

Abstract
Chronic symptoms indicating excess cortical excitability follow mild traumatic brain injury, particularly repetitive mild traumatic brain injury (rmTBI). Yet mechanisms underlying post-traumatic excitation/inhibition (E/I) ratio abnormalities may differ between the early and late post-traumatic phases. We therefore measured seizure threshold and cortical gamma-aminobutyric acid (GABA) and glutamate (Glu) concentrations, 1 and 6 weeks after rmTBI in mice. We also analyzed the structure of parvalbumin-positive interneurons (PVIs), their perineuronal nets (PNNs), and their electroencephalography (EEG) signature (gamma frequency band power). For mechanistic insight, we measured cortical oxidative stress, reflected in the reduced/oxidized glutathione (GSH/GSSG) ratio. We found that seizure susceptibility increased both early and late after rmTBI. However, whereas increased Glu dominated the E/I 1 week after rmTBI, Glu concentration normalized and the E/I was instead characterized by depressed GABA, reduced per-PVI parvalbumin expression, and reduced gamma EEG power at the 6-week post-rmTBI time point. Oxidative stress was increased early after rmTBI, where transient PNN degradation was noted, and progressed throughout the monitoring period. We conclude that GSH depletion, perhaps triggered by early Glu-mediated excitotoxicity, leads to late post-rmTBI loss of PVI-dependent cortical inhibitory tone. We thus propose dampening of Glu signaling, maintenance of redox state, and preservation of PVI inhibitory capacity as therapeutic targets for post-rmTBI treatment.
Keywords: concussion, repetitive mild traumatic brain injury, seizure
Introduction
Mild traumatic brain injury (mTBI; concussion), with an estimated incidence of 600 per 100,000 people, is the most common neurological injury globally. In the United States of America, up to 3 million people are treated for mTBI annually, approximately half of whom continue to suffer from a range of symptoms for months to years after injury (Daneshvar et al. 2011; Grandhi et al. 2017; McInnes et al. 2017; Nelson et al. 2019). Notably, mTBI is often recurrent, termed repetitive mild traumatic brain injury (rmTBI) (Castile et al. 2012; Guerriero et al. 2012; Theadom et al. 2015; Mannix et al. 2016), suggesting that concussion is associated with a risk of cumulative damage (Eisenberg et al. 2013; Howell et al. 2017)—this is particularly worrisome for those individuals engaged in contact sports or similar activities (Guerriero et al. 2012; Marar et al. 2012; Harmon et al. 2013; Mannix et al. 2016; Mez et al. 2017; Alosco et al. 2018; Howell et al. 2018).
The neurologic sequelae resulting from mTBI, and especially from rmTBI, include chronic pain (Andary et al. 1997; Grandhi et al. 2017), mood disorders (Panayiotou et al. 2010; Stein et al. 2019), and sleep disturbances (Viola-Saltzman and Watson 2012; Mantua et al. 2017) in the subacute post-traumatic period. Longitudinal studies also indicate increased epilepsy risk after mTBI, albeit only in the late (years to decades after injury), chronic, post-traumatic phase where the causal relationship between injury and epilepsy is less certain (Annegers et al. 1998; Lucke-Wold et al. 2015; Keret et al. 2017). Such post-traumatic symptoms are consistent with pathologic gain of cortical excitability, loss of cortical inhibition, or both (Yizhar et al. 2011; Luscher and Fuchs 2015; Trevelyan et al. 2015; Parker et al. 2016; Lissemore et al. 2018; Lanza and Ferri 2019). Given that postconcussion symptoms may present or persist weeks to months after injury (Rutherford et al. 1979; Rao et al. 2010; Cole and Bailie 2016; Biagianti et al. 2020), these likely reflect a slow, continuing biological process during which cortical excitation/inhibition (E/I) balance shifts toward excitation, as it does after more severe traumatic brain injury (TBI) (Hsieh et al. 2017; Hameed et al. 2019). Yet the pathophysiology underlying this post-traumatic progressive E/I derangement remains unknown.
In severe TBI animal models, loss of cortical inhibition and epileptogenesis are accompanied by cell death and pathologic loss of gamma-aminobutyric acid (GABA)ergic inhibitory interneurons—the plurality of which are parvalbumin-positive inhibitory interneurons (PVIs) whose fast-spiking nature and high metabolic demand renders them disproportionately vulnerable to post-traumatic metabolic challenges (Sohal et al. 2009; Cabungcal et al. 2013; Cantu et al. 2015; Hsieh et al. 2017; Hameed et al. 2019). Yet concussive brain injury, as evident in the mouse rmTBI model, occurs without immediate neuronal death (Khuman et al. 2011; Meehan 3rd et al. 2012), and the mechanistic link between rmTBI and the loss of cortical inhibition that presumably follows remains unclear.
In the absence of significant neuronal loss, both chronic postconcussive symptoms (Panayiotou et al. 2010; Viola-Saltzman and Watson 2012; Grandhi et al. 2017; Mantua et al. 2017; Stein et al. 2019) and the long-term increased risk of epilepsy after concussion (Annegers et al. 1998; Lucke-Wold et al. 2015) may result from post-traumatic cell dysfunction rather than cell death. For PVI circuitry, such dysfunction may be cell autonomous as reflected in per-cell PV expression. Alternatively, loss of the specialized extracellular matrix surrounding PVIs, the perineuronal nets (PNNs), may also impair the PVI inhibitory capacity, which is reliant on PNN-mediated maintenance of local redox state, among other essential PNN functions (Hsieh et al. 2017; Favuzzi et al. 2017; Suttkus et al. 2014; Sorg et al. 2016). Accordingly, we use the established mouse rmTBI concussion model, where neuronal death is absent (Meehan 3rd et al. 2012) to test whether PVI structure and function is nevertheless compromised in the weeks that follow injury. This insight may be critical for the design of effective treatment after injury. Similarly, to identify future therapeutic targets, we test whether causal contributors to PVI dysfunction such as glutamate (Glu)-mediated excitotoxicity (Behrens et al. 2007), increased oxidative stress (Hsieh et al. 2017), and degradation of the PNN surrounding this vulnerable interneuron population (Cabungcal et al. 2013) (all described in a range of more severe brain injuries) follow rmTBI.
Methods
Animal Preparation
Two hundred and twenty-two 2-month-old adult C57 black male mice (20–30 g) were randomized to receive sham or verum injury (N = 110 sham; 112 rmTBI). Mice were housed in standard cages in a temperature-controlled facility with a 12-h light/dark cycle and a continuous supply of water and food ad libitum. All animal procedures were approved by, and in accordance with the guidelines of, the Institutional Animal Care and Use Committee at Boston Children’s Hospital and the National Institutes of Health (NIH) Guide for the Care and Use of Laboratory Animals. All efforts were made to minimize the number of mice used in the present experiments.
Repetitive Mild Traumatic Brain Injury
The mouse rmTBI model was used as previously described, with modifications (Meehan 3rd et al. 2012; Mannix et al. 2013). Briefly, mice were anesthetized with 4% isoflurane for 45 s, placed prone on a delicate task wiper (Kimwipe; Kimberly-Clark), and secured by the tail. The dorsal surface of the cranium was placed flush beneath a hollow guide tube centered over the skull. A 54-g metal bolt was dropped from a height of 107 cm through the guide tube, impacting the dorsal aspect of the skull between the coronal and lambdoid sutures. At impact, the mouse’s head readily penetrated the Kimwipe, resulting in a rotational acceleration. Animals were subjected to one injury per day for five consecutive days. After each injury, animals were placed in a left lateral recumbent position, and loss of consciousness (LOC) was defined as the time from removal of anesthesia to spontaneous righting. All mice recovered in room air. A separate cohort was produced for each experiment. Sham-injured age-matched control mice underwent anesthesia but not concussive injury (Fig. 1).
Figure 1 .
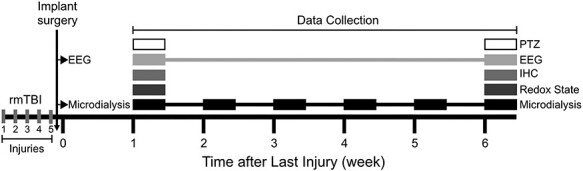
Timeline of rmTBI experiments and cohorts. Mice were exposed to five weight drop injuries over 5 days. Mice designated to microdialysis or EEG groups were implanted with hardware after the final injury. One week after the final injury animals were assigned to one of the following groups: microdialysis, EEG, 1-week HPLC for oxidative stress (GSH:GSSG), 1-week IHC, 1-week PTZ challenge, 6-week HPLC, 6-week IHC, and 6-week PTZ. Microdialysis measures were obtained weekly for 6 weeks. EEG was recorded once after final verum or sham injury and again 6 weeks after the last verum or sham injury.
Extracellular Glutamate and GABA Measurement
Guide Cannula Implantation
Approximately 1 h after the final injury, a guide cannula was surgically implanted into the barrel cortex of a cohort of mice (coordinates: −0.22 mm anterior-posterior (AP), 3.0 mm medial-lateral (ML), 1.0 mm dorsal-ventral (DV); Lein et al. 2007; N = 6 per sham and verum injury group). These coordinates were chosen based on pilot data and high PVI density in this region. The cannula was permanently fixed to the skull using anchoring screws and dental cement. Opioid analgesia (1 mg/kg buprenorphine sustained release; ZooPharm) was administered subcutaneously to provide 48-h postoperative coverage.
Microdialysis
Seven days after implantation, mice were anesthetized with 2–3.5% isoflurane, and a microdialysis probe (MD2255, Bioanalytical Systems, Inc.) was inserted into the guide cannula. The probe was perfused with artificial cerebrospinal fluid (119 mM NaCl, 2.5 mM KCl, 1 mM NaH2PO4, 2.5 mM CaCl2, 1.5 mM MgCl2, 11.0 mM glucose, pH 7.4) at 2 μL/min for 24 h prior to sample collection. Following equilibration, samples were collected for 30-min intervals and cooled to 4°C. Samples were analyzed using high-performance liquid chromatography (HPLC).
GABA and Glutamate HPLC
HPLC was performed as described previously (McNair et al. 2019). Briefly, working reagent was prepared fresh daily by diluting 1 mL of stock reagent (20 mg OPA, 1 mL methanol, 10 mg N-acetyl cysteine, 9 mL 0.5 M carbonate buffer) into 4 mL of 0.5 M carbonate buffer pH 10. Derivatization was performed by mixing 50 μL working reagent with 50 μL 1:10 sample diluted in water. Samples were injected using a refrigerated autosampler (System Gold 508; Beckman Coulter) into an HPLC system consisting of a chromatographic column (Atlantis dC18, 4.6 × 250 mm, 5 μm; Waters Corporation), and a fluorescence detector (2475 FLR Detector; Waters Corporation) with a detection wavelength of λex = 229 nm and λem = 450 nm. The mobile phase (100 mM Na2HPO4, 22% [v/v] methanol, 3.5% acetonitrile, pH adjusted to 6.75 with phosphoric acid) was pumped at 0.25 mL/min by a solvent delivery module (System Gold 125; Beckman Coulter) as described previously.
Glutathione (GSH) Measurement
Sample Preparation
Animals (N = 5 per sham and verum injury group) were sacrificed and fresh tissue microdissected and snap frozen. Frozen barrel cortex was homogenized (10% in phosphate buffered saline [PBS], pH 5.5), sonicated (15 s on ice), and an aliquot (20 μL) was removed to determine protein content. The remaining sonicate was added (4:1) to perchloric acid (0.4 N) and centrifuged (13 000 rpm, 60 min, 4°C) to remove protein and cellular debris. Sample preparation was done as rapidly as possible at 4°C and at a pH well below the 8.8 pKa of the thiol group of GSH. GSH is much more stable under these conditions, minimizing artificial conversion to glutathione disulfide (GSSG).
GSH/GSSG HPLC
Samples were analyzed by HPLC as previously described (Trivedi et al. 2014). Briefly, 10 μL of sample was injected onto a reverse-phase C8 column (ZORBAX Eclipse XDB-C8, 3 × 150 mm, 3.5 μm; Agilent Technologies Inc.) and measured with an electrochemical detector (CouloChem III; Thermo Scientific) running a boron-doped diamond analytical cell (Model 5040; Thermo Scientific) at an operating potential of 1500 mV. A dual mobile phase gradient elution was used to resolve analytes, consisting of a mobile phase containing 25 mM sodium phosphate and 2.1 mM 1-octanesulfonic acid, adjusted to pH 2.65 with phosphoric acid, with the second mobile phase (B) containing 50% acetonitrile. The system was run at a flow rate of 1 mL/min at ambient temperature with the following gradients: 0–8 min 0% B, 8–20 min, gradient to 30% B. The system equilibrated at 0% B from 25 to 36 min. Peak area analysis (32-Karat software v.8.0; Beckman Coulter) was based on standard curves generated for each compound. Samples were normalized against protein content.
Immunohistochemistry (IHC)
Perfusion of Cortical Tissues
Under deep anesthesia, mice (1 week, N = 4 per sham and verum injury group; 6 week, N = 6 per sham and verum injury group) were perfused transcardially with ice-cold PBS followed by 4% paraformaldehyde (PFA). Brain tissue was harvested and postfixed in 4% PFA for 24 h at 4oC before transferring into 30% sucrose. Cryopreserved brain tissue was then frozen in Tissue-Plus OCT Compound (Fisher Healthcare) and stored at −80°C for at least 24 h before sectioning. Free-floating cryosections (coronal, 30 μm, from bregma −1.23 mm through bregma −2.03 mm; Paxinos and Franklin 2013) were obtained at −20°C, washed briefly with PBS, and incubated with primary antibodies overnight at 4°C followed by Alexa Fluor-conjugated secondary antibodies for 1 h at room temperature (Hsieh et al. 2017).
Immunostaining
Anti-parvalbumin (PV) antibody (Marly) was used to investigate PVI. PNN integrity was studied using biotinylated Wisteria floribunda agglutinin (WFA) (MilliporeSigma), a plant lectin that binds to chondroitin sulfate proteoglycan chains of PNN (Hsieh et al. 2017). Anti-guinea pig Alexa Fluor 488 and Streptavidin Alexa 594 conjugate were used as secondary antibodies (Invitrogen). All perfusion, tissue fixation, and immunostaining procedures were carried out under the same conditions using the same batch of buffers to minimize variability between samples. Immunostained sections were mounted using Fluoromount medium (Southern Biotechnology), and images acquired on an Olympus Fluoview FV1000 confocal microscope.
Image Acquisition
Samples were first identified by fluorescence imaging under low power magnification (×10 objective). Image acquisition was carried out using the FV10-ASW software (v2.1 C), with the following parameters: 5% laser output, ×1 gain control, laser intensity between 500 and 700, offset between 10% and 15% such that signals were within the linear range. Individual channels were acquired sequentially.
Cell Count
Images were analyzed by ImageJ software as described previously (Hsieh et al. 2017; Hameed et al. 2019). Cells were counted for a standard area in the image of the barrel cortex across 8 images per animal. First, regions corresponding to layers II–VI, 400 μm wide in the middle of the imaging field were chosen. Then, by setting the same threshold for all images and applying filters for size (50–5000 μm2) and circularity (0–1.0), cells positively stained for PV, and/or WFA were identified above background signals. 4',6-diamidino-2-phenylindole (DAPI) counterstain was further used to visually confirm that all counted structures were cells. The number of cells identified above was then recorded. Results from rmTBI mice were normalized to sham controls.
PVI Intensity
Per condition background was defined by sampling noncell-containing area within a representative selection of pictures. This background was used as a threshold to determine the average intensity of positive cells per animal.
Coincidence Detection
Positive cells were defined as PV+ (above threshold) and/or PNN+ (above threshold). Threshold values for PV and PNN were applied to each channel and merged to create a single mask of positive cells. This mask was applied to each channel separately, and intensity values were calculated for PV and PNN.
Electroencephalography (EEG)
Mice (N = 6 per sham and verum injury group) were intraperitoneally implanted with wireless telemetry transmitters (PhysioTel ETA-F10; DSI, Data Sciences International) and 1-channel continuous video-EEG (sampling rate 1000 Hz) was recorded from active (left occipital lobe) and reference (right olfactory bulb) electrodes (Dhamne et al. 2017; Kelly et al. 2018; Purtell et al. 2018; Yuskaitis et al. 2018). Data were collected for 48 h with the first 24 h corresponding to an acclimation period such that only data from the last 24 h were analyzed. EEG analysis was performed at 1- and 6-week post-rmTBI time points.
Using a custom MATLAB algorithm (R2018a, MathWorks Inc.), raw time series 24-h EEG data were preprocessed to remove movement-related artifacts or electrical noise using a combination of amplitude thresholding and time windowing. The 24-h EEG was divided into nonoverlapping 30-s blocks, and a time–frequency decomposition was obtained for each block using continuous wavelet transform. Mean spectral power in the gamma frequency (30–80 Hz) band, normalized to total power in the 1–80 Hz band (Dhamne et al. 2017) was computed from these 30-s blocks for 3 longer time periods: the full 24 h, 12-h dark (7 AM–7 PM), and 12-h light (7 AM–7 PM) to study circadian fluctuations in gamma oscillations.
Pentylenetetrazol (PTZ) Challenge
Mice (1 week, N = 32 per sham and verum injury group; 6 weeks, N = 48 per sham and verum injury group) were administered PTZ (40 mg/kg, IP) and placed individually in a clear home cage for video monitoring for 10 min. Latencies to induced seizure onset was measured by recording the time to first myoclonic seizure (Racine stage 3) and first generalized tonic–clonic seizure (GTC; Racine stage 5). To minimize bias, each recording was scored offline by 2 blinded reviewers and a third independent reviewer reconciled disagreements between the 2 scorings (Dhamne et al. 2015; Gersner et al. 2015; Dhamne et al. 2017).
Data Processing and Statistical Analysis
Data were analyzed using GraphPad Prism 8.0.2 (GraphPad Software Inc.) with the significance level set at P < 0.05. Results were compared between sham and rmTBI groups at each time point and normalized to sham control as necessary per experiment. All data are presented as mean ± standard error of the mean (mean ± SEM).
HPLC
Mean Glu and GABA levels and Glu/GABA ratios, and ratios of GSH/GSSG were analyzed using 2-way analysis of variance with Sidak’s multiple comparisons tests.
IHC
PVI and PNN counts, and average intensity values were calculated from 4 slices (8 images) per animal, then compared using the Mann–Whitney U test. The percent of PVI+ compared with the number of PNN+ cells per animal was analyzed by Welch’s t-test. PVI+ and PNN+ cell population analyses were conducted by first sorting the data using frequency distribution, then compared between rmTBI and sham control group using Chi-square test.
EEG
Gamma power in the rmTBI group was normalized to sham and compared at 1- and 6-week time points using paired and unpaired Student’s t-tests.
PTZ Challenge
Latency to seizure onset was compared using Kaplan–Meier analysis and log-rank (Mantel–Cox) test.
Results
LOC After Sham and Verum Injury
Consistent with published data on the mouse rmTBI model (Mannix et al. 2014, 2016; Main et al. 2017; Shandra et al. 2019) verum injury resulted in ~18 s longer LOC compared with sham controls. Five-day averages for the sham and rmTBI groups were 35 ± 6 versus 53 ± 2 s (P < 0.01), respectively. Per day (days 1 to 5) mean LOC durations for sham or verum rmTBI groups were 29 ± 6 versus 56 ± 15, 37 ± 9 versus 52 ± 17, 39 ± 9 versus 54 ± 16, 34 ± 7 versus 50 ± 23, and 35 ± 8 versus 53 ± 21 seconds, respectively.
rmTBI Causes Early and Late Increases in Cortical Glu/GABA Ratio
rmTBI resulted in elevated extracellular Glu concentrations 1 week (rmTBI: 17.55 ± 1.72 μM, sham: 8.51 ± 0.18 μM; 106% increase, P < 0.0001) and 2 weeks (rmTBI: 11.92 ± 1.25 μM, sham: 8.56 ± 0.45 μM; 39% increase, P < 0.02) after final injury (Fig. 2A). However, Glu concentration normalized from the third week onward. In contrast to the early Glu excess, extracellular GABA remained stable during the first 3 weeks post-rmTBI and progressively declined thereafter (Fig. 2B), decreasing by 22% (rmTBI: 0.86 ± 0.07 μM, sham: 1.10 ± 0.04 μM; P < 0.05) at week 4, 26% (rmTBI: 0.79 ± 0.07 μM, sham: 1.07 ± 0.06 μM; P < 0.03) at week 5, and 47% (rmTBI: 0.56 ± 0.11 μM, sham: 1.04 ± 0.05 μM; P < 0.001) at week 6 after injury.
Figure 2 .
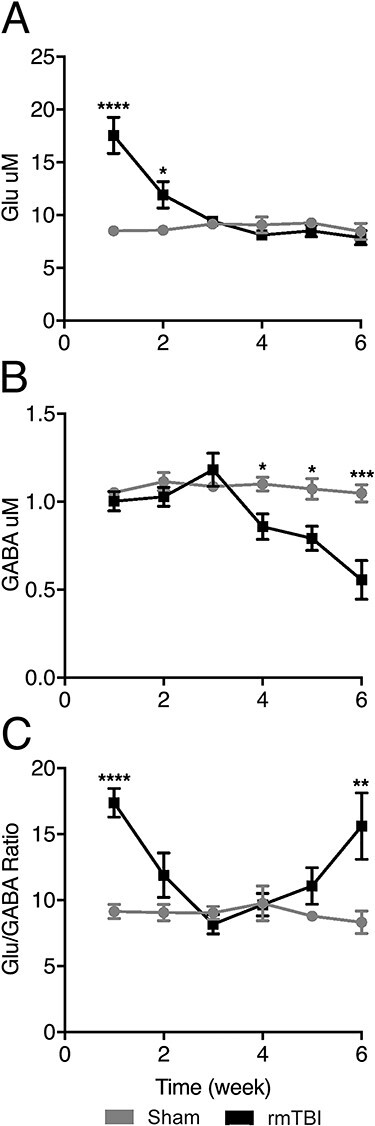
Extracellular levels of GABA and Glu in barrel cortex. GABA and Glu were measured weekly using microdialysis in rmTBI and sham-injured animals. (A) rmTBI increased extracellular Glu 1 and 2 weeks after injury. Glu concentration returned to baseline levels in week 3. (B) rmTBI decreased extracellular GABA at 4-, 5-, and 6-week post-rmTBI. (C) rmTBI caused 2 distinct shifts toward excitation at 1- and 6-week postinjury. The excitatory shift is driven by the excess Glu at 1 week and the paucity of GABA at 6 weeks after rmTBI. (Mean ± SEM, *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001).
The relative Glu to GABA ratio mirrored the changes in individual Glu and GABA levels. Injured mice had a significantly higher Glu/GABA ratio compared with controls (P < 0.0001) at 2 time points: 1 week after rmTBI, due to significantly increased extracellular Glu; and 6 weeks after final injury, this time driven by GABA decline (P < 0.003, Fig. 2C).
Our results thus demonstrate 2 distinct phases of neurotransmitter imbalance after rmTBI: (1) an acute phase characterized by increased extracellular Glu, followed by (2) a chronic phase characterized by a progressive decrease in GABA.
rmTBI Results in an Increase in Oxidative Stress
Oxidative stress is associated with progressive brain injury and is also a pathogenic regulator of PV expression as elevated levels are associated with PV loss (Cabungcal et al. 2013). Reduced and oxidized forms of the major intracellular antioxidant GSH were quantified to investigate the redox state of the barrel cortex.
Cortical GSH/GSSG ratios were reduced in injured mice at both one (19 ± 4.7% decrease, P < 0.01) and 6 weeks (38 ± 1.4% decrease, P < 0.0001) after injury, indicating persistent increased oxidative stress (Fig. 3).
Figure 3 .
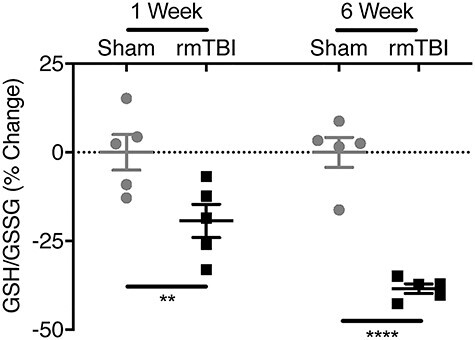
Intracellular redox status in barrel cortex. Reduced and oxidized glutathione (GSH and GSSG, respectively) were measured in barrel cortex using HPLC–electrochemical detection. The GSH/GSSG ratio was calculated to indicate redox state with lower ratios indicating increased oxidative stress. rmTBI increased oxidative stress at 1- and 6-week postinjury. (Bars indicate mean ± SEM, **P ≤ 0.01, ****P ≤ 0.0001).
rmTBI Causes Reversible Early PNN Loss, Followed by Progressive Loss of PV
Cortical PVI count increased by 29% over sham in injured mice (rmTBI: 129 ± 8%, normalized sham: 100 ± 3%; P < 0.03) 1 week after final injury, before falling to 60% of control levels (rmTBI: 60 ± 10%, normalized sham: 100 ± 3%; P < 0.01) 6-week post-rmTBI (Fig. 4A,B). Per cell PV intensity also declined by 42% (rmTBI: 58 ± 20%, normalized sham: 100 ± 8%; P < 0.05) in rmTBI mice 6 weeks after injury (Fig. 4A,C). PNN staining revealed a 17% decrease in PNN-enwrapped cells (rmTBI: 83 ± 2%, normalized sham: 100 ± 6%; P < 0.03) (Fig. 4A,D) and 38% PNN intensity loss (rmTBI: 63 ± 3%, normalized sham: 100 ± 5%; P < 0.03) in rmTBI mice (Fig. 4A,E) 1 week after injury. However, there was no difference in PNN staining between groups 6-week post-rmTBI.
Figure 4 .
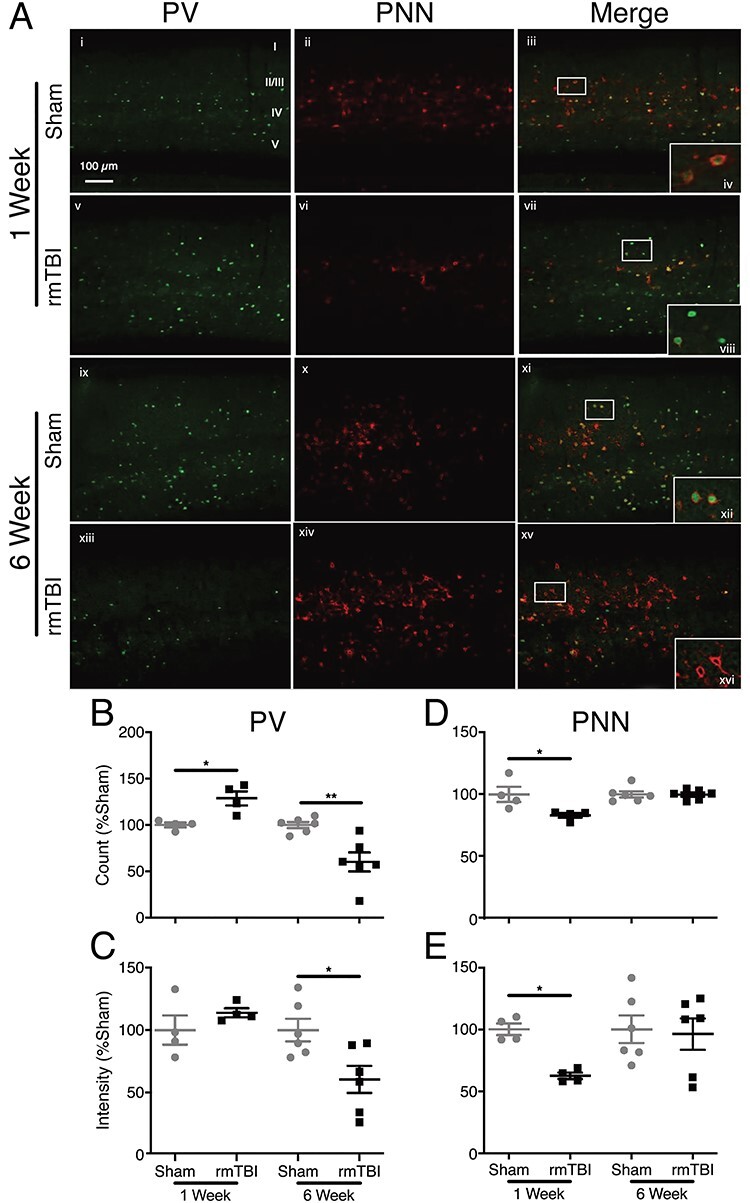
PV and PNN expression in barrel cortex. (A) IHC staining of barrel cortex. PV (green) and PNN (red). Callout top right healthy PVI enwrapped in healthy PNN. Callout second row PVI with degraded PNNs. Callout third row healthy PVI enwrapped in healthy PNN. Callout bottom “hollow” PNNs wrapping PVI that have lost PV expression. (B) There is an increase in PVI number 1 week after injury and a decrease in count 6 weeks after injury. (B) PV intensity does not change 1 week after injury; however, intensity decreases in injured mice at 6 weeks after rmTBI. (C) PNNs are reduced 1 week after injury and normalize by 6 weeks. (D) PNN intensity also decreases 1 week after rmTBI and normalizes 6 weeks after. (Bars indicate mean ± SEM, *P ≤ 0.05, **P ≤ 0.01).
To further investigate the relationship between these markers, we labeled all cells as either PV+, PNN+, or both, and computed the proportion of PNN+ cells that were also PV+. That is, we measured the percentage of PNN+ cells that were also PV+. While difference between rmTBI and sham control was absent 1 week after injury, 6 weeks after rmTBI, injured mice had a 19% reduction in the of PV+/PNN+ ratio (rmTBI: 81 ± 7%, sham: 100 ± 0.4%; P < 0.05, Fig. 5). When these cell populations were examined in their entirety (rather than as ratios), 1 week after injury, there was no change in PV expression (rmTBI: 929 ± 13, sham: 827 ± 15; Fig. 6A), whereas PNN decreased significantly (rmTBI: 702 ± 11, sham: 1319 ± 17; P < 0.0001, Fig. 6B). Six weeks after rmTBI, PV expression had shifted with 952 individual cells measuring 0–200 a.u. in injured mice compared with 75 in sham controls (P < 0.0001, Fig. 6C). PNN+ cells at 6 weeks had also shifted in the rmTBI group (P < 0.0001, Fig. 6D).
Figure 5 .
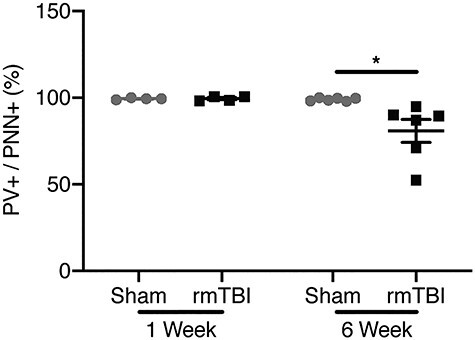
Per-cell expression of PV and PNN in barrel cortex. Percentage of PV+ cells per PNN+ cells per animal 1 and 6 weeks after verum or sham rmTBI. One week after injury, percent PV+/PNN+ cells in rmTBI animals is not different from sham animals. Six weeks after rmTBI, percentage of PV+ cell drops in mice exposed to verum rmTBI. (Bars indicate mean ± SEM, *P ≤ 0.05).
Figure 6 .
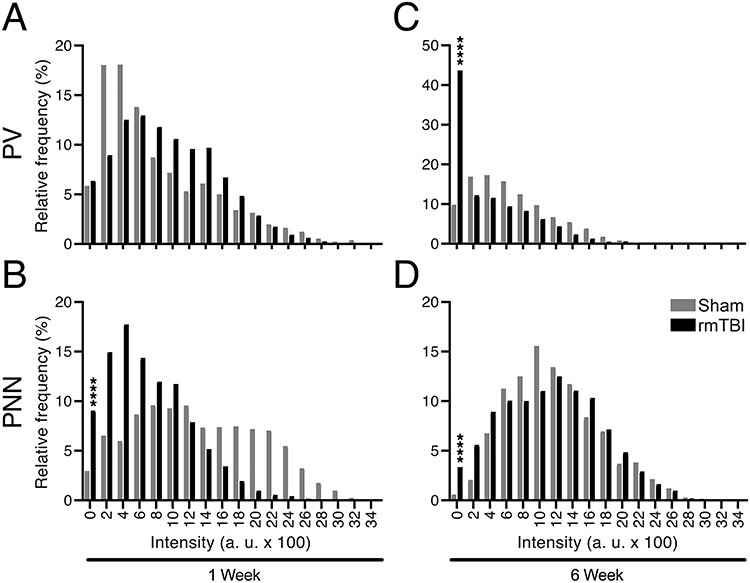
PV-positive and PNN-positive counts in the barrel cortex. Cells identified as PV+ and/or PNN+ were masked and PV and PNN fluorescent levels were measured separately for each masked area. Relative frequency of cells at given PV and PNN intensity levels were plotted. (A) One week after rmTBI relative percentage PV intensity values showed no significant difference between rmTBI and sham animals in the percentage of cells between 0 and 200 a.u. of all cells measured. However, a trend is observed toward a shift in intensity of PV cells in rmTBI group in bins 800–1800. (B) PNN intensity values at this time point decreased in rmTBI group, as 203 cells compared with 58 cells fell in the 0–200 a.u. category. (C) When PV+ cells were investigated 6 weeks after rmTBI, 952 cells fell between 0 and 200 a.u. in rmTBI animals compared with 75 cells in sham group. (D) PNN intensity values at this time point decreased as well in rmTBI group, as 95 cells compared with 14 cells fell in the 0–200 a.u. category (****P ≤ 0.0001).
Progressive Loss of Gamma EEG Power Follows rmTBI
A trend toward decreased cortical EEG gamma power (30–80 Hz) in the injured group was evident 6 weeks after rmTBI relative to sham controls, when EEG was analyzed in 24-h blocks (Fig. 7A). This trend was driven primarily by a significant reduction in power during the 12-h light phase when mice are less active (7 AM–7 PM), and where sham and injured groups clearly separate (P < 0.04, Fig. 7C). In contrast, difference in gamma power between groups is absent during the more active 12-h dark (7 AM–7 PM, Fig. 7B) portion of the circadian cycle.
Figure 7 .
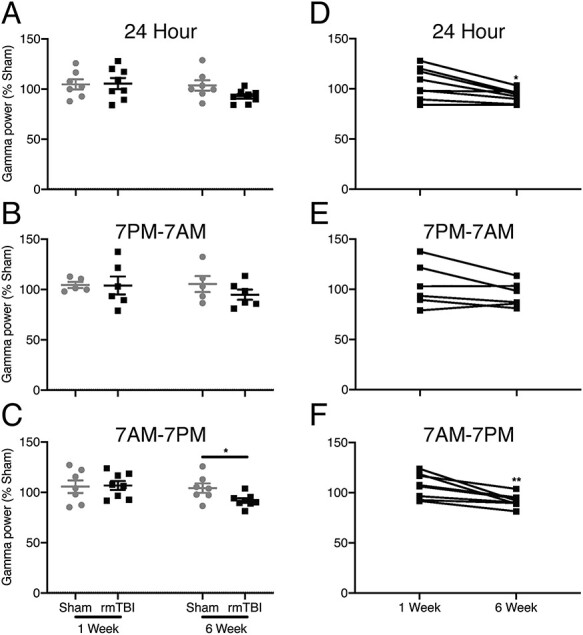
EEG gamma oscillations. (A) When tested across the full 24-h recording period, only a trend toward a decrease in gamma power 6 weeks after injury is evident (unpaired t-test, P = 0.066). (B) During the dark period from 7 PM to 7 AM when animals are more active no difference between sham- and verum-injured groups was found at either time point. (C) In contrast, during the light period from 7 PM to 7 AM when mice are less active and more likely to sleep, depression in gamma power in the verum, rmTBI group was evident 6 weeks after injury. (D) When individual mice were animals were followed longitudinally across both the 1- and 6-week time points, normalized (to sham) gamma power, as assessed over the 24-h period reliably declined 6 weeks after injury. (E) This difference in gamma power between initial and late recordings was absent in the active, dark, period 7 PM–7 AM, but (F) reliable when analysis was constrained to the inactive, light, period of 7 PM–7 AM. (Bars indicate mean ± SEM, *P ≤ 0.05, **P ≤ 0.01).
Longitudinal, paired analysis within the rmTBI group (normalized to sham control) between 1- and 6-week time points indicates that the loss of EEG gamma power is progressive. The loss of gamma power as a function of time is robust during the entire 24-h period (P < 0.02, Fig. 7D). Circadian variations in gamma power captured across the day (Fig. 7F) and night (Fig. 7E) cycles, indicate that the majority, but not the entirety, of gamma power occurs during sleep or inactivity as differences are strongly driven by the 12-h light (7 AM–7 PM) cycle (P < 0.01).
Induced Seizure Susceptibility Progressively Increases After rmTBI
There was a significant difference in the incidence of first myoclonus (Racine stage 3) after PTZ administration between injured mice and sham controls 1 week after rmTBI, with 21 of 30 (70%) injured mice experiencing a myoclonic seizure compared with 12 of 32 (38%) sham controls (P < 0.05, Fig. 8A). However, only 3 of 30 (10%) rmTBI animals and 3 of 32 (9%) sham mice progressed to GTC (not significant, Fig. 8B).
Figure 8 .
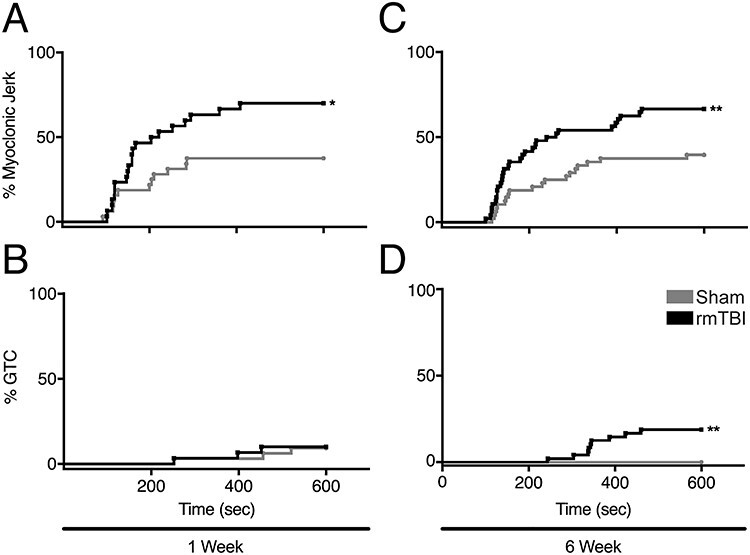
Latency to seizure induced by PTZ injection. (A) One week after rmTBI, 70% injured mice experienced a myoclonic seizure, compared with 38% uninjured control animals. Kaplan–Meier analysis displaying the incidence of first myoclonus in both groups revealed a difference between the curves. (B) Only 10% rmTBI animals and 9% sham mice had GTC at 1 week. (C) Six weeks after rmTBI, 67% mice reached Racine stage 3 in comparison to 40% uninjured sham animals. (D) However, 6 weeks after rmTBI, 19% mice had GTC, compared with 0% sham controls. (Mean ± SEM, *P ≤ 0.05, **P ≤ 0.01).
Progression to Racine stage 3 remained greater in the injured cohort (32/48; 67%) versus sham mice 6 weeks after rmTBI (19/48; 40%) (P < 0.01, Fig. 8C). Notably, however, a significantly increased incidence of GTC in injured mice (9/48; 19%) compared with sham controls (0/48; 0%) (P < 0.002, Fig. 8D) indicated progressively increase seizure susceptibility following injury.
Discussion
We demonstrate for the first time a neuronal basis for the pathologic E/I imbalance after rmTBI in mice. Our results indicate that an increase in cortical excitability after repetitive brain trauma is initially driven by an increase in extracellular Glu, which presumably drives PV expression and increases oxidative stress, resulting in a loss of PNNs 1 week after injury. This initial increase in Glu-mediated excitability is accompanied by GSH consumption, and subsequently followed by an inhibitory decline driven by loss of both PV and extracellular GABA. Notably, injured mice express a shift in the E/I balance toward excitation, both subacutely (driven by increased extracellular Glu) and chronically (driven by reduced extracellular GABA), signifying distinct mechanisms for the early and late stages of increased seizure susceptibility (and perhaps other postconcussive symptoms) following rmTBI.
In contrast to severe TBI where only progressive PVI and PNN loss has been recorded in animal models (Hsieh et al. 2017; Hameed et al. 2019), here we report a subacute increase in PV expression within PVIs and loss of PNNs surrounding PVIs after rmTBI. Greater PV expression 1 week after mild repetitive injury may be related to the increased extracellular Glu, as increased glutamatergic tone has been reported to drive activity-dependent PVI maturation, resulting in increased PV expression (Donato et al. 2013). It would follow that the increased PVI count observed at this time point is presumably a consequence of enhanced PV in cells that previously expressed PV below detectable levels, rather than de novo PVI formation.
This increase in extracellular Glu may well lead to excitotoxic neuronal injury, particularly to PVIs whose high baseline metabolic demand renders them disproportionately vulnerable to post-traumatic metabolic challenges (Sohal et al. 2009; Cantu et al. 2015; Hsieh et al. 2017; Hameed et al. 2019). Elevated glutamatergic tone has been shown to drive oxidative stress in PVIs (Behrens et al. 2007; Powell et al. 2012) and is associated with PNN degradation (Cabungcal et al. 2013). The removal of PNN from PVIs increases their sensitivity to further oxidative stress, thus creating a negative feedback loop that may ultimately lead to their dysfunction or death (Cabungcal et al. 2013). For example, iron-induced oxidative stress following mTBI might contribute to further redox imbalance, which, without adequate PNN to mitigate, would lower the threshold for exacerbated cellular damage upon repeated injury (Nisenbaum et al. 2014).
Interestingly, the increased PVI count 1 week after rmTBI did not correlate with a higher extracellular concentration of GABA, suggesting that the compensatory increases in PVI number and PV expression are insufficient to completely counterbalance the elevated extracellular Glu, hence the increase in E/I ratio and seizure susceptibility in the subacute postinjury phase—exhibited by decreased latency to myoclonic seizures after rmTBI. However, given that only a fraction of the injured mice progressed to GTC, PV upregulation may well have prevented generalization of the seizures.
Six weeks after rmTBI, a distinct second pathophysiology emerged, characterized by a sharp decline in extracellular GABA beginning 4 weeks after injury. Depressed GABA concentrations are accompanied by decreased per-cell PV expression, in stark contrast to the increase in PV expression seen 1 week after rmTBI where a concomitant increase in extracellular GABA was not observed. We propose that the depressed GABA in the late post-TBI phase result at least in part from the per-cell PV loss. Normal intracellular PV concentration does not necessitate fast PVI firing or enhanced GABA signaling. Rather, robust PV expression simply allows for the possibility of increased PVI firing frequency. This is due in part to the calcium-chelating property of PV that enables PVIs to depolarize and repolarize rapidly without sustaining injury that may lead to dysfunction or death (Volman et al. 2011; Steullet et al. 2017). Accordingly, we propose that the depressed per-cell PV density (at times to below detection by our methods), limits PVI firing capacity and thus directly relates to the depressed extracellular GABA levels and increased seizure susceptibility in the late post-rmTBI phase.
A loss of gamma EEG power accompanies the depressed GABA concentration and PV expression 6 weeks after injury. Local oscillations in the gamma range (30–80 Hz) in the cortical EEG are driven largely by the resonance of fast-spiking PVIs (Cardin et al. 2009; Carlen et al. 2012). Given that PV expression is significantly reduced after brain injury (Hsieh et al. 2017), and that cortical gamma EEG oscillations are driven by PVI circuits (Volman et al. 2011), our results suggest that the progressive decrease in cortical EEG gamma power reflects post-traumatic PVI dysfunction, thus supporting a direct relationship between the two, and utility of cortical gamma EEG as a biomarker that is representative of PVI-mediated cortical inhibitory tone.
While PNN loss in a high-Glu and low-GSH state indicates extracellular matrix degradation in the initial post-rmTBI state, eventual recovery suggests a certain stability of the new PV expression state as enhanced PNNs are associated with a loss of plasticity (Hensch 2005). This recovery in the context of high oxidative stress due to decreased GSH/GSSG may be a compensatory adjustment, but is nonetheless insufficient to arrest the PV loss in the chronic postconcussive state. On the contrary, the restored PNN may stabilize a pathologic state of depressed PV expression that corresponds to the observed decrease in extracellular GABA in the chronic post-rmTBI period.
Distinct PVI states emerged at the 2 time points examined in this experiment. One week after injury, cells with upregulated PV expression were present above levels of the control condition. We propose that these inhibitory interneurons upregulated their expression of PV as a result of activity-dependent maturation within the context of increased extracellular Glu. Concurrently, PNNs were degraded due to a Glu-mediated increase in oxidative stress. Loss or damage to PNNs leaves PVI vulnerable to persistent oxidative stress (Cabungcal et al. 2013), yet also more plastic (Beurdeley et al. 2012; Spatazza et al. 2013). This acute (PV+/PNN−) PVI state is indicative of increased activity-dependent plasticity at the cost of decreased structural stability. 6-week post rmTBI, PNN recovered, but PV expression had dropped below levels of detection by IHC, though the cells remained viable via DAPI. These PV−/PNN+ cells are structurally intractable with an impaired ability to sequester calcium, resulting in a reduced firing capacity. It is unlikely these cells are functionally similar to PV+/PNN+ PVIs.
The presence of each of these cell types within their environmental context in rmTBI creates a shift toward excitation in the E/I balance but for opposing reasons. Initially, the brain compensates for the increase in Glu by increasing PV expression in PVIs, which are more plastic without their nets. However, this is an incomplete compensation as PV+/PNN− cells are more sensitive to oxidative stress, and injured animals still showed increased seizure susceptibility. Ultimately, this compensation fails when PV expression falls to 60% of sham and PNN recover, dampening plasticity and firing rate and resulting in altered PVI activity and increased seizure susceptibility. These distinct states provide insight into the need for differing therapeutic strategies as a function of time from injury. Early-stage intervention may focus on Glu buffering and oxidative stress protection to prevent loss of PNN and unsustainable PVI maturation, whereas long-term therapies target interfering with GABA clearance in order to preserve some tone or mitigating oxidative stress to prevent further decline. To date, N-methyl-D-aspartate-type glutamate receptor (NMDAR) antagonism with memantine has been tested as an intervention after rmTBI (Mei et al. 2018). While neuroprotective, memantine decreases PV expression in PVI (Powell et al. 2012). The baseline reduction of GABA concentration and impaired PVI function in our study may explain why previous trials of memantine were incompletely effective at alleviating behavioral rmTBI symptoms (Mei et al. 2018). Specifically, memantine therapy reduced reactive astrocytosis after rmTBI but did not mitigate behavioral deficits. Since decreased PV expression in an already compromised system may worsen the chronic post-rmTBI condition, our results indicate that strategies other than NMDAR antagonism may have utility in this setting.
Our results identify mechanistic similarities between rmTBI and more severe brain injury. Among the explanations for the similarity may be reactive astrocytosis, which is a pathologic feature described after mouse rmTBI (Mannix et al. 2014). Reactive astrocytosis is associated with downregulation of the membrane transporter protein GLT-1, which is responsible for clearing excess Glu from the extracellular space. Both are also present shortly after TBI by lateral fluid percussion injury (Goodrich et al. 2013), and this paucity of GLT-1 presumably further impairs buffering of any increased Glu in the extracellular space. As in rmTBI, the susceptibility of PVI to injury is also present in the more severe TBI model (Hsieh et al. 2017). These commonalities suggest that the phenotype produced from rmTBI may have more serious consequences than initially anticipated. In particular, the PVI loss and increased seizure susceptibility in the rmTBI point to a mechanistic picture that is qualitatively similar across a range of TBI severities.
Increased seizure susceptibility after a minimum of 1 concussion has been documented in patients (Christensen et al. 2009; Hung et al. 2014; Mahler et al. 2015; Keret et al. 2017), with a relative risk of developing epilepsy approximately doubling after injury. These clinical data along with our finding of lowered seizure threshold after rmTBI indicate that postconcussive shift in the E/I balance, even if modest, can increase seizure susceptibility among other long-term consequences. Given that concussion is associated with increased vulnerability to future trauma (Fehily and Fitzgerald 2017), our results indicate that the successive injury may occur in the setting of already compromised inhibition that can predispose traumatized brains to worse outcomes even from mild future insult. This is analogous to the higher incidence of epilepsy in patients who suffer concussion and have a family history of epilepsy (Christensen et al. 2009). Preventative treatment modalities for post-traumatic epilepsy, even after mild repetitive brain injury, thus warrant consideration, particularly as about 30% of acquired epilepsy cases are intractable (Elliott et al. 2019).
Finally, while the temporal pattern of injury in our model (5 consecutive daily injuries) is distinct from typical human concussion patterns, we note that concussion is associated with increased vulnerability to future trauma (Fehily and Fitzgerald 2017). Our results thus indicate that compromised inhibition might predispose traumatized brains to worse outcomes upon successive mild injuries.
Conclusion
Repetitive concussion causes a progressive phenotype characterized by an initial Glu increase that is accompanied by GSH consumption and followed by a dysfunction of PVIs and loss of GABAergic inhibitory tone. This is in accordance with clinical observations indicating an increased risk of developing epilepsy after mild brain injury. From a therapeutic perspective, this progression of pathophysiology needs to be considered, as pharmacologic targets are likely to be distinct at the individual phases of the post-rmTBI syndrome. The PV loss and loss of extracellular GABA in the chronic post-rmTBI phase also provide mechanistic insight into the increased epilepsy risk that follows concussion in humans (Christensen et al. 2009; Hung et al. 2014; Mahler et al. 2015). In this regard, while direct GABA measurements are impractical in clinical practice, the loss of gamma power on EEG may serve as a biomarker for the loss of cortical inhibition and subtle epileptogenesis that follows concussion. Along similar lines, mapping inhibitory tone by gamma EEG measures may also enable markers of target engagement by therapeutics that are aimed to arrest or reverse the above-described process.
P.M. and N.H. contributed equally to this work. Authors’ Contributions: Study concept and design: P.M., N.H., R.M., T.H. and A.R. Data acquisition and analysis: all authors. Drafting the manuscript and figures: all authors
Funding
This work was part of the Football Players Health Study at Harvard, funded by a grant from the National Football League Players Association (NFLPA), NIH R01NS088583 (to A.R.) and Japan Society for the Promotion of Science (JSPS) (WPI-IRCN to T.K.H.)
Notes
We thank the Harvard Center for Biological Imaging for infrastructure and support, and the Animal Behavior and Physiology Core (CHB IDDRC U54HD090255). Authors thank Alisa Pasichnik for her assistance in seizure scoring.
Conflict of Interest: None declared.
References
- Alosco ML, Mez J, Tripodis Y, Kiernan PT, Abdolmohammadi B, Murphy L, Kowall NW, Stein TD, Huber BR, Goldstein LE, et al. 2018. Age of first exposure to tackle football and chronic traumatic encephalopathy. Ann Neurol. 83:886–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andary MT, Crewe N, Ganzel SK, Haines-Pepi C, Kulkarni MR, Stanton DF, Thompson A, Yosef M. 1997. Traumatic brain injury/chronic pain syndrome: a case comparison study. Clin J Pain. 13:244–250. [DOI] [PubMed] [Google Scholar]
- Annegers JF, Hauser WA, Coan SP, Rocca WA. 1998. A population-based study of seizures after traumatic brain injuries. N Engl J Med. 338:20–24. [DOI] [PubMed] [Google Scholar]
- Behrens MM, Ali SS, Dao DN, Lucero J, Shekhtman G, Quick KL, Dugan LL. 2007. Ketamine-induced loss of phenotype of fast-spiking interneurons is mediated by NADPH-oxidase. Science. 318:1645–1647. [DOI] [PubMed] [Google Scholar]
- Beurdeley M, Spatazza J, Lee HH, Sugiyama S, Bernard C, Di Nardo AA, Hensch TK, Prochiantz A. 2012. Otx2 binding to perineuronal nets persistently regulates plasticity in the mature visual cortex. J Neurosci Off J Soc Neurosci. 32:9429–9437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biagianti B, Stocchetti N, Brambilla P, Van Vleet T. 2020. Brain dysfunction underlying prolonged post-concussive syndrome: a systematic review. J Affect Disord. 262:71–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabungcal JH, Steullet P, Morishita H, Kraftsik R, Cuenod M, Hensch TK, Do KQ. 2013. Perineuronal nets protect fast-spiking interneurons against oxidative stress. Proc Natl Acad Sci U S A. 110:9130–9135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantu D, Walker K, Andresen L, Taylor-Weiner A, Hampton D, Tesco G, Dulla CG. 2015. Traumatic brain injury increases cortical glutamate network activity by compromising GABAergic control. Cereb Cortex. 25:2306–2320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardin JA, Carlen M, Meletis K, Knoblich U, Zhang F, Deisseroth K, Tsai LH, Moore CI. 2009. Driving fast-spiking cells induces gamma rhythm and controls sensory responses. Nature. 459:663–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlen M, Meletis K, Siegle JH, Cardin JA, Futai K, Vierling-Claassen D, Ruhlmann C, Jones SR, Deisseroth K, Sheng M, et al. 2012. A critical role for NMDA receptors in parvalbumin interneurons for gamma rhythm induction and behavior. Mol Psychiatry. 17:537–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castile L, Collins CL, McIlvain NM, Comstock RD. 2012. The epidemiology of new versus recurrent sports concussions among high school athletes, 2005-2010. Br J Sports Med. 46:603–610. [DOI] [PubMed] [Google Scholar]
- Christensen J, Pedersen MG, Pedersen CB, Sidenius P, Olsen J, Vestergaard M. 2009. Long-term risk of epilepsy after traumatic brain injury in children and young adults: a population-based cohort study. Lancet. 373:1105–1110. [DOI] [PubMed] [Google Scholar]
- Cole WR, Bailie JM. 2016. Neurocognitive and psychiatric symptoms following mild traumatic brain injury. In: Laskowitz D, Grant G, editors. Translational research in traumatic brain injury. Boca Raton (FL): CRC Press/Taylor and Francis Group. [PubMed] [Google Scholar]
- Daneshvar DH, Riley DO, Nowinski CJ, McKee AC, Stern RA, Cantu RC. 2011. Long-term consequences: effects on normal development profile after concussion. Phys Med Rehabil Clin N Am. 22:683–700 ix. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhamne SC, Ekstein D, Zhuo Z, Gersner R, Zurakowski D, Loddenkemper T, Pascual-Leone A, Jensen FE, Rotenberg A. 2015. Acute seizure suppression by transcranial direct current stimulation in rats. Ann Clin Transl Neurol. 2:843–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhamne SC, Silverman JL, Super CE, Lammers SHT, Hameed MQ, Modi ME, Copping NA, Pride MC, Smith DG, Rotenberg A, et al. 2017. Replicable in vivo physiological and behavioral phenotypes of the Shank3B null mutant mouse model of autism. Mol Autism. 8:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donato F, Rompani SB, Caroni P. 2013. Parvalbumin-expressing basket-cell network plasticity induced by experience regulates adult learning. Nature. 504:272–276. [DOI] [PubMed] [Google Scholar]
- Eisenberg MA, Andrea J, Meehan W, Mannix R. 2013. Time interval between concussions and symptom duration. Pediatrics. 132:8–17. [DOI] [PubMed] [Google Scholar]
- Elliott J, van Katwyk S, McCoy B, Clifford T, Potter BK, Skidmore B, Wells GA, Coyle D. 2019. Decision models for assessing the cost effectiveness of treatments for pediatric drug-resistant epilepsy: a systematic review of economic evaluations. Pharmacoeconomics. 37:1261–1276. [DOI] [PubMed] [Google Scholar]
- Favuzzi E, Marques-Smith A, Deogracias R, Winterflood CM, Sanchez-Aguilera A, Mantoan L, Maeso P, Fernandes C, Ewers H, Rico B. 2017. Activity-dependent gating of parvalbumin interneuron function by the perineuronal net protein Brevican. Neuron. 95:639–655.e610. [DOI] [PubMed] [Google Scholar]
- Fehily B, Fitzgerald M. 2017. Repeated mild traumatic brain injury: potential mechanisms of damage. Cell Transplant. 26:1131–1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gersner R, Ekstein D, Dhamne SC, Schachter SC, Rotenberg A. 2015. Huperzine A prophylaxis against pentylenetetrazole-induced seizures in rats is associated with increased cortical inhibition. Epilepsy Res. 117:97–103. doi: 10.1016/j.eplepsyres.2015.08.012. [DOI] [PubMed] [Google Scholar]
- Goodrich GS, Kabakov AY, Hameed MQ, Dhamne SC, Rosenberg PA, Rotenberg A. 2013. Ceftriaxone treatment after traumatic brain injury restores expression of the glutamate transporter, GLT-1, reduces regional gliosis, and reduces post-traumatic seizures in the rat. J Neurotrauma. 30:1434–1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grandhi R, Tavakoli S, Ortega C, Simmonds MJ. 2017. A review of chronic pain and cognitive, mood, and motor dysfunction following mild traumatic brain injury: complex, comorbid, and/or overlapping conditions? Brain Sci. 7:160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerriero RM, Proctor MR, Mannix R, Meehan WP 3rd. 2012. Epidemiology, trends, assessment and management of sport-related concussion in United States high schools. Curr Opin Pediatr. 24:696–701. [DOI] [PubMed] [Google Scholar]
- Hameed MQ, Hsieh TH, Morales-Quezada L, Lee HHC, Damar U, MacMullin PC, Hensch TK, Rotenberg A. 2019. Ceftriaxone treatment preserves cortical inhibitory interneuron function via transient salvage of GLT-1 in a rat traumatic brain injury model. Cereb Cortex. 29:4506–4518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harmon KG, Drezner JA, Gammons M, Guskiewicz KM, Halstead M, Herring SA, Kutcher JS, Pana A, Putukian M, Roberts WO. 2013. American Medical Society for Sports Medicine position statement: concussion in sport. Br J Sports Med. 47:15–26. [DOI] [PubMed] [Google Scholar]
- Hensch TK. 2005. Critical period mechanisms in developing visual cortex. Curr Top Dev Biol. 69:215–237. [DOI] [PubMed] [Google Scholar]
- Howell DR, Beasley M, Vopat L, Meehan WP 3rd. 2017. The effect of prior concussion history on dual-task gait following a concussion. J Neurotrauma. 34:838–844. [DOI] [PubMed] [Google Scholar]
- Howell DR, Lynall RC, Buckley TA, Herman DC. 2018. Neuromuscular control deficits and the risk of subsequent injury after a concussion: a scoping review. Sports Med. 48:1097–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh TH, Lee HHC, Hameed MQ, Pascual-Leone A, Hensch TK, Rotenberg A. 2017. Trajectory of parvalbumin cell impairment and loss of cortical inhibition in traumatic brain injury. Cereb Cortex. 27:5509–5524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung R, Carroll LJ, Cancelliere C, Cote P, Rumney P, Keightley M, Donovan J, Stalnacke BM, Cassidy JD. 2014. Systematic review of the clinical course, natural history, and prognosis for pediatric mild traumatic brain injury: results of the international collaboration on mild traumatic brain injury prognosis. Arch Phys Med Rehabil. 95:S174–S191. [DOI] [PubMed] [Google Scholar]
- Kelly E, Schaeffer SM, Dhamne SC, Lipton JO, Lindemann L, Honer M, Jaeschke G, Super CE, Lammers SH, Modi ME, et al. 2018. mGluR5 modulation of behavioral and epileptic phenotypes in a mouse model of tuberous sclerosis complex. Neuropsychopharmacology. 43:1457–1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keret A, Bennett-Back O, Rosenthal G, Gilboa T, Shweiki M, Shoshan Y, Benifla M. 2017. Posttraumatic epilepsy: long-term follow-up of children with mild traumatic brain injury. J Neurosurg Pediatr. 20:64–70. [DOI] [PubMed] [Google Scholar]
- Khuman J, Meehan WP 3rd, Zhu X, Qiu J, Hoffmann U, Zhang J, Giovannone E, Lo EH, Whalen MJ. 2011. Tumor necrosis factor alpha and Fas receptor contribute to cognitive deficits independent of cell death after concussive traumatic brain injury in mice. J Cereb Blood Flow Metab. 31:778–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanza G, Ferri R. 2019. The neurophysiology of hyperarousal in restless legs syndrome: hints for a role of glutamate/GABA. Adv Pharmacol. 84:101–119. [DOI] [PubMed] [Google Scholar]
- Lein ES, Hawrylycz MJ, Ao N, Ayres M, Bensinger A, Bernard A, Boe AF, Boguski MS, Brockway KS, Byrnes EJ, et al. 2007. Genome-wide atlas of gene expression in the adult mouse brain. Nature. 445:168–176. [DOI] [PubMed] [Google Scholar]
- Lissemore JI, Bhandari A, Mulsant BH, Lenze EJ, Reynolds CF 3rd, Karp JF, Rajji TK, Noda Y, Zomorrodi R, Sibille E, et al. 2018. Reduced GABAergic cortical inhibition in aging and depression. Neuropsychopharmacology. 43:2277–2284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucke-Wold BP, Nguyen L, Turner RC, Logsdon AF, Chen YW, Smith KE, Huber JD, Matsumoto R, Rosen CL, Tucker ES, et al. 2015. Traumatic brain injury and epilepsy: underlying mechanisms leading to seizure. Seizure. 33:13–23. [DOI] [PubMed] [Google Scholar]
- Luscher B, Fuchs T. 2015. GABAergic control of depression-related brain states. Adv Pharmacol. 73:97–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahler B, Carlsson S, Andersson T, Adelow C, Ahlbom A, Tomson T. 2015. Unprovoked seizures after traumatic brain injury: a population-based case-control study. Epilepsia. 56:1438–1444. [DOI] [PubMed] [Google Scholar]
- Main BS, Sloley SS, Villapol S, Zapple DN, Burns MP. 2017. A mouse model of single and repetitive mild traumatic brain injury. J Vis Exp JoVE. 124:e55713. doi: 10.3791/55713(2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mannix R, Berglass J, Berkner J, Moleus P, Qiu J, Andrews N, Gunner G, Berglass L, Jantzie LL, Robinson S, et al. 2014. Chronic gliosis and behavioral deficits in mice following repetitive mild traumatic brain injury. J Neurosurg. 121:1342–1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mannix R, Meehan WP 3rd, Pascual-Leone A. 2016. Sports-related concussions—media, science and policy. Nat Rev Neurol. 12:486–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mannix R, Meehan WP, Mandeville J, Grant PE, Gray T, Berglass J, Zhang J, Bryant J, Rezaie S, Chung JY, et al. 2013. Clinical correlates in an experimental model of repetitive mild brain injury. Ann Neurol. 74:65–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantua J, Henry OS, Garskovas NF, Spencer RMC. 2017. Mild traumatic brain injury chronically impairs sleep- and wake-dependent emotional processing. Sleep. 40:zsx062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marar M, McIlvain NM, Fields SK, Comstock RD. 2012. Epidemiology of concussions among United States high school athletes in 20 sports. Am J Sports Med. 40:747–755. [DOI] [PubMed] [Google Scholar]
- McInnes K, Friesen CL, MacKenzie DE, Westwood DA, Boe SG. 2017. Mild traumatic brain injury (mTBI) and chronic cognitive impairment: a scoping review. PLoS One. 12:e0174847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNair LF, Andersen JV, Aldana BI, Hohnholt MC, Nissen JD, Sun Y, Fischer KD, Sonnewald U, Nyberg N, Webster SC, et al. 2019. Deletion of neuronal GLT-1 in mice reveals its role in synaptic glutamate homeostasis and mitochondrial function. J Neurosci. 39:4847–4863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meehan WP 3rd, Zhang J, Mannix R, Whalen MJ. 2012. Increasing recovery time between injuries improves cognitive outcome after repetitive mild concussive brain injuries in mice. Neurosurgery. 71:885–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mei Z, Qiu J, Alcon S, Hashim J, Rotenberg A, Sun Y, Meehan WP 3rd, Mannix R. 2018. Memantine improves outcomes after repetitive traumatic brain injury. Behav Brain Res. 340:195–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mez J, Daneshvar DH, Kiernan PT, Abdolmohammadi B, Alvarez VE, Huber BR, Alosco ML, Solomon TM, Nowinski CJ, McHale L, et al. 2017. Clinicopathological evaluation of chronic traumatic encephalopathy in players of American football. JAMA. 318:360–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson LD, Temkin NR, Dikmen S, Barber J, Giacino JT, Yuh E, Levin HS, McCrea MA, Stein MB, Mukherjee P, et al. 2019. Recovery after mild traumatic brain injury in patients presenting to US level I trauma centers: a Transforming Research and Clinical Knowledge in Traumatic Brain Injury (TRACK-TBI) Study. JAMA Neurol. 76:1049–1059. doi: 10.1001/jamaneurol.2019.1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nisenbaum EJ, Novikov DS, Lui YW. 2014. The presence and role of iron in mild traumatic brain injury: an imaging perspective. J Neurotrauma. 31:301–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panayiotou A, Jackson M, Crowe SF. 2010. A meta-analytic review of the emotional symptoms associated with mild traumatic brain injury. J Clin Exp Neuropsychol. 32:463–473. [DOI] [PubMed] [Google Scholar]
- Parker RS, Lewis GN, Rice DA, McNair PJ. 2016. Is motor cortical excitability altered in people with chronic pain? A Systematic Review and Meta-Analysis. Brain Stimul. 9:488–500. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Franklin KBJ. 2013. Paxinos and Franklin’s the mouse brain in stereotaxic coordinates. Amsterdam: Elsevier/AP. [Google Scholar]
- Powell SB, Sejnowski TJ, Behrens MM. 2012. Behavioral and neurochemical consequences of cortical oxidative stress on parvalbumin-interneuron maturation in rodent models of schizophrenia. Neuropharmacology. 62:1322–1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purtell H, Dhamne SC, Gurnani S, Bainbridge E, Modi ME, Lammers SHT, Super CE, Hameed MQ, Johnson EL 3rd, Sahin M, et al. 2018. Electrographic spikes are common in wildtype mice. Epilepsy Behav. 89:94–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao V, Bertrand M, Rosenberg P, Makley M, Schretlen DJ, Brandt J, Mielke MM. 2010. Predictors of new-onset depression after mild traumatic brain injury. J Neuropsychiatry Clin Neurosci. 22:100–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutherford WH, Merrett JD, McDonald JR. 1979. Symptoms at one year following concussion from minor head injuries. Injury. 10:225–230. [DOI] [PubMed] [Google Scholar]
- Shandra O, Winemiller AR, Heithoff BP, Munoz-Ballester C, George KK, Benko MJ, Zuidhoek IA, Besser MN, Curley DE, Edwards GF, et al. 2019. Repetitive diffuse mild traumatic brain injury causes an atypical astrocyte response and spontaneous recurrent seizures. J Neurosci Off J Soc Neurosci. 39:1944–1963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohal VS, Zhang F, Yizhar O, Deisseroth K. 2009. Parvalbumin neurons and gamma rhythms enhance cortical circuit performance. Nature. 459:698–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorg BA, Berretta S, Blacktop JM, Fawcett JW, Kitagawa H, Kwok JC, Miquel M. 2016. Casting a wide net: role of perineuronal nets in neural plasticity. J Neurosci Off J Soc Neurosci. 36:11459–11468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spatazza J, Lee HH, Di Nardo AA, Tibaldi L, Joliot A, Hensch TK, Prochiantz A. 2013. Choroid-plexus-derived Otx2 homeoprotein constrains adult cortical plasticity. Cell Rep. 3:1815–1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein MB, Jain S, Giacino JT, Levin H, Dikmen S, Nelson LD, Vassar MJ, Okonkwo DO, Diaz-Arrastia R, Robertson CS, et al. 2019. Risk of posttraumatic stress disorder and major depression in civilian patients after mild traumatic brain injury: a TRACK-TBI study. JAMA Psychiatry. 76:249–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steullet P, Cabungcal JH, Coyle J, Didriksen M, Gill K, Grace AA, Hensch TK, LaMantia AS, Lindemann L, Maynard TM, et al. 2017. Oxidative stress-driven parvalbumin interneuron impairment as a common mechanism in models of schizophrenia. Mol. Psychiatry. 22:936–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suttkus A, Rohn S, Weigel S, Glockner P, Arendt T, Morawski M. 2014. Aggrecan, link protein and tenascin-R are essential components of the perineuronal net to protect neurons against iron-induced oxidative stress. Cell Death Dis. 5:e1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theadom A, Parmar P, Jones K, Barker-Collo S, Starkey NJ, KM MP, Ameratunga S, Feigin VL, Group BR . 2015. Frequency and impact of recurrent traumatic brain injury in a population-based sample. J Neurotrauma. 32:674–681. [DOI] [PubMed] [Google Scholar]
- Trevelyan AJ, Muldoon SF, Merricks EM, Racca C, Staley KJ. 2015. The role of inhibition in epileptic networks. J Clin Neurophysiol. 32:227–234. [DOI] [PubMed] [Google Scholar]
- Trivedi M, Shah J, Hodgson N, Byun HM, Deth R. 2014. Morphine induces redox-based changes in global DNA methylation and retrotransposon transcription by inhibition of excitatory amino acid transporter type 3-mediated cysteine uptake. Mol Pharmacol. 85:747–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viola-Saltzman M, Watson NF. 2012. Traumatic brain injury and sleep disorders. Neurol Clin. 30:1299–1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volman V, Behrens MM, Sejnowski TJ. 2011. Downregulation of parvalbumin at cortical GABA synapses reduces network gamma oscillatory activity. J Neurosci. 31:18137–18148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yizhar O, Fenno LE, Prigge M, Schneider F, Davidson TJ, O'Shea DJ, Sohal VS, Goshen I, Finkelstein J, Paz JT, et al. 2011. Neocortical excitation/inhibition balance in information processing and social dysfunction. Nature. 477:171–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuskaitis CJ, Jones BM, Wolfson RL, Super CE, Dhamne SC, Rotenberg A, Sabatini DM, Sahin M, Poduri A. 2018. A mouse model of DEPDC5-related epilepsy: neuronal loss of Depdc5 causes dysplastic and ectopic neurons, increased mTOR signaling, and seizure susceptibility. Neurobiol Dis. 111:91–101. [DOI] [PMC free article] [PubMed] [Google Scholar]