

Abstract
Motivation
The HLA system plays a pivotal role in both clinical applications and immunology research. Typing HLA genes in patient and donor is indeed required in hematopoietic stem cell and solid-organ transplantation, and the histocompatibility complex region exhibits countless genetic associations with immune-related pathologies. Since the discovery of HLA antigens, the HLA system nomenclature and typing methods have constantly evolved, which leads to difficulties in using data generated with older methodologies.
Results
Here, we present Easy-HLA, a web-based software suite designed to facilitate analysis and gain knowledge from HLA typing, regardless of nomenclature or typing method. Easy-HLA implements a computational and statistical method of HLA haplotypes inference based on published reference populations containing over 600 000 haplotypes to upgrade missing or partial HLA information: ‘HLA-Upgrade’ tool infers high-resolution HLA typing and ‘HLA-2-Haplo’ imputes haplotype pairs and provides additional functional annotations (e.g. amino acids and KIR ligands). We validated both tools using two independent cohorts (total n = 2500). For HLA-Upgrade, we reached a prediction accuracy of 92% from low- to high-resolution of European genotypes. We observed a 96% call rate and 76% accuracy with HLA-2-Haplo European haplotype pairs prediction. In conclusion, Easy-HLA tools facilitate large-scale immunogenetic analysis and promotes the multi-faceted HLA expertise beyond allelic associations by providing new functional immunogenomics parameters.
Availability and implementation
Easy-HLA is a web application freely available (free account) at: https://hla.univ-nantes.fr.
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
HLA genes from the major histocompatibility complex (MHC) encode a specific group of cell surface molecules mediating recognition of non-self-antigens by the immune system. HLA plays key roles in transplantation management and success. HLA matching between a patient and potential donors is essential in hematopoietic stem cell transplantation (HSCT) (Copelan, 2006; Loiseau et al., 2007) and solid-organ transplantations (Held et al., 1994). Donor-recipient compatibility is defined by the number of alleles shared across HLA-A, -B, -C, -DRB1 and -DQB1 genes. The chance of graft success is optimal when donor and recipient are fully compatible and have the lowest number of HLA alleles mismatches (Lee et al., 2007; Zachary and Leffell, 2016). The level of typing resolution is positively correlated with the probability of allele matching during donor search. Additionally, time restrictions in solid-organ transplantation from deceased donors often make HLA allele high-resolution typing impossible, and only intermediate resolution or even low-resolution genotyping may be available at the time of organ allocation. Beyond these major clinical impacts, HLA has been frequently associated with numerous immune-related pathologies (MacArthur et al., 2017; Tian et al., 2017; Vince et al., 2014).
MHC genomic region on chromosome 6 (6p21.3) is the most complex and polymorphic locus of the human genome (Howell et al., 2010). The MHC counts >220 genes (Horton et al., 2004), including 21 polymorphic HLA genes from the classical HLA class I (e.g. HLA-A, HLA-B and HLA-C) and HLA class II (e.g. HLA-DRB1 and HLA-DQB1). The HLA system comprises >22 000 described alleles (Robinson et al., 2015) (https://www.ebi.ac.uk/ipd/imgt/hla/stats.html). HLA alleles correspond to a specific sequence of HLA genes and can be considered as single nucleotide variants (including tetra-allelic ones as well as insertions and deletions) haplotypes. HLA haplotypes can be constructed from these HLA alleles; here, we consider the five main genes HLA-A∼B∼C∼DRB1∼DQB1 for haplotyping, and named the 5-gene haplotypes as the following example: A*34:02∼B*14:01∼C*08:02∼DRB1*04:05∼DQB1*03:02. The complexity of this region is not only due to its diversity but also to its linkage disequilibrium (LD). LD is defined as the non-random association of neighboring polymorphisms, i.e. the difference between the observed frequency of allele combinations (haplotypes) and the expected frequency under random transmission. LD and haplotype frequencies are shaped by selective pressure, genetic drift, non-random mating, recombination events and shared genetic effect between alleles (Ahmad et al., 2003; Goodin et al., 2018).
HLA typing techniques have considerably evolved over the years with a wide array of methods providing increasing levels of resolution (Erlich, 2012, Table 1). Historically, phenotyping was performed by detecting HLA proteins on cell surface with specific antibodies. These serology-based methods have progressively been replaced with DNA-based typing. Today, full-length HLA genes sequenced through Next-Generation Sequencing (NGS) provides the highest standard and resolution. In parallel with HLA typing methods, nomenclature has greatly evolved (Table 1), which nowadays significantly hampers retrospective analyses and HSCT compatible donor search from archived HLA datasets recorded in low/mid-resolutions with possibly some missing genes (e.g. HLA-C or HLA-DQB1) (Hurley et al., 2004). This major pitfall for clinics and biomedical research highlights the crucial need for high-resolution allele imputation from low or intermediate resolution in order to reduce allele ambiguity by simultaneously increasing genotype resolution and imputing unknown genes (Madbouly et al., 2014).
Table 1.
Common nomenclature reporting HLA types
| Name | Typing | Resolution | Nomenclature |
|---|---|---|---|
| Broad serology | Phenotyping (lymphocytotoxicity) | Low | B14 |
| Split serology | Phenotyping (lymphocytotoxicity) | Low | B64 B65 |
| First-field | Genotyping (PCR SSP) | Low | B*14 |
| NMDP code | Genotyping (PCR SSO) | Intermediate | 14:HUJ |
| Second-field | Genotyping (Sanger sequencing and/or NGS) | High | B*14:01 B*14:02 |
Note: HLA alleles nomenclature established by the World Health Organization Nomenclature Committee (http://hla.alleles.org/nomenclature/committee.html). Nomenclature is regularly updated. Here, we consider HLA-B*14:01:01 as an example. ‘NMDP codes’ allele codes narrow the list of alleles that must be considered at a given locus by eliminating some possibilities (e.g. B*14:HUJ means that the typing is either B*14:01 or B*14:02). ‘NMDP codes’ are implemented and updated by the NMDP (https://bioinformatics.bethematchclinical.org/hla-resources/allele-codes/allele-code-lists/allele-code-list-in-alphabetical-order/). PCR SSO: sequence specific oligonucleotide. PCR SSP: sequence specific primers (Howell et al., 2010).
Finally, many current typing technologies are not designed to deliver full-length HLA haplotypes. Knowledge of haplotype pairs can be particularly useful to determine if unrelated individuals have a chance to be haplo-identical in an HSCT clinical setting; and in research, haplotypes are necessary for functional annotations. Familial explorations can be performed to determine haplotypes from parental genotypes; however, this technique is expensive and challenging to implement as it requires access to relatives’ DNA. Beyond this family-based approach, computational haplotype inference based on probabilistic models from genotypic data has been proposed (Salem et al., 2005). Several methods for haplotype inference exist, from algorithms based on parsimony (Clark, 1990) or on likelihood [such as the Expectation-Maximization (EM) algorithms] (Excoffier and Slatkin, 1995) to Bayesian algorithms (Stephens and Donnelly, 2003). Overall, the most commonly used methods to compute HLA haplotypes are EM-based algorithms (Eberhard et al., 2013), which can accommodate several loci with an arbitrary number of alleles for a large number of individuals with ambiguous haplotypes (Eberhard et al., 2013; Salem et al., 2005). However, they show limited performance with small sample size and do not support haplotype determination from a unique individual. Moreover, results are dependent on inherent dataset characteristics: individuals genetic heterogeneity, number of loci and genotype resolution (Eberhard et al., 2013). These methods are not always straightforward or need powerful computation (Salem et al., 2005). Previously, a maximum likelihood-based HLA haplotype imputation technique was validated on several datasets for unrelated HSCT donor search (Gourraud et al., 2005). This method computes the most likely haplotype pair from HLA genotypes based on HLA genotype frequencies throughout donor transplant registries. Most of the reference haplotype frequencies come from the large reference population of the National Marrow Donor Program (NMDP). NMDP designates the US voluntary bone marrow donor registry. This registry has proposed several breakthroughs in the field of bone marrow transplantation by making available the large HLA haplotype database used in the current study, and also, by creating a specific nomenclature: ‘NMDP codes’. These codes allow to describe HLA typing with some allele ambiguity represented by two to five letter codes (Table 1).
Following this strategy, we developed Easy-HLA, a user-friendly web application designed to deliver a complete suite of HLA annotations (freely available through a secure connection at https://hla.univ-nantes.fr). From HLA genotypes and regardless of resolution level, Easy-HLA can statistically resolve HLA genotype ambiguity, and increase HLA data resolution and functional annotations. Easy-HLA facilitates the use of HLA data collected from both classical and historic laboratory procedures. In this article, we present our application and its validation using independent cohorts delivering optimized information for immunogenetic investigations.
2 Implementation
Easy-HLA is a web-based application suite designed to predict haplotypes from HLA genotypes. The input HLA genotypes can be entered with low/mid-resolution and/or can contain ambiguities, in a single request (one individual genotype) or batch mode (several individuals genotypes). Regarding security and data storage, the loaded data files are deleted immediately after analysis completion, and the output data files are safely conserved on our server for 1 week.
We implemented our tools with web scripting languages using PHP (Hypertext Preprocessor) combined with the pgSQL procedural language. The pgSQL language is used to interrogate the haplotype database and find haplotype pairs corresponding to the input genotype. PHP functions were designed to query multiple databases (serological identity, NMDP nomenclature equivalence) to translate the HLA nomenclature complexity. We used estimates from maximum likelihood-based statistical method to infer HLA haplotypes and subsequently predict unavailable HLA information.
2.1 Database
Easy-HLA main algorithm is based on HLA haplotype frequencies from a large reference population, these frequencies were obtained with a maximum likelihood-based HLA haplotype imputation technique previously validated (Gourraud et al., 2005). We stored our data in a PostgreSQL database. The core reference haplotype frequencies come from the NMDP published in 2013 for uses in clinical transplant and immunological research (Gragert et al., 2013). From the HLA genotypes of 6.59 million US subjects, the NMDP estimated high-resolution HLA haplotype frequencies in five ancestral populations using an EM algorithm (Schaid et al., 2002). The large sample size allows an accurate estimation of rare alleles and haplotype frequencies. The NMDP haplotype database thereby reports frequencies of over 600 000 haplotypes divided into five ancestral populations (African-Americans: 198 216; Asian and Pacific Islanders: 158 307; Europeans: 304 697; Hispanics: 220 020 and Native-Americans: 36 417). We completed this large dataset with RFGM (Registre France Greffe de Moelle), a French population database containing >16 000 haplotypes (Gourraud et al., 2015; Pappas et al., 2015). The user has the possibility to choose the best matching reference population with his/her input individual(s) ancestry among these six reference datasets.
2.2 Algorithm
From each HLA genotype, our algorithm enumerates each possible haplotype pair and computes the corresponding likelihood. Considering a diploid genotype (G) for three HLA genes (A, B and C) and two alleles per gene (upper and lower cases), we obtain four distinct theoretical haplotype pairs [or diplotypes, d1–4, Equation (1)]. We can generalize the computation of N theoretical diplotypes from a diploid genotype (G) for x genes (with heterozygous alleles) with the equation N = 2x−1.
Enumeration of diplotypes
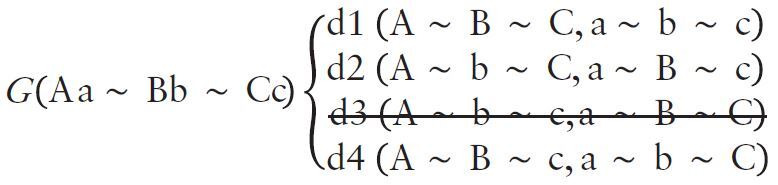 |
(1) |
Our algorithm is founded on a reference database of HLA haplotype frequencies (f) in different populations: haplotypes not reported in the reference dataset are removed from the haplotype list [a∼B∼C strikethrough in d3 in Equation (1)], resulting in n previously observed pairs of haplotypes (here, n = 3) and therefore reducing the space of haplotypes to explore.
We calculated genotypic frequencies from haplotype frequencies by following Hardy Weinberg's genetic distribution law. When a diplotype is homozygous, the likelihood (L) is the squared value of the haplotype frequency (f2). When the diplotype is heterozygous the likelihood (L) is:
Likelihood of an enumerated heterozygous diplotype
| (2) |
where f(A ∼ B ∼ C) and f(a ∼ b ∼ c) corresponds to the respective frequencies of each haplotype. The number of homozygous diplotypes is low (Gragert et al., 2013); therefore, we consider only the likelihood for heterozygous diplotypes.
When HLA genotypes are specified with allelic ambiguities (low-resolution) and/or untyped loci (incomplete genotype), multiple alternative diplotypes can be inferred. For allele ambiguity, the Easy-HLA imputation algorithm produces all possible HLA genotypes associated with the ambiguous input. Correspondingly, when a locus is missing [in our example, the HLA-B gene was not typed and is recorded as XX—Equation (3)], Easy-HLA generates all possible alleles for this missing gene (B, b and β). Equation (3) displays only haplotypes pairs reported in our reference database with a frequency above the user-defined threshold. Indeed, we do not show every possible theoretical haplotype pairs as many are not observed in our population datasets, and would therefore have a null estimated frequency.
Enumeration of diplotypes from an incomplete genotype with a missing locus using all compatible haplotypes present in our database
| (3) |
From an incomplete HLA genotype, Easy-HLA algorithm hence produces all possible diplotypes and then computes their corresponding likelihood. For each diplotype, a confidence measurement named post-probability (Post-P) is calculated as the ratio of likelihood of a particular diplotype (L(dt)) among the likelihood of all n possible diplotypes (L(di)):
Post-probability of each possible diplotype
| (4) |
where i is an index for enumerating the different diplotypes and n is the number of possible diplotypes. The post-probability of the most likely diplotype is then:
Post-probability of the most likely diplotype
| (5) |
The diplotype with the highest post-probability is by definition dependent of the haplotype frequencies in the reference dataset. When interpreting the output, one has to be cautious when top post-probabilities are close, as the real haplotype pair might then not always be the most likely.
From the likelihood of each predicted diplotype, Easy-HLA can then infer a high-resolution genotype for the incomplete or ambiguous input genotype. The likelihood (L) of the imputed high-resolution genotype is:
Likelihood of the imputed high-resolution genotype
| (6) |
where i is an index for enumerating the different diplotypes di, n is the number of possible diplotypes and L is the likelihood of a diplotype obtained from haplotype frequencies f.
2.3 Software presentation
Easy-HLA input is an HLA genotype for each gene (HLA-A, -B, -C, -DRB1 and -DQB1), accepting various levels of HLA nomenclature [Table 1—serology resolution, generic HLA genotyping obtained by molecular biology or codes gathering different HLA alleles (NMDP/MAC UI codes)], as well as missing or incomplete genotypes (Fig. 1). After logging into a personal account, the user has to enter a genotype and select the reference population matching his/her data among six: African-Americans, Asian/Pacific Islanders, Europeans, Hispanics, Native-Americans or French. Alternatively, it is possible to run a search on all combined populations, in that case the output does not provide any frequencies, but indicates the population in which the haplotypes are the most frequent. All data are securely collected, processed and stored. In batch mode, the user uploads a file containing the set of genotypes (automatically deleted after the imputation). In addition, the post-probability threshold (confidence value) should be chosen carefully as it impacts the call rate (chance to have a result) and output accuracy (see below and Fig. 2). On the user interface, a field is available to specify a frequency threshold to further restrict the list of possible diplotypes. Indeed, genotypes with a post-probability below the selected threshold are automatically excluded to prevent an over-representation of rare genotypes. Overall, Easy-HLA displays the different high-resolution genotypes with their likelihood and post-probability starting with the most probable one (Fig. 1A). Optionally, it is possible to select only the most probable high-resolution genotype (in batch mode). When our algorithm does not find a corresponding pair of haplotypes in the reference dataset for a given genotype, it cannot make a prediction and returns an information message: ‘No matching donor found with your selected criteria’. However, in the HLA-2-Haplo module, if one reported haplotype corresponds to half the given genotype, the algorithm infers the missing second haplotype to propose a haplotype pair solving the genotype.
Fig. 1.
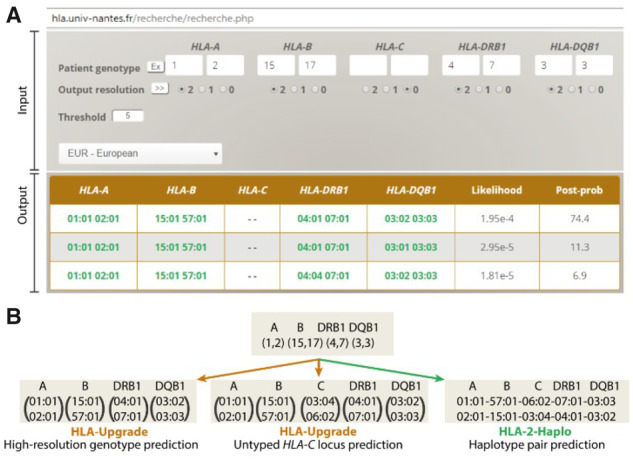
Easy-HLA software presentation. (A) Example of the single query mode. The patient genotype is entered for each gene in first or second-field, serology, NMDP codes or left empty. The user must choose the output resolution (2: second-field, 1: first-field, 0: empty), the post-probability threshold and reference population. The output table contains full mid to high-resolution genotypes with their respective likelihood and post-probability. (B) Easy-HLA delivers updated HLA information from low-resolution HLA typing. In this example, we start with a classical HLA serological genotype (A∼B∼DRB1∼DQB1). HLA-Upgrade statistically predicts high-resolution genotypes (left panel), and can also predict an untyped locus, such as HLA-C (middle panel). Finally, HLA-2-Haplo imputes the most likely haplotype pair. These predictions are all done in silico and as such prevent from additional genotyping in the laboratory
Fig. 2.

Validation of the HLA-Upgrade module in the European (EUR, left) and African-American (AFA, right) populations (post-probability threshold set at 0%). HLA-A∼B∼C∼DRB1∼DQB1 high-resolution genotypes were predicted from different gene combinations of first-field genotypes: HLA-A∼B∼DRB1 (blue), HLA-A∼B∼C∼DRB1 (red), HLA-A∼B∼C∼DRB1∼DQB1 (dark blue). (A) Prediction accuracy per locus. (B) Prediction accuracy according to genotype post-probability. (C) Call rate according to genotype post-probability. (D) Prediction accuracy according to call rate. (Color version of this figure is available at Bioinformatics online.)
The HLA-Upgrade module can predict a full 5-loci HLA-A, -B, -C, -DRB1 and -DQB1 genotyping at high-resolution level by statistically resolving missing, low-resolution or ambiguous typings such as NMDP codes. By updating HLA genotypes, HLA-Upgrade empowers the analysis of old cohorts or cohorts with a long delay of recruitment (Fig. 1B), for which HLA-C and HLA-DQB1 genes are often missing (only recently added in routine genotyping).
The HLA-2-Haplo module predicts the most likely haplotype pair from a given genotype (Fig. 1B) and provides their frequencies in different populations. HLA-2-Haplo can be a particularly useful tool to determine if unrelated individuals have a chance to be haplo-identical in an HSCT clinical setting and to provide Supplementary Material for research. To solve phasing ambiguity for a given HLA genotype, our algorithm compares the potential haplotype pairs with the previously reported haplotypes stored in our large reference database, and can impute the second haplotype if only one haplotype from the diplotype was previously observed. Three additional functions, only available in batch mode, are offered with this module: (i) HLA-expr delivers HLA-C predicted expression (based on allele specific mean HLA-C expression, see (Vince et al., 2016); (ii) HLA-AA provides HLA alleles amino acids and (iii) HLA-KIRlig indicates the KIR (Killer-cell immunoglobulin-like receptor) binding group (C1/C2 groups, Bw4/Bw6 or KIR2DL2 ligands) for each individual HLA allele.
3 Performance
3.1 HLA-Upgrade validation
We validated the HLA-Upgrade module using two independent cohorts of unrelated individuals with high-resolution (second-field) HLA genotyping for HLA-A∼B∼C∼DRB1∼DQB1 loci: (i) 1579 Europeans from the Nantes blood center and (ii) 917 individuals of African ancestry from Consortium on Asthma among African-ancestry Populations in the Americas (Barnes et al., 1996; Mathias et al., 2016). To evaluate the database exhaustiveness on the presence of haplotypes from both cohorts in the database, we tested HLA genotypes at high-resolution from both cohorts in HLA-Upgrade. We found 96.5% of the European cohort genotypes and 70.1% of the African-American cohort genotypes represented in the database. From these full high-resolution datasets, we simulated low-resolution HLA genotypes for both cohorts by creating 12 different situations based on four resolution levels [first-field, second-field, serology and NMDP simulated with correspondence table (https://www.ebi.ac.uk/ipd/imgt/hla/)] and on three levels of input loci (HLA-A∼B∼DRB1, HLA-A∼B∼C∼DRB1 and HLA-A∼B∼C∼DRB1∼DQB1). We used HLA-Upgrade to predict full HLA-A∼B∼C∼DRB1∼DQB1 high-resolution genotypes for each of the 12 simulated datasets, and defined accuracy as the percentage of correct predictions compared to the original HLA typing. We defined call rate as the number of predictions compared to the total number of expected predictions.
The resolution level impacts prediction accuracy, prediction is almost twice as good for intermediate-resolution (NMDP) and high-resolution (second-field) genotypes compared to low-resolution genotype (serology and first-field) (Supplementary Fig. S1). For the HLA-A∼B∼C∼DRB1∼DQB1 genotype, the prediction accuracy was 58.1%, 58.6%, 97.6% and 100% for first-field, serology, NMDP and second-field resolution level, respectively. Interestingly, split serology was as accurate as first-field HLA genotyping, meaning that the erroneous predictions are not based on split antigens. Second, results obtained from NMDP codes and second-field HLA typing did not differ drastically, emphasizing the typing precision of the high-definition sequence specific oligonucleotide. Similarly, the level of input loci impacts the prediction accuracy (Supplementary Fig. S1 and Fig. 2A): inputs lacking alleles from one gene (HLA-C or HLA-DQB1) resulted in a drop of accuracy. On average, HLA-C prediction was 20 points better when HLA-C was provided in input (Fig. 2A). We showed similar results from inputs lacking HLA-DQB1 in the African cohort. The prediction accuracy per locus in the European cohort from first-field resolution HLA-A∼B∼C∼DRB1∼DQB1 genotype inputs was 97% for HLA-A, 93% for HLA-B, 96% for HLA-C, 86% for HLA-DRB1 and 93% for HLA-DQB1 (Fig. 2A, left panel). As a comparison, the prediction accuracy per locus in the African-American cohort was 87%, 90%, 89%, 78% and 92% for HLA-A, -B, -C, -DRB1 and -DQB1, respectively (Fig. 2A, right panel). The prediction accuracy by locus for the European cohort was on average 10 points higher than for the African-derived cohort, probably because of the smaller sample size and larger HLA haplotype diversity in African-ancestry populations.
For each input level (3, 4 or 5 genes), prediction accuracy (Fig. 2B) and call rate (Fig. 2C) of the full genotype change almost linearly with an increasing post-probability threshold (confidence measure). For the 5-loci input level, accuracy increased from 58% to 92% in Europeans and from 35% to 64% in African-Americans for a post-probability threshold going from 0% (accepts everything) to 90% (includes genotypes with post-probability > 90%), respectively. On the contrary, call rate decreased from 100% to 20% in Europeans and from 100% to 10% in African-Americans for a threshold increasing from 0% to 90%, respectively. Correspondingly for the 3-loci input level, accuracy increased from 38% to 94% in Europeans and from 16% to 67% in African-Americans and call rate decreased from 100% to 3% in Europeans and from 100% to 0% in African-Americans for a post-probability threshold of 0% and 90%, respectively. Results are consistent with the additional validation presented in Supplementary Fig. S4.
The prediction accuracy increases as the call rate declines (Fig. 2D), exemplifying a challenging risk/benefit balance that limits error risk (increased accuracy) at the cost of a lower number of output results (low call rate). The post-probability parameter is therefore crucial for HLA-Upgrade prediction performances. By default, we recommend a post-probability threshold set at 10% for exploratory research with HLA-Upgrade, advocating for more results with a compromise on allele accuracy. At this threshold, we have a 100% call rate, but we eliminate genotypes with very low frequencies for European- and African-ancestry populations.
3.2 HLA-2-Haplo validation
We validated HLA-2-Haplo module using two independent cohorts with high-resolution HLA-A∼B∼C∼DRB1∼DQB1: (i) 273 European-ancestry (genotyping) and (ii) 116 African-ancestry individuals [HLA imputation from single nucleotide polymorphism (SNP) genotypes] (Barnes et al., 1996; Mathias et al., 2016). African-ancestry individuals were previously genome-wide SNP genotyped (Johnston et al., 2017) and we imputed HLA alleles for the 5-loci HLA-A, -B, -C, -DRB1 and -DQB1 with the HIBAG R package (Zheng et al., 2014). For each individual, we deduced haplotype pairs from parental HLA typing (hereditary familial study with parents/child trios). The method used is segregation analysis. Families were selected with an ascending minimum of one first degree relative (parents/children, Supplementary Fig. S6).
For each validation cohort, we predicted haplotypes with HLA-2-Haplo from the 5-HLA loci in high-resolution (Fig. 3). Similar to HLA-Upgrade, post-probability measures the confidence of predicted haplotypes based on frequency. Prediction accuracy ranged from 76% to 90% in Europeans and from 70% to 86% in African-Americans with a post-probability from 0% to 90%, respectively (Fig. 3A). For both cohorts, we observed a drop of call rate after the post-probability threshold reached 40% and down to 45% for the 90% post-probability threshold (Fig. 3B). Overall, prediction accuracy is relatively stable across call rates (Fig. 3C). For HLA-2-Haplo, we recommend a default post-probability threshold of 30%, where the call rate reaches a maximum whereas ambiguities with rare genotypes are minimal: call rate is then 99% and 96%, and accuracy is 70% and 76% for the African- and European-ancestry populations, respectively.
Fig. 3.
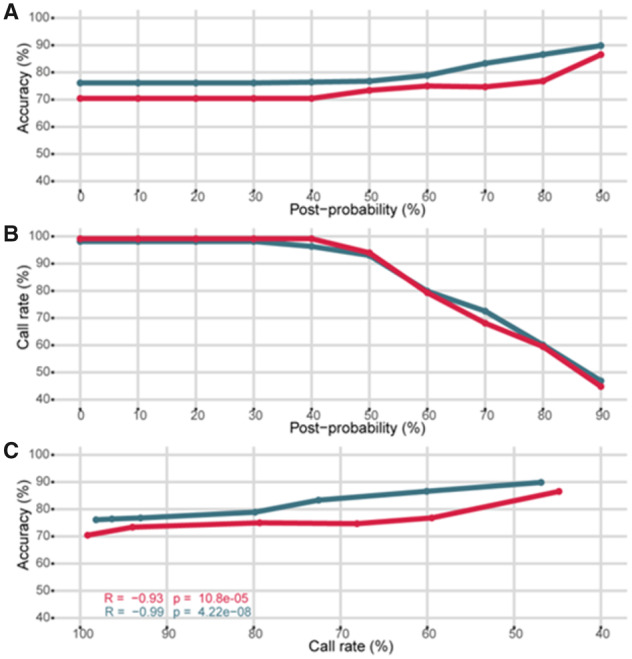
Validation of the HLA-2-Haplo module in the European (EUR, dark blue) and African-American (AFA, red) populations. (A) Accuracy of haplotypes pairs prediction according to the calculated post-probability. (B) Call rate of haplotypes pairs prediction according to the calculated post-probability. (C) Accuracy of haplotypes pairs prediction according to the call rate. (Color version of this figure is available at Bioinformatics online.)
Using the same validation cohorts, we compared performance of haplotype pair prediction between HLA-2-Haplo and the ‘haplo.stats’ R package (Schaid et al., 2002), a statistical tool based on a maximum likelihood method for haplotyping when linkage phase is ambiguous in cohort studies (‘haplo.em’ function). Unlike the usual EM tools, their algorithm considers more than two alleles per locus, accept missing allele, and use a ‘progressive insertion’: the algorithm progressively inserts batches of loci into haplotypes of growing lengths before iterating over the EM steps.
We tested the impact of different sampling sizes for input genotypes (10, 50, 100 or all cohort) on the prediction accuracy (Supplementary Fig. S2 and Table S1). Compared to HLA-2-Haplo predictions for Europeans (76% accuracy) and African-Americans (70% accuracy), the ‘haplo.stats’ predictions were very dependent of sampling size and accuracy was systematically lower than HLA-2-Haplo. ‘haplo.stats’ accurately predicted 22.6% (21.7–23.5) with 10 genotypes, 36.8% (36.4–37.1) with 100 and up to 46.1% (46.0–46.2) with the Europeans whole cohort (n = 273) and 14.9% (14.1–15.7) with 10 genotypes, 40.5% (40.3–40.7) with 100 and up to 42.4% (42.2–42.6) with the African-Americans whole cohort (n = 116). Our results are therefore ‘more reproducible’, in the sense that a given genotype will always output the same results no matter what other individual observations are in the dataset. This methodological characteristic explains why sample size does not impact prediction accuracy (70% for AFA and 76% for EUR) with HLA-2-Haplo.
3.3 Execution
To test our tools’ performance, we evaluated HLA-Upgrade runtime on under 48 conditions with a post-probability threshold of 0% and only the first result in output (Supplementary Fig. S3) including: two ancestral populations (European and African-American), four input file sizes (10 100, 1000 and 5000 genotypes), three resolutions (Split serological resolution, first-field and second-field) and two loci combinations (A∼B∼DRB1 and A∼B∼C∼DRB1∼DQB1).
HLA-Upgrade took ∼12 s to analyze files with 100-s field A∼B∼DRB1 genotypes of European ancestry, 8.6 min in first-field and 6.6 min in split serology. We observed a linear runtime progression with the different file sizes (Supplementary Fig. S3). First-field genotypes required a longer execution time than the other two resolutions, which can be explained by a larger number of possible haplotypes.
In addition to input file size and resolution, execution runtime was also impacted by the input level of missingness and the reference population database size. Indeed, a higher number of haplotypes to browse translates into increased runtime: runtime for A∼B∼DRB1 genotypes was 3-fold longer than for A∼B∼C∼DRB1∼DQB1 genotypes and Europeans (304 697 haplotypes in the reference database) took on average 3-fold longer than African-Americans (198 216 haplotypes in the reference database).
4 Discussion
Easy-HLA is a web application suite designed to facilitate large-scale immunogenetic analyses and gain knowledge from HLA typing, regardless of the variety of nomenclature or typing methods. In this report, we presented two tools, HLA-Upgrade and HLA-2-Haplo, based on a large HLA haplotype reference database. Our tools integrate published external data comprising a published set of haplotype frequencies estimated from bone marrow donor registries (>6.5 million individuals and 600 000 unique haplotypes) in order to facilitate an accurate interpretation of the input dataset.
HLA-Upgrade can successfully predict a full HLA-A, -B, -C, -DRB1 and -DQB1 high-resolution genotyping in different populations from low-resolution and/or partially known HLA typing. As expected, HLA-Upgrade performance positively correlates with HLA typing input resolution: when there is more uncertainty or missingness in input, prediction will be lower. Users must find a balance between highly confident results (high accuracy) and number of predicted genotypes (call rate). From our validation cohorts, we recommend a default post-probability threshold at 10%. At this threshold, from the first-field HLA-A∼B∼C∼DRB1∼DQB1 genotype, we predicted a high-resolution genotype with an accuracy of 78.5–92.3% and 85.5–96.9% per HLA locus and a call rate of 98.1% and 99.8% in African- and European-ancestry populations, respectively. We also validated HLA-Upgrade using the 1000 Genomes project HLA data and obtained consistent conclusions in Europeans, Africans, Hispanics and Asian-Pacific Islanders (Supplementary Fig. S4). We tested allele frequencies difference between imputed and non-imputed data: this shows a good correlation with a very limited loss of diversity toward frequent alleles (Supplementary Fig. S7). Currently, HLA-Upgrade outputs the post-probability (confidence measure) for the overall 5-loci prediction. For future perspectives, we plan to implement an allele post-probability and a locus post-probability to underline the high allelic variability and emphasize the impact of rare alleles amongst the different genotypes. As a proof-of-concept, we carried out a preliminary study to weight the accuracy by genotype frequencies. Indeed, we can consider that rare alleles should not carry the same weight as common alleles in our computation as they will skew the accuracy results. With weighting, our prediction is 43% point better for the A∼B∼DRB1 genotype and 30% point for the A∼B∼C∼DRB1∼DQB1 genotype. Weighting the accuracy computation with HLA genotype frequency considerably improved accuracy for HLA-Upgrade in individuals of European ancestry (see Supplementary Fig. S5), so this strategy is very promising.
The HLA-2-Haplo module accurately predicts haplotype pairs from HLA genotypes. From high-resolution input and a 30% post-probability threshold, we obtained a 99% and 96% call rate and 70% and 76% prediction accuracy for African- and European-ancestry populations, respectively. Importantly, HLA-2-Haplo systematically outperforms ‘haplo.stats’, a pre-existing HLA haplotyping R package, independently of sample size.
Both Easy-HLA inference tools rely on a statistical algorithm based on HLA haplotype frequencies from a large reference database (>600 000 haplotypes from five different ancestral populations). One major strength of our strategy is the size and diversity of our reference registry including the biggest published haplotype frequency database from the NMDP (Gragert et al., 2013) and the RFGM databases (Gourraud et al., 2015). However, these databases also convey most of Easy-HLA’s limitations: exhaustiveness (96.5% European and 70.1% African-American cohort genotypes are present in the database), population diversity coverage [mixed populations (REF)], typing errors, resolution level and HLA loci coverage. For example, the current haplotype frequency databases do not include HLA-DPB1. Our reference database samples HLA haplotypes from the USA and from France and therefore does not represent exhaustive HLA genetic diversity. However, we believe this bias is mostly compensated by the large size of the database, which allows both an accurate estimate of haplotype frequencies and the presence of many rare haplotypes, overall improving our predictions. Finally, Easy-HLA is flexible and our algorithms are fully compatible to evolve regularly with future database releases.
Here, we developed algorithms implemented in a user-friendly web application to facilitate the analysis and reveal the full details of HLA typing. Easy-HLA tools are of great interest both for biomedical research and clinical applications. First, HLA-Upgrade allows to update archived HLA cohorts recorded in low/mid-resolutions and/or with missing loci (such as HLA-C), which empowers the users to combine old and recent datasets to perform large immunogenetic association analyses with various immune-related pathologies. HLA-Upgrade could also be translated into clinics to assess HSCT compatibility between a patient and potential donors with low/mid-resolution. Second, HLA-2-Haplo predicts haplotypes that are the breeding-ground for further research investigations and for functional immunogenomic annotations: HLA-C expression (HLA-expr), amino acid equivalence (HLA-AA) and KIR ligand classification (HLA-KIRlig). Our tools are currently used in HSCT: first, for unrelated donors search using HLA-Upgrade to update an old typing in a donor database, and second, for haplo-identical transplantation using HLA-2-Haplo to predict haplotypes. Our tools have also been used in biomedical research: using HLA-Upgrade, we have updated two large solid-organ transplantation cohorts from low-resolution to high-resolution genotypes, hence empowering us to now carry out allele, haplotype and immunogenetic data (HLA-2-Haplo additional functionalities) associations with graft survival.
In conclusion, Easy-HLA (freely available online at: https://hla.univ-nantes.fr) facilitates large-scale analyses and promotes the multi-faceted HLA expertise beyond allelic associations. Our tool perfectly illustrates how computational and statistical modeling can relay and upgrade high-value experimental data to better enlighten clinical practice and sustain research.
Funding
The authors thank Labex IGO (ANR-11-LABX-0016-01) and IHU-CESTI for their support. Nicolas Vince has received funding from the European Union’s Horizon 2020 research and innovation program under the Marie Skłodowska-Curie grant agreement number 846520. dbGaP Study Accession: phs001123.v1.p1.
Conflict of Interest: none declared.
Supplementary Material
References
- Ahmad T. et al. (2003) Haplotype-specific linkage disequilibrium patterns define the genetic topography of the human MHC. Hum. Mol. Genet., 12, 647–656. [PubMed] [Google Scholar]
- Barnes K.C. et al. (1996) Linkage of asthma and total serum IgE concentration to markers on chromosome 12q: evidence from Afro-Caribbean and Caucasian populations. Genomics, 37, 41–50. [DOI] [PubMed] [Google Scholar]
- Clark A.G. (1990) Inference of haplotypes from PCR-amplified samples of diploid populations. Mol. Biol. Evol., 7, 111–122. [DOI] [PubMed] [Google Scholar]
- Copelan E.A. (2006) Hematopoietic stem-cell transplantation. N. Engl. J. Med., 354, 1813–1826. [DOI] [PubMed] [Google Scholar]
- Eberhard H.-P. et al. (2013) Comparative validation of computer programs for haplotype frequency estimation from donor registry data. Tissue Antigens, 82, 93–105. [DOI] [PubMed] [Google Scholar]
- Erlich H. (2012) HLA DNA typing: past, present, and future. Tissue Antigens, 80, 1–11. [DOI] [PubMed] [Google Scholar]
- Excoffier L., Slatkin M. (1995) Maximum-likelihood estimation of molecular haplotype frequencies in a diploid population. Mol. Biol. Evol., 12, 921–927. [DOI] [PubMed] [Google Scholar]
- Goodin D.S. et al. (2018) Highly conserved extended haplotypes of the major histocompatibility complex and their relationship to multiple sclerosis susceptibility. PLoS One, 13, e0190043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gourraud P.-A. et al. (2005) Inferred HLA haplotype information for donors from hematopoietic stem cells donor registries. Hum. Immunol., 66, 563–570. [DOI] [PubMed] [Google Scholar]
- Gourraud P.-A. et al. (2015) High-resolution HLA-A, HLA-B, and HLA-DRB1 haplotype frequencies from the French Bone Marrow Donor Registry. Hum. Immunol., 76, 381–384. [DOI] [PubMed] [Google Scholar]
- Gragert L. et al. (2013) Six-locus high resolution HLA haplotype frequencies derived from mixed-resolution DNA typing for the entire US donor registry. Hum. Immunol., 74, 1313–1320. [DOI] [PubMed] [Google Scholar]
- Held P.J. et al. (1994) The impact of HLA mismatches on the survival of first cadaveric kidney transplants. N. Engl. J. Med., 331, 765–770. [DOI] [PubMed] [Google Scholar]
- Horton R. et al. (2004) Gene map of the extended human MHC. Nat. Rev. Genet., 5, 889–899. [DOI] [PubMed] [Google Scholar]
- Howell W.M. et al. (2010) The HLA system: immunobiology, HLA typing, antibody screening and crossmatching techniques. J. Clin. Pathol., 63, 387–390. [DOI] [PubMed] [Google Scholar]
- Hurley C.K. et al. (2004) Hematopoietic stem cell donor registry strategies for assigning search determinants and matching relationships. Bone Marrow Transplant, 33, 443–450. [DOI] [PubMed] [Google Scholar]
- Johnston H.R. et al. (2017) Identifying tagging SNPs for African specific genetic variation from the African Diaspora Genome. Sci. Rep., 7, 46398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S.J. et al. (2007) High-resolution donor-recipient HLA matching contributes to the success of unrelated donor marrow transplantation. Blood, 110, 4576–4583. [DOI] [PubMed] [Google Scholar]
- Loiseau P. et al. (2007) HLA association with hematopoietic stem cell transplantation outcome: the number of mismatches at HLA-A, -B, -C, -DRB1, or -DQB1 is strongly associated with overall survival. Biol. Blood Marrow Transplant, 13, 965–974. [DOI] [PubMed] [Google Scholar]
- MacArthur J. et al. (2017) The new NHGRI-EBI catalog of published genome-wide association studies (GWAS Catalog). Nucleic Acids Res., 45, D896–D901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madbouly A. et al. (2014) Validation of statistical imputation of allele-level multilocus phased genotypes from ambiguous HLA assignments. Tissue Antigens, 84, 285–292. [DOI] [PubMed] [Google Scholar]
- Mathias R.A. et al. (2016) A continuum of admixture in the Western Hemisphere revealed by the African Diaspora genome. Nat. Commun., 7, 12522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pappas D.J. et al. (2015) Comparison of high-resolution human leukocyte antigen haplotype frequencies in different ethnic groups: consequences of sampling fluctuation and haplotype frequency distribution tail truncation. Hum. Immunol., 76, 374–380. [DOI] [PubMed] [Google Scholar]
- Robinson J. et al. (2015) The IPD and IMGT/HLA database: allele variant databases. Nucleic Acids Res., 43, D423–D431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salem R.M. et al. (2005) A comprehensive literature review of haplotyping software and methods for use with unrelated individuals. Hum. Genomics, 2, 39–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaid D.J. et al. (2002) Score tests for association between traits and haplotypes when linkage phase is ambiguous. Am. J. Hum. Genet., 70, 425–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens M., Donnelly P. (2003) A comparison of Bayesian methods for haplotype reconstruction from population genotype data. Am. J. Hum. Genet., 73, 1162–1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian C. et al. (2017) Genome-wide association and HLA region fine-mapping studies identify susceptibility loci for multiple common infections. Nat. Commun., 8, 599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vince N. et al. (2016) HLA-C Level is regulated by a polymorphic Oct1 binding site in the HLA-C promoter region. Am. J. Hum. Genet., 99, 1353–1358. DOI: 10.1016/j.ajhg.2016.09.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vince N. et al. (2014) HLA class I and KIR genes do not protect against HIV type 1 infection in highly exposed uninfected individuals with hemophilia A. J. Infect. Dis., 210, 1047–1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zachary A.A., Leffell M.S. (2016) HLA mismatching strategies for solid organ transplantation—a balancing act. Front. Immunol., 7, 575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng X. et al. (2014) HIBAG—HLA genotype imputation with attribute bagging. Pharmacogenomics J., 14, 192–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.