUUCUUAGCAA‐3′), ATF4 (Sh‐ATF4: 5′‐GCCUAGGUCUCUUAGAUGATT‐3′), and a negative control (NC1: 5′‐TTCTCCGAACGTGTCACGTTTC‐3′) were constructed in the LKD007 vector (sequence element: pLKD‐CMV‐EGFP‐2A‐Puro‐U6‐shRNAs). We also used the catalytically inactive mutant of Smurf2, C716A, to study the Smurf2 function, which was generated by introducing a Cys‐to‐Ala point mutation at AA 716. Lentiviruses for Smurf2 (OE‐Smurf2), C716A (OE‐C716A), PTX3 (OE‐PTX3), and UBB (HA‐UBB) overexpression were constructed with the GV367 vector (sequence element: Ubi‐MCS‐SV40‐EGFP‐IRES‐puromycin), and the negative control was named NC2. All vectors contained a tag, and the proteins were expressed as GFP fusion proteins; thus, the expression of the Smurf2, C716A, PTX3, and UBB proteins was regulated by the same promoters as GFP. Lentiviruses (109 TU/mL) and polybrene (5 μg/mL) were added to the medium at an MOI of 50 and incubated with the MSCs for 24 hours. The total amounts of transfected lentiviruses in the other groups were equalized by adding the NC lentiviruses. Then, sterile FACS was performed based on GFP expression.
Expression plasmid constructs, including full‐length pcDNA3.1(+)‐Flag‐Smurf2 (Flag‐Smurf2), pcDNA3.1(+)‐Flag‐C716A (Flag‐C716A), pcDNA3.1(+)‐Myc‐PTX3 (Myc‐PTX3), pcDNA3.1(+)‐HA‐Ubiquitin (HA‐UBB), pcDNA3.1(+)‐HA‐K48‐Ubiquitin (HA‐K48‐UBB), pcDNA3.1(+)‐HA‐K63‐Ubiquitin (HA‐K63‐UBB), and empty pcDNA3.1(+)‐vectors, were all constructed by and purchased from Obio Technology. A Lipofectamine 3000 Transfection Kit (Invitrogen) was used for transfection. Briefly, 293T cells were seeded into six‐well plates at a density of 3 × 105 cells/well. Each plasmid was used at a concentration of 2.5 μg/well and mixed with 5 μL of Lipofectamine 3000 Transfection Reagent (Invitrogen) and 5 μL of P3000 (Invitrogen) according to the manufacturer's instructions.
2.11. Protein profile assay
The supernatants of HUVECs that had been cocultured with HDMSCs or ASMSCs for 24 hours were collected. Then, the supernatants were analyzed using a Proteome Profiler Human Angiogenesis Array Kit (ARY007; R&D Systems) according to the manufacturer's instructions. For each assay, 500 μL of cell culture supernatant was used, and the cytokine optical densities were measured with HLI++mage software (Western Vision). Densitometry analysis of the signal intensities was performed to quantify differences in cytokine levels between ASMSCs and HDMSCs.
2.12. ELISA
The supernatants of HUVECs that had been cocultured with HDMSCs or ASMSCs for 24 hours were collected. The level of the PTX3 protein in the cell culture supernatants was quantified using a Human PTX3 Quantikine ELISA Kit (DPTX30; R&D Systems) according to the manufacturer's protocol.
2.13. Co‐immunoprecipitation (Co‐IP) and LC‐MS/MS
For immunoprecipitation (IP), the cells were washed with PBS, and protein lysates were obtained using IP protein lysis buffer (P0013; Beyotime). The supernatants were first incubated with antibodies against Smurf2 (12024; CST), PTX3 (373951; Santa Cruz Bio.), Flag (8146; CST), Myc (2276; CST), or their IgG controls (3452, 37988; CST) for 2 hours, followed by Protein G‐agarose beads (10004D; Thermo Fisher Scientific, Rockford, Illinois) overnight. After three washes with the IP protein lysis buffer, the samples were collected with SDT buffer (4% wt/vol SDS, 100 mM Tris/HCl, and 1 mM 1,4‐dithiothreitol [DTT], pH 7.6) and analyzed using liquid chromatography with tandem mass spectrometry (LC‐MS/MS) to identify the proteins interacting with Smurf2. The entire LC‐MS/MS procedure was performed by the Applied Protein Technology Company (Shanghai, China).
2.14. Dual‐luciferase reporter assay
The plasmids described below were all constructed by GeneChem. We scanned Smurf2 promoter regions spanning from −2000 to 0 bp of the transcription start site and predicted the potential binding sites for ATF4 with the JASPAR database (http://jaspar.genereg.net/). The ATF4 gene and Smurf2 promoter (WT‐Luc) were synthesized and inserted into the expression vectors GV219 (sequence element: CMV‐MCS‐SV40‐Neomycin) and GV238 (sequence element: MCS‐firefly luciferase), respectively. Smurf2 promoter mutants (Mutant‐Luc) with a mutation in the region containing the predicted ATF4 binding sites were also constructed in the GV238 vector. For the luciferase reporter assays, MSCs were seeded into 12‐well plates and transfected with the indicated plasmids using Lipofectamine 3000 Transfection Reagent (Invitrogen). The cells were collected 36 hours after transfection. The luciferase activity was measured using the Dual‐Luciferase Reporter Assay System (Promega, Madison, Wisconsin). Smurf2 promoter‐driven luciferase activity was normalized to the thymidine kinase Renilla activity.
2.15. Chromatin immunoprecipitation (ChIP)
ChIP assays were performed using the EZ‐Magna ChIP A/G Assay Kit (Millipore, Billerica, Massachusetts) according to the manufacturer's instructions. Briefly, cells were crosslinked with 1% formaldehyde at room temperature for 10 minutes. Then, the cells were collected and lysed with nuclear lysis buffer containing a protease inhibitor cocktail to isolate the nuclei. Sonication was performed to shear the chromatin and generate 200 to 1000 bp DNA fragments. The sheared chromatin was immunoprecipitated with primary antibodies against ATF4 (11815; CST), normal rabbit IgG (2729; CST), and anti‐RNA polymerase II. After the protein‐DNA crosslinks were reversed, the precipitated DNA was purified and subsequently analyzed using qRT‐PCR. Data were normalized to the input control. The ChIP‐qPCR primers for the Smurf2 promoter were forward, 5′‐CCCCCACGCTATTGTCCTTA‐3′, and reverse, 5′‐CCAGGAACCGCATGGTTTTT‐3′.
2.16. Statistical analysis
Data are presented as the means ± SD. One‐way ANOVA or a t test was used according to the type of data. Statistical analyses were conducted using SPSS software (SPSS, Inc). A P value of less than .05 was considered indicative of a significant difference.
3. RESULTS
3.1. ASMSCs exhibit a stronger ability to promote angiogenesis compared with HDMSCs
We obtained the hip synovium from patients with AS and OA patients during hip replacement surgery to detect the microvessel density in local lesions. A significantly greater number of CD31+ microvessels was observed in the synovial lesions of patients with AS than in the lesions of patients with OA (Figure 1A) as quantified by the area of CD31‐positive staining (Figure 1B) and vessel density (Figure 1C). To quantify CD31 expression in AS, we further analyzed CD31 mRNA levels in samples from all patients, and higher expression was detected in patients with AS (Supplementary Figure S1B). MSCs secrete various angiogenic factors to modulate angiogenesis, and their levels are abnormally increased under pathological conditions. 10 , 11 Then, we analyzed the correlation between angiogenesis and MSCs in patients with AS by performing IF staining for their markers, CD31 and CD105. Compared to patients with OA, patients with AS had more CD31‐positive stained vessels and higher CD105 expression (Supplementary Figure S1A). We cocultured HUVECs with MSCs in a Transwell system for 24 hours to investigate the different effects of HDMSCs and ASMSCs on vascular endothelial cells. Based on the results of the CCK‐8 assay, HUVEC proliferation was increased after coculturing with ASMSCs (Figure 1D). Wound healing assays (Figure 1E,F) and Transwell assays (Figure 1G,H) revealed that ASMSCs markedly increased HUVEC motility. The tube formation assay on Matrigel is an in vitro model of angiogenesis. 32 After culturing with ASMSCs, the HUVECs produced a greater number of capillary‐like structures than after culturing with HDMSCs (Figure 1I). Quantitative measurements indicated that the cell‐covered area (Figure 1J), total tube length (Figure 1K), total branch points (Figure 1L), and total loops (Figure 1M) were all significantly increased after coculturing with ASMSCs. Thus, ASMSCs enhance the angiogenic activities of endothelial cells.
FIGURE 1.
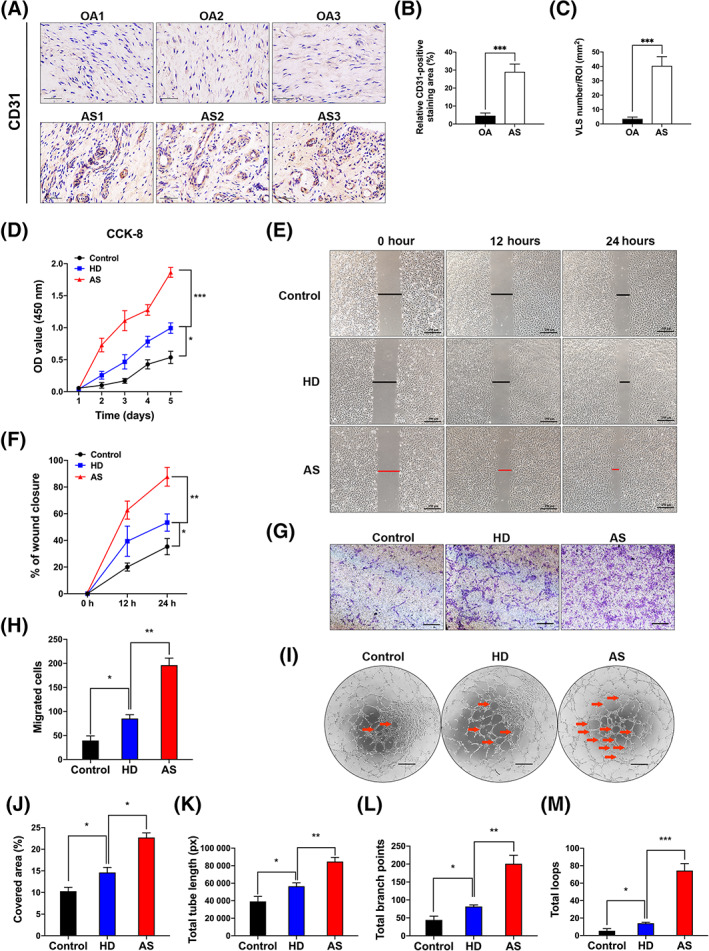
Mesenchymal stem cells in patients with ankylosing spondylitis (AS) (ASMSCs) exhibited a stronger ability to promote angiogenesis compared to MSCs in patients with healthy donors (HDMSCs). A, Representative images showing significantly higher levels of CD31 immunohistochemical staining in the hip synovium of patients with AS than in the hip synovium of patients with OA (n = 10 patients per group). Scale bar = 50 μm. B, The sections were scored by analyzing CD31 levels (brown staining) in high‐magnification (×400) digitized images. C, Vessel‐like structures (VLS) were defined by the presence of CD31‐positive structures with lumens. Quantification of CD31 expression and the VLS within the ROI revealed higher levels of both parameters in patients with AS than in patients with OA. D, Human umbilical vein endothelial cell (HUVEC) proliferation was substantially increased when these cells were cocultured with ASMSCs compared to when they were cocultured with HDMSCs, as measured using the CCK‐8 assay. E,F, Wound healing and G,H, Transwell assays revealed that ASMSCs increased the motility of HUVECs. Scale bar = 250 μm. I, Representative images of the tube formation assay on Matrigel using HUVECs cocultured with HDMSCs or ASMSCs. The red arrow indicates the vessel structures with a relatively complete vascular lumen, and the images were quantified by determining the (J) cell‐covered area, (K) total tube length, (L) total branch points, and (M) total loops. Scale bar = 400 μm. n = 10 different HDMSC or ASMSC lines per group. All data are presented as the means ± SD. *P < .05, **P < .01, ***P < .001
3.2. Upregulation of Smurf2 promotes angiogenesis mediated by ASMSCs
The UPS is involved in the pathogenesis of AS, 15 and the main components of the UPS are E1‐E2‐E3 ligases and ubiquitin proteins; thus, E3 ligases are the main components of the UPS. 16 From the results of our previous studies, we found that several E3 ligases were upregulated in AS patients. 28 , 29 , 33 Therefore, we tested the expression of these enzymes by qRT‐PCR and found that the mRNA levels of four E3 ligases were significantly upregulated in ASMSCs (Supplementary Figure S1D). Among these enzymes, Smurf2 expression was the most significantly upregulated (Supplementary Figure S1D; Figure 2A). Then, we found that consistent with the mRNA findings, Smurf2 protein levels were increased in ASMSCs (Figure 2B,C). We also investigated the expression of Smurf2 in patients with AS by performing IHC staining. Compared to that in patients with OA, noticeably higher Smurf2 expression was detected in the local lesions of patients with AS (Supplementary Figure S1C). The specificity of the Smurf2 antibody was verified by IF staining (Supplementary Figure S2C). We constructed lentiviruses to silence Smurf2 and overexpress Smurf2 in MSCs and inactive Smurf2 C716A mutants, which were named Sh‐Smurf2, OE‐Smurf2, and OE‐C716A, respectively, to examine the effect of Smurf2 on angiogenesis. Both HDMSCs and ASMSCs were effectively transfected with the lentivirus (Supplementary Figure S1F), and sterile FACS was performed to select positive cells transfected with the vector encoding GFP (Supplementary Figure S1E). The expression of Smurf2 was reduced to very low levels in cells transfected with the vector carrying the shRNA, and the overexpression of Smurf2 or C716A was observed in cells transfected with the overexpression vectors (Figure 2D,E).
FIGURE 2.
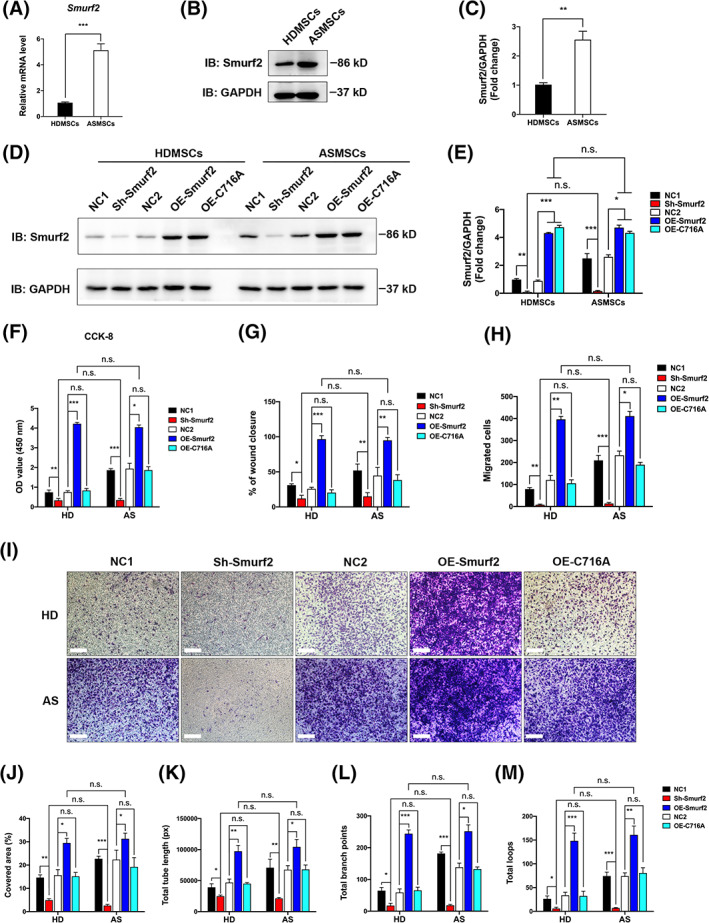
Upregulation of Smurf2 promoted angiogenesis by mesenchymal stem cells (MSCs) in patients with ankylosing spondylitis (ASMSCs). A‐C, Compared to those in the MSCs in patients with healthy donors (HDMSCs), the levels of both Smurf2 mRNA and protein were increased in ASMSCs. Lentiviruses were used to silence or overexpress Smurf2 or C716A (catalytically inactive mutant of Smurf2) in both HDMSCs and ASMSCs. D,E, The interference efficiency of shRNA targeting Smurf2 and the overexpression efficiency of OE‐Smurf2 and OE‐C716A were both verified by Western blot analyses. F‐I, In the CCK‐8 (fifth day), wound healing and Transwell assays, Smurf2 knockdown decreased human umbilical vein endothelial cell (HUVEC) proliferation and migration, whereas Smurf2 overexpression exerted the opposite effect, but C716A overexpression did not induce a similar phenotype. Scale bar = 250 μm. J‐M, Smurf2 knockdown in MSCs decreased HUVEC tube formation, whereas overexpression of Smurf2 in MSCs increased HUVEC tube formation, with no notable difference between HDMSCs and ASMSCs. However, overexpression of C716A did not exert a significant effect on the HDMSCs or ASMSCs. The cell‐covered area, total tube length, total branch points and total loops were quantified. n = 10 different HDMSC or ASMSC lines per group. All data are presented as the means ± SD. *P < .05, **P < .01, ***P < .001. n.s., not significant
Then, we cocultured HUVECs with the selected MSC population in which Smurf2 expression was modulated. After Smurf2 knockdown, HUVEC angiogenesis decreased, and no notable difference was detected between HUVECs cocultured with HDMSCs and HUVECs cocultured with ASMSCs (Figure 2F‐M; Supplementary Figure S2A,B). However, Smurf2 overexpression increased tube formation, whereas C716A overexpression did not exert a similar effect. The results of the CCK‐8 (Figure 2F), wound healing (Supplementary Figure S2A; Figure 2G), and Transwell migration assays (Figure 2H,I) were consistent with the results of the tube formation assay (Supplementary Figure S2B; Figure 2J‐M), indicating that elevated Smurf2 was likely the main cause of abnormal angiogenesis induced by ASMSCs, and the catalytically inactive C716A mutant lost pro‐angiogenic activity.
We performed a Matrigel plug assay in vivo to further confirm the regulatory effect of Smurf2 on angiogenesis. Images of H&E staining (Figure 3A) of the implants of the nude mice revealed that the implants in the HUVEC‐only group, HUVEC+ASMSCNC group, HUVEC+ASMSCOE‐Smurf2 group and HUVEC+ASMSCSh‐Smurf2 group all contained vessels and red blood cells, but the implants in the ASMSC‐only group did not. Compared with the HUVEC+ASMSCNC group, the HUVEC+ASMSCOE‐Smurf2 group exhibited many more newly formed blood vessels with clear lumens and borders, whereas the HUVEC+ASMSCSh‐Smurf2 group exhibited a significant decrease in the number of blood vessels. Microvessels were immunostained for the endothelial marker CD31 to verify the results obtained from the H&E‐stained sections. As shown in the images of IHC staining, the implants from the HUVEC+ASMSCOE‐Smurf2 group expressed CD31 at higher levels and contained many more smaller, tightly sealed capillaries compared with the implants from the HUVEC+ASMSCNC group, whereas the HUVEC+ASMSCSh‐Smurf2 group exhibited a decrease in these parameters, which was consistent with the H&E staining observations. We pretransfected the HUVECs with an empty lentivirus vector containing GFP to label and select these cells and then marked these HUVECs with GFP staining to distinguish them from mouse endothelial cells and MSCs. We performed IF staining for CD31 and GFP and observed the coexpression of these proteins. As shown in the images of IF staining, both the CD31 expression and vessel density were increased in the HUVEC+ASMSCOE‐Smurf2 group and decreased in the HUVEC+ASMSCSh‐Smurf2 group compared with those in the HUVEC+ASMSCNC group. As described in a previous report, 11 MSCs also exerted a significant positive effect on blood vessel formation, which was observed in the HUVEC‐only group and HUVEC+ASMSCNC group. Quantitative analysis of the density of the new blood vessels, which was defined as the area of CD31‐positive staining and VLS density, confirmed that Smurf2 expression in ASMSCs was important for the beneficial effect of HUVECs on angiogenesis (Figure 3B‐D). Taken together, elevated Smurf2 is essential for ASMSC‐induced angiogenesis.
FIGURE 3.
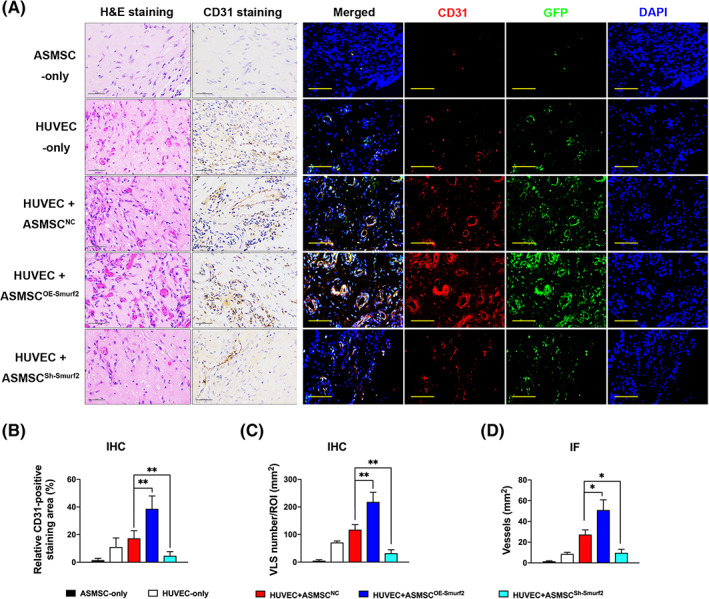
Matrigel plug assay in vivo. We pretransfected human umbilical vein endothelial cells (HUVECs) with an empty lentivirus vector expressing the GFP protein to label the HUVECs and then marked these HUVECs with GFP staining to distinguish them from mouse endothelial cells and mesenchymal stem cells (MSCs). A, H&E and CD31 IHC staining (scale bar = 50 μm) and IF staining (scale bar = 100 μm; red, CD31; green, GFP; blue, DAPI) illustrating varying blood vessel morphologies and quantities in the implants from the five groups. Vessel‐like structures (VLS) were defined by the presence of CD31‐positive structures with lumens. Stained sections contained VLS and red blood cells in the implants from the HUVEC‐only group, HUVEC+ASMSCNC group, HUVEC+ASMSCOE‐Smurf2 group and HUVEC+ASMSCSh‐Smurf2 group. Compared with the HUVEC+ASMSCNC group, the HUVEC+ASMSCOE‐Smurf2 group exhibited many more newly formed blood vessels with clear lumens and borders, whereas the HUVEC+ASMSCSh‐Smurf2 group exhibited significant decreases in these parameters. B‐D, Quantification of the angiogenic capacity is presented as the vessel density and percentage of the CD31‐stained area. n = 3 different ASMSC lines per group. All data are presented as the means ± SD. *P < .05, **P < .01
3.3. Smurf2 functions as an E3 ubiquitin ligase to ubiquitinate and degrade PTX3
We first performed co‐IP and LC‐MS/MS experiments to investigate the proteins that interact with Smurf2 in ASMSCs and are involved in angiogenesis and obtained 323 potential Smurf2‐interacting proteins (Supplementary Table S2). Then, we used a Proteome Profiler Human Angiogenesis Array Kit to identify the differentially altered levels of angiogenic factors between supernatants of HDMSCs or ASMSCs cocultured with HUVECs. Abnormal levels of several factors were observed in the supernatants of ASMSCs cocultured with HUVECs (Supplementary Figure S3A,B). EGF levels were increased and may have a role in the enhanced angiogenesis of ASMSCs. When EGF was blocked by a neutralizing antibody, the enhanced angiogenesis capacity of ASMSCs was impaired to some extent, but the difference from the HDMSCs group was still significant. The results of the CCK‐8 (Supplementary Figure S3D), wound healing (Supplementary Figure S3C,F), and Transwell migration assays (Supplementary Figure S3E,H) were consistent with the results of the tube formation assay (Supplementary Figure S3G,I‐L). Thus, EGF may not be the main factor inducing angiogenesis by ASMSCs.
Finally, using overlapping portions of two independent datasets, we identified PTX3 as a potential substrate of Smurf2 (Figure 4A). Then, an IF assay revealed the colocalization of Smurf2 and PTX3 in the cytoplasm of both HDMSCs and ASMSCs, and Smurf2 expression was increased, whereas PTX3 expression was decreased in ASMSCs compared with HDMSCs (Figure 4B,C). Furthermore, we conducted reciprocal co‐IP and Western blotting assays to confirm that Smurf2 and PTX3 interacted with each other in ASMSCs (Figure 4D). Compared with HDMSCs, the level of the PTX3 protein was decreased in ASMSCs, but an increased Smurf2‐PTX3 interaction was observed (Figure 4E,H). Smurf2, an E3 ubiquitin ligase, ubiquitinates and degrades proteins. 34 Therefore, we detected the level of ubiquitinated PTX3 in HDMSCs and ASMSCs. PTX3 is generated by the NF‐κB pathway, 35 and proteasome inhibitors inhibit the NF‐κB pathway. 36 We initially confirmed that MG132 and MG101 substantially inhibited endogenous PTX3 expression (Supplementary Figure S2D). Thus, we were only able to overexpress the HA‐UBB and attempt to promote the ubiquitination of endogenous PTX3 in the absence of proteasome inhibitors. Compared with those in HDMSCs, decreased PTX3 levels and increased PTX3 ubiquitination were observed in ASMSCs (Figure 4F). However, compared with that in HDMSCs, no significant change in the level of the PTX3 mRNA was detected in ASMSCs based on the qRT‐PCR results (Figure 4G). We further measured the level of secreted PTX3 in ASMSCs cocultured with HUVECs and observed a decrease in the extracellular level, as confirmed using ELISA (Figure 4I). We investigated Smurf2 and PTX3 expression in patients with AS using IF staining to directly examine whether the Smurf2‐PTX3‐angiogenesis pathway is involved in AS. A negative correlation between Smurf2 and PTX3 protein levels was observed. Compared to those in patients with OA, Smurf2 was expressed at higher levels, and PTX3 was expressed at lower levels in patients with AS (Figure 4J).
FIGURE 4.
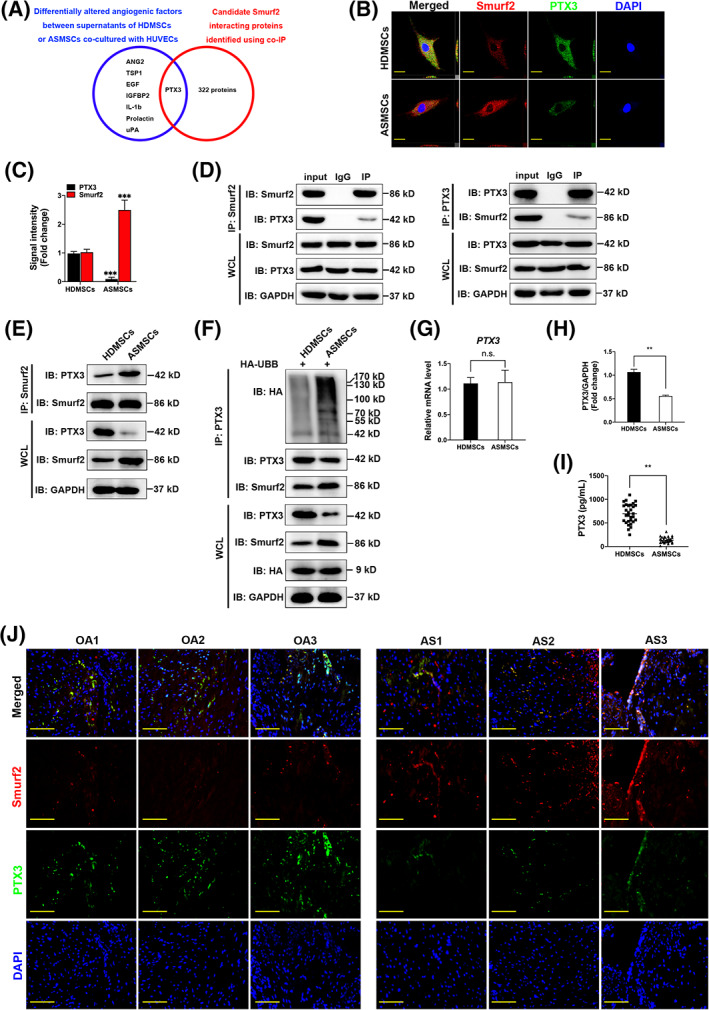
PTX3 is a Smurf2 target in mesenchymal stem cells (MSCs) in patients with ankylosing spondylitis (AS) (ASMSCs). A, Venn diagram showing the differentially altered angiogenic factors between supernatants of MSCs in patients with healthy donors (HDMSCs) or ASMSCs cocultured with human umbilical vein endothelial cells (HUVECs; blue), candidate Smurf2‐interacting proteins in ASMSCs identified in the co‐IP (red) and overlapping PTX3 datasets. B, Immunofluorescence (IF) staining showing the colocalization of Smurf2 and PTX3 both in HDMSCs and ASMSCs. Compared with HDMSCs, the expression of Smurf2 increased, and the expression of PTX3 decreased in ASMSCs (red, Smurf2; green, PTX3; blue, DAPI; scale bar = 20 μm). C, Quantitative analysis of the signal intensity in (B) (n = 10 different HDMSC or ASMSC lines per group). D, By detecting the protein expression of whole‐cell lysates (WCL) in each group, it was proven that the protein lysates enriched for the target protein by IP or IgG negative control were equivalent. Co‐IP mixtures were evaluated using Western blotting. Endogenous Smurf2 and PTX3 interacted with each other in ASMSCs (n = 10 different ASMSC lines per group). E,H, PTX3 and Smurf2 interacted with each other both in HDMSCs and ASMSCs. Compared with that in HDMSCs, the level of the PTX3 protein was decreased in ASMSCs, but the strength of the Smurf2‐PTX3 interaction was increased (n = 10 different HDMSC or ASMSC lines per group). F, The transfection efficiency of HA‐UBB was equal. After overexpression of HA‐UBB in MSCs, lower protein levels and increased ubiquitination of PTX3 were observed in ASMSCs compared to HDMSCs (n = 10 different HDMSC or ASMSC lines per group). G, Similar levels of the PTX3 mRNA were detected in HDMSCs and ASMSCs (n = 10 different HDMSC or ASMSC lines per group). I, PTX3 secretion into the supernatants of HDMSCs or ASMSCs cocultured with HUVECs was measured using an ELISA (n = 30 different HDMSC or ASMSC lines per group). J, Based on IF staining, the expression levels of Smurf2 and PTX3 were negatively correlated in patients with AS. Compared to those in patients with OA, Smurf2 was expressed at higher levels, whereas PTX3 was expressed at lower levels in patients with AS (red, Smurf2; green, PTX3; blue, DAPI; n = 10 patients per group; scale bar = 50 μm). All data are presented as the means ± SD. **P < .01, ***P < .001. n.s., not significant
We cotransfected the HA‐UBB, Myc‐PTX3, Flag‐Smurf2, and Flag‐C716A plasmids into 293T cells and observed that Flag‐Smurf2 significantly induced the ubiquitination of Myc‐PTX3, whereas Flag‐C716A did not exert a similar effect (Figure 5A). Furthermore, Smurf2 mediated both K63‐linked (Figure 5B) and K48‐linked ubiquitination of PTX3 (Figure 5C). We blocked the lysosomal and proteasomal degradation pathways in ASMSCs with the proteasome inhibitor MG132 and the lysosome inhibitor chloroquine to further determine whether PTX3 degradation was mediated by the proteasome or lysosomes. MG132 reversed the degradation of Myc‐PTX3 mediated by Flag‐Smurf2, whereas chloroquine had less of an effect (Figure 5D). Therefore, PTX3 degradation is mainly regulated by the UPS.
FIGURE 5.
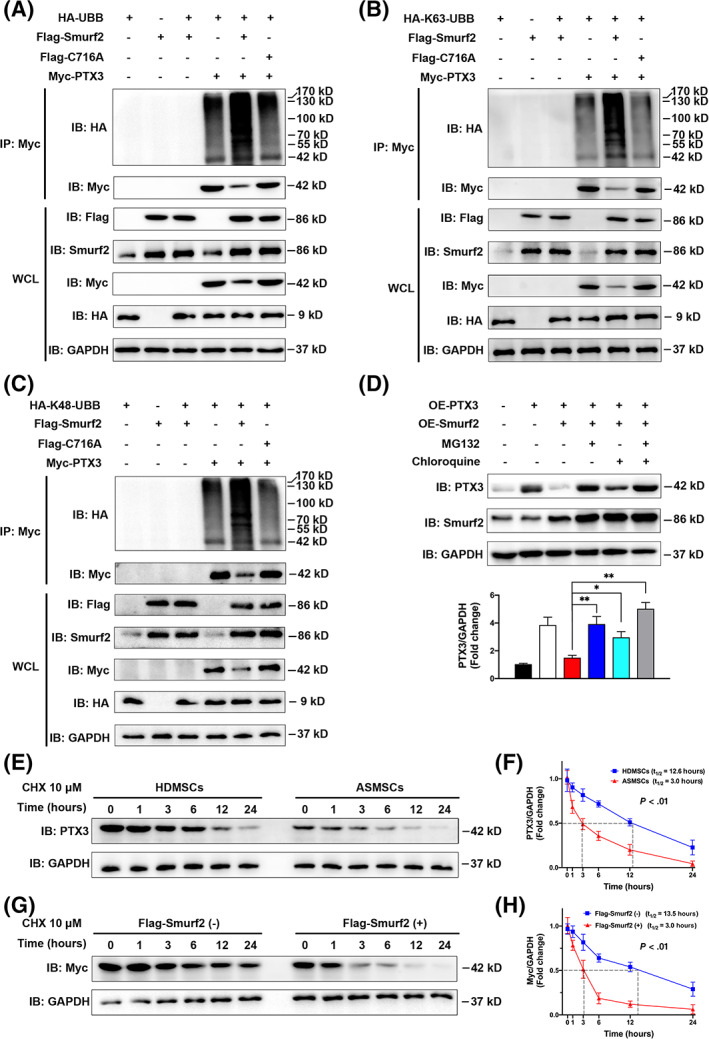
Smurf2 functioned as an E3 ubiquitin ligase to ubiquitinate and degrade PTX3. After coexpression of HA‐UBB, Flag‐Smurf2, Flag‐C716A, and Myc‐PTX3 in 293T cells for 36 hours, the cell lysates were harvested. A, Western blots showing that Flag‐Smurf2 significantly induced the ubiquitination of Myc‐PTX3, whereas Flag‐ C716A did not exert a significant effect. B,C, Flag‐Smurf2 significantly induced both the K63‐linked and K48‐linked ubiquitination of Myc‐PTX3 (n = 3 independent experiments). D, Myc‐PTX3 was transiently overexpressed in mesenchymal stem cells (MSCs) in patients with ankylosing spondylitis (ASMSCs) to avoid interfering with the NF‐κB pathway (n = 10 different ASMSC lines per group), and then the cells were treated for 12 hours with the proteasome inhibitor MG132 (10 μM) or lysosome inhibitor chloroquine (50 μM). Western blots showed that MG132 reversed the degradation of Myc‐PTX3 mediated by Flag‐Smurf2, whereas chloroquine exerted less of an effect. E, Protein lysates were prepared at the indicated time points after treatment with 10 μM CHX, and PTX3 protein levels were evaluated using Western blotting. Compared to that in MSCs in patients with healthy donors (HDMSCs), the half‐life of PTX3 was shorter in ASMSCs. F, Quantitative analysis of the rate of PTX3 degradation in HDMSCs and ASMSCs (n = 10 different HDMSC or ASMSC lines per group, P < .01). G, Myc‐PTX3 was transfected into 293T cells along with Flag‐Smurf2 or empty vectors. At 36 hours after transfection, protein lysates were prepared at the indicated time points after treatment with 10 μM CHX. The rate of Myc‐PTX3 degradation was higher when coexpressed with Flag‐Smurf2. H, Quantitative analysis of the rate of PTX3 degradation with Flag‐Smurf2 and empty vectors (n = 3 independent experiments, P < .01). All data are presented as the means ± SD. *P < .05, **P < .01
Next, we explored the half‐life of PTX3 in MSCs treated with the protein synthesis inhibitor CHX. Compared with that in HDMSCs, the half‐life of PTX3 was shorter in ASMSCs (Figure 5E,F). The discrepancy in the PTX3 half‐life between HDMSCs and ASMSCs disappeared when Smurf2 expression was modulated by either knockdown (Supplementary Figure S4A,B) or overexpression (Supplementary Figure S4C,D). Overexpression of the C716A mutant did not exert a significant effect on the rate of PTX3 degradation in either HDMSCs or ASMSCs (Supplementary Figure S4C,D). After Myc‐PTX3 was transfected into 293T cells, coexpression of Flag‐Smurf2 significantly decreased the half‐life of Myc‐PTX3 (Figure 5G,H), further supporting the hypothesis that Smurf2 degrades the PTX3 protein. The half‐life of Myc‐PTX3 was noticeably increased when the proteasomal degradation pathway was blocked (Supplementary Figure S4E,F) and slightly increased when the lysosomal degradation pathway was blocked (Supplementary Figure S4G,F). Based on these results, PTX3 degradation is mainly regulated by the proteasome. In summary, Smurf2 may function as an E3 ubiquitin ligase to ubiquitinate PTX3 on both the K63 and K48 ubiquitin chains but mainly induces PTX3 degradation by the UPS.
3.4. Elevated Smurf2 decreases PTX3 expression to modulate ASMSC‐mediated angiogenesis
First, we explored the effect of PTX3 on angiogenesis. We constructed lentiviruses to silence PTX3 expression in HDMSCs and overexpress PTX3 in ASMSCs and then cocultured HUVECs with the selected MSC populations. The expression of PTX3 was almost completely absent in cells transfected with the shRNA and overexpressed after lentiviral transfection (Supplementary Figure S5A). For HDMSCs, the proliferation (Supplementary Figure S5B), migration (Supplementary Figure S5C‐F), and tube formation (Supplementary Figure S5G‐K) capacities of HUVECs in the Sh‐PTX3 group were significantly higher than those in the NC1 group. Furthermore, compared with the NC2 group, the OE‐PTX3 group of ASMSCs substantially inhibited HUVEC proliferation, migration, and tube formation. The increased angiogenic capacity of ASMSCs was inhibited by modulating the PTX3 expression. Therefore, ASMSCs induced more angiogenesis because they expressed PTX3 at lower levels.
We further evaluated the Smurf2‐PTX3‐angiogenesis axis in MSCs. We performed functional angiogenesis assays after coculturing HUVECs with HDMSCs overexpressing Smurf2. Since PTX3 exerts its anti‐angiogenic functions by binding and blocking FGF2, 22 , 23 , 24 , 25 we treated the coculture system with an anti‐FGF2 antibody. Compared with the NC group, the group overexpressing Smurf2 exhibited increased proliferation (Figure 6A), wound healing (Figure 6B,C), migration (Figure 6D,E), and tube formation (Figure 6F‐J), whereas these changes were reversed by the anti‐FGF2 antibody. The anti‐FGF2 antibody decreased the angiogenic capacity of the OE‐Smurf2 group to the levels observed in the NC group. In summary, Smurf2 promotes angiogenesis in MSCs by modulating PTX3 expression.
FIGURE 6.
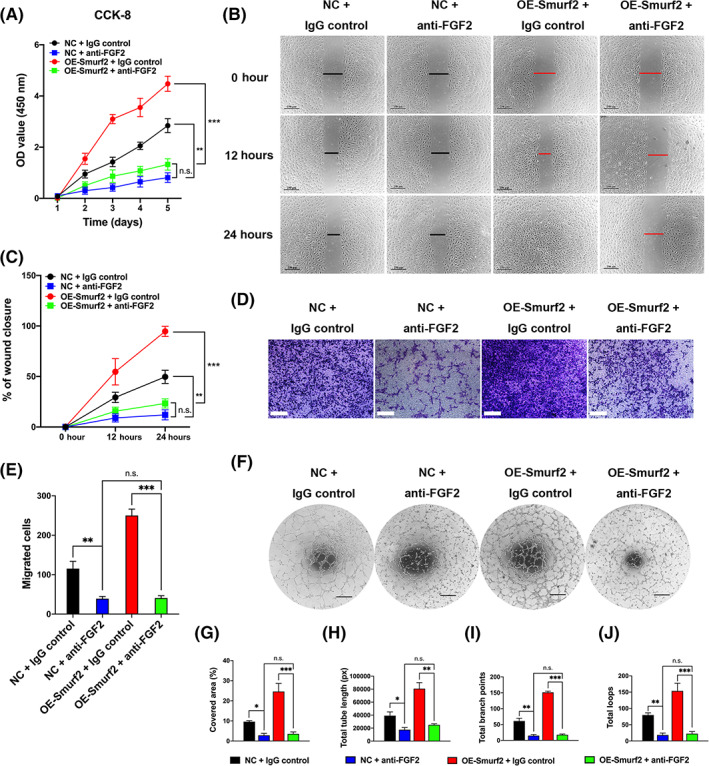
The Smurf2‐PTX3 axis modulates mesenchymal stem cell (MSC)‐mediated angiogenesis. We performed functional assays after coculturing human umbilical vein endothelial cells (HUVECs) with MSCs in patients with healthy donors (HDMSCs) overexpressing Smurf2. An FGF2 neutralizing antibody was added to the coculture system to block the anti‐angiogenic function of PTX3. A‐F, Compared with the NC group (NC + IgG control), the group overexpressing Smurf2 (OE‐Smurf2 + IgG control) exhibited increased proliferation (CCK‐8), wound healing, migration (Transwell) and tube formation, whereas these changes were reversed by the anti‐FGF2 antibody. The anti‐FGF2 antibody decreased the angiogenic capacity of the OESmurf2 group to the levels observed in the NC group, as shown in the NC + anti‐FGF2 group and OE‐Smurf2 + anti‐FGF2 group. G‐J, All tube formation assays quantified the cell‐covered area, total tube length, total branch points and total loops. All data are presented as the means ± SD (n = 10 different HDMSC lines per group). Scale bars = 250 μm in (B) and (D) and 400 μm in (F). *P < .05, **P < .01, ***P < .001. n.s., not significant
3.5. Regulation of Smurf2 by ATF4 activation in ASMSCs
The aforementioned results revealed Smurf2 upregulation in ASMSCs. We sought to identify the signaling pathway that may induce Smurf2 expression. HLA‐B27 is the genetic marker that predisposes people to AS, and the main hypothesis proposed to explain the role of HLA‐B27 in AS is that HLA‐B27 misfolding triggers endoplasmic reticulum (ER) stress. 37 We initially detected the levels of ER stress sensor proteins in ASMSCs, for example, inositol‐requiring enzyme 1 (IRE1), protein kinase R‐like endoplasmic reticulum kinase (PERK), ATF4 and ATF6, and observed significantly increased levels of the ATF4 mRNA and protein in ASMSCs compared with HDMSCs (Figure 7A; Supplementary Figure S6B,C). ER stress may increase the efficiency of protein degradation systems, such as the UPS, to eliminate excess misfolded proteins. 38 Therefore, we suspected that the induction of ER stress may upregulate E3 ubiquitin ligases such as Smurf2.
FIGURE 7.
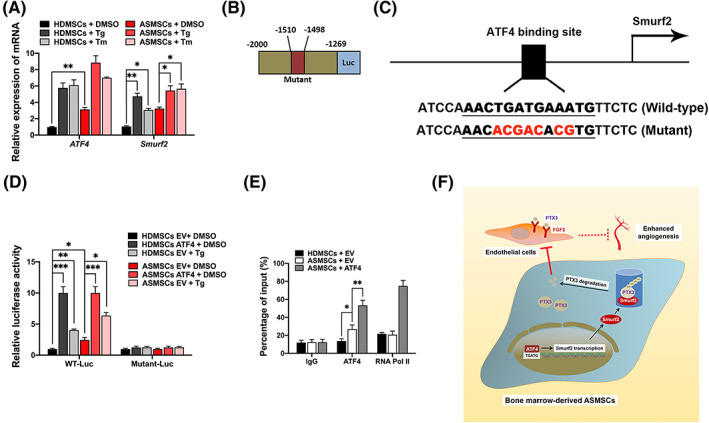
Regulation of Smurf2 by ATF4 activation in mesenchymal stem cells (MSCs) in patients with ankylosing spondylitis (ASMSCs). A, The altered cellular response may indicate an increased ability of E3 to ubiquitinate its substrates. 32 Compared to those in MSCs in patients with healthy donors (HDMSCs), the levels of both ATF4 mRNA and protein were increased in ASMSCs. Then, we induced pharmacologic ER stress in MSCs by treating them with tunicamycin (Tm) or thapsigargin (Tg), which are two ATF4 agonists. Smurf2 expression was induced by Tg and Tm in both ASMSCs and HDMSCs (n = 10 different HDMSC or ASMSC lines per group). Therefore, we speculated that Smurf2 might be a molecular target of ATF4. We examined the promoter region of Smurf2 and predicted a potential ATF4‐binding motif in the promoter region of the Smurf2 gene using JASPAR profiles to test this hypothesis. B,C, Schematic of the luciferase reporter plasmid constructs: WT‐Luc and Mutant‐Luc. D, Luciferase reporter assays further showed that ATF4 and Tg increased the transcriptional activity of this promoter; however, mutation of the motif substantially attenuated the transactivation activity of ATF4. E, ChIP assays in MSCs further showed that ATF4 overexpression activated the ATF4 binding site of the Smurf2 gene and promoted Smurf2 transcription (n = 10 different HDMSC or ASMSC lines per group). F, We concluded that Smurf2 transcription is regulated by ATF4‐induced ER stress and that Smurf2 negatively regulates PTX3 stability in ASMSCs and abnormally promotes angiogenesis through the PTX3‐FGF2 axis. All data are presented as the means ± SD. *P < .05, **P < .01, ***P < .001
First, we constructed lentiviruses to silence ATF4 expression in HDMSCs and ASMSCs and observed significant decreases in both Smurf2 mRNA and protein levels, whereas only the level of the PTX3 was decreased (Supplementary Figure S6A‐C). Based on this finding, the transcription of PTX3 may not be directly affected by ATF4, but its protein stability is potentially regulated by Smurf2, the transcriptional activation of which was modulated by ATF4. Then, we cocultured HUVECs with the selected MSC population that was transfected with Sh‐ATF4. After ATF4 knockdown, HUVEC angiogenesis decreased, and no notable difference was observed between HDMSCs and ASMSCs. The results of the CCK‐8 (Supplementary Figure S6D), wound healing (Supplementary Figure S6E,F), and Transwell migration assays (Supplementary Figure S6G,H) were consistent with the results of the tube formation assay (Supplementary Figure S6I‐M), indicating that ATF4 knockdown in MSCs negatively regulated HUVEC angiogenesis.
Then, we induced pharmacological ER stress in ASMSCs and HDMSCs with the ATF4 agonists tunicamycin (Tm) and thapsigargin (Tg), and Smurf2 expression was upregulated by Tm and Tg (Figure 7A). As ATF4 is a UPR transcription factor, 37 we speculated that Smurf2 transcription might be directly regulated by ATF4. We used JASPAR profiles to examine the promoter region of Smurf2 and test this hypothesis, and we identified a potential ATF4‐binding motif in the proximal promoter region of the Smurf2 gene. The predicted biding site was 5′‐AACTGATGAAATG‐3′, and the mutated sequence was constructed as follows: 5′‐AACacgacAcgTG‐3′ (Figure 7B,C). Luciferase reporter assays confirmed that ATF4 increased the transcriptional activity of the Smurf2 promoter; however, mutation of the motif substantially attenuated the transactivation mediated by ATF4 (Figure 7D). ChIP assays further showed that ATF4 overexpression activated the ATF4‐binding site of the Smurf2 gene and promoted Smurf2 transcription (Figure 7E). Therefore, ATF4 signaling may be an important mechanism leading to the upregulation of Smurf2. Overall, we concluded that Smurf2 transcription is regulated by ATF4‐induced ER stress and that Smurf2 negatively regulates PTX3 stability in ASMSCs and abnormally promotes angiogenesis through the PTX3‐FGF2 axis (Figure 7F).
4. DISCUSSION
According to recent studies, angiogenesis participates in the pathogenesis of AS, which in turn has important functional consequences in terms of inflammation and pathological osteogenesis. 1 , 2 , 3 , 4 , 5 , 6 , 39 , 40 However, the molecular mechanism of abnormal angiogenesis has seldom been investigated in patients with AS. In the present study, we identified Smurf2 as one of the key factors that promotes angiogenesis in ASMSCs by ubiquitinating and degrading PTX3. Smurf2 knockdown led to PTX3 upregulation, and the angiogenesis of MSCs was inhibited. Consistent with these findings, Smurf2 overexpression downregulated PTX3, and MSCs exhibited increased angiogenesis. Smurf2 might bind to PTX3 in the cytoplasm and load a K63‐linked and K48‐linked ubiquitin chain onto PTX3, resulting in the recognition and degradation of PTX3 by the proteasome. Therefore, Smurf2‐mediated ubiquitination and degradation of PTX3 promotes angiogenesis mediated by ASMSCs.
Based on accumulating evidence, several angiogenic factors are abnormally expressed in patients with AS and closely related to the BASDAI. 2 , 3 , 4 , 5 The effectiveness of an anti‐TNFα antibody is negatively correlated with angiogenesis in the synovial membrane. 39 , 40 In the present study, we performed IHC staining of the hip synovium from patients with AS and observed a significantly higher microvessel density in the lesions of patients with AS than in the lesions of patients with OA. Furthermore, we directly detected the CD31 mRNA level in the whole patient sample and observed higher levels of CD31 expression in patients with AS than in patients with OA. Thus, abnormal angiogenesis may be involved in the pathogenesis of AS. MSCs secrete a variety of pro‐angiogenic factors to maintain angiogenesis homeostasis, and dysregulated angiogenesis of MSCs is involved in inflammation and disrupted bone metabolism in patients with autoimmune diseases. 12 However, researchers have not determined whether ASMSCs are involved in the abnormal angiogenesis observed in patients with AS. CD105 expression in MSCs was positively correlated with CD31 expression, which is a marker of angiogenesis. This finding from an in vivo experiment revealed a strong association between ASMSCs and the abnormal angiogenesis observed in patients with AS. As shown in the presents study, ASMSCs effectively increased the proliferation, migration, and angiogenic tube formation of vascular endothelial cells. These findings provide evidence that abnormal angiogenesis occurs in patients with AS at the cellular level and provide novel insights into the pathogenesis of AS.
The UPS plays an important role in the pathogenesis of AS 15 and functions as a key component of the angiogenic switch in patients with various angiogenic pathologies. 14 Therefore, we detected the expression of several E3 ligase enzymes in ASMSCs and observed significantly upregulated Smurf2 expression at both the mRNA and protein levels. Smurf2 upregulation was observed in vivo in the local lesions of patients with AS, suggesting that its abnormal expression was related to AS pathogenesis. Our previous study and those of other scholars have reported that Smurf2 plays a central role in the osteogenic 31 , 41 and neuronal differentiation of MSCs. 42 In the present study, we attempted to explain the angiogenic effect of Smurf2 on MSCs. Smurf2 is overexpressed in various squamous cell carcinomas and mediates the protective ubiquitination of epidermal growth factor receptors (EGFRs), which are considered to play an important role in tumor‐associated angiogenesis and may be responsible for EGFR overexpression in certain tumors. 43 As shown in the study by Kong et al, Smurf2 ubiquitinates and degrades a dominant inhibitor of the helix‐loop‐helix transcription factor family, ID1, 44 and overexpression of ID1 facilitates tumor angiogenesis. 45 Based on these studies, Smurf2 inhibits angiogenesis by degrading ID1. Due to its many substrates, Smurf2 may exert different effects on angiogenesis in different pathological processes. MSCs have been reported to secrete various angiogenic factors that regulate the formation of vascular structures 8 ; however, few studies have examined the role of the UPS pathway for the selective degradation of proteins in regulating the secretion of these factors. In the present study, Smurf2, as an important mediator of the UPS, functions in MSCs to regulate angiogenesis. Smurf2 expression is increased in ASMSCs. Downregulation of Smurf2 alleviates the abnormal angiogenesis of ASMSCs, whereas upregulation of Smurf2 improves the angiogenesis of HDMSCs, and the catalytically inactive mutant of Smurf2, C716A, does not possess a similar function. Therefore, the angiogenesis‐modulating function of Smurf2 depends on its enzyme activity as an E3 ubiquitin ligase and lays the foundation for subsequent research on this enzyme. The abnormally elevated Smurf2 mediated angiogenesis in patients with AS suggested that its pathogenesis may depend on the proteasome. If the function of the proteasome is inhibited, abnormal ASMSC functions and enhanced angiogenesis may be impaired. Bortezomib, which is a proteasome inhibitor, is currently used to treat patients with multiple myeloma 46 and has also shown activity in patients with a variety of autoimmune diseases, such as systemic autoimmune diseases, inflammatory bowel disease, multiple sclerosis, and organ‐specific autoimmune diseases. 47 , 48 Our research provides support for the application of proteasome inhibitors in the treatment of AS.
The UPS is a highly regulated mechanism of protein degradation and turnover. E3 ligase enzymes dictate substrate specificity; therefore, identification of E3 ligase enzymes is key to studying the phenotypic mechanism regulating ubiquitination. 16 In the present study, our first step was to identify the E3 ligase enzyme Smurf2 in the regulatory pathway of abnormal angiogenesis mediated by ASMSCs. Next, we tried to determine the specific substrate of Smurf2 in this process. We overlapped two independent datasets to narrow the scope of the screen, and the differentially altered angiogenic factors between HDMSCs and ASMSCs were identified using a Proteome Profiler Human Angiogenesis Array Kit, whereas the potential interacting partners of Smurf2 in ASMSCs were identified using co‐IP and LC‐MS/MS. Finally, PTX3 levels were decreased in ASMSCs, and PTX3 interacted with Smurf2. EGF was also present at higher levels in the coculture system of HUVECs and ASMSCs, and EGF neutralization could partially inhibit the pro‐angiogenic ability of ASMSCs. However, abnormal ASMSC‐angiogenesis was completely reversed by modulating PTX3 expression. Thus, ASMSCs induce a higher level of angiogenesis mainly because they express PTX3 at lower levels. Nevertheless, there may still be two mechanisms that regulate ASMSC‐induced angiogenesis (ie, Smurf2‐PTX3‐FGF2 and higher expression of EGF). If blocking angiogenesis can represent a potential therapeutic option for AS patients, blocking Smurf2 and UPS may not be sufficient, and drugs that inhibit EGF‐related pathways may also be needed.
PTX3 is a soluble pattern recognition receptor with a high affinity for FGF2. The interaction of PTX3‐FGF2 exerts inhibitory effects on angiogenesis. 22 , 23 , 24 , 25 As mentioned above, studies have preliminarily reported that PTX3 may participate in the pathogenesis of AS. 26 , 27 In addition, MSC‐derived PTX3 was reported to dissolve clots and remodel the matrix, both of which are required for MSC involvement in wound healing. 49 Our study explored the specific role of the Smurf2‐PTX3 axis in regulating angiogenesis mediated by MSCs. PTX3, which strongly affects the regulation of angiogenesis by MSCs, was abundantly expressed in MSCs. The anti‐FGF2 antibody reversed the increased angiogenic capacity of Smurf2 overexpression, indicating that Smurf2 promotes angiogenesis in ASMSCs by modulating PTX3 expression. We conducted co‐IP, Western blotting, and IF staining to confirm that Smurf2 and PTX3 interacted with each other in the cytoplasm of MSCs. Compared with that in HDMSCs, the level of the PTX3 protein was decreased in ASMSCs, but an increased Smurf2‐PTX3 interaction was observed. A similar negative correlation between Smurf2 and PTX3 levels was also detected in tissue samples from patients with AS.
However, when we studied the endogenous Smurf2‐mediated ubiquitination of PTX3, we encountered some problems. Once a protein has been ubiquitinated, it is likely to be degraded by the UPS. Therefore, MG132 and other proteasome inhibitors are usually used to inhibit protein degradation by the UPS to better detect the ubiquitination signal. However, PTX3 is generated by the NF‐κB pathway, 35 and proteasome inhibitors such as MG132 and MG101 significantly promote IκBα accumulation and inhibit the NF‐κB pathway. 36 We confirmed that MG132 and MG101 substantially inhibit PTX3 expression, and we therefore transfected HA‐UBB into MSCs in an attempt to increase the ubiquitination of PTX3 for better detection. And increased PTX3 ubiquitination were observed in ASMSCs. Then, we exogenously cotransfected the HA‐UBB, Myc‐PTX3, Flag‐Smurf2, and Flag‐C716A plasmids into 293T cells and observed that Flag‐Smurf2 significantly induced the ubiquitination of Myc‐PTX3 by adding both the K63‐linked and K48‐linked ubiquitin chains, whereas Flag‐C716A did not exert a similar effect. We inhibited the lysosomal and proteasomal degradation pathways in ASMSCs and found that degradation of PTX3 was mainly regulated by the UPS. Moreover, compared with that in HDMSCs, the half‐life of PTX3 was shorter in ASMSCs. After modulating Smurf2 expression, PTX3 degradation was affected consistently. It would be expected that the C716A mutant should increase the protein stability of PTX3 by competing with endogenous Smurf2. However, overexpression of C716A slightly increased the half‐life of PTX3 in HDMSCs and ASMSCs, but the effect was not statistically significant. The key site that affects the E3 ligase activity of Smurf2 is lysine at position 716, but other positions including Try at position 459, 50 Trp at position 535, and Tyr at position 581 51 could also affect the enzyme activity of Smurf2. Specifically, as Ogunjimi et al reported, Smurf2 mutants Tyr581Ala (W581A), Trp535Ala (Y535A), and Trp535Asp (Y535D) also have impaired ubiquitin ligase activity and behave similarly to the mutant C716A, 51 and the mutation of Try at AA 459 on the ubiquitin binding surface interferes with Smurf2‐dependent degradation of RhoA and blocks polyubiquitylation. 50 These results suggested that there may be multiple active sites of Smurf2 that coregulate PTX3 ubiquitination and degradation, and further study is needed. Although the mechanisms by which UPS degradation negatively regulates NFκB activation are clearly understood, 52 our findings suggested that as one of the downstream proteins of the NFκB pathway, PTX3 is also precisely ubiquitinated and degraded. Thus, the regulation of PTX3 ubiquitination should be further studied, and further studies may explain the roles of PTX3 in various cellular activities. A limitation of our study is that we only identified the K63 and K48 ubiquitin chain linkages and did not explore the ubiquitination lysine site in PTX3.
Smurf2 overexpression plays key roles in several physiological and pathological processes, 17 , 18 , 19 , 20 , 21 , 53 but the mechanisms that control Smurf2 expression have not been clearly elucidated. MicroRNA‐322 and microRNA‐503 were identified as novel factors that regulate Smurf2 expression at the translational level. 54 Upstream transcription factor 2 was recently identified as a negative transcription factor for Smurf2 in a subtype of breast cancer. 55 However, little is understood about the mechanism positively regulating Smurf2 expression at the transcriptional level. In the present study, Smurf2 expression was upregulated in ASMSCs at both the mRNA and protein levels, and Smurf2 expression was mainly affected by regulation at the transcriptional level. Within the context of the pathogenesis of AS, we considered why Smurf2 expression was increased in ASMSCs. HLA‐B27 is the genetic marker that predisposes people to AS, and problems arise when HLA‐B27 tends to misfold, thus generating ER stress. The ER has quality control mechanisms in place that interact in a coordinated manner with the UPS to remove unfolded proteins. 56 Therefore, we suspected that ER stress may affect Smurf2 expression. We detected several ER stress sensor proteins in ASMSCs and observed higher ATF4 expression in ASMSCs than in HDMSCs. ATF4 knockdown in MSCs negatively regulated HUVEC functions. ATF4 is a key transcription factor that alters gene expression in cells in response to ER stress, which participates in the AS pathological process. 37 ATF4 was also reported as a key regulator of vascular endothelial growth factor expression in the local bone microenvironment, which contributes to increased bone angiogenesis. 57 , 58 In the present study, ATF4 expression was upregulated in patients with AS and promoted Smurf2 transcription to subsequently modulate the PTX3‐FGF2 pathway and angiogenesis. Starting from another mechanism, ATF4 was involved in regulating the pathogenesis of AS.
We induced pharmacological ER stress with two ATF4 agonists, Tg and Tm, and observed increased levels of the Smurf2 mRNA and protein. PTX3 expression was modulated only at the protein level. Based on the consistent changes in Smurf2 and ATF4 expression under ER stress, we further validated the transcriptional regulation of Smurf2 by ATF4 and identified the specific ATF4‐binding site in the Smurf2 gene. Our data further suggested that the UPS appears to be associated with ER stress in patients with AS and provided evidence that this crosstalk is involved in AS pathogenesis. However, the present study still has some limitations. First, the Lys ubiquitination site in PTX3 must be further explored. Second, the use of the MSC‐specific Smurf2 knockout system should be considered to better clarify the exact role of Smurf2 in MSCs in the future. Based on the possible mechanisms by which the MET 20 , 21 may be induced in MSCs upon Smurf2 depletion, the inducible system should be considered.
5. CONCLUSIONS
Based on the results of our present study, Smurf2 plays an important role in regulating ASMSC‐induced angiogenesis by mediating PTX3 ubiquitination and degradation. Smurf2 may represent a therapeutic target in AS treatment.
CONFLICT OF INTEREST
The authors declared no potential conflicts of interest.
AUTHOR CONTRIBUTIONS
M.M., W.Y., Z.C.: planned the project, performed the experiments, interpreted the data and drafted the article; P.W., H.L.: collected the human samples and isolated and cultured the MSCs; R.M., Y.J.: analyzed the data; Z.X., P.S.; revised the article and provided advice; H.S., Y.W.: supervised the study.
Supporting information
Supplementary Figure S1 (A) Representative images of IF staining for angiogenesis and MSC markers in the hip synovium of patients with AS and patients with OA (n = 10 patients per group). CD31 and CD105 were co‐expressed in patients with AS, indicating that ASMSCs may participate in the abnormal angiogenesis observed in patients with AS. Scale bar = 100 μm. (B) QRT‐PCR revealed significantly higher expression of the CD31 mRNA in the hip synovium of patients with AS than in the hip synovium of patients with OA (n = 10 patients per group). (C) Representative images showing significantly higher Smurf2 IHC staining in the hip synovium of patients with AS than in the hip synovium of patients with OA (n = 10 patients per group). Scale bar = 100 μm. (D) The expression of E3 ligases was upregulated in ASMSCs. QRTPCR showing upregulation of the mRNA levels of 4 E3 ligases, TRIM21, TRIM25, TRIM56 and Smurf2 in ASMSCs, and Smurf2 expression exhibited the most significant change (n = 10 different HDMSC or ASMSC lines per group). (E) Sterile FACS cell sorting was performed using cells transfected with the vector encoding GFP. After lentivirus transfection, the GFP‐expressing population of MSCs was 60.21%. Then, the transfected MSCs were selected with blank control MSCs (0.50%) for a background comparison. (F) Both HDMSCs and ASMSCs were effectively transfected with the lentivirus, for example, NC and Sh‐Smurf2. Scale bar = 500 μm. All data are presented as the means ± SD. *, P < 0.05. ***, P < 0.001.
Supplementary Figure S2 (A) and (B) Representative images of the wound healing assay and the tube formation assay of HUVECs co‐cultured with differently treated HDMSCs or ASMSCs. Smurf2 knockdown led to decreased HUVEC migration and tube formation, while Smurf2 overexpression exerted the opposite effect, and C716A overexpression did not produce similar phenotypes. Scale bars = 250 μm in (A) and 400 μm in (B). (C) IF staining for Smurf2 to verify the specificity of the antibody. Compared to that in the NC group, Smurf2 expression in the Sh‐Smurf2 group was knocked down by the lentivirus. The red fluorescence signal was substantially decreased, and the green fluorescence signal of PTX3 was increased (red, Smurf2; green, PTX3; blue, DAPI; n = 10 different ASMSC lines per group; scale bar = 20 μm). (D) MSCs were treated with proteasome inhibitors for 12 hours to block the UPS, and both MG132 and MG101 substantially inhibited PTX3 expression (n = 10 different HDMSC lines per group).
Supplementary Figure S3 (A) Identification of the differentially altered angiogenic factors between the supernatants of HDMSCs or ASMSCs co‐cultured with HUVECs. Quantification of the results obtained using the Human Angiogenesis Array Kit (ARY007; R&D Systems) showed that several factors displayed abnormal levels in the supernatants of ASMSCs co‐cultured with HUVECs, including angiopoietin‐2 (ANG2), EGF, IGFBP2, IL1β, prolactin, thrombospondin‐1 (TSP1), uPA and PTX3. Refer to the column number in (B) for the coordinates in (A). (C), (D), (E) and (G) The potential role of EGF in the abnormal angiogenesis of ASMSCs was explored by applying an EGF neutralizing antibody to modulate the EGF level. The increased angiogenesis capacity of ASMSCs was impaired by the anti‐EGF antibody to some extent, but the difference compared with the HDMSCs group was still significant (n = 10 different HDMSC or ASMSC lines per group). (F), (H), (I), (J), (K) and (L) The quantification of the results from the wound healing, migration and tube formation assays using HUVECs is shown. All data are presented as the means ± SD. Scale bars = 250 μm in (C) and (E), and 400 μm in (G). *, P < 0.05. **, P < 0.01.
Supplementary Figure S4 (A), (B), (C) and (D) The difference in the half‐life of PTX3 between HDMSCs and ASMSCs disappeared when Smurf2 expression was modulated by either silencing or overexpression. Overexpression of C716A did not exert a significant effect on the rate of PTX3 degradation in either HDMSCs or ASMSCs. n = 10 different HDMSC or ASMSC lines per group. (E), (F) and (G) Myc‐PTX3 was transiently overexpressed in ASMSCs, and protein lysates were prepared at the indicated time points after treatment with 10 μM CHX alone or in combination with the proteasome inhibitor MG132 (10 μM) or the lysosome inhibitor chloroquine (50 μM). Western blots showing that MG132 obviously inhibited the degradation of PTX3, while chloroquine exerted less of an effect on the degradation of PTX3. PTX3 degradation was mainly regulated by the proteasome. n = 10 different ASMSC lines per group. n.s., not significant.
Supplementary Figure S5 PTX3 silencing induced ASMSC‐mediated angiogenesis in vitro. (A) The silencing efficiency and overexpression efficiency of Sh‐PTX3 and OE‐PTX3, respectively, were verified using Western blotting analyses. (B), (C), (E) and (G) In the CCK‐8, wound healing, Transwell and tube formation assays, PTX3 knockdown led to increased HUVEC proliferation, migration and tube formation when co‐cultured with HDMSCs, while PTX3 overexpression decreased HUVEC proliferation, migration and tube formation when cocultured with ASMSCs. The increased angiogenic capacity of ASMSCs was reversed by modulating PTX3 expression. ASMSCs induced higher levels of angiogenesis because they expressed PTX3 at lower levels. (D), (F), (H), (I), (J) and (K) The quantification of the results from the wound healing, migration and tube formation assays is shown. All data are presented as the means ± SD. n = 10 different HDMSC or ASMSC lines per group. Scale bars = 250 μm in (C) and (E) and 400 μm in (G). *, P < 0.05. **, P < 0.01. ***, P < 0.001.
Supplementary Figure S6 (A) Expression of the Smurf2 and PTX3 mRNAs after modulation of ATF4 expression in HDMSCs and ASMSCs. QRT‐PCR showed that ATF4 knockdown decreased the expression of the Smurf2 mRNA but did not affect the expression of the PTX3 mRNA. (B) and (C) Western blots showing that ATF4 knockdown reduced the levels of the Smurf2 protein and increased the levels of the PTX3 protein in both HDMSCs and ASMSCs (n = 10 different HDMSC or ASMSC lines per group). (D), (E), (G) and (I) In the CCK‐8, wound healing, Transwell and tube formation assays, ATF4 knockdown led to decreased HUVEC proliferation, migration and tube formation when co‐cultured with both HDMSCs and ASMSCs. Thus, ATF4 knockdown negatively regulated HUVEC functions. (F), (H), (J), (K), (L) and (M) The quantification of the results from the wound healing, migration and tube formation assays is shown. All data are presented as the means ± SD. n = 10 different HDMSC or ASMSC lines per group. Scale bar = 250 μm in (C) and (E), and 400 μm in (G). *, P < 0.05. **, P < 0.01. ***, P < 0.001. n.s., not significant.
Supplementary Table S1 Primer sequences used for qRT‐PCR
Supplementary Table S2 Supplementary Table
ACKNOWLEDGMENTS
This study was supported by grants from the National Natural Science Foundation of China (81871750), the Natural Science Foundation of Guangdong Province (2018A030313232), the Medical Science and Technology Research Project of Guangdong Province (A2018292), the Health Welfare Fund Project of Futian District (FTWS2020078), and the Key Realm R&D Programme of Guangdong Province (2019B020236001). The authors thank Prof. Kaishun Hu (Medical Research Center, Sun Yat‐sen Memorial Hospital, SYSU, Guangzhou, China) for his guidance. In addition, Mengjun Ma would like to thank her husband, Ge Sun, for his care and support over the past years.
Ma M, Yang W, Cai Z, et al. SMAD‐specific E3 ubiquitin ligase 2 promotes angiogenesis by facilitating PTX3 degradation in MSCs from patients with ankylosing spondylitis. Stem Cells. 2021;39:581–599. 10.1002/stem.3332
Mengjun Ma, Wen Yang, and Zhaopeng Cai contributed equally to this study.
Funding information Health Welfare Fund Project of Futian District, Grant/Award Number: FTWS2020078; Key Realm R&D Programme of GuangDong Province, Grant/Award Number: 2019B020236001; Medical Science and Technology Research Project of GuangDong Province, Grant/Award Number: A2018292; National Natural Science Foundation of China, Grant/Award Number: 81871750; Natural Science Foundation of Guangdong Province, Grant/Award Number: 2018A030313232
Contributor Information
Yanfeng Wu, Email: wuyf@mail.sysu.edu.cn.
Huiyong Shen, Email: shenhuiy@mail.sysu.edu.cn.
DATA AVAILABILITY STATEMENT
All data, models, and code generated or used during the study appear in the submitted article.
REFERENCES
- 1. Appel H, Kuhne M, Spiekermann S, et al. Immunohistochemical analysis of hip arthritis in ankylosing spondylitis: evaluation of the bone‐cartilage interface and subchondral bone marrow. Arthritis Rheum. 2006;54(6):1805‐1813. [DOI] [PubMed] [Google Scholar]
- 2. Li XL, Lin TT, Qi CY, et al. Elevated serum level of IL‐33 and sST2 in patients with ankylosing spondylitis: associated with disease activity and vascular endothelial growth factor. J Invest Med. 2013;61(5):848‐851. [DOI] [PubMed] [Google Scholar]
- 3. Lin TT, Lu J, Qi CY, et al. Elevated serum level of IL‐27 and VEGF in patients with ankylosing spondylitis and associate with disease activity. Clin Exp Med. 2015;15(2):227‐231. [DOI] [PubMed] [Google Scholar]
- 4. Sari I, Kebapcilar L, Alacacioglu A, et al. Increased levels of asymmetric dimethylarginine (ADMA) in patients with ankylosing spondylitis. Intern Med. 2009;48(16):1363‐1368. [DOI] [PubMed] [Google Scholar]
- 5. Poddubnyy D, Conrad K, Haibel H, et al. Elevated serum level of the vascular endothelial growth factor predicts radiographic spinal progression in patients with axial spondyloarthritis. Ann Rheum Dis. 2014;73(12):2137‐2143. [DOI] [PubMed] [Google Scholar]
- 6. Baeten D, Demetter P, Cuvelier C, et al. Comparative study of the synovial histology in rheumatoid arthritis, spondyloarthropathy, and osteoarthritis: influence of disease duration and activity. Ann Rheum Dis. 2000;59(12):945‐953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Uccelli A, Moretta L, Pistoia V. Mesenchymal stem cells in health and disease. Nat Rev Immunol. 2008;8(9):726‐736. [DOI] [PubMed] [Google Scholar]
- 8. Bartaula‐Brevik S, Pedersen TO, Finne‐Wistrand A, et al. Angiogenic and immunomodulatory properties of endothelial and mesenchymal stem cells. Tissue Eng Part A. 2016;22(3–4):244‐252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hu X, Zhang L, Jin J, et al. Heparanase released from mesenchymal stem cells activates integrin beta1/HIF‐2alpha/Flk‐1 signaling and promotes endothelial cell migration and angiogenesis. Stem Cells. 2015;33(6):1850‐1862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Grellier M, Bordenave L, Amedee J. Cell‐to‐cell communication between osteogenic and endothelial lineages: implications for tissue engineering. Trends Biotechnol. 2009;27(10):562‐571. [DOI] [PubMed] [Google Scholar]
- 11. Premer C, Blum A, Bellio MA, et al. Allogeneic mesenchymal stem cells restore endothelial function in heart failure by stimulating endothelial progenitor cells. EBioMedicine. 2015;2(5):467‐475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mohanty ST, Kottam L, Gambardella A, et al. Alterations in the self‐renewal and differentiation ability of bone marrow mesenchymal stem cells in a mouse model of rheumatoid arthritis. Arthritis Res Ther. 2010;12(4):R149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Halpern JL, Kilbarger A, Lynch CC. Mesenchymal stem cells promote mammary cancer cell migration in vitro via the CXCR2 receptor. Cancer Lett. 2011;308(1):91‐99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Rahimi N. The ubiquitin‐proteasome system meets angiogenesis. Mol Cancer Ther. 2012;11(3):538‐548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wright C, Edelmann M, diGleria K, et al. Ankylosing spondylitis monocytes show upregulation of proteins involved in inflammation and the ubiquitin proteasome pathway. Ann Rheum Dis. 2009;68(10):1626‐1632. [DOI] [PubMed] [Google Scholar]
- 16. Glickman MH, Ciechanover A. The ubiquitin‐proteasome proteolytic pathway: destruction for the sake of construction. Physiol Rev. 2002;82(2):373‐428. [DOI] [PubMed] [Google Scholar]
- 17. Zuscik MJ, Rosier RN, Schwarz EM. Altered negative regulation of transforming growth factor beta signaling in scleroderma: potential involvement of SMURF2 in disease. Arthritis Rheum. 2003;48(7):1779‐1780. [DOI] [PubMed] [Google Scholar]
- 18. David D, Jagadeeshan S, Hariharan R, Nair A, Pillai R. Smurf2 E3 ubiquitin ligase modulates proliferation and invasiveness of breast cancer cells in a CNKSR2 dependent manner. Cell Div. 2014;9:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Li Y, Yang D, Tian N, et al. The ubiquitination ligase SMURF2 reduces aerobic glycolysis and colorectal cancer cell proliferation by promoting ChREBP ubiquitination and degradation. J Biol Chem. 2019;294(40):14745‐14756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Manikoth Ayyathan D, Koganti P, Marcu‐Malina V, et al. SMURF2 prevents detrimental changes to chromatin, protecting human dermal fibroblasts from chromosomal instability and tumorigenesis. Oncogene. 2020;39(16):3396‐3410. [DOI] [PubMed] [Google Scholar]
- 21. Blank M, Tang Y, Yamashita M, Burkett SS, Cheng SY, Zhang YE. A tumor suppressor function of Smurf2 associated with controlling chromatin landscape and genome stability through RNF20. Nat Med. 2012;18(2):227‐234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Camozzi M, Rusnati M, Bugatti A, et al. Identification of an antiangiogenic FGF2‐binding site in the N terminus of the soluble pattern recognition receptor PTX3. J Biol Chem. 2006;281(32):22605‐22613. [DOI] [PubMed] [Google Scholar]
- 23. Nicoli S, Presta M. The zebrafish/tumor xenograft angiogenesis assay. Nat Protoc. 2007;2(11):2918‐2923. [DOI] [PubMed] [Google Scholar]
- 24. Presta M, Camozzi M, Salvatori G, Rusnati M. Role of the soluble pattern recognition receptor PTX3 in vascular biology. J Cell Mol Med. 2007;11(4):723‐738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Rusnati M, Camozzi M, Moroni E, et al. Selective recognition of fibroblast growth factor‐2 by the long pentraxin PTX3 inhibits angiogenesis. Blood. 2004;104(1):92‐99. [DOI] [PubMed] [Google Scholar]
- 26. Zhang X, Ding W. Association of genetic variants in pentraxin 3 gene with ankylosing spondylitis. Med Sci Monit. 2016;22:2911‐2916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Surdacki A, Sulicka J, Korkosz M, et al. Blood monocyte heterogeneity and markers of endothelial activation in ankylosing spondylitis. J Rheumatol. 2014;41(3):481‐489. [DOI] [PubMed] [Google Scholar]
- 28. Liu W, Wang P, Xie Z, et al. Abnormal inhibition of osteoclastogenesis by mesenchymal stem cells through the miR‐4284/CXCL5 axis in ankylosing spondylitis. Cell Death Dis. 2019;10(3):188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Xie Z, Wang P, Li Y, et al. Imbalance between bone morphogenetic protein 2 and noggin induces abnormal osteogenic differentiation of mesenchymal stem cells in ankylosing spondylitis. Arthritis Rheumatol. 2016;68(2):430‐440. [DOI] [PubMed] [Google Scholar]
- 30. Xie Z, Wang P, Li J, et al. MCP1 triggers monocyte dysfunctions during abnormal osteogenic differentiation of mesenchymal stem cells in ankylosing spondylitis. J Mol Med (Berl). 2017;95(2):143‐154. [DOI] [PubMed] [Google Scholar]
- 31. Li J, Wang P, Xie Z, et al. TRAF4 positively regulates the osteogenic differentiation of mesenchymal stem cells by acting as an E3 ubiquitin ligase to degrade Smurf2. Cell Death Differ. 2019;26(12):2652‐2666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Arnaoutova I, Kleinman HK. In vitro angiogenesis: endothelial cell tube formation on gelled basement membrane extract. Nat Protoc. 2010;5(4):628‐635. [DOI] [PubMed] [Google Scholar]
- 33. Xie Z, Li J, Wang P, et al. Differential expression profiles of long noncoding RNA and mRNA of osteogenically differentiated mesenchymal stem cells in ankylosing spondylitis. J Rheumatol. 2016;43(8):1523‐1531. [DOI] [PubMed] [Google Scholar]
- 34. Koganti P, Levy‐Cohen G, Blank M. Smurfs in protein homeostasis, signaling, and cancer. Front Oncol. 2018;8:295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Liu Y, Yu C, Ji K, et al. Quercetin reduces TNF‐alpha‐induced mesangial cell proliferation and inhibits PTX3 production: involvement of NF‐kappaB signaling pathway. Phytother Res. 2019;33(9):2401‐2408. [DOI] [PubMed] [Google Scholar]
- 36. Matsuo Y, Sawai H, Ochi N, et al. Proteasome inhibitor MG132 inhibits angiogenesis in pancreatic cancer by blocking NF‐kappaB activity. Dig Dis Sci. 2010;55(4):1167‐1176. [DOI] [PubMed] [Google Scholar]
- 37. Colbert RA, DeLay ML, Klenk EI, et al. From HLA‐B27 to spondyloarthritis: a journey through the ER. Immunol Rev. 2010;233(1):181‐202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Colbert RA, Tran TM, Layh‐Schmitt G. HLA‐B27 misfolding and ankylosing spondylitis. Mol Immunol. 2014;57(1):44‐51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Tosovsky M, Bradna P, Andrys C, et al. The VEGF and BMP‐2 levels in patients with ankylosing spondylitis and the relationship to treatment with tumour necrosis factor alpha inhibitors. Acta Med Austriaca. 2014;57(2):56‐61. [DOI] [PubMed] [Google Scholar]
- 40. Baeten D, De Keyser F. The histopathology of spondyloarthropathy. Curr Mol Med. 2004;4(1):1‐12. [DOI] [PubMed] [Google Scholar]
- 41. Lin TH, Gibon E, Loi F, et al. Decreased osteogenesis in mesenchymal stem cells derived from the aged mouse is associated with enhanced NF‐kappaB activity. J Orthop Res. 2017;35(2):281‐288. [DOI] [PubMed] [Google Scholar]
- 42. Yu YL, Chou RH, Shyu WC, et al. Smurf2‐mediated degradation of EZH2 enhances neuron differentiation and improves functional recovery after ischaemic stroke. EMBO Mol Med. 2013;5(4):531‐547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ray D, Ahsan A, Helman A, et al. Regulation of EGFR protein stability by the HECT‐type ubiquitin ligase SMURF2. Neoplasia. 2011;13(7):570‐578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kong Y, Cui H, Zhang H. Smurf2‐mediated ubiquitination and degradation of Id1 regulates p16 expression during senescence. Aging Cell. 2011;10(6):1038‐1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Tsai CH, Yang MH, Hung AC, et al. Identification of Id1 as a downstream effector for arsenic‐promoted angiogenesis via PI3K/Akt, NF‐kappaB and NOS signaling. Toxicol Res (Camb). 2016;5(1):151‐159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Skrott Z, Cvek B. Linking the activity of bortezomib in multiple myeloma and autoimmune diseases. Crit Rev Oncol Hematol. 2014;92(2):61‐70. [DOI] [PubMed] [Google Scholar]
- 47. van Dam LS, Osmani Z, Kamerling SWA, et al. A reverse translational study on the effect of rituximab, rituximab plus belimumab, or bortezomib on the humoral autoimmune response in SLE. Rheumatology (Oxford). 2020;59(10):2734‐2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Fierabracci A. Proteasome inhibitors: a new perspective for treating autoimmune diseases. Curr Drug Targets. 2012;13(13):1665‐1675. [DOI] [PubMed] [Google Scholar]
- 49. Cappuzzello C, Doni A, Dander E, et al. Mesenchymal stromal cell‐derived PTX3 promotes wound healing via fibrin remodeling. J Invest Dermatol. 2016;136(1):293‐300. [DOI] [PubMed] [Google Scholar]
- 50. Ogunjimi AA, Wiesner S, Briant DJ, et al. The ubiquitin binding region of the Smurf HECT domain facilitates polyubiquitylation and binding of ubiquitylated substrates. J Biol Chem. 2010;285(9):6308‐6315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Ogunjimi AA, Briant DJ, Pece‐Barbara N, et al. Regulation of Smurf2 ubiquitin ligase activity by anchoring the E2 to the HECT domain. Mol Cell. 2005;19(3):297‐308. [DOI] [PubMed] [Google Scholar]
- 52. Xu H, You M, Shi H, Hou Y. Ubiquitin‐mediated NFkappaB degradation pathway. Cell Mol Immunol. 2015;12(6):653‐655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Zepp JA, Wu L, Qian W, et al. TRAF4‐SMURF2‐mediated DAZAP2 degradation is critical for IL‐25 signaling and allergic airway inflammation. J Immunol. 2015;194(6):2826‐2837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Cao S, Xiao L, Rao JN, et al. Inhibition of Smurf2 translation by miR‐322/503 modulates TGF‐beta/Smad2 signaling and intestinal epithelial homeostasis. Mol Biol Cell. 2014;25(8):1234‐1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Tan Y, Chen Y, Du M, et al. USF2 inhibits the transcriptional activity of Smurf1 and Smurf2 to promote breast cancer tumorigenesis. Cell Signal. 2019;53:49‐58. [DOI] [PubMed] [Google Scholar]
- 56. Thomaidou S, Zaldumbide A, Roep BO. Islet stress, degradation and autoimmunity. Diabetes Obes Metab. 2018;20(Suppl 2):88‐94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Longchamp A, Mirabella T, Arduini A et al. AMino acid restriction triggers angiogenesis via GCN2/ATF4 regulation of VEGF and H2S production. Cell 2018;173(1):117–129.e114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Zhu K, Jiao H, Li S, et al. ATF4 promotes bone angiogenesis by increasing VEGF expression and release in the bone environment. J Bone Miner Res. 2013;28(9):1870‐1884. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure S1 (A) Representative images of IF staining for angiogenesis and MSC markers in the hip synovium of patients with AS and patients with OA (n = 10 patients per group). CD31 and CD105 were co‐expressed in patients with AS, indicating that ASMSCs may participate in the abnormal angiogenesis observed in patients with AS. Scale bar = 100 μm. (B) QRT‐PCR revealed significantly higher expression of the CD31 mRNA in the hip synovium of patients with AS than in the hip synovium of patients with OA (n = 10 patients per group). (C) Representative images showing significantly higher Smurf2 IHC staining in the hip synovium of patients with AS than in the hip synovium of patients with OA (n = 10 patients per group). Scale bar = 100 μm. (D) The expression of E3 ligases was upregulated in ASMSCs. QRTPCR showing upregulation of the mRNA levels of 4 E3 ligases, TRIM21, TRIM25, TRIM56 and Smurf2 in ASMSCs, and Smurf2 expression exhibited the most significant change (n = 10 different HDMSC or ASMSC lines per group). (E) Sterile FACS cell sorting was performed using cells transfected with the vector encoding GFP. After lentivirus transfection, the GFP‐expressing population of MSCs was 60.21%. Then, the transfected MSCs were selected with blank control MSCs (0.50%) for a background comparison. (F) Both HDMSCs and ASMSCs were effectively transfected with the lentivirus, for example, NC and Sh‐Smurf2. Scale bar = 500 μm. All data are presented as the means ± SD. *, P < 0.05. ***, P < 0.001.
Supplementary Figure S2 (A) and (B) Representative images of the wound healing assay and the tube formation assay of HUVECs co‐cultured with differently treated HDMSCs or ASMSCs. Smurf2 knockdown led to decreased HUVEC migration and tube formation, while Smurf2 overexpression exerted the opposite effect, and C716A overexpression did not produce similar phenotypes. Scale bars = 250 μm in (A) and 400 μm in (B). (C) IF staining for Smurf2 to verify the specificity of the antibody. Compared to that in the NC group, Smurf2 expression in the Sh‐Smurf2 group was knocked down by the lentivirus. The red fluorescence signal was substantially decreased, and the green fluorescence signal of PTX3 was increased (red, Smurf2; green, PTX3; blue, DAPI; n = 10 different ASMSC lines per group; scale bar = 20 μm). (D) MSCs were treated with proteasome inhibitors for 12 hours to block the UPS, and both MG132 and MG101 substantially inhibited PTX3 expression (n = 10 different HDMSC lines per group).
Supplementary Figure S3 (A) Identification of the differentially altered angiogenic factors between the supernatants of HDMSCs or ASMSCs co‐cultured with HUVECs. Quantification of the results obtained using the Human Angiogenesis Array Kit (ARY007; R&D Systems) showed that several factors displayed abnormal levels in the supernatants of ASMSCs co‐cultured with HUVECs, including angiopoietin‐2 (ANG2), EGF, IGFBP2, IL1β, prolactin, thrombospondin‐1 (TSP1), uPA and PTX3. Refer to the column number in (B) for the coordinates in (A). (C), (D), (E) and (G) The potential role of EGF in the abnormal angiogenesis of ASMSCs was explored by applying an EGF neutralizing antibody to modulate the EGF level. The increased angiogenesis capacity of ASMSCs was impaired by the anti‐EGF antibody to some extent, but the difference compared with the HDMSCs group was still significant (n = 10 different HDMSC or ASMSC lines per group). (F), (H), (I), (J), (K) and (L) The quantification of the results from the wound healing, migration and tube formation assays using HUVECs is shown. All data are presented as the means ± SD. Scale bars = 250 μm in (C) and (E), and 400 μm in (G). *, P < 0.05. **, P < 0.01.
Supplementary Figure S4 (A), (B), (C) and (D) The difference in the half‐life of PTX3 between HDMSCs and ASMSCs disappeared when Smurf2 expression was modulated by either silencing or overexpression. Overexpression of C716A did not exert a significant effect on the rate of PTX3 degradation in either HDMSCs or ASMSCs. n = 10 different HDMSC or ASMSC lines per group. (E), (F) and (G) Myc‐PTX3 was transiently overexpressed in ASMSCs, and protein lysates were prepared at the indicated time points after treatment with 10 μM CHX alone or in combination with the proteasome inhibitor MG132 (10 μM) or the lysosome inhibitor chloroquine (50 μM). Western blots showing that MG132 obviously inhibited the degradation of PTX3, while chloroquine exerted less of an effect on the degradation of PTX3. PTX3 degradation was mainly regulated by the proteasome. n = 10 different ASMSC lines per group. n.s., not significant.
Supplementary Figure S5 PTX3 silencing induced ASMSC‐mediated angiogenesis in vitro. (A) The silencing efficiency and overexpression efficiency of Sh‐PTX3 and OE‐PTX3, respectively, were verified using Western blotting analyses. (B), (C), (E) and (G) In the CCK‐8, wound healing, Transwell and tube formation assays, PTX3 knockdown led to increased HUVEC proliferation, migration and tube formation when co‐cultured with HDMSCs, while PTX3 overexpression decreased HUVEC proliferation, migration and tube formation when cocultured with ASMSCs. The increased angiogenic capacity of ASMSCs was reversed by modulating PTX3 expression. ASMSCs induced higher levels of angiogenesis because they expressed PTX3 at lower levels. (D), (F), (H), (I), (J) and (K) The quantification of the results from the wound healing, migration and tube formation assays is shown. All data are presented as the means ± SD. n = 10 different HDMSC or ASMSC lines per group. Scale bars = 250 μm in (C) and (E) and 400 μm in (G). *, P < 0.05. **, P < 0.01. ***, P < 0.001.
Supplementary Figure S6 (A) Expression of the Smurf2 and PTX3 mRNAs after modulation of ATF4 expression in HDMSCs and ASMSCs. QRT‐PCR showed that ATF4 knockdown decreased the expression of the Smurf2 mRNA but did not affect the expression of the PTX3 mRNA. (B) and (C) Western blots showing that ATF4 knockdown reduced the levels of the Smurf2 protein and increased the levels of the PTX3 protein in both HDMSCs and ASMSCs (n = 10 different HDMSC or ASMSC lines per group). (D), (E), (G) and (I) In the CCK‐8, wound healing, Transwell and tube formation assays, ATF4 knockdown led to decreased HUVEC proliferation, migration and tube formation when co‐cultured with both HDMSCs and ASMSCs. Thus, ATF4 knockdown negatively regulated HUVEC functions. (F), (H), (J), (K), (L) and (M) The quantification of the results from the wound healing, migration and tube formation assays is shown. All data are presented as the means ± SD. n = 10 different HDMSC or ASMSC lines per group. Scale bar = 250 μm in (C) and (E), and 400 μm in (G). *, P < 0.05. **, P < 0.01. ***, P < 0.001. n.s., not significant.
Supplementary Table S1 Primer sequences used for qRT‐PCR
Supplementary Table S2 Supplementary Table
Data Availability Statement
All data, models, and code generated or used during the study appear in the submitted article.