

Abstract
Individuals with posttraumatic stress disorder (PTSD) are at increased risk for the development of various forms of dementia. Nevertheless, the neuropathological link between PTSD and neurodegeneration remains unclear. Degeneration of the human basal forebrain constitutes a pathological hallmark of neurodegenerative diseases, such as Alzheimer's and Parkinson's disease. In this seed‐based resting‐state (rs‐)fMRI study identifying as outcome measure the temporal BOLD signal fluctuation magnitude, a seed‐to‐voxel analyses assessed temporal correlations between the average BOLD signal within a bilateral whole basal forebrain region‐of‐interest and each whole‐brain voxel among individuals with PTSD (n = 65), its dissociative subtype (PTSD+DS) (n = 38) and healthy controls (n = 46). We found that compared both with the PTSD and healthy controls groups, the PTSD+DS group exhibited increased BOLD signal variability within two nuclei of the seed region, specifically in its extended amygdaloid region: the nucleus accumbens and the sublenticular extended amygdala. This finding is provocative, because it mimics staging models of neurodegenerative diseases reporting allocation of neuropathology in early disease stages circumscribed to the basal forebrain. Here, underlying candidate etiopathogenetic mechanisms are neurovascular uncoupling, decreased connectivity in local‐ and large‐scale neural networks, or disrupted mesolimbic dopaminergic circuitry, acting indirectly upon the basal forebrain cholinergic pathways. These abnormalities may underpin reward‐related deficits representing a putative link between persistent traumatic memory in PTSD and anterograde memory deficits in neurodegeneration. Observed alterations of the basal forebrain in the dissociative subtype of PTSD point towards the urgent need for further exploration of this region as a potential candidate vulnerability mechanism for neurodegeneration in PTSD.
Keywords: basal forebrain, dissociative subtype, extended amygdaloid area, local BOLD hemodynamic signal variability, neurodegenerative illnesses, posttraumatic stress disorder, resting‐state functional magnetic resonance imaging
In this seed‐based resting‐state (rs‐)fMRI study we investigated the role of the human basal forebrain in PTSD, as a putative neuroimaging marker of PTSD‐related neurodegeneration. We observed an increased hemodynamic BOLD signal variability within the basal forebrain of PTSD subtype dissociative group, which mimics previous reports of dysfunction and degeneration of the basal forebrain in early phases of neurodegenerative diseases such as Alzheimer's disease. Observed alterations of the basal forebrain in the dissociative subtype of PTSD point toward the urgent need for further exploration of this region as a potential candidate vulnerability mechanism for neurodegeneration in PTSD.
1. INTRODUCTION
Epidemiological studies in military and civilian populations of both sexes provide clinical and population‐level evidence for a link between posttraumatic stress disorder (PTSD) and increased risk for the development of various forms of dementia (Flatt, Gilsanz, Quesenberry, Albers, & Whitmer, 2018; Qureshi et al., 2010; Weiner et al., 2013; Yaffe et al., 2010). In a recent stratified, retrospective cohort study including a total of 181,093 male veterans 55 years or older, when compared with those without PTSD, veterans diagnosed with PTSD showed a nearly two‐fold increased risk of developing Alzheimer's disease (AD), frontotemporal dementia (FTD), Lewy Body dementia (LBD), vascular dementia (VaD), and senile dementia (SN) (Yaffe et al., 2010). These rates were replicated in another stratified, retrospective cohort study assessing the prevalence of AD, VaD, and other nonspecific dementia in 499,844 civilians of both sexes 60 years or older (Flatt et al., 2018). In addition, a longitudinal study including 7,280 civilians of both sexes 45 years or older, demonstrated a nearly four‐fold increased risk of developing Parkinson's disease (PD) in subjects diagnosed with PTSD (Chan et al., 2017).
Nevertheless, the neuropathological link between PTSD and dementia remains to be identified (Greenberg, Tanev, Marin, & Pitman, 2014). Several candidate etiopathogenetic mechanisms have been proposed due to their presence in both PTSD and neurodegenerative diseases. These include hippocampal atrophy, alterations of the hypothalamic–pituitary–adrenal (HPA)‐axis activity, and neuroendocrine‐immune interactions (Greenberg et al., 2014; Herbert et al., 2006; Pitman, 2010; Yaffe et al., 2010). However, careful analysis demonstrates that the mechanistic underpinnings of these processes in PTSD and neurodegenerative diseases are dissimilar and not associated (Greenberg et al., 2014; Pitman, 2010), pointing towards the need for identification of alternative mechanisms that may form, in part, the neurobiological underpinnings of PTSD‐related neurodegeneration.
Here, we investigate patterns of resting‐state activity of the basal forebrain in PTSD, a potential imaging marker of vulnerability for neurodegeneration in this population. Critically, the basal forebrain emerges as one of the first brain regions to show neuropathology in association with early asymptomatic or prodromal phases of neurodegenerative diseases such as AD (Arendt, Brückner, Morawski, Jäger, & Gertz, 2015; Mesulam, Shaw, Mash, & Weintraub, 2004; Nie et al., 2017; Schmitz et al., 2016; Schmitz et al., 2018; Teipel et al., 2014; Yi et al., 2016), PD (Bohnen & Albin, 2011; Zeighami et al., 2015), and LBD (Grothe et al., 2014). Emerging evidence supports the use of resting‐state fMRI as an imaging biomarker of neurodegenerative diseases (Cope et al., 2018; Hampton et al., 2020; Hohenfeld, Werner, & Reetz, 2018; Wisch et al., 2020) suggesting that in early, preclinical phases of neurodegenerative diseases, moment‐to‐moment variability in BOLD signal at rest may be related to the load of neurodegeneration associated neuropathology (Millar et al., 2020), and may signal either abnormalities in neurovascular coupling (Hillman, 2014; Solis, Hascup, & Hascup, 2020) or alterations in functional connectivity specifically associated with the basal forebrain (Chiesa et al., 2019; Wang, Belden, Hanser, Geddes, & Loui, 2020).
Although neurodegenerative diseases such as AD are robustly associated with alterations in the basal forebrain magnocellular cholinergic nuclei (Hampel et al., 2018; Mesulam, 2013; Schmitz et al., 2016), and PTSD in the basal forebrain extended amygdaloid region (Gerlicher, Tüscher, & Kalisch, 2019; Liberzon & Garfinkel, 2009), emerging evidence is suggesting that both conditions share structural and functional neuropathology affecting the extended amygdala nucleus accumbens (NAc) (Arendt et al., 2015; Lieberman, Gorka, Funkhouser, Shankman, & Phan, 2017; Zeighami et al., 2015).
Importantly, structural and functional alterations of basal forebrain extended amygdaloid region NAc in PTSD are subtype‐specific (Liberzon & Garfinkel, 2009; Lieberman et al., 2017). Specifically, alterations of the basal forebrain NAc are seldom observed in the classical PTSD presentation (Logue et al., 2018), with only one study reporting an increase in their BOLD signal among such patients showing severe hyperarousal symptoms (Pavic et al., 2003). By contrast, recurrent decreased activation or degeneration of EANAc nuclei has been reported among patients with the dissociative subtype of PTSD (PTSD+DS) (Felmingham et al., 2014; Green, Corsi‐Travali, & Neumeister, 2013; Lieberman et al., 2017; Ospina et al., 2019), a presentation apparent in ~14–30% of patients with PTSD (Stein et al., 2013; Steuwe, Lanius, & Frewen, 2012; Wolf et al., 2012), and characterized by symptoms of depersonalization and derealization (Frewen & Lanius, 2013; van Huijstee & Vermetten, 2018). Critically, cognitive dysfunction, similar in pattern, although of lesser magnitude to neurodegenerative conditions, is strongly associated with dissociative symptomatology, the hallmark presentation of this subtype of PTSD (Boyd et al., 2018).
Structural and functional pathology of the NAc in PTSD+DS is often reported in association with a plethora of signs and symptoms characterizing its presentation, such as blunted reward processing (Lieberman et al., 2017) associated with fear memory processes (Izquierdo, Furini, & Myskiw, 2016), as well as hedonic and motivational deficits, including anhedonia (Klawonn & Malenka, 2018; Olson, Kaiser, Pizzagalli, Rauch, & Rosso, 2018; Russo & Nestler, 2013) and apathy (Pineau, Marchand, & Guay, 2015; Sailer et al., 2008). These (Pineau et al., 2015; Sailer et al., 2008) are concurrent with heightened signs of fear and anxiety belonging to PTSD+DS presentation, such as more freezing and fewer vocalizations (Shackman & Fox, 2016).
Similarly, NAc structural and functional neuropathology underlying neurodegenerative diseases such as AD and PD is characterized by the severe neuronal loss (Lehéricy et al., 1989; Oyanagi et al., 1994) an aberrant reduction of the NAc's volume and shape (Carriere et al., 2014; Halabi et al., 2013; Pievani et al., 2013), decreased regional cerebral blood flow (rCBF) (Jeong et al., 2018) and decreased resting‐state functional connectivity (Wang et al., 2020). Anatomical studies of the NAc postmortem have further revealed pathophysiological markers of neurodegeneration such as aggregates of neurofibrillary tangles (Kawakami et al., 2014; Selden, Mesulam, & Geula, 1994). In a pattern similar to that observed for PTSD+DS, these abnormalities have been associated with underlying reward‐based cognitive and behavioral deficits, including apathy and aberrant fear memory in AD (Jeong et al., 2018; Nasrouei, Rattel, Liedlgruber, Marksteiner, & Wilhelm, 2020; Nobili et al., 2017) and PD (Carriere et al., 2014; Kinoshita, Tada, Muroi, Unno, & Ishii, 2015; Muhammed et al., 2016).
These observed commonalities in not only underlying disease pathogenesis but also the clinical presentation of reward deficits typically associated with dysfunctional EA activity across the dissociative subtype of PTSD and several neurodegenerative conditions, suggests that abnormal structure or functioning of the human basal forebrain EA nuclei, specifically the NAc, may predict vulnerability to neurodegeneration in PTSD, a pattern most likely to be observed in the dissociative subtype PTSD (Bohnen & Albin, 2011; Carriere et al., 2014; Grothe et al., 2014; Izquierdo et al., 2016; Jeong et al., 2018; Klawonn & Malenka, 2018; Liberzon & Garfinkel, 2009; Pineau et al., 2015; Ryle, Court, Smith, & Morris, 1975; Sailer et al., 2008; Schmitz et al., 2016; Zeighami et al., 2015).
Here, we examined the resting state activity of the basal forebrain in PTSD (n = 65), its dissociative subtype (PTSD+DS) (n = 38), and healthy controls (n = 46). Following the hypothesis stated above, we predicted that in the present study, we would observe an abnormal pattern of resting‐state activity of the basal forebrain extended amygdaloid area (EA), which would manifest as an abnormal increase in BOLD signal variability at the rest of the NAc in the PTSD+DS patient group.
2. METHODS
2.1. Participants
One‐hundred and forty‐nine age‐ and sex‐matched subjects were included in the study: 65 patients with a primary diagnosis of PTSD without the dissociative subtype (PTSD), 38 patients with a primary diagnosis of PTSD with the dissociative subtype (PTSD+DS), and 46 healthy control individuals. Of these, 83.5% of the patients with PTSD (PTSD‐DS and PTSD+DS) met the criteria for interpersonal childhood trauma according to responses on the Childhood Trauma Questionnaire (CTQ) (Bernstein et al., 1994; DiLillo et al., 2006). Participants were recruited from 2009 to 2016 via referrals from family physicians, mental health professionals, psychology/psychiatry clinics, community programs for traumatic‐stress survivors and posters/advertisements within the London, Ontario community.
A primary PTSD diagnosis was determined using the CAPS‐4 (n = 122) or CAPS‐5 (n = 27) (Clinician‐Administered PTSD Scale; CAPS‐4 cut‐off score 50) (Blake et al., 1995). As per standard methods, PTSD patients with the dissociative subtype were further required to score at least “2” on both the frequency and intensity scales assessing depersonalization and derealization symptoms (Nicholson et al., 2015). For each participant, comorbid Axis‐I disorders were assessed using the SCID (structured clinical interview for DSM‐IV Axis I disorders). A battery of questionnaires was also administered, including the beck depression inventory (BDI) (Beck, Guth, Steer, & Ball, 1997), the child trauma questionnaire (CTQ) (Bernstein et al., 1994), and the multiscale dissociative inventory (MDI) (Briere, Weathers, & Runtz, 2005). State reliving and depersonalization/derealization symptoms experienced during the resting state scan were assessed using the response to script driven imagery scale (RSDI) (Hopper, Frewen, van der Kolk, & Lanius, 2007; Mickleborough et al., 2011), adapted to resting state.
Exclusion criteria for all participants included the presence of metal implants that violated 3.0 T scanner safety regulations, a previous head injury associated with loss of consciousness, current or past history of neurological disorders, significant untreated medical illness, and pervasive developmental mental disorders. Patients with PTSD were excluded further if they met the criteria for current or past history of bipolar or psychotic disorders, or if patients had alcohol/substance dependency or abuse that had not sustained full remission for at least 6 months before study entry. Control participants were excluded if lifetime criteria were met for any Axis‐I psychiatric disorder.
All scanning took place at Robarts Research Institute's Center for Functional and Metabolic Mapping or at Lawson Health Research Institute in London, Ontario, Canada. The study was approved by the Research Ethics Board of the Western University of Canada. All participants provided written informed consent to partake in the study.
2.2. Data acquisition
Whole‐brain fMRI (functional magnetic resonance imaging) data were obtained using a 3.0 T scanner (Magnetom Tim Trio, Siemens Medical Solutions, Erlangen, Germany) with a 32‐channel phased‐array head coil where the participant's head was supported with foam padding. BOLD (blood‐oxygen level‐dependent) fMRI data were collected using a manufacturer's standard gradient‐echo planar imaging (EPI) pulse sequence (single‐shot, blipped‐EPI) with an interleaved slice acquisition order with the following parameters: time resolution (TR) = 3,000 ms, echo‐time (TE) = 20 ms, voxel size = 2 × 2 × 2 mm3, Field of View (FOV) = 192 × 192 × 128 mm3 (94 × 94 matrix, 64 contiguous slices), flip angle (FA) =90°. High‐resolution T1‐weighted anatomical images were also obtained (MP‐RAGE: 192 slices, voxel size = 1 × 1 × 1 mm3). Resting‐state data were obtained for 6 min according to standard methods (Bluhm et al., 2009; Fransson, 2005).
2.3. Resting‐state fMRI data preprocessing
Image pre‐processing was performed with CONN v.18.a toolbox (The Gabrieli Lab, McGovern Institute for Brain Research, Cambridge, MA) for MATLAB R2018a (Mathworks Inc., MA). The functional images for each subject were realigned to the mean fMRI image using a 6‐parameter rigid‐body transformation (6 time series containing the three translation and three rotation parameters over time for each subject and session) to correct for subject head motion. Moreover, in order to address microhead motion (<0.1 mm), an important source of spurious correlations in resting‐state functional connectivity analysis (Van Dijk, Sabuncu, & Buckner, 2012), further removal of motion artifacts was performed through the artifact detection tool (ART), as implemented in the CONN toolbox preprocessing routine, censoring procedure of volumes where high‐motion was detected, the so‐called “scrubbing” strategy (Power et al., 2014). Functional images were segmented simultaneously into gray matter, white matter, and cerebrospinal fluid (CSF) and normalized to the MNI space. The structural images were also segmented simultaneously into gray matter, white matter, and CSF and normalized to the MNI space. The functional images were then smoothed with a three‐dimensional isotropic Gaussian kernel of 8 mm FWHM (full‐width at half maximum). The smoothed functional images were subsequently denoised with the Compcor method, which regresses out of the data the white matter and CSF time courses. Smoothed and denoised functional images were bandpass‐filtered to reduce the signal‐to‐noise ratio using 0.012 and 0.1 Hz as the high‐pass and low‐pass frequency cut‐offs, respectively.
2.4. Seed‐based region of interest
The current study employed a stereotactic mask of the bilateral basal forebrain developed at the German Center for Neurodegenerative Diseases, Rostock, Germany, in conformity to the standard MNI152 template (1 mm), and designed after the coordinates and anatomical landmarks reported in (Teipel et al., 2005).
2.5. Seed‐based analysis
Connectivity analyses were performed using the CONN v.18.a toolbox (The Gabrieli Lab, McGovern Institute for Brain Research, Cambridge, MA) for MATLAB R2018a (Mathworks Inc., MA). Individual seed‐based connectivity (SBC) maps representing the level of functional connectivity between the seed located at the bilateral basal forebrain and every voxel in the brain were computed as the Fisher‐transformed bivariate correlation coefficient between the seed average BOLD time series and each whole‐brain voxel BOLD time series (Z‐values, the inverse hyperbolic tangent values of the correlation coefficient).
2.6. Second‐level analysis
Second‐level statistical analyses was performed using statistical parametric mapping software (SPM12, Wellcome Trust Center for Neuroimaging, London, UK: http:// www.fil.ion.ucl.ac.uk/spm) within MATLAB R2018a (Mathworks Inc., MA). The Fischer's Z‐transformed correlations between the seed BOLD time series (averaged across all voxels within the bilateral basal forebrain mask) and the time series of all other individual voxels in the brain were used to compute functional connectivity differences between groups. A one‐way analysis of variance (ANOVA) including three groups, PTSD, PTSD dissociative subtype (PTSD+DS) and healthy controls, was conducted for the second‐level analyses. As our groups were age‐ and sex‐matched, and exhibited an equivalent medication status (past and current), only motion parameters (6‐parameter rigid‐body transformation) were included as nuisance covariates in our analyses. To determine significant correlations, results were explored at the whole‐brain activation level at voxel‐level height‐threshold fwe‐corrected p‐value <.05 (Table 1).
TABLE 1.
Medication status previous to and at the scanning moment, absolute figures of patient per group
| PTSD | PTSD + DS | ||||
|---|---|---|---|---|---|
| Past | Present | Past | Present | ||
| Selective serotonin reuptake inhibitor (SSRI) | |||||
| Citalopram | 3 | 2 | 1 | 1 | |
| Fluoxetine | 4 | 1 | 2 | 3 | |
| Fluvoxamine | 1 | ‐ | ‐ | ‐ | |
| Paroxetine | 3 | ‐ | ‐ | ‐ | |
| Sertraline | 3 | 2 | 1 | ‐ | |
| Serotonin antagonist and reuptake inhibitor (SARI) | |||||
| Trazodone | 1 | 3 | 1 | 2 | |
| Serotonin‐norepinephrine reuptake inhibitor (SNRI) | |||||
| Venlafaxine | 7 | 5 | 1 | ‐ | |
| Norepinephrine‐dopamine reuptake inhibitor (NDRI) and nicotinic receptor antagonist | |||||
| Bupropion | 2 | 1 | 5 | 1 | |
| Reversible inhibitor of monoamine oxidase‐A (RIMA) | |||||
| Moclobemide | 1 | ‐ | ‐ | ‐ | |
| Tricyclic antidepressant (TCA) | |||||
| Amitriptyline | 3 | ‐ | ‐ | ‐ | |
| Protriptyline | 1 | ‐ | ‐ | ‐ | |
| Dopamine and serotonin receptors blocker | |||||
| Olanzapine | 1 | ‐ | ‐ | ‐ | |
| Quetiapine | 4 | 1 | 2 | ‐ | |
| Dopamine and serotonin antagonist | |||||
| Risperidone | 2 | 1 | ‐ | ‐ | |
| Dibenzoxazepine | |||||
| Loxapine | 1 | ‐ | ‐ | ‐ | |
| CYP3A4 inhibitor | |||||
| Haloperidol | ‐ | 1 | ‐ | ‐ | |
| Dopamine and norepinephrine reuptake inhibitor | |||||
| Methylphenidate | 1 | ‐ | ‐ | ‐ | |
| Serotonin, dopamine, and norepinephrine transporters inhibition or reversion | |||||
| Dextroamphetamine | 1 | ‐ | ‐ | ‐ | |
| Benzodiazepine | |||||
| Clonazepan | ‐ | 1 | 1 | 2 | |
| Diazepam | ‐ | 1 | ‐ | ‐ | |
| Lorazepam | 2 | 1 | 3 | 2 | |
| Temazepam | 3 | ‐ | ‐ | ‐ | |
| Triazolobenzodiazepine (TBZD) | |||||
| Alprazolam | 1 | ‐ | ‐ | ‐ | |
| Nonbenzodiazepine | |||||
| Zopiclone | 2 | 1 | 3 | 1 | |
Abbreviations: PTSD, nondissociative PTSD group; PTSD+DS, dissociative subtype PTSD group.
2.7. Demographic and psychological measures
Quantile‐Quantile plots demonstrated that participants' ages across all three groups were not normally distributed. Accordingly, a Kruskall‐Wallis test was performed to assess age differences across participant groups, and to ensure that groups were age‐matched. A Pearson's Chi‐square test assessed sex differences between groups, as well as differences between medication status, past and current.
When assessing potential between‐group differences in psychological measures, Levene's test of homogeneity of variances demonstrated that the principle of homogeneity of variances was violated in all tested measures. As such, a one‐way between‐groups analysis of variance (ANOVA), followed by post hoc Games‐Howell testing, was used to assess between‐group differences in total CAPS‐IV scores, averaged MDI scores for trait depersonalization, and derealization, state reliving, depersonalization, derealization and numbness RSDI scores, BDI and CTQ scores.
2.8. Correlational analysis
Using SPM12 multiple regression analyses, CAPS‐IV total scores, averaged MDI depersonalization and derealization scores, state reliving, depersonalization/derealization and numbness (during the resting state scan) RSDI scores, were assessed as predictors of basal forebrain connectivity among individuals in the PTSD and PTSD+DS groups.
3. RESULTS
The primary finding of our study was increased BOLD signal variability at rest within the seed region, the basal forebrain, specifically, at the extended amygdaloid area (EA) nuclei, the nucleus accumbens (NAc) and the sublenticular extended amygdala (SLEA), in the dissociative subtype PTSD group. No such pattern was observed in the comparison groups.
3.1. Between‐group analysis of clinical variables scores at the behavioral level
A one‐way between‐group ANOVA, followed by a post hoc Games‐Howell test given the heterogeneity of variance, was performed on CAPS, CTQ, and BID scores. For all three variables, the respective analysis yielded significant differences between all three groups. For the CAPS scores, the PTSD+DS group exhibited higher scores than the PTSD (p = .018) and the control (p < .001) groups. The PTSD group exhibited lower scores than the PTSD+DS group and higher scores than the control group only (p < .001). Likewise, for the CTQ variables, higher scores were observed in the PTSD+DS group as compared to the PTSD group (p = .002) and the control group (p < .001). The PTSD group exhibited lower scores than the PTSD+DS group and higher scores than the control group only (p < .001). This pattern of results was replicated for the BDI scores, where the post hoc Games‐Howell testing yielded higher scores in the PTSD+DS group as compared to the PTSD group (p = .001) and the control group (p < 0.001). The PTSD group exhibited higher scores than the control group only (p < 0.001).
A one‐way between‐group ANOVA performed on the averaged RSDI item assessing numbness symptoms during the resting state scan also violated the assumption of homogeneity of variance. Follow‐up post hoc Games‐Howell testing revealed a significant difference between the control and PTSD groups. Specifically, both the PTSD+DS (n = 38) (p < 0.001) and PTSD (n = 61) (p < .001) groups exhibited higher scores as compared to the control group (n = 45).
3.2. Within‐group results
Within‐group analyses revealed abundant, widespread connectivity of the bilateral basal forebrain with the frontal lobe in the control and PTSD groups, a pattern not present in the PTSD+DS group.
3.3. Within‐group results: Healthy controls
The control and PTSD groups showed a highly overlapping functional connectivity pattern. Analysis of healthy control data at the whole‐brain level (pfwe = 0.05) revealed increased connectivity of the bilateral basal forebrain with the bilateral nucleus accumbens (NAc) (controls MNI = ±8 6–12, p < 0.001, k = 1927).
3.4. Within‐group results: PTSD
In the PTSD group, a whole‐brain analysis (p fwe = .05) revealed increased functional connectivity of the bilateral basal forebrain with the bilateral NAc (MNI = ±8 6–12, pfwe < .001, k = 2,476). Moreover, increased connectivity with the left anterior cingulate cortex (MNI = −8 48 –4, p fwe < .001, k = 161), a cluster extending into the right anterior prefrontal cortex (MNI = 4 56 –4, p fwe = .002; MNI = 8 46 –8, p fwe = .004), was observed. Finally, increased connectivity was also observed between the bilateral basal forebrain and the right parahippocampal area (MNI = 24 –24 −12, p fwe = .003, k = 23).
3.5. Within‐group results: PTSD + DS
In the PTSD subtype dissociative patient group (PTSD+DS), a whole‐brain analysis (p fwe = .05) did not yield any significant results (Tables 2 and 3).
TABLE 2.
Demographics and clinical variables statistics
| (a) Demographics | |||
|---|---|---|---|
| Measure | PTSD | PTSD+DS | Control |
| M ± SD | M ± SD | M ± SD | |
| Age | 37.89 ± 12.04 | 40.55 ± 13.68 | 34.82 ± 11.96 |
| Sex | 43 F/22 M | 31 F/7 M | 32 F/14 M |
| CAPS‐4 | 67.3 ± 13.1 | 81.55 ± 13.12 | 0.9 ± 3.35 |
| CAPS‐5 | 34 ± 9.22 | 36.77 ± 7.85 | ‐ |
| CTQ | 58.35 ± 22.62 | 69.27 ± 18.64 | 32.33 ± 9.35 |
| BDI | 23.46 ± 8.04 | 32.85 ± 13.39 | 1.13 ± 2.05 |
| MDI DepDer | 7.92 ± 2.7 | 12.38 ± 4.1 | 5.22 ± 0.55 |
| RSDI DepDer | 3.48 ± 1.35 | 4.94 ± 2.07 | 2.67 ± 0.49 |
| RSDI reliving | 2.82 ± 1.23 | 3.26 ± 1.42 | 2.04 ± 0.3 |
| RSDI numbness | 1.60 ± 0.88 | 1.92 ± 0.93 | 1.11 ± 0.32 |
| (b) Clinical variables statistics | |||
|---|---|---|---|
| Variable | Levene's test | ANOVA | Games‐Howell M ± SE |
| CAPS‐4 | F(2,120) = 26.398; p < .001 | ||
|
PTSD and PTSD+DS PTSD and control PTSD+DS and control |
NA |
Mean diff = 11.55 ± 4.1; p = .018 Mean diff = 66.65 ± 1.9; p < .001 Mean diff = 78.2 ± 3.6; p < .001 |
|
| CTQ | F(2,137) = 19.41; p < .001 | NA | |
|
PTSD and PTSD+DS PTSD and control PTSD+DS and control |
Mean diff = 14.8 ± 4.3; p = .002 Mean diff = 22.15 ± 3.3; p < .001 Mean diff = 37 ± 3.4; p < .001 |
||
| BDI | F(2,135) = 23.4; p < .001 | NA | |
|
PTSD and PTSD+DS PTSD and control PTSD+DS and control |
Mean diff = 9.5 ± 2.5; p = .001 Mean diff = 22.2± 1.1; p < .001 Mean diff = 32 ± 2.2; p < .001 |
||
| RSDI numbness | F(2,129) = 22.23; p < .001 | NA | |
|
PTSD and PTSD+DS PTSD and control PTSD+DS and control |
Mean diff = 0.7 ± 1.97; p = .933 Mean diff = 0.56 ± 0.14; p < .001 Mean diff = 0.63 ± 0.16; p = .001 |
||
Note: aRSDI scores were not available for the whole sample.
Abbreviations: BDI, beck depression inventory; CAPS, clinician administered PTSD scale; CAPS‐5 assessed through independent 2‐sample t test; Control, age‐matched control group; CTQ, childhood trauma questionnaire; DEP, depersonalization; DER, derealization; M, mean; MDI, multiscale dissociation inventory; n, number of participants corresponding to a group; NA, not applicable; PTSD, nondissociative PTSD group; PTSD+DS, dissociative subtype PTSD group; RSDI, the responses to script‐driven imagery scale; SD, standard deviation; SE, standard error.
TABLE 3.
Whole‐brain P fwe = .05 corrected for multiple comparisons
| Contrast | Seed region | Target region | MNI x y z | p fwe | Cluster size | T‐score | Z‐score |
|---|---|---|---|---|---|---|---|
| Control |
Basal forebrain bilateral |
Striatum bilateral |
8 6 –12 −8 6 –12 |
<.001 <.001 |
1927 |
21.49 20.63 |
Inf Inf |
| PTSD | Basal forebrain bilateral | Striatum bilateral |
8 6 –12 −8 6 –12 |
<.001 <.001 |
2,476 | ‐ | ‐ |
|
Cingulate anterior left (BA 32) Frt med orbital right (BA 10) |
−8 48 –4 4 56 –4 8 46–8 |
<.001 0.002 0.004 |
161 |
6.48 5.9 5.77 |
6.06 5.58 5.46 |
||
|
Hippocampus R Parahippocampal right |
24 –24 –12 24 –26 –22 |
0.003 0.023 |
23 |
5.8 5.35 |
5.5 5.10 |
||
| PTSD+DS | Basal forebrain bilateral | No Suprathreshold | ‐ | ‐ | ‐ | ‐ | ‐ |
Note: Within‐group Basal Forebrain connectivity—Whole‐brain analysis pfwe .05.
Abbreviations: Inf, infinitum; MNI, Montreal Neurological Institute; PTSD, nondissociative PTSD group; PTSD+DS, dissociative subtype PTSD group.
3.6. Between‐group results: Effects of interest
A full‐factorial analysis at the whole‐brain level (p fwe = .05) revealed significant increased functional connectivity within the basal forebrain extended amygdaloid area (EA), notably, the bilateral SLEA (MNI = −18 0 –12, p fwe < .001, k = 299; MNI = 20 –2 −12, p fwe < .001, k = 192), a cluster extending bilaterally into the nucleus accumbens (NAc) (MNI = −10 6 –12, p fwe < .001; MNI = 12 8 –12, p fwe < .001) (Table 4).
TABLE 4.
Whole‐brain p fwe = .05 corrected for multiple comparisons
| Contrast | Seed region | Target region | MNI x y z | p fwe | Cluster size | F‐score | Z‐score |
|---|---|---|---|---|---|---|---|
| Effects of interest |
Basal forebrain Bilateral |
NAc‐SLEA left |
−18 0 –12 −10 6 –12 |
<.001 | 299 |
67.37 66.21 |
Inf Inf |
| NAc‐SLEA right |
20 –2 –12 12 8 –12 |
<.001 | 192 |
44.78 42.41 |
Inf 7.8 |
Note: Effects of interest Whole‐brain analysis p fwe = .05.
Abbreviations: Inf, infinitum; MNI, Montreal Neurological Institute; NAc, nucleus accumbens; PTSD, nondissociative PTSD group; PTSD+DS, dissociative subtype PTSD group; SLEA, sublenticular extended amygdala.
3.7. Between‐group results: Differential functional connectivity between PTSD, PTSD + DS, and controls
The whole‐brain (p fwe = .05) analysis revealed that when compared to the PTSD dissociative subtype (PTSD+DS), healthy controls exhibited a cluster of activations overlapping with the seed region, the basal forebrain, in its extended amygdaloid (EA) region. Here, our results suggest an increase in hemodynamic signal variability within the EA, with peaks localized in the left nucleus accumbens (NAc) (MNI = −8 6 –12, p fwe < .001, k = 292) and the right SLEA (MNI = 20 –2 −12, p fwe < .001, k = 137).
A strikingly similar result was observed when comparing the PTSD patient group to the PTSD+DS group. At whole‐brain (p fwe = .05) level analysis, we observed a cluster of activations also overlapping with the seed region, the basal forebrain, in its extended amygdaloid (EA) region. Again, our results suggest an increase in hemodynamic signal variability within the EA, with peaks localized in the left NAc (MNI = −10 6 –12, p fwe < .001, k = 312), a cluster extending ipsilaterally to the left SLEA (MNI = −18 –2 −12, p fwe < .001). Additionally, a third peak of activation was observed outside of the basal forebrain, in the right piriform cortex (MNI = 14 8–14, p fwe < .001, k = 228).
When compared with the PTSD patient group, healthy controls exhibited no suprathreshold clusters in the whole‐brain analysis. Similarly, when compared with healthy controls, the PTSD patient group showed no significant connectivity at the whole‐brain level analysis (Table 5).
TABLE 5.
Whole‐brain p fwe = .05 corrected for multiple comparisons
| Contrast | Seed region | Target region | MNI x y z | p fwe | Cluster size | T‐voxel | Z‐score |
|---|---|---|---|---|---|---|---|
| CTRL>PTSD+DS | Basal forebrain bilateral | NAc left | −8 6–12 | <.001 | 292 | 9.82 | Inf |
| SLEA right | 20 –2 –12 | <.001 | 137 | 7.4 | 6.81 | ||
| CTRL>PTSD |
Basal forebrain bilateral |
No suprathreshold | ‐ | ‐ | ‐ | ‐ | ‐ |
| PTSD+DS > CTRL |
Basal forebrain bilateral |
No suprathreshold | ‐ | ‐ | ‐ | ‐ | ‐ |
| PTSD+DS > PTSD |
Basal forebrain bilateral |
No suprathreshold | ‐ | ‐ | ‐ | ‐ | ‐ |
| PTSD>PTSD+DS |
Basal forebrain bilateral |
NAc‐SLEA left |
−10 6 –12 −18 –2 –12 |
<0.001 | 312 |
10.8 10.32 |
Inf Inf |
| PTSD > CTRL |
Basal forebrain bilateral |
No suprathreshold | ‐ | ‐ | ‐ | ‐ | ‐ |
Note: PTSD, PTSD + DS, Controls: Between‐group differences in connectivity. Whole‐brain analysis p fwe = .05.
Abbreviations: Inf, infinitum; MNI, Montreal Neurological Institute; NAc, nucleus accumbens; PTSD, nondissociative PTSD group; PTSD+DS, dissociative subtype PTSD group; SLEA, sublenticular extended amygdala.
3.8. Clinical variable correlations to functional connectivity within the PTSD + DS group
State emotional numbness, as measured by the RSDI during the resting state scan, predicted bilateral basal forebrain connectivity with the right SLEA (MNI = 16 –8 −20, p fwe = .004, k = 9) in the PTSD+DS group (Table 6).
TABLE 6.
ROI analysis p fwe = .05 corrected for multiple comparisons
| Clinical variable | Group | Seed region | Target region | MNI x y z | p fwe | Cluster size | Cluster size before ROI | t‐value | z‐value |
|---|---|---|---|---|---|---|---|---|---|
| RSDI numb |
PTSD+DS |
Basal forebrain bilateral | SLEA right | 16–8 –20 | 0.004 | 9 | 10 | 4.18 | 3.59 |
| SCID und. Somatoform disorder | PTSD+DS | Basal forebrain bilateral | Hypothalamus right | 2 –6 –8 | 0.013 | 62 | 120 | 4.20 | 3.67 |
| Hypothalamus left | −6 –2 –8 | 0.014 | 4.16 | 3.64 |
Note: Clinical variable prediction of basal forebrain connectivity in the PTSD + DS patient group.
Abbreviations: MNI, Montreal Neurological Institute; PTSD, nondissociative PTSD group; PTSD+DS, dissociative subtype PTSD group; SLEA, sublenticular extended amygdala.
4. DISCUSSION
4.1. Increased BOLD signal variability at rest in the basal forebrain extended amygdala nuclei of the dissociative subtype of PTSD
In the current study, the most significant findings emerged when comparing both the PTSD and the healthy control group to the PTSD+DS patient group. Specifically, our analysis revealed an increase in hemodynamic signal variability at rest of the basal forebrain extended amygdala (EA) in PTSD+DS patients. In SBC analysis, results encompassing the seed area are used to indirectly quantify the level of homogeneity or correlation of voxels within the seed. The observed increase in hemodynamic signal variability at rest observed here suggests that EA nuclei voxels are decorrelated from the remaining basal forebrain nuclei, a finding that argues against the notion that this region serves as a node or vertices supporting network connectivity among patients with this subtype.
There are several potential neurobiological explanations for this finding. For example, this increase in hemodynamic signal variability may have a purely hemodynamic origin, supporting an impairment in neurovascular coupling (Hillman, 2014; Liu, 2013), the process by which neuronal activity underlies changes in deoxyhemoglobin driven by localized changes in brain blood flow and oxygenation, ultimately constituting the BOLD signal detected in fMRI. Such patterns of abnormal blood flow and oxygenation may underlie neurodegenerative diseases such as AD, PD and FTD and are thought to arise from a complex bidirectional link between the accumulation and aggregation of amyloid‐B (AB) plaques and hyperphosphorylated tau tangles in the affected region, both preceding their appearance or being worsened by it (Park et al., 2020; Solis et al., 2020).
Alternatively, our results may represent neural factors associated with a disruption of resting‐state neuronal connectivity between adjacent nuclei of the basal forebrain, specifically, connectivity between the EA region NAc and SLEA nuclei, the nucleus basalis of Meynert, the diagonal band of Broca, the medial septal nucleus, and the substantia innominata. Here, it is well known that local node properties also induce alterations in remote functional connectivity (Deco et al., 2014). Changes in hemodynamic signal variability are concomitant with changes in functional connectivity, such that nodes with the greatest increase in signal variability also experience the greatest decrease in functional connectivity. Signal variability is highly associated with functional embedding of individual areas in large‐scale networks (Shafiei et al., 2019). As such, our observation of increased hemodynamic signal variability of the basal forebrain EA region NAc and SLEA nuclei among PTSD+DS patients may indicate the disconnection of these regions from the large‐scale networks in which they are normally embedded (Figures 1 and 2).
FIGURE 1.
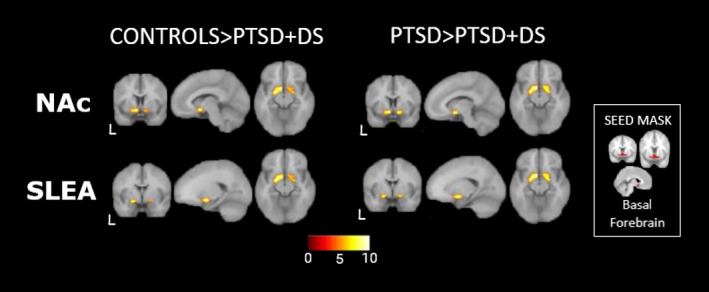
Between‐group comparison. Left: Controls>PTSD+DS, basal forebrain decreased resting state functional connectivity with left NAc (top) and right SLEA (bottom), basal forebrain nuclei integrating the mesolimbic dopaminergic reward circuitry. RIGHT: PTSD>PTSD+DS, basal forebrain decreased resting state functional connectivity with left NAc and left SLEA. Insert: Seed basal forebrain mask used in the analysis, allowing observation of the partial overlapping with significant peaks in NAc and SLEA. Abbreviations: PTSD, nondissociative PTSD group; PTSD+DS, dissociative subtype PTSD group; NAc, nucleus accumbens; SLEA, sublenticular extended amygdala; L: left hemisphere
FIGURE 2.
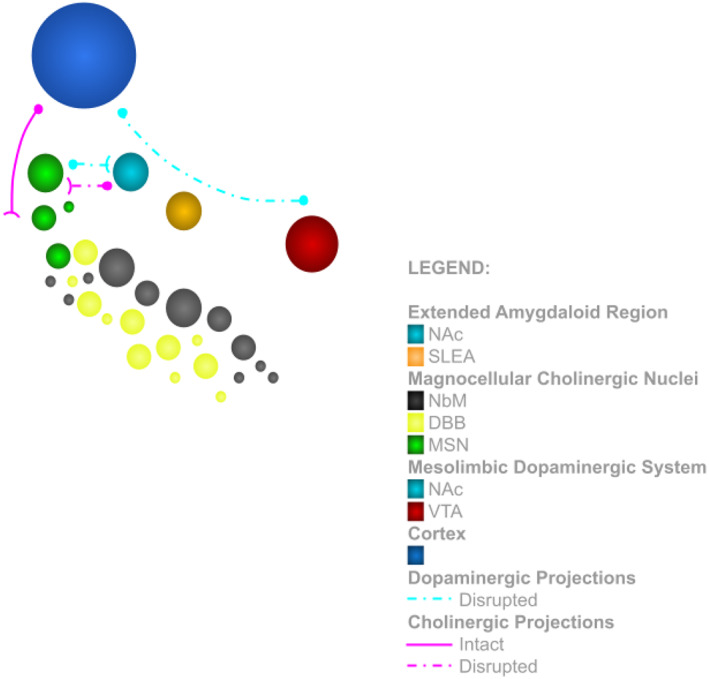
Schematic diagram of the basal forebrain in the dissociative subtype PTSD group, featuring the extended amygdaloid region (NAc and SLEA) and the magnocellular nuclei (NbM, DBB, and MSN), their reciprocal disrupted dopaminergic and cholinergic projections, the disrupted dopaminergic projections within the mesolimbic dopaminergic system (NAc and VTA) and the intact cholinergic projections to the cortex. Abbreviations: NAc, nucleus accumbens; SLEA, sublenticular extended amygdala; NbM, nucleus basalis of Meynert; DBB, diagonal band of Broca; MSN, medial septal nucleus; VTA, ventral tegmental area
From an etiopathogenetic standpoint, our results are in keeping with one main staging model of neurodegenerative diseases such as AD, which suggests an initial allocation of pathology in the basal forebrain, subsequently resulting in its dysfunction and degeneration (Mesulam et al., 2004; Schmitz et al., 2016). Only in later stages of the disease is it thought that this pathology spreads to other nodes or vertices of the basal forebrain projection pathways (Schmitz et al., 2018). As such, our results among patients with the dissociative subtype of PTSD are in keeping with the purported initial or prodromal stage of neurodegenerative diseases, where the presence of pathophysiology and neurodegeneration, is typically confined to the basal forebrain itself. Taken together, these findings provide a preliminary signal that abnormal neurovascular coupling or neuronal processes within the basal forebrain among individuals with PTSD+DS may serve as candidate neuroimaging markers of vulnerability to subsequent neurodegeneration in this population.
Longitudinal studies, as well as complementary neuropathological examination of postmortem brain tissue of PTSD+DS patients, will be necessary to clarify whether this local decorrelation is related to neurodegeneration‐related neuropathology in the basal forebrain in patients with this condition (Cordella et al., 2018; Seeley, Crawford, Zhou, Miller, & Greicius, 2009). Such investigation would also allow for monitoring of the progressive spread of putative neuropathology through other basal forebrain nuclei, or through other nodes of the remote functional networks constituting the NAc's and SLEA's spatially distinct projection systems (Cordella et al., 2018; Schmitz et al., 2018; Seeley et al., 2009) (Table 7).
TABLE 7.
Differences between PTSD and PTSD+DS phenotypes, in conformity with the new DSM‐V classification
| Symptom | PTSD | PTSD + DS |
|---|---|---|
| Aggressive/externalizing symptoms | X | X |
| Increased heart‐rate | X | X |
| Guilt/shame symptoms | X | X |
| Dysphoric anhedonic symptoms | X | X |
| Psychogenic amnesia | X | X |
| Depersonalization | X | |
| Derealization | X |
Abbreviations: PTSD, nondissociative PTSD group; PTSD+DS, dissociative subtype PTSD group.
4.2. Increased hemodynamic signal variability in core nodes of the reward processing circuitry: A link between traumatic memory in PTSD and declarative memory deficits (anterograde amnesia) in AD and PD?
Our findings point further to the disconnection of the NAc and the SLEA from the long‐range functional networks in which they are normally embedded in, including the network underlying the reward processing circuitry (Brown, Latimer, & Winn, 1996; Russo & Nestler, 2013; Sailer et al., 2008; Sharma et al., 2017; Waraczynski, Abbott, & Schultz, 2017).
Specifically, the NAc integrates the best‐characterized reward circuitry in the human brain, represented by its bidirectional connections with the ventral tegmental area (VTA) (Russo & Nestler, 2013). Importantly, impairment of the VTA‐NAc circuitry affects the reward‐based learning thought to underlie fear memory extinction in PTSD, a dysfunctionality that is believed to contribute to the development of persistent trauma memories (Brown et al., 1996; Korem, Lange, Hillard, & Akirav, 2017; Radke et al., 2019; van der Kolk, Hopper, & Osterman, 2001). By contrast, enhanced brain reward function is thought to play a central role in resilience to trauma (Vythilingam et al., 2009).
Reward sensitivity processes are also disproportionately affected in neurodegenerative diseases, including reward‐based memory processes (Nobili et al., 2017; Perry & Kramer, 2015). Remarkably, dysfunctional fear extinction processes are common to the presentation of AD (Nasrouei et al., 2020) and PD (Kinoshita et al., 2015). Prior to reinforcement and reversal learning, though, it is thought that coordinated activity of the hippocampus and the VTA‐NAc circuitry (Cordella et al., 2018; Nobili et al., 2017; Perry & Kramer, 2015) enhances memory encoding and consolidation thus playing a role in declarative memory formation (Miendlarzewska, Bavelier, & Schwartz, 2016; Setlow, 1997). Accordingly, disruption in this circuitry is thought to contribute to anterograde memory deficits observed in neurodegenerative diseases such as AD and PD with dementia (PD + D) (Braak & Braak, 1991; Braak & Del Tredici, 2017).
Further studies, however, are warranted to determine just how much overlap exists between the underlying dysfunction or degeneration of the VTA‐NAc circuitry that has been associated with abnormal fear memory processes in PTSD and in neurodegenerative conditions. Moreover, it will be critical to examine the potential role of the VTA‐NAc circuitry as a neurobiological link between the persistence of traumatic memory in PTSD (Korem et al., 2017) and declarative memory deficits, specifically anterograde amnesia, in neurodegeneration (Braak & Braak, 1991; Braak & Del Tredici, 2017).
4.3. The SLEA and threat‐related responses in PTSD + DS patient group
The SLEA is a cell column in the ventral forebrain comprised of neurons that represent an extension of the central and medial nucleus of the amygdala (CeA), connecting it to the bed nucleus of the stria terminalis (BSTL) (Paxinos & Mai, 2004). Because of this bridging role, the SLEA is implicated in stress‐induced behavior (Fudge et al., 2017) relevant for fear memory processes. Notably, the SLEA is thought to contribute to the instantiation of fear and anxiety states (Kovner et al., 2019; Shackman & Fox, 2016; Tillman et al., 2018), thus playing a central role in the evaluation of potentially threat‐related information. Additionally, the SLEA assists in the preparation of threat‐related defensive responses (Oler et al., 2017; Shackman & Fox, 2016).
The increase in hemodynamic signal variability in the SLEA in PTSD+DS group, observed in the present study may help to explain differences in threat‐related defensive responses observed in PTSD and in PTSD+DS. Specifically, whereas PTSD patients exhibit most frequently hyperarousal and active defensive responses, PTSD+DS is most frequently characterized by passive defensive responses such as emotional blunting and tonic immobility/freezing responses, responses likely stemming, in part, from hedonic and motivational deficits associated with dysfunctional reward‐based processes (Harricharan et al., 2016; Lanius et al., 2017; Olivé et al., 2018). To the best of our knowledge, however, the SLEA is not associated directly with the etiopathogenetic mechanisms underpinning neurodegenerative diseases.
4.4. Altered dopaminergic and “intact” cholinergic projection pathways
Our results point towards specific alterations in the dopaminergic projection pathways in the PTSD+DS patient group, where the EA region, including both the NAc and SLEA, forms a critical part of the dopaminergic mesolimbic system (Klawonn & Malenka, 2018; Volkow, Wise, & Baler, 2017). The emergence of dopaminergic dysfunction or loss in AD and PD pathogenesis has been extensively documented (Esposito, Di Matteo, & Di Giovanni, 2007; Martorana & Koch, 2014; McAuley, 2003; Murray et al., 1995). By contrast, these results suggest that the basal forebrain cholinergic nuclei, namely, the nucleus basalis of Meynert, the diagonal band of Broca, the medial septal nucleus, and the substantia innominata exhibit similar activity patterns across PTSD, PTSD+DS and healthy controls. Thus, we did not find direct evidence that PTSD is associated with one of the main etiopathogenetic mechanisms of several neurodegenerative diseases: the dysfunction and degeneration of the basal forebrain cholinergic pathways (Bohnen & Albin, 2011; Grothe et al., 2014; Hampel et al., 2018; Mesulam, 2004; Mesulam, 2013).
Nevertheless, in the absence of more comprehensive study, it is not possible to rule out entirely the potential impact of dopaminergic dysfunction upon cholinergic activity in PTSD. Here, previous studies indicate that the EA dopaminergic nuclei send direct modulatory projections to the basal forebrain cholinergic nuclei (Smiley, Subramanian, & Mesulam, 1999), which modulate indirectly cholinergic cortical excitability in AD patients (Martorana et al., 2009). Since cholinergic dysfunction and loss affects cortical structures in the advanced stages of neurodegenerative diseases (Mesulam, 2004; Schmitz et al., 2016), longitudinal studies may be necessary to examine any putative link between disrupted resting‐state connectivity of the basal forebrain extended amygdaloid area in PTSD+DS and potential cholinergic neuropathology in the cortex.
4.5. PTSD‐related neurodegeneration: A manifold, subtype‐, and disease‐specific condition
Epidemiological studies have provided clinical and population‐level evidence for a link between PTSD and increased risk for the development of various forms of dementia including AD, PD, Vascular Dementia, and FTD. The present findings reveal a pattern of dysfunction of the basal forebrain extended amygdala NAc in the PTSD dissociative subtype that is highly similar to patterns of dysfunction and degeneration of the NAc reported previously in AD and PD, providing a preliminary signal that dysfunction in this region may serve as a selective vulnerability mechanism to neurodegeneration among individuals with subtype.
Interestingly, other neurodegenerative diseases do not show a similar pattern of dysfunction or degeneration of the basal forebrain, including, to the best of our knowledge, FTD. Thus, efforts to elucidate the neurobiological and behavioral correlates of PTSD‐related neurodegeneration must consider not only differences in PTSD presentation, but also the likelihood of developing specific neurodegenerative disease: the etiopathogenetic mechanisms underpinning the increased risk of dementia in PTSD are many. In keeping with the National Institute of Mental Health (NIMH) Research Domain Criteria Initiative (RDoC) (Kozak & Cuthbert, 2016) strategic goals, which advocate the revision of subject inclusion criteria to correspond to the appropriate range of disease variance, attempts to examine the neurobiological and behavioral correlates of increased risk for dementia in specific PTSD presentations and specific neurodegenerative disease may result in more personalized treatment approaches.
5. CONCLUSION
Our results are novel in revealing an increase in hemodynamic signal variability in the basal forebrain extended amygdaloid region in PTSD. This result is in keeping with staging models of neurodegenerative diseases reporting allocation of neuropathology in early disease stages circumscribed to the basal forebrain. Our result may be associated with etiopathogenetic mechanisms such as neurovascular uncoupling, decreased connectivity in local‐ and large‐scale neural networks, or disrupted mesolimbic dopaminergic circuitry, acting indirectly upon the basal forebrain cholinergic projection pathways. These abnormalities may underpin reward‐related deficits representing a putative link between persistent traumatic memory in PTSD and anterograde memory deficits in neurodegeneration. Accordingly, we hypothesize that increased hemodynamic signal variability at the level of the basal forebrain extended amygdaloid region represents an important candidate neuroimaging marker predicting vulnerability to neurodegeneration in the dissociative subtype of PTSD. Given what appears to be the heightened risk of developing neurodegenerative conditions in PTSD, these suppositions must be urgently investigated.
6. LIMITATIONS AND FUTURE DIRECTIONS
The current study presents several limitations related primarily to our use of a retrospective dataset, whose original research goals, and experimental design were not the study of the neurobiological underpinnings of PTSD‐related neurodegeneration. Here, one of the main limitations of the current study is its cross‐sectional character where no longitudinal data were available to assess the relation between altered connectivity of basal forebrain extended amygdaloid region in PTSD+DS and the emergence of associated changes in neuropsychological functioning. Moreover, we did not have available data sampling the pathological hallmarks of neurodegeneration: CSF or blood tests aimed to detect the building up of plaques of misfolded proteins or neurofibrillary tangles (Clouston et al., 2019; Mohamed et al., 2018). Cytokine levels in the brain were also not available. Future studies are also needed to assess the basal forebrain nuclei‐specific connectivity, as well as the interplay between neurochemical systems and PTSD medications, such as psychotropic medication and novel antidepressants, that have been linked to increased dementia risk (Mawanda, Wallace, McCoy, & Abrams, 2017). Future research should be aimed at filling these critical gaps.
DISCLOSURE OF INTERESTS
The authors of the current study have no competing financial or non‐financial interests to report.
ACKNOWLEDGMENTS
The current study has as contract grant sponsors the Canadian Institutes for Military and Veteran Health Research, contract grant number W7714‐125626/001/SV, and the Canadian Institutes of Health Research, contract grant numbers 137150, 148784, and 97914.
The authors of the current study would like to thank Dr. Michel Grothe from the German Center for Neurodegenerative Diseases Rostock (DZNE) for sharing masks of the basal forebrain developed by his working group, as well as for his thorough explanation about the anatomy of the basal forebrain; Alfonso Nieto for technical guidance on the use of CONN toolbox, as well as for help in the interpretation of our results; and Prof. Marek M. Mesulam for key comments made during elaboration of the current article. We also thank the patients who contributed their data to this study and to members of Prof. Ruth Lanius' laboratory, Suzy Southwell and Nancy Mazza.
Olivé I, Makris N, Densmore M, McKinnon MC, Lanius RA. Altered basal forebrain BOLD signal variability at rest in posttraumatic stress disorder: A potential candidate vulnerability mechanism for neurodegeneration in PTSD. Hum Brain Mapp. 2021;42:3561–3575. 10.1002/hbm.25454
Funding information Canadian Institutes for Military and Veteran Health Research, Grant/Award Number: W7714‐125626/001/SV; Canadian Institutes of Health Research, Grant/Award Numbers: 137150, 148784, 97914
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available on request from the senior author RAL. The data are not publicly available due to them containing information that could compromise research participant privacy/consent.
REFERENCES
- Arendt, T. , Brückner, M. K. , Morawski, M. , Jäger, C. , & Gertz, H.‐J. (2015). Early neurone loss in Alzheimer's disease: Cortical or subcortical? Acta Neuropathologica Communications, 3, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck, A. T. , Guth, D. , Steer, R. A. , & Ball, R. (1997). Screening for major depression disorders in medical inpatients with the beck depression inventory for primary care. Behaviour Research and Therapy, 35, 785–791. [DOI] [PubMed] [Google Scholar]
- Bernstein, D. P. , Fink, L. , Handelsman, L. , Foote, J. , Lovejoy, M. , Wenzel, K. , … Ruggiero, J. (1994). Initial reliability and validity of a new retrospective measure of child abuse and neglect. The American Journal of Psychiatry, 151, 1132–1136. [DOI] [PubMed] [Google Scholar]
- Blake, D. D. , Weathers, F. W. , Nagy, L. M. , Kaloupek, D. G. , Gusman, F. D. , Charney, D. S. , & Keane, T. M. (1995). The development of a clinician‐administered PTSD scale. Journal of Traumatic Stress, 8, 75–90. [DOI] [PubMed] [Google Scholar]
- Bluhm, R. L. , Williamson, P. C. , Osuch, E. A. , Frewen, P. A. , Stevens, T. K. , Boksman, K. , … Lanius, R. A. (2009). Alterations in default network connectivity in posttraumatic stress disorder related to early‐life trauma. Journal of Psychiatry & Neuroscience, 34, 187–194. [PMC free article] [PubMed] [Google Scholar]
- Bohnen, N. I. , & Albin, R. L. (2011). The cholinergic system and Parkinson disease. Behavioural Brain Research, 221, 564–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyd, J. E. , Protopopescu, A. , O'Connor, C. , Neufeld, R. W. J. , Jetly, R. , Hood, H. K. , … McKinnon, M. C. (2018). Dissociative symptoms mediate the relation between PTSD symptoms and functional impairment in a sample of military members, veterans, and first responders with PTSD. European Journal of Psychotraumatology, 9, 1463794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braak, H. , & Braak, E. (1991). Neuropathological stageing of Alzheimer‐related changes. Acta Neuropathologica, 82, 239–259. [DOI] [PubMed] [Google Scholar]
- Braak, H. , & Del Tredici, K. (2017). Neuropathological staging of brain pathology in sporadic Parkinson's disease: Separating the wheat from the chaff. Journal of Parkinson's Disease, 7, S71–S85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briere, J. , Weathers, F. W. , & Runtz, M. (2005). Is dissociation a multidimensional construct? Data from the multiscale dissociation inventory. Journal of Traumatic Stress, 18, 221–231. [DOI] [PubMed] [Google Scholar]
- Brown, V. J. , Latimer, M. P. , & Winn, P. (1996). Memory for the changing cost of a reward is mediated by the sublenticular extended amygdala. Brain Research Bulletin, 39, 163–170. [DOI] [PubMed] [Google Scholar]
- Carriere, N. , Besson, P. , Dujardin, K. , Duhamel, A. , Defebvre, L. , Delmaire, C. , & Devos, D. (2014). Apathy in Parkinson's disease is associated with nucleus accumbens atrophy: A magnetic resonance imaging shape analysis. Movement Disorders, 29, 897–903. [DOI] [PubMed] [Google Scholar]
- Chan, Y.‐L. E. , Bai, Y.‐M. , Hsu, J.‐W. , Huang, K.‐L. , Su, T.‐P. , Li, C.‐T. , … Chen, M.‐H. (2017). Post‐traumatic stress disorder and risk of Parkinson disease: A Nationwide longitudinal study. The American Journal of Geriatric Psychiatry, 25, 917–923. [DOI] [PubMed] [Google Scholar]
- Chiesa, P. A. , Cavedo, E. , Grothe, M. J. , Houot, M. , Teipel, S. J. , Potier, M.‐C. , … INSIGHT‐preAD Study Group and the Alzheimer Precision Medicine Initiative (APMI) . (2019). Relationship between basal forebrain resting‐state functional connectivity and brain amyloid‐β deposition in cognitively intact older adults with subjective memory complaints. Radiology, 290, 167–176. [DOI] [PubMed] [Google Scholar]
- Clouston, S. A. P. , Deri, Y. , Diminich, E. , Kew, R. , Kotov, R. , Stewart, C. , … Luft, B. J. (2019). Posttraumatic stress disorder and total amyloid burden and amyloid‐β 42/40 ratios in plasma: Results from a pilot study of world trade center responders. Alzheimers Dement (Amst), 11, 216–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cope, T. E. , Rittman, T. , Borchert, R. J. , Jones, P. S. , Vatansever, D. , Allinson, K. , … Rowe, J. B. (2018). Tau burden and the functional connectome in Alzheimer's disease and progressive supranuclear palsy. Brain, 141, 550–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordella, A. , Krashia, P. , Nobili, A. , Pignataro, A. , La Barbera, L. , Viscomi, M. T. , … D'Amelio, M. (2018). Dopamine loss alters the hippocampus‐nucleus accumbens synaptic transmission in the Tg2576 mouse model of Alzheimer's disease. Neurobiology of Disease, 116, 142–154. [DOI] [PubMed] [Google Scholar]
- Deco, G. , Ponce‐Alvarez, A. , Hagmann, P. , Romani, G. L. , Mantini, D. , & Corbetta, M. (2014). How local excitation‐inhibition ratio impacts the whole brain dynamics. The Journal of Neuroscience, 34, 7886–7898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiLillo, D. , Fortier, M. A. , Hayes, S. A. , Trask, E. , Perry, A. R. , Messman‐Moore, T. , … Nash, C. (2006). Retrospective assessment of childhood sexual and physical abuse: A comparison of scaled and behaviorally specific approaches. Assessment, 13, 297–312. [DOI] [PubMed] [Google Scholar]
- Esposito, E. , Di Matteo, V. , & Di Giovanni, G. (2007). Death in the substantia nigra: A motor tragedy. Expert Review of Neurotherapeutics, 7, 677–697. [DOI] [PubMed] [Google Scholar]
- Felmingham, K. L. , Falconer, E. M. , Williams, L. , Kemp, A. H. , Allen, A. , Peduto, A. , & Bryant, R. A. (2014). Reduced amygdala and ventral striatal activity to happy faces in PTSD is associated with emotional numbing. PLoS One, 9, e103653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flatt, J. D. , Gilsanz, P. , Quesenberry, C. P. , Albers, K. B. , & Whitmer, R. A. (2018). Post‐traumatic stress disorder and risk of dementia among members of a health care delivery system. Alzheimers Dement, 14, 28–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fransson, P. (2005). Spontaneous low‐frequency BOLD signal fluctuations: An fMRI investigation of the resting‐state default mode of brain function hypothesis. Human Brain Mapping, 26, 15–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frewen, P. , & Lanius, R. (2013). Healing the traumatized self (1st ed.). New York: W. W Norton & Company. [Google Scholar]
- Fudge, J. L. , Kelly, E. A. , Pal, R. , Bedont, J. L. , Park, L. , & Ho, B. (2017). Beyond the classic VTA: Extended amygdala projections to DA‐striatal paths in the primate. Neuropsychopharmacology, 42, 1563–1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerlicher, A. M. V. , Tüscher, O. , & Kalisch, R. (2019). L‐DOPA improves extinction memory retrieval after successful fear extinction. Psychopharmacology, 236, 3401–3412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green, C. R. , Corsi‐Travali, S. , & Neumeister, A. (2013). The role of BDNF‐TrkB signaling in the pathogenesis of PTSD. Journal of Depression and Anxiety, 2013. 10.4172/2167-1044.S4-006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenberg, M. S. , Tanev, K. , Marin, M.‐F. , & Pitman, R. K. (2014). Stress, PTSD, and dementia. Alzheimers Dement, 10, S155–S165. [DOI] [PubMed] [Google Scholar]
- Grothe, M. J. , Schuster, C. , Bauer, F. , Heinsen, H. , Prudlo, J. , & Teipel, S. J. (2014). Atrophy of the cholinergic basal forebrain in dementia with Lewy bodies and Alzheimer's disease dementia. Journal of Neurology, 261, 1939–1948. [DOI] [PubMed] [Google Scholar]
- Halabi, C. , Halabi, A. , Dean, D. L. , Wang, P.‐N. , Boxer, A. L. , Trojanowski, J. Q. , … Seeley, W. W. (2013). Patterns of striatal degeneration in frontotemporal dementia. Alzheimer Disease and Associated Disorders, 27, 74–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hampel, H. , Mesulam, M.‐M. , Cuello, A. C. , Farlow, M. R. , Giacobini, E. , Grossberg, G. T. , … Khachaturian, Z. S. (2018). The cholinergic system in the pathophysiology and treatment of Alzheimer's disease. Brain, 141, 1917–1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hampton, O. L. , Buckley, R. F. , Manning, L. K. , Scott, M. R. , Properzi, M. J. , Peña‐Gómez, C. , … Schultz, A. P. (2020). Resting‐state functional connectivity and amyloid burden influence longitudinal cortical thinning in the default mode network in preclinical Alzheimer's disease. Neuroimage Clinical, 28, 102407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harricharan, S. , Rabellino, D. , Frewen, P. A. , Densmore, M. , Théberge, J. , McKinnon, M. C. , … Lanius, R. A. (2016). fMRI functional connectivity of the periaqueductal gray in PTSD and its dissociative subtype. Brain and Behavior: A Cognitive Neuroscience Perspective, 6, e00579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbert, J. , Goodyer, I. M. , Grossman, A. B. , Hastings, M. H. , de Kloet, E. R. , Lightman, S. L. , … Seckl, J. R. (2006). Do corticosteroids damage the brain? Journal of Neuroendocrinology, 18, 393–411. [DOI] [PubMed] [Google Scholar]
- Hillman, E. M. C. (2014). Coupling mechanism and significance of the BOLD signal: A status report. Annual Review of Neuroscience, 37, 161–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hohenfeld, C. , Werner, C. J. , & Reetz, K. (2018). Resting‐state connectivity in neurodegenerative disorders: Is there potential for an imaging biomarker? Neuroimage Clinical, 18, 849–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopper, J. W. , Frewen, P. A. , van der Kolk, B. A. , & Lanius, R. A. (2007). Neural correlates of reexperiencing, avoidance, and dissociation in PTSD: Symptom dimensions and emotion dysregulation in responses to script‐driven trauma imagery. Journal of Traumatic Stress, 20, 713–725. [DOI] [PubMed] [Google Scholar]
- Izquierdo, I. , Furini, C. R. G. , & Myskiw, J. C. (2016). Fear memory. Physiological Reviews, 96, 695–750. [DOI] [PubMed] [Google Scholar]
- Jeong, H. , Kang, I. , Im, J. J. , Park, J.‐S. , Na, S.‐H. , Heo, Y. , … Song, I.‐U. (2018). Brain perfusion correlates of apathy in alzheimer's disease. Dementia Neurocognitive Disorder, 17, 50–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawakami, I. , Hasegawa, M. , Arai, T. , Ikeda, K. , Oshima, K. , Niizato, K. , … Akiyama, H. (2014). Tau accumulation in the nucleus accumbens in tangle‐predominant dementia. Acta Neuropathologica Communications, 2, 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinoshita, K. , Tada, Y. , Muroi, Y. , Unno, T. , & Ishii, T. (2015). Selective loss of dopaminergic neurons in the substantia nigra pars compacta after systemic administration of MPTP facilitates extinction learning. Life Sciences, 137, 28–36. [DOI] [PubMed] [Google Scholar]
- Klawonn, A. M. , & Malenka, R. C. (2018). Nucleus accumbens modulation in reward and aversion. Cold Spring Harbor Symposia on Quantitative Biology, 83, 119–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korem, N. , Lange, R. , Hillard, C. J. , & Akirav, I. (2017). Role of beta‐catenin and endocannabinoids in the nucleus accumbens in extinction in rats exposed to shock and reminders. Neuroscience, 357, 285–294. [DOI] [PubMed] [Google Scholar]
- Kovner, R. , Fox, A. S. , French, D. A. , Roseboom, P. H. , Oler, J. A. , Fudge, J. L. , & Kalin, N. H. (2019). Somatostatin gene and Protein expression in the non‐human primate central extended amygdala. Neuroscience, 400, 157–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozak, M. J. , & Cuthbert, B. N. (2016). The NIMH research domain criteria initiative: Background, issues, and pragmatics. Psychophysiology, 53, 286–297. [DOI] [PubMed] [Google Scholar]
- Lanius, R. A. , Rabellino, D. , Boyd, J. E. , Harricharan, S. , Frewen, P. A. , & McKinnon, M. C. (2017). The innate alarm system in PTSD: Conscious and subconscious processing of threat. Current Opinion in Psychology, 14, 109–115. [DOI] [PubMed] [Google Scholar]
- Lehéricy, S. , Hirsch, E. C. , Cervera, P. , Hersh, L. B. , Hauw, J. J. , Ruberg, M. , & Agid, Y. (1989). Selective loss of cholinergic neurons in the ventral striatum of patients with Alzheimer disease. Proceedings of the National Academy of Sciences of the United States of America, 86, 8580–8584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberzon, I. , & Garfinkel, S. N. (2009). Functional neuroimaging in post‐traumatic stress disorder. In LeDoux J. E., Keane T., & Shiromani P. (Eds.), Post‐traumatic stress disorder (pp. 297–317). Totowa, NJ: Humana Press. [Google Scholar]
- Lieberman, L. , Gorka, S. M. , Funkhouser, C. J. , Shankman, S. A. , & Phan, K. L. (2017). Impact of posttraumatic stress symptom dimensions on psychophysiological reactivity to threat and reward. Journal of Psychiatric Research, 92, 55–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, T. T. (2013). Neurovascular factors in resting‐state functional MRI. NeuroImage, 80, 339–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logue, M. W. , van Rooij, S. J. H. , Dennis, E. L. , Davis, S. L. , Hayes, J. P. , Stevens, J. S. , … Morey, R. A. (2018). Smaller hippocampal volume in posttraumatic stress disorder: A multisite ENIGMA‐PGC study: Subcortical volumetry results from posttraumatic stress disorder consortia. Biological Psychiatry, 83, 244–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martorana, A. , & Koch, G. (2014). Is dopamine involved in Alzheimer's disease? Frontiers in Aging Neuroscience, 6, 252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martorana, A. , Mori, F. , Esposito, Z. , Kusayanagi, H. , Monteleone, F. , Codecà, C. , … Koch, G. (2009). Dopamine modulates cholinergic cortical excitability in Alzheimer's disease patients. Neuropsychopharmacology, 34, 2323–2328. [DOI] [PubMed] [Google Scholar]
- Mawanda, F. , Wallace, R. B. , McCoy, K. , & Abrams, T. E. (2017). PTSD, psychotropic medication use, and the risk of dementia among US veterans: A retrospective cohort study. Journal of the American Geriatrics Society, 65, 1043–1050. [DOI] [PubMed] [Google Scholar]
- McAuley, J. H. (2003). The physiological basis of clinical deficits in Parkinson's disease. Progress in Neurobiology, 69, 27–48. [DOI] [PubMed] [Google Scholar]
- Mesulam, M. (2004). The cholinergic lesion of Alzheimer's disease: Pivotal factor or side show? Learning & Memory, 11, 43–49. [DOI] [PubMed] [Google Scholar]
- Mesulam, M. , Shaw, P. , Mash, D. , & Weintraub, S. (2004). Cholinergic nucleus basalis tauopathy emerges early in the aging‐MCI‐AD continuum. Annals of Neurology, 55, 815–828. [DOI] [PubMed] [Google Scholar]
- Mesulam, M.‐M. (2013). Cholinergic circuitry of the human nucleus basalis and its fate in Alzheimer's disease. The Journal of Comparative Neurology, 521, 4124–4144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mickleborough, M. J. S. , Daniels, J. K. , Coupland, N. J. , Kao, R. , Williamson, P. C. , Lanius, U. F. , … Lanius, R. A. (2011). Effects of trauma‐related cues on pain processing in posttraumatic stress disorder: An fMRI investigation. Journal of Psychiatry & Neuroscience, 36, 6–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miendlarzewska, E. A. , Bavelier, D. , & Schwartz, S. (2016). Influence of reward motivation on human declarative memory. Neuroscience and Biobehavioral Reviews, 61, 156–176. [DOI] [PubMed] [Google Scholar]
- Millar, P. R. , Ances, B. M. , Gordon, B. A. , Benzinger, T. L. S. , Fagan, A. M. , Morris, J. C. , & Balota, D. A. (2020). Evaluating resting‐state BOLD variability in relation to biomarkers of preclinical Alzheimer's disease. Neurobiology of Aging, 96, 233–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohamed, A. Z. , Cumming, P. , Srour, H. , Gunasena, T. , Uchida, A. , Haller, C. N. , … Department of Defense Alzheimer's Disease Neuroimaging Initiative . (2018). Amyloid pathology fingerprint differentiates post‐traumatic stress disorder and traumatic brain injury. Neuroimage Clinical, 19, 716–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muhammed, K. , Manohar, S. , Ben Yehuda, M. , Chong, T. T.‐J. , Tofaris, G. , Lennox, G. , … Husain, M. (2016). Reward sensitivity deficits modulated by dopamine are associated with apathy in Parkinson's disease. Brain, 139, 2706–2721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray, A. M. , Weihmueller, F. B. , Marshall, J. F. , Hurtig, H. I. , Gottleib, G. L. , & Joyce, J. N. (1995). Damage to dopamine systems differs between Parkinson's disease and Alzheimer's disease with parkinsonism. Annals of Neurology, 37, 300–312. [DOI] [PubMed] [Google Scholar]
- Nasrouei, S. , Rattel, J. A. , Liedlgruber, M. , Marksteiner, J. , & Wilhelm, F. H. (2020). Fear acquisition and extinction deficits in amnestic mild cognitive impairment and early Alzheimer's disease. Neurobiology of Aging, 87, 26–34. [DOI] [PubMed] [Google Scholar]
- Nicholson, A. A. , Densmore, M. , Frewen, P. A. , Théberge, J. , Neufeld, R. W. , McKinnon, M. C. , & Lanius, R. A. (2015). The dissociative subtype of posttraumatic stress disorder: Unique resting‐state functional connectivity of basolateral and centromedial amygdala complexes. Neuropsychopharmacology, 40, 2317–2326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nie, X. , Sun, Y. , Wan, S. , Zhao, H. , Liu, R. , Li, X. , … Zhang, B. (2017). Subregional structural alterations in hippocampus and nucleus accumbens correlate with the clinical impairment in patients with Alzheimer's disease clinical Spectrum: Parallel combining volume and vertex‐based approach. Frontiers in Neurology, 8, 399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nobili, A. , Latagliata, E. C. , Viscomi, M. T. , Cavallucci, V. , Cutuli, D. , Giacovazzo, G. , … D'Amelio, M. (2017). Dopamine neuronal loss contributes to memory and reward dysfunction in a model of Alzheimer's disease. Nature Communications, 8, 14727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oler, J. A. , Tromp, D. P. M. , Fox, A. S. , Kovner, R. , Davidson, R. J. , Alexander, A. L. , … Fudge, J. (2017). Connectivity between the central nucleus of the amygdala and the bed nucleus of the stria terminalis in the non‐human primate: Neuronal tract tracing and developmental neuroimaging studies. Brain Structure & Function, 222, 21–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olivé, I. , Densmore, M. , Harricharan, S. , Théberge, J. , McKinnon, M. C. , & Lanius, R. (2018). Superior colliculus resting state networks in post‐traumatic stress disorder and its dissociative subtype. Human Brain Mapping, 39, 563–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson, E. A. , Kaiser, R. H. , Pizzagalli, D. A. , Rauch, S. L. , & Rosso, I. M. (2018). Anhedonia in trauma‐exposed individuals: Functional connectivity and decision‐making correlates. Biological Psychiatry: Cognitive Neuroscience and Neuroimaging, 3, 959–967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ospina, J. P. , Larson, A. G. , Jalilianhasanpour, R. , Williams, B. , Diez, I. , Dhand, A. , … Perez, D. L. (2019). Individual differences in social network size linked to nucleus accumbens and hippocampal volumes in functional neurological disorder: A pilot study. Journal of Affective Disorders, 258, 50–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oyanagi, K. , Makifuchi, T. , Ohtoh, T. , Chen, K. M. , Gajdusek, D. C. , Chase, T. N. , & Ikuta, F. (1994). The neostriatum and nucleus accumbens in parkinsonism‐dementia complex of Guam: A pathological comparison with Alzheimer's disease and progressive supranuclear palsy. Acta Neuropathologica, 88, 122–128. [DOI] [PubMed] [Google Scholar]
- Park, L. , Hochrainer, K. , Hattori, Y. , Ahn, S. J. , Anfray, A. , Wang, G. , … Iadecola, C. (2020). Tau induces PSD95‐neuronal NOS uncoupling and neurovascular dysfunction independent of neurodegeneration. Nature Neuroscience, 23, 1079–1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavic, L. , Gregurek, R. , Petrović, R. , Petrović, D. , Varda, R. , Vukusić, H. , & Crnković‐Marković, S. (2003). Alterations in brain activation in posttraumatic stress disorder patients with severe hyperarousal symptoms and impulsive aggressiveness. European Archives of Psychiatry and Clinical Neuroscience, 253, 80–83. [DOI] [PubMed] [Google Scholar]
- Paxinos, G. , & Mai, J. (2004). The human nervous system. Cambridge, MA: Elsevier. [Google Scholar]
- Perry, D. C. , & Kramer, J. H. (2015). Reward processing in neurodegenerative disease. Neurocase, 21, 120–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pievani, M. , Bocchetta, M. , Boccardi, M. , Cavedo, E. , Bonetti, M. , Thompson, P. M. , & Frisoni, G. B. (2013). Striatal morphology in early‐onset and late‐onset Alzheimer's disease: A preliminary study. Neurobiology of Aging, 34, 1728–1739. [DOI] [PubMed] [Google Scholar]
- Pineau, H. , Marchand, A. , & Guay, S. (2015). Specificity of cognitive and behavioral complaints in post‐traumatic stress disorder and mild traumatic brain injury. Behavioral Sciences (Basel), 5, 43–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitman, R. K. (2010). Posttraumatic stress disorder and dementia: What is the origin of the association? JAMA, 303, 2287–2288. [DOI] [PubMed] [Google Scholar]
- Power, J. D. , Mitra, A. , Laumann, T. O. , Snyder, A. Z. , Schlaggar, B. L. , & Petersen, S. E. (2014). Methods to detect, characterize, and remove motion artifact in resting state fMRI. NeuroImage, 84, 320–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qureshi, S. U. , Kimbrell, T. , Pyne, J. M. , Magruder, K. M. , Hudson, T. J. , Petersen, N. J. , … Kunik, M. E. (2010). Greater prevalence and incidence of dementia in older veterans with posttraumatic stress disorder. Journal of the American Geriatrics Society, 58, 1627–1633. [DOI] [PubMed] [Google Scholar]
- Radke, A. K. , Kocharian, A. , Covey, D. P. , Lovinger, D. M. , Cheer, J. F. , Mateo, Y. , & Holmes, A. (2019). Contributions of nucleus accumbens dopamine to cognitive flexibility. The European Journal of Neuroscience, 50, 2023–2035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russo, S. J. , & Nestler, E. J. (2013). The brain reward circuitry in mood disorders. Nature Reviews. Neuroscience, 14, 609–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryle, M. , Court, J. , Smith, T. , & Morris, R. (1975). Gonadotrophic stimulation of oestrogen synthesis by cultured immature mouse ovaries. The Journal of Endocrinology, 66, 225–232. [DOI] [PubMed] [Google Scholar]
- Sailer, U. , Robinson, S. , Fischmeister, F. P. S. , König, D. , Oppenauer, C. , Lueger‐Schuster, B. , … Bauer, H. (2008). Altered reward processing in the nucleus accumbens and mesial prefrontal cortex of patients with posttraumatic stress disorder. Neuropsychologia, 46, 2836–2844. [DOI] [PubMed] [Google Scholar]
- Schmitz, T. W. , Mur, M. , Aghourian, M. , Bedard, M.‐A. , Spreng, R. N. , & Alzheimer's Disease Neuroimaging Initiative . (2018). Longitudinal Alzheimer's degeneration reflects the spatial topography of cholinergic basal forebrain projections. Cell Reports, 24, 38–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz, T. W. , Nathan Spreng, R. , & Alzheimer's Disease Neuroimaging Initiative . (2016). Basal forebrain degeneration precedes and predicts the cortical spread of Alzheimer's pathology. Nature Communications, 7, 13249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeley, W. W. , Crawford, R. K. , Zhou, J. , Miller, B. L. , & Greicius, M. D. (2009). Neurodegenerative diseases target large‐scale human brain networks. Neuron, 62, 42–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selden, N. , Mesulam, M. M. , & Geula, C. (1994). Human striatum: The distribution of neurofibrillary tangles in Alzheimer's disease. Brain Research, 648, 327–331. [DOI] [PubMed] [Google Scholar]
- Setlow, B. (1997). The nucleus accumbens and learning and memory. Journal of Neuroscience Research, 49, 515–521. [DOI] [PubMed] [Google Scholar]
- Shackman, A. J. , & Fox, A. S. (2016). Contributions of the central extended amygdala to fear and anxiety. The Journal of Neuroscience, 36, 8050–8063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shafiei, G. , Zeighami, Y. , Clark, C. A. , Coull, J. T. , Nagano‐Saito, A. , Leyton, M. , … Mišic, B. (2019). Dopamine signaling modulates the stability and integration of intrinsic brain networks. Cerebral Cortex, 29, 397–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma, A. , Wolf, D. H. , Ciric, R. , Kable, J. W. , Moore, T. M. , Vandekar, S. N. , … Satterthwaite, T. D. (2017). Common dimensional reward deficits across mood and psychotic disorders: A connectome‐wide association study. The American Journal of Psychiatry, 174, 657–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smiley, J. F. , Subramanian, M. , & Mesulam, M. M. (1999). Monoaminergic‐cholinergic interactions in the primate basal forebrain. Neuroscience, 93, 817–829. [DOI] [PubMed] [Google Scholar]
- Solis, E. , Hascup, K. N. , & Hascup, E. R. (2020). Alzheimer's disease: The link between amyloid‐β and neurovascular dysfunction. Journal of Alzheimer's Disease, 76, 1179–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein, D. J. , Koenen, K. C. , Friedman, M. J. , Hill, E. , McLaughlin, K. A. , Petukhova, M. , … Kessler, R. C. (2013). Dissociation in posttraumatic stress disorder: Evidence from the world mental health surveys. Biological Psychiatry, 73, 302–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steuwe, C. , Lanius, R. A. , & Frewen, P. A. (2012). Evidence for a dissociative subtype of PTSD by latent profile and confirmatory factor analyses in a civilian sample. Depression and Anxiety, 29, 689–700. [DOI] [PubMed] [Google Scholar]
- Teipel, S. , Heinsen, H. , Amaro, E. , Grinberg, L. T. , Krause, B. , Grothe, M. , & Alzheimer's Disease Neuroimaging Initiative . (2014). Cholinergic basal forebrain atrophy predicts amyloid burden in Alzheimer's disease. Neurobiology of Aging, 35, 482–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teipel, S. J. , Flatz, W. H. , Heinsen, H. , Bokde, A. L. W. , Schoenberg, S. O. , Stöckel, S. , … Hampel, H. (2005). Measurement of basal forebrain atrophy in Alzheimer's disease using MRI. Brain, 128, 2626–2644. [DOI] [PubMed] [Google Scholar]
- Tillman, R. M. , Stockbridge, M. D. , Nacewicz, B. M. , Torrisi, S. , Fox, A. S. , Smith, J. F. , & Shackman, A. J. (2018). Intrinsic functional connectivity of the central extended amygdala. Human Brain Mapping, 39, 1291–1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Kolk, B. A. , Hopper, J. W. , & Osterman, J. E. (2001). Exploring the nature of traumatic memory. Journal of Aggression, Maltreatment & Trauma, 4, 9–31. [Google Scholar]
- Van Dijk, K. R. A. , Sabuncu, M. R. , & Buckner, R. L. (2012). The influence of head motion on intrinsic functional connectivity MRI. NeuroImage, 59, 431–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Huijstee, J. , & Vermetten, E. (2018). The dissociative subtype of post‐traumatic stress disorder: Research update on clinical and neurobiological features. Current Topics in Behavioral Neurosciences, 38, 229–248. [DOI] [PubMed] [Google Scholar]
- Volkow, N. D. , Wise, R. A. , & Baler, R. (2017). The dopamine motive system: Implications for drug and food addiction. Nature Reviews. Neuroscience, 18, 741–752. [DOI] [PubMed] [Google Scholar]
- Vythilingam, M. , Nelson, E. E. , Scaramozza, M. , Waldeck, T. , Hazlett, G. , Southwick, S. M. , … Ernst, M. (2009). Reward circuitry in resilience to severe trauma: An fMRI investigation of resilient special forces soldiers. Psychiatry Research, 172, 75–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, D. , Belden, A. , Hanser, S. B. , Geddes, M. R. , & Loui, P. (2020). Resting‐state connectivity of auditory and reward systems in Alzheimer's disease and mild cognitive impairment. Frontiers in Human Neuroscience, 14, 280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waraczynski, M. , Abbott, S. , & Schultz, A. V. (2017). CaV1.3 channel blockade in the extended amygdala has a delayed effect on the reward efficacy of medial forebrain bundle stimulation. Behavioural Brain Research, 317, 485–493. [DOI] [PubMed] [Google Scholar]
- Weiner, M. W. , Friedl, K. E. , Pacifico, A. , Chapman, J. C. , Jaffee, M. S. , Little, D. M. , … Carrillo, M. C. (2013). Military risk factors for Alzheimer's disease. Alzheimers Dement, 9, 445–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wisch, J. K. , Roe, C. M. , Babulal, G. M. , Schindler, S. E. , Fagan, A. M. , Benzinger, T. L. , … Ances, B. M. (2020). Resting state functional connectivity signature differentiates cognitively Normal from individuals who convert to symptomatic Alzheimer's disease. Journal of Alzheimer's Disease, 74, 1085–1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf, E. J. , Lunney, C. A. , Miller, M. W. , Resick, P. A. , Friedman, M. J. , & Schnurr, P. P. (2012). The dissociative subtype of PTSD: A replication and extension. Depression and Anxiety, 29, 679–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaffe, K. , Vittinghoff, E. , Lindquist, K. , Barnes, D. , Covinsky, K. E. , Neylan, T. , … Marmar, C. (2010). Posttraumatic stress disorder and risk of dementia among US veterans. Archives of General Psychiatry, 67, 608–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi, H.‐A. , Möller, C. , Dieleman, N. , Bouwman, F. H. , Barkhof, F. , Scheltens, P. , … Vrenken, H. (2016). Relation between subcortical grey matter atrophy and conversion from mild cognitive impairment to Alzheimer's disease. Journal of Neurology, Neurosurgery, and Psychiatry, 87, 425–432. [DOI] [PubMed] [Google Scholar]
- Zeighami. Y , Ulla, M., Iturria‐Medina, Y., Dadar, M., Zhang, Y., Larcher, K. M‐H, … Dagher, A. (2015). Network structure of brain atrophy in de novo Parkinson's disease. elife 4. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study are available on request from the senior author RAL. The data are not publicly available due to them containing information that could compromise research participant privacy/consent.