

Abstract
Background
Paroxysmal dyskinesias (PxDs) are characterized by involuntary movements and altered pre‐motor circuit activity. Causative mutations provide a means to understand the molecular basis of PxDs. Yet in many cases, animal models harboring corresponding mutations are lacking. Here we utilize the fruit fly, Drosophila, to study a PxD linked to a gain‐of‐function (GOF) mutation in the KCNMA1/hSlo1 BK potassium channel.
Objectives
We aimed to recreate the equivalent BK (big potassium) channel mutation in Drosophila. We sought to determine how this mutation altered action potentials (APs) and synaptic release in vivo; to test whether this mutation disrupted pre‐motor circuit function and locomotion; and to define neural circuits involved in locomotor disruption.
Methods
We generated a knock‐in Drosophila model using homologous recombination. We used electrophysiological recordings and calcium‐imaging to assess AP shape, neurotransmission, and the activity of the larval pre‐motor central pattern generator (CPG). We used video‐tracking and automated systems to measure movement, and developed a genetic method to limit BK channel expression to defined circuits.
Results
Neuronal APs exhibited reduced width and an enhanced afterhyperpolarization in the PxD model. We identified calcium‐dependent reductions in neurotransmitter release, dysfunction of the CPG, and corresponding alterations in movement, in model larvae. Finally, we observed aberrant locomotion and dyskinesia‐like movements in adult model flies, and partially mapped the impact of GOF BK channels on movement to cholinergic neurons.
Conclusion
Our model supports a link between BK channel GOF and hyperkinetic movements, and provides a platform to dissect the mechanistic basis of PxDs. © 2021 The Authors. Movement Disorders published by Wiley Periodicals LLC on behalf of International Parkinson and Movement Disorder Society
Keywords: Drosophila, BK channel, slowpoke, paroxysmal dyskinesia, central pattern generator, pre‐motor circuit, locomotion
Paroxysmal dyskinesias (PxDs) are characterized by intermittent attacks of dystonic, choreiform, and/or ballistic movements. 1 Distinct forms of PxD can be clinically differentiated based on the triggers for attacks. These include paroxysmal kinesigenic dyskinesia (triggered by sudden movement), paroxysmal exercise‐induced dyskinesia, and paroxysmal non‐kinesigenic dyskinesia (PNKD; often triggered by alcohol, caffeine, stress, and fatigue). 2 Here we focus on a PxD subtype whose neuropathological basis has not been investigated in an animal model: type‐3 PNKD (PNKD3; OMIM #609446).
PNKD3 is caused by autosomal dominant or de novo mutations in the KCNMA1 locus, which encodes the pore‐forming hSlo1 α‐subunit of the calcium (Ca2+)‐activated BK (big potassium) channel. 3 , 4 , 5 , 6 , 7 , 8 BK channels modulate neuronal excitability and action potential (AP) firing rate by contributing to the repolarization and afterhyperpolarization (AHP) phases of APs. 9 , 10 , 11 BK channels also limit neurotransmitter release at a variety of nerve terminals by driving inactivation of presynaptic voltage‐gated Ca2+ channels. 11 The first mutation linked to PNKD3 was a dominant missense mutation (1301A → G) in exon 10 of KCNMA1. This mutation results in the replacement of a negatively charged aspartic acid residue with a neutral glycine (D434G) in hSlo1, 4 and is associated with paroxysmal dystonic and choreiform movements of the mouth, tongue, and extremities. Electrophysiological analyses performed in non‐excitable cells indicate that D434G acts as a gain‐of‐function (GOF) mutation by increasing BK channel Ca2+ sensitivity, accelerating activation, and decelerating deactivation. 3 , 4 , 5 , 12 However, the impact of the D434G mutation on AP shape and neurotransmission in vivo has remained unclear. More fundamentally, this mutation has only been identified in a single multi‐generation family, 4 and there are currently no animal models of D434G or any other BK channel mutation linked to PNKD3. Demonstrating that equivalent mutations disrupt movement in non‐human models would strongly support a causative link between BK channel GOF and involuntary movements, and provide a platform for mechanistic studies.
We therefore generated a knock‐in Drosophila model of PNKD3. Utilizing this model, we provide in vivo evidence that the equivalent mutation to D434G in Drosophila alters AP waveforms and neurotransmitter release, disrupts structured activity of pre‐motor circuits, and perturbs coordinated movement in both larval and adult Drosophila. Furthermore, we partially map the impact of GOF BK channels on adult movement to cholinergic neurons. Our results provide independent support for the genetic linkage between hSlo1 D434G and PxD, and suggest a critical and conserved role for BK channels in the regulation of movement across distantly related bilateral species.
1. Materials and Methods
See Supplemental Information for details of Materials and Methods.
2. Results
2.1. Generation of a Drosophila Model of PNKD3
The D434 residue mutated in PNKD3 is located within the regulator of K+ conductance 1 (RCK1) domain of the channel 4 (Figure S1A), which contains binding sites for divalent cations and connects Ca2+‐binding to channel opening. 13 Consistent with its functional importance, 4 , 5 the D434 residue is highly conserved across Bilateria (Fig. 1A). Aspartic acid (D) appears fixed in Deuterostomes at equivalent positions to hSlo1 434, while in Protostome orthologs, including the Drosophila BK channel α‐subunit Slowpoke (SLO), a glutamic acid residue (E) is more prevalent (Fig. 1A). Importantly, mutating the murine equivalent of D434 (D369 in mSlo1) to E does not alter channel function over a broad range of Ca2+ concentrations, 5 consistent with the similar physiochemical properties of aspartic and glutamic acid. Since the above evidence supports functional conservation of this residue between humans and Drosophila, we used ends‐out homologous recombination to substitute the Drosophila residue orthologous to hSlo1 D434 (SLO E366) with glycine (Figs. 1B,C; S1B and S2). In parallel, we isolated corresponding controls harboring the genomically encoded E residue. As part of the homologous recombination process, both lines contain a 76 base pair (bp) sequence in a non‐conserved intronic region of slo that includes a single loxP site 14 (Fig. 1B). We isolated 10 E366G and four control alleles (Figures S1B,C, and S2), and outcrossed three of each to an isogenic iso31 strain for five generations to homogenize genetic background (see Materials and Methods). We term these alleles slo E366G and slo loxP, respectively.
FIG. 1.
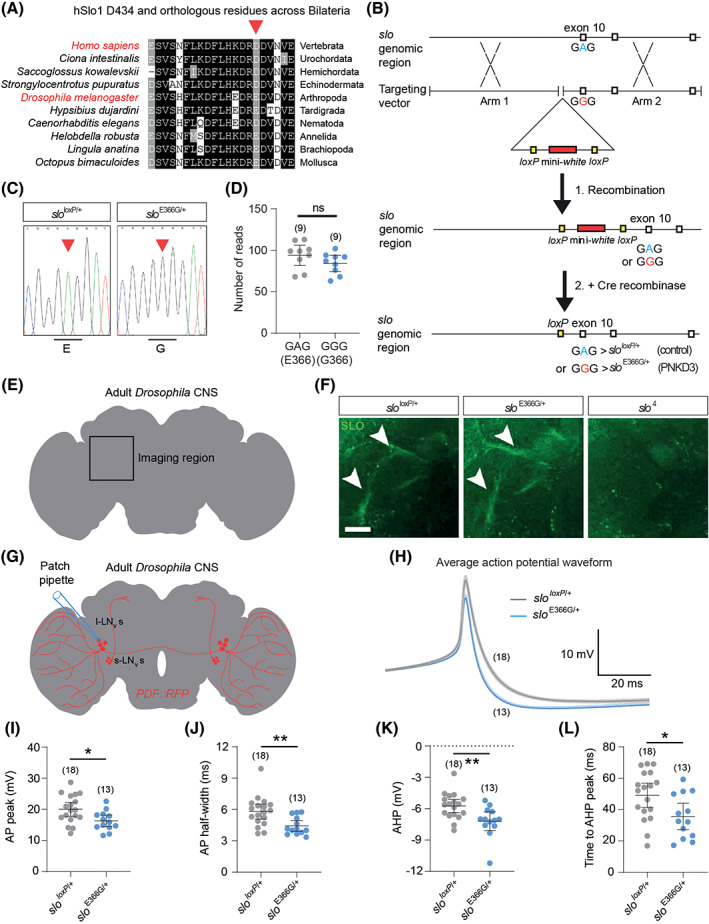
(A) Alignment of residues surrounding hSlo1 D434 (arrow) with orthologous BK α‐subunits from bilateral species spanning >540 million years of evolutionary divergence. (B) Schematic illustrating the procedure to generate the slo E366G and slo loxP alleles via ends‐out homologous recombination. The region surrounding exon 10 of the slo locus, which encodes the E366 residue, and corresponding targeting arms to induce homologous recombination, are shown. (C) Sanger sequence verification of the presence or absence of the A > G mutation in slo E366G/+ and slo loxP/+ flies via allele‐specific polymerase chain reaction (PCR). (D) RNAseq‐based quantification of slo + (GAG) and slo E366G (GGG) mRNAs from slo E366G/+ heterozygous head tissue. (E) Illustration of location of SLO‐positive axonal tracts (arrowheads in F) in the adult nervous system. CNS, central nervous system. (F) SLO channel expression in the slo loxP/+, slo E366G/+, and slo null (slo 4) backgrounds, imaged in region noted in (E). Scale, 20 μm. (G) Illustration showing morphology of large ventral lateral neurons (l‐LNvs) labeled with PDF promoter‐driven RFP (PDF::RFP) and location of patch‐clamp recording sites. (H) Average action potential (AP) waveforms in l‐LNvs. Darker and lighter shades show mean and standard error of the mean (SEM). (I–L) l‐LNv AP and afterhyperpolarization (AHP) parameters. Values of n are noted. Error bars: 95% confidence interval (CI). *P< 0.05, **P < 0.005, ***P < 0.0005, ns – P > 0.05, unpaired t‐test with Welch's correction (D, I, J, L), Mann–Whitney U test (K). [Color figure can be viewed at wileyonlinelibrary.com]
We combined RNAseq and immunofluorescent imaging to test whether the E366G mutation impacted slo mRNA stability or localization of the SLO channel. In slo E366G/+ heterozygotes, RNAseq revealed no significant difference in the number of mRNA reads from the slo locus containing either wild‐type (GAG; +) or mutant (GGG; E366G) codons in exon 10 (Fig. 1B,D). Nor was the overall expression of slo between slo E366G/+ and slo loxP/+ heterozygotes (our primary experimental genotypes – see below) significantly different (log2 fold‐change = 0.045; P = 0.16; q = 0.48; n = 9 independent biological replicates/genotype). Furthermore, immunostaining of SLO channels in the adult nervous system did not reveal any obvious difference in SLO localization within axonal tracts 15 between slo E366G/+ and slo loxP/+ flies (Fig. 1E–F). Hence, alterations in neuronal physiology and organismal behavior in slo E366G/+ flies likely derive from changes in BK channel activity as opposed to SLO expression. Interestingly, slo E366G homozygotes were lethal prior to adult eclosion from the pupal case, and slo E366G/E366G pupae exhibited morphological abnormalities (Figure S3), suggesting a profound and dose‐dependent impact of SLO E366G channels on organismal physiology and development. We therefore focus the remainder of our studies on slo E366G/+ heterozygotes, which are viable, fertile, and accurately model the dominant mode of inheritance of the hSlo1 D434G mutation in PNKD3. 4
2.2. Altered Action Potential Shape in sloE366G /+ Neurons
As noted above, BK channels play important roles in controlling neuronal excitability by modulating AP shape and the AHP. 10 To test how SLO E366G channels impact these neurophysiological properties, we performed ex vivo patch‐clamp recordings from adult large ventral lateral neurons (l‐LNvs) (Fig. 1G). These neurons are components of the Drosophila circadian clock network, and drive light‐dependent changes in arousal. 16 SLO channel expression in the l‐LNvs oscillates in a time‐dependent manner, with high SLO channel activity observed at Zetigeber Time (ZT) 18–20 (ie, during the night) and low activity at ZT6–8 (during the day). 17
Passive membrane properties of l‐LNvs were not altered in slo E366G/+ flies at ZT18–20 (Figure S4A,B). However, analysis of spontaneous APs revealed a significant reduction in mean AP amplitude and duration, as well as enhanced AHP amplitude and accelerated AHP kinetics, in slo E366G/+ l‐LNvs (Fig. 1H–L). These alterations appear to result from acute increases in SLO E366G channel expression rather than, for example, neurodevelopmental or homeostatic changes in l‐LNv excitability, since we did not observe any differences in AP or AHP properties at ZT6–8, when SLO expression is low 17 (Figure S5). Interestingly, while the D434G mutation has been hypothesized to increase neuronal firing rates by accelerating sodium channel recovery, 4 slo E366G/+ l‐LNvs did not exhibit alterations in either the rate of spontaneous firing (Figure S4C) or of higher frequency firing induced by +20 or +40 pA current injections (Figure S6) at ZT18–20, despite a narrowing of AP width (Fig. 1J).
2.3. SLO E366G Channels Reduce Neurotransmitter Release in a Ca2+‐Dependent Manner
In central presynaptic termini and neuromuscular junctions of diverse species, BK channels negatively tune neurotransmitter release by limiting activation of presynaptic voltage‐gated Ca2+ channels. 11 Thus, we next investigated whether SLO E366G channels influence neurotransmitter release.
To do so, we turned to a highly tractable glutamatergic synapse in Drosophila – the 3rd instar larval neuromuscular junction 18 (NMJ) (Fig. 2A). Neither the passive membrane properties of the muscle nor the morphology of motoneuron synapses were significantly different between slo E366G/+ and slo loxP/+ L3 larvae (Figure S7). We evoked postsynaptic excitatory junction potentials (EJPs) by severing and directly stimulating innervating motoneurons (Fig. 2A), and examined EJP amplitudes across a range of extracellular Ca2+ concentrations ([Ca2+]e). Since the probability of neurotransmitter release (P r) at the larval NMJ scales with AP‐evoked Ca2+ influx, 19 , 20 we used this paradigm to model the effect of SLO E366G channels on neurotransmitter release across a range of P r. At 1–3 mM [Ca2+]e (high Pr), we found no difference in EJP amplitudes between slo E366G/+ and slo loxP/+ larvae (Fig. 2B,D). In contrast, slo E366G/+ EJPs were significantly smaller compared to slo loxP/+ controls at 0.15–0.25 mM [Ca2+]e (low Pr) (Fig. 2C,D). This Ca2+‐dependent alteration in synaptic release was not accompanied by changes in the amplitude or frequency of spontaneously occurring miniature EJPs (mEJPs) (Fig. 2G–I), suggesting a presynaptic locus for this effect. To provide support for this premise, we examined short‐term plasticity in slo E366G/+ larvae over the same range of [Ca2+]e. The relationship between [Ca2+]e and short‐term plasticity is well established at this synapse. Low [Ca2+]e results in lower vesicle release per AP, smaller EJPs, and paired‐pulse facilitation (PPF). Conversely, increasing [Ca2+]e enhances vesicle fusion, increases EJP amplitudes, and reduces PPF. 21 , 22 We found that slo E366G/+ larvae displayed a significant increase in PPF at 0.15 mM [Ca2+]e, a non‐significant trend towards an increase at 0.25 mM [Ca2+]e, and no alteration at 1 or 3 mM [Ca2+]e (Fig. 2J–N), supporting the premise that presynaptic SLO E366G channels reduce neurotransmitter release at the NMJ at low [Ca2+]e.
FIG. 2.
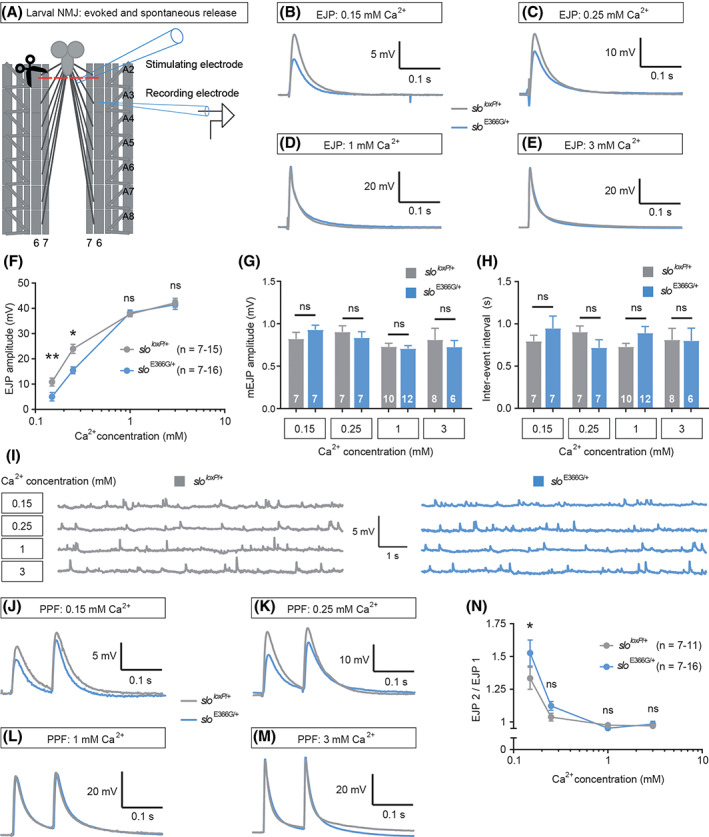
(A) Illustration of the electrophysiological protocol used in the larval preparation. A sharp intramuscular recording electrode records from abdominal segment A3 of the longitudinal body wall muscle 6. Motoneurons innervating the body wall muscles are severed just below the ventral nerve cord (VNC) and excitatory junction potentials (EJPs) are evoked by stimulating the severed end of the motoneurons innervating muscle 6, A3. Abdominal segments A2−A8 are shown. NMJ, neuromuscular junction. (B–E) Representative EJPs from slo loxP/+ and slo E366G/+ larvae at high and low [Ca2+]e. (F) Mean EJP amplitudes at various [Ca2+]e in slo loxP/+ and slo E366G/+ larvae (x‐axis shown as log10). (G–H) Mean mEJP amplitude (G) and inter‐event interval (H) across a range of [Ca2+]e. (I) Representative mEJPs from slo loxP/+ and slo E366G/+ larvae at different [Ca2+]e. (J–M) Representative paired‐pulse waveforms across a range of [Ca2+]e. PPF, paired‐pulse facilitation. (N) Paired‐pulse ratio shown as EJP2/EJP1 at various [Ca2+]e in slo loxP/+ and slo E366G/+ larvae (x‐axis shown as log10). Values of n are noted. Error bars: mean ± SEM. *P < 0.05, **P < 0.005, ns – P > 0.05, two‐way ANOVA with Sidak's multiple comparisons test (F, N). ns – P > 0.05, Mann–Whitney U test (G, H). [Color figure can be viewed at wileyonlinelibrary.com]
2.4. Perturbed Pre‐Motor Circuit Function in sloE366G /+ Larvae
Collectively, the above data suggest that while SLO E366G channels do not impact neurotransmission at the NMJ under physiological conditions, they may reduce neurotransmitter release at central synapses with an intrinsically lower Pr. 23 Thus, we next explored whether the activity of central pre‐motor circuits, which potentially harbor such synapses, were perturbed by expression of SLO E366G channels.
We focused on the larval central pattern generator (CPG) – an intrinsically active network that drives rhythmic excitation of motoneurons during foraging, 24 and which is located within the abdominal and thoracic segments of the ventral nerve cord (VNC). 25 We visualized CPG‐driven input to motoneuron dendrites in the VNCs of ex vivo larval brains by expressing a genetically encoded fluorescent Ca2+ sensor, GCaMP6m, 26 under the ok371‐Gal4 driver, which labels motoneurons in the abdominal segments of the VNC as well as other glutamatergic neurons (which can be distinguished based on location 27 ) (Fig. 3A–B). Fictive forward locomotion (CPG‐induced excitation of motoneurons that would drive locomotion) was visible as rhythmic waves of increased GCaMP6m fluorescence that moved from posterior to anterior motoneuron dendrites and cell bodies (Fig. 3C,E, and Video S1). The amplitude and frequency of posterior to anterior Ca2+ waves were reduced in slo E366G/+ VNCs (Fig. 3D,F–H), while the propagation of waves from segments A7–A4 occurred more rapidly (Fig. 3I). It was also possible to identify fictive turns. Dendritic Ca2+ spikes were usually concurrent between motoneurons on the left and right sides of the VNC. However, as described previously, 28 occasionally a spike in anterior segments occurred alongside a coincident trough in GCaMP6m fluorescence in the contralateral motoneuron dendrite (Fig. 3C, arrows). In behaving larvae this would result in unilateral anterior muscle contraction, facilitating a turn. Fictive turns were present in slo loxP/+ controls but were not observed in slo E366G/+ VNCs (Fig. 3C,J). These data strongly indicate that the activity of the CPG driving larval locomotion is perturbed by SLO E366G channels.
FIG. 3.
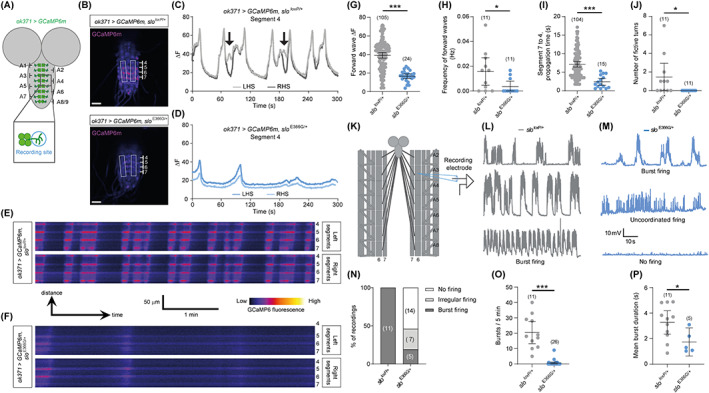
(A) Illustration of GCaMP6m‐labeled motoneuron cell bodies and dendritic regions in the ventral nerve cord (VNC) and location of recording area around motoneurons in the abdominal A4 segment of the VNC. (B) Representative images showing GCaMP6m‐labeled motoneuron cell bodies and dendrites in the VNCs of slo loxP/+ and slo E366G/+ larvae, and location of motoneuron dendrites on the left‐ (LHS) and right‐hand side (RHS) of abdominal segments 4–7. (C, D) Representative traces of GCaMP6m fluorescence over 300 seconds in slo loxP/+ (C) and slo E366G/+ (D) motoneuron dendrites within abdominal segment 4. Arrows in C indicate fictive turns, where the LHS and RHS motoneuron dendrites exhibit opposing patterns of excitation. (E, F) Line‐based kymographs showing rhythmic alterations in GCaMP6m fluorescence within dendritic domains of slo loxP/+ (E) and slo E366G/+ (F) motoneurons in abdominal segments 4–7. (G–J) Parameters of fictive locomotor patterns. Values of n are shown. Data are derived from n = 11 slo loxP/+ and slo E366G/+ larvae. (K) Illustration of the electrophysiological protocol used. Motoneuron axons innervating the body wall are left intact. Postsynaptic excitatory junction potentials (EJPs) are thus elicited via activation of motoneurons by the upstream larval central pattern generator (CPG). (L–M) Representative traces of spontaneous firing from slo loxP/+ (L) and slo E366G/+ (M) larvae. (N) Percentage of slo loxP/+ and slo E366G/+ larvae showing burst firing, irregular firing, or no firing. (O) Number of bursts in 5 minutes of recording. (P) Duration of bursts, only including recordings in which at least one burst occurred (derived from n = 11 larvae). Error bars: mean and 95% confidence interval (CI). *P < 0.05, ***P < 0.0005, Mann–Whitney U test (G–J, O) or unpaired t‐test with Welch's correction (P). [Color figure can be viewed at wileyonlinelibrary.com]
2.5. Aberrant Motoneuron Output and Locomotor Behavior in sloE366G /+ Larvae
To investigate the downstream consequences of CPG dysfunction in slo E366G/+ larvae, we determined whether patterned output from motoneurons was altered by recording intramuscular voltage changes in larval muscle segments innervated by intact motoneurons (Fig. 3K). Proprioceptive neurons in the larval body wall provide sensory feedback to the CPG and/or downstream pre‐motor circuits during movement, enhancing the frequency of contractive wave propagation and thus locomotor velocity. 29 Since we aimed to test how changes in the intrinsic activity of the CPG affected motoneuron output, independent of any potential alterations in sensory inputs, we limited proprioceptive feedback by inhibiting motoneuron‐induced muscle contractions via the voltage‐gated Ca2+ channel blocker nifedipine. 30 Consistent with previous studies, 31 , 32 we observed periodic bursts of high‐frequency EJPs in all slo loxP/+ control larvae (Fig. 3L,N). Similar bursts were observed in only a minority (5/26) of slo E366G/+ larvae (Fig. 3M–N), and when they did occur were less frequent and of shorter duration (Fig. 3O–P). Interestingly, prior work has shown that reducing SLO expression enhances the frequency of CPG‐induced motoneuron bursts. 33 Thus, loss and gain of BK channel function bidirectionally alter endogenous motoneuron firing in Drosophila larvae. Of the remaining slo E366G/+ larvae, 14/26 showed no firing activity during 5 minutes of recording, while 7/26 showed continuous, irregular activity that was not coordinated into bursts and quiescent periods (Fig. 3N). Therefore, motoneurons in slo E366G/+ larvae display abnormal patterns of spontaneous activity, consistent with dysfunction of the upstream movement‐driving CPG in slo E366G/+ larvae.
We next tested whether the above alterations in CPG function and patterned motoneuron output resulted in corresponding changes in larval locomotion. We measured locomotor activity in slo E366G/+ and slo loxP/+ larvae by video‐tracking 1 minute of movement across a flat agar plane (Fig. 4A). 34 In striking agreement with the above data, we found that both the total distance moved and number of turns initiated were substantially reduced in slo E366G/+ larvae (Figs. 4D–F and S8). These data provide proof‐of‐principle in an animal model that a BK channel GOF mutation associated with PNKD3 can alter locomotor control, and point to a central role for CPG dysfunction in generating these defects.
FIG. 4.
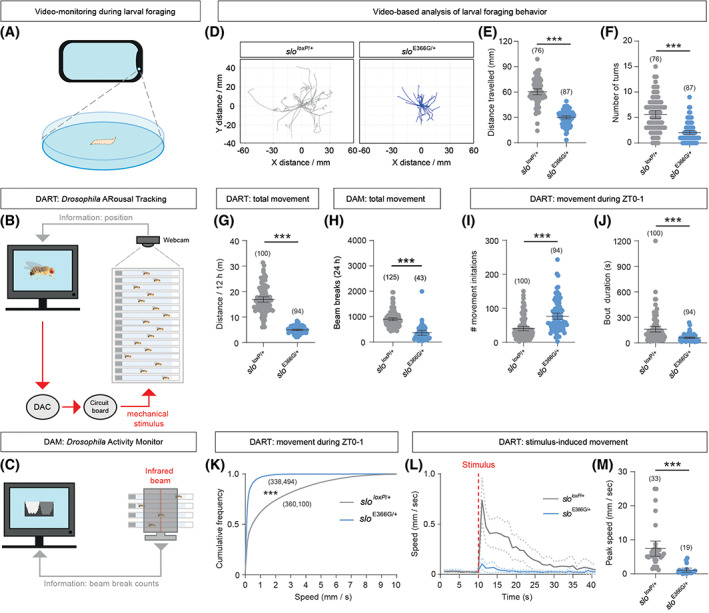
(A‐C) Schematics of phone‐based video‐tracking of Drosophila larvae foraging across an agar plate (A), the DART (Drosophila Arousal Tracking) video‐tracking system (B; DAC: digital to analog converter), and the DAM system (C), which counts breaks of an infrared beam bisecting a DAM monitor. (D) Overlaid traces of foraging paths traveled by individual slo loxP/+ (n = 26) and slo E366G/+ (n = 28) L3 larvae during 1 minute of free movement. (E, F) Mean distance traveled (E) and number of turns (F) over 1 minute between slo E366G/+ and slo loxP/+ L3 larvae. (G, H) Overall movement in adult male slo loxP/+ and slo E366G/+ flies measured by the DART (G) and DAM (Drosophila Activity Monitor) (H) systems. (I, J) Number (I) and duration (J) of movement bouts during 0–1 hours following lights‐on in slo loxP/+ and slo E366G/+ adult male flies. (K) Cumulative distribution of speeds in slo loxP/+ and slo E366G/+ males during 0–1 hours following lights‐on. Data are derived from 100 slo loxP/+ and 94 slo E366G/+ males. (L) Plot of locomotor speed in slo loxP/+ and slo E366G/+ males before and after an applied mechanical stimulus (red dotted line). (M) Peak speed in slo loxP/+ and slo E366G/+ males in the 1‐minute period following a mechanical stimulus. Values of n are shown. Error bars: mean and 95% confidence interval (CI). **P < 0.005, ***P < 0.0005, unpaired t‐test with Welch's correction (E, G), Mann–Whitney U test (F, H–J, M), or Kolmogorov–Smirnov test (K). [Color figure can be viewed at wileyonlinelibrary.com]
2.6. sloE366G /+ Adult Flies Exhibit Locomotor Dysfunction and Dyskinesia‐like Leg Twitches
Given that dyskinetic attacks in PNKD3 patients frequently involve involuntary movement of the extremities, 4 we were also interested in examining how the SLO E366G mutation impacted movement in adult flies – the limbed stage of the Drosophila life cycle. As noted above, slo E366G/+ adults are viable and fertile, albeit with reduced lifespan (Figure S9). While paroxysmal dystonia and chorea is co‐morbid with absence and generalized tonic–clonic seizures in some patients harboring the hSlo1 D434G mutation, we did not observe temperature‐ or bang‐induced seizures in slo E366G/+ adults, nor ether‐induced leg shaking. However, video recordings of slo E366G/+ adult flies revealed two clear movement‐related phenotypes. First, ~1/3 of slo E366G/+ flies (but no slo loxP/+ controls) exhibited spontaneous leg twitches across a 5‐minute period (Figure S10A,B; Video S2). Leg twitches in slo E366G/+ males were of median duration 4 seconds but could extend to >25 seconds, and were temporally clustered into dispersed bouts (Figure S10C–L), reminiscent of paroxysmal attacks in PNKD3 patients. 4 Second, slo E366G/+ males appeared to exhibit an overall reduction in locomotion (Video S2). To quantify this phenotype in more detail, we used an automated video‐tracking system called DART (Drosophila Arousal Tracking) 35 to monitor locomotion in adult male slo E366G/+ and slo loxP/+ flies over a 12‐hour period (Fig. 4B). slo E366G/+adult males indeed exhibited a robust reduction in distance traveled compared to controls (Figs. 4G and S11). Using a distinct, more high‐throughput, activity monitoring system (the Drosophila Activity Monitor; DAM 36 ) (Fig. 4C), we confirmed reduced locomotion in slo E366G/+ adult males (Fig. 4H) and found that such movement defects are apparent across the day/night cycle (Figure S12), showing that reduced movement in slo E366G/+ males is not an artefact caused by perturbed circadian rhythms. 37
Since male flies are inactive for much of the day, 38 we performed a more detailed analysis of movement during a period of normally heightened activity: 0–1 hours following lights‐on in 12 hours light: 12 hours dark conditions. During this time span, DART recordings revealed that slo E366G/+ males initiate movement more frequently than controls (Fig. 4I). However, the duration of locomotor bouts was shorter (Fig. 4J), and overall locomotor speeds significantly reduced (Fig. 4K), in slo E366G/+ males. Similar results were observed in slo E366G/+ adult females (Figure S13). We also used the DART system to apply a mechanical stimulus to quiescent slo E366G/+ and slo loxP/+ males (see Materials and Methods), and observed that the speed of stimulus‐induced movement was strongly reduced in slo E366G/+ males (Fig. 4L–M). Thus, the SLO E366G mutation impairs self‐driven movement in larval and adult Drosophila, and stimulus‐induced movement in adult Drosophila.
2.7. Cell‐Specific Induction of SLO E366G Channels Suggests a Pathogenic Role in Cholinergic Neurons
BK channels are broadly expressed in both neuronal and non‐neuronal tissues, 8 and the cell‐types in which GOF BK channels act to impact movement are unknown. To begin to identify such cell types, we developed a genetic system that allowed us to limit robust SLO expression to specific cell types of interest. This method is based upon a gene called dyschronic (dysc), orthologous to the human deaf‐blindness gene Whirlin/DFNB31. 15 , 39 dysc encodes a scaffold protein (DYSC) that promotes neuronal SLO channel expression. dysc loss‐of‐function (LOF) mutants exhibit greatly reduced neuronal SLO expression and arrhythmic patterns of locomotion across the day–night cycle, but no significant disruption in overall motor capacity. 15 If the SLO E366G mutation indeed causes GOF, we predicted that coincident homozygosity for the LOF dysc s168 allele would suppress movement defects in slo E336G/+ adults, since GOF BK channels would not be robustly expressed (Fig. 5A). Using the DAM system, we found that this was the case (Fig. 5B). We were then able to restore DYSC (and thus SLO E366G) expression in slo E366G/+, dysc s168 double mutants by combining a UAS‐dysc transgene with the UAS‐binding Gal4 transcription factor under control of a cell‐specific promoter. This approach yields robust SLO E366G expression in defined cell types, allowing us to test whether this causes movement defects. We found that global or pan‐neuronal (but not muscle‐specific) SLO E366G expression strongly reduced movement (Fig. 5C) and, furthermore, that inducing SLO E366G expression solely in cholinergic (but not glutamatergic, GABAergic, peptidergic, or insulinergic) neurons also reduced movement, albeit with lower penetrance relative to pan‐neuronal restoration (Fig. 5D). Thus, SLO E366G channels act in neurons to perturb adult movement, and this effect partially maps to cholinergic neurons.
FIG. 5.
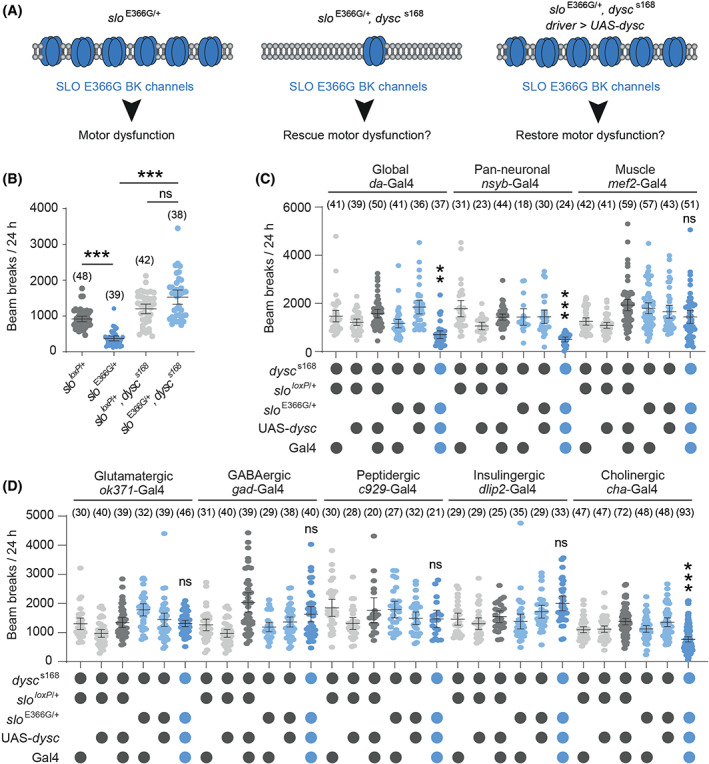
(A) Model of the dysc‐based system for indirect, cell‐specific control of SLO E366G expression. (B) Locomotor activity in slo E366G/+ and slo loxP/+ adult males with wild‐type or loss‐of‐function (dysc s168) dysc alleles. (C, D) Cell type‐specific restoration of DYSC in slo, dysc double mutants. Cell types associated with each promoter‐Gal4 driver are shown. Columns of blue circles denote the experimental genotypes in each dataset. Smaller dots in each graph represent measurements derived from individual adults. Values of n are noted. Error bars: mean and 95% confidence interval (CI). **P < 0.005, ***P < 0.0005, Kruskal−Wallis test with Dunn's post hoc test. [Color figure can be viewed at wileyonlinelibrary.com]
3. Discussion
PNKD3 has been linked to GOF mutations in the hSlo1 BK channel. 3 , 4 , 5 , 7 , 40 Yet, due to a lack of animal models of PNKD3, how pathologically enhanced BK channel activity impacts neuronal excitability, synaptic plasticity, and pre‐motor circuit activity in vivo has been unclear. In addition, whether equivalent mutations to hSlo1 D434G are capable of modifying movement in non‐human species has remained unexplored. Here we address these issues using a Drosophila knock‐in model of PNKD3.
Although highly homologous, Drosophila BK channels exhibit a lower Ca2+‐ and voltage‐sensitivity compared to their mammalian counterparts when expressed in Xenopus oocytes. 41 Furthermore, we have not directly measured BK channel currents in SLO‐expressing neurons of slo E366G/+ flies. Thus, the precise biophysical impact of the SLO E366G mutation on BK channel function remains to be fully determined. Nonetheless, several lines of evidence presented herein indicate that SLO E366G causes GOF, similarly to hSlo1 D434G, and enhances the net activity of BK channels in vivo. First, LOF mutations in mammalian and Drosophila BK channel α‐subunits result in a broadening of APs and a loss of the AHP current in BK channel‐expressing neurons. 33 , 42 However, in slo E366G/+ l‐LNv neurons, the reverse is true: AP width is narrowed and AHP amplitude is enhanced. Second, BK channel LOF enhances neurotransmitter release at the frog, Caenorhabditis elegans, and Drosophila, NMJs. 39 , 43 , 44 In contrast, we observed a reduction in neurotransmitter release solely at low [Ca2+]e at the slo E366G/+ larval NMJ. Third, the pronounced reduction in movement in slo E366G/+ adults was suppressed by loss of DYSC, a scaffold protein that binds SLO and promotes SLO expression in fly neurons. 15 Thus, while we cannot entirely rule out a neomorphic effect of the SLO E366G mutation, our data are consistent with SLO E366G acting as a dominant, GOF mutation. The specific impact of SLO E366G channels on neurotransmission at low [Ca2+]e leads us to speculate that the GOF effect of SLO E366G likely involves an increase in BK channel Ca2+‐sensitivity, similarly to the effects of an equivalent mutation to hSlo1 D434G in the murine BK channel α‐subunit. 5 Furthermore, the multifaceted defects in motor control combined with pre‐motor circuit dysfunction in slo E366G/+ larvae and adult flies support the genetic linkage between the hSlo1 D434G mutation and dyskinesia, 4 and demonstrate that BK channels play conserved roles in regulating movement across distantly related bilateral species.
Collectively, our analyses of neurotransmission at the larval NMJ, activity of the larval CPG, and the partial mapping of locomotor dysfunction in slo E366G/+ adult flies to cholinergic neurons suggests a model in which SLO E366G channels impair movement by at least two potentially overlapping mechanisms: first, by perturbing the intrinsic activity of CPGs that provide patterned excitatory input to motoneurons; and second, by reducing acetylcholine release from cholinergic pre‐motor circuits. Thus, while altered activity of cerebellar and basal ganglia‐thalamocortical circuits are common hallmarks of PxDs, 45 , 46 our findings suggest two particular pre‐motor circuits that will be interesting to examine in future vertebrate models of PNKD3. First, spinal CPG networks that transform descending command signals from brain regions such as the mesencephalic locomotor region into coordinated movements, 47 and second, striatal cholinergic interneurons (ChIs). This latter cell type is a particularly intriguing candidate for contributing to involuntary movements in PNKD3. BK channels in ChIs regulate AP repolarization, 42 and thus potentially impact neurotransmitter release from this cell type. Furthermore, acetylcholine release from ChIs modulates numerous striatal and neostriatal cell types via muscarinic and nicotinic acetylcholine receptors, including glutamatergic cortical afferents, GABAergic medium spiny output neurons, and GABAergic/dopaminergic interneurons; and aberrant patterns of striatal output are thought to contribute to involuntary movements in dystonia and dyskinesia. 48
However, it is important to note that our results suggest that SLO E366G channels act in more than one neural cell type to impair movement. Thus, it will be interesting to extend our cell‐specific mapping approach to examine the effect of expressing SLO E366G channels in other neuromodulatory circuits (dopaminergic, serotonergic, etc.), as well as specific pre‐motor neuropil domains in the Drosophila brain. It is also notable that, in contrast to slo E366G/+ flies, gross locomotor ability in PNKD3 patients is largely normal, with dyskinetic attacks generally triggered by alcohol, stress, or fatigue. 4 Such differences in phenotypic penetrance and severity due to orthologous Drosophila and human mutations may arise through a number of mechanisms, including the absence of BK channel β‐subunits in Drosophila (which modulate the biophysical impact of the D434G mutation 3 , 12 ), differences in the repertoire of ion channels contributing to AP shape and neurotransmitter release, divergent use of acetylcholine (Drosophila) and glutamate (vertebrates) as predominant excitatory neurotransmitters, and species‐specific variations in neural circuit architecture. Nonetheless, the robust motor defects observed in slo E366G/+ flies are an advantageous aspect of this model, since automated activity‐monitoring systems can now be deployed in concert with classical genetics to search for conserved genetic modifiers of motor dysfunction in this model. In contrast, involuntary movements in mouse models of inherited dyskinesia/dystonia frequently require stress or drug injections to induce their occurrence, 49 , 50 limiting their utility for high‐throughput phenotype‐based screens.
Our Drosophila model thus provides a rapid platform to define key neuronal subtypes and cellular pathways via which GOF BK channels alter motor control, and identify genetic perturbations capable of modifying phenotypic severity. It will also be interesting to utilize Drosophila models to study other KCNMA1/hSlo1 mutations linked to PNKD3, 6 , 7 as well as LOF mutations in KCNMA1 linked to ataxia. 51 , 52
Author Roles
Conceptualization: P.K., S.L., J.E.C.J. Methodology: P.K., E.B., K.‐F.C., A.P., S.L., J.E.C.J. Software: P.K. Validation: P.K., S.L., E.B. Formal Analysis: P.K., S.L., E.B., J.E.C.J. Investigation: P.K., S.L., E.B., K.‐F.C., S.L., J.E.C.J. Writing – Original Draft: J.E.C.J. Writing – Review and Editing: P.K., E.B., K.‐F.C., S.L., D.M.K., J.J.L.H., J.E.C.J. Visualization: P.K., S.L., E.B., J.E.C.J. Supervision: D.M.K., J.J.L.H., J.E.C.J. Project Administration: J.J.L.H., J.E.C.J. Funding Acquisition: P.K., D.M.K., J.J.L.H., J.E.C.J.
Supporting information
Appendix S1: Supplementary Information
Video S1 Video shows representative examples of waves of motoneuron excitation in the ex vivo larval ventral nerve cord (VNC) visualized via GCaMP6m driven by the glutamatergic neuron driver ok371‐Ga4. In the slo loxP/+ control VNC (left), repetitive waves of excitation traveling from posterior to anterior segments can clearly be observed. Unilateral motoneuron excitation in the upper segments can also be infrequently observed. In the slo E366G/+ VNC (right), both the magnitude and frequency of increases in motoneuron GCaMP6m fluorescence is reduced, indicative of perturbed input from the upstream central pattern generator.
Video S2 Video shows representative examples of three slo loxP/+ (left) or slo E366G/+ (right) adult male flies freely moving in glass tubes containing an agar‐sucrose food source. Tubes are horizontally placed. Note the more rapid speed and increased continuity of movement of slo loxP/+ compared to slo E366G/+ males. Subsequent zoom of the left‐hand slo E366G/+ male illustrates a bout of unilateral leg twitches, similar instances of which were frequently observed in slo E366G/+ but not slo loxP/+ flies.
Acknowledgments
We thank Prof. Kyunghee Koh (Thomas Jefferson University), Dr. Stefan Pulver (University of St. Andrews), and Prof. Justin Blau (New York University) for sharing Drosophila stocks; Dr. Srinivasa Rao Rao (University of Oxford) for help with larval video‐tracking analyses; Prof. Kirill Volynski, Prof. Henry Houlden, and Talya Goble (University College London) for comments on the manuscript; and Tom Lowe for help with video editing.
James J.L. Hodge and James E.C. Jepson jointly suprvised this work.
Patrick Kratschmer, Simon A. Lowe, and Edgar Buhl contributed equally to this work.
Relevant conflicts of interest/financial disclosures: None for all authors.
Funding agencies: This study was supported by a Wellcome Strategic Award (WT104033AIA) to D.M.K. and J.E.C.J.; a MRC New Investigator Grant (MR/P012256/1) to J.E.C.J.; a Wellcome PhD studentship award to P.K. (109003/Z/15/Z), a MRC Programme Grant to D.M.K. (MR/L01095X/1), and a Leverhulme Trust grant (RPG‐2016‐318) to J.J.L.H.
Contributor Information
James J.L. Hodge, Email: j.jepson@ucl.ac.uk, Email: james.hodge@bristol.ac.uk.
James E.C. Jepson, Email: j.jepson@ucl.ac.uk.
References
- 1. Erro R, Sheerin UM, Bhatia KP. Paroxysmal dyskinesias revisited: a review of 500 genetically proven cases and a new classification. Mov Disord 2014;29(9):1108–1116. 10.1002/mds.25933 [DOI] [PubMed] [Google Scholar]
- 2. Bhatia KP. Paroxysmal dyskinesias. Mov Disord 2011;26(6):1157–1165. 10.1002/mds.23765 [DOI] [PubMed] [Google Scholar]
- 3. Diez‐Sampedro A, Silverman WR, Bautista JF, et al. Mechanism of increased open probability by a mutation of the BK channel. J Neurophysiol 2006;96(3):1507–1516. 10.1152/jn.00461.2006 [DOI] [PubMed] [Google Scholar]
- 4. Du W, Bautista JF, Yang H, et al. Calcium‐sensitive potassium channelopathy in human epilepsy and paroxysmal movement disorder. Nat Genet 2005;37(7):733–738. 10.1038/ng1585 [DOI] [PubMed] [Google Scholar]
- 5. Yang J, Krishnamoorthy G, Saxena A, et al. An epilepsy/dyskinesia‐associated mutation enhances BK channel activation by potentiating Ca2+ sensing. Neuron 2010;66(6):871–883. 10.1016/j.neuron.2010.05.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zhang ZB, Tian MQ, Gao K, et al. De novo KCNMA1 mutations in children with early‐onset paroxysmal dyskinesia and developmental delay. Mov Disord 2015;30(9):1290–1292. 10.1002/mds.26216 [DOI] [PubMed] [Google Scholar]
- 7. Zhang G, Gibson RA, McDonald M, et al. A gain‐of‐function mutation in KCNMA1 causes dystonia spells controlled with stimulant therapy. Mov Disord 2020;35(10):1868–1873. 10.1002/mds.28138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bailey CS, Moldenhauer HJ, Park SM, et al. KCNMA1‐linked channelopathy. J Gen Physiol 2019;151(10):1173–1189. 10.1085/jgp.201912457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hu H, Shao LR, Chavoshy S, et al. Presynaptic Ca2+−activated K+ channels in glutamatergic hippocampal terminals and their role in spike repolarization and regulation of transmitter release. J Neurosci 2001;21(24):9585–9597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bean BP. The action potential in mammalian central neurons. Nat Rev Neurosci 2007;8(6):451–465. 10.1038/nrn2148 [DOI] [PubMed] [Google Scholar]
- 11. Griguoli M, Sgritta M, Cherubini E. Presynaptic BK channels control transmitter release: physiological relevance and potential therapeutic implications. J Physiol 2016;594(13):3489–3500. 10.1113/JP271841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lee US, Cui J. {beta} subunit‐specific modulations of BK channel function by a mutation associated with epilepsy and dyskinesia. J Physiol 2009;587(Pt 7):1481–1498. 10.1113/jphysiol.2009.169243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Xia XM, Zeng X, Lingle CJ. Multiple regulatory sites in large‐conductance calcium‐activated potassium channels. Nature 2002;418(6900):880–884. 10.1038/nature00956 [DOI] [PubMed] [Google Scholar]
- 14. Rong YS, Titen SW, Xie HB, et al. Targeted mutagenesis by homologous recombination in D. melanogaster . Genes Dev 2002;16(12):1568–1581. 10.1101/gad.986602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jepson JE, Shahidullah M, Lamaze A, et al. Dyschronic, a drosophila homolog of a deaf‐blindness gene, regulates circadian output and slowpoke channels. PLoS Genet 2012;8(4):e1002671. 10.1371/journal.pgen.1002671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Shang Y, Griffith LC, Rosbash M. Light‐arousal and circadian photoreception circuits intersect at the large PDF cells of the drosophila brain. Proc Natl Acad Sci U S A 2008;105(50):19587–19594. 10.1073/pnas.0809577105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Tabuchi M, Monaco JD, Duan G, et al. Clock‐generated temporal codes determine synaptic plasticity to control sleep. Cell 2018;175(5):1213–27 e18. 10.1016/j.cell.2018.09.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Harris KP, Littleton JT. Transmission, development, and plasticity of synapses. Genetics 2015;201(2):345–375. 10.1534/genetics.115.176529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Jan LY, Jan YN. Properties of the larval neuromuscular junction in Drosophila melanogaster . J Physiol 1976;262(1):189–214. 10.1113/jphysiol.1976.sp011592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Littleton JT, Stern M, Schulze K, et al. Mutational analysis of drosophila synaptotagmin demonstrates its essential role in Ca(2+)‐activated neurotransmitter release. Cell 1993;74(6):1125–1134. 10.1016/0092-8674(93)90733-7 [DOI] [PubMed] [Google Scholar]
- 21. Hallermann S, Heckmann M, Kittel RJ. Mechanisms of short‐term plasticity at neuromuscular active zones of drosophila. HFSP J 2010;4(2):72–84. 10.2976/1.3338710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kittel RJ, Wichmann C, Rasse TM, et al. Bruchpilot promotes active zone assembly, Ca2+ channel clustering, and vesicle release. Science 2006;312(5776):1051–1054. 10.1126/science.1126308 [DOI] [PubMed] [Google Scholar]
- 23. Ermolyuk YS, Alder FG, Henneberger C, et al. Independent regulation of basal neurotransmitter release efficacy by variable Ca(2)+ influx and Bouton size at small central synapses. PLoS Biol 2012;10(9):e1001396. 10.1371/journal.pbio.1001396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Clark MQ, Zarin AA, Carreira‐Rosario A, et al. Neural circuits driving larval locomotion in drosophila. Neural Dev 2018;13(1):6. 10.1186/s13064-018-0103-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Berni J, Pulver SR, Griffith LC, et al. Autonomous circuitry for substrate exploration in freely moving drosophila larvae. Curr Biol 2012;22(20):1861–1870. 10.1016/j.cub.2012.07.048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chen TW, Wardill TJ, Sun Y, et al. Ultrasensitive fluorescent proteins for imaging neuronal activity. Nature 2013;499(7458):295–300. 10.1038/nature12354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Mahr A, Aberle H. The expression pattern of the drosophila vesicular glutamate transporter: a marker protein for motoneurons and glutamatergic centers in the brain. Gene Expression Patterns 2006;6(3):299–309. 10.1016/j.modgep.2005.07.006 [DOI] [PubMed] [Google Scholar]
- 28. Pulver SR, Bayley TG, Taylor AL, et al. Imaging fictive locomotor patterns in larval drosophila. J Neurophysiol 2015;114(5):2564–2577. 10.1152/jn.00731.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hughes CL, Thomas JB. A sensory feedback circuit coordinates muscle activity in drosophila. Mol Cell Neurosci 2007;35(2):383–396. 10.1016/j.mcn.2007.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Klose MK, Dason JS, Atwood HL, et al. Peptide‐induced modulation of synaptic transmission and escape response in drosophila requires two G‐protein‐coupled receptors. J Neurosci 2010;30(44):14724–14734. 10.1523/JNEUROSCI.3612-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Fox LE, Soll DR, Wu CF. Coordination and modulation of locomotion pattern generators in drosophila larvae: effects of altered biogenic amine levels by the tyramine beta hydroxlyase mutation. J Neurosci 2006;26(5):1486–1498. 10.1523/JNEUROSCI.4749-05.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Imlach WL, Beck ES, Choi BJ, et al. SMN is required for sensory‐motor circuit function in drosophila. Cell 2012;151(2):427–439. 10.1016/j.cell.2012.09.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kadas D, Ryglewski S, Duch C. Transient BK outward current enhances motoneurone firing rates during drosophila larval locomotion. J Physiol 2015;593(22):4871–4888. 10.1113/JP271323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Rao SR, Olechnowicz SWZ, Kratschmer P, et al. Small animal video tracking for activity and path analysis using a novel open‐source multi‐platform application (AnimApp). Sci Rep 2019;9(1):12343. 10.1038/s41598-019-48841-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Faville R, Kottler B, Goodhill GJ, et al. How deeply does your mutant sleep? Probing arousal to better understand sleep defects in drosophila. Sci Rep 2015;5:8454. 10.1038/srep08454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Pfeiffenberger C, Lear BC, Keegan KP, et al. Locomotor activity level monitoring using the drosophila activity monitoring (DAM) system. Cold Spring Harbor Protoc 2010;2010(11):pdb prot5518. 10.1101/pdb.prot5518 [DOI] [PubMed] [Google Scholar]
- 37. Fernandez MP, Chu J, Villella A, et al. Impaired clock output by altered connectivity in the circadian network. Proc Natl Acad Sci U S A 2007;104(13):5650–5655. 10.1073/pnas.0608260104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Koh K, Joiner WJ, Wu MN, et al. Identification of SLEEPLESS, a sleep‐promoting factor. Science 2008;321(5887):372–376. 10.1126/science.1155942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Jepson JE, Shahidullah M, Liu D, et al. Regulation of synaptic development and function by the drosophila PDZ protein Dyschronic. Development 2014;141(23):4548–4557. 10.1242/dev.109538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Moldenhauer HJ, Matychak KK, Meredith AL. Comparative gain‐of‐function effects of the KCNMA1‐N999S mutation on human BK Channel properties. J Neurophysiol 2020;123(2):560–570. 10.1152/jn.00626.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wei A, Solaro C, Lingle C, et al. Calcium sensitivity of BK‐type KCa channels determined by a separable domain. Neuron 1994;13(3):671–681. 10.1016/0896-6273(94)90034-5 [DOI] [PubMed] [Google Scholar]
- 42. Bennett BD, Callaway JC, Wilson CJ. Intrinsic membrane properties underlying spontaneous tonic firing in neostriatal cholinergic interneurons. J Neurosci 2000;20(22):8493–8503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Robitaille R, Garcia ML, Kaczorowski GJ, et al. Functional colocalization of calcium and calcium‐gated potassium channels in control of transmitter release. Neuron 1993;11(4):645–655. 10.1016/0896-6273(93)90076-4 [DOI] [PubMed] [Google Scholar]
- 44. Wang ZW, Saifee O, Nonet ML, et al. SLO‐1 potassium channels control quantal content of neurotransmitter release at the C. elegans neuromuscular junction. Neuron 2001;32(5):867–881. 10.1016/s0896-6273(01)00522-0 [DOI] [PubMed] [Google Scholar]
- 45. Long Z, Xu Q, Miao HH, et al. Thalamocortical dysconnectivity in paroxysmal kinesigenic dyskinesia: combining functional magnetic resonance imaging and diffusion tensor imaging. Mov Disord 2017;32(4):592–600. 10.1002/mds.26905 [DOI] [PubMed] [Google Scholar]
- 46. Zhou B, Chen Q, Zhang Q, et al. Hyperactive putamen in patients with paroxysmal kinesigenic choreoathetosis: a resting‐state functional magnetic resonance imaging study. Mov Disord 2010;25(9):1226–1231. 10.1002/mds.22967 [DOI] [PubMed] [Google Scholar]
- 47. Grillner S, El Manira A. Current principles of motor control, with special reference to vertebrate locomotion. Physiol Rev 2020;100(1):271–320. 10.1152/physrev.00015.2019 [DOI] [PubMed] [Google Scholar]
- 48. Eskow Jaunarajs KL, Bonsi P, Chesselet MF, et al. Striatal cholinergic dysfunction as a unifying theme in the pathophysiology of dystonia. Prog Neurobiol 2015;127–128:91–107. 10.1016/j.pneurobio.2015.02.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Tan GH, Liu YY, Wang L, et al. PRRT2 deficiency induces paroxysmal kinesigenic dyskinesia by regulating synaptic transmission in cerebellum. Cell Res 2018;28(1):90–110. 10.1038/cr.2017.128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Mencacci NE. Bi‐allelic variants in TSPOAP1, encoding the active zone protein RIMBP1, cause autosomal recessive dystonia. https://wwwbiorxivorg/content/101101/20200524086215v1 2020 [DOI] [PMC free article] [PubMed]
- 51. Du X, Carvalho‐de‐Souza JL, Wei C, et al. Loss‐of‐function BK channel mutation causes impaired mitochondria and progressive cerebellar ataxia. Proc Natl Acad Sci U S A 2020;117(11):6023–6034. 10.1073/pnas.1920008117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Liang L, Li X, Moutton S, et al. De novo loss‐of‐function KCNMA1 variants are associated with a new multiple malformation syndrome and a broad spectrum of developmental and neurological phenotypes. Hum Mol Genet 2019;28(17):2937–2951. 10.1093/hmg/ddz117 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1: Supplementary Information
Video S1 Video shows representative examples of waves of motoneuron excitation in the ex vivo larval ventral nerve cord (VNC) visualized via GCaMP6m driven by the glutamatergic neuron driver ok371‐Ga4. In the slo loxP/+ control VNC (left), repetitive waves of excitation traveling from posterior to anterior segments can clearly be observed. Unilateral motoneuron excitation in the upper segments can also be infrequently observed. In the slo E366G/+ VNC (right), both the magnitude and frequency of increases in motoneuron GCaMP6m fluorescence is reduced, indicative of perturbed input from the upstream central pattern generator.
Video S2 Video shows representative examples of three slo loxP/+ (left) or slo E366G/+ (right) adult male flies freely moving in glass tubes containing an agar‐sucrose food source. Tubes are horizontally placed. Note the more rapid speed and increased continuity of movement of slo loxP/+ compared to slo E366G/+ males. Subsequent zoom of the left‐hand slo E366G/+ male illustrates a bout of unilateral leg twitches, similar instances of which were frequently observed in slo E366G/+ but not slo loxP/+ flies.