

Abstract
The use of ex vivo drug sensitivity testing to predict drug activity in individual patients has been actively explored for almost 50 years without delivering a generally useful predictive capability. However, extended failure should not be an indicator of futility. This is especially true in cancer research where ultimate success is often preceded by less successful attempts. For example, both immune- and genetic-based targeted therapies for cancer underwent numerous failed attempts before biological understanding, improved targets, and optimized drug development matured to facilitate an arsenal of transformational drugs. Similarly, the concept of directly assessing drug sensitivity of primary tumor biopsies—and the use of this information to help direct therapeutic approaches—has a long history with a definitive learning curve. In this review, we will survey the history of ex vivo testing as well as the current state of the art for this field. We will present an update on methodologies and approaches, describe the use of these technologies to test cutting-edge drug classes, and describe an increasingly nuanced understanding of tumor types and models for which this strategy is most likely to succeed. We will consider the relative strengths and weaknesses of predicting drug activity across the broad biological context of cancer patients and tumor types. This will include an analysis of the potential for ex vivo drug sensitivity testing to accurately predict drug activity within each of the biological hallmarks of cancer pathogenesis.
Introduction
The use of ex vivo drug sensitivity assays as diagnostic tools for clinical therapeutic decision making has an extensive and, sometimes, checkered history. This has driven some to conclude that the effort is futile, however, more recent approaches are showing substantial promise. Improved methodologies, readouts, and classes of drugs that can be tested have led to encouraging results as well as an expansion of the ways in which this concept can be used. In addition to the idea of using these assays to aid in clinical decision making for individual patients, more recent studies have also leveraged large data sets that integrate ex vivo drug sensitivity data with other data types, such as genomic, transcriptomic, and clinical information, to enable insights into biological mechanisms of drug response and identification of drug sensitivity signals that may have been missed without these agnostic approaches. Additionally, retrospective studies of contemporary classes of drugs tested with newer drug sensitivity assay technologies have repeatedly demonstrated high predictive power for clinical response. In the context of renewed excitement around the field of ex vivo drug sensitivity testing (including the formation of the Society for Functional Precision Medicine, SFPM, https://www.sfpm.io), it is worth revisiting the history of this field to further understand how updated approaches have facilitated improved use of these assays. It is also worth considering the domains of cancer biology and classes of therapeutics that are best suited for this type of testing and the areas where current technologies still may face challenges in accurately predicting drug activity. This can perhaps best be conceptualized by taking into consideration the capacity for current approaches to accurately model each of the biological hallmarks of cancer.
Historical Perspective
The concept of using ex vivo functional testing of primary tumor biopsies to evaluate and prioritize drug sensitivities has existed for at least 50 years. Over the past ~20 years, many have formed an opinion that this approach is not successful, and this has been at least partly driven by working group literature reviews(Burstein et al 2011, Samson et al 2004, Schrag et al 2004). It is worth revisiting these initial studies, the methodologies that were employed, tumor types that were studied, drugs that were evaluated, and the actual performance of the assays in those studies.
Assay readouts
A number of methodologies have been employed for ex vivo drug sensitivity testing. Early studies involved cellular disassociation of tumor biopsies, exposure of unselected cells to a single concentration of each drug for a short period (typically 1 hour), washing of the cells and plating into a semi-solid media. After 2 weeks, cells that expanded to form colonies were manually enumerated with brightfield microscopy(Salmon et al 1978, Von Hoff et al 1983, Von Hoff et al 1991, Von Hoff et al 1990). In other cases, single-cell suspensions from disassociated tumors were placed into culture and efficacy of a small panel of drugs was evaluated by dye exclusion assays and manual enumeration of viable cells in drug-treated versus control conditions(Brower et al 2008, Cortazar et al 1997, Gallion et al 2006, Gazdar et al 1990, Mi et al 2008, Shaw et al 1993, Wilbur et al 1992). Typically, this required expansion of the initial tumor biopsy, which occurred over ~1–12 weeks in culture. Importantly, due to the timing of such cell line expansion assays, clinical implementation of results was not typically executed at the clinical stage of tumor biopsy acquisition, but only at the time of a subsequent disease relapse. In addition, as expanded on below, the drugs tested in these early studies were not targeted to molecular abnormalities of the tumors, hence, would have been less likely to strongly determine response.
A number of variations on these methods have been employed, such as more automated measurements of cell viability through use of ATP content quantification from lysed cells(Cree et al 2007, Kurbacher et al 1998, Ugurel et al 2006) or assessment of cell metabolic activity (MTT) at the assay endpoint(Wu et al 2008, Xu et al 1999) as well as the use of tritiated thymidine to measure proliferative cells in semi-solid media(Joo et al 2009, Kern et al 1985, Kern & Weisenthal 1990, Loizzi et al 2003). Some studies used alternative, in vivo assays such as surgical implantation of tumor pieces into kidneys of immunocompetent CD2F1 mice and excision of tumors after short in vivo treatment courses (~5 days)(Maenpaa et al 1995).
Characterization of Assayed Cells
One major issue for ex vivo drug sensitivity testing has been the characterization of the extent to which tumor cells maintain a variety of biological properties found in the original tumor. The technical capacity to perform detailed validation studies of these ex vivo cells was often limited in earlier studies. In many cases, validation of tumor fidelity was limited to cytologic/morphologic stains (e.g. Hematoxylin/Eosin, Wright-Giemsa, Papanicolaou, peroxidase, dye exclusion, etc.)(Brower et al 2008, Cortazar et al 1997, Gallion et al 2006, Gazdar et al 1990, Mi et al 2008, Shaw et al 1993, Wilbur et al 1992), the capacity for culture surfaces, such as polypropylene(Wilbur et al 1992), or media/suspension formats, such as soft agar to exclude non-tumor cell growth(Joo et al 2009, Kern et al 1985, Kern & Weisenthal 1990, Loizzi et al 2003, Von Hoff et al 1991, Von Hoff et al 1990), and the favoring of tumor versus non-tumor cells for in vivo growth(Maenpaa et al 1995). Initial validation studies had been performed for some assays using microscopy, karyotyping, cytologic stains, or clonogenicity of cells in immunocompromised mice (summarized in(Von Hoff et al 1983)). However, most of these readouts would not be considered sufficient by current standards to validate tumor fidelity, and we now know that many of these validation techniques do not truly distinguish tumor from non-tumor cells. Often, these studies were carried out only for initial assay development, but not performed for the cases being tested for actual clinical decision-making (or only for a small fraction, such as 5% of cases(Von Hoff et al 1983)). Clearly, as this field has moved forward, the utilization of technological improvements in cellular and molecular biology to facilitate rigorous and high-throughput validation of ex vivo tumor cell biology—from the standpoint of genetics, cell state, and heterogeneity of cell populations—has been important.
Tumor Types
Initial studies in the field of ex vivo drug sensitivity testing were confined to a relatively small number of solid tumor types – primarily breast(Brower et al 2008, Kern et al 1985, Kern & Weisenthal 1990, Mi et al 2008, Von Hoff et al 1983, Von Hoff et al 1990, Xu et al 1999), ovarian(Cree et al 2007, Gallion et al 2006, Joo et al 2009, Kurbacher et al 1998, Loizzi et al 2003, Maenpaa et al 1995, Salmon et al 1978, Von Hoff et al 1983, Von Hoff et al 1991, Von Hoff et al 1990), lung (non-small cell and small cell)(Cortazar et al 1997, Gazdar et al 1990, Kern et al 1985, Kern & Weisenthal 1990, Shaw et al 1993, Von Hoff et al 1983, Von Hoff et al 1990, Wilbur et al 1992), melanoma(Kern et al 1985, Kern & Weisenthal 1990, Ugurel et al 2006, Von Hoff et al 1983, Von Hoff et al 1990), and colorectal(Kern et al 1985, Kern & Weisenthal 1990, Von Hoff et al 1983, Von Hoff et al 1990, Wu et al 2008) cancer, with a small number of cases of other tumor types. Many tumor types were not tested in significant numbers in any of these studies due to a lack of success of long-term culture of these cells. This included most subsets of hematologic malignancy, although some of the earliest studies did focus on multiple myeloma(Salmon et al 1978). Since these initial studies, it has become clear that successful culture of malignant cells that maintain fidelity to the original tumor requires substantially different approaches and culture conditions from one tumor lineage to another. In addition, revised protocols favoring shorter term assays that do not involve expansion of tumor cells, media additives that ensure better maintenance of tumor cell biology, and strategies that enable retention or add back of microenvironmental tumor features, have facilitated new assay formats and enabled inclusion of previously excluded tumor types.
Furthermore, many studies tested only metastatic lesions and, in these instances, the studies typically tested a single metastatic lesion. In the intervening period since the performance of these initial studies, we have developed a much clearer understanding of the clonal evolution of tumors between primary and metastatic sites, and we also now have a fuller understanding of the biological divergence that occurs between distinct metastatic sites. With this knowledge in mind, it is probably unsurprising that an ex vivo drug sensitivity test on a single metastatic lesion was not always predictive for overall response of the patient.
Drugs Tested and Definitions of Drug Activity
Importantly, the drugs that were tested in these early studies were relatively few in number and were restricted to classes of chemotherapy drugs with broad toxicity profiles. Drug classes tested included RNA polymerase inhibitors, alkylating agents, DNA-damaging agents, topoisomerase inhibitors, antimetabolites, and microtubule-disruptive drugs. These drugs rarely elicit durable responses in patients with advanced disease (the populations being tested in these studies), and the window of response between most sensitive and least sensitive cases was narrow, creating difficulties in distinguishing and clinically correlating exceptional responses. Many newer classes of agents were not yet available for testing at the time of these studies, and as will be covered later in this review, it is becoming clear that not all drug classes can be equally assessed using ex vivo drug sensitivity testing – indeed the format for the assay must be considered in the context of the mechanism of action of the drug (discussed below in greater detail).
The definition for activity of a drug is also an important consideration. One approach involved assays that focused on extreme drug resistance (EDR) to identify therapeutic strategies unlikely to provide clinical benefit(Eltabbakh et al 1998, Joo et al 2009, Kern 1998, Mehta et al 2001, Nagourney 2000, Soslow et al 2000). These EDR assays have seen extensive clinical use and include formats that have been commercialized. However, most studies focused on identification of sensitive cases and in many initial studies, the definition of an active drug was a reduction of 50% tumor cell growth compared with control conditions. Although some studies attempted to rank order drug activity, many studies considered drugs to be active once they surpassed this 50% threshold. This presents one key question that still confronts the field today – how best to interpret ex vivo drug activity in a way that will most accurately translate into clinical activity.
Reported Activity
In spite of all of the technical challenges faced by early iterations of ex vivo drug sensitivity testing, there were signals in most studies that response rates were significantly improved in the drug sensitivity-guided arm. Improvement in response to drug treatment was typically transient, which most likely represented the best possible outcome for many of the cases tested for the given drug regimens utilized. However, these results pointed to an initial promising concept that has since been improved with more advanced readouts, the advent of newer drug classes, greater rigor in characterizing cellular inputs and outputs, and careful consideration of the biology being modeled.
In this respect, it is worth revisiting the potential of ex vivo drug sensitivity testing. Early conclusions that these approaches were not yet suitable for wide-scale implementation in their current state was certainly warranted. However, conjecture that these approaches cannot work seems premature, especially given the technical advances seen in current approaches that have facilitated improvements in both rigor and biological knowledge.
The Present: Current State of Assay Development
Current approaches to ex vivo testing of drug sensitivity have evolved to include numerous formats across different tumor types. These more advanced platforms harness the use of distinct media formulations, testing in two- as well as three-dimensions, inclusion of multiple cell types, various readouts for measuring phenotypic responses in aggregate cell populations as well as in single-cells, measurements of drug impact on specific cellular pathways and processes, and in vivo approaches. A review in 2015(Friedman et al 2015) provided an excellent overview of many of these techniques. Here, we provide an updated view and consider the relative merits of these approaches for accurately assessing drug activity within each biological hallmark of cancer. This will include discussion of the path forward for implementation of and improvements to the field of ex vivo drug sensitivity testing.
Direct Phenotypic Measurements
Many different approaches have built upon the early strategies described above, and have made important changes to assay format and readout. At a basic level, there are still similarities in approach – tumor biopsies (liquid or solid) are obtained from patients, and cells are exposed to drugs with readout of functional impact of drugs on cell phenotype. However, many efforts have been made to shorten the time to assay completion which enables testing on non-expanded cells, thereby eliminating the chance for tumor cells to drift and/or non-tumor cells to overtake the culture. Furthermore, additional updating of culturing conditions and implementation of cutting-edge engineering technologies have also helped to better mimic the native tumor microenvironment.
Two Dimensions
Two-dimensional cultures have taken on numerous formats over the past 10–15 years. These have included strategies for both hematologic and solid tumors and formats have diverged between the tumor types. For hematologic malignancies, two-dimensional liquid cultures have most commonly been performed in “basic” media conditions that forgo most media supplements in favor of minimal essential components to maintain cell viability, either in the form of fetal bovine serum(Tyner et al 2013) or minimal mixtures of recombinant protein supplements(Pemovska et al 2013). This has been done in an attempt to protect the cell’s phenotypic responses from undue influence from external stimuli and preserve drug responses that are most determined by cellular intrinsic features of the tumor.
Alternative methods have incorporated exogenous growth factors to maintain higher cell viability and/or recapitulate certain elements of the tumor microenvironment over the course of short-term culture. These factors are sometimes derived from conditioned media of stromal cells or from recombinant proteins. In certain cases, rigorous comparisons have been made between these different media conditions. Results have shown that careful selection of media conditions can improve efficiency of successful culture for certain cancer types, such as ovarian tumors, as well as retaining the molecular and histopathologic landscape of the original tumor in long-term culture(Ince et al 2015). Other studies have used this approach to model the impact on drug response of a matrix of microenviornmental factors ((Watson et al 2018) and other similar examples described in more detail below). In studies of hematologic cancers that have tested different media compositions, data have indicated that many classes of drugs exhibited similar results between the different conditions, however, substantial differences in activity were seen with certain drug classes with some drug families, such as topoisomerase inhibitors, BCL2 inhibitors, and many kinase inhibitors, that exhibited better tumor cell killing in basic conditions versus JAK kinase inhibitors, that were more active in supplemented conditions(Karjalainen et al 2017). Changes in basic biology, such as changes in BCL2 family member usage and dependence were also observed from one media condition to another. These findings point to a very important concept for the field of ex vivo drug sensitivity testing – different classes of drugs may require customized strategies for testing to ensure results that are most indicative of in vivo biology and predictive of clinical response, and emphasize the need to capture the related in vivo biology and patient response.
Alternative media supplements have also been shown to promote the length of time that primary leukemia cells can be maintained ex vivo without undergoing spontaneous differentiation. Indeed, the functional determinants of hematopoietic stem cell (HSC) self-renewal play an important role in ex vivo culturing and screening of primary hematopoietic cells. Key regulators of HSC self-renewal, such as JARID1B, JHDMLF, MSI2, and PROX1, influence hematopoietic stem cell renewal in both mouse and human ex vivo model systems(Cellot et al 2013, Deneault et al 2009, Hope et al 2010). A number of studies have demonstrated that antagonists of aryl hydrocarbon receptors (AHR) such as SR1-4(Boitano et al 2010, Bouchez et al 2011) and agonists of NOTCH ligand(Delaney et al 2010) have the ability to support expansion of human long-term HSCs in vivo and in vitro. High-throughput chemical screens helped identify small molecules, such as UM171 and UM729, with the ability to synergize with AHR antagonists in inhibiting leukemia stem cell (LSC) differentiation and supporting LSC activity in vitro(Fares et al 2014, Pabst et al 2014). The mechanism of action of UM171 is thought to be through effects on pro- and anti-inflammatory and detoxification networks. These networks mediate self-renewal and support hematopoietic stem cell (HSC) expansion, which are controlled by NFKB activation and protein C receptor (PROCR)-dependent reactive oxygen species (ROS) detoxification(Chagraoui et al 2019). Using a shRNA library screen, MacPherson and colleagues identified members of the HBO1 protein complex that were also critical regulators of LSC maintenance(MacPherson et al 2020). They developed a highly potent small molecule inhibitor of HBO1 and demonstrated that the compound was a competitive analogue of acetyl-CoA.
For solid tumors, different media conditions have been determined to facilitate expansion of solid tumor cells in a way that maintains good fidelity of both genetic and cell state. This strategy has been termed “conditional reprogramming” and involves co-culture of tumor biopsy cells on a feeder layer of irradiated fibroblasts as well as the addition of small-molecule inhibitors of RHO kinase (ROCK). Recent iterations of this demonstrate that some tumor types don’t require the fibroblast feeder layer and that the key to continued proliferation is the ROCK inhibitor. These updated media conditions, combined with miniaturization strategies have facilitated limited expansion of primary solid tumor cells that remain comparable to the original tumor and can also enable testing of much larger panels of agents, including the newest classes of drugs. Importantly, patient cells propagated with these growth conditions have been validated against original tumor tissue by short tandem repeat profiling, comparative genomic hybridization, and spectral karyotyping(Liu et al 2017, Liu et al 2012, Suprynowicz et al 2012, Yuan et al 2012). Additional approaches not dependent on a ROCK inhibitor have been identified for a number of tumor lineages that also retain tumor fidelity. Indeed, similar to hematologic malignancies, there is an expanding literature base on diverse culture conditions to propagate a variety of solid tumor types (for example(Ince et al 2015)), indicating a need to evaluate the strategies that are most successful at recapitulating the in vivo tumor biology and clinical response. Strategies that can retain or enable adding back of microenvironmental features of in vivo tumor biology might be expected to exhibit the best predictive power.
In addition to the use of two-dimensional cultures as a tool to understand drug sensitivity at the level of individual patient samples and drugs, many studies have taken a population perspective to understand the patterns of sensitivity across broad panels of drugs and the manner by which these sensitivity patterns correlate with genetic, transcriptomic, and clinical features in hematologic malignancies(Dietrich et al 2018, Frismantas et al 2017, Lee et al 2018, Pemovska et al 2013, Tyner et al 2018, Tyner et al 2013). Studies of a similar nature in solid tumors have also incorporated a combination of two-dimensional, three-dimensional, and in vivo patient-derived models to integrate with genomics across a variety of solid tumor types(Brodin et al 2019, Friedman et al 2017, Pauli et al 2017, Saeed et al 2019, Saeed et al 2017), and in some cases sensitivity to RNAi-mediated gene knockdown has been included in the analyses to help understand and prioritize drug sensitivity patterns(Moser et al 2014, Tyner et al 2009, Xu et al 2018). Analytical tools to assist with such studies, such as the open source Breeze platform for quality control and analysis of high-throughput drug sensitivity data, will assuredly help with these large-scale analyses in future studies(Potdar et al 2020).
Three-Dimensional Semi-Solid Cultures
In 1966, Bradley and Metcalf developed the first ex vivo assay performed on murine hematopoietic cell isolates which was able to assess the number of the colony forming units (CFU) or colony forming cells (CFC) in a given sample. This clonogenic assay, which measures the differentiation and maturation of CFCs helped to characterize the granulocyte-macrophage lineage (GM-CFU) and also identified a key growth factor called colony stimulating factor (CSF)(Bradley & Metcalf 1966). Pike and Robinson were able to adopt the assay for growing human bone marrow cell colonies in agar‐gel medium(Pike & Robinson 1970) and Norman Iscove developed a second-generation technique that utilized methylcellulose(Iscove et al 1974) with or without supplemental growth factors. This assay is still used extensively today, and the development of these ex vivo bone marrow colony formation assays has been instrumental for understanding normal hematopoiesis as well as for assessment of leukemia cell clonogenic potential. While colony formation assays have been used to assess the impact of drug exposure on leukemia cell growth and clonogenicity, this technique has not readily been transformed into a high throughput method for drug sensitivity assessment.
A conceptually related technique for expansion and testing of solid tumor cells in semi-solid media – organoids – has been broadly used to expand solid tumor cells ex vivo for many different experimental purposes, including drug sensitivity evaluation. Following dissociation of primary biopsies, cells are plated into polymerized Matrigel with a variety of recombinant and small-molecule additives, including agonists of EGF and WNT signaling and inhibitors of TGFB and ROCK pathways(Sato et al 2011, Sato et al 2009). Alternative approaches have included culture of cells in collagen matrices that incorporate an air-liquid interface with some of the same culture additives, such as WNT pathway agonists(Ootani et al 2009). Heterogeneous cell mixtures, such as tumor cells with fibroblasts have been tested in this air-liquid interface system to show changes on drug sensitivity with cell co-cultures compared with tumor-only organoids(Prina-Mello et al 2018). Organoid technology has also been miniaturized, taking advantage of a simplified geometry that seeds cells around the rim of the wells (mini-rings), and this has facilitated more automated, high-throughput screening of larger collections of compounds in a shorter, more clinically-relevant timeframe(Phan et al 2019). Organoid protocols, with numerous modifications have been used to study normal developmental biology of numerous tissue types, to generate genetically engineered in vitro models that recapitulate common genetic lesions, such as mutation of KRAS or loss of TP53, APC, and SMAD4, and to establish organoid cultures from primary tumors derived from the head and neck, pancreas, breast, colorectal, and rectal regions. Recent reviews of the organoid field have summarized these advances (Clevers & Tuveson 2019, Neal & Kuo 2016, Tuveson & Clevers 2019).
Some of the studies that derived organoids from primary tumors have provided evidence that non-tumor cell types, such as tumor-infiltrating immune cells and cancer-associated fibroblasts, can be maintained within the expanded organoids. These multi-cellular models are able to recapitulate certain elements of the tumor microenvironment and can facilitate studying the impact of perturbations on the tumor microenvironment as well as the tumor cells(Neal et al 2018). These findings have been mirrored in some of the hematopoietic primary culture systems, where non-tumor cells such as macrophages and T-cells have been shown to play an important influencing role in drug responses and have facilitated the ex vivo analysis of additional classes of agents, such as those that act on immune cells(Carey et al 2017, Edwards et al 2019, Edwards et al 2018, Kuusanmaki et al 2020, Majumder et al 2019). A related approach leveraged microarray technology to create a matrix of pairwise combinations of recombinant extracellular matrix proteins with soluble growth factors and cytokines(Smith et al 2019) and showed some of these factors could influence breast cancer cell response to tyrosine kinase inhibitors in a disease subset specific way(Watson et al 2018). While these findings have collectively opened up a new avenue of research for ex vivo tumor models, they have also created a need for updated readouts that incorporate single-cell granularity and multi-parameter phenotypes. This increase in sophistication of readouts will facilitate both the distinguishing of tumor from non-tumor cells in scoring response, and will also allow for scoring of desirable phenotypic impacts on the non-tumor cells (e.g. promotion of T-cell activation states). Furthermore, as with two-dimensional culture, different formulations of media components are under investigation to determine if additional phenotypic states and tumor cell heterogeneity reflective of the parent tumor can be obtained in organoid cultures.
Induced pluripotent stem cells (iPSC) have also been used in conjunction with organoid techniques to produce disease models from cancers such as colorectal carcinoma that exhibit organoid architecture, cellular composition, and pathway activation states that maintain high fidelity to the biology of the in vivo tumor state(Crespo et al 2017). A variety of other tissue types have been used in conjunction with iPSC technology to derive high fidelity models (reviewed in(Papapetrou 2016)), and these techniques promise to offer robust model systems for future ex vivo cancer drug screening.
3D Bio-printed models and Organotypic Cultures
In some of the most recent strategies, three-dimensional bio-printing technologies have been integrated with primary cell and organoid techniques to add a variety of biologically important features to primary patient ex vivo models. These approaches have enabled the creation of spatial architecture and physical properties, such as fluid flow, that resemble the native tumor microenvironment. Testing has been performed on media, or bioink, with varying levels of components such as gelatin and alginate to show that different bioink formulations can impact on bio-printed tissue phenotypes(Jiang et al 2019).
Additional components, such as laminin/peptide additives to matrigel(Schmidt et al 2019), as well as infiltrating cell types, such as fibroblasts(Amann et al 2014), have been incorporated into semi-solid matrices to interact with tumor cells in a way that mimics the in vivo biology of the tumor. These approaches have been used to create ex vivo niches that resemble tumor microenvironments. In some cases, these strategies have integrated cellular components from multiple organs, such as liver, heart, and lung(Skardal et al 2017). The bone marrow microenvironment of hematologic malignancies has also been modeled in three-dimensional scaffolds made of components such as fibrin gels or zirconium oxide and containing purified cells such as mesenchymal stromal cells and/or hematopoietic stem progenitor cells that were shown to preserve a population of HSPCs giving rise to multi-lineage differentiated blood cells(Chou et al 2020, Sieber et al 2018). In one recent study, a multi-cellular lung organoid that exhibited air sac structures, production of lung surfactant protein, and angiogenesis was used in a multi-cellular organoid platform to mimic lung colonization of tumor cells, and drugs that impact on metastasis and angiogenesis were successfully tested in this assay(Ramamoorthy et al 2019). Another study demonstrated scaffold-free bio-printing with defined architecture to model tumor-stromal interactions, including multiple stromal cell types, native ECM deposition, and self-organized vasculature, which allowed evaluation of tumor phenotypes including proliferation, signaling and migration in response to extrinsic signals and therapies(Langer et al 2019). Reviews of these three-dimensional bio-printing techniques have been recently published(Ashok et al 2020, Brancato et al 2020, Fan et al 2019, Schneeberger et al 2017).
Related approaches have also been developed to more closely model the in vivo tumor microenvironment by culturing thin slices of tumor biopsies in their native state (organotypic cultures), which have enabled retention of native states of cellular heterogeneity, spatial architecture, and have shown high rates of predictivity of clinical responses(Hirt et al 2014, Kenny et al 2015, Majumder et al 2015, Nagourney et al 2012, Ridky et al 2010, Vaira et al 2010).
Evolving Readouts
Over the history of ex vivo drug sensitivity testing, readouts have evolved dramatically. As described above, initial strategies required laborious and subjective manual quantification under a microscope, which later gave way to automated strategies that measure thymidine incorporation, ATP content, or metabolic activity of cells. In recent years, new instrumentation and biological tools have facilitated dramatic updates to ex vivo readouts. These include the incorporation of high-throughput flow cytometry(Kuusanmaki et al 2020, Majumder et al 2019, Teh et al 2020) and imaging analysis(Jacob et al 2016), both of which enable quantification of specific phenotypes on a single-cell level. These new techniques can then be combined with the multitude of high-quality antibodies that are now available to organize cells into discrete biological populations as well as antibodies that can accurately distinguish between specific cell states (proliferation, death, activation) and/or measure drug impact on specific pathways (signaling, apoptosis, etc.). Together they have enabled the identification of cell sub-populations that demonstrate pathway resistance to drug exposure and have facilitated important discoveries, such as the manner by which certain drugs and drug classes act preferentially on cells from specific lineages (both healthy and malignant), while sparing others that can give rise to resistance and relapse. These assays also offer the possibility of assessing the effects of specific interactions (e.g. tumor-fibroblast or tumor-immune) on drug response.
Single cell analyses have also been applied in a number of formats involving microfluidic chips measuring a variety of readouts including buoyant mass of cells. These techniques can reveal single-cell heterogeneity of bulk tumor cells and can be used as a readout of viability of single cells from tumors (bulk hematopoietic tumors or circulating tumor cells (CTSs) from solid tumors) after exposure to therapeutic agents. Modified media and culturing conditions, such as suspension culture to mimic the biology of circulating cells, has been shown to improve readouts from CTCs. Other groups have also harvested circulating tumor cells (CTCs) from patient blood and established cultures of these CTCs that have enabled drug sensitivity assessments(Brouzes et al 2009, Stevens et al 2016, Yu et al 2014).
Other new technologies have also facilitated the direct measurement of pathway states, which have included pathways critical to apoptosis. One prominent method that has been used in this way has been BH3 profiling, which measures the propensity of cells to undergo apoptosis in a manner that distinguishes dependence on specific BCL2 family members(Certo et al 2006, Deng et al 2007, Letai 2008). This technique can be done for cells in a static or dynamic state(Montero & Letai 2016, Montero et al 2015) and has been modified to be performed in aggregate with plate-reader technology or in single cells with flow cytometry(Ryan & Letai 2013). BH3 profiling has been used to successfully predict response to a wide variety of cancer therapeutics and tumor types, and it was a pivotal technique for the successful development and implementation of inhibitors of BCL2 family members for lymphoid and myeloid leukemias(Del Gaizo Moore et al 2007, Deng et al 2007, Konopleva et al 2016, Pan et al 2014, Touzeau et al 2016).
In vivo models
A variety of in vivo models have also been utilized to functionally assess drug response in a way that will lead to new biological understanding and clinically predictive therapeutic strategies. One approach has been to utilize immunocompromised(Hudson et al 1998) mice as a vessel to expand primary tumor cells and interrogate response to in vivo dosing (reviewed in(Hidalgo et al 2014, Lai et al 2017, Meyerrose et al 2003, Shultz et al 2007)). A variety of mouse strains now exist that are immuno-deficient due to specific genetic mechanisms, leading to a range of immune compromised states. In addition, some of these strains have been engineered to express humanized components of the tumor microenvironment, such as expression of specific human growth factors and cytokines that help support engraftment and growth of specific cellular lineages(Feuring-Buske et al 2003, Ito et al 2002). Injection of tumor cells into these mice has also been done in varying formats, ranging from sub-cutaneous, intravenous or intrafemoral injections for hematologic malignancies or to model bone metastases(Wu et al 1998), injection into sub-cutaneous ossicles with humanized bone marrow microenvironment(Reinisch et al 2016), and orthotopic injections (reviewed in(Hoffman 2015)). Cumulatively, these different mouse strains and injection protocols have enabled patient-derived xenografts (PDXs) to model a wide variety of tumor types and a multitude of different drug classes and drug combinations have been tested using this approach. However, the throughput of using patient-derived xenograft models for assessment of drug sensitivity is far less than any of the ex vivo approaches described above and some studies have used these high-fidelity models to support expanded screening ex vivo, thereby, revealing important drug sensitivity vulnerabilities in disease of high unmet medical need, such as TCF3-HLF rearranged acute lymphoblastic leukemia(Fischer et al 2015). Further not all tumors adapt to in vivo growth in murine models. Indeed, the ability to grow as PDXs correlates with tumor aggressiveness. Unfortunately, due to the aggressiveness of the tumors many patients are deceased prior to being able to perform predictive drug screens(McAuliffe et al 2015).
In an effort to increase the throughput of in vivo drug assessment testing, and also to perform such testing in tumor native environments, alternative approaches have been developed to inject capsules containing segregated libraries of drugs directly into primary tumors in immune competent or compromised mouse models. Micro-dosing technology limits the spread of each drug to the immediate local region, which can be measured along with assessment of drug impact on tumor cell phenotypes upon resection of the tumor and immuno-staining of the region surrounding the injected capsule(Jonas et al 2015, Klinghoffer et al 2015). This technology has been successfully demonstrated in primary tumors in mice, and has been shown to reveal tumor adaptation(Jonas et al 2016) and impact on immune microenvironment(Frazier et al 2017). Clinical trials in humans are currently ongoing.
Hallmarks of Ex Vivo Drug Testing – Future Directions
Hanahan, Weinberg, and Coussens contributed foundational thought pieces around essential biological hallmarks of cancer(Hanahan & Coussens 2012, Hanahan & Weinberg 2011). These biological driver categories provide a useful framework from which to consider the various formats of ex vivo drug sensitivity testing, and the strategies that are best suited to evaluate drug sensitivity within each hallmark (Figure 1). It is useful to group these ten hallmarks into four categories based on the feasibility of assessing activity of drugs targeting these processes by various ex vivo platforms. In particular, it is key to note that capturing of some of these hallmarks involve the measurement of tumor cell phenotypes, while others depend on heterogenous cell culture systems with phenotypic measurement of non-tumor cells, such as stromal cells and/or immune cells. Strategies that can accurately model both tumor intrinsic and diverse aspects of microenvironmental biology will offer powerful tools for understanding complex interactions between drugs that target these various aspects of tumor biology.
Figure 1.
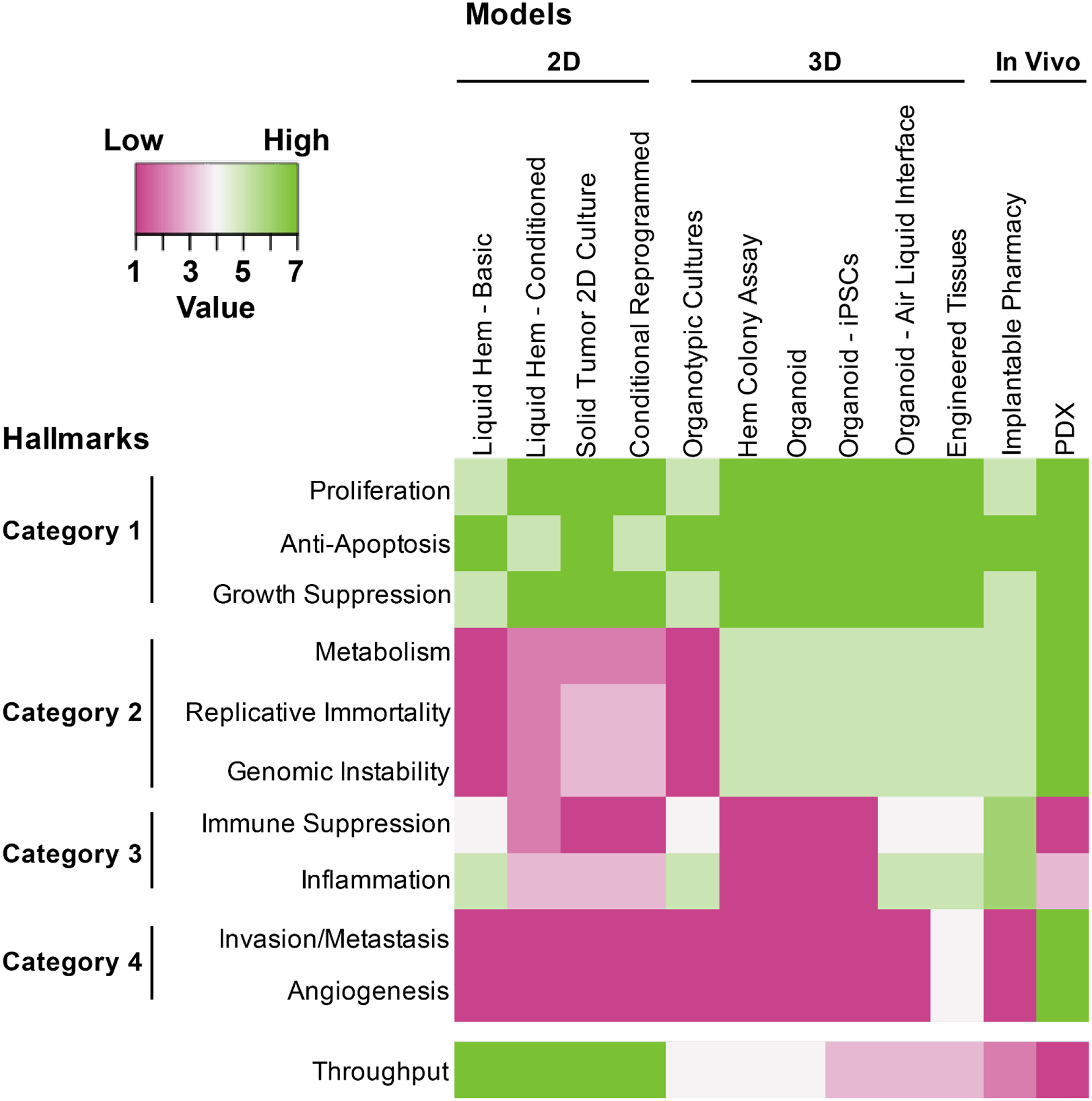
The 10 hallmarks of cancer are organized into 4 categories based on feasibility of various ex vivo platforms to accurately assess sensitivity to drugs targeting those hallmarks. In Category 1, most platforms are highly capable of directly assessing cell proliferation and apoptosis or, at least, assessing biomarkers of these phenotypic states. The Category 2 hallmarks of metabolism, replicative immortality, and genomic instability are better modeled with approaches that enable robust cell proliferation and/or that can readout phenotypes beyond short term cultures. Hallmarks in Category 3 can be best modeled with assays that include heterogeneous cell mixtures that are derived from or mimic the native tumor state. In this way, some of the basic models may actually perform better than models with greater perturbations on the tumor biopsy, however, these heterogeneous cell mixtures may be added back with some of the sophisticated engineered models. Finally, Category 4 hallmarks may be the most challenging, but can be modeled using bioengineered 3-dimensional approaches or patient-derived xenograft models. In general, the 3-dimensional and in vivo approaches are able to accurately model a larger number of hallmarks, though it is worth noting that these strategies currently do not have the throughput of the more basic approaches.
Category 1: (1) Proliferation, (2) Anti-Apoptosis, & (3) Growth Suppression
Drugs targeting cellular proliferation (e.g. tyrosine kinase inhibitors), pathways that regulate apoptosis (e.g. BCL2 family inhibitors), and checkpoints on cell division (e.g. cyclin-dependent kinase inhibitors) have been shown to read out effectively in all formats for ex vivo drug sensitivity screening. Since these drugs target the cellular phenotypes that are most directly assessed with techniques that measure relative numbers of viable cells and there are also accurate biomarkers for these pathways and cellular processes, these hallmarks can dependably be measured using ex vivo drug sensitivity testing. Indeed, there are many examples of ex vivo platforms that have accurately measured sensitivity of these classes of drugs within clinical or genetic subsets with known response patterns, in retrospective correlative studies, and in growing numbers of prospective case studies. It is worth noting that pathways governing cell growth and death can be modulated by cell extrinsic signals from the microenvironment. In this respect, some of the approaches summarized below in category 3 may also be useful for ensuring the most accurate measurements of these category 1 hallmarks.
Category 2: (4) Metabolism, (5) Replicative Immortality & (6) Genomic Instability
The hallmarks of metabolism, replicative immortality, and genomic instability can be targeted by drugs, which impact directly on phenotypes of tumor cells that can be directly measured. However, the specific phenotypes are often not as easily or quickly measured through quantification of viable cells. In this way, these three hallmarks are also well assessed by ex vivo drug sensitivity assays, though, the readouts that are needed to accurately quantify drug activity may be more nuanced and, in some cases, may require biomarkers that are tailored to the specific hallmark (or specific drug). For example, metabolic inhibitors may induce differentiation rather than cell death, requiring markers of cell maturation state and assays to measure metabolic states or directly measure metabolites could be very useful for this hallmark and drug class. Drugs that target replicative immortality or genomic instability may be measured by cell death/viability more easily, though the time-frame to this endpoint may require culturing methods that can maintain cells in a proliferative state for longer periods of time.
Category 3: (7) Immune Suppression & (8) Inflammation
The assessment of immune suppression or inflammation in primary tumors has been attempted in several ex vivo assays. Clearly, the assays must retain a heterogeneity of cells for these platforms to be effective and, in many cases, the readouts will need to be uniquely tailored to the mode of action of the drugs (e.g. discrete measurements of T-cell activity for immune checkpoint inhibitors; assessment of secreted or cell contact-mediated factors from cancer-associated fibroblasts, etc.). Immune, inflammatory, and stromal cells are frequently lost or diluted through sequential passage making early ex vivo analysis of these processes important, or necessitating a means by which immune or other stromal cells can be re-introduced into ex vivo models, such as with bio-printed tissues.
Category 4: (9) Invasion/Metastasis & (10) Angiogenesis
The hallmarks of invasion/metastasis and angiogenesis are probably the most challenging to readout for ex vivo drug sensitivity assays, and most platforms are probably not capable of accurately assessing these facets of tumor biology, although recent studies of engineered tissues have made great progress in modeling these facets of tumor biology(Langer et al 2019, Ramamoorthy et al 2019). Clearly, these hallmarks can be measured using some of the in vivo models touched on in this review, such as patient-derived xenograft or other mouse models. Much exciting work remains to model these hallmarks using some of the more advanced, engineered three-dimensional assay technologies.
Acknowledgements
The Knight Cancer Institute at Oregon Health & Science University is supported by a Cancer Center Support Grant from the National Cancer Institute (3P30CA069533). R.C.S. is supported by the National Cancer Institute (R01 grants CA196228 and CA186241), U01 CA224012, and U54 CA209988. G.B.M. received support from the National Cancer Institute P50 CA217685, P50 CA098258, UO1 CA 2197842, SAC11052 from the Susan G. Komen Breast Cancer Foundation, BCRF-19-110 from the Breast Cancer Research Foundation, 545152 from the Ovarian Cancer Research Alliance, and a kind gift from the Sheldon and Miriam Adelson Medical Research Foundation. J.W.G. received grants from the Susan G Komen Foundation (SAC190012), the Murdock Charitable Trust, the Prospect Creek Foundation, the Brenden-Colson Center for Pancreatic Care, the National Institutes of Health (U54 HG008100) and the National Cancer Institute (U2C CA233280, U54 CA209988, U01 CA195469, U01 CA195469, R44CA224994). J.W.T. received grants from the Mark Foundation for Cancer Research, the Silver Family Foundation, the V Foundation for Cancer Research, the Gabrielle’s Angel Foundation for Cancer Research, and the National Cancer Institute (1R01CA183947, 1U01CA217862, 1U54CA224019).
Disclosure Statement
G.B.M. is SAB member or Consultant with AstraZeneca, Chrysallis Biotechnology, ImmunoMET, Ionis, Lilly, PDX Pharmaceuticals, Signalchem Lifesciences, Symphogen, Tarveda, has stock options with Catena Pharmaceuticals, ImmunoMet, SignalChem, Tarveda has licensed technology to Myriad Genetics and Nanostring, and has sponsored research with Nanostring and Ionis. J.W.G. received research support from, Danaher/Cepheid, PDX Pharmaceuticals and Thermo Fisher Scientific (formerly FEI), Zeiss, Quantitative Imaging and Micron. He has licensed technology to Abbott Diagnostics, and he has ownership positions in PDX Pharmaceuticals, KromaTid, and Convergent Genomics. J.W.T. has received research support from Agios, Aptose, Array, AstraZeneca, Constellation, Genentech, Gilead, Incyte, Janssen, Petra, Seattle Genetics, Syros, and Takeda.
References
- Amann A, Zwierzina M, Gamerith G, Bitsche M, Huber JM, et al. 2014. Development of an innovative 3D cell culture system to study tumour--stroma interactions in non-small cell lung cancer cells. PLoS One 9(3): e92511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashok A, Choudhury D, Fang Y, Hunziker W. 2020. Towards manufacturing of human organoids. Biotechnol Adv 39(107460. [DOI] [PubMed] [Google Scholar]
- Boitano AE, Wang J, Romeo R, Bouchez LC, Parker AE, et al. 2010. Aryl hydrocarbon receptor antagonists promote the expansion of human hematopoietic stem cells. Science 329(5997): 1345–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouchez LC, Boitano AE, de Lichtervelde L, Romeo R, Cooke MP, Schultz PG. 2011. Small-molecule regulators of human stem cell self-renewal. Chembiochem 12(6): 854–7 [DOI] [PubMed] [Google Scholar]
- Bradley TR, Metcalf D. 1966. The growth of mouse bone marrow cells in vitro. Aust J Exp Biol Med Sci 44(3): 287–99 [DOI] [PubMed] [Google Scholar]
- Brancato V, Oliveira JM, Correlo VM, Reis RL, Kundu SC. 2020. Could 3D models of cancer enhance drug screening? Biomaterials 232(119744. [DOI] [PubMed] [Google Scholar]
- Brodin BA, Wennerberg K, Lidbrink E, Brosjo O, Potdar S, et al. 2019. Drug sensitivity testing on patient-derived sarcoma cells predicts patient response to treatment and identifies c-Sarc inhibitors as active drugs for translocation sarcomas. Br J Cancer 120(4): 435–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brouzes E, Medkova M, Savenelli N, Marran D, Twardowski M, et al. 2009. Droplet microfluidic technology for single-cell high-throughput screening. Proc Natl Acad Sci U S A 106(34): 14195–200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brower SL, Fensterer JE, Bush JE. 2008. The ChemoFx assay: an ex vivo chemosensitivity and resistance assay for predicting patient response to cancer chemotherapy. Methods Mol Biol 414(57–78 [DOI] [PubMed] [Google Scholar]
- Burstein HJ, Mangu PB, Somerfield MR, Schrag D, Samson D, et al. 2011. American Society of Clinical Oncology clinical practice guideline update on the use of chemotherapy sensitivity and resistance assays. J Clin Oncol 29(24): 3328–30 [DOI] [PubMed] [Google Scholar]
- Carey A, Edwards DKt, Eide CA, Newell L, Traer E, et al. 2017. Identification of Interleukin-1 by Functional Screening as a Key Mediator of Cellular Expansion and Disease Progression in Acute Myeloid Leukemia. Cell reports 18(13): 3204–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cellot S, Hope KJ, Chagraoui J, Sauvageau M, Deneault E, et al. 2013. RNAi screen identifies Jarid1b as a major regulator of mouse HSC activity. Blood 122(9): 1545–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Certo M, Del Gaizo Moore V, Nishino M, Wei G, Korsmeyer S, et al. 2006. Mitochondria primed by death signals determine cellular addiction to antiapoptotic BCL-2 family members. Cancer Cell 9(5): 351–65 [DOI] [PubMed] [Google Scholar]
- Chagraoui J, Lehnertz B, Girard S, Spinella JF, Fares I, et al. 2019. UM171 induces a homeostatic inflammatory-detoxification response supporting human HSC self-renewal. PLoS One 14(11): e0224900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou DB, Frismantas V, Milton Y, David R, Pop-Damkov P, et al. 2020. On-chip recapitulation of clinical bone marrow toxicities and patient-specific pathophysiology. Nat Biomed Eng [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clevers H, Tuveson DA. 2019. Organoid Models for Cancer Research. Annual Review of Cancer Biology 3(223–34 [Google Scholar]
- Cortazar P, Gazdar AF, Woods E, Russell E, Steinberg SM, et al. 1997. Survival of patients with limited-stage small cell lung cancer treated with individualized chemotherapy selected by in vitro drug sensitivity testing. Clin Cancer Res 3(5): 741–7 [PubMed] [Google Scholar]
- Cree IA, Kurbacher CM, Lamont A, Hindley AC, Love S, Group TCAOCT. 2007. A prospective randomized controlled trial of tumour chemosensitivity assay directed chemotherapy versus physician’s choice in patients with recurrent platinum-resistant ovarian cancer. Anticancer Drugs 18(9): 1093–101 [DOI] [PubMed] [Google Scholar]
- Crespo M, Vilar E, Tsai SY, Chang K, Amin S, et al. 2017. Colonic organoids derived from human induced pluripotent stem cells for modeling colorectal cancer and drug testing. Nat Med 23(7): 878–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Gaizo Moore V, Brown JR, Certo M, Love TM, Novina CD, Letai A. 2007. Chronic lymphocytic leukemia requires BCL2 to sequester prodeath BIM, explaining sensitivity to BCL2 antagonist ABT-737. J Clin Invest 117(1): 112–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delaney C, Heimfeld S, Brashem-Stein C, Voorhies H, Manger RL, Bernstein ID. 2010. Notch-mediated expansion of human cord blood progenitor cells capable of rapid myeloid reconstitution. Nat Med 16(2): 232–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deneault E, Cellot S, Faubert A, Laverdure JP, Frechette M, et al. 2009. A functional screen to identify novel effectors of hematopoietic stem cell activity. Cell 137(2): 369–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng J, Carlson N, Takeyama K, Dal Cin P, Shipp M, Letai A. 2007. BH3 profiling identifies three distinct classes of apoptotic blocks to predict response to ABT-737 and conventional chemotherapeutic agents. Cancer Cell 12(2): 171–85 [DOI] [PubMed] [Google Scholar]
- Dietrich S, Oles M, Lu J, Sellner L, Anders S, et al. 2018. Drug-perturbation-based stratification of blood cancer. J Clin Invest 128(1): 427–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards DKt, Watanabe-Smith K, Rofelty A, Damnernsawad A, Laderas T, et al. 2019. CSF1R inhibitors exhibit antitumor activity in acute myeloid leukemia by blocking paracrine signals from support cells. Blood 133(6): 588–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards VD, Sweeney DT, Ho H, Eide CA, Rofelty A, et al. 2018. Targeting of colony-stimulating factor 1 receptor (CSF1R) in the CLL microenvironment yields antineoplastic activity in primary patient samples. Oncotarget 9(37): 24576–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eltabbakh GH, Piver MS, Hempling RE, Recio FO, Lele SB, et al. 1998. Correlation between extreme drug resistance assay and response to primary paclitaxel and cisplatin in patients with epithelial ovarian cancer. Gynecol Oncol 70(3): 392–7 [DOI] [PubMed] [Google Scholar]
- Fan H, Demirci U, Chen P. 2019. Emerging organoid models: leaping forward in cancer research. J Hematol Oncol 12(1): 142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fares I, Chagraoui J, Gareau Y, Gingras S, Ruel R, et al. 2014. Cord blood expansion. Pyrimidoindole derivatives are agonists of human hematopoietic stem cell self-renewal. Science 345(6203): 1509–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feuring-Buske M, Gerhard B, Cashman J, Humphries RK, Eaves CJ, Hogge DE. 2003. Improved engraftment of human acute myeloid leukemia progenitor cells in beta 2-microglobulin-deficient NOD/SCID mice and in NOD/SCID mice transgenic for human growth factors. Leukemia 17(4): 760–3 [DOI] [PubMed] [Google Scholar]
- Fischer U, Forster M, Rinaldi A, Risch T, Sungalee S, et al. 2015. Genomics and drug profiling of fatal TCF3-HLF-positive acute lymphoblastic leukemia identifies recurrent mutation patterns and therapeutic options. Nature genetics 47(9): 1020–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frazier JP, Bertout JA, Kerwin WS, Moreno-Gonzalez A, Casalini JR, et al. 2017. Multidrug Analyses in Patients Distinguish Efficacious Cancer Agents Based on Both Tumor Cell Killing and Immunomodulation. Cancer Res 77(11): 2869–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman AA, Letai A, Fisher DE, Flaherty KT. 2015. Precision medicine for cancer with next-generation functional diagnostics. Nat Rev Cancer 15(12): 747–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman AA, Xia Y, Trippa L, Le LP, Igras V, et al. 2017. Feasibility of Ultra-High-Throughput Functional Screening of Melanoma Biopsies for Discovery of Novel Cancer Drug Combinations. Clin Cancer Res 23(16): 4680–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frismantas V, Dobay MP, Rinaldi A, Tchinda J, Dunn SH, et al. 2017. Ex vivo drug response profiling detects recurrent sensitivity patterns in drug-resistant acute lymphoblastic leukemia. Blood 129(11): e26–e37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallion H, Christopherson WA, Coleman RL, DeMars L, Herzog T, et al. 2006. Progression-free interval in ovarian cancer and predictive value of an ex vivo chemoresponse assay. Int J Gynecol Cancer 16(1): 194–201 [DOI] [PubMed] [Google Scholar]
- Gazdar AF, Steinberg SM, Russell EK, Linnoila RI, Oie HK, et al. 1990. Correlation of in vitro drug-sensitivity testing results with response to chemotherapy and survival in extensive-stage small cell lung cancer: a prospective clinical trial. J Natl Cancer Inst 82(2): 117–24 [DOI] [PubMed] [Google Scholar]
- Hanahan D, Coussens LM. 2012. Accessories to the crime: functions of cells recruited to the tumor microenvironment. Cancer Cell 21(3): 309–22 [DOI] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. 2011. Hallmarks of cancer: the next generation. Cell 144(5): 646–74 [DOI] [PubMed] [Google Scholar]
- Hidalgo M, Amant F, Biankin AV, Budinska E, Byrne AT, et al. 2014. Patient-derived xenograft models: an emerging platform for translational cancer research. Cancer Discov 4(9): 998–1013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirt C, Papadimitropoulos A, Mele V, Muraro MG, Mengus C, et al. 2014. “In vitro” 3D models of tumor-immune system interaction. Adv Drug Deliv Rev 79–80(145–54 [DOI] [PubMed] [Google Scholar]
- Hoffman RM. 2015. Patient-derived orthotopic xenografts: better mimic of metastasis than subcutaneous xenografts. Nat Rev Cancer 15(8): 451–2 [DOI] [PubMed] [Google Scholar]
- Hope KJ, Cellot S, Ting SB, MacRae T, Mayotte N, et al. 2010. An RNAi screen identifies Msi2 and Prox1 as having opposite roles in the regulation of hematopoietic stem cell activity. Cell Stem Cell 7(1): 101–13 [DOI] [PubMed] [Google Scholar]
- Hudson WA, Li Q, Le C, Kersey JH. 1998. Xenotransplantation of human lymphoid malignancies is optimized in mice with multiple immunologic defects. Leukemia 12(12): 2029–33 [DOI] [PubMed] [Google Scholar]
- Ince TA, Sousa AD, Jones MA, Harrell JC, Agoston ES, et al. 2015. Characterization of twenty-five ovarian tumour cell lines that phenocopy primary tumours. Nat Commun 6(7419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iscove NN, Sieber F, Winterhalter KH. 1974. Erythroid colony formation in cultures of mouse and human bone marrow: analysis of the requirement for erythropoietin by gel filtration and affinity chromatography on agarose-concanavalin A. J Cell Physiol 83(2): 309–20 [DOI] [PubMed] [Google Scholar]
- Ito M, Hiramatsu H, Kobayashi K, Suzue K, Kawahata M, et al. 2002. NOD/SCID/gamma(c)(null) mouse: an excellent recipient mouse model for engraftment of human cells. Blood 100(9): 3175–82 [DOI] [PubMed] [Google Scholar]
- Jacob T, Agarwal A, Ramunno-Johnson D, O’Hare T, Gonen M, et al. 2016. Ultrasensitive proteomic quantitation of cellular signaling by digitized nanoparticle-protein counting. Sci Rep 6(28163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang T, Munguia-Lopez JG, Gu K, Bavoux MM, Flores-Torres S, et al. 2019. Engineering bioprintable alginate/gelatin composite hydrogels with tunable mechanical and cell adhesive properties to modulate tumor spheroid growth kinetics. Biofabrication 12(1): 015024. [DOI] [PubMed] [Google Scholar]
- Jonas O, Landry HM, Fuller JE, Santini JT Jr., Baselga J, et al. 2015. An implantable microdevice to perform high-throughput in vivo drug sensitivity testing in tumors. Science translational medicine 7(284): 284ra57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonas O, Oudin MJ, Kosciuk T, Whitman M, Gertler FB, et al. 2016. Parallel in-vivo assessment of drug phenotypes at various time points during systemic BRAF inhibition reveals tumor adaptation and altered treatment vulnerabilities. Clin Cancer Res [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joo WD, Lee JY, Kim JH, Yoo HJ, Roh HJ, et al. 2009. Efficacy of taxane and platinum-based chemotherapy guided by extreme drug resistance assay in patients with epithelial ovarian cancer. J Gynecol Oncol 20(2): 96–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karjalainen R, Pemovska T, Popa M, Liu M, Javarappa KK, et al. 2017. JAK1/2 and BCL2 inhibitors synergize to counteract bone marrow stromal cell-induced protection of AML. Blood 130(6): 789–802 [DOI] [PubMed] [Google Scholar]
- Kenny HA, Lal-Nag M, White EA, Shen M, Chiang CY, et al. 2015. Quantitative high throughput screening using a primary human three-dimensional organotypic culture predicts in vivo efficacy. Nat Commun 6(6220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kern DH. 1998. Heterogeneity of drug resistance in human breast and ovarian cancers. Cancer J Sci Am 4(1): 41–5 [PubMed] [Google Scholar]
- Kern DH, Drogemuller CR, Kennedy MC, Hildebrand-Zanki SU, Tanigawa N, Sondak VK. 1985. Development of a miniaturized, improved nucleic acid precursor incorporation assay for chemosensitivity testing of human solid tumors. Cancer Res 45(11 Pt 1): 5436–41 [PubMed] [Google Scholar]
- Kern DH, Weisenthal LM. 1990. Highly specific prediction of antineoplastic drug resistance with an in vitro assay using suprapharmacologic drug exposures. J Natl Cancer Inst 82(7): 582–8 [DOI] [PubMed] [Google Scholar]
- Klinghoffer RA, Bahrami SB, Hatton BA, Frazier JP, Moreno-Gonzalez A, et al. 2015. A technology platform to assess multiple cancer agents simultaneously within a patient’s tumor. Science translational medicine 7(284): 284ra58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konopleva M, Pollyea DA, Potluri J, Chyla B, Hogdal L, et al. 2016. Efficacy and Biological Correlates of Response in a Phase II Study of Venetoclax Monotherapy in Patients with Acute Myelogenous Leukemia. Cancer Discov 6(10): 1106–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurbacher CM, Cree IA, Bruckner HW, Brenne U, Kurbacher JA, et al. 1998. Use of an ex vivo ATP luminescence assay to direct chemotherapy for recurrent ovarian cancer. Anticancer Drugs 9(1): 51–7 [DOI] [PubMed] [Google Scholar]
- Kuusanmaki H, Leppa AM, Polonen P, Kontro M, Dufva O, et al. 2020. Phenotype-based drug screening reveals association between venetoclax response and differentiation stage in acute myeloid leukemia. Haematologica 105(3): 708–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai Y, Wei X, Lin S, Qin L, Cheng L, Li P. 2017. Current status and perspectives of patient-derived xenograft models in cancer research. J Hematol Oncol 10(1): 106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langer EM, Allen-Petersen BL, King SM, Kendsersky ND, Turnidge MA, et al. 2019. Modeling Tumor Phenotypes In Vitro with Three-Dimensional Bioprinting. Cell reports 26(3): 608–23 e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SI, Celik S, Logsdon BA, Lundberg SM, Martins TJ, et al. 2018. A machine learning approach to integrate big data for precision medicine in acute myeloid leukemia. Nat Commun 9(1): 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letai AG. 2008. Diagnosing and exploiting cancer’s addiction to blocks in apoptosis. Nat Rev Cancer 8(2): 121–32 [DOI] [PubMed] [Google Scholar]
- Liu X, Krawczyk E, Suprynowicz FA, Palechor-Ceron N, Yuan H, et al. 2017. Conditional reprogramming and long-term expansion of normal and tumor cells from human biospecimens. Nat Protoc 12(2): 439–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Ory V, Chapman S, Yuan H, Albanese C, et al. 2012. ROCK inhibitor and feeder cells induce the conditional reprogramming of epithelial cells. Am J Pathol 180(2): 599–607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loizzi V, Chan JK, Osann K, Cappuccini F, DiSaia PJ, Berman ML. 2003. Survival outcomes in patients with recurrent ovarian cancer who were treated with chemoresistance assay-guided chemotherapy. Am J Obstet Gynecol 189(5): 1301–7 [DOI] [PubMed] [Google Scholar]
- MacPherson L, Anokye J, Yeung MM, Lam EYN, Chan YC, et al. 2020. HBO1 is required for the maintenance of leukaemia stem cells. Nature 577(7789): 266–70 [DOI] [PubMed] [Google Scholar]
- Maenpaa JU, Heinonen E, Hinkka SM, Karnani P, Klemi PJ, et al. 1995. The subrenal capsule assay in selecting chemotherapy for ovarian cancer: a prospective randomized trial. Gynecol Oncol 57(3): 294–8 [DOI] [PubMed] [Google Scholar]
- Majumder B, Baraneedharan U, Thiyagarajan S, Radhakrishnan P, Narasimhan H, et al. 2015. Predicting clinical response to anticancer drugs using an ex vivo platform that captures tumour heterogeneity. Nat Commun 6(6169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majumder MM, Leppa AM, Hellesoy M, Dowling P, Malyutina A, et al. 2019. Multi-parametric single cell evaluation defines distinct drug responses in healthy hematological cells that are retained in corresponding malignant cell types. Haematologica [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAuliffe PF, Evans KW, Akcakanat A, Chen K, Zheng X, et al. 2015. Ability to Generate Patient-Derived Breast Cancer Xenografts Is Enhanced in Chemoresistant Disease and Predicts Poor Patient Outcomes. PLoS One 10(9): e0136851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta RS, Bornstein R, Yu IR, Parker RJ, McLaren CE, et al. 2001. Breast cancer survival and in vitro tumor response in the extreme drug resistance assay. Breast Cancer Res Treat 66(3): 225–37 [DOI] [PubMed] [Google Scholar]
- Meyerrose TE, Herrbrich P, Hess DA, Nolta JA. 2003. Immune-deficient mouse models for analysis of human stem cells. Biotechniques 35(6): 1262–72 [DOI] [PubMed] [Google Scholar]
- Mi Z, Holmes FA, Hellerstedt B, Pippen J, Collea R, et al. 2008. Feasibility assessment of a chemoresponse assay to predict pathologic response in neoadjuvant chemotherapy for breast cancer patients. Anticancer Res 28(3B): 1733–40 [PubMed] [Google Scholar]
- Montero J, Letai A. 2016. Dynamic BH3 profiling-poking cancer cells with a stick. Mol Cell Oncol 3(3): e1040144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montero J, Sarosiek KA, DeAngelo JD, Maertens O, Ryan J, et al. 2015. Drug-induced death signaling strategy rapidly predicts cancer response to chemotherapy. Cell 160(5): 977–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moser R, Xu C, Kao M, Annis J, Lerma LA, et al. 2014. Functional kinomics identifies candidate therapeutic targets in head and neck cancer. Clin Cancer Res 20(16): 4274–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagourney RA. 2000. Correlation between extreme drug resistance assay and response to primary paclitaxel and cisplatin in patients with epithelial ovarian cancer. Gynecol Oncol 76(1): 143. [DOI] [PubMed] [Google Scholar]
- Nagourney RA, Blitzer JB, Shuman RL, Asciuto TJ, Deo EA, et al. 2012. Functional profiling to select chemotherapy in untreated, advanced or metastatic non-small cell lung cancer. Anticancer Res 32(10): 4453–60 [PubMed] [Google Scholar]
- Neal JT, Kuo CJ. 2016. Organoids as Models for Neoplastic Transformation. Annu Rev Pathol 11(199–220 [DOI] [PubMed] [Google Scholar]
- Neal JT, Li X, Zhu J, Giangarra V, Grzeskowiak CL, et al. 2018. Organoid Modeling of the Tumor Immune Microenvironment. Cell 175(7): 1972–88 e16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ootani A, Li X, Sangiorgi E, Ho QT, Ueno H, et al. 2009. Sustained in vitro intestinal epithelial culture within a Wnt-dependent stem cell niche. Nat Med 15(6): 701–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pabst C, Krosl J, Fares I, Boucher G, Ruel R, et al. 2014. Identification of small molecules that support human leukemia stem cell activity ex vivo. Nat Methods 11(4): 436–42 [DOI] [PubMed] [Google Scholar]
- Pan R, Hogdal LJ, Benito JM, Bucci D, Han L, et al. 2014. Selective BCL-2 inhibition by ABT-199 causes on-target cell death in acute myeloid leukemia. Cancer Discov 4(3): 362–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papapetrou EP. 2016. Patient-derived induced pluripotent stem cells in cancer research and precision oncology. Nat Med 22(12): 1392–401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pauli C, Hopkins BD, Prandi D, Shaw R, Fedrizzi T, et al. 2017. Personalized In Vitro and In Vivo Cancer Models to Guide Precision Medicine. Cancer Discov 7(5): 462–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pemovska T, Kontro M, Yadav B, Edgren H, Eldfors S, et al. 2013. Individualized systems medicine strategy to tailor treatments for patients with chemorefractory acute myeloid leukemia. Cancer Discov 3(12): 1416–29 [DOI] [PubMed] [Google Scholar]
- Phan N, Hong JJ, Tofig B, Mapua M, Elashoff D, et al. 2019. A simple high-throughput approach identifies actionable drug sensitivities in patient-derived tumor organoids. Commun Biol 2(78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pike BL, Robinson WA. 1970. Human bone marrow colony growth in agar-gel. J Cell Physiol 76(1): 77–84 [DOI] [PubMed] [Google Scholar]
- Potdar S, Ianevski A, Mpindi JP, Bychkov D, Fiere C, et al. 2020. Breeze: an integrated quality control and data analysis application for high-throughput drug screening. Bioinformatics [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prina-Mello A, Jain N, Liu B, Kilpatrick JI, Tutty MA, et al. 2018. Culturing substrates influence the morphological, mechanical and biochemical features of lung adenocarcinoma cells cultured in 2D or 3D. Tissue Cell 50(15–30 [DOI] [PubMed] [Google Scholar]
- Ramamoorthy P, Thomas SM, Kaushik G, Subramaniam D, Chastain KM, et al. 2019. Metastatic Tumor-in-a-Dish, a Novel Multicellular Organoid to Study Lung Colonization and Predict Therapeutic Response. Cancer Res 79(7): 1681–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinisch A, Thomas D, Corces MR, Zhang X, Gratzinger D, et al. 2016. A humanized bone marrow ossicle xenotransplantation model enables improved engraftment of healthy and leukemic human hematopoietic cells. Nat Med 22(7): 812–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridky TW, Chow JM, Wong DJ, Khavari PA. 2010. Invasive three-dimensional organotypic neoplasia from multiple normal human epithelia. Nat Med 16(12): 1450–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan J, Letai A. 2013. BH3 profiling in whole cells by fluorimeter or FACS. Methods 61(2): 156–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saeed K, Ojamies P, Pellinen T, Eldfors S, Turkki R, et al. 2019. Clonal heterogeneity influences drug responsiveness in renal cancer assessed by ex vivo drug testing of multiple patient-derived cancer cells. Int J Cancer 144(6): 1356–66 [DOI] [PubMed] [Google Scholar]
- Saeed K, Rahkama V, Eldfors S, Bychkov D, Mpindi JP, et al. 2017. Comprehensive Drug Testing of Patient-derived Conditionally Reprogrammed Cells from Castration-resistant Prostate Cancer. Eur Urol 71(3): 319–27 [DOI] [PubMed] [Google Scholar]
- Salmon SE, Hamburger AW, Soehnlen B, Durie BG, Alberts DS, Moon TE. 1978. Quantitation of differential sensitivity of human-tumor stem cells to anticancer drugs. N Engl J Med 298(24): 1321–7 [DOI] [PubMed] [Google Scholar]
- Samson DJ, Seidenfeld J, Ziegler K, Aronson N. 2004. Chemotherapy sensitivity and resistance assays: a systematic review. J Clin Oncol 22(17): 3618–30 [DOI] [PubMed] [Google Scholar]
- Sato T, Stange DE, Ferrante M, Vries RG, Van Es JH, et al. 2011. Long-term expansion of epithelial organoids from human colon, adenoma, adenocarcinoma, and Barrett’s epithelium. Gastroenterology 141(5): 1762–72 [DOI] [PubMed] [Google Scholar]
- Sato T, Vries RG, Snippert HJ, van de Wetering M, Barker N, et al. 2009. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature 459(7244): 262–5 [DOI] [PubMed] [Google Scholar]
- Schmidt SK, Schmid R, Arkudas A, Kengelbach-Weigand A, Bosserhoff AK. 2019. Tumor Cells Develop Defined Cellular Phenotypes After 3D-Bioprinting in Different Bioinks. Cells 8(10) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneeberger K, Spee B, Costa P, Sachs N, Clevers H, Malda J. 2017. Converging biofabrication and organoid technologies: the next frontier in hepatic and intestinal tissue engineering? Biofabrication 9(1): 013001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrag D, Garewal HS, Burstein HJ, Samson DJ, Von Hoff DD, et al. 2004. American Society of Clinical Oncology Technology Assessment: chemotherapy sensitivity and resistance assays. J Clin Oncol 22(17): 3631–8 [DOI] [PubMed] [Google Scholar]
- Shaw GL, Gazdar AF, Phelps R, Linnoila RI, Ihde DC, et al. 1993. Individualized chemotherapy for patients with non-small cell lung cancer determined by prospective identification of neuroendocrine markers and in vitro drug sensitivity testing. Cancer Res 53(21): 5181–7 [PubMed] [Google Scholar]
- Shultz LD, Ishikawa F, Greiner DL. 2007. Humanized mice in translational biomedical research. Nat Rev Immunol 7(2): 118–30 [DOI] [PubMed] [Google Scholar]
- Sieber S, Wirth L, Cavak N, Koenigsmark M, Marx U, et al. 2018. Bone marrow-on-a-chip: Long-term culture of human haematopoietic stem cells in a three-dimensional microfluidic environment. J Tissue Eng Regen Med 12(2): 479–89 [DOI] [PubMed] [Google Scholar]
- Skardal A, Murphy SV, Devarasetty M, Mead I, Kang HW, et al. 2017. Multi-tissue interactions in an integrated three-tissue organ-on-a-chip platform. Sci Rep 7(1): 8837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith R, Devlin K, Kilburn D, Gross S, Sudar D, et al. 2019. Using Microarrays to Interrogate Microenvironmental Impact on Cellular Phenotypes in Cancer. J Vis Exp 147) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soslow RA, Slomovitz BM, Saqi A, Baergen RN, Caputo TA. 2000. Tumor suppressor gene, cell surface adhesion molecule, and multidrug resistance in Mullerian serous carcinomas: clinical divergence without immunophenotypic differences. Gynecol Oncol 79(3): 430–7 [DOI] [PubMed] [Google Scholar]
- Stevens MM, Maire CL, Chou N, Murakami MA, Knoff DS, et al. 2016. Drug sensitivity of single cancer cells is predicted by changes in mass accumulation rate. Nat Biotechnol 34(11): 1161–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suprynowicz FA, Upadhyay G, Krawczyk E, Kramer SC, Hebert JD, et al. 2012. Conditionally reprogrammed cells represent a stem-like state of adult epithelial cells. Proc Natl Acad Sci U S A 109(49): 20035–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teh CE, Gong JN, Segal D, Tan T, Vandenberg CJ, et al. 2020. Deep profiling of apoptotic pathways with mass cytometry identifies a synergistic drug combination for killing myeloma cells. Cell Death Differ [DOI] [PMC free article] [PubMed] [Google Scholar]
- Touzeau C, Ryan J, Guerriero J, Moreau P, Chonghaile TN, et al. 2016. BH3 profiling identifies heterogeneous dependency on Bcl-2 family members in multiple myeloma and predicts sensitivity to BH3 mimetics. Leukemia 30(3): 761–4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuveson D, Clevers H. 2019. Cancer modeling meets human organoid technology. Science 364(6444): 952–55 [DOI] [PubMed] [Google Scholar]
- Tyner JW, Deininger MW, Loriaux MM, Chang BH, Gotlib JR, et al. 2009. RNAi screen for rapid therapeutic target identification in leukemia patients. Proc Natl Acad Sci U S A 106(21): 8695–700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyner JW, Tognon CE, Bottomly D, Wilmot B, Kurtz SE, et al. 2018. Functional genomic landscape of acute myeloid leukaemia. Nature 562(7728): 526–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyner JW, Yang WF, Bankhead A, Fan G, Fletcher LB, et al. 2013. Kinase Pathway Dependence in Primary Human Leukemias Determined by Rapid Inhibitor Screening. Cancer Res 73(1): 285–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ugurel S, Schadendorf D, Pfohler C, Neuber K, Thoelke A, et al. 2006. In vitro drug sensitivity predicts response and survival after individualized sensitivity-directed chemotherapy in metastatic melanoma: a multicenter phase II trial of the Dermatologic Cooperative Oncology Group. Clin Cancer Res 12(18): 5454–63 [DOI] [PubMed] [Google Scholar]
- Vaira V, Fedele G, Pyne S, Fasoli E, Zadra G, et al. 2010. Preclinical model of organotypic culture for pharmacodynamic profiling of human tumors. Proc Natl Acad Sci U S A 107(18): 8352–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Von Hoff DD, Clark GM, Stogdill BJ, Sarosdy MF, O’Brien MT, et al. 1983. Prospective clinical trial of a human tumor cloning system. Cancer Res 43(4): 1926–31 [PubMed] [Google Scholar]
- Von Hoff DD, Kronmal R, Salmon SE, Turner J, Green JB, et al. 1991. A Southwest Oncology Group study on the use of a human tumor cloning assay for predicting response in patients with ovarian cancer. Cancer 67(1): 20–7 [DOI] [PubMed] [Google Scholar]
- Von Hoff DD, Sandbach JF, Clark GM, Turner JN, Forseth BF, et al. 1990. Selection of cancer chemotherapy for a patient by an in vitro assay versus a clinician. J Natl Cancer Inst 82(2): 110–6 [DOI] [PubMed] [Google Scholar]
- Watson SS, Dane M, Chin K, Tatarova Z, Liu M, et al. 2018. Microenvironment-Mediated Mechanisms of Resistance to HER2 Inhibitors Differ between HER2+ Breast Cancer Subtypes. Cell Syst 6(3): 329–42 e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilbur DW, Camacho ES, Hilliard DA, Dill PL, Weisenthal LM. 1992. Chemotherapy of non-small cell lung carcinoma guided by an in vitro drug resistance assay measuring total tumour cell kill. Br J Cancer 65(1): 27–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu B, Zhu JS, Zhang Y, Shen WM, Zhang Q. 2008. Predictive value of MTT assay as an in vitro chemosensitivity testing for gastric cancer: one institution’s experience. World J Gastroenterol 14(19): 3064–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu TT, Sikes RA, Cui Q, Thalmann GN, Kao C, et al. 1998. Establishing human prostate cancer cell xenografts in bone: induction of osteoblastic reaction by prostate-specific antigen-producing tumors in athymic and SCID/bg mice using LNCaP and lineage-derived metastatic sublines. Int J Cancer 77(6): 887–94 [DOI] [PubMed] [Google Scholar]
- Xu C, Nikolova O, Basom RS, Mitchell RM, Shaw R, et al. 2018. Functional Precision Medicine Identifies Novel Druggable Targets and Therapeutic Options in Head and Neck Cancer. Clin Cancer Res 24(12): 2828–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu JM, Song ST, Tang ZM, Jiang ZF, Liu XQ, et al. 1999. Predictive chemotherapy of advanced breast cancer directed by MTT assay in vitro. Breast Cancer Res Treat 53(1): 77–85 [DOI] [PubMed] [Google Scholar]
- Yu M, Bardia A, Aceto N, Bersani F, Madden MW, et al. 2014. Cancer therapy. Ex vivo culture of circulating breast tumor cells for individualized testing of drug susceptibility. Science 345(6193): 216–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan H, Myers S, Wang J, Zhou D, Woo JA, et al. 2012. Use of reprogrammed cells to identify therapy for respiratory papillomatosis. N Engl J Med 367(13): 1220–7 [DOI] [PMC free article] [PubMed] [Google Scholar]