

Abstract
Pancreatic ductal adenocarcinoma (PDAC) is a highly aggressive disease with a 5-year survival rate of <10%. The tumour microenvironment (TME) of PDAC is characterized by excessive fibrosis and deposition of extracellular matrix, termed desmoplasia. This unique TME leads to high interstitial pressure, vascular collapse and low nutrient and oxygen diffusion. Together, these factors contribute to the unique biology and therapeutic resistance of this deadly tumour. To thrive in this hostile environment, PDAC cells adapt by using non-canonical metabolic pathways and rely on metabolic scavenging pathways such as autophagy and macropinocytosis. Here, we review the metabolic pathways that PDAC use to support their growth in the setting of an austere TME. Understanding how PDAC tumours rewire their metabolism and use scavenging pathways under environmental stressors might enable the identification of novel therapeutic approaches.
ToC Blurb
Pancreatic ductal adenocarcinoma (PDAC) has a unique tumour microenvironment. Notably, PDAC can reprogramme metabolism in response to this microenvironment. This Review discusses metabolism in pancreatic cancer, including insights into mechanisms and processes as well as the potential therapeutic applications.
Pancreatic ductal adenocarcinoma (PDAC) is a lethal disease with a 5- year survival rate of less than 10%1. This poor prognosis is due to a number of factors, including lack of early detection methods and an intense therapeutic resistance2. Over the past decades, multiple studies have identified the recurrent genetic alterations associated with PDAC development and progression3,4. These alterations include the oncogenic activation of KRAS, which occurs in >90% of patients, and inactivation of tumor suppressor genes such as TP53, SMAD4, and CDKN2A3. Oncogenic activation of KRAS is an early event in the progression to PDAC, whereas deletion or inactivation of TP53 occurs at the later stages. Other mutations have been identified in PDAC patients; these include but are not limited to GNAS5, RNF436, KDM6A7, and BRAF8,9. The functional consequences of these mutations have been validated using genetically engineered mouse models (GEMMs) and human cell line models10,11,12. Unfortunately, the deep understanding of PDAC genetics has not yet translated into effective therapies in this disease.
Another characteristic feature of PDAC is its distinctive tumour microenvironment (TME). The PDAC TME is composed of an abundant and dense fibrous stroma (desmoplasia), consisting of extracellular matrix (ECM) such as collagen and hyaluronic acid13. Indeed, it is common for desmoplasia to constitute the bulk of the tumour mass while PDAC tumour cells often make up only a small fraction in both human and animal models14. Also, a heterogeneous cellular population is present in the TME that includes cancer-associated fibroblasts (for example, pancreatic stellate cells (PSCs), a specialized type of fibroblast similar to hepatic stellate cells)15, immune cells, endothelial cells and neurons. The dense stroma promotes the collapse of the tumour vasculature, resulting in poor oxygen diffusion (hypoxia)16. Indeed, PDAC is amongst the most hypoxic of human tumours17,18. Although there are technical challenges in assessing the absolute amounts of metabolites in the TME, the collapse of the tumour vasculature and the nutrient competition between different cell types within the TME likely lead to a decrease in nutrient availability. Indeed, attempts at measuring metabolites in PDAC compared with normal pancreas in mouse models and human PDAC, as well as in tumour interstitial fluid show varying degrees of nutrient limitation19,20.
To survive and thrive in this hostile environment, PDAC cells can reprogramme metabolism in response to nutrient and oxygen fluctuations in the TME21,22. Understanding how PDAC rewires metabolism could provide insights into its pathogenesis and ultimately enable the development of improved therapies. In this Review, we will emphasize three major areas of metabolism in pancreatic cancer: central carbon metabolism and fuel source utilization, nutrient scavenging mechanisms, and metabolic crosstalk in the complex TME. Lastly, we will provide an overview of the current advances in identifying and targeting metabolic dependencies in pancreatic cancers.
Genetic alterations and metabolism
Alterations in tumour metabolism have been recognized as a hallmark of cancer23. Indeed, to support unconstrained proliferation, cancer cells have increased requirements for biomass, energy and macromolecules. Numerous studies have identified the role of oncogenic mutations in dysregulating cellular metabolism in PDAC and other malignancies24. In PDAC, oncogenic KRAS is linked to an increase in glucose uptake and shunting toward anabolic pathways, glutamine reprogramming and reactive oxygen species (ROS) control25,26. Also, to deal with scarcity or imbalance of nutrient availability, oncogenic KRAS is associated with the activation of unique metabolic scavenging pathways27. In 2020, it was shown that oncogenic KRAS can also reprogramme the metabolism of PDAC by non-cell intrinsic factors, such as upregulating cytokines, and promoting immune cell infiltration that drives tumour metabolic alterations in a PDAC mouse model28,29.
Importantly, other mutations, amplifications, or deletions that occur concurrently with oncogenic KRAS also alter PDAC metabolism in distinct ways. These include mutations in the G-protein αs (such as GNAS), TP53, PTEN and MYC. GNAS-activating mutations drive a shift in the utilization of lipids and increase fatty acid oxidation, promoting PDAC tumour progression30. The transcription factor MYC is considered a master regulator of many metabolic processes in many cancers including PDAC31,32,33. Multiple studies have established that MYC is an important component of PDAC progression in model systems through both extrinsic and intrinsic factors34,35,36. The tumor suppressor p53, which is frequently mutated in PDAC development, controls cellular metabolism by inducing the expression of different transcriptional programmes such as the autophagy network37 and by controlling ROS through the induction of the p53-target TIGAR (TP53-induced glycolysis and apoptosis regulator)38,39. Additionally, it has been demonstrated that restoration of wild-type p53 in a PDAC mouse model induces accumulation of α-ketoglutarate, leading to increased chromatin accessibility that ultimately lead to tumour suppression40. Lastly, the tumour suppressor PTEN, a lipid phosphatase, regulates the activity of the phosphoinositide-3-kinase (PI3K)–AKT pathway, which is involved in many metabolic processes including glucose metabolism, redox control and de novo lipid synthesis41. PTEN expression is commonly lost in PDAC, leading to hyperactive PI3K–AKT signalling that cooperates with mutant KRAS to promote tumour initiation and progression in multiple PDAC models42,43,44,45. In summary, oncogenic KRAS in concert with other genetic and/or genomic events has a critical role in driving metabolic shifts in PDAC tumours.
Fuel source utilization
Glucose.
The glucose molecule stores high levels of chemical energy and is a major source of energy in the cell46,24. During glycolysis, glucose undergoes a series of reactions that end up in the production of energy in the form of ATP and two molecules of pyruvate. Pyruvate can be transported to the mitochondria to feed into the tricarboxylic acid (TCA) cycle (described in the next section) or converted to lactate in the cytosol. Indeed, Otto Warburg in the 1920s observed that cancer cells tend to undergo glycolysis even in the presence of oxygen, producing high levels of lactate47. Importantly, glucose can be diverted from glycolysis into side branches that produce key metabolites needed for biomass, these include: the pentose phosphate pathway, which is responsible for the production of the cofactor nicotinamide adenine dinucleotide phosphate (NADPH), as well as for the production of ribose 5-phosphate, an essential intermediate of ribose synthesis48; and the hexosamine biosynthetic pathway, responsible for the synthesis of uridine diphosphate N-acetyl glucosamine (UDP-GlcNAc), an important substrate for protein glycosylation49. Glycolytic intermediates can also be utilized as precursors for the serine–glycine–one-carbon metabolism pathway, supporting nucleotide synthesis, methylation reactions, and lipid synthesis50,51.
As glucose metabolism has so many roles in cellular metabolism, it is not surprising that oncogenic signalling (such as KRAS or MYC) drives increased uptake and downstream utilization of glucose to support tumour growth. Through an integrated metabolomics and transcriptomic analysis of a mouse model of PDAC, it was demonstrated that oncogenic KRASG12D enhances glucose utilization in PDAC through the transcriptional upregulation of glucose transporter GLUT1 and the enzymes hexokinase 1, hexokinase 2, phosphofructokinase 1, and lactate dehydrogenase A25. The transcriptional upregulation of these genes leads to an overall increase in glycolytic flux. Additionally, KRASG12D increased the expression of glutamine-fructose-6-phosphate transaminase 1, the first and rate-limiting step of the hexosamine biosynthetic pathway, and increased flux through this pathway25. It was also shown that KRASG12D increased flux of glucose into the non-oxidative arm of pentose phosphate pathway by increasing the expression of the rate-limiting enzymes ribulose-5-phosphate-3-epimerase and ribose-5-phosphate isomerase. Importantly, genetic inhibition of the hexosamine biosynthetic pathway or pentose phosphate pathway in vitro and in vivo reduced PDAC growth25. Further studies both in vitro and in vivo systems demonstrated these KRAS-driven metabolic reprogrammings were orchestrated by the RAF–MEK–ERK pathway which led to MYC upregulation52. Indeed, the response to MEK inhibition in PDAC correlates with a decrease in MYC and ribose-5-phosphate isomerase levels52. Lastly, it was shown that in addition to the transcriptional changes that oncogenic KRAS induces in cancer cells, it can also directly interact with effectors from the glycolytic pathway. The KRAS 4A isoform directly interacts with hexokinase 1, leading to enhanced glycolytic flux both in vitro and in vivo53. Taken together, these studies demonstrated that oncogenic KRAS induces metabolic reprogramming that drives glucose usage for anabolic reactions (Fig. 1A).
Figure 1. Metabolic alterations in pancreatic cancers.

22(A) In pancreatic cancer cells, oncogenic KRASG12D upregulates the uptake of glucose and enhances glycolysis. Additionally, glycolytic intermediates are shunted into the pentose phosphate pathway (PPP) and the hexosamine biosynthesis pathway. Ultimately, this metabolic rewiring produces metabolites required for the synthesis of DNA and/or RNA and substrates for glycosylation. (B) In pancreatic cancers, the non-essential amino acid glutamine is metabolized by a non-canonical pathway, whereby glutamine-derived glutamate is converted into aspartate. Aspartate is transported into the cytosol where it undergoes a series of metabolic reactions that ultimately lead to increased NADPH production contributing to redox balance. Abbreviations: HK, Hexokinase; GFAT, Glucosamine—fructose-6-phosphate aminotransferase isomerizing; GlcNAc, Uridine diphosphate N-acetylglucosamine; Gln, Glutamine; Glu, Glutamate; Ala, Alanine; Asp, Aspartate; Cit, Citrate; Suc, Succinate; Fum, Fumarate; Mal, Malate; OAA, Oxaloacetic acid.
Glutamine.
Mitochondria are critical for providing both cellular energy and the anabolic substrates required for cellular growth and homeostasis54. To do so, mitochondria maintain enzymes and metabolite pools distinct from other organelles within the cell. Because many of these metabolites cannot be directly transported across mitochondrial membranes, biochemical reactions are compartmentalized within the mitochondria55. Indeed, the mitochondria play an important part in the synthesis of ATP through the activity of the TCA cycle and oxidative phosphorylation. A major input into the TCA cycle is the entry of pyruvate into the mitochondria through the mitochondrial pyruvate carrier56. However, other substrates such as fatty acids and certain amino acids such as glutamine can feed into the TCA cycle. Multiple groups have demonstrated that mitochondrial metabolism is critical for tumour growth in multiple types of cancer including PDAC57,58 (discussed later).
Glutamine is a non-essential amino acid that is highly abundant in human plasma and has multiple roles in cellular metabolism59,60. The carbon skeleton of glutamine is important to the contribution of TCA cycle intermediates. In the first step of glutaminolysis, glutamine is converted into glutamate by the glutaminase enzyme. Classically, glutamine-derived glutamate is then converted into alpha-ketoglutarate through a deamination reaction catalyzed by glutamate dehydrogenase (GLUD1). Then, α-ketoglutarate enters the TCA cycle replenishing the metabolic intermediates, a process that generates ATP through the production of NADH and FADH2.
Besides the role of glutamine in the TCA cycle, glutamine has other biosynthetic roles in cell metabolism. For example, the amine portion of glutamine can serve as a nitrogen donor for the synthesis of other amino acids, hexosamines, as well as supports the de novo synthesis of pyrimidines and purines61. Glutamine also contributes to the production of the cofactor NADPH and glutathione, both of which have roles in cellular redox balance60,62. Given its abundance in the plasma and critical roles in multiple metabolic pathways, is not surprising that many studies have established that cancer cells rely on glutamine for growth and survival.
In PDAC, glutamine has been shown to be critical for redox homeostasis through a non-canonical pathway63. Mechanistically, this process occurs by the conversion of glutamate to α-ketoglutarate and aspartate in the mitochondria by the mitochondrial aspartate aminotransferase (GOT2). Then, aspartate is transported into the cytoplasm where it is subsequently metabolized by cytosolic GOT1, malate dehydrogenase (MDH1), and malic enzyme (ME1). Ultimately, these metabolic reactions produce pyruvate and increased reducing power in the form of NADPH. Genetic inhibition of key enzymes (GOT1, MDH1, and ME1) leads to a decrease in PDAC growth both in vivo and in vitro63 (Fig. 1B). This study demonstrated that the metabolism of glutamine in PDAC cells depend more on the transamination reactions of GOT1 and GOT2 than GLUD1. Along these lines, Raho and colleagues showed that the mitochondrial uncoupling protein 2 (UCP2) catalyzes the efflux of glutamate-derivate aspartate in human PDAC64. Genetic inhibition of UCP2 disrupts redox balance in vitro and reduces tumor growth in a xenograft PDAC model64.
In 2019, it was demonstrated that PDAC requires the glutamate ammonia ligase to synthesize glutamine and enable efficient growth in glutamine-deprived environments. Indeed, the ablation of glutamate ammonia ligase reduced PDAC tumour growth in an autochthonous model model65. Similarly, it was demonstrated in human PDAC that the SLC1A5 gene has a novel variant (SLC1A5_var) that is localized in the inner membrane of the mitochondria and acts as a glutamine transporter66. This variant of SLC1A5 is dependent on the transcription factor HIF-2α and ectopic expression of this transporter in human PDAC lines increased mitochondrial respiration, glutathione synthesis, and promoted chemotherapy resistance in vitro66. In addition, inhibition of SLC1A5_var resulted in a reduction in tumour growth in a PDAC xenograft mouse model. In summary, glutamine has multiple roles in PDAC metabolism, suggesting that targeting the acquisition and utilization of this amino acid could be a promising therapeutic strategy (see later). However, in contrast to its many roles in supporting tumour growth, glutamine depletion has been shown to increase the metastatic potential of PDAC in preclinical models. Indeed, it was shown that glutamine deprivation drives the epithelial-to-mesenchymal transition (EMT) signature both in vitro and in vivo through upregulation of the transcription factor Slug1 in a process dependent on the ATF4 and the MEK–ERK signaling cascade67. Thus, it will be critical to thoroughly evaluate the effect of inhibiting distinct aspects of glutamine metabolism on PDAC growth and/or metastasis.
Lysosomal nutrient scavenging
The TME of PDAC is characterized by high stromal content, hypoxia and low nutrients compared to adjoining pancreatic tissue19,68. To adapt to these conditions, PDAC relies on the use of lysosome nutrient scavenging pathways such as autophagy and macropinocytosis to provide molecular substrates that can support cellular homeostasis under stressful conditions27,69,70,71. PDAC upregulates transcriptional programmes that orchestrate lysosomal biogenesis and autophagy induction through the function of the MiT–TFE family of transcription factors— MITF, TFE3 and TFEB72. In PDAC, these transcription factors are constitutively active in the nucleus, leading to transcriptional activation of genes in the autophagy–lysosome network. Inhibition of MiT–TFE proteins leads to a metabolic crisis due to the impairment of both autophagy flux and macropinocytosis72. These findings demonstrated that nutrient scavenging is a mechanism of survival and progression in PDAC. Further investigation is necessary to understand how the various aspects of the autophagy–lysosome network are coordinated upon environmental stressors such as nutrient starvation and hypoxia.
PDAC requires autophagy to support tumour growth
Macroautophagy (hereafter referred to as autophagy) is a well-conserved catabolic process that sequesters organelles and macromolecules and targets them for lysosomal degradation to maintain cellular homeostasis and survival during periods of starvation or stress71,73. Initially, autophagy was thought to be a non-selective process, however, studies have shown that autophagy can be highly selective74. The molecular mechanism of autophagy is tightly coordinated by more than 30 autophagy-related (ATG) proteins that are responsible for the autophagy cascade and are broken down into four major steps: initiation and nucleation of the autophagosome; autophagosome closure; autophagosome–lysosome fusion; lysosomal degradation and recycling (Fig. 2).
Figure 2. Pancreatic cancers rely on autophagy and lysosomal scavenging.
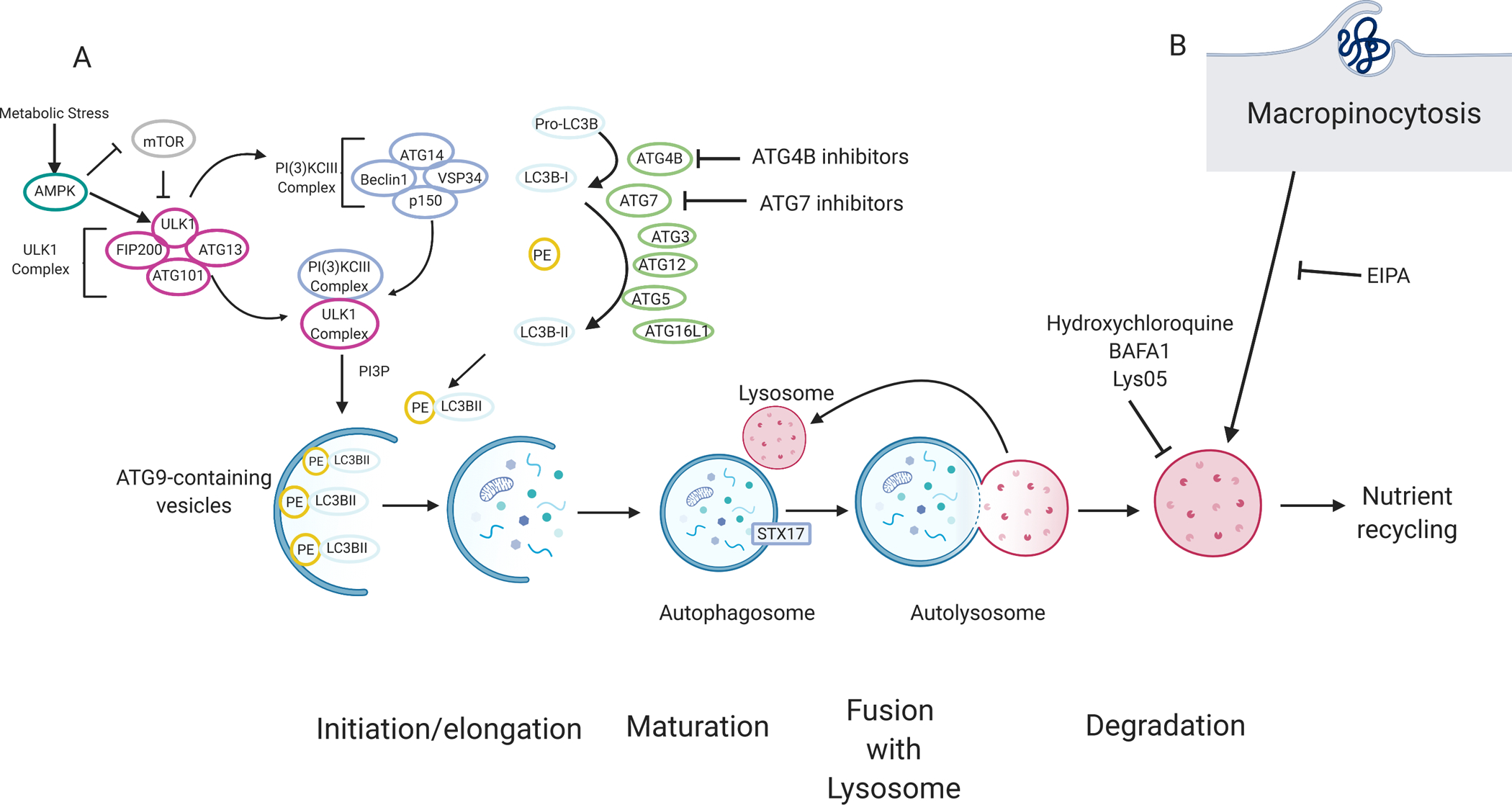
(A)70The molecular mechanism of autophagy can be categorized by four main stages: initiation and/or elongation, closure, fusion with the lysosome, and degradation. Nutrient starvation is a major trigger for autophagy activation through mTOR and AMPK. A series of protein–lipid interactions convert pro-LC3B into LC3B-I by ATG4B. LC3B is subsequently conjugated into phosphatidylethanolamine (PE), producing LC3B-II, via ubiquitin conjugation-like E1-E2-E3 series enzymes ATG7, ATG3, and ATG12-ATG5-ATG16L1 respectively. LC3B-II is associated with the autophagosome and acts as a docking site for autophagy cargo receptors. Cargo, such as damaged proteins or organelles are sequestered in a double membrane vesicle called an autophagosome. Ultimately, the autophagosome fuses with the lysosome, and the content is degraded. Degraded products are shuttled back into the cytoplasm where they can be used in various metabolic pathways. Autophagy and lysosomal degradation can be inhibited by compounds such as hydroxychloroquine, Lys05, and bafilomycin (BAFA1). Other compound tools targeting other modulators of autophagy such as ATG4B inhibitors are currently being tested. (B) Macropinocytosis, a non-selective endocytic pathway, allows extracellular content, such as albumin, to be degraded by the lysosome and can provide amino acids and other metabolites to the cell. Macropinocytosis can be inhibited by EIPA (5-(N-ethyl-N-isopropyl)-Amiloride).
A decade ago, our lab demonstrated that PDAC has elevated levels of basal autophagy and inhibition of autophagy either by RNA interference or pharmacologically using chloroquine (an inhibitor of lysosomal acidification), impaired mitochondrial metabolism, decreased proliferation in vitro, and reduced tumour growth in vivo75. The importance of autophagy in PDAC tumorigenesis was further confirmed by crossing the autochthonous PDAC mouse model with a conditional knockout of the essential autophagy gene, Atg5 (Atg5L/L). This study, along with others, supported the critical role of autophagy in the progression of PDAC76,77,78.
To further investigate how autophagy supports the progression of PDAC in vivo, a new GEMM PDAC model was developed that enables the acute and reversible inhibition of autophagy under temporal and spatial control using a combination of the Cre-Lox and Dox-inducible systems79. This model utilized a dominant negative mutant of ATG4B (ATG4BC74A), a cysteine protease essential for the conjugation of LC3 with phosphatidylethanolamine, which potently inhibits autophagy. The ATG4B mutant was crossed into the well-established pancreatic cancer ‘KPC’ (LSLKrasG12D/+; Trp53L/+;Ptf1Cre) mouse model. Here, it was shown that autophagy inhibition could decrease the growth of established tumours in an autochthonous mouse model. An additional key finding was the fact that autophagy promotes PDAC growth through both cell-autonomous and non-autonomous mechanisms. Inhibition of autophagy in the host (while intact in the tumour cells) impaired tumour growth, likely in part through disruption of a metabolic crosstalk (see later)79. Furthermore, these studies also demonstrated an important role in the antitumour response to autophagy inhibition
Although progress has been made in the study of the role of autophagy in PDAC, little is known about the molecular substrates that autophagy provides under environmental stress. While autophagy inhibition decreases oxidative phosphorylation in PDAC, the metabolic causes of this phenotype have not been fully identified75. For example, it has been shown that in a Kras-driven lung cancer mouse model, that autophagy-deficient cells (Atg7−/−) were more susceptible to nutrient starvation than wild-type cells. It was further demonstrated that, in this context, autophagy is critical to maintaining nucleotide pools under starvation conditions, to support cellular survival80. In another study using an LKB1-deficient lung cancer model in which the essential autophagy gene Atg7 was conditionally deleted, it was demonstrated that autophagy-deficient cells increase de novo fatty acid synthesis to maintain lipid levels and support energy production through fatty acid oxidation81. These findings suggest that autophagy is an important mechanism to ensure metabolic homeostasis in other types of cancers during periods of stress.
Non-metabolic role of autophagy–lysosome in PDAC
Pancreatic cancers are characterized by a lack of response to immunotherapies such as anti PD-1 and anti-CTL4A and multiple reasons have been proposed to explain the immunosuppressive PDAC TME82,83. Human and murine PDAC tumours have been shown to have decreased expression of major histocompatibility complex (MHC) class I molecule on the plasma membrane, which is further reduced in metastatic lesions in the liver84,85. In 2020, it was demonstrated in human and murine PDAC that MHC-I is targeted for degradation through an autophagy-dependent mechanism using the autophagy cargo receptor NBR1 to induce immune evasion86. Genetic inhibition of autophagy machinery or pharmacological inhibition (chloroquine) restored the surface levels of MHC-I. Autophagy inhibition led to an increase in anti-tumour T cell activation, infiltration and cytotoxicity in a syngeneic orthotopic transplant PDAC model. Lastly, autophagy inhibition synergizes with dual immune checkpoint blockade therapy in a syngeneic PDAC model86. Taken together, these findings demonstrate a molecular mechanism by which autophagy enhances immune evasion and provides further rationale for targeting the autophagy–lysosomal system in PDAC.
Macropinocytosis
Macropinocytosis, a nutrient-scavenging pathway that has been identified as an essential mechanism to support pancreatic cancer growth69n, is a non-selective endocytic programme that can uptake content from extracellular fluid through large vesicles known as macropinosomes. PDAC cells can uptake extracellular protein (such as serum albumin) from the extracellular fluid, and this content is then degraded by the lysosome, providing amino acids that eventually fuel central carbon metabolism87. This mechanism can support survival and growth of PDAC and other Ras-driven tumours under nutrient-limiting conditions68.
It has been known for many years that oncogenic RAS induces macropinocytosis87. Moreover, the activity of other effectors such as the PI3K signalling pathway and small GTPases, Rac1 and Cdc42 are required88. Using a short-interfering RNA screen to comprehensively identify regulators of micropinocytosis, it was demonstrated that the vacuolar ATPase (V-ATPase) is a critical effector of Ras-dependent macropinocytosis89. This study supports a model in which oncogenic KRAS promotes the translocation of the v-ATPase to the plasma membrane through the activity of protein kinase A and Rac1. In another study, it was demonstrated that human PDAC xenografts have decreased levels of amino acids, such as glutamine, at their core90, which correlates with the amount of macropinocytosis. This work provides a model whereby epidermal growth factor receptor signaling regulates glutamine deprivation-induced macropinocytosis through the activation of the p21 activated kinase. Using an unbiased functional proteomic screen in the doxycycline-inducible KRAS mouse model of PDAC, Yao and colleagues identified syndecan 1 as a novel effector of macropinocytosis in PDAC91. Furthermore, Hobbs and colleagues demonstrated that PDAC cell lines harbouring KRASG12R activating mutations are unable to induce macropinocytosis as this mutant fails to interact with p110α, an essential effector of KRAS signalling. It was further shown that PDAC cells harboring KRASG12R have distinct drug sensitivity profiles compared to those harbouring KRASG12D or KRASG12V 92. Understanding the metabolic and signalling differences between distinct mutant alleles of KRAS could help stratify patients with PDAC for appropriate therapies.
Taken together, these studies suggest that macropinocytosis could be a potential target for PDAC. As autophagy and macropinocytosis both converge at the lysosome, it will be important to determine whether there are any compensatory scavenging mechanisms under stress when either of these pathways are inhibited. Understanding how macropinocytosis and autophagy coordinate in response to nutrient deprivation, hypoxia, or other TME stressors will be critical to understanding the metabolic scavenging response in PDAC.
Tumor–stroma crosstalk
Cancer-associated fibroblasts (CAFs) are a predominant cellular population within the TME of PDAC93,94. Several years ago, it was demonstrated that pancreatic CAFs can secrete metabolites to support cellular metabolism in PDAC cells in vitro and in vivo95. Through a combination of biochemical and metabolic assays, it was shown that CAFs provide alanine to pancreatic cancer cells to support tumour growth in PDAC. Further characterization utilizing stable-isotope tracing and flux analysis demonstrated that alanine has pleiotropic roles in the metabolism of PDAC cells by contributing to the TCA cycle, synthesis of proteinogenic amino acids, de novo synthesis of fatty acids and transamination reactions. Interestingly, pancreatic cancer cells selectively express the mitochondrial glutamic-pyruvic transaminase 2, creating a unique metabolic situation whereby alanine is used to supply mitochondrial pyruvate to fuel the TCA cycle while allowing cytosolic pyruvate to be utilized to maintain cytosolic NAD+ regeneration96. This metabolic crosstalk between PDAC cells and CAFs was characterized in a panel of PDAC cell lines and xenograft models demonstrating that this interaction is enabled by specific transporters96. Further analyses demonstrated that PDAC requires the neutral amino acid transporter SLC38A2 to uptake alanine96. On the other hand, CAFs utilize SLC1A4 to secrete alanine. Loss of SLC38A2 causes an unrecoverable metabolic crisis and impairs PDAC growth. Disruption of SLC38A2 in non-transformed cells was dispensable, suggesting that targeting this transporter could be promising in PDAC therapy. This work highlights the importance of dissecting the metabolic niches that occur between tumour cells and their environment.
Another example of how CAFs can interact with PDAC cells demonstrated that CAFs can secrete specific lipids to support PDAC progression97. Auciello and colleagues showed that CAFs can secrete lipids such as lysophosphatidylcholines, which support biomass production in PDAC cells through a mechanism dependent on autotaxin–lysophosphatidic acid signalling. Inhibition of autotaxin signalling, both genetically and pharmacologically, leads to a decrease in PDAC tumour progression in vivo97. Another study demonstrated that CAFs not only can support the metabolism and progression of PDAC, they can also contribute to chemotherapy resistance. CAFs secrete the pyrimidine, deoxycytidine, which acts as an inhibitor of the effects of the antimetabolite gemcitabine in human PDAC cells in vitro98,99 A similar mechanism was shown by the Lyssiotis group, who demonstrated that tumour-associated macrophages in the TME of PDAC secreted deoxycytidine, which inhibits the uptake and incorporation of gemcitabine both in vitro and in vivo100. The pharmacological or genetic ablation of tumour-associated macrophages led to an increase in gemcitabine response in vivo using a syngeneic PDAC model100. In addition to metabolism, CAFs can also secrete cytokines and chemokines such as CXCL12, IL-6, and LIF that support PDAC progression101,102,103. In another report, Francescone and colleagues identified Netrin G1 (NetG1) as an important player in PDAC progression104. They showed that NetG1+CAFs support PDAC growth under nutrient deprivation through secretion of glutamine. Interestingly, genetic inhibition ablation or neutralization of NetG1 in CAFs increase anti-tumor function of natural killer (NK) and decrease tumor burden104.
It has been shown that human PDAC tumors have high neuronal innervation, which correlates with poor prognosis105,106. In this regard, multiple studies have demonstrated the protumorigenic effects of neurotrophic factors or neurotransmitters secreted by neurons in PDAC107,108,109,110. However, these studies have not investigated the metabolic contribution of neurons to PDAC. Banh and colleagues demonstrated a novel metabolic crosstalk where peripheral axons release serine to support PDAC growth and survival111. Indeed, the exogenous serine (Ser) was critical to support the growth of ~40% of human PDAC lines that do not express PHGDH, an important enzyme in the Ser biosynthesis pathway. Interestingly, Ser appeared dispensable for the canonical metabolic roles that have been described in cancer. Depletion of Ser selectively reduced mRNA translation rates on specific Ser codons. This allowed the translation and secretion of particular proteins with a favorable codon mix, such as nerve growth factor (NGF), thereby increasing neuronal innervation in the setting of low Ser levels. To evaluate the role of Ser depletion in tumor innervation, the authors injected human PDAC lines into the pancreata of mice and exposed them to a nutrient-rich diet or a Ser-Gly free diet. Strikingly, the Ser-Gly free diet induced increased innervation in PDAC tumors. Pharmacological inhibition of neuronal innervation by LOXO-101, an FDA-approved inhibitor of the NGF receptor, decreased tumor burden in Ser-Gly deprivation in Ser-dependent PDAC111. In summary, this study demonstrated a novel metabolic crosstalk between neuronal axons and PDAC that could lead to novel therapeutic opportunities111.
In summary, the role of stroma in the progression of PDAC is an area of extensive research as these cells can have both tumorigenic and anti-tumour effects. The implementation of single-cell analyses is beginning to provide insights into the heterogeneous role of CAFs and other cell types in the TME of multiple types of cancers, including PDAC94. Although not in pancreatic cancer models, additional types of nutrient sharing and/or competitions have been identified that affect nutrient viability112,113. With the increased understanding of the microbiome in cancer114,115 as well as the multitude of cell types in the complex TME of PDAC116,117,118,119, new insights into tumor-stroma crosstalk are anticipated.
Targeting metabolic liabilities
lysosomal scavenging in pancreatic cancers
Having demonstrated the importance of autophagy in the progression of pancreatic cancer, multiple clinical trials have begun to evaluate the use of the lysosomal inhibitor, hydroxychloroquine for PDAC treatment. Although hydroxychloroquine treatment demonstrated only minimal activity as a monotherapy, the combination of hydroxycholoroquine with standard chemotherapy (gemcitabine and nab-paclitaxel (GA)) markedly improved response rates in randomized trials of localized and metastatic PDAC120,121,122,123. In the past few years, through various approaches, several groups found that inhibition of either MEK or ERK in pancreatic cancers increases the level of autophagy in preclinical experimental models124,125,126. Treatment with hydroxychloroquine and MEK and/or ERK inhibitors showed synergistic responses in various models of pancreatic cancer including patient-derived xenografts. Moreover, the treatment of hydroxychloroquine and a MEK inhibitor (Trametinib) reduced the tumour burden in a single patient, leading to the development of multiple clinical trials to test this approach in patients with PDAC (NCT04145297; NCT03825289; NCT04132505)127,128,129. Given the preclinical results of combining autophagy inhibition with immune checkpoint inhibitors86, clinical trials are also in development to assess these combinations.
Targeting glutamine metabolism in pancreatic cancers
The reliance of PDAC on glutamine and the availability of clinical-grade inhibitors to glutaminase, a key enzyme that converts glutamine to glutamate, provided a strong rationale to study this approach. Although short-term in vitro assays showed suppression of growth, efficacy was lost with prolonged treatment through compensatory mechanisms130. Moreover, the translation of these findings to in vivo PDAC mouse models (including autochthonous and allograft models) failed to reduce tumour growth. Proteomic and metabolomic analyses showed that PDAC cells globally rewire their metabolism in response to this metabolic perturbation130. By studying these compensatory metabolic shifts, co-targeting approaches were determined. Indeed, combining the glutaminase inhibitor with buthionine sulfoximine to induce redox stress, produced a significant decrease in tumour growth in a nude mouse model. This study demonstrated the rapid rewiring that PDAC cells undergo upon metabolic perturbation and that understanding these adaptations can lead to effective therapeutic combinations130.
Previously, there have been attempts to target glutamine utilization in human cancers by using the glutamine antagonist, 6-diazo-5-oxo-l-norleucine (DON), which broadly inhibits all of the metabolic reactions that require glutamine131. However, these clinical studies failed, in a large part, due to gastrointestinal toxicities and poor pharmacological properties of the drug131. A study by Sharma et al. demonstrated that DON treatment decreased tumor growth of pancreatic cancer cells in an athymic nude mouse model132. Interestingly, DON treatment enhanced the response to anti-PD1 treatment in a syngeneic PDAC mouse model. Another study from Leone et al. developed a compound called JHU083, a prodrug version of DON, with better pharmacological properties and less toxicity than DON133. In this study, there were robust effects seen in multiple syngeneic tumour models (colon cancer, lymphoma, and melanoma), demonstrating that glutamine blockade resulted in tumour growth decreases and a better immune response when combined with immune checkpoint blockade133,134. The role of this new drug has not been tested in PDAC, however it might provide an opportunity to overcome the rapid metabolic compensatory response observed upon glutaminase inhibition, given its broad mechanism of action. Conversely, concerns remain regarding the potential toxicities of inhibiting all glutamine-dependent reactions. Further studies are required to validate this approach.
Targeting mitochondrial metabolism in PDAC
Mitochondria are critical organelles that have key roles in a myriad of metabolic processes135. In PDAC, it has been demonstrated that upon the ablation of KRAS using the iKRAS model (described earlier) a small population of pancreatic cancer cells survive25. Through a series of metabolic and transcriptomic analyses, it was shown that cells that survive genetic Kras extinction were less dependent on glycolysis, but are highly sensitive to inhibition of oxidative phosphorylation (OXPHOS)136. However, it will be critical to carefully assess any potential toxicity with this approach given the importance of OXPHOS in normal cells57.
Other aspects of the mitochondria biology have been explored in pancreatic cancers. For example, Yu and colleagues demonstrated that genetic or pharmacological ablation of dynamin-related protein-1 by the small-molecule Mdivi-1 or overexpression of mitofusin-2 stabilized mitochondrial fusion, and increased survival in a PDAC autochthonous mouse model138. Additionally, MacVicar, Ohba and colleagues demonstrated that YME1L, a mitochondrial inner membrane protease, is responsible for rewiring the mitochondrial proteome under hypoxia, a well-documented feature of pancreatic cancers139. Genetic inhibition of YME1L reduced tumour progression in a PDAC xenograft model, suggesting YME1L as a potential target. Collectively, these findings suggest that mitochondria function and metabolism might provide therapeutic targets in PDAC. Indeed, the concept of targeting different aspects of mitochondrial biology has been studied in the clinic134. For example, devimistat (CPI-613), a lipoate analogue that inhibits the activity of pyruvate dehydrogenase and α-ketoglutarate dehydrogenase has shown some anti-tumour activity in the clinic in early phase trials in pancreatic cancer and acute myeloid leukemia (AML)140,141,142,143. Early findings have demonstrated that CPI-613 treatment in combination with a standard chemotherapy regimen for PDAC (FOLFIRINOX), demonstrated a good therapeutic index, and might have activity in this disease144. Currently, CPI-613 is in a phase III trial for the clinical assessment of metastatic pancreatic cancers (NCT03504423)144 and acute myeloid leukemia (NCT03504410).145
The role of cysteine in pancreatic cancers
Cysteine is an essential amino acid for pancreatic cancer progression146. The uptake of cystine (the oxidized dimer of cysteine) occurs through the system xCT (Slc7a11), which mediates the exchange of intracellular glutamate for extracellular cystine, serving as a precursor for glutathione synthesis. Boutaina and colleagues deleted SLC7A11 in a panel of human-derived PDAC lines147. They demonstrated that SLC7A11 was important to retain the intracellular levels of glutathione147. Depletion of cysteine induces ferroptosis, a form of cell death resulting from excessive accumulation of lipid ROS in human PDAC lines in vitro147,148. Further investigation using the KPC model and a dual recombinase system showed that systemically targeting Slc7a11 increased ferroptosis in the tumours and extended survival of mice compared to control149. Treatment with cyst(e)inase150, an enzyme that depletes cysteine, also reduced the tumour progression and induced ferroptosis in vivo149. These studies provide a strong rationale for the use of specific inhibitors of the SLC7A11 system in PDAC.
Functional genomic screens as a tool to uncover metabolic dependencies in PDAC.
Studying the in vivo metabolism of PDAC have been challenging due to the complex TME and the heterogenous cellular populations within it. The application of isotope tracing experiments in multiple cancers (e.g., non-small-cell lung cancers) have been informative to delineate differences in fuel source utilization between in vivo versus in vitro systems151,152,153. However, despite the information obtained from these studies, the metabolic pathways that are required for the progression of PDAC in vivo or what metabolic pathways are required in different hosts (e.g. immune competent or immune compromised animals) have not been fully elucidated. To address this gap, two groups, independently performed the first in vivo metabolism-focused CRISPR screens in PDAC to determine the pathways that are essential for PDAC tumor growth154,155. Both studies demonstrated a striking similarity between in vivo and in vitro metabolic dependencies, indicating that standard 2D culture is a reliable system to study tumor metabolism. Interestingly, the heme biosynthesis pathway was identified as a major metabolic dependency in vivo154,155. Biancur and colleagues demonstrated that although pooled in vitro screens are an excellent method for globally studying metabolic dependencies, they have certain limitations such as dependencies being masked by intercellular nutrient sharing. Additionally, they demonstrated the benefits of using other in vitro systems such as 3-dimensional (3D) growth in the case of metabolic-cell signaling cross-talk154. Squalene synthase (FDFT1) was a significant metabolic dependency in vivo and in 3D culture through its regulation of PI3K/AKT signaling154. There was a more potent dependency in syngeneic models, suggesting a potential role of the immune system. Indeed, genetic or pharmacological inhibition of Fdft1 decreased tumor growth and increased infiltration of CD8+ T cells, suggesting Fdft1 as a potential target in PDAC155. In a similar fashion, Zhu and colleagues showed the essential autophagy gene Atg7 was an important mediator of immune evasion in PDAC155. Together, both studies provide an important resource for the field. We anticipate that these types of approaches will be more widely employed in the future and will allow characterization of metabolic dependencies in distinct genetic drivers or in other relevant situations, such as metastasis.
Conclusions
Pancreatic cancer is considered one of the most lethal malignancies. Despite the advances achieved during the past decades in terms of genetics and tumour biology, many gaps in the pathobiology of this disease remain. The field of tumour metabolism has provided key insights into the understanding of the mechanism by which pancreatic cancers survive and thrive in the TME, and how they become resistant to conventional therapy. It is important to highlight that other metabolites such as fatty acids157, cholesterol158,159 and other amino acids such as proline160 have been identified to support PDAC growth and progression. Future studies will certainly elucidate additional fuel sources for PDACs.
A current challenge in the metabolism field broadly is the lack of proper tools to deconvolute the metabolic contributions of the multiple cellular populations within the TME. Although some studies have implemented the use of stable isotope labeling in the in vivo settings151,152,153, these cannot decipher how tumour cells interact with other cellular populations. Specifically, in PDAC the tumour bulk is often comprised of non-PDAC cells (fibroblast, immune cells, neurons and others)13,161. Thus, understanding the metabolic contribution of each cell population in the complex PDAC ecosystem will provide important insights. A potential approach to overcome this limitation is to investigate the incorporation of labeled fuel sources into biomass or proteins, which are more stable molecules than metabolites162. Similarly, future studies will need to accurately assess the metabolic compartmentalization that occurs in different cellular organelles such as the mitochondria158 and the lysosome. Traditionally, methodologies to study metabolism focus on measuring changes in whole-cell metabolite pools, which fail to consider the compartmentalization of mitochondrial metabolic processes and metabolites. Rapid immunoprecipitation tools for metabolic profiling of both mitochondria163,164,165 and lysosomes,166,167 have been developed. These new technologies could elucidate key findings in tumour metabolism, not only in PDAC but in other types of cancers as well as in normal physiology. We anticipate that the use of in vivo functional genetic screens, improved in vitro culture systems (e.g.,organoids, co-cultures)168, and metabolomic imaging will provide a better understanding of the metabolic plasticity of tumours temporally and spatially. Such studies hold promise to improve therapeutic and diagnostic approaches.
Key point:
Pancreatic ductal adenocarcinoma is characterized by hypoxia, low nutrients, high interstitial pressure and desmoplasia.
To survive and thrive in this hostile environment, PDAC cells reprogramme their metabolism.
PDAC cells also utilize lysosomal scavenging pathways (e.g., autophagy and macropinocytosis) to support and maintain metabolic homeostasis.
Other cellular populations present at the tumor microenvironment of PDAC such as cancer-associated fibroblasts, neurons and immune cells can support PDAC growth in nutrient-limiting conditions.
Targeting the metabolic vulnerabilities of PDAC could provide new therapeutic interventions.
Acknowledgments
We apologize for the omission of any primary citations. We thank members of the Kimmelman lab for helpful discussion and thoughtful suggestions. The authors are supported by National Cancer Institute Grants, R35CA232124, P01CA117969, P30CA016087, and the Lustgarten Foundation, and SU2C to A.C.K. and HHMI Gilliam Fellowships for Advanced Study to J.E.R.
Footnotes
Competing interests
A.C.K. has financial interests in Vescor Therapeutics and is an inventor on patents pertaining to KRAS regulated metabolic pathways, redox control pathways in pancreatic cancer, targeting GOT1 as a therapeutic approach, and the autophagic control of iron metabolism. A.C.K. is on the scientific advisory board of Rafael/Cornerstone Pharmaceuticals and is a consultant for Deciphera and Abbvie. The other author declares no competing interests.
Related links
References
- 1.Siegel RL, Miller KD & Jemal A Cancer statistics, 2019. CA: A Cancer Journal for Clinicians 69, 7–34 (2019). [DOI] [PubMed] [Google Scholar]
- 2.Kleeff J et al. Pancreatic cancer. Nature Reviews Disease Primers 2, 1–22 (2016). [DOI] [PubMed] [Google Scholar]
- 3.Waddell N et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature 518, 495–501 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Raphael BJ et al. Integrated Genomic Characterization of Pancreatic Ductal Adenocarcinoma. Cancer Cell 32, 185–203.e13 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hollstein PE & Shaw RJ GNAS shifts metabolism in pancreatic cancer. Nature Cell Biology 20, 740–741 (2018). [DOI] [PubMed] [Google Scholar]
- 6.Hosein AN, Dangol G, Okumura T, Abou-Elkacem L & Maitra A Loss of Rnf43 to accelerate KRAS-mediated neoplasia in a clinically relevant genetically engineered mouse model of pancreatic adenocarcinoma. JCO 38, 733–733 (2020). [Google Scholar]
- 7.Watanabe S et al. Loss of KDM6A characterizes a poor prognostic subtype of human pancreatic cancer and potentiates HDAC inhibitor lethality. Int J Cancer 145, 192–205 (2019). [DOI] [PubMed] [Google Scholar]
- 8.Seghers A-K, Cuyle P-J & Van Cutsem E Molecular Targeting of a BRAF Mutation in Pancreatic Ductal Adenocarcinoma: Case Report and Literature Review. Targ Oncol 15, 407–410 (2020). [DOI] [PubMed] [Google Scholar]
- 9.Connor AA et al. Integration of Genomic and Transcriptional Features in Pancreatic Cancer Reveals Increased Cell Cycle Progression in Metastases. Cancer Cell 35, 267–282.e7 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Aguirre AJ et al. Activated Kras and Ink4a/Arf deficiency cooperate to produce metastatic pancreatic ductal adenocarcinoma. Genes Dev. 17, 3112–3126 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hingorani SR et al. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell 7, 469–483 (2005). [DOI] [PubMed] [Google Scholar]
- 12.Tuveson DA et al. Endogenous oncogenic K-ras(G12D) stimulates proliferation and widespread neoplastic and developmental defects. Cancer Cell 5, 375–387 (2004). [DOI] [PubMed] [Google Scholar]
- 13.Hosein AN, Brekken RA & Maitra A Pancreatic cancer stroma: an update on therapeutic targeting strategies. Nature Reviews Gastroenterology & Hepatology 17, 487–505 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Feig C et al. The pancreas cancer microenvironment. Clin. Cancer Res 18, 4266–4276 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ho WJ, Jaffee EM & Zheng L The tumour microenvironment in pancreatic cancer — clinical challenges and opportunities. Nature Reviews Clinical Oncology 1–14 (2020) doi: 10.1038/s41571-020-0363-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Olive KP et al. Inhibition of Hedgehog Signaling Enhances Delivery of Chemotherapy in a Mouse Model of Pancreatic Cancer. Science 324, 1457–1461 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Koong AC et al. Pancreatic tumors show high levels of hypoxia. International Journal of Radiation Oncology*Biology*Physics 48, 919–922 (2000). [DOI] [PubMed] [Google Scholar]
- 18.Hollinshead KER et al. Respiratory Supercomplexes Promote Mitochondrial Efficiency and Growth in Severely Hypoxic Pancreatic Cancer. Cell Rep 33, 108231 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sullivan MR et al. Quantification of microenvironmental metabolites in murine cancers reveals determinants of tumor nutrient availability. eLife 8, e44235 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kamphorst JJ et al. Human pancreatic cancer tumors are nutrient poor and tumor cells actively scavenge extracellular protein. Cancer Res. 75, 544–553 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lyssiotis CA & Kimmelman AC Metabolic Interactions in the Tumor Microenvironment. Trends in Cell Biology 27, 863–875 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Halbrook CJ & Lyssiotis CA Employing Metabolism to Improve the Diagnosis and Treatment of Pancreatic Cancer. Cancer Cell 31, 5–19 (2017). [DOI] [PubMed] [Google Scholar]
- 23.Ward PS & Thompson CB Metabolic reprogramming: a cancer hallmark even warburg did not anticipate. Cancer Cell 21, 297–308 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pavlova NN & Thompson CB The Emerging Hallmarks of Cancer Metabolism. Cell Metabolism 23, 27–47 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ying H et al. Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell 149, 656–670 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nagdas S et al. Drp1 Promotes KRas-Driven Metabolic Changes to Drive Pancreatic Tumor Growth. Cell Rep 28, 1845–1859.e5 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Perera RM & Bardeesy N Pancreatic Cancer Metabolism: Breaking It Down to Build It Back Up. Cancer Discov 5, 1247–1261 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hou P et al. Tumor Microenvironment Remodeling Enables Bypass of Oncogenic KRAS Dependency in Pancreatic Cancer. Cancer Discov 10, 1058–1077 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dey P et al. Oncogenic KRAS-Driven Metabolic Reprogramming in Pancreatic Cancer Cells Utilizes Cytokines from the Tumor Microenvironment. Cancer Discov 10, 608–625 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Patra KC et al. Mutant GNAS drives pancreatic tumorigenesis by inducing PKA-mediated SIK suppression and reprogramming lipid metabolism. Nat Cell Biol 20, 811–822 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stine ZE, Walton ZE, Altman BJ, Hsieh AL & Dang CV MYC, Metabolism, and Cancer. Cancer Discov 5, 1024–1039 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wirth M, Mahboobi S, Krämer OH & Schneider G Concepts to Target MYC in Pancreatic Cancer. Mol Cancer Ther 15, 1792–1798 (2016). [DOI] [PubMed] [Google Scholar]
- 33.Blake DR et al. Application of a MYC degradation screen identifies sensitivity to CDK9 inhibitors in KRAS-mutant pancreatic cancer. Sci Signal 12, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sodir NM et al. Myc instructs and maintains pancreatic adenocarcinoma phenotype. Cancer Discov (2020) doi: 10.1158/2159-8290.CD-19-0435. [DOI] [PubMed] [Google Scholar]
- 35.Muthalagu N et al. Repression of the Type I Interferon Pathway Underlies MYC- and KRAS-Dependent Evasion of NK and B Cells in Pancreatic Ductal Adenocarcinoma. Cancer Discov 10, 872–887 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bhattacharyya S et al. Acidic fibroblast growth factor underlies microenvironmental regulation of MYC in pancreatic cancer. J. Exp. Med 217, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kenzelmann Broz D et al. Global genomic profiling reveals an extensive p53-regulated autophagy program contributing to key p53 responses. Genes Dev 27, 1016–1031 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vousden KH & Ryan KM p53 and metabolism. Nature Reviews Cancer 9, 691–700 (2009). [DOI] [PubMed] [Google Scholar]
- 39.Cheung EC et al. Dynamic ROS Control by TIGAR Regulates the Initiation and Progression of Pancreatic Cancer. Cancer Cell (2020) doi: 10.1016/j.ccell.2019.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Morris JP et al. α-Ketoglutarate links p53 to cell fate during tumour suppression. Nature 573, 595–599 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hoxhaj G & Manning BD The PI3K–AKT network at the interface of oncogenic signalling and cancer metabolism. Nature Reviews Cancer 20, 74–88 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ying H et al. PTEN Is a Major Tumor Suppressor in Pancreatic Ductal Adenocarcinoma and Regulates an NF-κB–Cytokine Network. Cancer Discov 1, 158–169 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hill R et al. PTEN Loss Accelerates KrasG12D-Induced Pancreatic Cancer Development. Cancer Res 70, 7114–7124 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Baer R et al. Pancreatic cell plasticity and cancer initiation induced by oncogenic Kras is completely dependent on wild-type PI 3-kinase p110α. Genes Dev. 28, 2621–2635 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wu C-YC et al. PI3K Regulation of RAC1 is Required for KRAS-induced Pancreatic Tumorigenesis in Mice. Gastroenterology 147, 1405–1416.e7 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Palm W & Thompson CB Nutrient acquisition strategies of mammalian cells. Nature 546, 234–242 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Warburg O On the Origin of Cancer Cells. Science 123, 309–314 (1956). [DOI] [PubMed] [Google Scholar]
- 48.Patra KC & Hay N The pentose phosphate pathway and cancer. Trends Biochem. Sci 39, 347–354 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Akella NM, Ciraku L & Reginato MJ Fueling the fire: emerging role of the hexosamine biosynthetic pathway in cancer. BMC Biology 17, 52 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Newman AC & Maddocks ODK One-carbon metabolism in cancer. Br. J. Cancer 116, 1499–1504 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yang M & Vousden KH Serine and one-carbon metabolism in cancer. Nature Reviews Cancer 16, 650–662 (2016). [DOI] [PubMed] [Google Scholar]
- 52.Santana-Codina N et al. Oncogenic KRAS supports pancreatic cancer through regulation of nucleotide synthesis. Nat Commun 9, 4945 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Amendola CR et al. KRAS4A directly regulates hexokinase 1. Nature 576, 482–486 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Spinelli JB & Haigis MC The multifaceted contributions of mitochondria to cellular metabolism. Nat. Cell Biol 20, 745–754 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cunningham CN & Rutter J 20,000 picometers under the OMM: diving into the vastness of mitochondrial metabolite transport. EMBO Rep. e50071 (2020) doi: 10.15252/embr.202050071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bricker DK et al. A mitochondrial pyruvate carrier required for pyruvate uptake in yeast, Drosophila, and humans. Science 337, 96–100 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sullivan LB & Chandel NS Mitochondrial reactive oxygen species and cancer. Cancer Metab 2, 17 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Vasan K, Werner M & Chandel NS Mitochondrial Metabolism as a Target for Cancer Therapy. Cell Metabolism 0, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Altman BJ, Stine ZE & Dang CV From Krebs to clinic: glutamine metabolism to cancer therapy. Nat. Rev. Cancer 16, 619–634 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cluntun AA, Lukey MJ, Cerione RA & Locasale JW Glutamine Metabolism in Cancer: Understanding the Heterogeneity. Trends Cancer 3, 169–180 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kurmi K & Haigis MC Nitrogen Metabolism in Cancer and Immunity. Trends in Cell Biology 30, 408–424 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Harris IS & DeNicola GM The Complex Interplay between Antioxidants and ROS in Cancer. Trends in Cell Biology 0, (2020). [DOI] [PubMed] [Google Scholar]
- 63.Son J et al. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature 496, 101–105 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Raho S et al. KRAS-regulated glutamine metabolism requires UCP2-mediated aspartate transport to support pancreatic cancer growth. Nature Metabolism 2, 1373–1381 (2020). [DOI] [PubMed] [Google Scholar]
- 65.Bott AJ et al. Glutamine Anabolism Plays a Critical Role in Pancreatic Cancer by Coupling Carbon and Nitrogen Metabolism. Cell Rep 29, 1287–1298.e6 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yoo HC et al. A Variant of SLC1A5 Is a Mitochondrial Glutamine Transporter for Metabolic Reprogramming in Cancer Cells. Cell Metab. 31, 267–283.e12 (2020). [DOI] [PubMed] [Google Scholar]
- 67.Recouvreux MV et al. Glutamine depletion regulates Slug to promote EMT and metastasis in pancreatic cancer. J Exp Med 217, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kamphorst JJ et al. Human Pancreatic Cancer Tumors Are Nutrient Poor and Tumor Cells Actively Scavenge Extracellular Protein. Cancer Res 75, 544–553 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zhang Y & Commisso C Macropinocytosis in Cancer: A Complex Signaling Network. Trends Cancer 5, 332–334 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Santana-Codina N, Mancias JD & Kimmelman AC The Role of Autophagy in Cancer. Annu Rev Cancer Biol 1, 19–39 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kimmelman AC & White E Autophagy and Tumor Metabolism. Cell Metab. 25, 1037–1043 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Perera RM et al. Transcriptional control of autophagy-lysosome function drives pancreatic cancer metabolism. Nature 524, 361–365 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Amaravadi RK, Kimmelman AC & Debnath J Targeting Autophagy in Cancer: Recent Advances and Future Directions. Cancer Discov 9, 1167–1181 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Mancias JD & Kimmelman AC Mechanisms of Selective Autophagy in Normal Physiology and Cancer. J. Mol. Biol 428, 1659–1680 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Yang S et al. Pancreatic cancers require autophagy for tumor growth. Genes Dev. 25, 717–729 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Yang A et al. Autophagy Is Critical for Pancreatic Tumor Growth and Progression in Tumors with p53 Alterations. Cancer Discov (2014) doi: 10.1158/2159-8290.CD-14-0362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Rosenfeldt MT et al. p53 status determines the role of autophagy in pancreatic tumour development. Nature 504, 296–300 (2013). [DOI] [PubMed] [Google Scholar]
- 78.Elliott IA et al. Lysosome inhibition sensitizes pancreatic cancer to replication stress by aspartate depletion. PNAS 116, 6842–6847 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Yang A et al. Autophagy Sustains Pancreatic Cancer Growth through Both Cell-Autonomous and Nonautonomous Mechanisms. Cancer Discov 8, 276–287 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Guo JY et al. Autophagy provides metabolic substrates to maintain energy charge and nucleotide pools in Ras-driven lung cancer cells. Genes Dev. 30, 1704–1717 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Bhatt V et al. Autophagy modulates lipid metabolism to maintain metabolic flexibility for Lkb1-deficient Kras-driven lung tumorigenesis. Genes Dev. 33, 150–165 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Binnewies M et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nature Medicine 24, 541–550 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Pylayeva-Gupta Y, Lee KE, Hajdu CH, Miller G & Bar-Sagi D Oncogenic Kras-induced GM-CSF production promotes the development of pancreatic neoplasia. Cancer Cell 21, 836–847 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ryschich E et al. Control of T-Cell–Mediated Immune Response by HLA Class I in Human Pancreatic Carcinoma. Clin Cancer Res 11, 498–504 (2005). [PubMed] [Google Scholar]
- 85.Pommier A et al. Unresolved endoplasmic reticulum stress engenders immune-resistant, latent pancreatic cancer metastases. Science 360, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Yamamoto K et al. Autophagy promotes immune evasion of pancreatic cancer by degrading MHC-I. Nature 1–6 (2020) doi: 10.1038/s41586-020-2229-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Commisso C et al. Macropinocytosis of protein is an amino acid supply route in Ras-transformed cells. Nature 497, 633–637 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Amyere M et al. Constitutive macropinocytosis in oncogene-transformed fibroblasts depends on sequential permanent activation of phosphoinositide 3-kinase and phospholipase C. Mol. Biol. Cell 11, 3453–3467 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ramirez C, Hauser AD, Vucic EA & Bar-Sagi D Plasma membrane V-ATPase controls oncogenic RAS-induced macropinocytosis. Nature 576, 477–481 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lee S-W et al. EGFR-Pak Signaling Selectively Regulates Glutamine Deprivation-Induced Macropinocytosis. Developmental Cell 50, 381–392.e5 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Yao W et al. Syndecan 1 is a critical mediator of macropinocytosis in pancreatic cancer. Nature 568, 410–414 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Hobbs GA et al. Atypical KRASG12R Mutant Is Impaired in PI3K Signaling and Macropinocytosis in Pancreatic Cancer. Cancer Discov 10, 104–123 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Sahai E et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat. Rev. Cancer (2020) doi: 10.1038/s41568-019-0238-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Helms E, Onate MK & Sherman MH Fibroblast Heterogeneity in the Pancreatic Tumor Microenvironment. Cancer Discov (2020) doi: 10.1158/2159-8290.CD-19-1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Sousa CM et al. Pancreatic stellate cells support tumour metabolism through autophagic alanine secretion. Nature 536, 479–483 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Parker SJ et al. Selective alanine transporter utilization creates a targetable metabolic niche in pancreatic cancer. Cancer Discov (2020) doi: 10.1158/2159-8290.CD-19-0959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Auciello FR et al. A stromal lysolipid-autotaxin signaling axis promotes pancreatic tumor progression. Cancer Discov (2019) doi: 10.1158/2159-8290.CD-18-1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Shukla SK et al. MUC1 and HIF-1alpha Signaling Crosstalk Induces Anabolic Glucose Metabolism to Impart Gemcitabine Resistance to Pancreatic Cancer. Cancer Cell 32, 71–87.e7 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Dalin S et al. Deoxycytidine Release from Pancreatic Stellate Cells Promotes Gemcitabine Resistance. Cancer Res 79, 5723–5733 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Halbrook CJ et al. Macrophage-Released Pyrimidines Inhibit Gemcitabine Therapy in Pancreatic Cancer. Cell Metab. 29, 1390–1399.e6 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Shi Y et al. Targeting LIF-mediated paracrine interaction for pancreatic cancer therapy and monitoring. Nature 569, 131–135 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Garcia PE et al. Differential Contribution of Pancreatic Fibroblast Subsets to the Pancreatic Cancer Stroma. Cellular and Molecular Gastroenterology and Hepatology 0, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Hwang RF et al. Cancer-Associated Stromal Fibroblasts Promote Pancreatic Tumor Progression. Cancer Res 68, 918–926 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Francescone R et al. Netrin G1 promotes pancreatic tumorigenesis through cancer associated fibroblast driven nutritional support and immunosuppression. Cancer Discov (2020) doi: 10.1158/2159-8290.CD-20-0775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Ceyhan GO et al. Pancreatic neuropathy results in ‘neural remodeling’ and altered pancreatic innervation in chronic pancreatitis and pancreatic cancer. Am J Gastroenterol 104, 2555–2565 (2009). [DOI] [PubMed] [Google Scholar]
- 106.Liebl F et al. The impact of neural invasion severity in gastrointestinal malignancies: a clinicopathological study. Ann Surg 260, 900–907; discussion 907–908 (2014). [DOI] [PubMed] [Google Scholar]
- 107.Biankin AV et al. Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature 491, 399–405 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Renz BW et al. β2 adrenergic-neurotrophin feed-forward loop promotes pancreatic cancer. Cancer Cell 33, 75–90.e7 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Sinha S et al. PanIN Neuroendocrine Cells Promote Tumorigenesis via Neuronal Cross-talk. Cancer Res 77, 1868–1879 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Saloman JL et al. Ablation of sensory neurons in a genetic model of pancreatic ductal adenocarcinoma slows initiation and progression of cancer. Proc Natl Acad Sci U S A 113, 3078–3083 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Banh RS et al. Neurons Release Serine to Support mRNA Translation in Pancreatic Cancer. Cell 183, 1202–1218.e25 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Kedia-Mehta N & Finlay DK Competition for nutrients and its role in controlling immune responses. Nature Communications 10, 2123 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Ron-Harel N et al. T Cell Activation Depends on Extracellular Alanine. Cell Rep 28, 3011–3021.e4 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Geller LT et al. Potential role of intratumor bacteria in mediating tumor resistance to the chemotherapeutic drug gemcitabine. Science 357, 1156–1160 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Riquelme E et al. Tumor Microbiome Diversity and Composition Influence Pancreatic Cancer Outcomes. Cell 178, 795–806.e12 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Bapat AA, Hostetter G, Von Hoff DD & Han H Perineural invasion and associated pain in pancreatic cancer. Nature Reviews Cancer 11, 695–707 (2011). [DOI] [PubMed] [Google Scholar]
- 117.Demir IE, Friess H & Ceyhan GO Nerve-cancer interactions in the stromal biology of pancreatic cancer. Front. Physiol 3, (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Faulkner S, Jobling P, March B, Jiang CC & Hondermarck H Tumor Neurobiology and the War of Nerves in Cancer. Cancer Discov 9, 702–710 (2019). [DOI] [PubMed] [Google Scholar]
- 119.Bressy C et al. LIF Drives Neural Remodeling in Pancreatic Cancer and Offers a New Candidate Biomarker. Cancer Res. 78, 909–921 (2018). [DOI] [PubMed] [Google Scholar]
- 120.Wolpin BM et al. Phase II and pharmacodynamic study of autophagy inhibition using hydroxychloroquine in patients with metastatic pancreatic adenocarcinoma. Oncologist 19, 637–638 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Boone BA et al. Safety and Biologic Response of Pre-operative Autophagy Inhibition with Gemcitabine in Patients with Pancreatic Adenocarcinoma. Ann Surg Oncol 22, 4402–4410 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Karasic TB et al. Effect of Gemcitabine and nab-Paclitaxel With or Without Hydroxychloroquine on Patients With Advanced Pancreatic Cancer: A Phase 2 Randomized Clinical Trial. JAMA Oncol 5, 993–998 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Zeh H et al. A Randomized Phase II Preoperative Study of Autophagy Inhibition With High-Dose Hydroxychloroquine and Gemcitabine/Nab-Paclitaxel in Pancreatic Cancer Patients. Clin Cancer Res (2020) doi: 10.1158/1078-0432.CCR-19-4042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Bryant KL et al. Combination of ERK and autophagy inhibition as a treatment approach for pancreatic cancer. Nat. Med 25, 628–640 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Kinsey CG et al. Protective autophagy elicited by RAF→MEK→ERK inhibition suggests a treatment strategy for RAS-driven cancers. Nat. Med 25, 620–627 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Lee C-S et al. MAP kinase and autophagy pathways cooperate to maintain RAS mutant cancer cell survival. Proc. Natl. Acad. Sci. U.S.A 116, 4508–4517 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.University of Utah. A Phase I Trial of Ulixertinib (BVD-523) and Hydroxychloroquine in Patients With Advanced MAPK-Mutated Gastrointestinal Adenocarcinomas. https://clinicaltrials.gov/ct2/show/NCT04145297 (2020).
- 128.University of Utah. THREAD: A Phase I Trial of Trametinib and Hydroxychloroquine in Patients With Advanced Pancreatic Cancer. https://clinicaltrials.gov/ct2/show/NCT03825289 (2020).
- 129.M.D. Anderson Cancer Center. Binimetinib Plus Hydroxychloroquine in KRAS Mutant Pancreatic Cancer. https://clinicaltrials.gov/ct2/show/NCT04132505 (2019).
- 130.Biancur DE et al. Compensatory metabolic networks in pancreatic cancers upon perturbation of glutamine metabolism. Nat Commun 8, 15965 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Lemberg KM, Vornov JJ, Rais R & Slusher BS We’re Not “DON” Yet: Optimal Dosing and Prodrug Delivery of 6-Diazo-5-oxo-L-norleucine. Mol Cancer Ther 17, 1824–1832 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Sharma NS et al. Targeting tumor-intrinsic hexosamine biosynthesis sensitizes pancreatic cancer to anti-PD1 therapy. J. Clin. Invest 130, 451–465 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Leone RD et al. Glutamine blockade induces divergent metabolic programs to overcome tumor immune evasion. Science 366, 1013–1021 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Oh M-H et al. Targeting glutamine metabolism enhances tumor specific immunity by modulating suppressive myeloid cells. J. Clin. Invest (2020) doi: 10.1172/JCI131859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Spinelli JB & Haigis MC The multifaceted contributions of mitochondria to cellular metabolism. Nat. Cell Biol 20, 745–754 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Viale A et al. Oncogene ablation-resistant pancreatic cancer cells depend on mitochondrial function. Nature 514, 628–632 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.MacVicar T et al. Lipid signalling drives proteolytic rewiring of mitochondria by YME1L. Nature 575, 361–365 (2019). [DOI] [PubMed] [Google Scholar]
- 138.Yu M et al. Mitochondrial fusion exploits a therapeutic vulnerability of pancreatic cancer. JCI Insight 5, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.MacVicar T et al. Lipid signalling drives proteolytic rewiring of mitochondria by YME1L. Nature 575, 361–365 (2019). [DOI] [PubMed] [Google Scholar]
- 140.Zachar Z et al. Non-redox-active lipoate derivates disrupt cancer cell mitochondrial metabolism and are potent anticancer agents in vivo. J Mol Med 89, 1137 (2011). [DOI] [PubMed] [Google Scholar]
- 141.Retter AS Translational assessment of the efficacy of CPI-613 against pancreatic cancer in animal models versus patients with stage IV disease. JCO 30, 3075–3075 (2012). [Google Scholar]
- 142.Alistar A et al. A phase 1 study of first-in-class agent CPI-613 in combination with FOLFIRINOX for metastatic pancreatic cancer. Lancet Oncol 18, 770–778 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Lycan TW et al. A Phase II Clinical Trial of CPI-613 in Patients with Relapsed or Refractory Small Cell Lung Carcinoma. PLoS One 11, e0164244 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Philip PA et al. A Phase III open-label trial to evaluate efficacy and safety of CPI-613 plus modified FOLFIRINOX (mFFX) versus FOLFIRINOX (FFX) in patients with metastatic adenocarcinoma of the pancreas. Future Oncology 15, 3189–3196 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Rafael Pharmaceuticals Inc. Phase III Multicenter Open-Label Randomized Trial to Evaluate Efficacy and Safety of CPI-613® (Devimistat) in Combination With High Dose Cytarabine and Mitoxantrone (CHAM) Compared to High Dose Cytarabine and Mitoxantrone (HAM) Therapy and Control Sub-groups: Combination of Mitoxantrone, Etoposide and Cytarabine (MEC) and Combination of Fludarabine, Cytarabine, and Filgrastim (FLAG) in Older Patients (≥ 50 Years) With Relapsed/Refractory Acute Myeloid Leukemia (AML). https://clinicaltrials.gov/ct2/show/NCT03504410 (2020). [Google Scholar]
- 146.Harris IS & DeNicola GM The Complex Interplay between Antioxidants and ROS in Cancer. Trends Cell Biol. 30, 440–451 (2020). [DOI] [PubMed] [Google Scholar]
- 147.Daher B et al. Genetic ablation of the cystine transporter xCT in PDAC cells inhibits mTORC1, growth, survival and tumor formation via nutrient and oxidative stresses. Cancer Res (2019) doi: 10.1158/0008-5472.CAN-18-3855. [DOI] [PubMed] [Google Scholar]
- 148.Stockwell BR, Jiang X & Gu W Emerging Mechanisms and Disease Relevance of Ferroptosis. Trends in Cell Biology 30, 478–490 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Badgley MA et al. Cysteine depletion induces pancreatic tumor ferroptosis in mice. Science 368, 85–89 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Cramer SL et al. Systemic depletion of L-cyst(e)ine with cyst(e)inase increases reactive oxygen species and suppresses tumor growth. Nat. Med 23, 120–127 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Hui S et al. Glucose feeds the TCA cycle via circulating lactate. Nature 551, 115–118 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Faubert B et al. Lactate Metabolism in Human Lung Tumors. Cell 171, 358–371.e9 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Maher EA et al. Metabolism of [U-13 C]glucose in human brain tumors in vivo. NMR Biomed 25, 1234–1244 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Biancur DE et al. Functional Genomics Identifies Metabolic Vulnerabilities in Pancreatic Cancer. Cell Metabolism 33, 199–210.e8 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Zhu XG et al. Functional Genomics In Vivo Reveal Metabolic Dependencies of Pancreatic Cancer Cells. Cell Metabolism 33, 211–221.e6 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Biancur DE et al. Functional Genomics Identifies Metabolic Vulnerabilities in Pancreatic Cancer. Cell Metab (2020) doi: 10.1016/j.cmet.2020.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Bian Y, Yu Y, Wang S & Li L Up-regulation of fatty acid synthase induced by EGFR/ERK activation promotes tumor growth in pancreatic cancer. Biochemical and Biophysical Research Communications 463, 612–617 (2015). [DOI] [PubMed] [Google Scholar]
- 158.Guillaumond F et al. Cholesterol uptake disruption, in association with chemotherapy, is a promising combined metabolic therapy for pancreatic adenocarcinoma. Proc. Natl. Acad. Sci. U.S.A 112, 2473–2478 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Oni TE et al. SOAT1 promotes mevalonate pathway dependency in pancreatic cancer. J Exp Med 217, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Olivares O et al. Collagen-derived proline promotes pancreatic ductal adenocarcinoma cell survival under nutrient limited conditions. Nature Communications 8, 16031 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Lyssiotis CA & Kimmelman AC Metabolic Interactions in the Tumor Microenvironment. Trends Cell Biol. 27, 863–875 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Lau AN et al. Dissecting cell type-specific metabolism in pancreatic ductal adenocarcinoma. eLife 9, e56782 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Chen WW, Freinkman E, Wang T, Birsoy K & Sabatini DM Absolute Quantification of Matrix Metabolites Reveals the Dynamics of Mitochondrial Metabolism. Cell 166, 1324–1337.e11 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Chen WW, Freinkman E & Sabatini DM Rapid immunopurification of mitochondria for metabolite profiling and absolute quantification of matrix metabolites. Nat Protoc 12, 2215–2231 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Bayraktar EC et al. MITO-Tag Mice enable rapid isolation and multimodal profiling of mitochondria from specific cell types in vivo. Proc Natl Acad Sci U S A 116, 303–312 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Zoncu R et al. mTORC1 Senses Lysosomal Amino Acids Through an Inside-Out Mechanism That Requires the Vacuolar H+-ATPase. Science 334, 678–683 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Abu-Remaileh M et al. Lysosomal metabolomics reveals V-ATPase- and mTOR-dependent regulation of amino acid efflux from lysosomes. Science 358, 807–813 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Tuveson D & Clevers H Cancer modeling meets human organoid technology. Science 364, 952–955 (2019). [DOI] [PubMed] [Google Scholar]