

Abstract
Aging has largely been defined by analog measures of organ and organismal dysfunction. This has led to the characterization of aging processes at the molecular and cellular levels that underlie these gradual changes. However, current knowledge does not fully explain the growing body of data emerging from large epidemiological, systems biology, and single cell studies of entire organisms pointing to varied rates of aging between individuals (different functionality and lifespan), across lifespan (asynchronous aging), and within an organism at the tissue and organ levels (aging mosaicism). Here we consider these inhomogeneities in the broader context of the rate of aging and from the perspective of underlying cellular changes. These changes reflect genetic, environmental, and stochastic factors that cells integrate to adopt new homeostatic, albeit less functional, states, such as cellular senescence. In this sense, cellular aging can be viewed, at least in part, as a quantal process we refer to as “digital aging”. Nevertheless, analog declines of tissue dysfunction and organ failure with age could be the sum of underlying digital events. Importantly, cellular aging, digital or otherwise, is not uniform across time or space within the organism or between organisms of the same species. Certain tissues may exhibit earliest signs of cellular aging, acting as drivers for organismal aging as signals from those driver cells within those tissues may accelerate the aging of other cells locally or even systemically. Advanced methodologies at the systems level and at the single cell level are likely to continue to refine our understanding to the processes of how cells and tissues age and how the integration of those processes leads to the complexities of individual, organismal aging.
Introduction
Evolutionary theories of aging have sought to explain “why” we age, with concepts evolving over the decades from genetic and deterministic to more physiological and adaptive theories.1 Among the most widely accepted theories currently are those involving mutation accumulation, antagonistic pleiotropy and physiological trade-offs between the mortal soma and the immortal germline (Box 1).2,3,4 Such theories provide a basis for understanding aging as a result of processes whose manifestations are subject to evolutionary selection for reasons other than aging traits themselves. However, these theories shed little light on variations of the rate of aging within individuals of a species (Figure 1). In this Perspective, we will discuss challenges to the studies of aging and recent findings that highlight features of organismal aging that pertain to how individuals rather than species age.
Box 1: Evolutionary Theories of Aging.
As the renowned geneticist and Neo-Darwinian evolutionary theorist, Theodosius Dobzhansky, noted by the title of his 1973 essay, “Nothing in biology makes sense except in the light of evolution.”84 This is particularly germane to the biology of aging since all modern evolutionary theories of aging begin by positing that aging traits are largely if not entirely outside of evolutionary selective pressure. This is based on the principle that the forces of natural selection diminish with age because of the myriad of causes of extrinsic mortality in the wild. Thus, the challenge has been to ascribe the features of aging to evolutionary processes that could, at least in theory, account for their existence. Early theories centered around the idea that aging is genetically programmed and that aging is somehow adaptive for the species. This notion has been increasingly discounted as evidence has failed to provide support and as subsequent theories have suggested plausible accounts for aging phenotypes based upon processes that are under the influence of natural selection and those that are not.
The “mutation accumulation theory”, attributed to Medawar,2 postulates that mutations accumulate in the germline over generations, mutations that are largely if not entirely insignificant during development, growth, and early adulthood (and thus outside of evolutionary selection) but which manifest themselves as deleterious in the post-reproductive period. This theory posits that the expression of these deleterious genes either increases with age or is switched on later in life. To date, few examples have been reported that support this pattern. In some senses, the “antagonistic pleiotropy” theory of Williams expands upon this idea of late-life deleterious effects of gene expression,3 but posits that there is no increase over time or switch later in life because the expression of these genes early in life has actually been selected for based on their beneficial effects. Hence, the pleiotropy is the different traits in early and late life, and the antagonism is the beneficial versus detrimental effects of those traits. Williams himself proposed a theoretical example: a mutation may favor calcification of bone during development but in a different “somatic environment” (i.e., the aged soma) leads to calcification of the connective tissues of arteries. The “disposable soma” theory of Kirkwood takes a more physiological perspective to propose that an organism is able to shift allocation of its own resources to either reproduction or to processes that maintain the soma (e.g., growth, homeostasis, nutrient storage, etc.).4 In that sense, it is not a purely genetic theory as those that came before and does not invoke genomic mutations or the effects of specific alleles to explain cell and tissue senescence. While previous theories certainly set aside the germline as a specific case separate from the soma in terms of aging, the disposable soma theory directly incorporates the germline as a critical component of the equation about allocation of resources. The theory rests upon the notion of physiological trade-offs in the context of broader genetic programs that support somatic maintenance and the failure of those programs that accounts for aging phenotypes. This failure varies from species to species depending upon the environment and the need to allocate resources to the soma versus the germline. Of course, these are all theories that will evolve with increasing evidence and further understanding of the relationship between genetic programs, the response of those programs to environmental influences, and the susceptibility to functional decline in later life that we recognize as aging.
Figure 1. Lifespan as an integration of rates of aging at multiple levels.
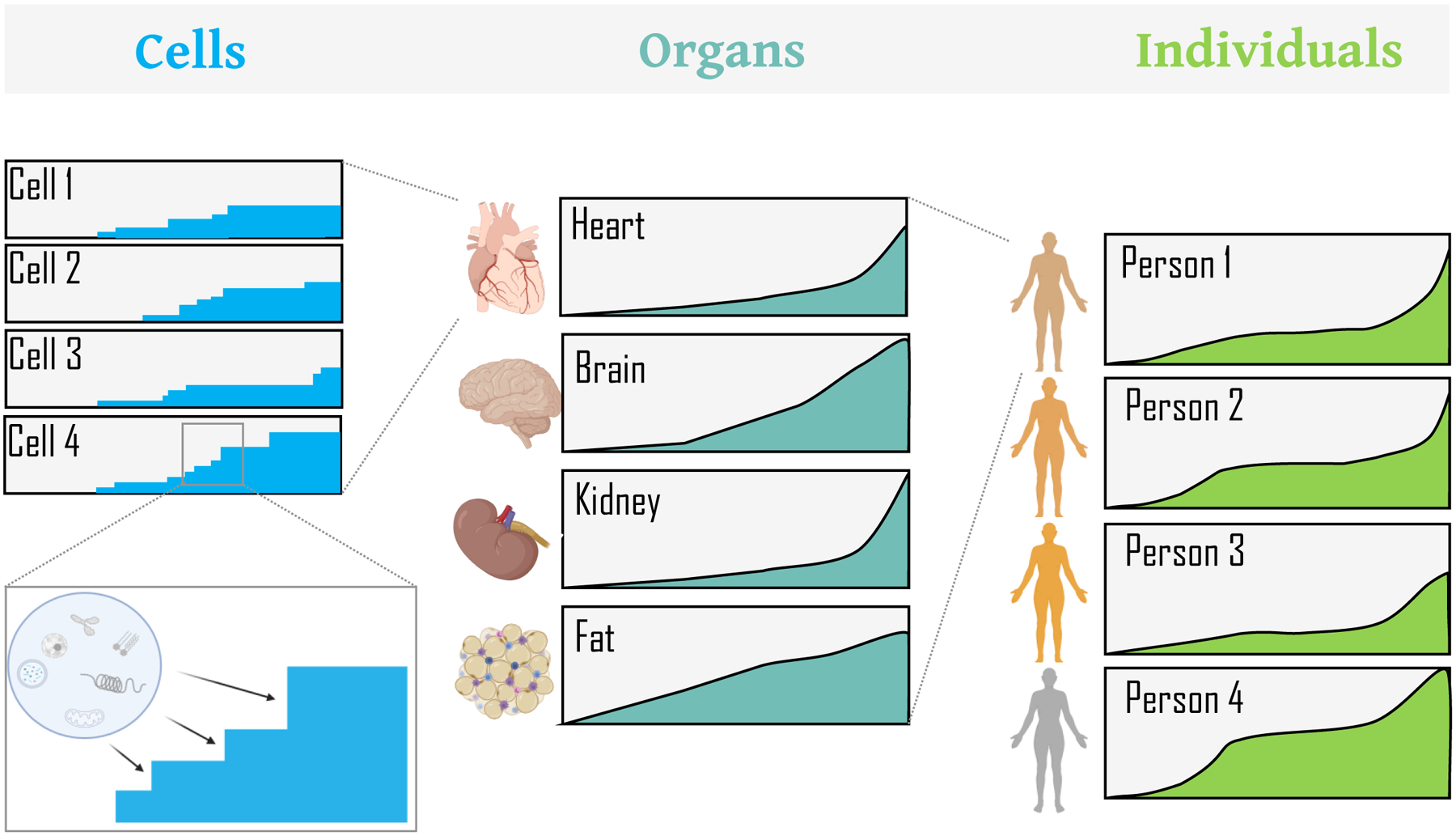
Graphical representation of rates of aging with boxes representing aging in individual cells, organs, and persons. Cellular stress results in distinct homeostatic cellular states. These “digital” cellular states (as described later in the text) are integrated to give rise to distinct rates of aging across organs, ultimately leading to analog functional readouts that differ among organs and thus among individuals. The x-axis represents time/age, the y-axis magnitude of change for a given box.
Throughout, we will refer to “aging” as the underlying processes that lead to the cellular, tissue, and organismal phenotypes associated with that phase of metazoan life known, in the vernacular, as “old age”. Specifically, these manifestations are the hallmark of that phase of life that follows the period of development and postnatal growth, that follows a period of relative homeostasis during adult life associated with reproductive maturity, and that is characterized primarily by a decline in structure and function. This third phase is the raison d’etre for the field of geroscience,5 and is also characterized in humans by a dramatic increase in incidence of age-related diseases, including cancer, cardiovascular diseases, and neurodegenerative diseases. Although the manifestations of aging are a feature of later life, their underlying cellular and molecular bases certainly begin earlier and, arguably, at least for some molecular changes, even during development.6 However, such molecular events can be identified as potential underpinnings of aging phenotypes only in retrospect, and all of these occur even in organisms with negligible senescence.7 As such, no unified theory of the molecular basis of cellular aging has emerged, primarily because of the myriad and interacting contributions,8 and we do not attempt to define “aging”, its potential causes, or when the underlying changes begin (Box 2). We focus primarily on how the manifestations of aging, at the organismal level, reflect variants in the rate of tissue aging across space and time, and how they may arise from features of cellular aging that might not have been predicted from first principles.
Box 2: What is Aging?
Few questions evoke greater debate and disagreement in the field of the biology of aging than “What is aging”. This is followed closely in terms of contentiousness by the highly related questions, “What causes aging?” and “When does aging begin?” For the purposes of this Perspective, we have chosen to focus on phenotypes of aging as enumerated in the text. But is this aging? This is a semantic issue, but a central one. For example, is Alzheimer’s disease the clinical syndrome of dementia, or is Alzheimer’s disease the pathology of amyloid plaques and neurofibrillary tangles? Clearly it is possible to have a syndrome of dementia without these specific pathological changes, and clearly there can be widespread pathology without the clinical syndrome. As such, for sporadic Alzheimer’s disease (unlike the rare genetic forms), the “cause” and the “onset” remain enigmatic. For aging, the definition is even more problematic, and answers to questions about cause and onset are even more elusive for one simple reason – aging is universal (or nearly so).
Clearly, phenotypes of aging arise as a result of underlying molecular processes, but unlike genetic diseases that are unequivocally due to genetic mutations, there is no obvious or comparable starting point. Rather, we consider the processes that lead to cellular aging phenotypes to be ones of system failures, failures that can arise from dysfunction of diverse and interconnected organelles and other subcellular structures, which in turn can arise from diverse and interconnected molecular defects at the levels of DNA, RNA, protein, and metabolites. This perspective maps onto the notion of interacting “layers” of aging that has been proposed.85 The identification of proximal causes of aging phenotypes is tractable. The search for “the cause” of aging is akin to the search for the beginning of a circle. The search for “the inception” of aging is akin to the search for the onset of homeostasis. So, we have taken the easy way out and avoided these questions (for now)
Lifespan versus aging
One of the reasons for a limited understanding of the determinants of individual aging is that the broader research field of aging and longevity has relied heavily on studies in model organisms in which lifespan, and not aging itself, has been the predominant focus and primary benchmark. Studies of the determinants of lifespan within a species have been highly productive because of the numerous available short-lived model systems,9,10 as well as the relative ease of the assay, namely counting the number of animals that are alive versus the number that are dead. This has led to a growing list of genes, drugs, and environmental factors (including diet) that can influence median and maximal lifespan of certain species.11,12,13
By contrast, the identification of the determinants that lead to an acceleration of aging in one member of a species and the slowing of aging in another has proved to be more challenging. While lifespan is a measure that incorporates the impacts of aging, the determinants of longevity are distinct from those that influence the rate at which an organism ages.14 Furthermore, lifespan studies are conducted in laboratory settings that allow for prolonged periods of “protected aging”,15 just as human societies do, thereby masking many increased risks of dying as a result of functional declines with age that would be evident in the wild. Therefore, it is possible for measured lifespan to be uncoupled, at least to a large extent, from the consequences of aging – recording of the death of a person provides no information as to how functional that person was over the previous decades. While there is clearly an interrelationship between aging and longevity, they are not inextricably linked.16 Evidence in C. elegans has provided support for this idea by demonstrating that genetic interventions that extend lifespan may do so without comparably altering the relevant aging phenotypes.17 As such, it is imperative that quantitative measures of aging itself be increasingly used – so called biomarkers – in order to understand the complex processes that underlie the manifestations of organismal aging. In addition, quantitative measures of aging will allow more rapid study of interventions to slow the rate of aging of long-lived species, including humans, in which longevity studies are extraordinarily difficult and expensive. This, then, leads to the concept of the rate of aging.
Rates of aging
Studies of the rate of aging have been entwined with the study of variations in lifespan across species. The notion of differences of rates of aging among species is related to the “rate of living” theory of aging, which posits that the maximal lifespan of a species is determined, inversely, by its metabolic rate.18 According to this theory, species with high metabolic rate age rapidly and die sooner, whereas species with low metabolic rate age more slowly and die later. While generally correlated, there are many exceptions, notably flying species, such as birds and bats, and a wide variety of eusocial animals. Within the animal kingdom, maximal lifespans vary by more than five orders of magnitude,19 with the current world-record holder being a 507-year-old quahog clam.20 Assuming that nearly all animals in a protected environment will age before dying, it is almost tautological that members of a species whose maximal lifespan is 100 days age more rapidly than those from a species whose maximal lifespan is 100 years. By contrast, comparisons of rates of aging among individuals within a given species shine a light on variations that are independent of the maximal lifespan, and the genetic determinants thereof, of that species. Within any given species, there will be individuals that age more rapidly and those that age more slowly. In humans, this is brought into sharp relief in older populations. For example, among centenarians, for whom a main focus has been their relative propensity to avoid diseases of aging and age-related declines, there is a tremendous variability in functional status.21 Furthermore, even for genetically identical organisms maintained in identical environments, there remains a large variability of aging trajectories (presumably related to different rates of aging) as determined by lifespan.22 Identifying which factors (especially if not genetic and not environmental) determine individual rates of aging remains a major challenge to the field. However, equally interesting and provocative is the question as to whether there are different rates of aging within a single individual, and what factors determine those rates (Figure 1). We will expand on these questions below.
At the level of an individual, a rate of aging can be determined by the rate of change of virtually any anatomic structure or physiological process (e.g., bone density, renal function, etc., for humans), as there are almost no aspects of anatomy or physiology that do not exhibit declines as an individual ages.23 It is from such observations that the notion of biological age as opposed to chronological age emerges – measurement of any such biomarker can identify an individual as being either biologically older or younger than the mean for members of the species of the same chronological age. Clearly, readily obtainable physiological measures or molecular biomarkers are essential to studies of biological age. A vast number of biomarkers, ranging from DNA methylation,24,25 to telomere length,26,27 to plasma protein composition,28 to facial morphologies,29 have been used to calculate biological age. Of course, any one of these measures produces a snapshot, whereas it is the changes of these biomarkers over time that reveal trajectories and rates of aging. With such measures of age-related changes, composite scores can be generated to provide an overall rate of organismal aging to predict, for example, health trajectory or life expectancy.
Based on the idea that measuring changes in biomarkers from one age to another allows for the calculation of a rate of aging, there are several key aspects to those measurements that will determine how relevant that calculated rate is for members of a species. A central feature is the definition of the starting point. It is essential to know that the biomarker is no longer changing along the trajectory of development and growth but has reached some steady state as a starting point, particularly when only two ages are used to assess the rate of change. As a trivial example, if height were used as a biomarker in humans, a starting point of age 5 would lead to the erroneous conclusion, regardless of what endpoint was used, that height increases during aging whereas we know that height in fact decreases over the decades of old age. This point highlights a second key aspect of measurements of rates of change of biomarkers, and that is that the accuracy of the rate, and especially changes in rate, will increase as the number of longitudinal measurements increase. In fact, as discussed below, some of the recent findings that shed light on unexpected temporal patterns of aging were discerned only because of multiple, serial biomarker measurements. Finally, caution should be exercised using biomarkers measured immediately prior to death, particularly in humans, because of the many confounding aspects of comorbidities that might yield highly variable results and thus skew trajectories.
Variations in the rate of aging in an individual across time and space
Using serial changes in biomarkers, whether molecular, anatomical, or physiological, allows for a definition of a rate of aging for an individual. This also allows for an assessment of whether that rate of aging is constant over time, or at least monotonic. In general, the decline of tissue function is commonly illustrated as gradual and steady,23 perhaps with a more rapid decline toward the end of life. However, is this how aging actually proceeds? Ultrastructural studies in C. elegans almost two decades ago hinted that it might not.30 The authors noted not only vastly different rates of aging of tissues and cells in individual worms, which they ascribed largely to stochastic events, but the onset of degenerative changes was neither linear nor synchronized. Mathematical modeling of longevity curves in flies also suggest at least 2 phases of aging with a sharp transition in between states characterized by intestinal degeneration.31 This discontinuous behavior of aging is becoming clearly evident in recent large-scale “omics” studies in mice and humans. Transcriptional changes in 17 organs across 10 ages from neonatal to 30-month-old-mice show very different trajectories of aging with varied onset and amplitudes of changes.32 In humans, a meta-analysis of >300 brains and 17 brain regions showed broad non-linear changes in gene expression which varied across brain regions with age.33 Similarly, DNA methylation, which shows a remarkable correlation with aging across species, was reported to be 24 times faster during the first few years of life compared to after puberty.34 Inflection points in DNA methylation indicative of non-monotonous changes were also observed in a 20-year follow-up Swedish aging cohort of 845 individuals around ages 70 and 85.35 A recent study of some 3,000 proteins in blood of over 4,000 healthy people concluded that markers of aging do not in fact suggest at monotonic aging process, but instead show periods of accelerated change and periods of slower change.28 Peripheral blood mononuclear cells exhibit two distinct peaks of rapid epigenomic changes in humans, one around 40 years of age and another around 60–70 years of age.36 Likewise, a longitudinal study of the transcriptome and DNA methylation state of skin in 121 humans between 21 and 76 years of age revealed 4 distinct “phases of aging” highlighted by the activity of different biological processes.37 Interestingly, functional MRI measurements of brain connectivity seem to support these asynchronous molecular measurements, showing non-linear changes and transition points around ages 40 and 70–75 years of age.38
In addition to these variations in rates of organismal aging over time, the above studies also provide clear evidence for the lack of uniformity of underlying aging process across space, namely among tissues in an individual (Figure 2). Undoubtedly, for any given species, there are certain tissues and organs whose functions decline earliest and most rapidly. For example, in humans the cardiovascular system exhibits a much greater functional decline with age than the gastrointestinal system,23 at least based on physiological parameters measured, as noted above, on absolute scales. In Drosophila, dysfunction of the gut and skeletal muscle appear to be primary drivers of aging and mortality.39,40,41,42 As mentioned above, transcriptomic studies strongly agree that tissues, organs, and even sub-regions within an organ such as the brain age at different rates.32,33 This is consistent with findings from studies in rats of organ-specific changes in transcriptome and proteome with age.43 In addition, tissues within human and mouse individuals show unique DNA methylation rates.34,25. Thus, spatially, there is clear heterogeneity of aging rates among tissues, and the aging of members of one species may be driven by functional loss of tissues that are different from those in another species. However, is it the case that, within a species, the relative rates of aging of different tissues and organs are the same from one individual to another? In one pioneering human study, a multi-omics analysis of aging biomarkers suggested that different individuals manifest different “ageotypes”, meaning that the tissue that ages most rapidly differs from one person to another.44 Therefore, for any given species, although there may be certain organs and tissues that tend to be the most susceptible to functional decline with age, which is the actual driver of organismal aging for any given member of that species is highly variable.
Figure 2: Asynchronous aging of different tissues and organs.
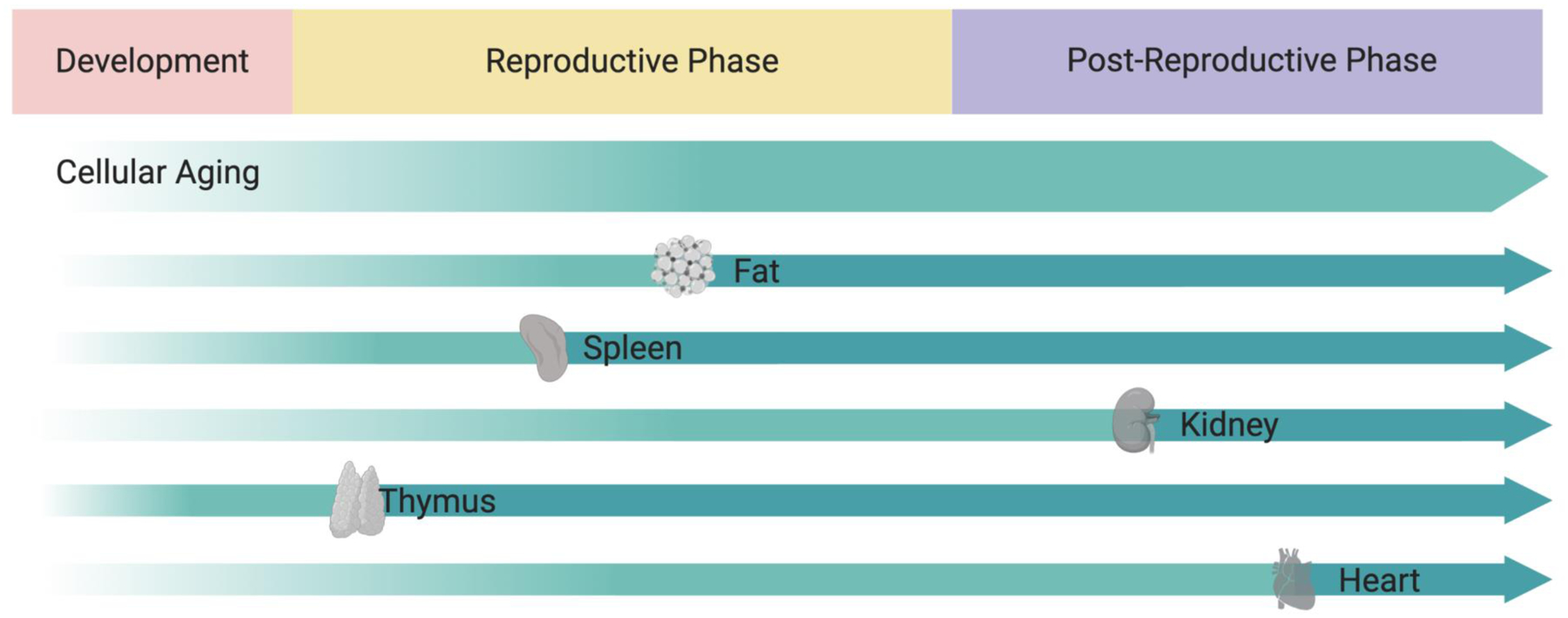
Differential rates of aging at the cellular level lead to organ aging at different rates and at different stages of life. Aging may start early in life in every tissue but, different tissues may age faster than other. The different rates of aging between tissues depicted here for illustrative purposes are based on transcriptomic changes across 17 organs in aging mice as described recently (Schaum, Lehallier, Hahn et al., Nature 2020).
If the most rapidly aging tissues are the main drivers of organismal aging, then it naturally begs the question as to what are intrinsic changes to the cellular and extracellular matrix constituents that drive the aging of those tissues. Within any tissue, changes in the cellular composition could be a result of changes in proportions of resident cells (such as, for example, an increase in the proportion of fibroblasts or resident macrophages) or to intrinsic changes to subpopulations without any change in cellular proportions. Analysis of bulk populations of cells may fail to reveal such a distinction, but single cell analysis has the potential to reveal the relative contribution of these two distinct, but of course not mutually exclusive, processes of tissue aging. In a groundbreaking single cell study, RNA isolated from individual cardiomyocytes of young and old mice showed broad mosaicism in gene expression among cells within individual mice and increased differences in expression between cells.45. Isotope-based dating of DNA and protein at the ultrastructural level in various tissues in mice revealed extensive cellular mosaicism within aging tissues.46 Cells of the same type have vastly different ages based on DNA dating (including postmitotic cells), some as old as the animal, and subcellular structures within individual neurons or pancreatic beta cells have different ages based on protein dating. These observations are supported by a rapidly growing number of single cell RNA sequencing studies showing greater cell-to-cell variability in gene expression in human T cells and pancreatic cells, as well as differences in cellular mutation rates with age.47,48 A recent mouse aging cell atlas (Tabula muris senis) further underlines this finding across 23 tissues and organs.49 In addition, the atlas, as well as the related tissue RNA bulk study,32 demonstrate a change of composition of the cellular constituents. Notably, multiple tissues exhibited an increase in the numbers and percentages of pro-inflammatory immune cells as a function of age, a conclusion independently drawn in single cell transcriptomic surveys of 7 tissues in aging rats and 3 tissues in aging mice.50,51
Digital aging versus analog aging
With regard to intrinsic changes to cells as a driver of tissue aging, a particular cell type in a tissue could change gradually over time in a process of “analog” aging, analogous to how we characterize organismal aging. Indeed, in the scRNA-Seq study mentioned above, parenchymal cells in multiple tissues exhibited progressive changes in gene expression with age.49 However, at the cellular level, the possibility of a certain amount “digital aging” needs also to be considered, digital in the sense of discrimination by discrete units, not necessarily binary units (Figure 3). Whereas the progression of tissue aging suggests an analog process with such measures as physiological function, gene expression levels, and protein aggregates changing gradually, we know that there are at least some discrete state transitions of cells in a tissue that can underlie this apparent analog decline. For example, for the thymus, heart, and other tissues,52,53 there a progressive decline in cell number with age due to apoptotic cell death. The death of a cell can be considered one such digital readout – cells are either alive or dead. Likewise, cellular senescence is another digital cellular state in the sense that cells can be classified by various markers as being either senescent or non-senescent.54 The extent to which the age-dependent phenotype of a tissue is due to the burden of senescent cells provides for another example of a gradual processes being determined by the relative levels of cells in one state or another. Indeed, based on scRNA-Seq data, the proportion of cells expressing the senescence marker p16 more than doubled in a number of tissues of old mice compared to young mice in multiple tissues of mouse.49 None of these transitions occurs instantaneously, and there are clearly intermediate states. Nevertheless, such cellular states are routinely determined as discrete and quantal rather than continuous. Even in the case of senescence where multiple different senescent states have been defined,55 they are still modeled as discrete. With this model taken to its extreme, the analog decline of the function of a tissue can be modeled as a result of the gradual accumulation of cells that have transitioned from a functional to one or more dysfunctional states. Further single cell studies will clearly delineate the relative contribution of analog versus digital cellular aging as drivers of tissue aging.
Figure 3. Digital aging.
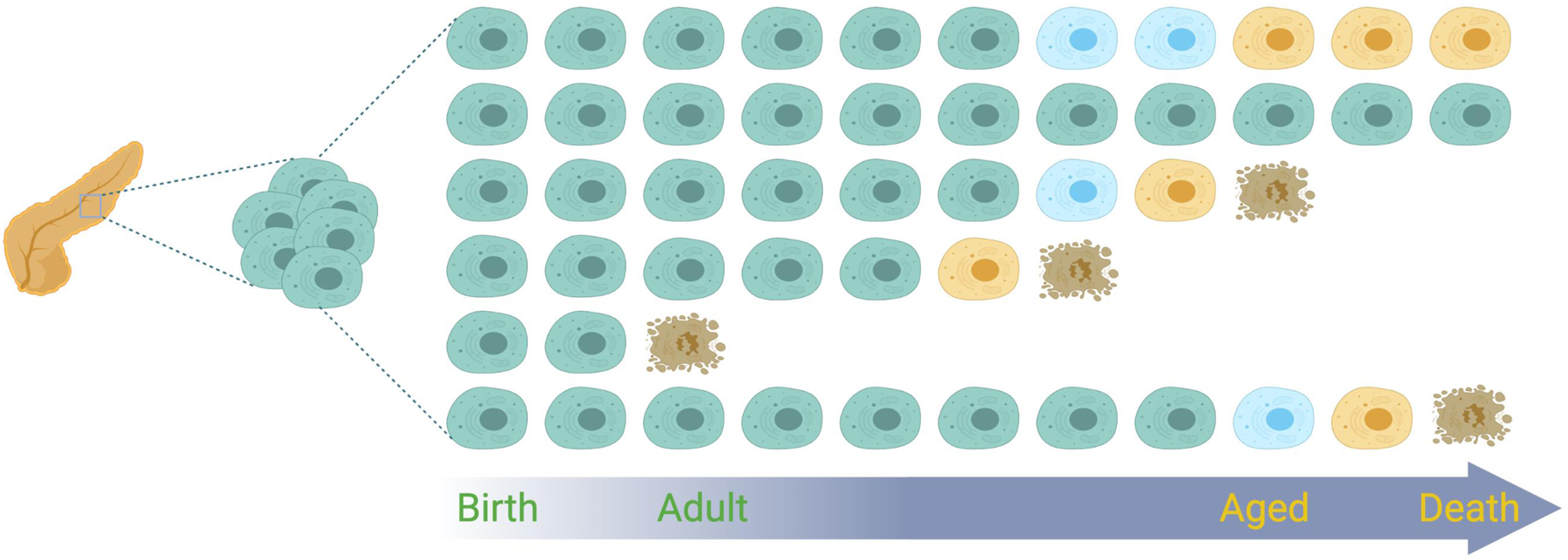
Cells from the same organ, born at the same time, show distinct temporal patterns of functional decline. These functional changes can be viewed as occurring in digital steps corresponding to discreet states of dysfunction (blue, yellow), resulting in different homeostatic organ states over time. Cells do not necessarily have to follow the same sequences of digital steps and acquire the same levels of dysfunction before they die, shown here as the adoption of brown, apoptotic features.
This notion of aging by quantal steps can clearly be extended to the subcellular world to explore proximal causes of age-related changes in cellular phenotypes. Proteins can be appropriately folded or mis-folded; RNA can be correctly spliced or mis-spliced; DNA can have single base changes. The extent to which cellular aging is due to digital versus analog changes at the molecular level is certain to depend of which cellular phenotype is under consideration in the same way that different tissues may exhibit age-related changes as a result of digital cellular aging to different degrees.
Likewise, it will be interesting to consider whether interventions that extend lifespan or healthspan act by shifting the balance between digital states at the cellular level. As an example, senolytic therapy is clearly aimed to enhance healthspan by cells in a particular state, not all cells in an aged tissue.56 Aging is associated with changes in the population of immune cells infiltrates,51 and caloric restriction can reverse those trends.50 This is not an effect on the aging states of the parenchymal cells but certainly results in age-related changes at the tissue level as a consequence of cell composition. Whereas transcriptional signature changes that are common among various lifespan extension interventions,57 it remains to be determined by single cell analysis whether these changes reflect cell composition changes of a tissue as opposed to distributed gene expression changes among resident cells.
Is aging contagious?
Given the complex heterogeneities of cell and tissue aging in any single individual and the notion of the most rapidly aging tissues being the driver of the aging of that organism, do those more rapidly aging tissues accelerate the aging of other tissues in the body? Does the aging of one cell affect the age of another cell? Is aging contagious? The notion of the aging process “spreading” from one cell to another is highlighted, again, by the field of cellular senescence. The secretome of senescent cells has been shown to induce senescence of neighboring cells.58 In that sense, there can be cellular leaders that accelerate aging of other cells in the tissue.
The notion of cell-to-cell spreading of cellular dysfunction is of course not limited to the biology of senescence. This is becoming an increasingly recognized phenomenon in the pathogenesis of neurodegenerative diseases.59 In many diseases, including Alzheimer’s disease, Huntington’s disease, and Parkinson’s disease, a cardinal feature of the pathology is intracellular aggregation of proteins. While seemingly a cell-intrinsic phenomenon, one of the curious features of the pathology of these diseases is the apparent spread of the cellular abnormalities to anatomically connected brain regions. The general concept of this kind of spreading proteinopathy from one cell to another, locally, arise from the biology of prions and prion diseases.60 Of course, some prions are truly contagious, in the sense of being transmissible between individuals or across species, but the spread within the central nervous system of an individual suggests cell-to-cell spread.61 As with senescence, this phenomenon could represent the conversion of cells from one state (free from aggregates) to another (aggregate-laden) since protein aggregation can be self-propagating. As protein aggregation is one of the key features of cellular aging, it is intriguing to consider the possibility of aged cells achieving a sufficiently dysfunctional state as a result of protein aggregation, then conferring an “aging signal” to nearby cells through non-cell autonomous regulation of proteostasis.62
If aging is indeed contagious, is the spread restricted to neighboring cells or might it spread to distant tissues via the systemic circulation? Based on early work from our laboratories that ushered in a new era of the use of the technique of parabiosis in aging research, it is clear that systemic factors originating from distant tissues in the body are able to either promote or reverse cell and tissue aging phenotypes.63,64,65 These findings, as well as many follow up studies,66,67,68 including the demonstration that plasma infusions alone are sufficient to exert these effects,65,69 have unequivocally demonstrated that factors in the blood are able to communicate information from one or more source tissues to other tissues throughout the body. These could potentially accelerate, delay, or even reverse the rate of aging of other tissues in the body. Indeed, single cell RNAseq studies of brain endothelial cell aging showed that infusion of aged plasma can accelerate while young plasma can reverse aging as a function of the transcriptome.70 These studies highlight the fact that cellular aging does not occur independently of influences that are both local and systemic.
Aging and disease: Chicken and egg?
Together, these finding highlight another emerging concept in aging research. It has long been known that the major risk factor for the vast majority of chronic diseases is age itself.71 Without a doubt, the aging process renders cells and tissues more susceptible to the pathophysiological processes that underlie these diseases. However, the data of the acceleration of aging by blood-borne factors also leads to the converse question: Is having a chronic disease a major risk factor for accelerated aging? That is, are diseases risk factors for aging? As a disease engulfs a tissue, it is likely to lead to local and systemic changes that, themselves, may promote dysfunction on neighboring and distant cells. Epidemiological data does suggest that individuals with various chronic diseases such as cancer, diabetes, and HIV/AIDS are more likely to develop co-morbidities later in life,72,73,74 consistent with the notion that one disease (or its treatment) may accelerate aging processes more generally, thus rendering individuals more susceptible to other diseases, just as aging itself does. In this sense, and related to the notion of the spread of aging described above, aging begets aging. How this process is modulated within tissues and across tissues to prevent an exponential increase in the rate of aging over time may also relate to the ability of cells to adopt new, relatively stable homeostatic states even in the face of extrinsic stresses.
Targeting cellular and tissue drivers of organismal aging, or their means of spreading their aging messages to nearby and distant cells and tissues, holds the promise of extending healthspan by slowing the overall rate of aging. If successful, this would not only slow the structural and functional declines that are the quintessential features of the organismal aging, but also would reduce the incidence of age-related diseases. Pharmacologic and dietary interventions that extend lifespan in model organisms and may do so by slowing aging rates are increasingly being developed for testing in humans.75 However, it is likely that, for humans, a considerable degree of personalized approaches will be necessary. Even among highly genetically similar strains of yeast or mice, dietary restriction is equally likely to shorten lifespan as to extend lifespan.76,77,78 A more thorough understanding of the molecular processes of aging, the cellular basis of tissue aging, and the local and systemic communication networks that can accelerate or decelerate the rate of aging will certainly increase the number and diversity interventions to extend healthspan. Furthermore, the ability of interventions not only to slow the rate of aging but actually to rejuvenate cells and tissues seemingly by reversing aging processes expands the spectrum of healthspan-promoting treatments.79,80,81,82,83 Coupled with technological advances to measure aging phenotypes and rates of aging at the cellular, tissue, and organismal levels, this will also likely increase the pace of the translation from the laboratory to the clinic.
Conclusion
As the number of distinct measures and biomarkers of aging increases and as the technologies to quantify them develop, the picture that emerges of the patterns of aging will continue to become increasingly more complex. Clearly, these temporal patterns cannot be discerned from lifespan studies and distinct patterns of aging among different tissues (as illustrated in Figure 1) cannot be discerned from samples of whole model organisms. Likewise, individual patterns of aging cannot be understood from pooled samples. We posit that the increased complexity that has and will continue to emerge will redirect attention away from singular and linear “causes” of aging and toward a more unified model of both intrinsic (e.g., oxidative damage, protein aggregation, genomic instability) and extrinsic (e.g. inflammation, hypoxia, nutritional deprivation) stresses impinging on cells whose responses are dictated by genetically determined homeostatic response. Such a model provides a framework for incorporating findings that highlight phenotypes of aging that deviate from models of uniform and synchronized processes throughout an organism.
REFERENCES
- 1.Flatt T, Partridge L (2018) Horizons in the evolution of aging. BMC Biol, 16: 93- [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Medawar PB (1952) An Unsolved Problem of Biology. H. K. Lewis, London. [Google Scholar]
- 3.Williams GC (1957) Pleiotropy, natural selection, and the evolution of senescence. Evolution, 11: 398–411. [Google Scholar]
- 4.Kirkwood TB (1977) Evolution of ageing. Nature, 270: 301–304. [DOI] [PubMed] [Google Scholar]
- 5.Kennedy BK, Berger SL, Brunet A, Campisi J, Cuervo AM, Epel ES, Franceschi C, Lithgow GJ, Morimoto RI, Pessin JE, Rando TA, Richardson A, Schadt EE, Wyss-Coray T, Sierra F (2014) Geroscience: linking aging to chronic disease. Cell, 159: 709–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gladyshev VN (2020) The ground zero of organismal life and aging. Trends Mol Med, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rose MR (2009) Adaptation, aging, and genomic information. Aging (Albany NY), 1: 444–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gladyshev VN (2016) Aging: progressive decline in fitness due to the rising deleteriome adjusted by genetic, environmental, and stochastic processes. Aging Cell, 15: 594–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tissenbaum HA, Guarente L (2002) Model organisms as a guide to mammalian aging. Dev Cell, 2: 9–19. [DOI] [PubMed] [Google Scholar]
- 10.Piper MDW, Partridge L (2018) Drosophila as a model for ageing. Biochim Biophys Acta Mol Basis Dis, 1864: 2707–2717. [DOI] [PubMed] [Google Scholar]
- 11.Singh PP, Demmitt BA, Nath RD, Brunet A (2019) The genetics of aging: A vertebrate perspective. Cell, 177: 200–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gonzalez-Freire M, Diaz-Ruiz A, Hauser D, Martinez-Romero J, Ferrucci L, Bernier M, de Cabo R (2020) The road ahead for health and lifespan interventions. Ageing Res Rev, 59: 101037- [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fontana L, Partridge L, Longo VD (2010) Extending healthy life span--from yeast to humans. Science, 328: 321–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hayflick L (2007) Entropy explains aging, genetic determinism explains longevity, and undefined terminology explains misunderstanding both. PLoS Genet, 3: e220- [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Goodell MA, Rando TA (2015) Stem cells and healthy aging. Science, 350: 1199–1204. [DOI] [PubMed] [Google Scholar]
- 16.Brett JO, Rando TA (2014) Alive and well? Exploring disease by studying lifespan. Curr Opin Genet Dev, 26: 33–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bansal A, Zhu LJ, Yen K, Tissenbaum HA (2015) Uncoupling lifespan and healthspan in Caenorhabditis elegans longevity mutants. Proc Natl Acad Sci U S A, 112: E277–E286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pearl R (1928) The Rate of Living. University of London Press, London., [Google Scholar]
- 19.Rando TA (2006) Stem cells, ageing and the quest for immortality. Nature, 441: 1080–1086. [DOI] [PubMed] [Google Scholar]
- 20.Butler PG, Wanamaker AD, Scourse JD, Richardson CA, Reynolds DJ (2013) Variability of marine climate on the North Icelandic Shelf in a 1357-year proxy archive based on growth increments in the bivalve Artica islandica. Palaeogeogr Palaeoclimatol Palaeoecol, 373: 141–151. [Google Scholar]
- 21.Ailshire JA, Beltran-Sanchez H, Crimmins EM (2015) Becoming centenarians: disease and functioning trajectories of older US Adults as they survive to 100. J Gerontol A Biol Sci Med Sci, 70: 193–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kirkwood TB, Feder M, Finch CE, Franceschi C, Globerson A, Klingenberg CP, LaMarco K, Omholt S, Westendorp RG (2005) What accounts for the wide variation in life span of genetically identical organisms reared in a constant environment? Mech Ageing Dev, 126: 439–443. [DOI] [PubMed] [Google Scholar]
- 23.Khan SS, Singer BD, Vaughan DE (2017) Molecular and physiological manifestations and measurement of aging in humans. Aging Cell, 16: 624–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bocklandt S, Lin W, Sehl ME, Sanchez FJ, Sinsheimer JS, Horvath S, Vilain E (2011) Epigenetic predictor of age. PLoS ONE, 6: e14821- [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stubbs TM, Bonder MJ, Stark AK, Krueger F, von MF, Stegle O, Reik W (2017) Multi-tissue DNA methylation age predictor in mouse. Genome Biol, 18: 68- [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Heidinger BJ, Blount JD, Boner W, Griffiths K, Metcalfe NB, Monaghan P (2012) Telomere length in early life predicts lifespan. Proc Natl Acad Sci U S A, 109: 1743–1748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Steenstrup T, Kark JD, Verhulst S, Thinggaard M, Hjelmborg JVB, Dalgard C, Kyvik KO, Christiansen L, Mangino M, Spector TD, Petersen I, Kimura M, Benetos A, Labat C, Sinnreich R, Hwang SJ, Levy D, Hunt SC, Fitzpatrick AL, Chen W, Berenson GS, Barbieri M, Paolisso G, Gadalla SM, Savage SA, Christensen K, Yashin AI, Arbeev KG, Aviv A (2017) Telomeres and the natural lifespan limit in humans. Aging (Albany NY), 9: 1130–1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lehallier B, Gate D, Schaum N, Nanasi T, Lee SE, Yousef H, Moran LP, Berdnik D, Keller A, Verghese J, Sathyan S, Franceschi C, Milman S, Barzilai N, Wyss-Coray T (2019) Undulating changes in human plasma proteome profiles across the lifespan. Nat Med, 25: 1843–1850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen W, Qian W, Wu G, Chen W, Xian B, Chen X, Cao Y, Green CD, Zhao F, Tang K, Han JD (2015) Three-dimensional human facial morphologies as robust aging markers. Cell Res, 25: 574–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Herndon LA, Schmeissner PJ, Dudaronek JM, Brown PA, Listner KM, Sakano Y, Paupard MC, Hall DH, Driscoll M (2002) Stochastic and genetic factors influence tissue-specific decline in ageing C. elegans. Nature, 419: 808–814. [DOI] [PubMed] [Google Scholar]
- 31.Tricoire H, Rera M (2015) A new, discontinuous 2 phases of aging model: Lessons from Drosophila melanogaster. PLoS ONE, 10: e0141920- [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schaum N, Lehallier B, Hahn O, Palovics R, Hosseinzadeh S, Lee SE, Sit R, Lee DP, Losada PM, Zardeneta ME, Fehlmann T, Webber JT, McGeever A, Calcuttawala K, Zhang H, Berdnik D, Mathur V, Tan W, Zee A, Tan M, Pisco AO, Karkanias J, Neff NF, Keller A, Darmanis S, Quake SR, Wyss-Coray T (2020) Ageing hallmarks exhibit organ-specific temporal signatures. Nature, 583: 596–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Isildak U, Somel M, Thornton JM, Donertas HM (2020) Temporal changes in the gene expression heterogeneity during brain development and aging. Sci Rep, 10: 4080- [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Horvath S (2013) DNA methylation age of human tissues and cell types. Genome Biol, 14: R115- [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li X, Ploner A, Wang Y, Magnusson PK, Reynolds C, Finkel D, Pedersen NL, Jylhava J, Hagg S (2020) Longitudinal trajectories, correlations and mortality associations of nine biological ages across 20-years follow-up. Elife, 9: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Marquez EJ, Chung CH, Marches R, Rossi RJ, Nehar-Belaid D, Eroglu A, Mellert DJ, Kuchel GA, Banchereau J, Ucar D (2020) Sexual-dimorphism in human immune system aging. Nat Commun, 11: 751- [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Holzscheck N, Sohle J, Kristof B, Gronniger E, Gallinat S, Wenck H, Winnefeld M, Falckenhayn C, Kaderali L (2020) Multi-omics network analysis reveals distinct stages in the human aging progression in epidermal tissue. Aging (Albany NY), 12: 12393–12409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Edde M, Leroux G, Altena E, Chanraud S (2020) Functional brain connectivity changes across the human life span: From fetal development to old age. J Neurosci Res, [DOI] [PubMed] [Google Scholar]
- 39.Biteau B, Hochmuth CE, Jasper H (2008) JNK activity in somatic stem cells causes loss of tissue homeostasis in the aging Drosophila gut. Cell Stem Cell, 3: 442–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Clark RI, Salazar A, Yamada R, Fitz-Gibbon S, Morselli M, Alcaraz J, Rana A, Rera M, Pellegrini M, Ja WW, Walker DW (2015) Distinct shifts in microbiota composition during Drosophila aging impair intestinal function and drive mortality. Cell Rep, 12: 1656–1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Demontis F, Perrimon N (2010) FOXO/4E-BP signaling in Drosophila muscles regulates organism-wide proteostasis during aging. Cell, 143: 813–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Owusu-Ansah E, Song W, Perrimon N (2013) Muscle mitohormesis promotes longevity via systemic repression of insulin signaling. Cell, 155: 699–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ori A, Toyama BH, Harris MS, Bock T, Iskar M, Bork P, Ingolia NT, Hetzer MW, Beck M (2015) Integrated transcriptome and proteome analyses reveal organ-specific proteome deterioration in old rats. Cell Syst, 1: 224–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ahadi S, Zhou W, Schussler-Fiorenza Rose SM, Sailani MR, Contrepois K, Avina M, Ashland M, Brunet A, Snyder M (2020) Personal aging markers and ageotypes revealed by deep longitudinal profiling. Nat Med, 26: 83–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bahar R, Hartmann CH, Rodriguez KA, Denny AD, Busuttil RA, Dolle ME, Calder RB, Chisholm GB, Pollock BH, Klein CA, Vijg J (2006) Increased cell-to-cell variation in gene expression in ageing mouse heart. Nature, 441: 1011–1014. [DOI] [PubMed] [Google Scholar]
- 46.Arrojo E Drigo, Lev-Ram V, Tyagi, Ramachandra R, Deerinck, Bushong E, Phan S, Orphan V, Lechene C, Ellisman MH, Hetzer MW (2019) Age mosaicism across multiple scales in adult tissues. Cell Metab, 30: 343–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Martinez-Jimenez CP, Eling N, Chen HC, Vallejos CA, Kolodziejczyk AA, Connor F, Stojic L, Rayner TF, Stubbington MJT, Teichmann SA, de la Roche M, Marioni JC, Odom DT (2017) Aging increases cell-to-cell transcriptional variability upon immune stimulation. Science, 355: 1433–1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Enge M, Arda HE, Mignardi M, Beausang J, Bottino R, Kim SK, Quake SR (2017) Single-cell analysis of human pancreas reveals transcriptional signatures of aging and somatic mutation patterns. Cell, 171: 321–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tabula Muris Consortium (2020) A single-cell transcriptomic atlas characterizes ageing tissues in the mouse. Nature, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ma S, Sun S, Geng L, Song M, Wang W, Ye Y, Ji Q, Zou Z, Wang S, He X, Li W, Esteban CR, Long X, Guo G, Chan P, Zhou Q, Belmonte JCI, Zhang W, Qu J, Liu GH (2020) Caloric restriction reprograms the single-cell transcriptional landscape of rattus norvegicus aging. Cell, 180: 984–1001. [DOI] [PubMed] [Google Scholar]
- 51.Kimmel JC, Penland L, Rubinstein ND, Hendrickson DG, Kelley DR, Rosenthal AZ (2019) Murine single-cell RNA-seq reveals cell-identity- and tissue-specific trajectories of aging. Genome Res, 29: 2088–2103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Goronzy JJ, Weyand CM (2005) T cell development and receptor diversity during aging. Curr Opin Immunol, 17: 468–475. [DOI] [PubMed] [Google Scholar]
- 53.Tower J (2015) Programmed cell death in aging. Ageing Res Rev, 23: 90–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gorgoulis V, Adams PD, Alimonti A, Bennett DC, Bischof O, Bishop C, Campisi J, Collado M, Evangelou K, Ferbeyre G, Gil J, Hara E, Krizhanovsky V, Jurk D, Maier AB, Narita M, Niedernhofer L, Passos JF, Robbins PD, Schmitt CA, Sedivy J, Vougas K, von ZT, Zhou D, Serrano M, Demaria M (2019) Cellular senescence: Defining a path forward. Cell, 179: 813–827. [DOI] [PubMed] [Google Scholar]
- 55.Hernandez-Segura A, de Jong TV, Melov S, Guryev V, Campisi J, Demaria M (2017) Unmasking transcriptional heterogeneity in senescent cells. Curr Biol, 27: 2652–2660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Robbins PD, Jurk D, Khosla S, Kirkland JL, LeBrasseur NK, Miller JD, Passos JF, Pignolo RJ, Tchkonia T, Niedernhofer LJ (2020) Senolytic drugs: Reducing senescent cell viability to extend health span. Annu Rev Pharmacol Toxicol, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tyshkovskiy A, Bozaykut P, Borodinova AA, Gerashchenko MV, Ables GP, Garratt M, Khaitovich P, Clish CB, Miller RA, Gladyshev VN (2019) Identification and application of gene expression signatures associated with lifespan extension. Cell Metab, 30: 573–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Acosta JC, Banito A, Wuestefeld T, Georgilis A, Janich P, Morton JP, Athineos D, Kang TW, Lasitschka F, Andrulis M, Pascual G, Morris KJ, Khan S, Jin H, Dharmalingam G, Snijders AP, Carroll T, Capper D, Pritchard C, Inman GJ, Longerich T, Sansom OJ, Benitah SA, Zender L, Gil J (2013) A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat Cell Biol, 15: 978–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Davis AA, Leyns CEG, Holtzman DM (2018) Intercellular spread of protein aggregates in neurodegenerative disease. Annu Rev Cell Dev Biol, 34: 545–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Prusiner SB (2013) Biology and genetics of prions causing neurodegeneration. Annu Rev Genet, 47: 601–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Guo JL, Lee VM (2014) Cell-to-cell transmission of pathogenic proteins in neurodegenerative diseases. Nat Med, 20: 130–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Morimoto RI (2020) Cell-nonautonomous regulation of proteostasis in aging and disease. Cold Spring Harb Perspect Biol, 12: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Conboy IM, Conboy MJ, Wagers AJ, Girma ER, Weissman IL, Rando TA (2005) Rejuvenation of aged progenitor cells by exposure to a young systemic environment. Nature, 433: 760–764. [DOI] [PubMed] [Google Scholar]
- 64.Brack AS, Conboy MJ, Roy S, Lee M, Kuo CJ, Keller C, Rando TA (2007) Increased Wnt signaling during aging alters muscle stem cell fate and increases fibrosis. Science, 317: 807–810. [DOI] [PubMed] [Google Scholar]
- 65.Villeda SA, Luo J, Mosher KI, Zou B, Britschgi M, Bieri G, Stan TM, Fainberg N, Ding Z, Eggel A, Lucin KM, Czirr E, Park JS, Couillard-Despres S, Aigner L, Li G, Peskind ER, Kaye JA, Quinn JF, Galasko DR, Xie XS, Rando TA, Wyss-Coray T (2011) The ageing systemic milieu negatively regulates neurogenesis and cognitive function. Nature, 477: 90–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ruckh JM, Zhao JW, Shadrach JL, van WP, Rao TN, Wagers AJ, Franklin RJ (2012) Rejuvenation of regeneration in the aging central nervous system. Cell Stem Cell, 10: 96–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Salpeter SJ, Khalaileh A, Weinberg-Corem N, Ziv O, Glaser B, Dor Y (2013) Systemic regulation of the age-related decline of pancreatic beta-cell replication. Diabetes, 62: 2843–2848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Katsimpardi L, Litterman NK, Schein PA, Miller CM, Loffredo FS, Wojtkiewicz GR, Chen JW, Lee RT, Wagers AJ, Rubin LL (2014) Vascular and neurogenic rejuvenation of the aging mouse brain by young systemic factors. Science, 344: 630–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Castellano JM, Mosher KI, Abbey RJ, McBride AA, James ML, Berdnik D, Shen JC, Zou B, Xie XS, Tingle M, Hinkson IV, Angst MS, Wyss-Coray T (2017) Human umbilical cord plasma proteins revitalize hippocampal function in aged mice. Nature, 544: 488–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Chen MB, Yang AC, Yousef H, Lee D, Chen W, Schaum N, Lehallier B, Quake SR, Wyss-Coray T (2020) Brain endothelial cells are exquisite sensors of age-related circulatory cues. Cell Rep, 30: 4418–4432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Niccoli T, Partridge L (2012) Ageing as a risk factor for disease. Curr Biol, 22: R741–R752. [DOI] [PubMed] [Google Scholar]
- 72.Keating NL, Norredam M, Landrum MB, Huskamp HA, Meara E (2005) Physical and mental health status of older long-term cancer survivors. J Am Geriatr Soc, 53: 2145–2152. [DOI] [PubMed] [Google Scholar]
- 73.Kirkman MS, Briscoe VJ, Clark N, Florez H, Haas LB, Halter JB, Huang ES, Korytkowski MT, Munshi MN, Odegard PS, Pratley RE, Swift CS (2012) Diabetes in older adults: a consensus report. J Am Geriatr Soc, 60: 2342–2356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.High KP, Brennan-Ing M, Clifford DB, Cohen MH, Currier J, Deeks SG, Deren S, Effros RB, Gebo K, Goronzy JJ, Justice AC, Landay A, Levin J, Miotti PG, Munk RJ, Nass H, Rinaldo CR Jr., Shlipak MG, Tracy R, Valcour V, Vance DE, Walston JD, Volberding P (2012) HIV and aging: state of knowledge and areas of critical need for research. A report to the NIH Office of AIDS Research by the HIV and Aging Working Group. J Acquir Immune Defic Syndr, 60 Suppl 1: S1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Longo VD, Antebi A, Bartke A, Barzilai N, Brown-Borg HM, Caruso C, Curiel TJ, de CR, Franceschi C, Gems D, Ingram DK, Johnson TE, Kennedy BK, Kenyon C, Klein S, Kopchick JJ, Lepperdinger G, Madeo F, Mirisola MG, Mitchell JR, Passarino G, Rudolph KL, Sedivy JM, Shadel GS, Sinclair DA, Spindler SR, Suh Y, Vijg J, Vinciguerra M, Fontana L (2015) Interventions to slow aging in humans: Are we ready? Aging Cell, 14: 497–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Schleit J, Johnson SC, Bennett CF, Simko M, Trongtham N, Castanza A, Hsieh EJ, Moller RM, Wasko BM, Delaney JR, Sutphin GL, Carr D, Murakami CJ, Tocchi A, Xian B, Chen W, Yu T, Goswami S, Higgins S, Holmberg M, Jeong KS, Kim JR, Klum S, Liao E, Lin MS, Lo W, Miller H, Olsen B, Peng ZJ, Pollard T, Pradeep P, Pruett D, Rai D, Ros V, Singh M, Spector BL, Vander WH, An EH, Fletcher M, Jelic M, Rabinovitch PS, MacCoss MJ, Han JD, Kennedy BK, Kaeberlein M (2013) Molecular mechanisms underlying genotype-dependent responses to dietary restriction. Aging Cell, 12: 1050–1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Rikke BA, Liao CY, McQueen MB, Nelson JF, Johnson TE (2010) Genetic dissection of dietary restriction in mice supports the metabolic efficiency model of life extension. Exp Gerontol, 45: 691–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Liao CY, Rikke BA, Johnson TE, Diaz V, Nelson JF (2010) Genetic variation in the murine lifespan response to dietary restriction: from life extension to life shortening. Aging Cell, 9: 92–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Rando TA, Chang HY (2012) Aging, rejuvenation, and epigenetic reprogramming: resetting the aging clock. Cell, 148: 46–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Villeda SA, Plambeck KE, Middeldorp J, Castellano JM, Mosher KI, Luo J, Smith LK, Bieri G, Lin K, Berdnik D, Wabl R, Udeochu J, Wheatley EG, Zou B, Simmons DA, Xie XS, Longo FM, Wyss-Coray T (2014) Young blood reverses age-related impairments in cognitive function and synaptic plasticity in mice. Nat Med, 20: 659–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wyss-Coray T (2016) Ageing, neurodegeneration and brain rejuvenation. Nature, 539: 180–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ocampo A, Reddy P, Martinez-Redondo P, Platero-Luengo A, Hatanaka F, Hishida T, Li M, Lam D, Kurita M, Beyret E, Araoka T, Vazquez-Ferrer E, Donoso D, Roman JL, Xu J, Rodriguez EC, Nunez G, Nunez DE, Campistol JM, Guillen I, Guillen P, Izpisua Belmonte JC (2016) In vivo amelioration of age-associated hallmarks by partial reprogramming. Cell, 167: 1719–1733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Sarkar TJ, Quarta M, Mukherjee S, Colville A, Paine P, Doan L, Tran CM, Chu CR, Horvath S, Qi LS, Bhutani N, Rando TA, Sebastiano V (2020) Transient non-integrative expression of nuclear reprogramming factors promotes multifaceted amelioration of aging in human cells. Nat Commun, 11: 1545- [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Dobzhansky T (1973) Nothing in biology makes sense except in the light of evolution. Amer Biol Teacher, 35: 125–129. [Google Scholar]
- 85.Zhang R, Chen HZ, Liu DP (2015) The four layers of aging. Cell Syst, 1: 180–186. [DOI] [PubMed] [Google Scholar]