

Abstract
Cellular and humoral response to acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) infections is on focus of research. We evaluate herein the feasibility of expanding virus‐specific T cells (VST) against SARS‐CoV‐2 ex vivo through a standard protocol proven effective for other viruses. The experiment was performed in three different donors' scenarios: (a) SARS‐CoV‐2 asymptomatic infection/negative serology, (b) SARS‐CoV‐2 symptomatic infection/positive serology, and (c) no history of SARS‐CoV‐2 infection/negative serology. We were able to obtain an expanded VST product from donors 1 and 2 (1.6x and 1.8x increase of baseline VST count, respectively) consisting in CD3 + cells (80.3% and 62.7%, respectively) with CD4 + dominance (60% in both donors). Higher numbers of VST were obtained from the donor 2 as compared to donor 1. T‐cell clonality test showed oligoclonal reproducible peaks on a polyclonal background for both donors. In contrast, VST could be neither expanded nor primed in a donor without evidence of prior infection. This proof‐of‐concept study supports the feasibility of expanding ex vivo SARS‐CoV‐2‐specific VST from blood of convalescent donors. The results raise the question of whether the selection of seropositive donors may be a strategy to obtain cell lines enriched in their SARS‐CoV‐2‐specificity for future adoptive transfer to immunosuppressed patients.
Keywords: adoptive immunotherapy, COVID‐19, lymphocyte expansion, respiratory virus, SARS‐CoV‐2, third‐party donors, virus‐specific T cells
1. INTRODUCTION
The coronavirus disease 2019 (COVID‐19) pandemic is causing an enormous health and economic global impact worldwide. Solid tumor or oncohematological patients who develop COVID‐19 are at higher risk of mortality, 1 , 2 surely due to their hampered humoral and cellular immune response against SARS‐CoV‐2. 3 , 4 Adaptive T cell immune responses are increasingly recognized as key factor controlling viral clearance, severity of COVID‐19, and humoral immune responses. 5 A deep understanding of T cell responses to SARS‐CoV‐2 is critical to improve our assessment of the probability of SARS‐CoV‐2 reinfection, COVID‐19 prognosis, the estimation of general population immunity, as well as for guiding vaccine development.
Although immunoglobulin G (IgG) and M (IgM) seroconversion rate after COVID‐19 is common, it can vary from 80% to 100% in symptomatic and 15% to 95% in asymptomatic patients after SARS‐CoV‐2 infection. 6 , 7 , 8 , 9 , 10 Moreover, the humoral response is not always achieved, and the presence of IgG can be as short as few days or weeks after recovery in nearly 40% of patients. 6 In contrast, strong and long‐lasting CD4 and CD8 T cell responses 5 , 11 have been observed in nearly all infected cases, highlighting their predominant role in SARS‐CoV‐2 control and clearance.
Currently, there are several ways to asses T cell response to SARS‐CoV‐2 infection such as ex vivo T cell flow cytometry and ELISPOT‐based assays. 11 , 12 However, limitations of these tests include expertise in interpretation, lack of consensus in the standardization of the methodology, and low burden of circulating SARS‐CoV‐2‐specific T cells (<0.1% of total lymphocyte subset in peripheral blood). 12 Furthermore, T cell cross‐reactivity observed with other common seasonal human coronavirus could be regarded as a limitation since it may overestimate the real immunity in the general population. 5 , 13 This issue is further aggravated by the current lack of evidence that such cross‐reactivity could confer functional protection against SARS‐CoV‐2. 14
Innovative approaches, such as ex vivo virus‐specific T cells (VST) expansion used for cytomegalovirus (CMV) and Epstein Barr virus (EBV), 15 , 16 may be of value for SARS‐CoV‐2 since it could offer a great number of VST against SARS‐CoV‐2 for clinical or laboratory research purposes. 17
We present herein a proof‐of‐concept experiment to investigate the feasibility of expanding SARS‐CoV‐2 VST ex vivo. We report the results of extending the applicability of optimized VST expansion protocol against CMV and EBV to the SARS‐CoV‐2 and discuss about the potential implications of our findings.
2. MATERIALS AND METHODS
2.1. Donor selection
For the study purpose, we selected two healthcare workers with PCR‐confirmed SARS‐CoV‐2 infection on 11 and 26 March 2020, respectively, and a volunteer healthy donor. All three volunteer donors were previously registered in REDOCEL, a database with high‐resolution typed blood donors Human Leukocyte Antigen (HLA) system consenting to be contacted whether cellular product donations were required for adoptive therapies. This donor bank is enriched with young individuals carrying common Spanish HLA Class I/II alleles, which facilitates HLA matching for future patients. To carry out our proof‐of‐concept experiments, donors were selected in order to included three different immunologic profiles: two donors with PCR‐confirmed history of SARS‐CoV‐2 infection, one of which was asymptomatic with no seroconversion (donor 1), while the other was mildly symptomatic and had detectable circulating IgG and IgM antibodies (donor 2), and a third donor without previous history of infection or seroconversion (donor 3).
Peripheral blood mononuclear cells (PBMCs) were obtained from 50 mL blood donations on 12 May 2020 after written informed consent. Cell processing and ex vivo expansion experiments were conducted at the GMP facility in Hospital Universitario y Politécnico La Fe. Summary of the entire protocol applied is described in Figure 1. This project has been approved by the ethical committee and institutional review board (registration number 2020‐123‐1).
FIGURE 1.
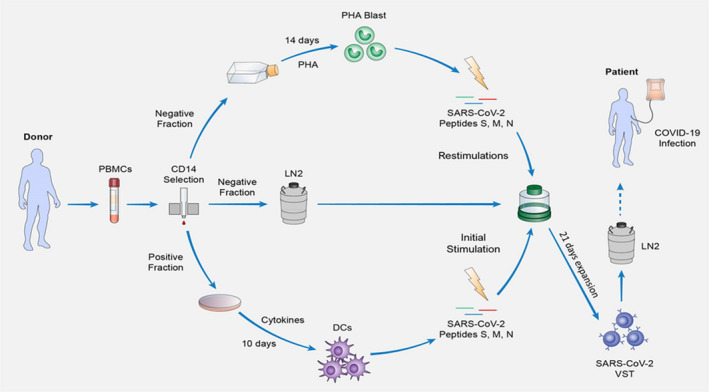
Expansion protocol. Schematic representation of the 31‐day expansion process of SARS‐CoV‐2 VST. The protocol starts with a density gradient separation of the blood sample donation to obtain peripheral mononuclear cells (PBMCs) that will be magnetically separated into CD14 positive and negative fractions. The CD14 positively selected monocytes are plated in petri dishes and stimulated with a cocktail of cytokines for a period of 10 days in order to differentiate them into DCs. From the CD14 negative fraction, a portion is cultivated in T75 flasks for 14 days in the presence phytohemagglutinin‐P (PHA) to induce lymphocyte blasts (PHA‐blasts), followed a 30 Gy gamma‐irradiation and cryopreservation until their usage as antigen presenting cells, while the rest of the negative fraction is cryopreserved until the start of the T cell culture. Once DCs are differentiated, this portion of the negative fraction is thawed and seeded on G‐Rex for an initial stimulation with SARS‐CoV‐2‐peptide loaded DCs (day 0), followed by 2 restimulations using peptide‐loaded PHA‐Blasts (day 7 and 14). On the last day of expansion (day 21), the culture is sampled for characterization and cells are harvested, aliquoted, cryopreserved, and stored in liquid nitrogen until their use for adoptive transfer into HLA‐matched severe COVID‐19 patients
2.2. HLA typing
HLA‐A, ‐B, ‐C, ‐DRB1, and ‐DQB1 loci typing of the blood donors included in the experiments was performed in the Histocompatibility Laboratory of the Valencia Transfusion Center by next generation sequencing (NGS) using commercially available reagents (GenDX, Utrecht, The Netherlands) and a MiniSeq platform (Illumina).
2.3. Diagnostic and serologic testing for SARS‐CoV‐2
SARS‐CoV‐2 diagnostic was performed by real‐time reverse transcriptase‐polymerase chain reaction (rRT‐PCR) by detection gene E and gene N SARS‐CoV‐2 Realtime PCR Kit (Vircell, Spain). Serologic testing was performed by qualitative determination by chemiluminescence immunoassay (CLIA) Maglumi 2019‐nCoV IgG, Maglumi 2019‐nCoV IgM (SNIBE Diagnostic, Shenzhen, China), and COVID‐19 VIRCLIA IgG, COVID‐19 VIRCLIA (IgM and IgA) (Vircell).
2.4. Clinical laboratory tests
The analytical panel designed to evaluate the hematological changes related to SARS‐CoV‐2 infection included absolute lymphocyte and platelet counts, lymphocyte subpopulations analysis, as well as the serum levels of C‐reactive protein, lactate dehydrogenase, D‐dimers, fibrinogen, ferritin, cardiac troponin, and IL‐6. Serum C‐reactive protein (CRP) and Ferritin were measured by an immunoturbidimetric assay (Roche Hitachi), IL‐6, and Procalcitonin by “ECLIA” method and LDH activity with the IFCC reference procedure.
2.5. Neutralization assay protocol
In the donors with PCR confirmed COVID‐19 history, the neutralization capacity of circulating antibodies against the spike protein of SARS‐CoV‐2 was assessed using a vesicular stomatitis virus pseudotyped with the SARS‐CoV‐2 Spike protein. Experiments were performed as previously described 18 with the exception that all tests were done in triplicate using fourfold antibody dilutions ranging from 1:20 to 1:20,480. The dose resulting in 50% neutralization was calculated using a three parameter logistic regression with the drc package in R using the LL3 function.
2.6. Generation of DCs
Donor‐derived dendritic cells (DCs) were generated from freshly PBMCs isolated by gradient centrifugation with Ficoll–Paque (Lymphoprep StemCell Technologies) from 50 mL of anticoagulated (acid‐citrate‐dextrose) venous blood. After separating the PBMCs, monocytes were isolated by immunomagnetic bead positive selection using the CliniMACS CD14 Reagent (Miltenyi Biotec, Germany) according to the manufacturer's instructions. At this point, 50% of the CD14 negative fraction was cryopreserved with 10% DMSO + 90% human AB serum in polystyrene cryovials and kept at –80°C until its further usage.
The CD14 + selected monocytes were matured for 10 days using a cocktail of cytokines into monocyte‐derived DCs. Briefly, cells were seeded in petri dishes at 37°C and 5% CO2 in IMDM (Lonza Walkersville, USA) in a concentration of 1 × 106 cells/mL and differentiated for the first 5 days in the presence of 1000 IU/mL Granulocyte‐Macrophage Colony‐stimulating factor (GM‐CSF) and interleukin (IL)‐4, with the media changed and cytokines supplemented every other day. On day 5, cells were washed and reseeded at the same concentration in IMDM media supplemented with 1 ug/mL Prostaglandin E2 (Sigma Aldrich), 400 IU/mL tumor necrosis factor‐alpha (TNFα), 1000 IU/mL IL‐1B, and 1000 IU/mL IL‐6 (all cytokines were manufactured by Miltenyi, unless otherwise stated). On day 7, IMDM was changed and cytokines supplemented at the same concentration as day 5. Finally, on day 10, DCs were harvested, counted, and gamma‐irradiated with 30 Gy of 137Cs before being peptide‐pulsed and used as antigen‐presenting cells in the first culture stimulation.
2.7. Generation of PHA blasts
The noncryopreserved CD14 negative fraction was used to generate PHA blasts required for repeated cycles of culture restimulation. Briefly, cells were culture for 7 days in T75 cm2 flasks at concentration of 1 × 106 cells/mL in RPMI 1640 (Sigma) supplemented with 10% Fetal Bovine Serum (FBS, Thermo Fisher) and in the presence of PHA (Mitogen Phytohemagglutinin‐P) at 1 mg/mL (Sigma). On day 7, cells were washed, counted, and reseeded at 1 × 106 cells/mL in RPMI 1640 supplemented with 10% Fetal Bovine Serum and in the presence of 100 IU/mL IL‐2 (Miltenyi). From days 7 to 14, cells were counted every other day and cellular concentration adjusted with fresh IL‐2 supplemented media. The stimulated blasts were then harvested, gamma‐irradiated with 30 Gy of 137Cs, and cryopreserved until further use.
2.8. Generation of SARS‐CoV‐2‐specific T cell lines
After the DCs differentiation, the previously cryopreserved CD14 negative fraction was thawed and cells seeded in a gas‐permeable rapid expansion cultureware (G‐Rex) system (Wilson Wolf Manufacturing) at a density of 1 × 106/cm2 in X‐VIVO™ 15 Medium (Lonza), supplemented with 5% human AB serum (obtained from AB donors pooled plasma) and 2 mM L‐glutamine (Lonza). The irradiated matured DCs were pulsed for 2 hours in RPMI 1640 with 1 µg/mL of proteins S, protein M, and protein N PepTivator SARS‐CoV‐2 (Miltenyi). These PepTivators are composed by a pool of lyophilized peptides, consisting mainly of 15‐mer sequences with 11 amino acids overlap, covering the immunodominant sequence domains of the surface (or spike) glycoprotein (S), covering the complete sequence of the membrane glycoprotein (M) and the complete sequence of the nucleocapsid phosphoprotein (N) of SARS‐CoV‐2 (GenBank MN908947.3, Protein QHD43416.1). DCs were then washed with RPMI and co‐cultured in the G‐Rex at 37°C, in 5% CO2 humidified atmosphere, at a ratio of 10:1 PBMC/DC for a period of 7 days. On day 7, cells were counted, fresh media supplemented with 20 IU/mL of IL‐2 was added and culture underwent a first restimulation with peptide‐pulsed PHA‐blast. The previously transformed and irradiated autologous blasts were thawed and loaded for 2 hours in RPMI with 1 µg/mL of each PepTivator. After a washing step, incubated PHA‐blasts were added into the G‐Rex at a ratio of 4:1 PBMC/PHA‐blast. From days 7 to 14 fresh media with 20 IU/mL IL‐2 was added to the culture, and on day 14 a similar restimulation step with loaded PHA‐blasts was performed. From days 14 to 21, the IL‐2 concentration was increased to 50 IU/mL. On day 21, VST were harvested, aliquoted, and cryopreserved. Cellular product characterization was performed by culture sampling at days 14 and 21 of culture.
2.9. T cell clonality testing
T cell clonality was assessed in ex vivo blood samples and in the final expanded cellular product using a commercial kit (Master Diagnostica) via multiplexed amplification of the TCR γ locus using standardized primer sets and qualitative interpretation of fragment size distributions by capillary electrophoresis according to the BIOMED‐2 protocol 19 , 20
2.10. Flow cytometry analysis
Flow cytometry analyses were performed on days 0, 14, and 21 of cell expansion. Briefly, cells were resuspended in RPMI in Falcon round‐bottom 5 mL FACS tubes at a concentration of 1 × 106 cells/mL and stimulated with 1 µg/mL of the specific peptides. About 10 ng/mL of PMA (phorbol 12‐myristate 13‐acetate) and 1 µM Ionomycin (Sigma Aldrich) were used for a positive control, and DMSO at the concentration contained in the PepTivator was used as a negative control. After 2 hours of stimulation, 10 μg Brefeldin A (Sigma‐Aldrich) was added to the tubes and kept overnight at 37°C in a 5% CO2 humidified atmosphere. Cells were then harvested, washed, permeabilized using Cytofix/Cytoperm (Becton Dickinson, San Jose, CA, USA), and stained with specific combinations of the following fluorochrome‐conjugated antibodies: CD3‐FITC, CD4‐PECy7, CD4‐BB700, CD8‐APCH7, CD19‐PeCy7, CD56‐APC, CD14‐V450, interferon gamma (IFNγ)‐PE and TNFα‐APC (BD Biosciences). For the analysis of degranulation and assessing cytotoxic potential, day 21 expanded lymphocytes were restimulated in vitro with 1 mg/mL of the corresponding peptide pool in the presence of antihuman CD107a‐BB700 antibody for 1 hour at 37°C in a humidified 5% CO2 atmosphere. DMSO was added as a negative control for spontaneous CD107a expression. After 1 hour, 1.2 µL/mL Monensin (2mM stock solution) and 10 μg Brefeldin A was added. Cells were then harvested, washed, and stained for additional surface molecules. In all staining, dead cells were excluded using the Fixable Viability Stain 510 (Becton Dickinson) and doublets were excluded by FSC‐A versus FSC‐H gating. Data were acquired on a FACSCanto II cytometer (Becton Dickinson) and analyzed using the FlowJo software (Tree Star, version 10, USA). A response was considered to be positive if the peptide stimulation was at least twice the background of the DMSO control sample from the same donor and higher than 0.15%. 21 A donor was considered to have a detectable T cell memory to SARS‐CoV‐2, if a positive response was detectable to any of the SARS‐CoV‐2 PepTivators.
2.11. Sterility testing and cryopreservation
Sterility of the expanded cells was assessed at the end of the culture by direct Gram staining, by testing for aerobic/anaerobic bacteria contamination using BacT/ALERT FA/FN Plus detection media (Biomerieux Diagnostics, USA) or fungal/mycobacterial contamination using BACTEC MYCO/F LYTIC (BD) medium and mycoplasma contamination was also ruled out by qPCR.
Expanded cells were aliquoted in polystyrene cryovials and cryopreserved with 10% DMSO + 90% human AB serum using a passive freezing container (Mr Frosty, Thermo Fischer Scientific) before being transfer into a liquid nitrogen tank.
3. RESULTS
3.1. Donor SARS‐CoV‐2 infection history
Clinical characteristics of each donor are detailed in Table 1. Donor 1 is a 45‐year‐old male with a history of SARS‐CoV‐2 infection after close contact with a COVID‐19 patient. He did not develop any clinical symptoms at any time. SARS‐CoV‐2 PCR positivity was confirmed at several time points in nasopharyngeal swab specimens, with the first negative rRT‐PCR occurring 42 days after the infectious contact. Serological testing at different time points (1, 2, and 4 months after the first positive PCR) was consistently negative.
TABLE 1.
Donors characteristics
| Parameter | Donor 1 | Donor 2 | Donor 3 | |
|---|---|---|---|---|
| Age (years) | 45 | 41 | 30 | |
| Sex | Male | Male | Male | |
| Ethnicity | Caucasian | Caucasian | Caucasian | |
| HLA class I/II | HLA‐A |
01:01:01 02:01:01 |
02:01:01 ‐ |
11:01 ‐ |
| HLA‐B |
18:01:01 35:02:01 |
49:01:01 51:01:01 |
08:01:01:01 55:01:01 |
|
| HLA‐C |
04:01:01 07:01:01 |
06:02:01:02 15:02:01:01 |
03:03:01:01 12:03:01:01 |
|
| HLA‐DRB1 |
03:01:01G 11:04:01 |
11:01:01 13:02:01 |
03:01:01 14:54:01 |
|
| HLA‐DQB1 |
02:01:01G 03:01P |
03:01P 06:04 |
02:01:01 05:03:01 |
|
| SARS‐CoV‐2 infection | Symptoms | No | Mild | No |
| rRT‐PCR swab test | Positive | Positive | NA | |
| Hospitalization requirement | No | No | NA | |
|
Time from infectious contact to negative rRT‐PCR (days) |
42 | 23 | NA | |
| Status at blood donation | Time since negative rRT‐PCR (days) | 47 | 26 | NA |
| Symptoms | No | No | No | |
| rRT‐PCR swab test | Negative | Negative | Negative | |
| Serologic testing | Negative | IgG+ | Negative | |
| Neutralizing Abs | No | Yes | NT | |
| T cell memory | Yes | Yes | No | |
Description of the relevant characteristics of each donor, their previous history in relation with SARS‐CoV‐2 infection and status at blood donation.
Abbreviations: NA, not applicable; NT, not tested.
Donor number 2 is a 41‐year‐old male with general mild symptoms, including myalgia and night‐sweat but without pneumonia, with a disease duration of 23 days from the first positive PCR to negativity. This donor's serological testing was consistently positive for anti‐SARS‐CoV‐2 IgG at several time points (1, 2, and 4 months after the first positive PCR).
Donor 3 is a 30‐year‐old male with no prior history of close contact with infected individuals and no symptoms of SARS‐CoV‐2 infection since the outbreak of the COVID‐19 pandemic in Spain, therefore representing individuals with no previous contact with this virus.
3.2. Donor immune characterization at blood donation
At the time of blood donation (3 months after the first positive PCR for donors 1 and 2), all donors were asymptomatic, and the absence of RNA viral shedding was confirmed via a swab test. Results of the analytical panel assessing COVID‐19 related laboratory changes at the time of donation were within the reference intervals. Additionally, the negativity of SARS‐CoV‐2 RNA in the blood samples was confirmed by rRT‐PCR prior to the expansions. At the time of donation, neutralizing antibody testing showed no neutralization activity at a serum dilution of 1:20 for donor 1 and 50% neutralizing activity at a dilution of 1:48 (SE ± 5.01) for donor 2 (Figure 2A).
FIGURE 2.
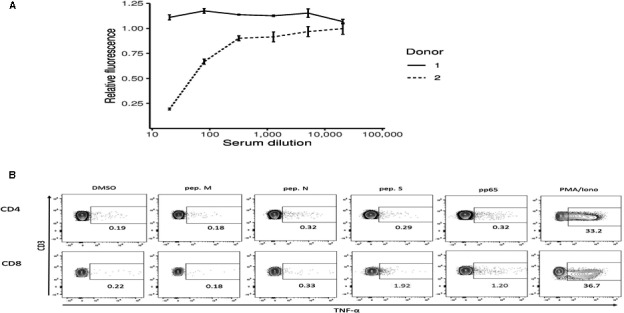
Donors characterization. (A) Neutralization capacity of circulating antibodies against the spike protein of SARS‐CoV‐2 assessed using a vesicular stomatitis virus pseudotyped with the SARS‐CoV‐2 Spike protein in both convalescent SARS‐CoV‐2 donors. (B) Example of the flow cytometry assessment of the anti‐SARS‐CoV‐2 T cell memory in donor 2. TNFα was selected as the read‐out, DMSO used as negative control and the CMV pp65 peptides and PMA/Ionomycin as positives controls
Assessment of T cell memory against SARS‐CoV‐2 was performed by flowcytometry in ex vivo fresh PBMCs samples measuring intracellular TNFα production in response to the different SARS‐CoV‐2 PepTivators (Figure 2B). CD8 + T cell responses were observed in both convalescent donors 1 and 2 for overlapping peptides of protein S (1.6% and 1.7%, respectively). However, both CD4 and CD8 responses for protein M and N could not reach sufficient levels to be deemed positive according to our defined threshold. In donor number 3, and in agreement with the absence of contact history with this new virus strain, no responses were detected to any of the SARS‐CoV‐2 peptides in neither the CD4 nor CD8 subpopulations.
3.3. Results of SARS‐CoV‐2 VST expansion
T cell expansion and cell cultures were analyzed for cell number growth, cellular content, specificity according to cytokine production (day 14), and cytotoxic potential (day 21), as detailed in Table 2.
TABLE 2.
Expansion results
| Parameter | Donor 1 | Donor 2 | Donor 3 | |||||
|---|---|---|---|---|---|---|---|---|
| Day 7 | Fold expansion | 0.76x | 0.63x | 0.36x | ||||
| Day 14 | Fold expansion | 1.6x | 1.8x | 0.96x | ||||
|
Specificity (% TNFα/IFNγ) |
CD4+ | Pep. M + N+S | 1.4 | 16.7 | 0 | |||
| CD8+ | Pep. M + N+S | 0 | 27.0 | 0 | ||||
| Day 21 | Fold expansion | 1.6x | 1.2x | |||||
|
Product identity (% of total) |
CD3+ | 89.3 | 62.7 | |||||
| CD4+ (% of CD3) | 60.1 | 58.6 | ||||||
| CD8+ (% of CD3) | 35.0 | 37.4 | ||||||
| CD56+ | 2.23 | 33.9 | ||||||
| CD19+ | 2.59 | 0.33 | ||||||
| CD14+ | 5.42 | 0.22 | ||||||
|
Cytotoxic Potential (% CD107a) |
CD4+ | Pep. M | 3.3 | 5.0 | ||||
| Pep. N | 0 | 0 | ||||||
| Pep. S | 12.8 | 20.4 | ||||||
| CD8+ | Pep. M | 0 | 12.7 | |||||
| Pep. N | 0 | 0 | ||||||
| Pep. S | 15.1 | 30.0 | ||||||
| Sterility | OK | OK | ||||||
Summary of the cultures cell growth number, composition, and specificity. For donor number 3, culture was ended on day 14 because no SARS‐CoV‐2 specificity could be identified.
In terms of cellular growth, we observed an expected decrease in total cell number during the first 7 days of culture, corresponding to the death of non‐SARS‐CoV‐2‐specific (by‐stander) cells. After the second stimulation with loaded autologous PHA‐blasts, cell cultures from donors 1 and 2 expanded, reaching at day 14 a higher cell number than at the start of culture (1.6x and 1.8x increase, respectively). For donor 3, a marked decreased occurred from day 0 to day 7, with some cell number recovery until day 14 but never reaching the initial cell count and with a progressive decreased viability of the remaining cells.
On day 14, cultures were analyzed for their specificity to the SARS‐CoV‐2‐peptides. Flow cytometry after stimulation with PepTivators M, N, and S, assessed TNFα and/or IFNγ‐production in order to quantify the bulk responses to the viral peptides (Figure 3A). For both donors 1 and 2, CD4 + SARS‐CoV‐2‐specific T cells could be identified (1.4% and 16.7%, respectively). For donor 2, 27% of the CD8 + T cells also responded to the viral antigens, representing the majority of the polyfunctional cells secreting TNFα and IFNγ simultaneously, suggesting their multifunctional effector potential. For donor number 3, besides the progressive decrease in cell numbers, T cells did not respond differently to the DMSO‐negative control and the SARS‐CoV‐2 peptides, but only to the PMA/Ionomycin positive control, assuring the assay validity. For this reason, the cell culture was considered as not specific and was terminated at this time point.
FIGURE 3.
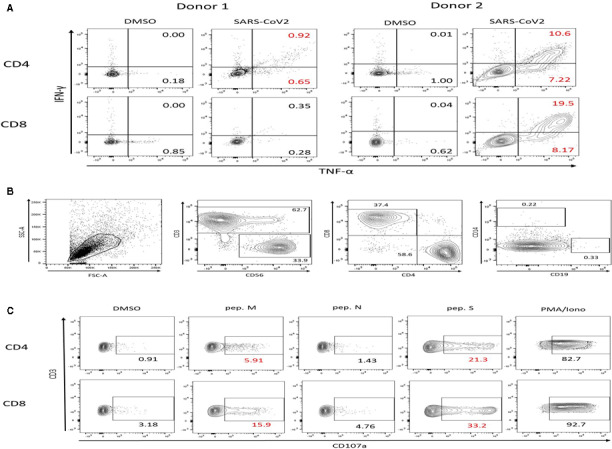
Cell cultures analysis. (A) Example of the flow cytometry analysis of T cell specificity at day 14 of culture for both donors 1 and 2. Cell responses to SARS‐CoV‐2 (M, N, and S) peptides were assessed by intracellular TNFα and IFNγ staining after overnight antigen restimulation. (B) Specific NK cells flow cytrometry analysis showing an important NK cell expansion (33.9%) in donor 2. (C) The responses were measured separately to the different viral PepTivators in order to discern the magnitude of response and expansion elicited to the epitopes of the protein M, N, and S. Cell frequencies are shown after removing the background
The end culture products (day 21) for both donors 1 and 2 mainly consisted of CD3 + cells (80.3% and 62.7%, respectively) and with a CD4 + dominance. Interestingly, in the culture from our serology positive donor 2, an important NK cell population expanded (CD56 + CD3‐; 33.9% of cells), in contrast with the culture from donor 1, where only 2.2% of CD56 + CD3− could be detected (Figure 3B). At this time point of the culture, the cytotoxic potential of the expanded VST was evaluated by measuring cell degranulation through CD107a staining. The responses were measured separately for each viral PepTivator in order to discern the magnitude of response and expansion elicited to the epitopes of the M, N and S proteins (Figure 3C). For both donors 1 and 2, strong degranulation responses were observed to overlapping peptide‐pool spanning the protein S from SARS‐CoV‐2 and in both CD4+ (12.8% and 20.4%) and CD8+ (15.1% and 30%) subpopulations, respectively. Additional CD4 responses to PepTivator M were identified for both donors 1 and 2 (3.3% and 5%). In terms of CD8 + T cell population responding to the membrane glycoprotein for SARS‐CoV‐2, responses could be detected in donor 1, yet these did not reach greater than twice the signal the background (our set criteria to be deemed positive, DMSO 3.23%, pep. M 5.77%). In contrast, a clear positive population of 12.7% CD107a + CD8+ T cells responded to this peptide after subtracting the background.
An additional T‐cell clonality study comparing samples from day 0 and day 21 supported cell expansion numbers and flowcytometry data. After expansion, multiple reproducible peaks on a polyclonal background could be detected in both donor 1 and donor 2, suggesting that the expanded cellular product is enriched for oligoclonal antigen‐specific T cells. The intensity of these peaks was more prominent for donor 2, which is also in accordance with the higher percentage of SARS‐CoV‐2 VSTs observed by flowcytometry (Figure 4).
FIGURE 4.
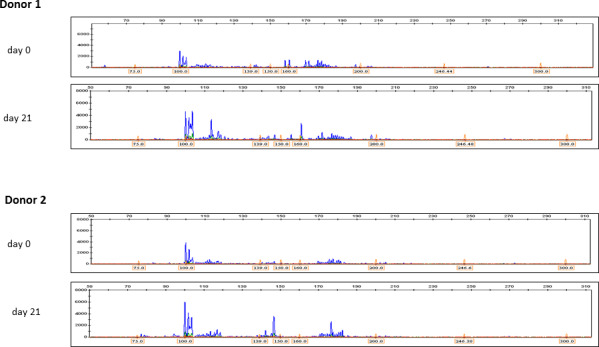
Clonality study. T cell clonality assessment via multiplexed amplification of the TCR γ locus in ex vivo blood samples (day 0) and in the final expanded cellular product (day 21) for both donor 1 and donor 2. Multiple reproducible peaks/bands could be found in donor 1 (seronegative and in which expansion of SARS‐Cov2‐specific was markedly inferior), suggesting the presence of multiple clones. For donor 2 (seropositive in which an effective expansion was observed), multiple reproducible peaks/bands were observed, suggesting an oligoclonal product
4. DISCUSSION
This proof‐of‐concept experiment shows that VST expansion against SARS‐CoV‐2 is feasible under GMP conditions and can achieve a nearly twofold increase in the number of this VST from patients with symptomatic and asymptomatic PCR‐confirmed SARS‐CoV‐2 infection. We observed a predominance of CD4 + T cells in the final VST product. The SARS‐CoV‐2 specificity of the expanded product was analyzed by flow cytometry showing a robust TNFα and IFNγ polarization after exposure to protein S, M, and N. Clonality studies of the final product also confirmed the presence of clonal VST against SARS‐CoV‐2 in both cases.
The two SARS‐CoV‐2 infected donors presented herein exhibited different patterns of VST expansion. We observed a higher number of expanded SARS‐CoV‐2 VST in donor 2 (symptomatic) in comparison to donor 1 (asymptomatic), suggesting that symptomatic donors could be more suitable for expansion. In contrast, we did not observe SARS‐CoV‐2 VST expansion in donor 3 (no prior history or contact with COVID‐19). The fact that donor 2 had neutralizing antibodies suggests a stronger T cell response and consequently a high number of expanded VST. Our observation is supported by prior findings, where the neutralizing antibody titers significantly correlated with the numbers of SARS‐CoV‐2 VST. 5 , 22 This suggests that a robust T cell response is required to achieve specific antibody response to SARS‐CoV‐2.
CD4 + T cells predominate in the final expanded VST product (60% in both donors). This fact is in line with prior reports where SARS‐CoV‐2‐specific CD4 + T cells predominate over CD8 + T cells. 5 , 12 , 23 However, we found a stronger cytokine response to protein S in CD8 + T cells in comparison to CD4 + T cells in both donors, as measured by the fraction of CD107a staining. This is very similar to influenza virus infection, where viral surface hemagglutinin elicits mostly CD4 + T cell responses, whereas the majority of CD8 + T cell responses are specific to viral internal proteins. 23 But a clear understanding of epitope sensitivity and HLA‐restrictions from the CD4 + and CD8 + T cells providing a protective immunity against SARS‐CoV‐2 is still lacking and merits further research. Another intriguing observation was the high percentage (33%) of NK cells obtained from donor 2 in the expanded VST product. Adaptive NK cells could also play a protective role against this new emerging virus. 24 , 25
Different approaches exist to obtain VST. One consists of isolating these T‐cells from whole blood or leukapheresis products with virus reactive cells using an automated device capturing IFNγ‐secreting cells. This approach has been already assayed using overlapping peptides of SARS‐CoV‐2 in COVID‐19 convalescent donors, but the reduced specific T cell number along with the presence of other nonspecific T cells in the final product represents important limitations. 26 To assure their clinical efficacy, HLA testing of donors and recipients is required in order to match HLA restrictions of the selected cells or even confirming the specificity of the IFNγ + cells. As an alternative, the application to SARS‐CoV‐2 of expansion protocols initially designed for CMV, EBV, and adenovirus (16) is feasible and leads to a highly specific VST product against SARS‐CoV‐2.
Donor 2 donation yielded a higher amount of SARS‐COV‐2 VST and could be considered as the most suitable donor. A starting bulk PBMC product with no detectable (by cytometry) CD4 memory and only 1% of CD8 + anti‐SARS‐CoV‐2 T cells could be enriched into a cellular product with a specificity of around 25% CD4 + and 42% CD8 + protein S and M specific VSTs. Although the PepTivator used herein did not include all SARS‐CoV‐2 proteins, we were able to expand enough specific SARS‐CoV‐2 VST. Although it is not clear which is the optimal dose of VST, it has been shown that an adoptive transfer of doses as low as 104 VST/Kg can expand in vivo and control viral replication in a high proportion of cases with a low incidence of graft versus host disease. 27 For clinical purposes, we can speculate that with a standard donation of 450 mL of whole blood could give rise to sufficient cells to treat five (70 Kg) COVID‐19 patients, assuming a starting cautious infusion dose of 0.5 × 106 VST/Kg.
Finally, we would like to highlight that the ex vivo expanded SARS‐COV‐2 VST product has several potential utilities for COVID‐19 research. First, it offers a higher number of VST. Second, it could help to identify the SARS‐CoV‐2 relevant antigens for T cell recognition for vaccine development. Third, it could be used to elucidate the immune profile of a protective T cell immunity against SARS‐CoV‐2. And at last, it could be useful to study the HLA restrictions from T cell receptors recognizing SARS‐CoV‐2 infected cells in order to optimize adoptive cellular therapies17
The limitations of the current experiments comprise the low number of donors included, the low blood volume used (50 mL) for expansion, and the fact that the peptides used during the expansion do not cover all SARS‐CoV‐2 proteins. It remains to be determined if the use of overlapping peptides spanning all SARS‐CoV‐2 proteins can improve protocol performance, increasing the number of expanded VST.
5. CONCLUSIONS
In this proof‐of‐concept study, we conclude that it is possible to expand ex vivo SARS‐CoV‐2 VSTs from convalescent donors of SARS‐CoV‐2 infection, using a standard protocol to obtain virus‐specific VSTs. Contrariwise, this approach was not successful to prime de novo responses to SARS‐CoV‐2 peptides in an uninfected healthy donor.
CONFLICT OF INTEREST
The author(s) declare that they have no conflict of interests.
AUTHOR CONTRIBUTIONS
Conceptualization and design, MG, C.A‐G., and JLP; Methodology C.A‐G., JM, C.F‐G., and VL; Formal Analysis, DP, MPC, MDG., EMG‐B., and CA; Investigation, AS, PS, I.G‐S., A.B‐R., AL, and I.L; Resources, AP, LL, JS, and CA; Review & Editing, JDLR, RG, and MAS; Funding Acquisition, GS; Project Administration, GS, JLP All authors conducted the experiments in their field of expertise and participated in the revision of the manuscript. All authors read and approved the final manuscript.
INSTITUTIONAL REVIEW BOARD STATEMENT
The study was conducted according to the guidelines of the Declaration of Helsinki, and approved by the Institutional Review Board (or Ethics Committee) of Hospital Universitario y Politécnico La Fe (protocol code: 2020‐123‐1 and date of approval: March 27, 2020)
INFORMED CONSENT STATEMENT
Informed consent was obtained from all subjects involved in the study.
Guerreiro M, Aguilar‐Gallardo C, Montoro J, et al. Adoptive transfer of ex vivo expanded SARS‐CoV‐2‐specific cytotoxic lymphocytes: A viable strategy for COVID‐19 immunosuppressed patients?. Transpl Infect Dis. 2021;23:e13602. 10.1111/tid.13602
Manuel Guerreiro and Cristóbal Aguilar‐Gallardo contributed equally to this work.
Funding information
The neutralization antibody assay was supported by Valencian government grant Covid_19‐SCI as well as the Spanish National Research Council grants CSIC‐COV19‐082 and CSIC‐COV‐19‐104 to RG.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available on request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.
REFERENCES
- 1. Piñana JL, Martino R, García‐García I, et al. Risk factors and outcome of COVID‐19 in patients with hematological malignancies. Exp Hematol Oncol. 2020;9:21. 10.1186/s40164-020-00177-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lee LYW, Cazier J‐B, Angelis V, et al. COVID‐19 mortality in patients with cancer on chemotherapy or other anticancer treatments: a prospective cohort study. Lancet (London, England). 2020;395:1919‐1926. 10.1016/S0140-6736(20)31173-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Solodky ML, Galvez C, Russias B, et al. Lower detection rates of SARS‐COV2 antibodies in cancer patients versus health care workers after symptomatic COVID‐19. Ann Oncol Off J Eur Soc Med Oncol. 2020;31:1087‐1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Au L, Boos LA, Swerdlow A, et al. Cancer, COVID‐19, and antiviral immunity: the CAPTURE study. Cell. 2020;183:4‐10. 10.1016/j.cell.2020.09.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Grifoni A, Weiskopf D, Ramirez SI, et al. Targets of T cell responses to SARS‐CoV‐2 coronavirus in humans with COVID‐19 disease and unexposed individuals. Cell. 2020;181:1489‐1501.e15. 10.1016/j.cell.2020.05.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Long Q‐X, Tang X‐J, Shi Q‐L, et al. Clinical and immunological assessment of asymptomatic SARS‐CoV‐2 infections. Nat Med. 2020;26:1200‐1204. 10.1038/s41591-020-0965-6 [DOI] [PubMed] [Google Scholar]
- 7. Wellinghausen N, Plonné D, Voss M, Ivanova R, Frodl R, Deininger S. SARS‐CoV‐2‐IgG response is different in COVID‐19 outpatients and asymptomatic contact persons. J Clin Virol Off Publ Pan Am Soc Clin Virol. 2020;130:104542. 10.1016/j.jcv.2020.104542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hung I‐N, Cheng V‐C, Li X, et al. SARS‐CoV‐2 shedding and seroconversion among passengers quarantined after disembarking a cruise ship: a case series. Lancet Infect Dis. 2020;20:1051‐1060. 10.1016/S1473-3099(20)30364-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Xiao T, Wang Y, Yuan J, et al. Early viral clearance and antibody kinetics of COVID‐19 among asymptomatic carriers. medRxiv. 2020. 10.1101/2020.04.28.20083139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Pancrazzi A, Magliocca P, Lorubbio M, et al. Comparison of serologic and molecular SARS‐CoV 2 results in a large cohort in Southern Tuscany demonstrates a role for serologic testing to increase diagnostic sensitivity. Clin Biochem. 2020;84:87‐92. 10.1016/j.clinbiochem.2020.07.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Le Bert N, Tan AT, Kunasegaran K, et al. SARS‐CoV‐2‐specific T cell immunity in cases of COVID‐19 and SARS, and uninfected controls. Nature. 2020;584:457‐462. 10.1038/s41586-020-2550-z [DOI] [PubMed] [Google Scholar]
- 12. Sekine T, Perez‐Potti A, Rivera‐Ballesteros O, et al. Robust T cell immunity in convalescent individuals with asymptomatic or mild COVID‐19. Cell. 2020;183:158‐168.e14. 10.1016/j.cell.2020.08.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mateus J, Grifoni A, Tarke A, et al. Selective and cross‐reactive SARS‐CoV‐2 T cell epitopes in unexposed humans. Science. 2020;370:89‐94. 10.1126/science.abd3871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. de Vries RD. SARS‐CoV‐2‐specific T‐cells in unexposed humans: presence of cross‐reactive memory cells does not equal protective immunity. Signal Transduct Target Ther. 2020;5:224. 10.1038/s41392-020-00338-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Saglio F, Hanley PJ, Bollard CM. The time is now: moving toward virus‐specific T cells after allogeneic hematopoietic stem cell transplantation as the standard of care. Cytotherapy. 2014;16:149‐159. 10.1016/j.jcyt.2013.11.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. O'Reilly RJ, Prockop S, Hasan AN, Koehne G, Doubrovina E. Virus‐specific T‐cell banks for “off the shelf” adoptive therapy of refractory infections. Bone Marrow Transplant. 2016;51:1163‐1172. 10.1038/bmt.2016.17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Barrett AJ, Prockop S, Bollard CM. Virus‐specific T cells: broadening applicability. Biol Blood Marrow Transplant. 2018;24:13‐18. 10.1016/j.bbmt.2017.10.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gozalbo‐Rovira R, Gimenez E, Latorre V, et al. SARS‐CoV‐2 antibodies, serum inflammatory biomarkers and clinical severity of hospitalized COVID‐19 patients. J Clin Virol. 2020;131:104611. 10.1016/j.jcv.2020.104611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. van Dongen JJM, Langerak AW, Brüggemann M, et al. Design and standardization of PCR primers and protocols for detection of clonal immunoglobulin and T‐cell receptor gene recombinations in suspect lymphoproliferations: report of the BIOMED‐2 Concerted Action BMH4‐CT98‐3936. Leukemia. 2003;17:2257‐2317. 10.1038/sj.leu.2403202 [DOI] [PubMed] [Google Scholar]
- 20. Langerak AW, Groenen P, Brüggemann M, et al. EuroClonality/BIOMED‐2 guidelines for interpretation and reporting of Ig/TCR clonality testing in suspected lymphoproliferations. Leukemia. 2012;26:2159‐2171. 10.1038/leu.2012.246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. McNeil LK, Price L, Britten CM, et al. A harmonized approach to intracellular cytokine staining gating: Results from an international multiconsortia proficiency panel conducted by the Cancer Immunotherapy Consortium (CIC/CRI). Cytometry A. 2013;83:728‐738. 10.1002/cyto.a.22319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ni L, Ye F, Cheng M‐L, et al. Detection of SARS‐CoV‐2‐specific humoral and cellular immunity in COVID‐19 convalescent individuals. Immunity. 2020;52:971‐977.e3. 10.1016/j.immuni.2020.04.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Peng Y, Mentzer AJ, Liu G, et al. Broad and strong memory CD4(+) and CD8(+) T cells induced by SARS‐CoV‐2 in UK convalescent individuals following COVID‐19. Nat Immunol. 2020;21:1336‐1345. 10.1038/s41590-020-0782-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Soleimanian S, Yaghobi R. Harnessing memory NK cell to protect against COVID‐19. Front Pharmacol. 2020;11:1309. 10.3389/fphar.2020.01309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Golchin A. Cell‐based therapy for severe COVID‐19 patients: clinical trials and cost‐utility. Stem Cell Rev Reports. 2020;17(1):56‐62. 10.1007/s12015-020-10046-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Leung W, Soh TG, Linn YC, et al. Rapid production of clinical‐grade SARS‐CoV‐2 specific T cells. Adv Cell Gene Ther. 2020;3(4):e101. 10.1002/acg2.101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Cobbold M, Khan N, Pourgheysari B, et al. Adoptive transfer of cytomegalovirus‐specific CTL to stem cell transplant patients after selection by HLA‐peptide tetramers. J Exp Med. 2005;202:379‐386. 10.1084/jem.20040613 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study are available on request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.