

Abstract
Background
Limited data exist on the disease course of neurofibromatosis type 2 (NF2) to guide clinical trial design.
Methods
A prospective database of patients meeting NF2 diagnostic criteria, reviewed between 1990 and 2020, was evaluated. Follow-up to first vestibular schwannoma (VS) intervention and death was assessed by univariate analysis and stratified by age at onset, era referred, and inheritance type. Interventions for NF2-related tumors were assessed. Cox regression was performed to determine the relationship between individual factors from time of diagnosis to NF2-related death.
Results
Three hundred and fifty-three patients were evaluated. During 4643.1 follow-up years from diagnosis to censoring, 60 patients (17.0%) died. The annual mean number of patients undergoing VS surgery or radiotherapy declined, from 4.66 and 1.65, respectively, per 100 NF2 patients in 1990-1999 to 2.11 and 1.01 in 2010-2020, as the number receiving bevacizumab increased (2.51 per 100 NF2 patients in 2010-2020). Five patients stopped bevacizumab to remove growing meningioma or spinal schwannoma. 153/353 (43.3%) had at least one neurosurgical intervention/radiation treatment within 5 years of diagnosis. Patients asymptomatic at diagnosis had longer time to intervention and better survival compared to those presenting with symptoms. Those symptomatically presenting <16 and >40 years had poorer overall survival than those presenting at 26-39 years (P = .03 and P = .02, respectively) but those presenting between 16 and 39 had shorter time to VS intervention. Individuals with de novo constitutional variants had worse survival than those with de novo mosaic or inherited disease (P = .004).
Conclusion
Understanding disease course improves prognostication, allowing for better-informed decisions about care.
Keywords: ependymoma, Manchester criteria, meningioma, neurofibromatosis type 2, vestibular schwannoma
Key Points.
Large single-center follow-up study of 353 NF2 patients over a 30-year time period.
Data collated provide clinicians with valuable prognostic information related to disease progression and survival that can inform care and guide clinical trial design.
Importance of the Study.
We describe the natural history of NF2 from diagnosis to first therapeutic intervention for vestibular schwannoma (VS), as well as the subsequent clinically managed disease course observed in a high-volume multidisciplinary specialist clinic. Survival trends and patterns of intervention utilized over time are documented according to age at symptom onset, era referred, and inheritance type.
Key observations are the improved outcomes (better survival and longer time period to intervention) for those asymptomatic at diagnosis, with inherited disease, and large NF2 deletions. Over time, the number of patients requiring more invasive neurosurgical measures to treat VS has decreased as the use of bevacizumab has increased. Time to invasive intervention has also significantly increased.
Improved knowledge of disease course helps guide future clinical trial design and allows patients and clinicians to make better-informed decisions about care, including treatment modality and timing.
Neurofibromatosis type 2 (NF2) (OMIM #101000) is a rare autosomal dominant tumor predisposition syndrome typified by bilateral vestibular schwannomas (VS), as well as a range of other tumors both in the central and peripheral nervous system. These tumors include cranial, spinal, peripheral nerve, and intradermal schwannomas, cranial and spinal meningiomas, and intrinsic central nervous system (CNS) tumors, usually spinal ependymomas. These are summarized in the Manchester criteria.1
Historically small numbers of NF2 patients have been managed within specialty silos including Neurosurgery, ENT, Clinical Genetics, and Neurology. A number of relatively small studies (n = 17-147) with limited follow-up of NF2 patients have assessed progression of hearing loss and VS,2–5 meningioma growth,6–9 or both.10 Only small pediatric studies have looked more comprehensively at NF2 natural history encompassing numerous tumor types.11
Latterly, multidisciplinary clinics have emerged providing highly specialized care. With the development of drug therapy in NF2, there is an urgent need to assess natural history to design and power clinical trials. Although bevacizumab has shown efficacy in treating schwannoma disease,12–15 there is little benefit seen in treating meningiomas.16 The benefit in children also appears more marginal for VS.14,15
Materials and Methods
Patient Inclusion
The prospectively curated Manchester NF2 database, available to senior author (D.G.E.), was reviewed with the purpose of describing NF2 disease course over a 30-year period (January 1990-February 2020), since the inception of multidisciplinary follow-up as recommended in national guidance.17 Patients included fulfilled at least Manchester criteria for NF21 had a proven pathogenic constitutional variant in the NF2 gene or identical somatic NF2 variants between two anatomically distinct tumors.18 Molecular testing was available from a blood sample on all patients using next-generation sequencing of all NF2 exons and multiplex ligation-dependent probe amplification (MLPA),18 although some pathogenic variants were previously found by Sanger sequencing. Four patients were refuted as having NF2 despite meeting diagnostic criteria as they had different NF2 variants in separate tumors and 2 had LZTR1 germline pathogenic variants.18 These patients have remained in the cohort for analysis on the basis of meeting clinical diagnostic criteria.
Surveillance Protocol
As per Evans et al.’s consensus statement,17 patients were seen yearly and underwent full neurological, ophthalmology, and audiological assessment. Initial MRI scan (head and spine) occurred between 10 and 12 years or earlier in severely affected families. MRI brain was performed every 2 years for patients <20 years and every 3 years for older patients who were asymptomatic without tumors. Once tumors were identified, MRIs were performed annually until the individual growth rate was established. Spinal MRI imaging was performed every 2-3 years unless no spinal tumors were present initially. In these cases, the frequency of scanning was usually reduced, unless clinically indicated, to every 5 years.
Follow-Up and Data Collection
Patients were followed up at least annually until death or leaving the Manchester service. Patients were censored at death or last clinical follow-up. Dates of surgical intervention for VS, cranial meningioma, spinal tumors, and radiotherapy were recorded. Hearing data were available on a subset of patients, categorized by era of diagnosis. Details of their most recent audiological assessment were included. Hearing was evaluated by a patient’s unaided optimal word discrimination score (ODS) using monosyllabic words in their better hearing ear and then, if they scored <50%, their aided (including cochlear or auditory brainstem implants [ABI]) sentence score at 70 dBHL.
Treatment with bevacizumab (nationally commissioned since 2010) for schwannomas with ≥4mm linear diameter or ≥60% volumetric growth over the preceding averaged 12-month period was documented. Treatment indications, set out by Morris et al.,14 represent the accepted protocol for treatment within the UK’s nationalized health care system. An imminent threat to neurological function, for example, brainstem compression, is a further indication, but progressive hearing loss without tumor growth is not. Intravenous bevacizumab was given as a 5 mg/kg infusion every 2 weeks, or a 7.5 mg/kg infusion every 3 weeks, for at least 6 months. Failure of tumor control at 6 months (≥20% growth) was the main indication to cease treatment. Major complications, for example, bleeding diathesis/major clot, also led to treatment cessation and evidence of renal impairment often led to dose reductions or drug holidays. A discussion was had at 24 months on whether to trial stopping treatment in those that responded.
Statistical Analysis
Kaplan-Meier analysis was used as a means of univariate analysis to assess: (i) cumulative incidence from date of diagnosis to date of first VS surgical/radiation intervention and (ii) cumulative survival from date of diagnosis to death. Patients were stratified by age at onset (asymptomatic at diagnosis or first related symptoms <16, 16-25, 26-39, or ≥40 years), era diagnosed (diagnosis ≤1998, 1999-2008, or ≥2009), and type of inheritance (inherited, constitutional de novo, de novo mosaic, or de novo with unproven inheritance pathogenic variant). Kaplan-Meier analysis was also performed according to mutation type and NF2 genetic severity score.19P values <.05 were regarded as statistically significant.
Stratification of patients according to era diagnosed has been used to assess NF2 outcomes previously.20 Changes in services available to NF2 patients have occurred in the UK over time. From the late 1990s (era 2), an increasing number of patients with NF2 were managed in four English regional specialty centers, of which Manchester was one. Post-2010 (era 3), highly specialized commissioning meant that all NF2 treatment in England was overseen by these centers. Advancement through these defined eras reflects practice change including improved imaging diagnostics and the introduction of noninvasive treatment options, for example, bevacizumab (era 3).
Regarding inheritance type, a patient was deemed to have inherited disease if they had a clinically affected parent and a confirmed pathogenic NF2 germline variant. Constitutional de novo disease was identified in those with no prior family history and a disease-causing NF2 germline variant. De novo mosaic variants were confirmed in those with identical NF2 variants detected in ≥2 tissue samples or allele frequencies of <40% in blood. The unproven de novo category represents those with a de novo germline pathogenic variant and an unconfirmed family history.
Cox regression was performed to determine the relationship between individual factors (univariately) from time of diagnosis to NF2-related death. Multivariate Cox regression was also performed for those factors found to be statistically significant in the univariate analysis. Statistical analyses were performed in SPSS version 23.021 and Stata version 14.22
Ethics
Ethics approval for the NF2 database was granted by the Central Manchester Ethics committee “Molecular Characterization of Neurofibromatosis, Schwannomatosis and Related Disorders” (IRAS Project ID: 36817).
Results
A total of 353 patients meeting NF2 criteria received at least one follow-up visit after initial assessment. There were 172 males (48.7%) and 181 females (51.3%). Mean age at NF2 diagnosis was 28.9 years (range = 0.2-86, median = 22). There were 4643.1 follow-up years from diagnosis (range = 0.1-41.5, median = 12.1) to censoring during which 60 patients died at a mean age of 45.2 years (range = 21.4-87.5, median = 42.1). Cumulative survival at 10 years was 93.3% (95% CI 90.4-96.2) and at 20 years was 73.7% (95% CI 66.6-80.8). Demographics by age of onset, era diagnosed, and inheritance are provided in Tables 1-3, respectively. Univariate/multivariate Cox regression analysis outcomes are shown in Table 4.
Table 1.
Demographics of Patients Subdivided by the Age at Which They First Displayed NF2-Related Symptoms
| Age of First NF2-Related Symptoms (Y) | P Value | P Value for Trend (Excluding Asymptomatic) | |||||
|---|---|---|---|---|---|---|---|
| Asymptomatic | <16 | 16-25 | 26-39 | ≥40 | |||
| Number | 67 | 91 | 66 | 64 | 65 | n/a | n/a |
| Truncating mutation, n (%) | 7 (10.5) | 38 (41.8) | 17 (25.8) | 2 (3.1) | 1 (1.5) | <.001 | <.001 |
| Died, n (%) | 2 (3.0) | 23 (25.3) | 13 (19.7) | 10 (15.6) | 12 (18.5) | .006 | .207 |
| Died within 15 y of diagnosis, n (%) | 1a (1.5) | 11 (12.1) | 6 (9.1) | 5 (7.8) | 7 (10.8) | .182 | .678 |
| Mean age at death, y | 33.1 | 31.9 | 37.5 | 55.3 | 72.7 | <.001 | <.001 |
| Most common presenting symptoms, n (%) | |||||||
| 1 | n/a | Vision loss, 21 (23.1) | Hearing loss, 36 (54.5) | Hearing loss, 40 (62.5) | Hearing loss, 35 (53.9) | ||
| 2 | n/a | Hearing loss, 19 (20.9) | Weakness/wasting, seizures, headache, tinnitus, 4 (6.1) | Pain, 5 (7.8) | Pain, 5 (7.7) | ||
| 3 | n/a | Weakness/ wasting, 14 (15.4) | Weakness/ wasting, 3 (4.7) | Weakness/wasting, headaches, poor balance, 4 (6.2) |
Abbreviations: n, number; %, percentage.
aSuicide death.
Table 2.
Demographics of Patients With a Clinical NF2 Diagnosis, Subdivided by Era of Diagnosis
| Year of Diagnosis of NF2 | P Value for Trend | ||||
|---|---|---|---|---|---|
| Era 1 <1998, n (%) | Era 21999-2008, n (%) | Era 3 2009-February 2020, n (%) | Total, n (%) | ||
| Number of patients | 113 | 115 | 125 | 353 | |
| Male | 55 (48.7) | 60 (52.2) | 57 (45.6) | 172 (48.7) | .618 |
| Female | 58 (51.3) | 55 (47.8) | 68 (54.4) | 181 (51.3) | |
| Median age diagnosis (y) | 24 | 20 | 27 | 23 | .175 |
| Bilateral VS | 104 (92.0) | 108 (93.9) | 94 (75.2) | 307 (86.7) | <.001 |
| Asymptomatic at diagnosis | 14 (12.3) | 24 (20.9) | 29 (23.2) | 67 (18.9) | .036 |
| No. with VS surgery/RTR in first 10 y | 72 (64.0) | 59 (51.3) | 39 (31.2) | 170 (48.3) | <.001 |
| Died | 39 (33.6) | 18 (15.7) | 3 (2.4) | 60 (16.7) | <.001 |
| Died within 10 y | 6 (5.3) | 10 (8.7) | 3 (2.4) | 19 (5.4) | .295 |
| Mean age at death (y) | 45.4 | 38.3 | 84.4 | 45.2 | .534 |
| Meningioma | 67 (59.7) | 52 (44.4) | 47 (36.8) | 166 (46.6) | .001 |
| Ependymoma | 31 (27.2) | 31 (27.0) | 25 (20.0) | 87 (24.6) | .178 |
| Bevacizumab | 13 (11.4) | 23 (21.7) | 22 (19.2) | 58 (17.5) | .219 |
| Meningioma surgery | 24 (20.2) | 17 (14.8) | 12 (11.2) | 53 (15.3) | .012 |
| Spinal surgery | 21 (18.4) | 18 (15.7) | 9 (7.2) | 48 (13.6) | .010 |
| Full constitutional Path variant | 82 (72.8) | 84 (73.0) | 60 (48.0) | 226 (64.1) | <.001 |
| Mosaic | 17 (14.9) | 21 (18.3) | 36 (28.8) | 74 (20.9) | .009 |
| De novo unknown probable mosaic | 13 (11.5) | 9 (7.8) | 27 (21.6) | 49 (13.9) | .021 |
| NF2 excluded (n) | 1 | 1 | 2 | 4 |
Abbreviations: n, number; %, percentage; VS, vestibular schwannoma; RTR, radiotherapy-related.
Table 3.
Demographics of Patients With a Clinical NF2 Diagnosis, Subdivided by Mode of Inheritance
| Inheritance Type | P Value | ||||
|---|---|---|---|---|---|
| De Novo Constitutional, n (%) | De Novo Mosaic, n (%) | De Novo Unknown, n (%) | Inherited, n (%) | ||
| Number | 106 | 74 | 49 | 120 | n/a |
| Female | 46 (43.4) | 47 (63.5) | 29 (59.2) | 59 (49.2) | .037 |
| Bilateral VS | 104 (98.1) | 55 (74.3) | 38 (77.6) | 109 (90.9) | <.001 |
| Median age diagnosis (y) | 19 | 35 | 48 | 16 | <.001 |
| VS surgery/RTR in first 10 y | 71 (67.0) | 39 (52.7) | 15 (30.6) | 42 (34.7) | <.001 |
| Died | 28 (26.4) | 7 (9.5) | 6 (12.2) | 19 (15.8) | .015 |
| Died within 10 y | 11 (10.4) | 2 (2.7) | 1 (2.0) | 5 (4.1) | .056 |
| Mean age at death (y) | 37.2 | 54.4 | 63.3 | 47.9 | .003 |
| Meningioma | 61 (57.5) | 39 (52.7) | 20 (40.8) | 46 (38.0) | .018 |
| Ependymoma | 43 (40.6) | 13 (17.6) | 3 (6.1) | 28 (23.3) | <.001 |
| Bevacizumab | 25 (23.5) | 5 (6.8) | 1 (2.0) | 27 (22.5) | <.001 |
| Meningioma surgery | 24 (22.6) | 10 (13.5) | 4 (8.2) | 15 (12.5) | .063 |
| Spinal surgery | 21 (19.8) | 11 (14.9) | 3 (6.1) | 13 (10.7) | .085 |
Abbreviations: n, number; %, percentage; VS, vestibular schwannoma; RTR, radiotherapy-related.
Four disputed diagnoses have been removed.
Table 4.
Univariate and Multivariate Cox Regression Analysis for NF2-Related Deaths—Unadjusted Estimates
| Univariate | Multivariate | ||||||
|---|---|---|---|---|---|---|---|
| Hazard Ratio | 95% Confidence Interval | Hazard Ratio | 95% Confidence Interval | ||||
| Gender | Male | Ref | Ref | ||||
| Female | 1.06 | 0.55 | 2.05 | 1.25 | 0.63 | 2.47 | |
| Age at diagnosis | 0.98 | 0.95 | 1.001 | ||||
| Age at symptoms | <16 | Ref | Ref | ||||
| 16-25 | 0.56 | 0.25 | 1.22 | 0.51 | 0.22 | 1.20 | |
| 26-39 | 0.23 | 0.08 | 0.68 | 0.34 | 0.10 | 1.16 | |
| >40 | 0.27 | 0.06 | 1.18 | 0.44 | 0.09 | 2.18 | |
| Asymptomatic | 0.09 | 0.01 | 0.64 | 0.16 | 0.02 | 1.32 | |
| Inherit type | Constitutional | Ref | Ref | ||||
| Inherited | 0.36 | 0.17 | 0.76 | 0.61 | 0.24 | 1.53 | |
| Mosaic | 0.47 | 0.18 | 1.26 | 0.83 | 0.21 | 3.24 | |
| Unknown | 0.12 | 0.02 | 0.89 | 0.16 | 0.02 | 1.47 | |
| Genetic severity | Severe | Ref | Ref | ||||
| Mild | 0.25 | 0.12 | 0.56 | 0.66 | 0.23 | 1.89 | |
| Moderate | 0.23 | 0.10 | 0.52 | 1.42 | 0.46 | 4.40 | |
| Bilateral VS | No | Ref | |||||
| Yes | 3.28 | 0.45 | 23.96 | ||||
| Meningioma | No | Ref | Ref | ||||
| Yes | 2.39 | 1.12 | 5.09 | 0.96 | 0.44 | 2.58 | |
| Ependymoma | No | Ref | Ref | ||||
| Yes | 2.76 | 1.44 | 5.31 | 1.59 | 0.74 | 3.44 | |
| Bevacizumab | No | Ref | |||||
| Yes | 0.61 | 0.21 | 1.72 | ||||
| Spinal operation | No | Ref | Ref | ||||
| Yes | 2.29 | 1.12 | 4.67 | 1.59 | 0.72 | 3.52 | |
| Meningioma operation | No | Ref | |||||
| Yes | 1.40 | 0.67 | 2.91 |
Abbreviation: Ref, reference, worst category used.
The Manchester NF2 service has reviewed an increasing number of patients with the mean number of patients on follow-up increasing from 103 in 1990-1999 to 247 in 2010-2020 (increase of 140%). The annual mean number of patients requiring a VS operation, surgery for spinal tumors, and radiotherapy declined (from 4.66, 1.75, and 1.65 individuals, respectively, per 100 NF2 patients between 1990 and 1999 to 2.11, 0.85, and 1.01 individuals per 100 NF2 patients, respectively, between 2010 and 2020), whereas the number receiving bevacizumab increased (Supplementary Table 1). In 2010, there was a decrease in those undergoing VS surgery and radiotherapy coinciding with the national funding of bevacizumab (Supplementary Figure 1). The annual mean number receiving bevacizumab between 2010 and 2020 was 2.51 per 100 NF2 patients (total of 62 patients).
Age of Onset of First NF2-Related Symptoms
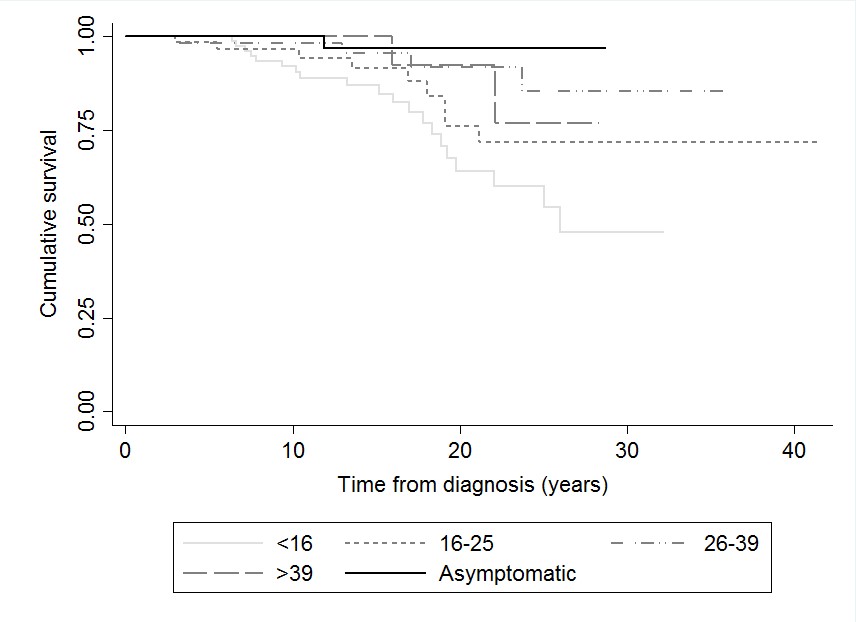
Patients displayed NF2-related symptoms at a variety of ages (Table 1). However, 67 (19.0%) were asymptomatic at diagnosis, having undergone, in most instances, a predictive genetic test. Asymptomatic patients had significantly better survival compared to symptomatic patients in all age groups at 25 years’ follow-up, apart from symptomatic patients presenting between 26 and 39 (P = .058 [Fig. 1a]). Two deaths were recorded in the asymptomatic group, one from suicide at 22 years of age and the other from cardiac arrest following a seizure aged 38 years. Additionally, asymptomatic individuals had a significantly longer time to first intervention compared to those with symptoms, regardless of age at presentation (P < .01 [Fig. 1b]). Thirteen patients asymptomatic at presentation (13/67, 19.4%) required a VS operation/radiation treatment, with their first intervention occurring at a mean of 6.1 years after diagnosis (range = 0.02-16.3, median = 3.6).
Fig. 1.

Kaplan-Meier curves demonstrating: a, overall survival by age of onset (censored at 25 years). b, time to first VS intervention (VS operation/radiotherapy) by age of onset. c, time to first VS intervention (VS operation/radiotherapy) by era (censored at 11 years). d, overall survival by inheritance type. e, time to first VS intervention (VS operation/radiotherapy) by inheritance type. f, overall survival by severity classification.
Ninety-one patients (25.8%) were symptomatic <16. In this group, vision loss was the most common presenting symptom (21/91, 23.1%). A range of etiologies were identified, including optic nerve meningioma (n = 5), orbital meningioma (n = 3), posterior subcapsular lenticular opacities (n = 3), epiretinal membrane (n = 3), retinal hamartoma (n = 2), optic nerve glioma (n = 2), optic atrophy (n = 1) as well as chorioretinal scarring (n = 1) and a retinal detachment caused by a tumor of undocumented pathology (n = 1). 41.8% of symptomatic patients <16 had a truncating NF2 variant. When the 23 deaths in the <16 group were examined in more detail, the proportion of those with a truncating variant increased to 65.2% (15/23). In addition, 19/23 (82.6%) had confirmed meningioma (mean = 2.39 meningioma per patient, median = 2) and 13/23 (56.5%) had ependymoma. All patients that died had bilateral VS (16 (69.6%) required surgical intervention, 7 (30.4%) underwent radiotherapy and 3 (13.0%) received bevacizumab), as well as spinal disease (mean = 16.4 large spinal tumors per patient, median = 12, 1 patient unknown).
Eleven patients <16 (12.1%) died within 15 years of diagnosis. Although a similar proportion of those symptomatic ≥40 died within 15 years (12.3%), mean age at death was of course younger (P value for trend <.001) (Table 1). However, the death rate was not raised significantly (25.3% <16 years vs 18.5% ≥40 years [χ 2 = 1.01, P = .315]).
Regarding time to first VS intervention (first VS operation or radiotherapy), those who presented at 16-25, required intervention at a significantly earlier time point compared to all groups apart from the 26-39 age group (Fig. 1b).
Hearing Data
Hearing data were available for 203 patients, stratified by era of diagnosis. The proportion of patients with no hearing has significantly declined over time (log-rank χ 2 = 12.77, P = .0004 between those diagnosed prior to 2000 vs those ≥2010). The P value for trend comparing ODS category 1 (>70%) to all other categories is significant with P < .001 (Supplementary Table 2).
Era Diagnosed
Table 2 provides detailed demographics of those diagnosed in three different eras. For the Kaplan-Meier analysis, those diagnosed prior to 1988 were removed and follow-up was censored at 11 years in an effort to eliminate survival bias. There was, however, no meaningful survival difference between different eras.
Those diagnosed most recently (>2008) did not require VS intervention (surgically or via radiotherapy) for a longer time period compared to those diagnosed <1998 (Fig. 1c, [log-rank χ 2 = 3.91, P = .048 between those diagnosed >2008 and <1998]). Bevacizumab was introduced at the beginning of era 3.
In era 3, there was a significant increase in the identification of asymptomatic individuals (χ 2 = 3.98, P = .046) and those mosaic for NF2 (χ 2 = 5.72, P = .017) compared to era 1.
Mode of Inheritance
Those with de novo constitutional variants had significantly worse survival (Fig. 1d) than those with de novo mosaic variants or inherited disease (log-rank χ 2 = 4.31 [P = .038] and 8.42 [P = .004], respectively). Individuals with constitutional variants, as well as mosaic disease, had a shorter time to VS intervention compared to those with inherited NF2 (log-rank χ 2 = 38.0 [P < .001] and 10.75 [P = .001], respectively [Fig. 1e]).
A greater proportion of those with de novo constitutional variants developed meningioma (56.6%) and ependymoma (40.6%), and subsequently required surgery (Table 3).
Genetic Severity and Mutation Type
Survival was reduced in those with classified severe genetic19 disease (P < .0001 [Fig. 1f]), but there was no discernible difference between those with mild or moderate disease. Those with truncating variants had poorer survival compared to those with large deletions (log-rank χ 2 = 12.25, P < .0001). No significant differences in time to intervention were observed between the defined severity groups.
Those with splice site variants trended toward a shorter time period to intervention compared to those with truncating variants and large deletions, but this was not significant (log-rank χ 2 = 3.41, P = .065 and log-rank χ 2 = 3.29, P = .07, respectively).
Fig. 2 shows the time course from diagnosis of 3 males with the same birth year diagnosed with NF2 between 1991 and 1994. This shows the milder course of an individual with a missense variant compared to the moderate course with a large deletion and severe course with a truncating variant.
Fig. 2.

Disease course in 3 males with full constitutional pathogenic variants in NF2 all born in the same calendar year. Patient A has an inherited missense variant c.1604T>C, p.leu535pro, patient B inherited a large NF2 deletion from intron 1 to exon 10, and patient C had a de novo constitutional truncating (nonsense) variant c.1219C>T p.Gln407*. Patient A is still well and asymptomatic in 2020 with no operations or radiation treatment. Patient B has had bilateral VS surgery but is otherwise well, whereas patient C died in 2004 aged 23 years. Treatment highlighted in bold italics. Abbreviations: VS, vestibular schwannoma; ABI, auditory brainstem implant; ONM, optic nerve meningioma.
Time to First Surgical/Radiation Intervention
A total of 94/353 (26.6%) had their first surgical/radiation treatment of VS within 2 years of diagnosis and 121/353 (34.3%) within 5 years. For a spinal schwannoma or meningioma operation, this was 18/353 (5.1%) and 34/353 (9.6%), respectively, although 12 had an operation before meeting NF2 criteria. Overall, 153/353 (43.3%) had at least one neurosurgical operation/radiation treatment within 5 years. Although bevacizumab has been very successful in treating VS, 3 patients had to have the treatment stopped to remove growing meningioma and 2 for spinal schwannomas.
Cause of Death
Of the 60 patients who died, 25 (41.7%) died from NF2 tumor progression or burden. The majority of these individuals (12/25, 48%) were <16 when they first had NF2-related symptoms. Five deaths occurred post-operatively (8.3%). Ten other deaths occurred due to NF2-related disease (NF2 malignancy, eg, sarcoma and ependymoma, fall, seizures due to meningioma, suicide, and pneumonia/septicemia in end-stage NF2). There were 20 non-NF2–related deaths. Seven (35%) died from cancer. Those presenting <16 were significantly more likely to have a NF2-related death compared to those ≥40 years (log-rank χ 2 = 16.1, P < .0001). A Kaplan-Meier plot demonstrating age at symptoms for NF2-related deaths is provided in the Supplementary material (Supplementary Figure 2).
Variables Contributing to NF2-Related Death
A number of factors were found to significantly affect time to NF2-related death through univariate Cox regression (Table 4). Those asymptomatic at presentation had reduced risk (HR 0.09, 95% CI 0.01-0.64), as did those presenting between 26 and 39 years (HR 0.23, 95% CI 0.08-0.68) compared to those presenting <16. Those with inherited disease had a reduced likelihood of NF2-related death compared to constitutional disease (HR 0.36, 95% 0.17-0.76), whereas those with meningioma and ependymoma had increased risk (HR 2.39, 95% CI 1.12-5.09 and HR 2.76, 95% CI 1.44-5.31). Subsequent multivariate analysis did not demonstrate significance for these findings after adjusting for other factors.
Discussion
Trend in Service Provision
Since its inception, the Manchester NF2 service has managed an increasing number of patients with clinically and/or molecularly confirmed NF2. Over that time, there have been ground-breaking developments in patient care, including the development of multidisciplinary team clinics,15 tailored therapy options, for example, bevacizumab to treat growing schwannoma or cystic ependymoma,5,14 and an improvement in genetic testing technology and capacity.23
The nature of patient management has subsequently evolved (Supplementary Figure 1), with those diagnosed more recently benefiting from specialist clinic input, and often a more conservative approach to tumor burden (Table 2). With this comes a reduction in associated surgical and radiotherapy-related morbidity. There has additionally been an increasing number of asymptomatic and mosaic individuals identified, allowing for further insight into these subgroups.
Influence of Age of Symptom Onset
Vision loss was the most common presenting symptom in those <16 (occurring in 23.1%). This is a novel finding. Hearing loss was also a common presentation (19/91, 20.9%). Vision loss secondary to ophthalmic complications, including amblyopia, retinal hamartoma, optic nerve meningioma, and juvenile posterior subcapsular lenticular opacities, has previously been described24–26 in pediatric NF2 populations, but not as the most commonly encountered presenting symptom. This is important when considering timing of ophthalmic surveillance in NF2 patients. Perhaps it can be argued that regular ophthalmological and auditory assessments should be considered from early childhood.1 In later age groups, the more “classical” symptoms related to VS (hearing loss, tinnitus, and imbalance) predominate.
A trend toward comparatively worse survival from date of diagnosis in both early (<16) and later (≥40) onset disease was observed compared to individuals presenting in middle age groups and those who were asymptomatic. It is intuitive that those presenting younger are likely to have a more progressive disease course. Previous literature reports an increased relative risk per year decrease in age at diagnosis.27 Poor survival in this subgroup may be linked to genotype. 41.8% of individuals presenting <16 with NF2 in our service had a confirmed constitutional truncating variant—a higher proportion than that seen in other age groups. The proportion of patients with a truncating variant is higher in those that died in this subgroup (65.2% [15/23]). These individuals (n = 23) had significant lifetime disease (see the section “Results”). Truncating variants are known to cause more burdensome NF2,19 resulting in a greater quantity of NF2-related tumors and disease-related mortality.27,28 As evidenced in this cohort, for those <16 who died, genotype appears to be the most significant contributory factor.
A more marginal response to bevacizumab observed in children could be an additional factor influencing prognosis.14,15 In Renzi et al.’s study, only 2/17 (11.8%) children (median age 15.8 years) on bevacizumab for VS achieved a radiological response.15 Plotkin et al. also suggest that pediatric patients have a limited response in terms of tumor shrinkage. However, there may be benefits to quality of life in terms of disease control (the absence of VS growth and preservation of hearing).29 Studied cohorts remain small, and as such, the role of bevacizumab in the pediatric NF2 population has not yet been fully realized. In this cohort, only 3/23 individuals <16 who subsequently died received bevacizumab. Two of these patients had a radiological response. Therefore, poor response to bevacizumab is unlikely to be a strong contributor to survival in our pediatric population.
For those ≥40, comparatively poorer survival cannot be attributed solely to genotypic factors (1.5% have constitutional truncating variant). Of the 12 individuals that died, 9 had mild and 3 had moderate disease (3/12 [25%] had meningioma, 3/12 [25%] had ependymoma, and 7/12 [58.3%] had spinal tumors). When cause of death is examined in more detail (Supplementary Table 3), 9/12 (75%) are not directly linked to NF2 sequelae (P < .0001—compared to ≤16) but rather associated with other age-related diseases (eg, solid organ malignancy, renal failure, dementia).
Those presenting in middle age groups (16-39) proceeded to VS intervention more quickly than others. Neurosurgical outcomes in pediatric populations are reported to have a low complication rate, with comparable results to adult series,30 and as such prompter intervention in adults compared to children is unlikely to be secondary to expected poorer outcomes or risk of procedure-related morbidity. It is instead likely to be related to the variation in presenting symptomatology across different age groups. Those presenting in young/middle adulthood are more likely to have established VS at diagnosis and require more immediate intervention. In pediatric populations, a watch-and-wait approach is more likely to be suitable when monitoring VS disease. This would particularly be the case for our cohort, where, for example, a significant proportion of those presenting <16 (23.1%) had vision loss related to early-onset ophthalmological complications. Slower growing tumors or atypical patterns of NF2 disease are reported to be common for those presenting at an older age, particularly, in the sixth or seventh decade. Such groups of patients may well therefore not require interventions for some time, if at all.31
Patients Asymptomatic at Presentation
Overall, patients who were asymptomatic (n = 67) at presentation did significantly better both with regard to survival and time to intervention compared to other patients. In terms of disease progression, these individuals were more likely to have mild/moderate disease in the long term (mild = 15, moderate = 46, severe = 6). Unsurprisingly, the vast majority of these individuals had inherited disease (inherited = 63, mosaic = 2, de novo = 2) and were diagnosed at a mean age of 17.6 years (median = 13, range = 1 month-66 years). Although screening behavior was not specifically assessed, it stands to reason that individuals are likely to do better if they can undertake screening measures17 prior to developing NF2-related symptoms. Any tumors, if present, would not have reached the threshold of causing clinical concern and would therefore not require immediate intervention.
Mutation type is a factor to consider in patients with predominantly inherited NF2. The majority had large deletions (n = 20/67, 29.9%), which have been previously reported to have prognostic advantage over truncating variants.18,19 Eighteen (26.8%) had a splice donor site variant. In total, 12/67 (17.9%) had a truncating variant (frameshift = 6, nonsense = 6 [1 of these had mosaic disease]). All the individuals with truncating variants, as expected, had classified “severe” disease.
Mode of Inheritance
Those with de novo constitutional variants had significantly worse survival than those with de novo mosaic variants or inherited disease. Over half of the de novo constitutional variants were truncating (55/106 [51.9%]), which indicates a poorer prognosis. These individuals are recognized to be less likely to reproduce secondary to disease severity.32 The majority of those with mosaic disease (63/74 [85.14%]) were classified as mild.19
Individuals with de novo constitutional and mosaic variants had a shorter time to VS intervention compared to those with inherited NF2, who, as previously discussed, are more likely to be asymptomatic at presentation. It therefore follows reason that those with inherited NF2 are likely to have a significantly longer time to treatment and will be more suited to a watch-and-wait management approach.
Mutation Type
Previously described genotype-phenotype correlations are congruous with this study’s observations, for example, those with truncating variants have a poorer disease prognosis than those with large NF2 deletions.20 However, those with truncating variants and large NF2 deletions trended toward a shorter time to VS intervention than those with splice site variants (P = .065 and P = .07, respectively).
Individuals with splice site variants have been previously reported to have lower mortality compared to those with truncating variants20 (not demonstrated in our cohort, P = .077) but time to VS intervention has not been previously examined. Patients with NF2 splice site variants exhibit phenotypic diversity, even within single families.20,33,34 Given the splice site variants in this cohort were heterogenous, it is difficult to draw firm conclusions about the apparent longer time to intervention in this subgroup of patients, and indeed, it was not a significant association. There were no other significant differences between other mutation types.
Era Effect
There was no obvious effect of improved survival in more recent eras. Much of the published era effect20 is likely to be secondary to multidisciplinary management introduction that has been shown to improve survival.17,20,27 Manchester, however, has used a multidisciplinary format for many years, and indeed was a specialist center in the original study.27 Furthermore, as the impact of bevacizumab will have helped some individuals diagnosed prior to 2009, longer follow-up in specialist centers will be required to assess any improved survival related to this. It was encouraging to note that time to intervention ≥2009 was significantly increased (Fig. 1c), which is likely bevacizumab-related.
Hearing Data
Analysis of the hearing data available demonstrated a significant difference between eras, with an improvement seen particularly post-2010 (era 3). This could, in part, be due to the length of follow-up of those diagnosed <2000. An increased use of cochlear implants in the last 10 years has brought more individuals into category 3 (ODS < 50%; aided > 50%), as opposed to ABI use in earlier cohorts which is equivalent to category 4 (ODS < 50%; aided < 50%). It is difficult to assess the effect of bevacizumab, which may well have positively contributed to this domain, but a detailed analysis is beyond the scope of this work.
Time to First Surgical/Radiation Intervention
For consideration of a new drug for use at diagnosis of NF2, if the primary outcome measure was avoidance of a CNS operation or radiation treatment, around 43% would be expected to have undergone such treatment within 5 years of diagnosis. Although other primary outcome measures such as progression-free survival of a target tumor (<20% volumetric growth) or improvement of symptoms can be considered, these may not be the most useful outcome measures in NF2. NF2 patients are often asymptomatic from growing tumors, and most meningiomas do not cause symptoms until a late stage. It is also important to consider that although a target tumor may respond to a drug, it may not treat other NF2-related tumors, as is the case with bevacizumab and meningiomas.14,16 This will be very relevant if a drug has activity on both meningioma and schwannoma.
Strengths and Limitations
A strength of this study is the robustness of the data collected, helped by the multidisciplinary nature of the NF2 clinic setting which allows for the majority of clinical data to be held centrally in patient notes and the NF2 database. Centrally coordinated care is a great asset for patients, and also for clinicians, who want to study patient management and outcomes holistically. Of course, some gaps in information collected are inevitable due to the large service area covered by the Manchester NF2 clinic, as some patients are lost to follow-up or transfer their care to another center.
The decision to assess time to first VS surgical or radiation intervention as a measure of outcomes in patients can be dependent on a number of factors, and is not necessarily a completely objective endpoint when measuring disease severity and progression. In the UK, the use of multidisciplinary-based assessment in a small number of specialist centers does limit variability in management somewhat, as does the national commissioning and guidelines for specialist drugs like bevacizumab. Proton radiotherapy is also governed by special committee in the UK and has been rarely used in our center (1 patient). Inevitably, however, the decision to intervene and the nature in which to do so can vary depending on patient and physician preference, which is a nuance in care that is difficult to capture in an objective fashion.
Conclusion
This study represents the largest, to our knowledge, single-center follow-up study of NF2 patients over a 30-year time period. Rich insights have been gained into the care provided to these patients, the development of treatment practices over time, and how prognostic outcomes vary across certain patient demographics.
There are implications for clinicians working in the NF2 clinic, in terms of the prognostic information that can be used to inform patients about their disease and decisions about their care. Such information is also useful for those wanting to coordinate drug treatment trials. For example, it can inform trial design to focus potential new treatments on the NF2 subgroups most at risk of disease progression and poorer survival and can help assess the impact on expected surgical/radiotherapy intervention rates.
Supplementary Material
{kind=link}
Acknowledgments
The authors wish to acknowledge NHS England for their support of the National NF2 Service. They also thank the English Highly Specialised Commissioning service for funding Manchester as one of the four centers to manage NF2 in England since 2010.
Funding
D.G.E., S.K.L., E.F.H., and M.J.S. are supported by the Manchester National Institute for Health Research (NIHR) Biomedical Research Centre (IS-BRC-1215-20007).
Conflict of interest statement
The authors have no conflicts of interest.
Authorship statement
D.G.E. had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis; Study concept/design: D.G.E.; Data acquisition: D.G.E.; Analysis and interpretation of data: C.F., E.F.H., and D.G.E.; Drafting of manuscript: C.F. and D.G.E.; Critical revision of manuscript: C.F., A.T.K., S.A.R., C.H.-W., S.K.L., S.R.F., O.N.P., E.S., O.M.T., R.D.L., S.S., J.-P.K., G.V., C.M., S.K., M.J.S., M.G.M., E.F.H., and D.G.E.; Statistical analysis: C.F., E.F.H., and D.G.E.; Supervision: D.G.E.
References
- 1. Evans DG, Huson SM, Donnai D, et al. A clinical study of type 2 neurofibromatosis. Q J Med. 1992;84(304):603–618. [PubMed] [Google Scholar]
- 2. Emmanouil B, Houston R, May A, et al. Progression of hearing loss in neurofibromatosis type 2 according to genetic severity. Laryngoscope. 2019;129(4):974–980. [DOI] [PubMed] [Google Scholar]
- 3. deTorres AT, Brewer CC, Zalewski CK, et al. Audiologic natural history of small volume cochleovestibular schwannomas in neurofibromatosis type 2. Otol Neurotol. 2018;39(3):357–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Peyre M, Bernardeschi D, Sterkers O, Kalamarides M. Natural history of vestibular schwannomas and hearing loss in NF2 patients. Neurochirurgie. 2018;64(5):342–347. [DOI] [PubMed] [Google Scholar]
- 5. Plotkin SR, Merker VL, Muzikansky A, Barker FG 2nd, Slattery W 3rd. Natural history of vestibular schwannoma growth and hearing decline in newly diagnosed neurofibromatosis type 2 patients. Otol Neurotol. 2014;35(1):e50–e56. [DOI] [PubMed] [Google Scholar]
- 6. Slattery WH 3rd, Fisher LM, Iqbal Z, Oppenhiemer M. Vestibular schwannoma growth rates in neurofibromatosis type 2 natural history consortium subjects. Otol Neurotol. 2004;25(5):811–817. [DOI] [PubMed] [Google Scholar]
- 7. Nowak A, Dziedzic T, Czernicki T, Kunert P, Marchel A. Clinical course and management of intracranial meningiomas in neurofibromatosis type 2 patients. Neurol Neurochir Pol. 2015;49(6):367–372. [DOI] [PubMed] [Google Scholar]
- 8. Evers S, Verbaan D, Sanchez E, Peerdeman S. 3D Volumetric measurement of neurofibromatosis type 2-associated meningiomas: association between tumor location and growth rate. World Neurosurg. 2015;84(4):1062–1069. [DOI] [PubMed] [Google Scholar]
- 9. Goutagny S, Bah AB, Henin D, et al. Long-term follow-up of 287 meningiomas in neurofibromatosis type 2 patients: clinical, radiological, and molecular features. Neuro Oncol. 2012;14(8):1090–1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Dirks MS, Butman JA, Kim HJ, et al. Long-term natural history of neurofibromatosis type 2-associated intracranial tumors. J Neurosurg. 2012;117(1):109–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ruggieri M, Gabriele AL, Polizzi A, et al. Natural history of neurofibromatosis type 2 with onset before the age of 1 year. Neurogenetics. 2013;14(2):89–98. [DOI] [PubMed] [Google Scholar]
- 12. Plotkin SR, Stemmer-Rachamimov AO, Barker FG 2nd, et al. Hearing improvement after bevacizumab in patients with neurofibromatosis type 2. N Engl J Med. 2009;361(4):358–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Plotkin SR, Merker VL, Halpin C, et al. Bevacizumab for progressive vestibular schwannoma in neurofibromatosis type 2: a retrospective review of 31 patients. Otol Neurotol. 2012;33(6):1046–1052. [DOI] [PubMed] [Google Scholar]
- 14. Morris KA, Golding JF, Axon PR, et al. ; UK NF2 Research Group . Bevacizumab in neurofibromatosis type 2 (NF2) related vestibular schwannomas: a nationally coordinated approach to delivery and prospective evaluation. Neurooncol Pract. 2016;3(4):281–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Renzi S, Michaeli O, Salvador H, et al. Bevacizumab for NF2-associated vestibular schwannomas of childhood and adolescence. Pediatr Blood Cancer. 2020;67(5):e28228. [DOI] [PubMed] [Google Scholar]
- 16. Nunes FP, Merker VL, Jennings D, et al. Bevacizumab treatment for meningiomas in NF2: a retrospective analysis of 15 patients. PLoS One. 2013;8(3):e59941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Evans DG, Baser ME, O’Reilly B, et al. Management of the patient and family with neurofibromatosis 2: a consensus conference statement. Br J Neurosurg. 2005;19(1):5–12. [DOI] [PubMed] [Google Scholar]
- 18. Evans DG, King AT, Bowers NL, et al. ; English Specialist NF2 Research Group . Identifying the deficiencies of current diagnostic criteria for neurofibromatosis 2 using databases of 2777 individuals with molecular testing. Genet Med. 2019;21(7):1525–1533. [DOI] [PubMed] [Google Scholar]
- 19. Halliday D, Emmanouil B, Pretorius P, et al. Genetic severity score predicts clinical phenotype in NF2. J Med Genet. 2017;54(10):657–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hexter A, Jones A, Joe H, et al. ; English Specialist NF2 Research Group . Clinical and molecular predictors of mortality in neurofibromatosis 2: a UK national analysis of 1192 patients. J Med Genet. 2015;52(10):699–705. [DOI] [PubMed] [Google Scholar]
- 21. IBM Corp. Released 2015. IBM SPSS Statistics for Windows, Version 23.0. Armonk, NY: IBM Corp. [Google Scholar]
- 22. StataCorp. 2015. Stata Statistical Software: Release 14. College Station, TX: StataCorp LP. [Google Scholar]
- 23. Evans DG, Hartley CL, Smith PT, et al. ; English Specialist NF research group . Incidence of mosaicism in 1055 de novo NF2 cases: much higher than previous estimates with high utility of next-generation sequencing. Genet Med. 2020;22(1):53–59. [DOI] [PubMed] [Google Scholar]
- 24. Halliday D, Emmanouil B, Vassallo G, et al. ; English NF2 research group . Trends in phenotype in the English paediatric neurofibromatosis type 2 cohort stratified by genetic severity. Clin Genet. 2019;96(2):151–162. [DOI] [PubMed] [Google Scholar]
- 25. Evans DG, Birch JM, Ramsden RT. Paediatric presentation of type 2 neurofibromatosis. Arch Dis Child. 1999;81(6):496–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ruggieri M, Iannetti P, Polizzi A, et al. Earliest clinical manifestations and natural history of neurofibromatosis type 2 (NF2) in childhood: a study of 24 patients. Neuropediatrics. 2005;36(1):21–34. [DOI] [PubMed] [Google Scholar]
- 27. Baser ME, Friedman JM, Aeschliman D, et al. Predictors of the risk of mortality in neurofibromatosis 2. Am J Hum Genet. 2002;71(4):715–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Baser ME, Kuramoto L, Woods R, et al. The location of constitutional neurofibromatosis 2 (NF2) splice site mutations is associated with the severity of NF2. J Med Genet. 2005;42(7):540–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Plotkin SR, Duda DG, Muzikansky A, et al. Multicenter, prospective, phase II and biomarker study of high-dose bevacizumab as induction therapy in patients with neurofibromatosis type 2 and progressive vestibular schwannoma. J Clin Oncol. 2019;37(35):3446–3454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. MacNally SP, Rutherford SA, King AT, et al. Outcome from surgery for vestibular schwannomas in children. Br J Neurosurg. 2009;23(3):226–231. [DOI] [PubMed] [Google Scholar]
- 31. Goutagny S, Bah AB, Parfait B, Sterkers O, Kalamarides M. Neurofibromatosis type 2 in the elderly population: clinical and molecular features. Am J Med Genet A. 2013;161A(4):667–670. [DOI] [PubMed] [Google Scholar]
- 32. Evans DG, Huson SM, Donnai D, et al. A genetic study of type 2 neurofibromatosis in the United Kingdom. I. Prevalence, mutation rate, fitness, and confirmation of maternal transmission effect on severity. J Med Genet. 1992;29(12):841–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Mautner VF, Baser ME, Kluwe L. Phenotypic variability in two families with novel splice-site and frameshift NF2 mutations. Hum Genet. 1996;98(2):203–206. [DOI] [PubMed] [Google Scholar]
- 34. Kluwe L, MacCollin M, Tatagiba M, et al. Phenotypic variability associated with 14 splice-site mutations in the NF2 gene. Am J Med Genet. 1998;77(3):228–233. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.