

Abstract
Huntington’s disease pathogenesis involves a genetic gain-of-function toxicity mechanism triggered by the expanded HTT CAG repeat. Current therapeutic efforts aim to suppress expression of total or mutant huntingtin, though the relationship of huntingtin’s normal activities to the gain-of-function mechanism and what the effects of huntingtin-lowering might be are unclear. Here, we have re-investigated a rare family segregating two presumed HTT loss-of-function (LoF) variants associated with the developmental disorder, Lopes-Maciel-Rodan syndrome (LOMARS), using whole-genome sequencing of DNA from cell lines, in conjunction with analysis of mRNA and protein expression. Our findings correct the muddled annotation of these HTT variants, reaffirm they are the genetic cause of the LOMARS phenotype and demonstrate that each variant is a huntingtin hypomorphic mutation. The NM_002111.8: c.4469+1G>A splice donor variant results in aberrant (exon 34) splicing and severely reduced mRNA, whereas, surprisingly, the NM_002111.8: c.8157T>A NP_002102.4: Phe2719Leu missense variant results in abnormally rapid turnover of the Leu2719 huntingtin protein. Thus, although rare and subject to an as yet unknown LoF intolerance at the population level, bona fide HTT LoF variants can be transmitted by normal individuals leading to severe consequences in compound heterozygotes due to huntingtin deficiency.
Introduction
Huntington’s disease (HD) (OMIM #143100) is a neurodegenerative disorder caused by an unstable CAG repeat expansion in the Huntington’s disease gene (Huntingtin; HTT) (1). Worsening motor, cognitive and behavioral changes typically become evident in the adult years, though clinical signs can also appear in juveniles (2). Although the details of the mechanism by which the CAG expansion mutation elicits this distinctive but complex progressive disease process are not yet known, genetic findings have provided criteria that guide its investigation and point to the operation of sequential components (3,4).
HTT CAG repeat sizes represent a continuum of normal (<27 units), high normal/intermediate (27-35 units), reduced penetrance (36-39 units) and full penetrance (>39 units) allele lengths (5). In the latter range, CAG repeat length is inversely correlated with the age at onset of clinical signs. This CAG expansion mutation is unusual in that it acts as a true dominant. The shorter CAG repeat allele present in typical heterozygous HD individuals does not contribute to disease onset and, in rare HD individuals with two expanded CAG repeats, neither the second (shorter) expanded allele nor the absence of a normal range allele contributes to disease onset (6,7), indicative of a gain-of-function disease mechanism. A recent large genome-wide association study seeking genetic modifiers of HD has revealed that the timing of disease onset is due to the length of the expanded CAG repeat, not of the encoded polyglutamine segment in the huntingtin protein. Moreover, this rate is influenced by variants in several genes involved DNA maintenance, as well as by variants in other genes that may play an indirect role in DNA stability or may act in a biologically unrelated manner (3,8).
These genetic observations support a proposed two-component model of HD and other unstable repeat disorders, in which the inherited expanded CAG repeat further expands somatically until a critical length threshold is reached, thereby triggering a distinct toxicity process in vulnerable target cells that results in cell dysfunction and loss (4). This model offers two different opportunities to interfere with or circumvent HD pathogenesis, if the mechanisms underlying each component can be delineated. Evidence for the rate-to-threshold component points to somatic instability of the expanded repeat, which is an area of active investigation, previously of interest in mouse models (9,10), that is now gaining traction in humans based on the identities of the DNA maintenance modifier genes with variants that produce the effect of slowing or hastening HD onset (3,8,11–17). The consequent HD toxicity-trigger mechanism provides a second opportunity for intervention, but there are currently both too few firm clues to its nature and too many possibilities for cellular processes that may be involved since the mutation has been reported to elicit a wide selection of potentially harmful phenotypes: aberrant gene regulation and differentiation, abnormal cellular metabolism and energetics, altered cell motility and adhesion (18). Currently, potential treatments being tested in clinical trials to slow the toxicity process in symptomatic subjects involve suppressing the expression of either total huntingtin or mutant huntingtin in an allele-specific manner (19,20). However, it remains uncertain what relationship the gain-of-function disease mechanism has to huntingtin’s normal activity(ies) or what the effects might be of suppressing huntingtin levels.
Huntingtin is a 350 kDa HEAT-repeat protein with a unique clamping mechanism enabled by its modular domain structure (21–23). It is expressed ubiquitously, with the highest levels in nervous system tissues, and physically interacts with hundreds of other proteins (24,25), as huntingtin participates in a wide variety of cellular processes, the most studied of which include synaptic vesicular trafficking, transcriptional regulation, signaling, mitochondrial function, and autophagy (18). The powerful genetic approach of investigating huntingtin’s role through loss-of-function (LoF) mutations has been pursued most extensively in the mouse. Heterozygote mice, with null and hypomorphic alleles, are viable, fertile and of normal appearance but homozygotes and compound heterozygote mutation mice display a range of variably penetrant developmental phenotypes, including death in utero, postnatally or in adulthood (26–30). Less is known concerning huntingtin function in humans, although we have reported a family segregating a t(4;12) balanced translocation that breaks HTT within intron 40 (31,32). This null allele does not express huntingtin and does not produce any apparent abnormal phenotypes in the two heterozygous individuals who have only one intact HTT allele (32). Recently, a rare family was reported in which two putative HTT LoF mutations were suggested to be the cause in compound heterozygotes of a rare congenital disorder featuring variable neurodevelopmental deficits: decreased muscle tone, spasticity of the limbs, dystonia, stereotypic hand movements, epilepsy and severe near-sightedness (LOMARS; OMIM #617435) (33,34). In the original report of this family, clinical testing by whole exome sequence analysis identified two different HTT DNA sequence changes inherited from the apparently normal carrier parents by their affected compound heterozygote children but not by an unaffected child. The paternal HTT variant c.4469+1G>A (NCBI Reference Sequence: NM_002111.8; dbSNP: rs1060505027) was reported to change the splice donor site in intron 34, and therefore to be likely to result in abnormal gene splicing. The maternal HTT variant was reported to change a highly conserved amino acid in huntingtin from phenylalanine (Phe) to leucine (Leu) (NP_002102.4: p.Phe2719Leu). However, no functional analyses were carried out to support the putative impact of these variants on huntingtin. Here, we have re-investigated this family through comprehensive whole-genome sequencing (WGS) of DNA from their tissue culture cell lines, combined with assessment of functional impact on RNA and protein expression. Our findings clarify confusion concerning these HTT variants in public databases, reaffirm they are the cause of the LOMARS phenotype and reveal each to be a hypomorphic mutation, resulting in severe deficiency of huntingtin in compound heterozygotes.
Results
Confirmation of inheritance of HTT variants as the genetic cause of the congenital developmental syndrome in compound heterozygotes
The LOMARS family pedigree, comprising developmentally normal parents and 3-year-old son along with his affected siblings, a 12-year-old sister (sibling #1), 2-year-old brother (sibling #3) and deceased brother who died at age 7 (sibling #2), is depicted in Figure 1A, as described in the original family report (33,34). Fibroblast cell cultures were established from skin-biopsy of both parents and the living affected son (sibling #3) and a lymphoblastoid cell line (LCL) was generated from blood cells of the affected daughter (sibling #1). In order to confirm and extend the previous results of clinical exome sequencing, we performed whole-genome sequence (WGS) analysis of the cell line DNAs. Variants were annotated with the Variant Effect Predictor (VEP) of Ensembl and were then filtered to eliminate those that were of low quality, failed Mendelian transmission, were predicted not to change protein structure or behavior or were missense variants predicted to be tolerated/benign (see Materials and Methods). Several candidate genes were either homozygous or compound heterozygous for variants that passed all filters, but all of these genes, except HTT, were discounted based upon comparisons of allele frequency and homozygosity in the Genome Aggregation Database (gnomAD) (see Materials and Methods). Therefore, this comprehensive WGS analysis confirmed that the reported HTT variants are the most likely genetic cause of the developmental syndrome in this family. However, they also revealed discrepancies in how both variants have been recorded in public databases. Consequently, to directly confirm the sites of these two causative variants, we performed Sanger sequencing of polymerase chain reaction (PCR) amplification products from the genomic DNA of the affected daughter (Fig. 1B).
Figure 1 .
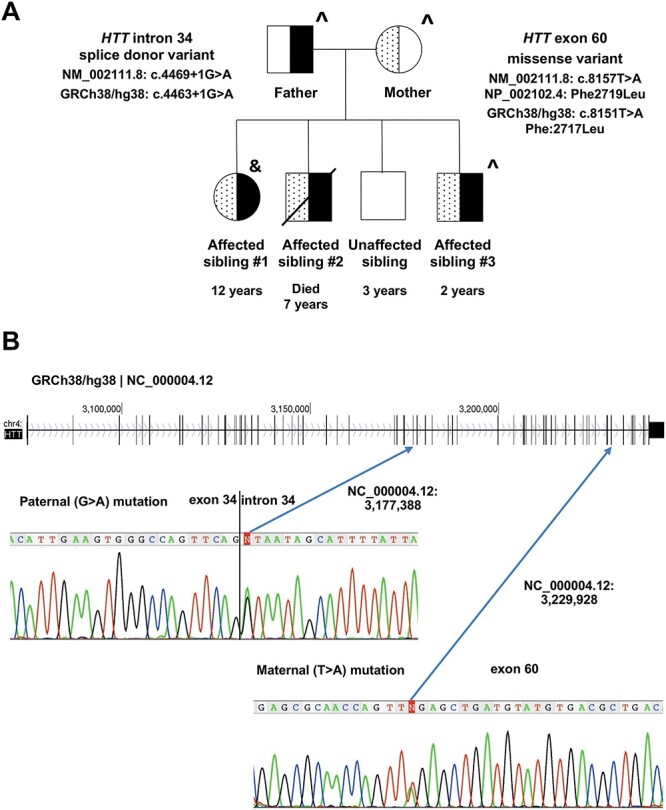
HTT variants in a LOMARS family. (A). Pedigree showing members of a LOMARS family. Age shown is at the time of initial clinical examination. Each of the two surviving siblings, the younger male sibling #3 and older female sibling #1, as well as deceased compound heterozygous male sibling #2, harbor mutant alleles from the asymptomatic heterozygous parents. The dotted half-symbol shading denotes a carrier of the HTT missense variant, which is annotated relative to the reference sequences shown next to the mother’s symbol. The solid half-symbol shading indicates a carrier of the HTT splice site variant, which is annotated relative to the reference sequences shown next to the father’s symbol. Also depicted, is a phenotypically normal male sibling. Fibroblast cell cultures were generated from skin-biopsy (^) and an LCL line was generated from a blood sample (&). (B). The ~ 185 kb of chromosome 4 spanned by HTT is depicted in a genome-view to show the locations of its 67 exons (vertical lines). Below the schematic are the Sanger sequencing chromatograms and genomic DNA nucleotide base calls (heterozygous position is denoted by ‘N’) for sibling #1 to indicate (arrows) the locations of the paternal splice site variant and the maternal missense variant, annotated using the GRCh38/hg38 | NC_000004.12 reference sequence. The correct annotations for each mutation, relative to the genome build and mRNA (NM) and protein (NP) reference sequences, are shown next to the father and mother’s symbols in the pedigree in panel A.
Confusion in the database recording of the positions of these two HTT variants results in part from mislabeling of the site of the missense change in the original report as c.8156T>A despite the ClinVar submission quoted in that same report being correct (c.8157T>A (p.Phe2719Leu)). However, a second ClinVar entry (NM_002111.8(HTT): c.8156T>A (p.Phe2719Tyr)) was subsequently generated that incorrectly describes the same variant and it has been propagated to an incorrect dbSNP entry (rs1060505028). A second source of confusion results from a difference of two codons in the size of the HTT CAG repeat between the human genome reference sequence (GRCh38/hg38; NC_000004.12) and the transcript reference sequence (NCBI Reference Sequence NM_002111.8), which has resulted in an erroneous ClinVar entry for the splice site variant (NM_002111.8(HTT):c.4463+1G>A rather than c.4469+1G>A). To correct these discrepancies, the accurate annotations against the appropriate reference sequences are shown on the pedigree in Figure 1A.
Compound HTT variant heterozygotes exhibit decreased levels of huntingtin
We reasoned that if these rare HTT variants indeed represent deleterious mutations, the levels of the encoded huntingtin proteins may be decreased. Immunoblot analysis of protein extracts from the fibroblast cell lines revealed lower levels of huntingtin in the heterozygous parents, compared with the controls (Fig. 2A), suggesting an effect of both variant alleles. Consistent with this interpretation, the affected son (sibling #3) displayed a far lower level of huntingtin than either the parents or the controls. This striking effect of compound heterozygosity for these HTT variants was confirmed in the LCL extract from the affected daughter (sibling #1) in comparison to age-at-collection matched LCL controls (Fig. 2B). Although both parents have one normal HTT allele, the fibroblasts of the mother showed slightly more normalized huntingtin signal than the father’s (Fig. 2A), suggesting that the paternal c.4469+1G>A splice donor variant might have a more severe impact on huntingtin expression than the maternal c.8157T>A missense variant.
Figure 2 .
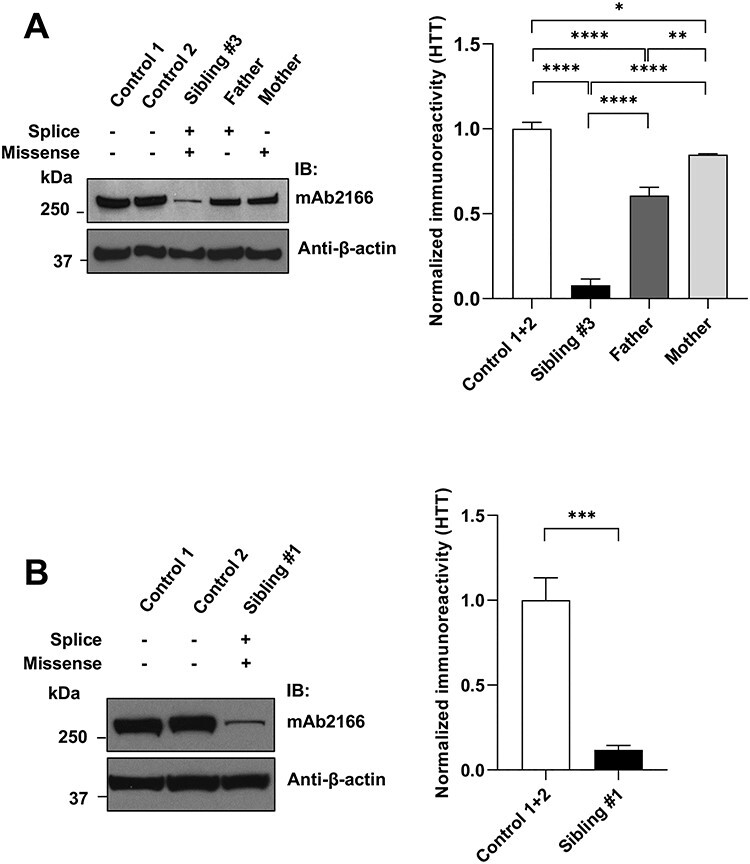
Huntingtin levels in LOMARS family member cultured cells. (A). Left panel shows representative images of immunoblots of fibroblast cell extracts from two different control lines (wild-type HTT homozygotes), the LOMARS father (wild-type HTT and splice site HTT variant), mother (wild-type HTT and missense HTT variant) and sibling #3 (splice site HTT variant and missense HTT variant), probed with mAb2166 to reveal huntingtin bands (top) and with β-actin (bottom) as loading control. Right panel shows the quantification of immunoblots (n = 2), plotted as the huntingtin/β-actin densitometry signal normalized to the mean ratio of the two control samples. Asterisks indicate level of statistical significance (ANOVA post-hoc Tukey’s HSD multiple comparisons test); *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. (B). Left panel shows representative image of immunoblot of LCL extracts from two controls (wild-type HTT homozygotes), and from LOMARS sibling #1 (splice site HTT variant and missense HTT variant). Right panel shows the quantification of immunoblots (n = 2), plotted as the huntingtin/β-actin densitometry signal normalized to the mean ratio of the two control samples. Asterisk indicates level of statistical significance (ANOVA post-hoc Tukey’s HSD multiple comparisons test); ***P < 0.001.
Paternal splice donor site variant causes exon 34 mis-splicing and decreased HTT mRNA
To examine the HTT mRNA produced from these variant alleles, we designed 10 different primer pairs spanning different regions of the HTT transcript (NCBI Reference Sequence: NM_002111.8; Supplementary Material, Table S1) to detect any potential alternative splice products by real-time-PCR (RT-PCR). Amplification from the LCL RNA of sibling #1 with primer pair #4, which spans the location of exon 34, yielded a faint alternative product of lower than expected molecular weight that we presumed to represent skipping of this exon due to the c.4469+1G>A splice donor site variant inherited on the paternal allele (data not shown). Sanger sequencing confirmed that this alternative PCR product lacked the entirety of exon 34 (Fig. 3A), as RNA splicing joined exon 33 directly to exon 35. None of the primer pairs covering other regions of the transcript showed any alternative product bands, but Sanger sequencing of RT-PCR product from exon 60 (primer pair #10) revealed dramatically lower expression of the paternal allele relative to the maternally inherited c.8157T>A missense allele (Fig. 3B).
Figure 3 .
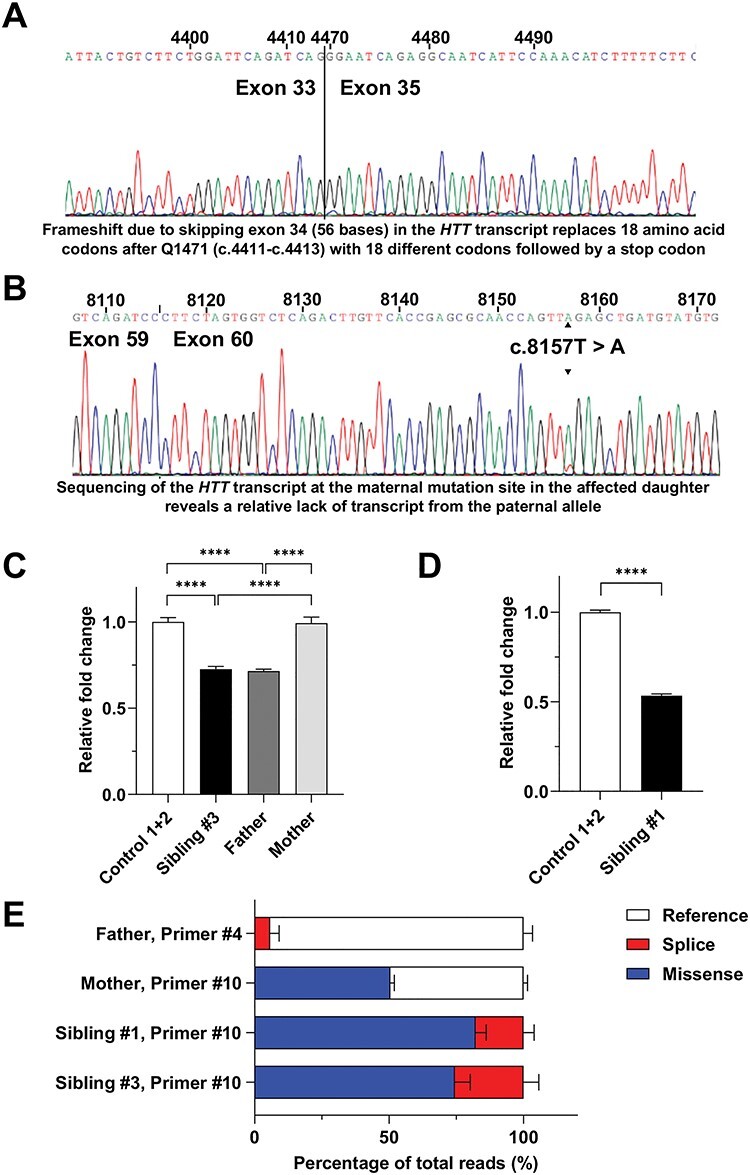
Downregulation of HTT mRNA by the splice site variant. (A). Sanger sequencing chromatogram and HTT nucleotide base calls for the LOMARS sibling #1 alternative product generated by primer pair #4, which reveals that the paternal allele has mis-spliced mRNA, skipping exon 34 (vertical line). Above is the nucleotide numbering from HTT transcript reference sequence NM_002111.8. As noted, below the chromatogram, the frameshift due to skipping exon 34 (56 bases) in the HTT transcript replaces 18 amino acid codons after glutamine Q1471 (c.4411-c.4413) with 18 different codons followed by a stop codon. (B). Sanger sequencing chromatogram and HTT nucleotide base calls for the LOMARS sibling #1 product generated by primer pair #10, which reveals that the paternal allele thymine (T) nucleotide is of a lower signal than the maternal allele adenine (A) nucleotide. Above, the nucleotide numbering is from HTT transcript reference sequence NM_002111.8. As noted, therefore, sequencing of the HTT transcript at the maternal mutation site in the affected daughter reveals a relative lack of transcript from the paternal allele. (C). The bar plot summarizes the mean difference (3 experiments) in primer pair #9 qRT-PCR product for fibroblast HTT mRNA for LOMARS father, mother and sibling #3, normalized to ACTB (β-actin) mRNA, as relative difference (change) compared with control value (controls 1 and 2 mean) (unshaded). Both sibling #3 (black bar) and father (dark grey bar), have significantly lower HTT mRNA compared with mother fibroblast mRNA (light grey bar). Statistical significance ****P < 0.0001; Tukey’s HSD multiple comparisons test. (D). The bar plot summarizes the mean difference (3 experiments) in primer pair #9 qRT-PCR product for LCL HTT mRNA for LOMARS sibling #1, normalized to ACTB (β-actin) mRNA, as relative difference (change) compared with that of the control LCL. Sibling #1 (shaded bar) has lower level of HTT mRNA (~50%), compared with control value (controls 1 and 2 mean) (unshaded). Statistical significance ****P < 0.0001; Tukey’s HSD multiple comparisons test. (E). The bar plot summarizes the MiSeq HTT read counts from primer pair #4 or primer pair #10 RT-PCR products (2 experiments) observed for fibroblast cells of the LOMARS father, mother, and sibling #3 and LCL of sibling #1, as indicated, showing the proportion that is expressed from the wild-type (reference), paternal splice-variant (splice) or maternal missense (missense) allele. The reference sequence used was human huntingtin CDS, from NCBI GenBank accession: BC172756.1 This reveals the predominance of non-splice mutant transcript, either normal wild-type (unshaded) or missense (blue) allele, compared with that of the splice variant allele (red). Note that mother fibroblast cells express ~50% wild-type HTT (unshaded) and 50% missense variant HTT transcript (blue).
Maternal c.8157T>A missense variant is not associated with decreased HTT mRNA
To investigate HTT mRNA expression in a more quantitative manner, we first focused on HTT mRNA expression, assessed using quantitative RT-PCT (qRT-PCR) with primer pair #9 spanning exon 55-57. For the series of family fibroblast RNAs, this qRT-PCR assay revealed a higher level of HTT mRNA product from the mother’s cells, which showed similar to control fibroblast, than cells from either the father or sibling #3, which showed comparable but relatively lower expression (Fig. 3C). As an additional confirmation, the total HTT mRNA in the LCLs of sibling #1 showed an ~ 50% decrease compared with control LCL (Fig. 3D).
A direct comparison of the effect of the two variant alleles on mRNA expression relative to the wild-type allele was achieved by MiSeq sequencing of RT-PCR products that span the sites of the two pathogenic variants from the father and mother, produced respectively from primer pairs #4 and #10. As expected from the previous findings, the vast majority of the product from the father’s RNA derived from his wild-type allele (Fig. 3E), due to the reduced expression of the splice variant allele. Notably, immunoblots using huntingtin antibodies covering various regions of the protein did not reveal any fragment that could be expressed by the mis-spliced mRNA, which is predicted to encode an ~ 163 kDa product due to a frameshift after Q1471, leading to 18 unrelated amino acids and termination at a TGA stop codon (Supplementary Material, Fig. S1). In contrast, the mother’s cells expressed equal amounts of RNA from the wild-type and missense variant alleles, indicating that only the paternal splice site variant results in reduced HTT mRNA expression. This conclusion was confirmed in the RNAs from sibling #1 and sibling #3, using MiSeq analysis of the RT-PCR product from primer pair #10, most of which derived from the maternal missense variant allele.
Taken together, these RT-PCR assays confirmed exon-skipping and reduced levels of paternal c.4469+1G>A splice donor variant HTT mRNA but a lack of impact of the maternal c.8157T>A missense variant on the mRNA level. However, the levels of huntingtin in cells from the affected siblings are too low to be explained by the effect of the paternal splicing variant allele alone.
Maternal missense variant huntingtin p.(F2719L) exhibits decreased half-life
Huntingtin is a predominantly HEAT/HEAT-like repeat solenoid, with a secondary structure that involves extensive HEAT-repeat mediated contacts (21–23) in which the Phe2719 residue is highly conserved, even in the sea urchin (Strongylocentrotus purpuratus) and sea anemone (Nematostella vectensis) (Fig. 4A). We therefore evaluated whether the variant leucine substitution Leu2719 may alter the stability of the protein, resulting in a decrease in huntingtin level. We utilized sensitive, selective reaction monitoring targeted mass spectrometry (SRM-MS) assays, demonstrated to quantify huntingtin species in an allele-specific manner, using extracts from the fibroblast cell series (Fig. 4B). These employed three pan-peptides, which were previously tested and shown to yield quantitative data (35), and two newly designed allele-specific peptides to detect peptides harboring either Phe2719 or Leu2719, respectively (Supplementary Material, Table S2). The pan-peptide assays, as suggested by our immunoblot analysis, detected more huntingtin in the fibroblast extracts from the mother, than from the father or sibling #3 (data not shown). As shown in Figure 4B, the variant allele-specific peptide assays demonstrated that, as expected, the father’s extracts contained only Phe2719 huntingtin. The mother’s fibroblast cells, despite equal levels of c.8157T>A variant and wild-type mRNA, expressed predominantly Phe2719 huntingtin, with a lower amount of Leu2719 huntingtin, suggesting reduced huntingtin stability due to the missense change. As expected based on the father’s results, sibling #3 expressed only Leu2719 huntingtin, from the maternal variant allele, thereby accounting for the low level of huntingtin signal detected in this compound heterozygote in the fibroblast immunoblot analysis (Fig. 2A).
Figure 4 .
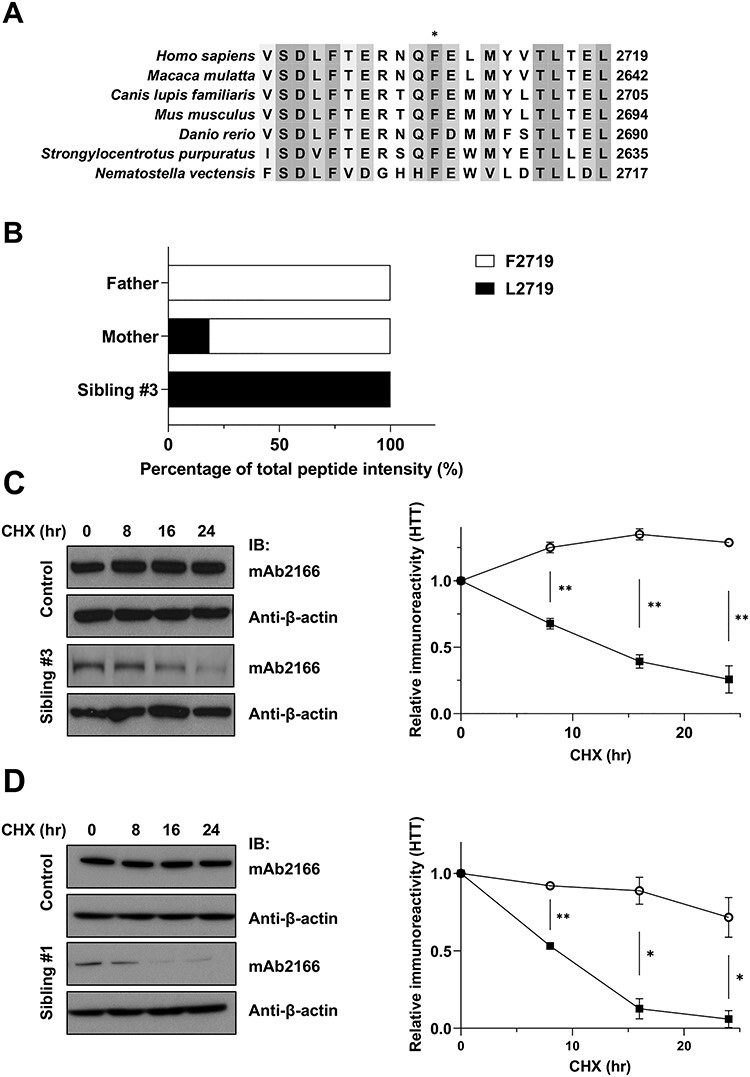
Reduced half-life of huntingtin with Phe2719Leu missense variant. (A). The multiple sequence alignment (Clustal Omega) compares the human huntingtin amino acid sequence in the region of the LOMARS family Phe2719Leu variant (*), with huntingtins predicted from the DNA sequences of the HTT orthologs of Homo sapiens, Macaca mulatta, Canis lupis familiaris, Mus musculus, Danio rerio, S. purpuratus and N. vectensis. Shading denotes the relative conservation of amino acid identity, across these organisms, from not conserved (unshaded) to completely conserved (darkest). (B). The bar plot summarizes the targeted mass spectrometry determined relative abundance of fibroblast cell allele-specific huntingtin peptides for LOMARS family father, mother and sibling #3, shown as proportion of the total peptides (NQFELMYVTLTELR + NQLELMYVTLTELR) that were wild-type Phe2719 (F2719) NQFELMYVTLTELR (unshaded) and missense variant Leu2719 (L2719) NQLELMYVTLTELR (shaded). Father fibroblast cells had only Phe2719 huntingtin, as expected, whereas sibling #3 fibroblast cells had exclusively missense Leu2719 huntingtin, which was also present in mother’s fibroblast cells, albeit at a lower level than wild-type Phe2719 huntingtin. (C). Left panel shows representative images of immunoblots of control and LOMARS sibling #3 fibroblast cell extracts, probed for huntingtin (mAb2166) and β-actin (β -actin), prepared at times after CHX treatment (0, 8, 16 and 24 h). Right panel shows a bar plot summarizing densitometric quantification of mean huntingtin-band immunoblot signal, relative to β-actin signal, at 0, 8, 16 and 24 h post CHX treatment, as determined from 2 experiments, revealing that Leu2719 missense variant huntingtin turnover (filled square) (estimated ½-life of 12.4 h) in sibling #3 cells is significantly faster than that of wild-type Phe2719 huntingtin (unfilled circle) expressed in control cells, with statistically significant differences between means at the 8, 16 and 24-hour time points (**P < 0.01). (D). Left panel shows representative images of immunoblots of control and LOMARS sibling #1 LCL extracts, probed for huntingtin (mAb2166) and β-actin (β -actin), prepared at times after CHX treatment (0,8,16 and 24 h). Right panel shows a bar plot summarizing densitometric quantification of mean huntingtin-band immunoblot signal, relative to β-actin signal, at 0, 8, 16 and 24 h post CHX treatment, as determined from 2 experiments, revealing that Leu2719 missense variant huntingtin (filled square) turnover (estimated ½-life of 6.8 h) in sibling #3 cells is significantly faster than that of wild-type Phe2719 huntingtin (>24 h) (unfilled circle) expressed in control cells, with statistically significant differences between means at the 8, 16 and 24 h time points (*P < 0.05, **P < 0.01).
To test the hypothesis of reduced stability of Leu2719 huntingtin, we performed cycloheximide (CHX) protein synthesis inhibition-chase experiments, quantified by relative signal detected in immunoblot analysis. There was a substantial decrease in huntingtin signal in fibroblast cells of sibling #3, compared with the signal in the parental cells with their predominant expression of Phe2719 huntingtin (Fig. 4C). This reduced stability of Leu2719 huntingtin was confirmed in the LCLs of sibling #1, which showed a much more rapid decrease of huntingtin compared with a control LCL (Fig. 4D). The half-life of Leu2719 huntingtin was estimated (from curve fitting) to be about 12.4 h in sibling #3 fibroblast cells and about 6.8 h in the sibling #1 LCL cells. This is rapid relative to the half-life of normal huntingtin, which could not be estimated in our 24 h experiment but has been determined to be longer than 24 h in LCL and around 48 h in fibroblasts (36,37).
To be certain that the reduced stability of Leu2719 huntingtin is not dependent on the special genetic background of this family but is rather a property of the altered protein, we generated the cDNAs called Q23-HTT-F2719 and Q23-HTT-L2719 in pcDNA3.1, encoding Phe2719 and Leu2719 huntingtin, each with a 23-glutamine segment at the amino-terminus. Each cDNA was transfected into an HEK293T HTT null cell line not expressing huntingtin due to inactivating-mutation generated using CRISPR/Cas9 technology (see Materials and Methods) (Supplementary Material, Fig. S2A). At 24 h post transfection, CHX treatment was initiated and cells were subsequently harvested at multiple time points (Supplementary Material, Fig. S2B). Indeed, we observed a significantly increased degradation rate for the Leu2719 huntingtin, relative to initial levels, at the 24 h time point as well as a lower initial level of the Leu2719 protein, for the same amount of transfected cDNA expression vector. A direct comparison of turnover rate yielded a half-life of 15.2 h for Leu2719 huntingtin but > 24 h for the Phe2719 huntingtin thereby confirming the dramatic change in huntingtin stability caused by altering this single conserved amino acid residue.
HTT intolerance to LoF mutation in heterozygotes
Our results demonstrate that effects on production and stability of huntingtin due to compound heterozygosity for the paternal HTT c.4469+1G>A splice donor variant and the maternal HTT c.8157T>A missense variant underlie the developmental disorder in this family. Each is an HTT hypomorph mutation that decreases the level of huntingtin, with the splice site mutation and its impact at the mRNA level causing a more severe reduction than the missense mutation and its increased rate of Leu2719 huntingtin turnover. The presence of these variants in the heterozygous carrier parents indicates that as the heterozygous null HTT allele in the t(4;12) subjects described previously, both hypomorph alleles are capable of transmission through the germline and neither causes an abnormal phenotype when heterozygous with a wild-type HTT allele. This implies that there should be many individuals heterozygous for damaging HTT variants in population-based DNA variation databases. However, when we examined the gnomAD (version 2.1.1) dataset, we found that there were far fewer HTT LoF variants than expected, suggesting that these suffer negative selection in humans. Compared with an expectation of ~ 159 high-quality, high-confidence HTT LoF variants, there were only 19 such variants, all of extremely low frequency, not observed as homozygous and not reported to be associated with a clinical phenotype in the ClinVar database (Supplementary Material, Table S3), corresponding to a probability of being LoF intolerant (pLI) of 1 (38). Notably, HTT was previously reported to show frequent frameshift variants in the CAG repeat region, often in the homozygous state (39) but the most recent analysis of gnomAD has judged these to be alignment artifacts (40). Recently, Karczewski et al. (38) have statistically evaluated the significance of the constraint, as judged by high-quality LoF variants using o/e (observed/expected) with lower and upper confidence limits and conservatively place HTT (o/e = 0.12 (0.08—0.18)) in the second decile of LoF intolerance among all human genes (LoF o/e upper bound fraction = 0.18). In contrast, HTT missense variants are far more frequent (1404 in gnomAD), only slightly underrepresented (o/e = 0.81 (0.78-0.85)), suggesting that in most cases these alterations are not damaging in a manner that elicits negative selection. Therefore, based on population data, HTT is highly intolerant to damaging LoF mutation, which belies the expectation from the observation that such alleles are observed in the parents and can be transmitted to their children.
Discussion
The rarity of HTT LoF mutations in humans, now highlighted by the evidence of population-intolerance to such variants in heterozygous individuals, has hindered HTT genotype–phenotype investigations to elucidate the physiologic limits of HTT expression. This knowledge is needed foremost to evaluate distinct alternate genetic hypotheses of the mechanism by which the expanded CAG repeat initiates toxicity in vulnerable target HD neurons. By corollary, it is then needed both to guide the design of interventions to delay the onset of HD that are based on interfering with this mechanism, either by lowering/silencing HTT expression or by specifically interfering with the mechanism in target cells, and to manage expectations for the outcomes that may be associated with such interventions in HD individuals.
There are now three inherited damaging LoF HTT mutations shown unequivocally to decrease huntingtin levels in human cells: the NM_002111.8: c.4469+1G>A splice donor variant that results in aberrant (exon 34) splicing and severely reduced mRNA; the NM_002111.8: c.8157T>A NP_002102.4: Phe2719Leu missense variant in which the substitution of a leucine for the conserved phenylalanine in the carboxyl-terminal region results in abnormally rapid turnover of the Leu2719 protein; and the previously reported t(4;12) chromosome translocation that transects HTT within intron 40, thereby bisecting the gene (32). The lack of any discernible abnormal phenotype in those heterozygous for any one of these rare mutations argues strongly against haploinsufficiency. The 50% level of huntingtin from their other HTT allele is evidently sufficient, as it is in the mouse, to support development into a normal adult. But, consistent with mouse studies, at some level <50% the amount of huntingtin is obviously insufficient, such that compound heterozygosity for the HTT hypomorph mutations described here is associated with variably penetrant neurodevelopmental abnormalities and premature death (33,34).
The clinical phenotype of HD individuals, with their two HTT alleles, one or both of which carry the CAG repeat expansion mutation in typical heterozygotes or in expanded allele homozygotes, respectively, is characteristic and is not exhibited by individuals heterozygous for an HTT LoF mutation. Nor does the progressive HD disorder at all resemble the dramatic neurodevelopmental syndrome exhibited by the three siblings who are HTT NM_002111.8: c.4469+1G>A/NM_002111.8:c.8157T>A compound heterozygotes. The lack of equivalency between the phenotypes produced by the HTT CAG expansion mutation and by HTT LoF mutation provides strong evidence to reject post-somatic-expansion toxicity-trigger mechanisms that involve either a simple or a dominant-negative loss of HTT function, in which one or both HTT alleles are functionally inactivated, respectively. The human genetic data instead favor a simple gain-of-function for the CAG repeat length-dependent mechanism that elicits toxicity in vulnerable HD target cells.
Formally, a toxic gain-of-function in HD may occur at the level of the HTT DNA, the mRNA or the huntingtin protein, although the latter has received the most attention (41–45). Indeed, studies focused on effects of mutant huntingtin in comparison to wild-type huntingtin have revealed a variety of differences, including altered localization in neural progenitor cells of the ventricular zone in fetal development (46), but it is not clear what, if any, relevance such potential differences have to the occurrence of HD after decades of life with no clinically detectable abnormality. Toxicity due to expanded RNA has been observed (42,47) and many reports document abnormal phenotypes produced by huntingtin or by polyglutamine-fragment both of which can disrupt a host of cellular processes ranging from chromatin remodeling and regulation of gene transcription to energy metabolism to cell adhesion and more (18), but in general the underlying molecular mechanisms remain obscure. However, two of our findings in particular now provide new routes to investigate each of the formal hypotheses for the CAG repeat length-dependent HD trigger-mechanism. Because NM_002111.8:c.8157T>A mutant HTT mRNA is stable but the encoded NP_002102.4:Leu2719 huntingtin is rapidly degraded, it becomes possible to test the hypothesis (with a CAG repeat expanded c.8157T>A HTT allele) that the toxicity mechanism comprises some feature imposed by the CAG repeat expansion on the DNA or the RNA, rather than at the level of an impact of elongated polyglutamine on the mutant huntingtin product. Second, the NM_002111.8:c.4469+1G>A splice donor variant clearly results in rapid turnover of HTT mRNA, making this single base change a validated target for testing whether the toxicity mechanism may simply be at the level of an impact of the unstable expanded repeat at the HTT locus itself, instead of at the level of either the RNA or the protein products.
Overall, our findings concerning these huntingtin hypomorph variants contrast directly with the accrued data from sequenced clinical and research genomes which argue that HTT is highly constrained. It is strikingly intolerant to heterozygous LoF mutation, based upon observation of relatively few individuals with such alleles. Although HTT does not show a similar lack of missense variants, as demonstrated by the Phe2719Leu change a small minority of these may also be damaging. Indeed, compound heterozygosity for two HTT missense variants (NP_002102.4, p.Phe705Leu and p.Thr1262Met) has been suggested to be associated with neurodevelopmental abnormalities in another reported LOMARS family (48). However, given the lack of phenotype in heterozygotes carrying any of these variants, there is no evidence that huntingtin’s role in neurodevelopment is involved in the selective removal of deleterious HTT LoF mutation alleles from the population. Indeed, the question of the source of this purifying selection is both intriguing and important and deserves a focused research effort to identify it.
Materials and Methods
Cultured LOMARS family cell lines
This study was approved by Massachusetts General Hospital (MGH) institutional review board. All subjects agreed to informed consent form. Primary dermal fibroblasts were collected from three subjects: sibling #3, father, and mother, and the fibroblast control lines used in qRT-PCR and CHX inhibition-chase are one male subject (control 1) and one female subject (control 2) from the same cohort. Skin biopsy was taken from sibling #3 and father and mother during clinical examination. Sample tissue was washed in antibiotic/antimycotic medium and subcutaneous and adipose tissue was removed. Next, the tissue was subjected to dispase digestion. Afterward, the dermis was separated from epidermis, and dermis tissue was minced before being subjected to collagenase digestion. After subsequent centrifugation and resuspension in supplemented medium, primary culture was seeded, and media were replaced every day until confluent. Cultures are maintained at 37°C in 5% CO2 incubator. Epstein–Barr-virus (EBV)-transformed LCL line was generated from sibling #1 as described previously (REF). In addition, two control LCL lines were used for qRT-PCR and family blot: one female subject (control 1), and one male subject (control 2), which was also used as control in CHX inhibition-chase immunoreactivity assay. Briefly, peripheral blood mononuclear cells were isolated from patient blood sample through Ficoll-Hypaque method and gradient centrifugation protocol. After treatment with cyclosporin A, cells were infected with aliquot of EBV supernatant harvested from B95-8 marmoset cell line and cultured at 37°C in 5% CO2.
Fibroblasts were grown in a monolayer at 37°C in D-MEM media (Invitrogen) with high glucose and L-glutamine and supplemented with 10% FBS (Sigma) and 1% penicillin–streptomycin (Invitrogen). Trypsin–ethylenediaminetetraacetic acid (EDTA) 0.05% (Invitrogen) was used to detach cells from culture plate. LCL were grown in RPMI-1640 media (Sigma) with L-glutamine and sodium bicarbonate and supplemented with 10% FBS (Sigma) and 1% penicillin–streptomycin (Invitrogen). HEK293T based HTT null cells lines were generated (see below) and maintained in Dulbecco’s Modified Eagle’s Medium (Invitrogen, 11 995-073) supplemented with 1% Penicillin–Streptomycin (Gibco, 15 140 122) and 10% FBS (Sigma, 12306C) at 37°C in a humidified 5% CO2 atmosphere.
WGS analysis
Genomic DNA was extracted from cultured primary dermal fibroblasts from sibling #3, the father, and mother, and from the LCL generated from sibling #1, using the AutoGen system. WGS was performed by the Broad Institute Genomic Services Platform to an average coverage of 37x. Variants were annotated with the VEP of the Ensembl (http://useast.ensembl.org/Tools/VEP). These variants were filtered to identify those consistent with the disease in the two affected children. First, low-quality variants were eliminated. Variants with a read depth of < 10 and a genotype quality of < 20 for all samples were removed. This resulted in a reduction from 13 260 591 variants pre-filter to 11 924 220 post filter (~10%). Second, variants unlikely to have a detrimental effect were removed (i.e. coding and non-coding variants predicted to be harmless or unlikely to change protein behavior; missense variants predicted to be both ‘tolerated’ by Sorting Intolerant From Tolerant (SIFT) and ‘benign’ by PolyPhen2). Then, variants were removed if their genetic transmission was not consistent with the family, whereas candidate genes were chosen if they exhibited an inheritance pattern consistent with a compound heterozygous or a homozygous model of disease. Allele frequencies for the candidate gene variants and the number of homozygous individuals carrying these variants were obtained from the gnomAD. Regions of homozygosity were analyzed with bcftools (version 1.9). These analyses eliminated inherited potentially damaging compound heterozygote or homozygote variants at several loci (CRYBG3, MROH7, OR10C1, OR52D1 and OR5V1) not mentioned in the original report (33,34) based upon their minor allele frequencies and observed number of homozygous normal individuals in the gnomAD v2.1.1; GRCh37/hg19 (https://gnomad.broadinstitute.org/gene/ENSG00000197386?dataset=gnomad_r2). In addition, sibling #1 and sibling #3 are homozygous for a single variant in C11orf40 and a single variant in the Fc receptor like-2 (FCRL2) gene. Both parents are heterozygous for these variants. Analysis of these regions with the bcftools suggests that these variants are contained in regions of autozygosity in this consanguineous family (https://www.omim.org/entry/617435). The variant seen in FCRL2, (rs138710224) has an observed allele frequency 0.0027 with three individuals homozygous (https://gnomad.broadinstitute.org/variant/1-157738309-G-T) for this variant. Although the variant seen in C11orf40 (insertion of AG after hg38 chr11:4592707) is not reported in gnomAD, a very similar variant, rs67037861, is observed with a high allele frequency (0.3098) and with 9193 homozygous individuals seen. Both of these variants result in the truncation of the 217 amino acids C11orf40 reading frame by 17 amino acids. The two previously reported HTT variants passed all criteria and are correctly annotated relative to the genome, transcript and protein reference sequences: HTT intron 34 splice donor variant is NM_002111.8: c.4469+1G>A and GRCh38/hg38: c.4463+1G>A, whereas the HTT exon 60 missense variant is NM_002111.8: c.8157T>A and NP_002102.4: Phe2719Leu and GRCh38/hg38: c.8151T>A Phe:2717Leu.
Immunoblot analysis
To determine huntingtin protein levels and to detect any potential truncated products, immunoblot analyses were performed on fibroblast cell and LCL whole cell lysates. Cells were lysed in radioimmunoprecipitation assay (RIPA) buffer (Pierce) with protease inhibitor cocktail tablet (Roche). Protein was quantified with bicinchoninic acid (BCA) assay (Pierce) and subjected to SDS-PAGE on a 10% Bis-Tris gel (Invitrogen). Protein was transferred at 100 V for 72 min at 4°C. Nitrocellulose membranes were blotted with mAb2166 (Millipore Sigma) as the primary antibody in 1:5000 dilution in 3% milk-TBST. A 1:10000 dilution of horseradish peroxidase-conjugated secondary antibody was used and chemiluminescence was captured on autoradiography film and developed. Additionally, beta-actin (Santa Cruz), GAPDH (Santa Cruz), and beta-tubulin (Cell Signaling) was probed with antibody as loading control. ImageJ/Fiji rectangle tool was used to quantify immunoreactivity against background, and the final values reflects ratios of mAb2166 signal to loading control (beta-actin, beta-tubulin, GAPDH), normalized to the mean of control ratios. Statistical analyses and graphs were generated using GraphPad Prism.
PCR amplification and qRT-PCR amplification
PCR primers for regions of HTT, including the regions surveyed for the splice and missense mutations, were designed using NCBI Primer design tool, which uses Primer3 and BLAST. A total of 10 primer pairs were designed to span exon–exon junctions of HTT mRNA (accession NM_002111.8) using human refSeq mRNA as database to detect primer specificity. PCR products, for the 10 primer pairs, were validated by agarose gel and confirmed by Sanger sequencing. Each yielded a single PCR product, except for primer pair #4 flanking exon 34, which also produced an alternative fainter band, which was confirmed by Sanger sequencing as the paternal allele mis-spliced RNA. For RT-PCR, total RNA was purified from fibroblast and LCL cells using RNeasy Plus kit (Qiagen) and cDNA synthesis was performed using SuperScript IV First-Strand Synthesis system (Thermo Scientific). Quantitative real-time PCR (qRT-PCR) was performed using LightCycler 480 SYBR Green I Master kit on Roche LightCycler 480 instrument. Among the 10 primer sets, primer pair #9 was found to be optimal for quantifying HTT mRNA in qRT-PCR experiments, because the double-stranded DNA melting curve, assessed by graphing the derivative of fluorescence over the temperature (−dF/dT), was a single peak, lacking any shoulder (data not shown). Beta-actin (ACTB) mRNA levels were used for assay normalization. Fold change for a sample is expressed as 2-ΔΔCt, where ΔΔCt = ΔCTE—ΔCTC. ΔCTE is defined as difference between mean Ct value of gene (HTT) over the mean Ct of the housekeeping gene (ACTB) in the experimental condition, parents and sibling #3 fibroblast or sibling #1 LCL. ΔCTC is defined as difference between mean Ct value of gene (HTT) over the mean Ct value of the housekeeping gene (ACTB) in the control fibroblast or LCL lines. Statistical analyses and graphs were generated using GraphPad Prism.
Illumina MiSeq analysis
We generated allelic ratios of HTT mRNA expression by deep-sequencing of RT-PCR amplification products generated by primer pair #4 for the paternal splice region and primer pair #10 for the maternal missense variant. RT-PCR products, from two independent experiments, were subjected to Illumina MiSeq sequencing platform at the Massachusetts General Hospital Center for Computational and Integrative Biology DNA Core Facility (Cambridge, MA). Samples were prepared as paired-end Tru-Seq compatible Illumina library, which was sequenced from both ends. Illumina compatible barcodes were ligated onto each sample, and library product size and quantification were used as quality control measure. Libraries were normalized and pooled in equimolar concentrations for multiplexed sequencing for 2 × 150 base-pair run. Data were analyzed, de-multiplexed, and entered into an automated de novo analytical pipeline for the detection of CRISPR variants from NGS reads. Amplicons were assembled via UltraCycler v1.0 de novo assembler (Brian Seed and Huajun Wang, unpublished). Briefly, low-frequency reads were filtered out (between 10-100), high-frequency sequence clusters (HFSCs) were generated, low mate-pair HFSCs were filtered out, and forward and reverse reads were merged to form the final analysis. The independent .seq format file reads were aligned to HTT CDS sequence of NCBI GenBank accession: BC172756.1 using Clustal Omega 1.2.0 multiple sequence alignment to identify wild-type allele reads and low-frequency mutations. Total aligned reads were>80 000 for fibroblast (father, mother and sibling #3) and>40 000 reads for LCL (sibling #1) products. For our purposes, instead of detecting low-frequency mutations, we wanted to quantify high-frequency allele proportions, so we analyzed the data further. Of the total aligned reads, the proportion that was variant or wild-type was determined manually, using Genestudio. First, the reads perfectly matching the reference sequence were quantified and annotated as ‘reference’. Then, reads that contained any single nucleotide variant (SNV), insertion/deletion, misalignment, or reads without consensus base-pair calls, were curated manually. Reads containing the expected missense or splice mutation without any other mutations were quantified and annotated as ‘splice mutant,’ if the sequence perfectly matched HTT∆34 expected sequence, or ‘missense mutant’ if they contained NM_002111.8: c.8157T>A variant base pair. In the case of sibling #3 primer pair #10 data, the reads matching reference were annotated as ‘splice mutant’ because they belonged to the paternal allele. Reads that contained any other mutations (indels, SNVs) or non-consensus base-pairs were annotated according to the rest of the sequence (missense, splice, reference) if they were high frequency, but all low-frequency reads (< 1% allele frequency) were omitted from the analysis. Using the final annotations and read quantification, sequence reads pertaining to the splice, mutant, or reference allele were expressed as a percentage of total aligned sequence reads in each run.
Targeted mass spectrometry
Fibroblast cells of father, mother and sibling #3 were lysed with RIPA buffer (Pierce) and subjected to sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE) on a 10% Bis-Tris polyacrylamide gel (Invitrogen). TCA precipitation was performed on the whole lysate to concentrate the protein amount. Total protein was stained with GelCode Blue Safe Protein Stain (Thermo Scientific). Recombinant human huntingtin with 23-glutamine polyglutamine segment, purified from Sf9 insect cells was also run as a standard for protein size (data not shown). The region corresponding to the standard was excised (~1.0 cm width × 0.5 cm height) from a single gel lane and sent to the Taplin Mass Spectrometry Core Facility (Harvard Medical School). Each prepared gel slice was trypsinized as previously described (21) and the samples were dried down in a vacuum centrifuge. Samples were dissolved in 30 μL of 5% acetonitrile in 0.1% trifluoroacetic acid prior to mass spectrometry analysis. Tryptic AQUA peptides were used as spike-in to monitor pan [N-terminal (VLLGEEEALEDDSESR), middle (VLISQSTEDIVLSR), and carboxyl-terminal (SLLVVSDLFTER)], which were previously used (REF), and missense allele-specific [normal (NQFELMYVTLTELR) and mutant (NQLELMYVTLTELR)] huntingtin which were obtained from previous data-dependent acquisition performed at Taplin HMS core (previously published), in each sample. A 5.0 μL aliquot was injected into LTQ Orbitrap Velos Pro ion-trap mass spectrometer (Thermo Fisher Scientific, Waltham, MA), and a single SRM analysis run was performed at resolution of 60 000, with collision energy of 35%, obtaining full MS scan at mass tolerances of 50 ppm in MS1 and 1 Da in MS2. Raw data files were analyzed using Xcalibur software (Thermo) with extracted ion chromatograms comparing the AQUA peptide and the native peptide. The most intense fragment ion of the top selected ion produced from the fragmentation (CID fragmentation) of the peptides was used as targeted mass spectra value. Correction for static and variable modifications was performed using a protein verification search. The ratio of MS value for native over AQUA peptide was used as representative for the absolute measurement of the peptide amount in the sample. To remove any signal background, we took the measurement for missense mutant NQLELMYVTLTELR peptide in the father sample (data not shown) and subtracted that number from all samples, because there is no missense mutation in father. Finally, the peptide measurement of each NQLELMYVTLTELR (mutant) and NQFELMYVTLTELR (wild-type) and was converted to percentage of total (NQLELMYVTLTELR + NQFELMYVTLTELR) peptide and summarized in graph shown in Figure 4B.
CHX chase experiments
Fibroblast cells and LCL were seeded in fresh media at a density of 2.5 million in 5 ml total culture volume and grown overnight before being treated with CHX at 30 μg/ml. At time points (fibroblast, LCL: 0, 8, 16, and 24 h; HEK293T: 0, 4, 8, and 24 h) cells were induced and harvested at the same end point. Whole lysate was extracted with RIPA (Pierce), and protein was quantified using a bicinchoninic acid assay (Thermo). In each lane of SDS-PAGE on 10% Bis-Tris polyacrylamide (Invitrogen), 50 μg of lysate was probed with mAB2166 and beta-actin antibody as loading control. ImageJ/Fiji rectangle tool was used to quantify immunoreactivity against background, and the final graph reflects normalized amount to loading control immunoreactivity. Results of densitometry are shown on the graph as a ratio of mAb2166 signal to beta-actin signal normalized to the 0 h time point. The data points reflect the turnover of huntingtin relative to turnover of beta actin. Statistical analyses and graphs were generated using GraphPad Prism.
HEK293T_HTT null cell line and Q23-HTT-F2719 and Q23-HTT-L2719 cDNA transfection
An HEK293T huntingtin null cell line, called HEK293T_HTT null, was generated by using CRISPR/Cas9 to remove the first exon and upstream promoter region of HTT (hg38 Chr4:3073768-3 075 356). Briefly, gRNA sequences targeting upstream and downstream of exon 1 (caccgCAGAGCGCAGAGAATGCGCG and caccgGGCCTGTCCTGAATTCACCG respectively) were independently cloned into the HF-PX459 plasmid (Addgene #118632) and co-transfected into cells using Lipofectamine 3000 (ThermoFisher). 48 h later, cells were selected for positive transfection by adding puromycin (3 ug/ml final). After 9 days in culture, including a passage to multiple wells, DNA was extracted from a single well and PCR was used to test for a large deletion between the two gRNA sequences (~1580 bp) by using Qiagen LongRange PCR kit (forward: 5’-TGAGGCCAACCTTACTCCCT and reverse: 5’-AACGCCTGCAGACCAACTTA). Once validated as positive for the deletion, cells were plated sparsely into a 96-well plate to produce wells seeded with a single cell and these clones were expanded. PCR amplification analysis confirmed that clonal line #14, named here HEK293_HTT null, had HTT deleted, and immunoblot analysis with Millipore mAb2166 confirmed that full-length or any fragment of huntingtin was not detected in SDS-PAGE analysis of protein lysate (Supplementary Material, Fig. S2A).
The HEK293T_HTT null cells were transfected with cDNAs expressing recombinant human huntingtin (23-glutamine segment) with normal Phe2719 and the Leu2719 variant. The HTT cDNA mammalian expression plasmids were generated in modified pcDNA3 vector as follows. The original cloning region of pcDNA3 vector (Invitrogen, Carlsbad, CA) was swapped with a region containing; 1X FLAG, 6X histidine tag, TEV protease recognition site, and several restriction enzyme sites, including HindIII, BamHI, XhoI, SacII and ApaI. Full-length HTT cDNA, from previously reported human huntingtin pALHDQ23 insect cell expression vector (21), was inserted between BamHI and SacII of the modified pcDNA3 vector. The huntingtin Leu2719 cDNA variant was generated by site-directed mutagenesis (GenScript Piscataway, NJ, USA). The huntingtin Phe2719 and Leu2719 expressing cDNA plasmids were transfected into HEK293T_HTT null cells using Lipofectamine 3000 (Invitrogen, L3000001), according to the manufacturer’s instructions. At 24 h post transfection, the cells were treated with CHX at 30 μg/ml and were harvested at each of several time points.
Statistical analysis
All statistical analysis was performed in GraphPad Prism 8. One-way analysis of variance (ANOVA) was performed to assess statistical significance of differences of means (immunoblot, qRT-PCR). The results of multiple comparison test are shown on each graph, noted with bar between groups and notation of adjusted P value in Tukey’s honestly significant difference (HSD) test. For CHX protein turnover assays, we first tested the difference of means at each time point (paired Student’s t-test, two-tailed). Then, to estimate the expected half-life of huntingtin, we assumed first-order decay kinetics and utilized GraphPad function of one-phase decay model, which follows the formula: Y = (Y0—Plateau) × exp(−K × X) + Plateau. For each assay, we constrained the plateau to be above 0, because we cannot have a negative value of protein amount and generated curve using least squares regression, giving an extrapolated rate constant (K), plateau, and half-life. We used extrapolated half-life values to compare between control and experimental groups. For brevity, only groups significantly different in comparisons were depicted with asterisks (****P < 0.0001, ***0.0001 < P < 0.001, **0.001 < P < 0.01, *0.01 < P < 0.05). All graphs show mean value as single point and standard deviation as error bars.
Constraint analysis
A total of 141 456 samples represented in gnomAD v2.1.1 with either exome or genome sequences were analyzed for HTT LoF variants, namely those that are most likely to disrupt coding or splicing regions. Missense variants and regulatory region variants, which also may influence huntingtin level and activity, were not considered, because their effects are difficult to predict. Of 147 potential HTT LoF variants, 113 were not included because they cluster as indels in low complexity repeat regions, mostly in exon 1, and represent spurious variant calls, whereas 14 others were removed because they lacked supporting exome data. The 20 remaining LoF variants are either splice site disruptions or premature stop codon gain due to frameshift. One of these was then removed as being of low confidence (frameshift 10 amino acid truncation in exon 67), leaving 19 high-confidence SNV most likely to represent true huntingtin LoF. These 19 high-confidence LoF variants (Supplementary Material, Table S3) were used in the gnomAD v2.1.1 constraint calculations. Briefly, gnomAD calculates the pLI), developed with ExAC (49), as well as the more recent o/e score, which also includes a 90% confidence interval metric. The denominator (i.e. ‘expected’ number of LoF SNVs) is the sum of the probability of observing a single-nucleotide mutation resulting in LoF multiplied by the number of chromosomes sampled, which is 251 496 (125 748 exomes X 2 chromosomes per exome). The mutation probability is based on trinucleotide context adjusted for sequencing depth and average methylation status. All InDels are excluded from constraint calculations because of the lack mutation rate models for InDels, which precludes a calibration for the expected count of InDels per gene.
Author Contributions
R.J., Y.L., D.B., K.C., B.S., J.L., R.C., D.L., J.R., T.G. and J.S.M. designed and conducted experiments and performed data analysis. I.S.S., M.E.M., J.F.G., J.-M.L., D.H., R.L. and S.K. contributed to the design of the study, and R.J., M.E.M., J.F.G. and I.S.S. were involved in writing the manuscript, which was reviewed by all the authors.
Supplementary Material
Acknowledgments
We thank the members of the Seong laboratory for suggestions and discussions. We are grateful to Ross Tomaino (Associate Director Taplin Mass Spectrometry Core Facility) for mass spectrometry. This work was supported by National Institutes of Health National Institute of Neurological Disorders and Stroke grants R01 NS079651 and R03 NS108028 (I.S.S.), R01 NS091161 (J.F.G.) and R01 NS105709 (J.-M.L.) and the CHDI Foundation Inc.
Conflict of Interest Statement. J.F.G. is a Scientific Advisory Board member and has a financial interest in Triplet Therapeutics, Inc. His NIH-funded project is using genetic and genomic approaches to uncover other genes that significantly influence when diagnosable symptoms emerge and how rapidly they worsen in Huntington’s Disease. The company is developing new therapeutic approaches to address triplet repeat disorders such Huntington’s disease, myotonic dystrophy and spinocerebellar ataxias. J.F.G. has also advised Wave Life Sciences USA, Inc. His interests were reviewed and are managed by Massachusetts General Hospital and Partners HealthCare in accordance with their conflict of interest policies. J.P. is Chief Scientific Officer of Global Gene Corp. The remaining authors have no conflicts of interest to declare.
Contributor Information
Roy Jung, Molecular Neurogenetics Unit, Center for Genomic Medicine, Massachusetts General Hospital, Boston, MA 02114, USA; Department of Neurology, Harvard Medical School, Boston, MA 02114, USA.
Yejin Lee, Molecular Neurogenetics Unit, Center for Genomic Medicine, Massachusetts General Hospital, Boston, MA 02114, USA; Department of Neurology, Harvard Medical School, Boston, MA 02114, USA.
Douglas Barker, Molecular Neurogenetics Unit, Center for Genomic Medicine, Massachusetts General Hospital, Boston, MA 02114, USA; Department of Neurology, Harvard Medical School, Boston, MA 02114, USA.
Kevin Correia, Molecular Neurogenetics Unit, Center for Genomic Medicine, Massachusetts General Hospital, Boston, MA 02114, USA; Department of Neurology, Harvard Medical School, Boston, MA 02114, USA.
Baehyun Shin, Molecular Neurogenetics Unit, Center for Genomic Medicine, Massachusetts General Hospital, Boston, MA 02114, USA; Department of Neurology, Harvard Medical School, Boston, MA 02114, USA.
Jacob Loupe, Molecular Neurogenetics Unit, Center for Genomic Medicine, Massachusetts General Hospital, Boston, MA 02114, USA; Department of Neurology, Harvard Medical School, Boston, MA 02114, USA.
Ryan L Collins, Molecular Neurogenetics Unit, Center for Genomic Medicine, Massachusetts General Hospital, Boston, MA 02114, USA; Medical and Population Genetics Program, The Broad Institute of M.I.T. and Harvard, Cambridge, MA, 02142, USA; Program in Bioinformatics and Integrative Genomics, Division of Medical Sciences, Harvard Medical School, Boston, MA 02114, USA.
Diane Lucente, Molecular Neurogenetics Unit, Center for Genomic Medicine, Massachusetts General Hospital, Boston, MA 02114, USA; Department of Neurology, Harvard Medical School, Boston, MA 02114, USA.
Jayla Ruliera, Molecular Neurogenetics Unit, Center for Genomic Medicine, Massachusetts General Hospital, Boston, MA 02114, USA; Department of Neurology, Harvard Medical School, Boston, MA 02114, USA.
Tammy Gillis, Molecular Neurogenetics Unit, Center for Genomic Medicine, Massachusetts General Hospital, Boston, MA 02114, USA; Department of Neurology, Harvard Medical School, Boston, MA 02114, USA.
Jayalakshmi S Mysore, Molecular Neurogenetics Unit, Center for Genomic Medicine, Massachusetts General Hospital, Boston, MA 02114, USA; Department of Neurology, Harvard Medical School, Boston, MA 02114, USA.
Lance Rodan, Division of Genetics and Genomics, Boston Children's Hospital, Boston, MA 02115, USA; Department of Neurology, Boston Children's Hospital, Harvard Medical School, MA 02115, USA.
Jonathan Picker, Division of Genetics and Genomics, Boston Children's Hospital, Boston, MA 02115, USA; Department of Child and Adolescent Psychiatry, Boston Children's Hospital, Harvard Medical School, MA 02115, USA.
Jong-Min Lee, Molecular Neurogenetics Unit, Center for Genomic Medicine, Massachusetts General Hospital, Boston, MA 02114, USA; Department of Neurology, Harvard Medical School, Boston, MA 02114, USA.
David Howland, CHDI Management/CHDI Foundation Inc., Princeton, NJ 08540, USA.
Ramee Lee, CHDI Management/CHDI Foundation Inc., Princeton, NJ 08540, USA.
Seung Kwak, CHDI Management/CHDI Foundation Inc., Princeton, NJ 08540, USA.
Marcy E MacDonald, Molecular Neurogenetics Unit, Center for Genomic Medicine, Massachusetts General Hospital, Boston, MA 02114, USA; Department of Neurology, Harvard Medical School, Boston, MA 02114, USA; Medical and Population Genetics Program, The Broad Institute of M.I.T. and Harvard, Cambridge, MA, 02142, USA.
James F Gusella, Molecular Neurogenetics Unit, Center for Genomic Medicine, Massachusetts General Hospital, Boston, MA 02114, USA; Medical and Population Genetics Program, The Broad Institute of M.I.T. and Harvard, Cambridge, MA, 02142, USA; Department of Genetics, Blavatnik Institute, Harvard Medical School, Boston, MA, 02115, USA.
Ihn Sik Seong, Molecular Neurogenetics Unit, Center for Genomic Medicine, Massachusetts General Hospital, Boston, MA 02114, USA; Department of Neurology, Harvard Medical School, Boston, MA 02114, USA.
References
- 1. The Huntington's Disease Collaborative Research Group (1993) A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. Cell, 72, 971–983. [DOI] [PubMed] [Google Scholar]
- 2. Roos, R.A. (2010) Huntington's disease: a clinical review. Orphanet J. Rare Dis., 5, 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Genetic Modifiers of Huntington’s Disease, C (2019) CAG repeat not Polyglutamine length determines timing of Huntington's disease onset. Cell, 178, 887–900.e814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kaplan, S., Itzkovitz, S. and Shapiro, E. (2007) A universal mechanism ties genotype to phenotype in trinucleotide diseases. PLoS Comput. Biol., 3, e235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hendricks, A.E., Latourelle, J.C., Lunetta, K.L., Cupples, L.A., Wheeler, V., MacDonald, M.E., Gusella, J.F. and Myers, R.H. (2009) Estimating the probability of de novo HD cases from transmissions of expanded penetrant CAG alleles in the Huntington disease gene from male carriers of high normal alleles (27-35 CAG). Am. J. Med. Genet. A, 149a, 1375–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cubo, E., Martinez-Horta, S.I., Santalo, F.S., Descalls, A.M., Calvo, S., Gil-Polo, C., Muñoz, I., Llano, K., Mariscal, N., Diaz, D. et al. (2019) Clinical manifestations of homozygote allele carriers in Huntington disease. Neurology, 92, e2101–e2108. [DOI] [PubMed] [Google Scholar]
- 7. Lee, J.M., Ramos, E.M., Lee, J.H., Gillis, T., Mysore, J.S., Hayden, M.R., Warby, S.C., Morrison, P., Nance, M., Ross, C.A. et al. (2012) CAG repeat expansion in Huntington disease determines age at onset in a fully dominant fashion. Neurology, 78, 690–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Genetic Modifiers of Huntington’s Disease, C (2015) Identification of genetic factors that modify clinical onset of Huntington's disease. Cell, 162, 516–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kovalenko, M., Dragileva, E., St Claire, J., Gillis, T., Guide, J.R., New, J., Dong, H., Kucherlapati, R., Kucherlapati, M.H., Ehrlich, M.E. et al. (2012) Msh2 acts in medium-spiny striatal neurons as an enhancer of CAG instability and mutant huntingtin phenotypes in Huntington's disease knock-in mice. PLoS One, 7, e44273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Pinto, R.M., Dragileva, E., Kirby, A., Lloret, A., Lopez, E., St Claire, J., Panigrahi, G.B., Hou, C., Holloway, K., Gillis, T. et al. (2013) Mismatch repair genes Mlh1 and Mlh3 modify CAG instability in Huntington's disease mice: genome-wide and candidate approaches. PLoS Genet., 9, e1003930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Long, J.D., Lee, J.M., Aylward, E.H., Gillis, T., Mysore, J.S., Abu Elneel, K., Chao, M.J., Paulsen, J.S., MacDonald, M.E. and Gusella, J.F. (2018) Genetic modification of Huntington disease acts early in the prediagnosis phase. Am. J. Hum. Genet., 103, 349–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ciosi, M., Maxwell, A., Cumming, S.A., Hensman Moss, D.J., Alshammari, A.M., Flower, M.D., Durr, A., Leavitt, B.R., Roos, R.A.C., team, T.-H. et al. (2019) A genetic association study of glutamine-encoding DNA sequence structures, somatic CAG expansion, and DNA repair gene variants, with Huntington disease clinical outcomes. EBioMedicine, 48, 568–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kennedy, L., Evans, E., Chen, C.M., Craven, L., Detloff, P.J., Ennis, M. and Shelbourne, P.F. (2003) Dramatic tissue-specific mutation length increases are an early molecular event in Huntington disease pathogenesis. Hum. Mol. Genet., 12, 3359–3367. [DOI] [PubMed] [Google Scholar]
- 14. Swami, M., Hendricks, A.E., Gillis, T., Massood, T., Mysore, J., Myers, R.H. and Wheeler, V.C. (2009) Somatic expansion of the Huntington's disease CAG repeat in the brain is associated with an earlier age of disease onset. Hum. Mol. Genet., 18, 3039–3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Flower, M., Lomeikaite, V., Ciosi, M., Cumming, S., Morales, F., Lo, K., Hensman Moss, D., Jones, L., Holmans, P., Investigators, T.-H. et al. (2019) MSH3 modifies somatic instability and disease severity in Huntington's and myotonic dystrophy type 1. Brain, 142, 1876–1886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Goold, R., Flower, M., Moss, D.H., Medway, C., Wood-Kaczmar, A., Andre, R., Farshim, P., Bates, G.P., Holmans, P., Jones, L. et al. (2019) FAN1 modifies Huntington's disease progression by stabilizing the expanded HTT CAG repeat. Hum. Mol. Genet., 28, 650–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lee, J.M., Chao, M.J., Harold, D., Abu Elneel, K., Gillis, T., Holmans, P., Jones, L., Orth, M., Myers, R.H., Kwak, S. et al. (2017) A modifier of Huntington's disease onset at the MLH1 locus. Hum. Mol. Genet., 26, 3859–3867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Saudou, F. and Humbert, S. (2016) The biology of Huntingtin. Neuron, 89, 910–926. [DOI] [PubMed] [Google Scholar]
- 19. Leavitt, B.R., Kordasiewicz, H.B. and Schobel, S.A. (2020) Huntingtin-lowering therapies for Huntington disease: a review of the evidence of potential benefits and risks. JAMA Neurol., 77, 764–772. [DOI] [PubMed] [Google Scholar]
- 20. Tabrizi, S.J., Leavitt, B.R., Landwehrmeyer, G.B., Wild, E.J., Saft, C., Barker, R.A., Blair, N.F., Craufurd, D., Priller, J., Rickards, H. et al. (2019) Targeting Huntingtin expression in patients with Huntington's disease. N. Engl. J. Med., 380, 2307–2316. [DOI] [PubMed] [Google Scholar]
- 21. Vijayvargia, R., Epand, R., Leitner, A., Jung, T.-Y., Shin, B., Jung, R., Lloret, A., Singh Atwal, R., Lee, H., Lee, J.-M. et al. (2016) Huntingtin's spherical solenoid structure enables polyglutamine tract-dependent modulation of its structure and function. Elife, 5, e11184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Guo, Q., Huang, B., Cheng, J., Seefelder, M., Engler, T., Pfeifer, G., Oeckl, P., Otto, M., Moser, F., Maurer, M. et al. (2018) The cryo-electron microscopy structure of huntingtin. Nature, 555, 117–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Jung, T., Shin, B., Tamo, G., Kim, H., Vijayvargia, R., Leitner, A., Marcaida, M.J., Astorga-Wells, J., Jung, R., Aebersold, R. et al. (2020) The Polyglutamine expansion at the N-terminal of Huntingtin protein modulates the dynamic configuration and phosphorylation of the C-terminal HEAT domain. Structure, 28, 1035–1050 e1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Harjes, P. and Wanker, E.E. (2003) The hunt for huntingtin function: interaction partners tell many different stories. Trends Biochem. Sci., 28, 425–433. [DOI] [PubMed] [Google Scholar]
- 25. Shirasaki, D.I., Greiner, E.R., Al-Ramahi, I., Gray, M., Boontheung, P., Geschwind, D.H., Botas, J., Coppola, G., Horvath, S., Loo, J.A. et al. (2012) Network organization of the huntingtin proteomic interactome in mammalian brain. Neuron, 75, 41–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. White, J.K., Auerbach, W., Duyao, M.P., Vonsattel, J.P., Gusella, J.F., Joyner, A.L. and MacDonald, M.E. (1997) Huntingtin is required for neurogenesis and is not impaired by the Huntington's disease CAG expansion. Nat. Genet., 17, 404–410. [DOI] [PubMed] [Google Scholar]
- 27. Zeitlin, S., Liu, J.P., Chapman, D.L., Papaioannou, V.E. and Efstratiadis, A. (1995) Increased apoptosis and early embryonic lethality in mice nullizygous for the Huntington's disease gene homologue. Nat. Genet., 11, 155–163. [DOI] [PubMed] [Google Scholar]
- 28. Murthy, V., Tebaldi, T., Yoshida, T., Erdin, S., Calzonetti, T., Vijayvargia, R., Tripathi, T., Kerschbamer, E., Seong, I.S., Quattrone, A. et al. (2019) Hypomorphic mutation of the mouse Huntington's disease gene orthologue. PLoS Genet., 15, e1007765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Duyao, M.P., Auerbach, A.B., Ryan, A., Persichetti, F., Barnes, G.T., McNeil, S.M., Ge, P., Vonsattel, J.P., Gusella, J.F. and Joyner, A.L. (1995) Inactivation of the mouse Huntington's disease gene homolog Hdh. Science, 269, 407–410. [DOI] [PubMed] [Google Scholar]
- 30. Nasir, J., Floresco, S.B., O'Kusky, J.R., Diewert, V.M., Richman, J.M., Zeisler, J., Borowski, A., Marth, J.D., Phillips, A.G. and Hayden, M.R. (1995) Targeted disruption of the Huntington's disease gene results in embryonic lethality and behavioral and morphological changes in heterozygotes. Cell, 81, 811–823. [DOI] [PubMed] [Google Scholar]
- 31. McKeown, C., Read, A.P., Dodge, A., Stecko, O., Mercer, A. and Harris, R. (1987) Wolf-Hirschhorn locus is distal to D4S10 on short arm of chromosome 4. J. Med. Genet., 24, 410–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ambrose, C.M., Duyao, M.P., Barnes, G., Bates, G.P., Lin, C.S., Srinidhi, J., Baxendale, S., Hummerich, H., Lehrach, H., Altherr, M. et al. (1994) Structure and expression of the Huntington's disease gene: evidence against simple inactivation due to an expanded CAG repeat. Somat. Cell Mol. Genet., 20, 27–38. [DOI] [PubMed] [Google Scholar]
- 33. Rodan, L.H., Cohen, J., Fatemi, A., Gillis, T., Lucente, D., Gusella, J. and Picker, J.D. (2016) A novel neurodevelopmental disorder associated with compound heterozygous variants in the huntingtin gene. Eur. J. Hum. Genet., 24, 1826–1827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Rodan, L.H., Cohen, J., Fatemi, A., Gillis, T., Lucente, D., Gusella, J. and Picker, J.D. (2016) A novel neurodevelopmental disorder associated with compound heterozygous variants in the huntingtin gene. Eur. J. Hum. Genet., 24, 1833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Shin, A., Shin, B., Shin, J.W., Kim, K.-H., Atwal, R.S., Hope, J.M., Gillis, T., Leszyk, J.D., Shaffer, S.A., Lee, R. et al. (2017) Novel allele-specific quantification methods reveal no effects of adult onset CAG repeats on HTT mRNA and protein levels. Hum. Mol. Genet., 26, 1258–1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Persichetti, F., Carlee, L., Faber, P.W., McNeil, S.M., Ambrose, C.M., Srinidhi, J., Anderson, M., Barnes, G.T., Gusella, J.F. and MacDonald, M.E. (1996) Differential expression of normal and mutant Huntington's disease gene alleles. Neurobiol. Dis., 3, 183–190. [DOI] [PubMed] [Google Scholar]
- 37. Wu, P., Lu, M.X., Cui, X.T., Yang, H.Q., Yu, S.L., Zhu, J.B., Sun, X.L. and Lu, B. (2016) A high-throughput-compatible assay to measure the degradation of endogenous Huntingtin proteins. Acta Pharmacol. Sin., 37, 1307–1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Karczewski, K.J., Francioli, L.C., Tiao, G., Cummings, B.B., Alföldi, J., Wang, Q., Collins, R.L., Laricchia, K.M., Ganna, A., Birnbaum, D.P. et al. (2020) The mutational constraint spectrum quantified from variation in 141,456 humans. Nature, 581, 434–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Saleheen, D., Natarajan, P., Armean, I.M., Zhao, W., Rasheed, A., Khetarpal, S.A., Won, H.-H., Karczewski, K.J., O’Donnell-Luria, A.H., Samocha, K.E. et al. (2017) Human knockouts and phenotypic analysis in a cohort with a high rate of consanguinity. Nature, 544, 235–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Minikel, E.V., Karczewski, K.J., Martin, H.C., Cummings, B.B., Whiffin, N., Rhodes, D., Alföldi, J., Trembath, R.C., van Heel, D.A., Daly, M.J. et al. (2020) Evaluating drug targets through human loss-of-function genetic variation. Nature, 581, 459–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Seong, I.S., Woda, J.M., Song, J.-J., Lloret, A., Abeyrathne, P.D., Woo, C.J., Gregory, G., Lee, J.-M., Wheeler, V.C., Walz, T. et al. (2010) Huntingtin facilitates polycomb repressive complex 2. Hum. Mol. Genet., 19, 573–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Marti, E. (2016) RNA toxicity induced by expanded CAG repeats in Huntington's disease. Brain Pathol., 26, 779–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Gray, M., Shirasaki, D.I., Cepeda, C., André, V.M., Wilburn, B., Lu, X.H., Tao, J., Yamazaki, I., Li, S.H., Sun, Y.E. et al. (2008) Full-length human mutant huntingtin with a stable polyglutamine repeat can elicit progressive and selective neuropathogenesis in BACHD mice. J. Neurosci., 28, 6182–6195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Priya, S.B. and Gromiha, M.M. (2019) Structural insights into the aggregation mechanism of huntingtin exon 1 protein fragment with different polyQ-lengths. J. Cell. Biochem., 120, 10519–10529. [DOI] [PubMed] [Google Scholar]
- 45. Sathasivam, K., Neueder, A., Gipson, T.A., Landles, C., Benjamin, A.C., Bondulich, M.K., Smith, D.L., Faull, R.L., Roos, R.A., Howland, D. et al. (2013) Aberrant splicing of HTT generates the pathogenic exon 1 protein in Huntington disease. Proc. Natl. Acad. Sci. U. S. A., 110, 2366–2370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Barnat, M., Capizzi, M., Aparicio, E., Boluda, S., Wennagel, D., Kacher, R., Kassem, R., Lenoir, S., Agasse, F., Braz, B.Y. et al. (2020) Huntington's disease alters human neurodevelopment. Science, 369, 787–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Schilling, J., Broemer, M., Atanassov, I., Duernberger, Y., Vorberg, I., Dieterich, C., Dagane, A., Dittmar, G., Wanker, E., van Roon-Mom, W. et al. (2019) Deregulated splicing is a major mechanism of RNA-induced toxicity in Huntington's disease. J. Mol. Biol., 431, 1869–1877. [DOI] [PubMed] [Google Scholar]
- 48. Lopes, F., Barbosa, M., Ameur, A., Soares, G., de Sá, J., Dias, A.I., Oliveira, G., Cabral, P., Temudo, T., Calado, E. et al. (2016) Identification of novel genetic causes of Rett syndrome-like phenotypes. J. Med. Genet., 53, 190–199. [DOI] [PubMed] [Google Scholar]
- 49. Lek, M., Karczewski, K.J., Minikel, E.V., Samocha, K.E., Banks, E., Fennell, T., O'Donnell-Luria, A.H., Ware, J.S., Hill, A.J., Cummings, B.B. et al. (2016) Analysis of protein-coding genetic variation in 60,706 humans. Nature, 536, 285–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.