

Abstract
COVID-19 is a disease that puts most of the world on lockdown and the search for therapeutic drugs is still ongoing. Therefore, this study used in silico screening to identify natural bioactive compounds from fruits, herbaceous plants, and marine invertebrates that are able to inhibit protease activity in SARS-CoV-2 (PDB: 6LU7). We have used extensive screening strategies such as drug likeliness, antiviral activity value prediction, molecular docking, ADME, molecular dynamics (MD) simulation, and MM/GBSA. A total of 17 compounds were shortlisted using Lipinski’s rule in which 5 compounds showed significant predicted antiviral activity values. Among these 5, only 2 compounds, Macrolactin A and Stachyflin, showed good binding energy of −9.22 and −8.00 kcal/mol, respectively, within the binding pocket of the Mpro catalytic residues (HIS 41 and CYS 145). These two compounds were further analyzed to determine their ADME properties. The ADME evaluation of these 2 compounds suggested that they could be effective in developing therapeutic drugs to be used in clinical trials. MD simulations showed that protein–ligand complexes of Macrolactin A and Stachyflin with the target receptor (6LU7) were stable for 100 nanoseconds. The MM/GBSA calculations of Mpro–Macrolactin A complex indicated higher binding free energy (−42.58 ± 6.35 kcal/mol). Dynamic cross-correlation matrix (DCCM) and principal component analysis (PCA) on the residual movement in the MD trajectories further confirmed the stability of Macrolactin A bound state with 6LU7. In conclusion, this study showed that marine natural compound Macrolactin A could be an effective therapeutic inhibitor against SARS-CoV-2 protease (6LU7). Additional in vitro and in vivo validations are strongly needed to determine the efficacy and therapeutic dose of Macrolactin A in biological systems.
Supplementary Information
The online version contains supplementary material available at 10.1007/s12010-021-03608-7.
Keywords: Coronavirus, Treatment, Molecular docking, MD simulation, ADMET, MM/GBSA
Introduction
The 2003 SARS-CoV outbreak caused 10% fatality [1], MERS-CoV produced 35% fatality [2], and now, SARS-CoV2 is responsible for 2.35% fatalities [3]. The experiences gained from managing previous novel coronavirus infections in healthcare facilities were associated with the lower fatalities of SARS-CoV2 [4, 5]. However, the virulence of SARS-CoV2 leads to national-based quarantine and standard operating procedures which were the only feasible approach to break the chain of infections while the entire world patiently waited for an effective vaccine against SARS-CoV2 [6]. SARS-CoV2 has a high RNA polymerase mutation rate that made this pathogen-resistant against antiviral drugs and, thus, increases the chances of re-infection [7–9]. Experience from using lopinavir/ritonavir and remdesivir during SARS-CoV led to identical treatment measures for SARS-CoV-2; however, the efficacy of these drugs is not as expected and patients in critical conditions have slimmer chances for recovery [10–12]. Despite the availability of vaccines, a recent survey showed that resistance in taking these vaccines was due to their unknown halal status and cost. The study recommended that the governments should subsidize the cost as for developing countries, these vaccines might not be affordable [11]. Moreover, healthcare facilities are restricted to developed areas, while rural communities rely on herbs to treat diseases and infections. Previous studies indicated that natural products were effective against coronavirus, coxsackievirus, dengue virus, enterovirus, hepatitis virus, herpes simplex virus, human immunodeficiency virus (HIV), and respiratory syncytial virus symptoms [13]. Unfortunately, technical barriers and certifications favor the use of modern medicine which led to natural compounds being gradually out of favor [14]. Recent studies showed that compounds from Andrographis paniculate were effective against SARS-CoV2 [15]. At the end of 2020, various news outlets such as Bloomberg, the business times, and straits times reported that Thailand has approved A. paniculate’s usage for the patient in the lower stage of COVID-19 infections. Antiviral research targets the inhibition of various virus parts such as spike proteins, reverse transcriptase, integrase, RNA, and protease enzyme (e.g., Mpro and 3CLpro) [16]. In silico screening and identification is low-cost, efficient, brief, and virtual to quantify activity relationships between target biomolecules such as DNA, RNA and proteins, and inhibitors from synthetic and natural sources during drug discovery [17, 18]. Research on SARS-CoV-2 identified 29 proteins comprising of structural, spike, envelope, membrane, nucleocapsid, non-structural, and adjunct proteins. Among all, an encoding protease was determined to be responsible for the process of making about 16 of these 29 proteins [19].
The macrolactin compounds are known to have a broad range of pharmacological activities including antiviral, antitumor, antibacterial, antiangiogenic, neuro-protective, antiproliferative activities, intestinal bowel disease protecting, and bone-remodeling activities [20]. The macrolactins are polyene macrolides made up of a 24-membered lactone ring containing conjugated double bonds [21]. So far, six macrolactins (A-F) have been chemically characterized, and only macrolactins A and E have been prepared by total synthesis [22]. Macrolactin A showed selective antibacterial activity, inhibited B16-F10 murine melanoma cancer cells in vitro assays, showed significant inhibition of mammalian Herpes simplex viruses (types I and II), and protected T-lymphoblast cells against human HIV viral replication [21].
Briefly, it can be said that the present study aimed to identify bioactive compounds from natural aquatic and terrestrial sources using in silico screening via molecular docking and molecular dynamics (MD) simulations [23]. This analysis could aid in predicting the suitability and efficacy of the compounds based on the stability of physiochemical properties and the protein–ligand complexes between selected inhibitors and SARS-CoV-2 viral protease.
Result and Discussion
Screening Process and Molecular Docking Analysis
The current research target was to identify potential molecules for SARS-CoV2 treatment that are abundant in the Asia-Pacific zone (i.e., India). A thorough literature survey was conducted to select candidate compounds that are available in the selected region, have proven antimicrobial activity and other pharmacological attributes, and are cost-effective. Natural compounds (n = 30) (Table 1) with antioxidant, antimicrobial, antiviral, and anticancer properties were identified from plants and animals. These compounds were screened using Lipinski’s rule against Mpro to assess the binding activity following published methods [54–57]. Among the 30 compounds, 17 possessed suitable drug-likeness properties (Supplementary Table S1). These compounds were then selected and proceeded with structure-based antiviral activity prediction using the PASS online server. The software gave a probability active (Pa Score) that ranged from 0 to 1, while previous studies showed that a value of >0.3 can be considered active [58]. A total of 5 compounds were predicted to possess the antiviral activity score (Pa) of 0.6 (60%) (Supplementary Table S2). These 5 compounds were further investigated for molecular docking study to identify a possible SARS-CoV2 Mpro inhibitor. Two compounds, namely, Macrolactin A (PUBCHEM ID: 6451096) and Stachyflin (PUBCHEM ID: 493326), showed low binding energy (−9.22 to −8.00 kcal/mol) which indicated effectiveness (Supplementary Table S3). These top 2 lead inhibitory compounds were further analyzed to assess their ADME properties (Table 2). Both of the compounds showed good physicochemical parameters where almost all the parameters were in suggested ranges. Then, they were further investigated by performing MD simulations which showed Macrolactin A and Stachyflin formed a stable protein–ligand complex. Finally, MM-GBSA analysis was carried out and Macrolactin A was the best inhibitor of the Mpro. Various literatures have reported different compounds that possess binding energy ranges from −7.0 to −8.5 kcal/mol, which might be a key to inhibit SARS-CoV2 infection [59, 60]. The overall flow for screening naturally occurring compounds against Mpro is depicted in Fig. 1. Macrolactin A showed binging energy of −9.22 kcal/mol to SARS-CoV-2 Mpro(PDB ID: 6LU7) protein and exhibited good pharmacokinetic property and could be a treatment to SARS-CoV-2 infection.
Table 1.
List of the molecules from natural products library
| Sl no. | Molecules | Source | PUBCEM ID | Pharmacological activity | Lipinski filtration result | Reference |
|---|---|---|---|---|---|---|
| 1 | CHRYSOPHANIC ACID | Rhubarb (Rheum rhabarbarum) | 10208 | Anticancer, antioxidant, neuroprotection, antibacterial, antiviral, regulating blood lipids | Pass | [24] |
| 2 | DIDEMNINS | Sea squirts (Aplidium albicans) | 123844 | Antiviral, antitumor, and immunosuppressive activity | Fail | [25] |
| 3 | EUDISTOMIN | Ascidians (Ritterella sigillinoides) | 3009848 | Anticancer, antibacterial | Pass | [26] |
| 4 | MANGIFERIN | Mango (Mangifera indica) | 5281647 | Antioxidant activity, antidiabetic, antitumor, lipometabolism regulating, cardioprotective, anti-hyperuricemic, anti-allergic, anti-inflammatory | Fail | [27] |
| 5 | MORIN | Almond (Prunus dulcis) | 5281670 | Cardiovascular disease, diabetic mellitus, neurodegenerative disease, cancer and anti-inflammatory | Pass | [28] |
| 6 | PHOMASETIN | Marine Fungi (Fusarium heterosporum) | 54693801 | Antiviral | Fail | [29] |
| 7 | SINAPIC ACID | Strawberry (Fragaria ananassa L.) | 637775 | Antioxidant, anti-inflammatory, anticancer, antimutagenic, antiglycemic, neuroprotective, and antibacterial | Pass | [30] |
| 8 | TRYPTOQUIVALINE | Marine Fungi (Neosartorya takakii) | 108075 | Antibacterial | Fail | [31] |
| 9 | AVAROL | Sponge (Dysidea avara) | 72185 | Antitumor, antiviral, anti-inflammatory | Fail | [32] |
| 10 | CLADOSIN C | Deep sea fungus (Cladosporium sphaerospermum) | 136845977 | Antiviral | Pass | [33] |
| 11 | ELLAGITANNIN | Pomegranate fruit (P. granatum L.) | 101601927 | Anti-inflammatory, anticancer, antioxidant and antimicrobial (antibacterial, antifungal and antiviral) | Fail | [34] |
| 12 | FUCOIDAN | Brown seaweed | 92023653 | Antiviral and antitumor activity | Pass | [35] |
| 13 | MARMELOSIN OR IMPERATORIN | Bael (Aegle marmelos) | 10212 | Antioxidant, antiproliferative, apoptotic, anticancer and antiviral, | Pass | [36] |
| 14 | NORDIHDROGUAIARETIC ACID | Desert shrub (Larrea tridentate) | 4534 | Anti-inflammation and antioxidant activity | Pass | [37] |
| 15 | PHENYLHEPTATRIYNE | Weed (Bidens pilosa L.) | 77981 | Cardio protective, anti-allergic, vein protective | Pass | [38] |
| 16 | STACHYFLIN | Stachybotrys sp. | 493326 | Antiviral | Pass | [39] |
| 17 | BROMELAIN | Pineapple (Ananas comosus) | 44263865 | Improvement of gastrointestinal tract related discomforts, anti-tumor, anti-inflammation, antimicrobial | Fail | [40, 41] |
| 18 | CLARITHROMYCIN | Synthesis from erythromycin which is derived from actinomycete (Streptomyces erythreus) | 84029 | Antimicrobial activity | Fail | [42] |
| 19 | EPICATECHIN | Tea (Camellia sinensis) | 72276 | Anti-hyperlipidaemic, anti-inflammatory, antioxidative, anticarcinogenic, and cytoprotective | Pass | [43] |
| 20 | HESPERIDIN | Orange (Citrus sinensis) | 10621 | Antihyperlipidemic, cardioprotective,antihypertensive, antidiabetic, anti-oxidant | Fail | [44] |
| 21 | MARMESIN | Bael (Aegle marmelos) | 334704 | Anticancer, anti-oxidant | Pass | [36] |
| 22 | PETROSIN | Sponge (Petrosia seriata) | 3009290 | Antiviral | Fail | [45] |
| 23 | RETROJUSTICIDIN B | Asian Flax (Linum austriacum) | 460888 | Antiviral | Pass | [46] |
| 24 | TACHYPLESIN | Horseshoe crab | 16129721 | Anticancer and antimicrobial | Fail | [47] |
| 25 | CAPSAICIN | Chili peppers (Capsicum annuum) | 1548943 | Analgesic, anti-obesity, anti-pruritic, anti-inflammatory, anti-apoptotic, anti-cancer, anti-oxidant, and neuro-protective | Pass | [48] |
| 26 | CURCUMIN | Turmeric (Curcuma longa) | 969516 | Anti-inflammatory, antioxidant, chemopreventive, chemotherapeutic, Antimicrobial activity | Pass | [49] |
| 27 | EQUISETIN | Fusarium equisti | 54684703 | Antimicrobial activity | Fail | [50] |
| 28 | LAMELLARIN | Mollusk Lamellaria | 461035 | Antiviral | Fail | [51] |
| 29 | MACROLACTIN-A | Marine Bacillus subtilis | 6451096 | Antimicrobial activity | Pass | [52] |
| 30 | RETUSIN | Cocobolo wood (Dalbergia retusa) | 5352005 | Anti-emetic effect, anti-tyrosinase activity, antioxidant | Pass | [53] |
Table 2.
ADME prediction of Macrolactin A and Stachyflin
| Properties and functions | Predictive results | Recommended range | |
|---|---|---|---|
| Macrolactin A | Stachyflin | ||
| Molecular weight (Da) | 402.53 | 385.502 | 130–725 |
| #Stars | 1 | 0 | 0–5 |
|
SASA (total solvent accessible surface area) |
710.02 | 592.553 | 300–1000 |
| Dipole | 0 | 0 | 1.0–12.5 |
| Donor H-bond | 3 | 3 | 0–6.0 |
| Acceptor H-bond | 7.1 | 5.7 | 2.0–20.0 |
|
QPlogPo/w (predicted octanol/water coefficient) |
3.704 | 2.619 | −2–6.5 |
|
QPlogS (predicted aqueous solubility) |
3.062 | −4.597 | −6.5–0.5 |
|
QPlogkhsa (binding prediction to human serum albumin) |
0 | 0.47 | −3–1.2 |
|
QplogBB (predicted blood brain/blood partition coefficient) |
−2.125 | −1.02 | −3.0–1.2 |
| No. of metabolites | 6 | 4 | 1–8 |
| % Human oral absorption | 94.59 | 84.45 |
> 80% is high < 25% is poor |
Fig. 1.

Virtual screening strategy of naturally occurring compounds against Mpro as inhibitor
Molecular docking is considered an important computational tool in the drug discovery process. It is performed to select the potential inhibitor compounds and study the different bond formations in the protein–ligand complex at the binding site. Mpro belongs to a protease class of proteins and which play an important role in the replication process of many viruses [61]. The catalytic site in SARS-CoV-2 main protease Mpro is formed of two amino acids: His41 and Cys145. These two amino acids formed a catalytic dyad [62] which serves in the maturation of the virus. Thus, it was considered an active target site in the protein for the inhibitors.
Docking interactions of these two naturally occurring compounds (Macrolactin A and Stachyflin) with the binding site residues are shown in Figs. 3 and 4 and Table S4. Macrolactin A (Fig. 2) interacted with the residues HIS 41, MET 49, PHE 140, LEU 141, ASN 142, GLY 143, SER 144, CYS 145, HIS 163, HIS 164, MET 165, GLU 166, ARG 188, and GLN 189 in the binding site of Mpro to form the docking complex (Fig. 3). Macrolactin A showed good binding affinity (−9.22 kcal/mol) with 0.175 μM, forming 3 hydrogen bonds, 9 Van der Waals interactions, hydrophobic interactions, carbon–hydrogen bond, and alkyl interactions (Fig. 3). Stachyflin (Fig. 2) also showed good binding affinity (−8.00 kcal/mol) with 1.37 μM with the residues THR 25, LEU 27, HIS 41, MET 49, LEU 141, ASN 142, GLY 143, SER 144, CYS 145, HIS 163, HIS 164, MET 165, GLU 166, ARG 188, and GLN 189 of Mpro active site. Stachyflin formed two hydrogen bonds, 9 Van der Waals interactions, hydrophobic interactions, π donor-hydrogen bonds, and alkyl and π-Alkyl interactions (Fig. 4). This result was in agreement with a previously published report that candidate and lead compounds formed the highest number of hydrogen with GLU 166 [63]. Previous studies also showed that natural compounds can interact with the catalytic site of Mpro proteases at HIS41 and CYS145 [57, 64]. In the docking study, it was observed that the earlier reported catalytic dyad HIS 41 and CYS145 did not involve hydrogen bonding with Macrolactin A; instead Van der Waals interactions took place and contributed to the overall binding on the ligand at the active site of Mpro. Previous research indicated that binding energy of −7.0 kcal/mol or less could be effective against SARS-CoV-2 which causes COVID-19 [59]. Studies reported that polyphenols like broussochalcone A, papyriflavonol A, 3′-(3-methylbut-2-enyl)-3′,4′,7-trihydroxyflavane, broussoflavan A, kazinol F, and kazinol J had good interaction with the catalytic residues of His41 and Cys145 of Mpro and exhibited good binding affinity −7.6 to −8.2 kcal/mol. These binding affinity values with the SARS-CoV-2 main protease Mpro are similar to our findings with Macrolactin A and Stachyflin compound [65]. Previous studies reported that the inhibitor N3 docks in the active binding site of 6LU7 and forms hydrogen bonds with THR190, GLN189, GLU166, HIS 164, PHE 140, and GLY 143 [66]. As mentioned previously, Thailand approved the use of A. paniculate for mild COVID-19 cases. Phytochemicals from A. paniculate also formed hydrophobic interactions and hydrogen bonding interactions with different residues THR24 to GLN192 [15]. The binding energy of the co-crystallized N3 inhibitor was previously reported to be −7.6 kcal/mol [63]. The binding energy of andrographolide (−6.26 kcal/mol) and dihydroxy dimethoxy flavone (−6.23 kcal/mol) from A. paniculate were better compared to hydroxychloroquine (−5.47 kcal/mol) [15]. Amentoflavone, a natural compound synthesized by plants, formed 3 hydrogen bonds and numerous hydrophobic interactions with 6LU7 had binding energy of −10.2 kcal/mol in a previous study [63]. Although all the three compounds showed comparatively good binding energies, the physicochemical properties and stability of the docking complexes needed to be tested.
Fig. 3.

Molecular docking displaying the interaction of Macrolactin A with 6LU7 protein. A 3D ligand interaction representing the docked pose in the binding site. B 2D plot of the ligand interaction with the amino acid residues
Fig. 4.

Molecular docking interaction of Stachyflin with 6LU7 protein. A 3D ligand interaction representing the docked pose in the binding site. B 2D plot of the ligand interaction with the amino acid residues
Fig. 2.

2D structures of top two selected hit compounds that required the lowest binding energies during the protein–ligand formation
ADME Property Analysis
Evaluations of physicochemical properties via ADME analysis are essential for assessing the efficacy of potential drug candidates and predict the drug-likeness properties. The calculated ADME properties and predicted physicochemical properties of the two-hit compounds are given in Table 2. All the analyzed parameters like molecular weight, SASA, hydrogen bond donor and acceptor, QPlogPo/w, QPlogS, QPlogkhsa, QplogBB, and number of metabolites of the two top compounds were in the recommended range. The higher percentage (%) of human oral absorption (94.59 and 84.45) of both compounds signifies the reduction of the amount of drug to attain the needed pharmacological effect. The drug-likeness scores of both the compounds were in the recommended range except dipole moment. Macrolactin A was the best compound; however, further derivatization in molecular structure could improve pharmacokinetic descriptors generally. Our percentage (%) of human oral absorption for values (94.59 and 84.45%) were comparable with andrographolide (77.65%) and dihydroxy dimethoxy flavone (93.829%) from A. paniculate and hydroxychloroquine (93.21%) [15].
Molecular Dynamics (MD) Simulation
The MD simulation (MDS) was performed for Mpro-Macrolactin A, and Mpro-Stachyflin docking complex with 100 ns simulation time and results were analyzed for root mean square deviation (RMSD), root mean square fluctuation (RMSF), and the number of hydrogen bonding. RMSD calculation of Mpro–Macrolactin A complex is more stable throughout simulation as compared to the Mpro–Stachyflin docking complex (Fig. 5). The Mpro when bound to compound Macrolactin A showed RMSD (C-alpha atom of Mpro) values between 2 and 3 Å with an average value of 2.8 Å. However, the Stachyflin–Mpro complex showed RMSD (C-alpha atom of Mpro) value between 2 and 3.5 Å with an average of 3.06 Å. The overall RMSD of the compound Macrolactin A throughout the 100 ns of simulation remained uniform, and hence, these results confirmed the stability of the protein–ligand complex, while in the case of Stachyflin slight increase in the RMSD value was observed after 50 ns of simulation which increases up to 4 Å. This shows that the protein–ligand complex was less stable which supported previous studies [59, 66]. RMSF results revealed the Cα of Mpro bound to Macrolactin A has an average RMSF value of 1.48 Å which depicted fewer fluctuations in the complex structure (Fig. 6). However, residues GLY 23 (1.71Å), LEU 50 (3.69Å), SER 139 (1.76Å), ASN 142 (1.81Å), ARG 222 (2.02Å), ASP 245 (1.81Å), GLY 278 (2.40Å), and CYS 300 (2.94Å) have shown slight fluctuations of the Macrolactin A–Mpro complex during the simulation, while, in case of Stachyflin bound Cα of Mpro exhibited an RMSF value of 1.77 Å, which is higher than Mpro bound Macrolactin A. The residues GLY 23 (1.5Å), ASP 48 (3.48 Å), GLN 127 (2.24 Å), TYR 154 (2.60Å), THR 169 (3.22 Å), and GLY 278 (2.85Å) exhibit higher fluctuations during simulations. Interactions of hydrogen bonds between ligands (Macrolactin A and Stachyflin) and Mpro through the 100 ns of MD simulations were evaluated (Fig. 7). Analysis of the results showed that the compound Macrolactin A formed 3 hydrogen bonds on average, during 100 ns of simulation with the Mpro. On the other hand, Stachyflin formed 1 hydrogen bond. This result confirmed the strong inhibition of Mpro by the Macrolactin A compound in the MD system and also showed similar molecular docking results of 3 hydrogen bonding within active site residues in Mpro protein. Hence, MD simulation analysis of Mpro–Macrolactin A complex and Mpro-Stachyflin showed better binding interaction in the pocket of Mpro. The protein–ligand total contacts timeline diagram is prepared to study the intermolecular interactions between Mpro residues and Macrolactin A compound (Fig. 8A) after performing 100 ns of MD simulation. The top panel showed a maximum of 9 specific contacts formed between the protein and the ligand throughout simulations and the bottom panel shows that the residues ASN 142, TYR 154, GLU 166, and GLN 189 showed crucial interactions with the ligand which is represented by a darker shade. On the other hand, Stachyflin formed a maximum of 6 numbers of contacts with only GLU 166 as significant contacts (Fig. 8B). Thus we may conclude that the compound Macrolactin A is successful in making good interactions with the binding site residues of Mpro than Stachyflin and is involved in the stabilizations in the complex. Mpro protein interactions with the ligand Macrolactin A and Stachyflin were plotted as stacked bar chart plot (Fig. 9), which was mainly categorized into four types: hydrogen bonds, hydrophobic, ionic, and water bridges. In the case of Macrolactin A–Mpro protein complex, water bridges play followed by hydrogen bonds and hydrophobic interactions play a very important role in interactions. The amino acids ASN 142, GLU 166, and GLN 189 exhibited hydrogen bond contact with the protein, while in the case of Stachyflin the amino acids LEU50 and GLU166 form hydrogen bond interactions. This indicated that the Macrolactin A–Mpro protein complex was stable throughout the simulation time. The conformational strains that might destabilize the complex were studied via ligand torsions dynamics, and detailed information was obtained. The ligand torsions dynamics for the rotatable bonds present in the Macrolactin A compound are plotted in Fig. 10, while for Stachyflin they were added to Supplementary Figure S1. The plot showed the simulation trajectory of the ligand from 0.00 to 100 ns and the conformational evolution of rotatable bonds (RB). Rotatable bond torsions were plotted in 2D and the bonds were color-coded. Dial plots showed the conformation of the torsion throughout the simulation. The beginning of the simulation is plotted in the center of the radial plot, while the time evolution is plotted radially outwards. The probability density of the torsions is shown in bar plots that summarized the information obtained from the dial plots. The values on the left Y-axis of the charts are expressed in kcal/mol. The compound Macrolactin A in total showed 20 rotatable bonds and the dial plots clearly showed that the rotatable bonds rotate around −180° to 180°, whereas, in Stachyflin, only two rotatable bonds are observed. This showed that the ligand Macrolactin A has more flexibility to bind and maintain a stable conformation in the active site residue of the protein.
Fig. 5.

Plot of root mean square deviations (RMSD) (C-alpha atom of Mpro) values, during 100 ns molecular dynamics simulation of Mpro in complex with A Macrolactin A and B Stachyflin
Fig. 6.

Plot of root mean square fluctuations (RMSF) values, during 100 ns MD simulation of Mpro in complex with A Macrolactin A and B Stachyflin
Fig. 7.

Plot of number of hydrogen bonding interactions, during 100 ns MD simulation of Mpro in complex with A Macrolactin A and B Stachyflin
Fig. 8.

Analysis of total contacts timeline formed between Mpro residues and A Macrolactin A and B Stachyflin during MD simulation. Darker shades correspond to a higher number of contacts.
Fig. 9.

Protein–ligand stacked bar chart plot of the interactions formed between ligand and protein residues during 100 ns molecular dynamics simulation of Mpro in complex with A Macrolactin A and B Stachyflin. Abscissa represents the amino acid number and ordinate represents interactions fraction
Fig. 10.
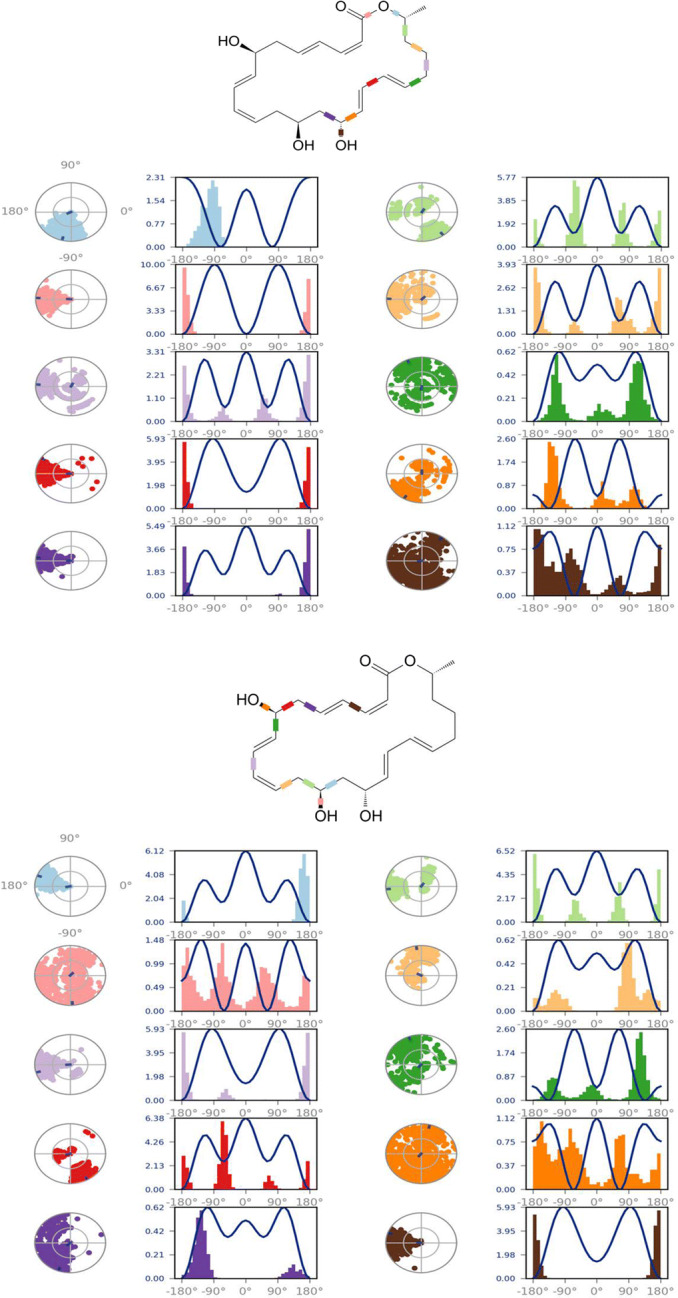
Analysis of the torsional degree of freedom for the rotatable bonds present in the Macrolactin A during 100 ns of MD simulation
Molecular Mechanics Generalized Born Surface Area (MM-GBSA) Calculations
MM-GBSA is a widely used technique to accurately calculate the binding free energy of protein–ligand binding affinities [67]. Utilizing the MD simulation trajectory, the binding free energy along with other contributing energy in form of MM-GBSA was determined for each Mpro (PDB ID: 6LU7) complex with the 2 ligands. The results (Table 2) suggested that the maximum contribution to ΔGbind in the stability of the simulated complexes was due to ΔGbindCoulomb, ΔGbindvdW, and ΔGbindLipo, while ΔGbindCovalent and ΔGbindSolvGB contributed to the instability of the corresponding complexes. The Mpro–Macrolactin A docked complexes showed comparatively higher binding free energy (−42kcal/mol), compared to other docking complexes of SARS CoV-2 Mpro-Stachyflin (−38.35kcal/mol). The binding energy obtained from MM-GBSA was found to be significantly lower than the binding free energy obtained from molecular docking using Autodock, which signifies stronger protein–ligand binding. These results supported the potential of screened compounds in inhibiting Mpro and showed the efficiency in binding to the selected protein and the ability to form stable protein–ligand complexes Table 3.
Table 3.
Binding free energy components for the docking complexes of 6LU7 protein with ligands calculated by MM-GBSA analysis
| Compound code | MM-GBSA (kcal/mol) | |||||
|---|---|---|---|---|---|---|
| ΔGbind | ΔGbindLipo | ΔGbindvdW | ΔGbindCoulomb | ΔGbindSolvGB | ΔGbindCovalent | |
| Macrolactin A | −42.58 ± 6.35 | −12.24 ± 1.23 | −36.13 ± 2.17 | −13.83 ± 5.54 | 19.73 ± 2.78 | 2.97 ± 1.90 |
| Stachyflin | −38.35 ± 8.40 | −11.65 ± 2.08 | −29.78 ± 6.17 | −13.31 ± 8.69 | 16.30 ± 7.42 | 1.83 ± 1.28 |
The analysis of the trajectories of the first and the last frame displayed significant differences in the conformation of Macrolactin A bound 6LU7 (Fig. 11). It was observed that at the beginning of the simulation, the ligand Macrolactin A was outward from the binding site (Fig. 10, red, 0 ns) whereas, at the final stage of 100 ns simulation, the ligand moved more inside the binding cavity (Fig. 12, blue, 100 ns). Apart from the movement of the ligand (around 270° rotation) toward binding cavity and stable fitting, that conformed into a more stabilized helical turn at GLY11-GLU14 and LEU227-TYR237 in 6LU7. These conformational variances were absent before simulation and visibly clear from the last trajectory of 100 ns dynamics. This entire structural evidence (Fig. 10) of ligand movement and conformational changes within the protein was captured from the MMGBSA calculations of the first and the last trajectories. The binding energy in MMGBSA calculations performed rigorous statistical sampling from first to the last trajectory. Therefore, the rotational event of the ligand as displayed in Fig. 11 is reproducible and the pose prediction corroborated the findings of MMGBSA calculations.
Fig. 11.

Position and movement of Macrolactin A at the binding site before simulation (red, 0 ns) and after simulation (blue, 100 ns). Conformational variances between first and last frame of MD simulation trajectories after 100 ns
Fig. 12.

DCCM plot (A), highly correlated dynamic domains of 6LU7 (B) and PCA (C) of global (PC1) and local motion (PC2) components of 6LU7 at Macrolactin bound A state from MD trajectories. Colors indicating the clustering of trajectories having statistically converged (red and gray: highly converged) (blue, cyan, and green: moderately converged) and (yellow and smoky red: less converged) into conformational space
Dynamic Cross-Correlation Matrix (DCCM) and Principal Component Analysis (PCA) of the MD Simulation Trajectories
DCCM was generated in order to analyze the correlative motion of structural domains to attain a stable conformation of 6LU7 after Macrolactin A binding from MD trajectories (Fig. 12A). The correlation scores on the central mean line (blue) displayed four distinct blocks having a high correlation of movement of amino acids in 6LU7 (Fig. 12A). The domain D1 comprised of residues 61–90 conforming into three distinct β-sheets and an extended loop (green, Fig. 12B), whereas, D2 domain having the highest cross-correlation between residues 95–165 conforming into more flexible extended loops and four β-sheets (red, Fig. 12B), while D3 and D4 domains conforming into α-helices from 245–265 (cyan) and 270–280 (purple) residues, respectively. Therefore, the dynamics cross-correlation matrix enables the domain conformations into better stability of 6LU7 at Macrolactin A bound state. DCCM analysis also corroborates with the RMSF of C-α backbone of 6LU7 (discussed in the previous section) of Macrolactin A bound state with moderate to fewer fluctuations of respective amino acid residues confirming good stable structure. Similarly, Piao and coworkers (2019) reported the domain conformation and stability of N-PDZ and E-PBM proteins by their correlation function from DCCM analysis [68].
PCA displayed the contribution of principal dynamic global motion (PC1) and local dynamic behavior (PC2) from the MD trajectories having the second-highest variance of 6LU7 with Macrolactin A bound complex (Fig. 12C). The eigenvalues (covariance) were plotted in the PCA contour plot indicating the motion magnitude as well as directions of residues in MD trajectories. It was observed that Macrolactin A bound 6LU7 showed large movement toward positive eigenvectors and the majority of the domain movements were contributed by global slow motion (PC1) and conforming into the more stable conformation of 6LU7 depicting the significant binding of Macrolactin A thus corroborating MMGBSA result. Moreover, PCA revealed the final MD trajectories after 100 ns seemed to be highly converged into stable geometry (Fig. 12C). Therefore, it may be suggested that Macrolactin A binds strongly and giving a stable conformation of C-α backbone of 6LU7.
The natural compound Macrolactin A showed excellent interaction with the active site residues along with the surrounding residues of Mpro. The MD simulations and MMGBSA binding energy calculations also showed it as a potential inhibitor candidate with good stability of the docking complex. Besides this, the compound also showed good pharmacokinetic parameters. The identified inhibitors open up the possibilities for designing potential drug molecule which selectively act as an inhibitor against SARS-CoV2 Mpro. In the present study, we have found a novel natural inhibitor against SARS-CoV2 main protease by targeting the core pocket of the protein active site.
Materials and Methods
Drug Likeness Profiling of Selected Natural Compounds
Virtual screening of the naturally occurring compounds (n = 30) for drug likeliness properties was carried out according to Lipinski’s rule of five [69]. The DruLiTo stand-alone software (http://www.niper.gov.in/pi_dev_tools/DruLiToWeb/DruLiTo_index.html) was implemented for the drug-likeness screening [70]. The compounds were selected from the literature based on cost, sustainable harvest, and year-round availability in India (Table S1). The drug-likeness compounds were then predicted for antiviral activity in silico using the PASS online server (http://www.pharmaexpert.ru/passonline/predict.php). The predicted antiviral activity score (Pa) ranges between 0 and 1, where the value 1 is considered the best antiviral activity, and zero stand for no antiviral activity (Table S4). The ADME properties of the selected lead compounds were calculated using a QikProp module in Schrodinger suite [71, 72]. Results from ADME properties like SASA, QPlogPo/w, QPlogS, QPlogkhsa, QplogBB, No. of Metabolites, and % human oral absorption were compared with a recommended range of values provided in the manual. The QikProp generated descriptors which were used to perform ADMET predictions or drug-likeness parameter (indicated by # stars). The #stars parameter described the QikProp pharmacokinetic properties that fall outside the optimum range of values for 95% of known drugs within the ConMedNP library [73–75].
Preparation of Ligands and Receptors
The Ligand was prepared using the LigPrep tool in the Maestro module of Schrodinger [72, 76]. Epik was used and Optimized Potential Liquid Simulations (OPLS3) force field (pH 7.0 ± 2.0, allowing 32 stereoisomers per ligand) were applied for optimization and energy minimization for each ligand compound [77]. The receptor protein crystal structures of the Mpro in PDB format (PDB ID: 6LU7, Resolution: 2.16 Å) were obtained from the Protein Data Bank (www.pdb.org) [78]. The PDB structure and the protein–ligand complexes were prepared using the Protein Preparation Wizard in the Schrödinger suite [76]. The water molecules in all protein structures were deleted before the missing residues and side chains were corrected using a Prime module [79]. The protein and the protein–ligand complexes were subjected to geometry refinement using OPLS2005 force field.
Molecular Docking
The crystal structure of Mpro (PDB ID: 6LU7) was used for molecular docking studies. All the protein–ligand docking complexes were performed using Autodock 4.2 [80–82]. The catalytic site of Mpro (His41 and Cys145) was chosen as binding sites for docking analysis [78].
The protein was kept rigid, while the natural compound structures (ligands) were kept flexible. Polar hydrogen and gasteiger charges were added to the Mpro protein (Mpro) and ligands. AutoDock Tools (v.1.5.6) of the MGL software package was used to prepare PDBQT files of the ligands and proteins. Lamarckian genetic algorithm (LGA) method was used to analyze the protein–ligand docking complexes. The grid box size was set to 22.5 Å, with a grid point spacing of 0.375Å, centered on x = −13.01, y =18.361, and z = 71.031Å. The binding interactions of the docking complexes were analyzed and its 3D and 2D interaction plots were analyzed using Discovery studio visualizer [83]. Only molecular docking complexes with the least binding energies were considered for further study.
MD Simulations
The top inhibitory compounds Macrolactin A and Stachyflin were further analyzed by molecular dynamics simulation under a Linux environment using the Desmond modules of the Schrodinger [67, 84]. The SPC (simple point charge) water box solvent model was used and the OPLS2005 force field was applied for the protein–ligand docking complexes. An orthorhombic periodic boundary box (X, Y, and Z-axis) conditions were set up at 10 Å distances to specify the shape and size of the protein–ligand docking complex. Counter ions (14 Na+, and 18 Cl− ions) were added to neutralize charges and then 0.15 M NaCl salt concentrations were added to make the system close to mimic the human physiological condition. The MD simulation was carried out for 100 ns at NPT (isothermal-isobaric ensemble, constant temperature, pressure, and the number of particles) ensemble temperature of 300k and 1.01325 bar of pressure. The Desmond simulation interaction diagram tool of Maestro was used to generate the simulation interaction diagram. Root-mean-square deviation (RMSD), root mean square fluctuation (RMSF), number of H-bonds, total contacts timeline, and protein–ligand interactions were recorded throughout the simulation data and were analyzed to validate our findings in molecular docking.
Molecular Mechanics Generalized Born Surface Area (MM-GBSA) Calculations:
The binding free energy calculation of the protein–ligand docking complexes was calculated by using the Prime-MM/GBSA and OPLS2005 force field [66, 67, 76] according to the procedures described elsewhere [85]. Prime MM-GBSA method calculates the binding free energy as follows:
where ∆Gbinding = total binding free energy and Gdocking complex, Gprotein, and Gligand are the free energies of the docking complex, protein, and ligand, respectively. The obtained results were presented as mean ± standard deviation (SD).
Dynamic Cross-Correlation Matrix (DCCM) and Principal Component Analysis (PCA) of the MD Simulation Trajectories
In order to analyze the domain correlations, a dynamic cross-correlation matrix (DCCM) was generated across all Cα-atoms for Macrolactin A and 6LU7 complex during the MD simulation of 100 ns. PCA was performed to extract the fast and slow motions of the trajectories during 100 ns simulation. A covariance matrix was generated to calculate the PCA for global slow motion and local fast motion of the contributing amino acid residues from each MD trajectory as described elsewhere [68]. The DCCM and PCA were done using trj_essential_dynamics script of Schrodinger [71, 72, 84].
Conclusions
Virtual screening of 30 natural compounds resulted in the identification of Macrolactin A as a lead compound for further in vitro and in vivo studies, and ultimately as a treatment of COVID-19. The molecular docking study showed that Macrolactin A strongly interacted with the key amino acids (His41 and Cys145) at the active site of SARS-CoV-2 Main protease (Mpro). The docking score of −9.22 kcal/mol was good and 3 hydrogen bonds and several other interactions were formed [23]. ADME and drug-likeness revealed that it possesses favorable physicochemical properties and provides a favorable characteristic according to Lipinski’s rule of five and could be used in developing a drug. The molecular dynamic simulation studies showed that the compound is thermodynamically feasible and binds stably with the active site of SARS-CoV-2 Mpro. MD simulations showed that bounds in Mpro–Macrolactin A complex were strong and stable throughout 100 ns. MM-GBSA analysis showed ΔGbind of −42.58 kcal/mol. Contacts timeline analysis of Macrolactin A exhibited that HIS 41, ASN 142, TYR 154, GLU 166, and GLN 189 were the crucial interactions. Thus, Macrolactin A could inhibit the protease’s catalytic activity and be a potential drug candidate against SARS-CoV2.
However, slight modification via derivatization might be required to improve its percentage of human oral. To study the conformational dynamics changes in the structure of SARS-CoV-2 Mpro protein due to the inhibitory activity of Macrolactin A, DCCM and PCA were performed. The DCCM and PCA showed better conformational stability in the catalytic site of SARS-CoV-2 main protease Mpro during Macrolactin A bound state. Overall, Macrolactin A was the most promising compound to be used against SARS-CoV-2. This compound is produced by soil bacteria Bacillus amylolique faciens and was first isolated in 1989 [66]. This could make producing this compound on an industrial scale very easy and cost-effective due to the ease of culturing bacillus species. It has been previously reported to possess strong antifungal [86] and antibacterial activity even against vancomycin-resistant enterococci and methicillin-resistant Staphylococcus aureus [87]. It is important to note that this is the first study to illustrate the potential of Macrolactin A for SARS-CoV-2 treatment and we hope the current research work will expedite the further biochemical study of the reported molecule. It has also been fully synthesized which make it even more accessible to the world [88].
Supplementary Information
Supplementary Materials: The following are available online, Table S1. Lipinski Filtered compounds with molecular properties; Table S2. Predicted Antiviral activity values of the selected drug-likeness compounds.; Table S3. Binding energies (kcal/mol) of selected natural bioactive compounds with SARS-CoV-2 Mpro and their interactions with the binding site amino acid residues. Fig S2. Analysis of total contacts timeline formed between Mpro protein residues and Stachyflin during MD simulation. Darker shades correspond to a higher number of contacts and Stachyflin.
(DOCX 133 kb)
(DOCX 16 kb)
(DOCX 17 kb)
(DOCX 14 kb)
Author Contribution
Conceptualization, KKB, TS, MKP, and SP; methodology, KKB, AG, BR, and DB; formal analysis, TS, KKB, BR, and AG; investigation SP, TS, KKB, and MKP; writing—original draft preparation, KKB, SP, HIS, BPD, and BRN; writing—review and editing, DB,AG, AJ, and HAE. All authors have read and agreed to the published version of the manuscript.
Data Availability
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Declarations
Ethics Approval and Consent to Participate
Not applicable. The manuscript does not contain data collected from humans or animals.
Consent for Publication
Not applicable. The manuscript does not contain any individual person’s data
Competing Interests
The authors declare no competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Kaushik Kumar Bharadwaj and Tanmay Sarkar contributed equally to this work.
References
- 1.Lee N, Allen Chan KC, Hui DS, Ng EKO, Wu A, Chiu RWK, Wong VWS, Chan PKS, Wong KT, Wong E, Cockram CS, Tam JS, Sung JJY, Lo YMD. Effects of early corticosteroid treatment on plasma SARS-associated coronavirus RNA concentrations in adult patients. Journal of Clinical Virology. 2004;31(4):304–309. doi: 10.1016/j.jcv.2004.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.de Groot RJ, Baker SC, Baric RS, Brown CS, Drosten C, Enjuanes L, Fouchier RAM, Galiano M, Gorbalenya AE, Memish ZA, Perlman S, Poon LLM, Snijder EJ, Stephens GM, Woo PCY, Zaki AM, Zambon M, Ziebuhr J. Middle East respiratory syndrome coronavirus (MERS-CoV): Announcement of the Coronavirus Study Group. Journal of virology. 2013;87(14):7790–7792. doi: 10.1128/JVI.01244-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Worldometer (2020). Coronavirus Update (Live)_ 7,405,051 cases and 416,626 deaths from COVID-19 virus pandemic - Worldometer. Reported Cases and Deaths by Country, Territory, or Conveyance.
- 4.Gautret P, Lagier JC, Parola P, Hoang VT, Meddeb L, Mailhe M, Doudier B, Courjon J, Giordanengo V, Vieira VE, Tissot Dupont H, Honoré S, Colson P, Chabrière E, la Scola B, Rolain JM, Brouqui P, Raoult D. Hydroxychloroquine and azithromycin as a treatment of COVID-19: results of an open-label non-randomized clinical trial. International Journal of Antimicrobial Agents. 2020;56(1):105949. doi: 10.1016/j.ijantimicag.2020.105949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lu R, Zhao X, Li J, Niu P, Yang B, Wu H, Wang W, Song H, Huang B, Zhu N, Bi Y, Ma X, Zhan F, Wang L, Hu T, Zhou H, Hu Z, Zhou W, Zhao L, Chen J, Meng Y, Wang J, Lin Y, Yuan J, Xie Z, Ma J, Liu WJ, Wang D, Xu W, Holmes EC, Gao GF, Wu G, Chen W, Shi W, Tan W. Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. The Lancet. 2020;395(10224):565–574. doi: 10.1016/S0140-6736(20)30251-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kumaravel SK, Subramani RK, Jayaraj Sivakumar TK, Madurai Elavarasan R, Manavalanagar Vetrichelvan A, Annam A, Subramaniam U. Investigation on the impacts of COVID-19 quarantine on society and environment: Preventive measures and supportive technologies. 3 Biotech. 2020;10(9):393. doi: 10.1007/s13205-020-02382-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Elena SF, Sanjuán R. Adaptive value of high mutation rates of RNA viruses: Separating causes from consequences. Journal of Virology. 2005;79(18):11555–11558. doi: 10.1128/JVI.79.18.11555-11558.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sahin AR, Erdogan A, Agaoglu PM, Dineri Y, Cakirci AY, Senel ME, et al. 2019 Novel coronavirus (COVID-19) Outbreak: A review of the current literature. Eurasion Journal of Medicine and Oncology. 2020;4(1):1–7. doi: 10.14744/ejmo.2020.12220. [DOI] [Google Scholar]
- 9.Mohapatra DD, Pattnaik S, Panda S. In vitro detected hly II cytotoxin in a strain of Staphylococcus aureus (BM S-2) and plant-derived aromatic components: A molecular docking study. Applied Biochemistry and Biotechnology. 2021;193(6):1639–1653. doi: 10.1007/s12010-021-03510-2. [DOI] [PubMed] [Google Scholar]
- 10.Cao B, Wang Y, Wen D, Liu W, Wang J, Fan G, Ruan L, Song B, Cai Y, Wei M, Li X, Xia J, Chen N, Xiang J, Yu T, Bai T, Xie X, Zhang L, Li C, Yuan Y, Chen H, Li H, Huang H, Tu S, Gong F, Liu Y, Wei Y, Dong C, Zhou F, Gu X, Xu J, Liu Z, Zhang Y, Li H, Shang L, Wang K, Li K, Zhou X, Dong X, Qu Z, Lu S, Hu X, Ruan S, Luo S, Wu J, Peng L, Cheng F, Pan L, Zou J, Jia C, Wang J, Liu X, Wang S, Wu X, Ge Q, He J, Zhan H, Qiu F, Guo L, Huang C, Jaki T, Hayden FG, Horby PW, Zhang D, Wang C. A trial of lopinavir–ritonavir in adults hospitalized with severe Covid-19. New England Journal of Medicine. 2020;382(19):1787–1799. doi: 10.1056/nejmoa2001282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang, N., Shang, J., Jiang, S., & Du, L. (2020). Subunit vaccines against emerging pathogenic human coronaviruses. Frontiers in Microbiology., 11. 10.3389/fmicb.2020.00298. [DOI] [PMC free article] [PubMed]
- 12.Mukherjee D, Garai S, Lahiri D, Nag M. Effects of SARS-COV-2 viral infection on cancer patients: A biological and statistical study during the COVID-19 outbreak. American Journal of Applied Bio-Technology Research. 2020;1(1):16–24. doi: 10.15864/ajabtr.1302. [DOI] [Google Scholar]
- 13.Lin LT, Hsu WC, Lin CC. Antiviral natural products and herbal medicines. Journal of traditional and complementary medicine. 2014;4(1):24–35. doi: 10.4103/2225-4110.124335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Harvey AL, Edrada-Ebel R, Quinn RJ. The re-emergence of natural products for drug discovery in the genomics era. Nature reviews drug discovery. 2015;14(2):111–129. doi: 10.1038/nrd4510. [DOI] [PubMed] [Google Scholar]
- 15.Lam TTY, Jia N, Zhang YW, Shum MHH, Jiang JF, Zhu HC, Tong YG, Shi YX, Ni XB, Liao YS, Li WJ, Jiang BG, Wei W, Yuan TT, Zheng K, Cui XM, Li J, Pei GQ, Qiang X, Cheung WYM, Li LF, Sun FF, Qin S, Huang JC, Leung GM, Holmes EC, Hu YL, Guan Y, Cao WC. Identifying SARS-CoV-2-related coronaviruses in Malayan pangolins. Nature. 2020;583(7815):282–285. doi: 10.1038/s41586-020-2169-0. [DOI] [PubMed] [Google Scholar]
- 16.Reeves JD, Piefer AJ. Emerging drug targets for antiretroviral therapy. Drugs. 2005;65(13):1747–1766. doi: 10.2165/00003495-200565130-00002. [DOI] [PubMed] [Google Scholar]
- 17.Chen X, Ung CY, Chen Y. Can an in silico drug-target search method be used to probe potential mechanisms of medicinal plant ingredients? Natural Product Reports. 2003;20(4):432–444. doi: 10.1039/b303745b. [DOI] [PubMed] [Google Scholar]
- 18.Gorgulla C, Boeszoermenyi A, Wang ZF, Fischer PD, Coote PW, Das KMP, et al. An open-source drug discovery platform enables ultra-large virtual screens. Nature. 2020;580(7805):663–668. doi: 10.1038/s41586-020-2117-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Adejoro IA, Babatunde DD, Tolufashe GF. Molecular docking and dynamic simulations of some medicinal plants compounds against SARS-CoV-2: an in silico study. Journal of Taibah University for Science. 2020;14(1):1563–1570. doi: 10.1080/16583655.2020.1848049. [DOI] [Google Scholar]
- 20.Chen J, Liu T, Wei M, Zhu Z, Liu W, Zhang Z. Macrolactin a is the key antibacterial substance of Bacillus amyloliquefaciens D2WM against the pathogen Dickeya chrysanthemi. European Journal of Plant Pathology. 2019;155(2):393–404. doi: 10.1007/s10658-019-01774-3. [DOI] [Google Scholar]
- 21.Gustafson K, Roman M, Fenical W. The macrolactins, a novel class of antiviral and cytotoxic macrolides from a deep-sea marine bacterium. Journal of the American Chemical Society. 1989;111(19):7519–7524. doi: 10.1021/ja00201a036. [DOI] [Google Scholar]
- 22.Kim E-N, Gao M, Choi H, Jeong G-S. Marine microorganism-derived macrolactins inhibit inflammatory mediator effects in LPS-induced macrophage and microglial cells by regulating BACH1 and HO-1/Nrf2 signals through inhibition of TLR4 activation. Molecules (Basel, Switzerland) 2020;25(3):656. doi: 10.3390/molecules25030656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bharadwaj, K. K., Sarkar, T., Ghosh, A., Baishya, D., Rabha, B., Panda, M., et al. (2021). Nature potential for COVID-19: Targeting SARS-CoV-2 Mpro inhibitor with bioactive compound. 10.26434/chemrxiv.14112515.v2.
- 24.Xie L, Tang H, Song J, Long J, Zhang L, Li X. Chrysophanol: A review of its pharmacology, toxicity and pharmacokinetics. Journal of Pharmacy and Pharmacology. 2019;71(10):1475–1487. doi: 10.1111/jphp.13143. [DOI] [PubMed] [Google Scholar]
- 25.Lee J, Currano JN, Carroll PJ, Joullié MM. Didemnins, tamandarins and related natural products. Natural Product Reports. 2012;29(3):404–424. doi: 10.1039/C2NP00065B. [DOI] [PubMed] [Google Scholar]
- 26.Jin H, Zhang P, Bijian K, Ren S, Wan S, Alaoui-Jamali MA, Jiang T. Total synthesis and biological activity of marine alkaloid Eudistomins Y1–Y7 and their analogues. Marine Drugs. 2013;11(5):1427–1439. doi: 10.3390/md11051427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sarkar T, Chakraborty R. Formulation, physicochemical analysis, sustainable packaging-storage provision, environment friendly drying techniques and energy consumption characteristics of mango leather production: A review. Asian Journal of Water, Environment and Pollution. 2018;15(3):79–92. doi: 10.3233/AJW-180046. [DOI] [Google Scholar]
- 28.Venu Gopal J. Morin Hydrate: Botanical origin, pharmacological activity and its applications: A mini-review. Pharmacognosy Journal. 2013;5(3):123–126. doi: 10.1016/j.phcgj.2013.04.006. [DOI] [Google Scholar]
- 29.Roy BG. Potential of small-molecule fungal metabolites in antiviral chemotherapy. Antiviral Chemistry and Chemotherapy. 2017;25(2):20–52. doi: 10.1177/2040206617705500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen C. Sinapic acid and its derivatives as medicine in oxidative stress-induced diseases and aging. Oxidative Medicine and Cellular Longevity. 2016;2016:3571614–3571610. doi: 10.1155/2016/3571614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gomes NM, Bessa LJ, Buttachon S, Costa PM, Buaruang J, Dethoup T, et al. Antibacterial and Antibiofilm activities of tryptoquivalines and meroditerpenes isolated from the marine-derived fungi Neosartorya paulistensis, N. laciniosa, N. tsunodae, and the soil fungi N. fischeri and N. siamensis. Marine Drugs. 2014;12(2):822–839. doi: 10.3390/md12020822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sladic D, Gasic MJ. Reactivity and biological activity of the marine sesquiterpene hydroquinone avarol and related compounds from sponges of the Order Dictyoceratida. Molecules. 2006;11(1):1–33. doi: 10.3390/11010001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wu G, Sun X, Yu G, Wang W, Zhu T, Gu Q, Li D. Cladosins A–E, hybrid Polyketides from a deep-sea-derived fungus, Cladosporium sphaerospermum. Journal of Natural Products. 2014;77(2):270–275. doi: 10.1021/np400833x. [DOI] [PubMed] [Google Scholar]
- 34.Kong KW, Mat-Junit S, Aminudin N, Ismail A, Abdul-Aziz A. Antioxidant activities and polyphenolics from the shoots of Barringtonia racemosa (L.) Spreng in a polar to apolar medium system. Food Chemistry. 2012;134(1):324–332. doi: 10.1016/j.foodchem.2012.02.150. [DOI] [Google Scholar]
- 35.Wang, Y., Xing, M., Cao, Q., Ji, A., Liang, H., & Song, S. (2019). Biological activities of fucoidan and the factors mediating its therapeutic effects: A review of recent studies. Marine Drugs, 17(3). 10.3390/md17030183. [DOI] [PMC free article] [PubMed]
- 36.Sarkar T, Salauddin M, Chakraborty R. In-depth pharmacological and nutritional properties of bael (Aegle marmelos): A critical review. Journal of Agriculture and Food Research. 2020;2:100081. doi: 10.1016/j.jafr.2020.100081. [DOI] [Google Scholar]
- 37.Manda G, Rojo AI, Martínez-Klimova E, Pedraza-Chaverri J, Cuadrado A. Frontiers in Pharmacology. 2020. Nordihydroguaiaretic acid: From herbal medicine to clinical development for cancer and chronic diseases. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Meltzer EO, Ratner PH, McGraw T. Oral phenylephrine HCl for nasal congestion in seasonal allergic rhinitis: A randomized, open-label, placebo-controlled study. The journal of allergy and clinical immunology. In practice. 2015;3(5):702–708. doi: 10.1016/j.jaip.2015.05.007. [DOI] [PubMed] [Google Scholar]
- 39.Motohashi Y, Igarashi M, Okamatsu M, Noshi T, Sakoda Y, Yamamoto N, Ito K, Yoshida R, Kida H. Antiviral activity of stachyflin on influenza A viruses of different hemagglutinin subtypes. Virology Journal. 2013;10(1):118. doi: 10.1186/1743-422X-10-118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rathnavelu V, Alitheen Banu N, Sohila S, Kanagesan S, Ramesh R. Potential role of bromelain in clinical and therapeutic applications (Review) Biomed Rep. 2016;5(3):283–288. doi: 10.3892/br.2016.720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sarkar T, Salauddin M, Hazra SK, Chakraborty R. The impact of raw and differently dried pineapple (Ananas comosus) fortification on the vitamins, organic acid and carotene profile of dairy rasgulla (sweetened cheese ball) Heliyon. 2020;6(10):e05233. doi: 10.1016/j.heliyon.2020.e05233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kondoh K, Hashiba M, Baba S. Inhibitory activity of clarithromycin on biofilm synthesis with Pseudomonas aeruginosa. Acta oto-laryngologica. Supplementum. 1996;525:56–60. [PubMed] [Google Scholar]
- 43.Bernatova I. Biological activities of (−)-epicatechin and (−)-epicatechin-containing foods: Focus on cardiovascular and neuropsychological health. Biotechnology Advances. 2018;36(3):666–681. doi: 10.1016/j.biotechadv.2018.01.009. [DOI] [PubMed] [Google Scholar]
- 44.Man M-Q, Yang B, Elias PM. Benefits of hesperidin for cutaneous functions. Evidence-Based Complementary and Alternative Medicine. 2019;2019:2676307–2676319. doi: 10.1155/2019/2676307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Thawabteh, A., Juma, S., Bader, M., Karaman, D., Scrano, L., Bufo, S. A., & Karaman, R. (2019). The biological activity of natural alkaloids against herbivores, cancerous cells and pathogens. Toxins, 11(11). 10.3390/toxins11110656. [DOI] [PMC free article] [PubMed]
- 46.Sagar KS, Chang C-C, Wang W-K, Lin J-Y, Lee S-S. Preparation and anti-HIV activities of retrojusticidin B analogs and azalignans. Bioorganic & medicinal chemistry. 2004;12(15):4045–4054. doi: 10.1016/j.bmc.2004.05.036. [DOI] [PubMed] [Google Scholar]
- 47.Vernen, F., Harvey, P. J., Dias, S. A., Veiga, A. S., Huang, Y.-H., Craik, D. J., Lawrence, N., & Troeira Henriques, S. (2019). Characterization of tachyplesin peptides and their cyclized analogues to improve antimicrobial and anticancer properties. International Journal of Molecular Sciences, 20(17). 10.3390/ijms20174184. [DOI] [PMC free article] [PubMed]
- 48.Basith, S., Cui, M., Hong, S., & Choi, S. (2016). Harnessing the therapeutic potential of capsaicin and its analogues in pain and other diseases. Molecules, 21(8). 10.3390/molecules21080966. [DOI] [PMC free article] [PubMed]
- 49.De R, Kundu P, Swarnakar S, Ramamurthy T, Chowdhury A, Nair GB, Mukhopadhyay AK. Antimicrobial activity of curcumin against <em>Helicobacter pylori</em> Isolates from India and during infections in mice. Antimicrobial Agents and Chemotherapy. 2009;53(4):1592–1597. doi: 10.1128/AAC.01242-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jiang, M., Chen, S., Li, J., & Liu, L. (2020). The biological and chemical diversity of tetramic acid compounds from marine-derived microorganisms. Marine Drugs, 18(2). 10.3390/md18020114. [DOI] [PMC free article] [PubMed]
- 51.Fukuda T, Ishibashi F, Iwao M. Lamellarin alkaloids: Isolation, synthesis, and biological activity. The Alkaloids. Chemistry and biology. 2020;83:1–112. doi: 10.1016/bs.alkal.2019.10.001. [DOI] [PubMed] [Google Scholar]
- 52.Arbsuwan N, Payoungkiattikun W, Sirithorn P, Daduang S, Jangpromma N, Dhiravisit A, Hahm YT, Neubert LK, Klaynongsruang S. Purification and characterization of macrolactins and amicoumacins from Bacillus licheniformis BFP011: A new source of food antimicrobial substances. CyTA - Journal of Food. 2018;16(1):50–60. doi: 10.1080/19476337.2017.1337047. [DOI] [Google Scholar]
- 53.Pshenichnyuk SA, Elkin YN, Kulesh NI, Lazneva EF, Komolov AS. Low-energy electron interaction with retusin extracted from Maackia amurensis: towards a molecular mechanism of the biological activity of flavonoids. Physical Chemistry Chemical Physics. 2015;17(26):16805–16812. doi: 10.1039/C5CP02890F. [DOI] [PubMed] [Google Scholar]
- 54.Joshi, R. S., Jagdale, S. S., Bansode, S. B., Shankar, S. S., Tellis, M. B., Pandya, V. K., Chugh, A., Giri, A. P., & Kulkarni, M. J. (2020). Discovery of potential multi-target-directed ligands by targeting host-specific SARS-CoV-2 structurally conserved main protease. Journal of Biomolecular Structure and Dynamics, 1–16. 10.1080/07391102.2020.1760137. [DOI] [PMC free article] [PubMed]
- 55.Enmozhi, S. K., Raja, K., Sebastine, I., & Joseph, J. (2020). Andrographolide as a potential inhibitor of SARS-CoV-2 main protease: An in silico approach. Journal of Biomolecular Structure and Dynamics, 1–7. 10.1080/07391102.2020.1760136. [DOI] [PMC free article] [PubMed]
- 56.Das, S., Sarmah, S., Lyndem, S., & Singha Roy, A. (2020). An investigation into the identification of potential inhibitors of SARS-CoV-2 main protease using molecular docking study. Journal of Biomolecular Structure and Dynamics, 1–11. 10.1080/07391102.2020.1763201. [DOI] [PMC free article] [PubMed]
- 57.Bharadwaj, S., Azhar, E. I., Kamal, M. A., Bajrai, L. H., Dubey, A., Jha, K., Yadava, U., Kang, S. G., & Dwivedi, V. D. (2020). SARS-CoV-2 Mpro inhibitors: Identification of anti-SARS-CoV-2 Mpro compounds from FDA approved drugs. Journal of Biomolecular Structure and Dynamics, 1–16. 10.1080/07391102.2020.1842807. [DOI] [PMC free article] [PubMed]
- 58.Tiwari V. Novel hybrid antiviral VTRRT-13V2.1 against SARS-CoV2 main protease: retro-combinatorial synthesis and molecular dynamics analysis. Heliyon. 2020;6(10):e05122. doi: 10.1016/j.heliyon.2020.e05122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Prasanth, D. S. N. B. K., Murahari, M., Chandramohan, V., Panda, S. P., Atmakuri, L. R., & Guntupalli, C. (2020). In silico identification of potential inhibitors from Cinnamon against main protease and spike glycoprotein of SARS CoV-2. Journal of Biomolecular Structure and Dynamics, 1–15. [DOI] [PMC free article] [PubMed]
- 60.Vivek-Ananth, R. P., Rana, A., Rajan, N., Biswal, H. S., & Samal, A. (2020). In silico identification of potential natural product inhibitors of human proteases key to SARS-CoV-2 infection. arXiv preprint arXiv:2006.00652. [DOI] [PMC free article] [PubMed]
- 61.Chen J. Pathogenicity and transmissibility of 2019-nCoV—A quick overview and comparison with other emerging viruses. Microbes and Infection. 2020;22(2):69–71. doi: 10.1016/j.micinf.2020.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mirza MU, Froeyen M. Structural elucidation of SARS-CoV-2 vital proteins: Computational methods reveal potential drug candidates against main protease, Nsp12 polymerase and Nsp13 helicase. Journal of pharmaceutical analysis. 2020;10(4):320–328. doi: 10.1016/j.jpha.2020.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Juárez-Saldívar, A., Lara-Ramírez, E. E., Reyes-Espinosa, F., Paz-González, A. D., Villalobos-Rocha, J. C., & Rivera, G. (2020). Ligand-based and structured-based in silico repurposing approaches to predict inhibitors of SARS-CoV-2 Mpro protein. Scientia Pharmaceutica, 88(4). 10.3390/scipharm88040054.
- 64.Mondal, P., Natesh, J., Abdul Salam, A. A., Thiyagarajan, S., & Meeran, S. M. (2020). Traditional medicinal plants against replication, maturation and transmission targets of SARS-CoV-2: Computational investigation. Journal of Biomolecular Structure and Dynamics, 1–18. 10.1080/07391102.2020.1842246. [DOI] [PMC free article] [PubMed]
- 65.Ghosh, R., Chakraborty, A., Biswas, A., & Chowdhuri, S. (2020). Identification of polyphenols from Broussonetia papyrifera as SARS CoV-2 main protease inhibitors using in silico docking and molecular dynamics simulation approaches. Journal of Biomolecular Structure and Dynamics, 1–14. 10.1080/07391102.2020.1802347. [DOI] [PMC free article] [PubMed]
- 66.Choudhary MI, Shaikh M, Tul-Wahab A, Ur-Rahman A. In silico identification of potential inhibitors of key SARS-CoV-2 3CL hydrolase (Mpro) via molecular docking, MMGBSA predictive binding energy calculations, and molecular dynamics simulation. Plos one. 2020;15(7):e0235030. doi: 10.1371/journal.pone.0235030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Prime, Schrödinger (2020).
- 68.Piao, L., Chen, Z., Li, Q., Liu, R., Song, W., Kong, R., & Chang, S. (2019). Molecular dynamics simulations of wild type and mutants of SAPAP in complexed with Shank3. International Journal of Molecular Sciences, 20(1). 10.3390/ijms20010224. [DOI] [PMC free article] [PubMed]
- 69.Lipinski CA. Drug-like properties and the causes of poor solubility and poor permeability. Journal of Pharmacological and Toxicological Methods. 2000;44(1):235–249. doi: 10.1016/S1056-8719(00)00107-6. [DOI] [PubMed] [Google Scholar]
- 70.Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings1PII of original article: S0169-409X(96)00423-1. The article was originally published in Advanced Drug Delivery Reviews 23 (1997) 3. Advanced Drug Delivery Reviews. 2001;46(1):3–26. doi: 10.1016/S0169-409X(00)00129-0. [DOI] [PubMed] [Google Scholar]
- 71.Schrödinger (2015). Qikprop 4.4 user manual. Retrieved from http://gohom.win/ManualHom/Schrodinger/Schrodinger_2015-2_docs/qikprop/qikprop_user_manual.pdf
- 72.Schrodinger Release 2020 QikProp, LLC New York, NY.
- 73.Duffy EM, Jorgensen WL. Prediction of properties from simulations: Free energies of solvation in hexadecane, octanol, and water. Journal of the American Chemical Society. 2000;122(12):2878–2888. doi: 10.1021/ja993663t. [DOI] [Google Scholar]
- 74.Jorgensen WL, Duffy EM. Prediction of drug solubility from Monte Carlo simulations. Bioorganic & Medicinal Chemistry Letters. 2000;10(11):1155–1158. doi: 10.1016/S0960-894X(00)00172-4. [DOI] [PubMed] [Google Scholar]
- 75.Jorgensen WL, Duffy EM. Prediction of drug solubility from structure. Advanced Drug Delivery Reviews. 2002;54(3):355–366. doi: 10.1016/S0169-409X(02)00008-X. [DOI] [PubMed] [Google Scholar]
- 76.Schrödinger Suite 2020 Protein Preparation Wizard, LLC New York, NY.
- 77.Harder E, Damm W, Maple J, Wu C, Reboul M, Xiang JY, Wang L, Lupyan D, Dahlgren MK, Knight JL, Kaus JW, Cerutti DS, Krilov G, Jorgensen WL, Abel R, Friesner RA. OPLS3: A force field providing broad coverage of drug-like small molecules and proteins. Journal of Chemical Theory and Computation. 2016;12(1):281–296. doi: 10.1021/acs.jctc.5b00864. [DOI] [PubMed] [Google Scholar]
- 78.Jin Z, Du X, Xu Y, Deng Y, Liu M, Zhao Y, et al. Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature. 2020;582(7811):289–293. doi: 10.1038/s41586-020-2223-y. [DOI] [PubMed] [Google Scholar]
- 79.Jacobson MP, Pincus DL, Rapp CS, Day TJF, Honig B, Shaw DE, Friesner RA. A hierarchical approach to all-atom protein loop prediction. Proteins: Structure, Function, and Bioinformatics. 2004;55(2):351–367. doi: 10.1002/prot.10613. [DOI] [PubMed] [Google Scholar]
- 80.Forli S, Huey R, Pique ME, Sanner MF, Goodsell DS, Olson AJ. Computational protein–ligand docking and virtual drug screening with the AutoDock suite. Nature Protocols. 2016;11(5):905–919. doi: 10.1038/nprot.2016.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, Olson AJ. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. Journal of Computational Chemistry. 2009;30(16):2785–2791. doi: 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Bikadi Z, Hazai E. Application of the PM6 semi-empirical method to modeling proteins enhances docking accuracy of AutoDock. Journal of Cheminformatics. 2009;1(1):15. doi: 10.1186/1758-2946-1-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Biovia D. BIOVIA Discovery Studio 2017 R2: A comprehensive predictive science application for the life sciences. 2017. [Google Scholar]
- 84.Schrödinger Release 2020–1: Desmond molecular dynamics system, D. E. Shaw Research, Maestro-Desmond Interoperability Tools New York, NY.
- 85.Lee, K. E., Bharadwaj, S., Yadava, U., & Kang, S. G. (2020). Computational and in vitro investigation of (-)-epicatechin and proanthocyanidin B2 as inhibitors of human matrix metalloproteinase 1. Biomolecules, 10(10). 10.3390/biom10101379. [DOI] [PMC free article] [PubMed]
- 86.Salazar F, Ortiz A, Sansinenea E. A strong antifungal activity of 7-O-Succinyl Macrolactin A vs Macrolactin A from Bacillus amyloliquefaciens ELI149. Current microbiology. 2020;77(11):3409–3413. doi: 10.1007/s00284-020-02200-2. [DOI] [PubMed] [Google Scholar]
- 87.Kim DH, Kim HK, Kim KM, Kim CK, Jeong MH, Ko CY, Moon KH, Kang JS. Antibacterial activities of macrolactin a and 7-O-succinyl macrolactin a from Bacillus polyfermenticus KJS-2 against vancomycin-resistant enterococci and methicillin-resistant Staphylococcus aureus. Archives of Pharmacal Research. 2011;34(1):147–152. doi: 10.1007/s12272-011-0117-0. [DOI] [PubMed] [Google Scholar]
- 88.Smith A, Ott G. Total synthesis of (−)-macrolactin A. Journal of the American Chemical Society. 1996;118(51):13095–13096. doi: 10.1021/ja963543a. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(DOCX 133 kb)
(DOCX 16 kb)
(DOCX 17 kb)
(DOCX 14 kb)
Data Availability Statement
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.