

Abstract
SARS‐CoV‐2 (severe acute respiratory syndrome coronavirus 2) is an emerging respiratory pathogen that has rapidly spread in human populations. Severe forms of infection associate cytokine release syndrome and acute lung injury due to hyperinflammatory responses even though virus clearance is achieved. Key components of inflammation include immune cell recruitment in infected tissues, a step which is under the control of endothelial cells. Here, we review endothelial cell responses in inflammation and infection due to SARS‐CoV‐2 together with phenotypic and functional alterations of monocytes, T and B lymphocytes with which they interact. We surmise that endothelial cells function as an integrative and active platform for the various cells recruited, where fine tuning of immune responses takes place and which provides opportunities for therapeutic intervention.
Keywords: COVID‐19, inflammation, immunomodulation
Abbreviations
- ACE2
angiotensin converting enzyme 2
- BAL
bronchoalveolar lavage
- cryo‐EM
cryoelectron microscopy
- DAMPS
damage associated molecular patterns
- EC
endothelial cells
- EVs
extracellular vesicles
- HCoV
common‐cold human coronavirus
- HLA
Human Leukocyte Antigens
- ICU
intensive care unit
- MERS
Middle East respiratory syndrome
- MHC
Major Histocompatibility Complex
- NOS
nitrogen oxide species
- NP
nucleocapside protein
- PRR
pattern recognition receptor
- ROS
reactive oxygen species
- SARS‐CoV‐2
severe acute respiratory syndrome coronavirus 2
- scRNAseq
single cell RNA seq
- S‐RBD
receptor binding domain of spike protein
- TEM
transendothelial migration
- TLR
toll‐like receptors
- TMPRSS2
trans‐membrane Protease Serine 2
- WPB
Weibel–Palade bodies
1. INTRODUCTION
The recent outbreak of a severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) infection and the resulting disease, COVID‐19, has caused significant morbidity and mortality in all countries with more than 2.6 million deaths and over 110 million infected people (Johns Hopkins Coronavirus Resource Center) as of March 2021. SARS‐CoV‐2 shares multiple similarities with SARS‐CoV, 1 , 2 and this is illustrated by similarities between their spike proteins. 3 An important target protein expressed on host cell membranes with a key role in COVID‐19 infection is Angiotensin Converting Enzyme 2 (ACE2). In the steady‐state ACE2 has a role in regulating blood pressure but in the COVID‐19 setting, ACE2 becomes a portal for viral entry to the cell. Upon the viral spike protein priming by the trans‐membrane protease serine 2 (TMPRSS2), ACE2‐mediated SARS‐CoV‐2 infection of the cell was shown by cryoelectron microscopy. SARS‐CoV‐2 spike protein directly binds to ACE2 4 , 5 , 6 with an even higher binding affinity than the spike protein of SARS‐CoV. 5 The importance of ACE2 in infection was demonstrated by a model of overexpression leading to more severe disease in mice. 7 Further studies (reviewed in 8 ) confirmed the critical role of ACE2 in allowing SARS‐CoV‐2 to enter host cells.
Many organs targeted by SARS‐CoV‐2 infection express ACE2, including the alveoli of the lung, which are covered with ACE2‐expressing epithelial cells. However, ACE2 expression is not restricted to epithelial cells. It is present in multiple extrapulmonary tissues including heart, kidneys, upper airways, the intestine, and blood vessels. 8 , 9 , 10 , 11 , 12 Expression of ACE2 is particularly detected on arterial smooth muscle cells as well as both arterial and venous endothelial cells. 12 Dysfunction of the endothelium contributes prominently to the pathophysiology of COVID‐19 and has been recently reviewed, with intricate disruptions of clotting, permeability, vascular tone, and angiogenesis. 13 , 14 , 15 , 16 In this review, we focus on the involvement of endothelial cells (EC) in inflammatory and immune responses to infection with SARS‐CoV‐2, together with alterations in T and B‐lymphocytes, and monocytes, which are major cell types interacting at focal areas of cell recruitment and immune tuning in the vessels of infected tissues.
1.1. Relevance of endothelial cell immune responses in SARS‐CoV‐2 infection
Different immune cell types converge at the barrier formed by EC. The innate and adaptive activities of endothelial cells, alongside the functions of assembled immune cells at a focal point in capillaries, create potential for the exchange of signals and dynamic fine‐tuning of immune responses. Endothelial dysregulation in patients with COVID‐19 has been strongly supported by the case reports of patients with endotheliitis in distinct vascular beds of different organs (lung, small intestine, heart, liver, and kidney) and evidence has been provided for direct viral infection of the EC and diffuse endothelial inflammation. 16
The ability of SARS‐CoV‐2 to infect human blood vessels was demonstrated in a study of capillary organoids developed from human induced pluripotent stem cells. 17 The validity of using such organoids to model vessels was underlined by their resemblance to human capillaries characterized by a lumen, a CD31+ endothelial lining, coverage by PDGFR+ pericytes and the presence of a basal membrane. 18 This was especially important regarding the mechanism of infection by SARS‐CoV‐2 because the size of infectious viral particles has been estimated as 80‐100 nm, 6 therefore even in the absence of tissue damage, traffic of the virus into the affected organs may occur after infection of vascular endothelial cells.
Evidence for modifications to the endothelium following infection has been demonstrated by assessing circulating biomarkers of endothelial function in COVID‐19 patients. 19 Both soluble E‐selectin and Angiopoietin‐2 were highly increased in a cohort of forty patients and whole blood gene expression analysis showed that increased E‐selectin associated with the degree of severity of disease in hospitalized patients. These data argue for altered endothelial function due to SARS‐CoV‐2 infection and in view of the role of the endothelium in immunoregulation, altered endothelial function may contribute to COVID‐19 disease. Historically, the endothelium was mainly considered as a barrier covering a vast surface area within the individual, the endothelial lining of blood vessels alone has been estimated as 350 m2. 20 However, it is not a homologous structure: macrovascular endothelium overlies arteries and veins while the microvascular endothelium covers arterioles, capillaries, and venules. As well as their barrier function between the circulation and the tissues, EC from different sites have functional specializations according to their tissue localization and activation status. The heterogeneity of EC types observed in different organs and in different vascular beds underlies the non‐uniformity of endothelial functions. 21 Recent data have pointed to the role of EC in the maintenance of immunological homeostasis and expression of viral, cytokine and toll‐like‐receptors allows EC to sense infectious threats and rapidly respond to inflammatory stimuli.
In the steady state, the constitutive but low level of expression of Human Leukocyte Antigens (HLA) molecules, co‐stimulatory and adhesion molecules permit endothelial maintenance of barrier function with minimal risk of initiating blood cell activation. However, under inflammatory conditions, increased expression of HLA and adhesion molecules (eg, ICAM‐1, VCAM‐1) promotes circulating immune cell recruitment and lengthened interactions with the endothelium that may ultimately contribute to transendothelial migration (TEM) mediated by three steps: rolling, activation and arrest. The molecular pathways involved in TEM vary according to the type of cell recruited, the vascular bed as well as the inflammatory signal, and TEM may be either paracellular or transcellular. 22 Because the expression of individual receptors and/or ligands varies according to the vascular bed and the inflammatory response, they play an active role in TEM. The endothelium also controls TEM by concentrating adhesion molecules and actin in the membrane surrounding leukocytes as they migrate (reviewed in 22). Yang et al reported that ICAM‐1 expression controlled transcellular TEM and that it was increased by over‐expression of ICAM‐1 23 and the role of endothelial ICAM‐1 expression in TEM was corroborated by van Buul et al. 24 However, because of the exceptional thinness of lung capillaries TEM may not follow the schema described above. 25
Increased expression of HLA and adhesion molecules permits EC to present antigen to CD8 and CD4 T lymphocytes although naïve T cell activation by human EC is obstructed by the lack of CD80 and CD86 co‐stimulatory molecule expression, this results in selective activation of memory T lymphocytes. 26 Endothelial cell activation also promotes maturation of memory CD8 into cytotoxic T lymphocytes, some of which are specific for EC targets 27 and the differentiation of Th17 CD4 in response to EC production of IL‐6. 28 Amplification of the Th1 subset by EC has been observed in vitro and in vivo in a model of vascular allograft transplantation. 29 , 30 The complement cascade has also been implicated in EC mediated T lymphocyte activation after triggering of the NLRP3 inflammasome and endothelial production of IL‐18. 31
1.2. Altered function of ECs in SARS‐CoV‐2 infection
Several studies of SARS‐CoV‐2 infection have pointed out its unique inflammatory response in vitro, in animal models and in patient samples. 32 The orientation of the immune response is associated with different clinical outcomes and COVID‐19 disease appears to be bi‐phasic with an initial stage of infection followed by an aggressive and sustained cytokine storm. Localization at the interface between the circulation and the tissues assigns a key role in signaling blood borne pathogens to EC, mediated by expression of PRR and DAMPS. Moreover, expression of cytokine receptors allows EC to rapidly react to inflammatory mediators and to amplify inflammation by increasing adhesion molecule expression. In this context, both micro and macro‐vascular EC produce high levels of pro‐inflammatory cytokines and immune‐cell attracting chemokines after activation either by other pro‐inflammatory cytokines; antibody‐binding; danger receptor activation or cellular interactions. 33 , 34 , 35 The co‐stimulatory molecule PD‐L1 is expressed by EC in the steady‐state and is highly increased in the presence of inflammation. However PD‐L1 may have a broader role than reported in immune regulatory responses, since PD‐L1 ligation has been recently implicated in endothelium activation and permeability to leukocytes. 36 The adhesion molecule E‐selectin, also involved in EC permeability, is only expressed by activated cells and is increased by inflammatory cytokines.
Although there is limited information concerning the effect of SARS‐Cov‐2 infection on endothelial permeability, existing data from both two and three‐dimensional models of the human blood‐brain barrier suggest disruption of the endothelium in the presence of the spike protein subunits. The impaired barrier function was simultaneous to increased expression of adhesion molecules and production of pro‐inflammatory cytokines and occurred without any change in endothelial viability. 37 However endothelial apoptosis has been reported after histological analysis of endothelial cells in the small intestine of a patient with COVID‐19 disease. 16 When EC viability in patients with Covid 19 was monitored by studying circulating CD146+ EC, significantly fewer apoptotic cells were detected than in healthy controls. Also, a positive correlation between the number of copies of SARS‐CoV‐2 RNA in the cellular fraction and the proportion of apoptotic circulating endothelial cell progenitors was observed in patients with severe COVID‐19, these data may suggest modified endothelial cell turnover in order to repair vascular damage in SARS‐CoV‐2 infection. 38
Production of extracellular vesicles (EVs) by endothelial cells has been examined after SARS‐CoV‐2 infection, and Krishnamacary et al reported that the contents of circulating EVs in plasma differ according to the severity of disease. The results of this study reiterate the importance of certain factors already described, for example ACE2 was present in circulating EVs, and factors indicating endothelial perturbation (TNF superfamily and IL‐6 family proteins) were higher in EVs from patients with severe and moderate disease. 39 Extracellular vesicles from patients with severe disease were enriched with a protein marker of macrophages, CD163, in comparison with EVs from patients with moderate disease. Extracellular vesicles containing such cargo may contribute to the inflammation associated with COVID‐19. Moreover, the same authors report that human pulmonary microvascular ECs underwent more cell death when exposed to EVs from patients with severe disease compared to EVs from asymptomatic patients. Finally, markers of endothelial activation were also increased in circulating EVs from patients, including Tissue Factor and von Willebrand Factor.
Endothelial cells play an important role in the thromboinflammatory as well as the coagulation response. They are characterized by their ability to synthesize Weibel–Palade bodies (WPB), specialized storage vesicles containing von Willebrand Factor, P‐selectin, Angiopoetin‐2, and chemokines implicated in these responses. In the context of SARS‐Cov‐2 infection, modified thrombotic and coagulation responses have been extensively reported (Reviewed in 13 and 40).
1.3. Cytokine profiles and endothelial cells in SARS‐CoV‐2 infected patients
Severely flawed type I IFN responses have been recently linked to high blood viral load in patients with severe and critical COVID‐19 versus mild to moderate disease. 41 A further study of a cohort of 63 patients with COVID‐19 reported the association between circulating IP10 levels and different degrees of disease severity as well as the ability of IP10 measures to discriminate severe from moderate and moderate from low levels of illness. This association between IP10 and disease severity was also reported in an early study of a COVID‐19 patient cohort in China. 42 IP10 levels also correlated with IL‐6 and IL‐10, however neither IL‐6 nor IL‐10, selectively associated with different degrees of disease severity. Thus, the flawed IFN type 1 response correlated with an excessive NF‐kB driven inflammatory response associated with heightened TNFα and IL‐6 levels. The high serum levels of IL‐6 and TNFα measured upon admission to hospital have been validated as predictors of disease severity and death independently of demographics, range of comorbidities, hypoxia and common laboratory inflammation markers. 43 Interestingly, this study examined both protein and transcriptional levels of IL‐6 in peripheral blood, IL‐6 protein was hugely elevated, but this increase was not reproduced at the transcriptional level. IL‐6 inducible genes were also highly increased (IL‐6R, STAT‐3). The authors pointed out that the discrepancy between RNA and protein levels may be due to IL‐6 and TNFα originating from non‐circulating cells such as endothelial cells. 41 The concentration of IL‐6 in blood did not correspond with the expression level in monocytes. 44
IL‐6 has been repeatedly associated with the COVID‐19‐associated cytokine storm with highly elevated circulating levels detected by different laboratories. Endothelial cells in the steady state secrete IL‐6 and this is a recurrent response of EC to activating stimuli; constitutive IL‐6 production by endothelial cells is increased under inflammatory conditions,
by interaction with non‐HLA matched PBMC, by antibody binding 45 and by TLR mediated activation. 36 , 46 Secretion of IL‐6 is amplified following EC activation in vitro and in vivo in models of organ transplantation. 28 , 29 Up‐regulation of IL‐6 and chemokines implicated in the recruitment of neutrophils, monocytes, T lymphocytes and natural killer cells were reported in serum samples from patients as well as increased chemokine gene expression in post‐mortem lung biopsy material. 32 This indicates that SARS‐CoV‐2 infection activates existing mechanisms of cellular recruitment within tissues. Increased CCL2 was associated with low numbers of circulating inflammatory monocytes in infected patients. 41 Since it has been reported that CCL2 production by EC is downstream of IL‐6 signaling 47 , 48 the observation that treatments with an anti‐IL‐6R monoclonal antibody are effective in COVID‐19 infected patients may be a result of the disruption of inflammatory mechanisms activated by IL‐6. 49 , 50 Microvascular EC also secrete RANTES and this is further increased by endothelial activation, 35 leukocyte recruitment by RANTES may contribute to their localization at particular sites of endothelial activation (resumed in Figure 1). Finally, in an IL‐6‐rich inflammatory setting, EC differentiation of pro‐inflammatory CD4 T subsets (Th17 and Th1) is promoted in vivo and in vitro whereas inhibition of IL‐6 interaction with the IL‐6R by an anti‐IL‐6R monoclonal antibody inhibited Th17 differentiation and biased HLA‐DR dependent CD4+ differentiation towards a CD54‐dependent Treg expansion. 28 , 29 The level of IL‐6 production may control whether the environment is more permissive to Treg or to pro‐inflammatory T‐CD4+ differentiation by the endothelial cell (see Figure 1).
FIGURE 1.
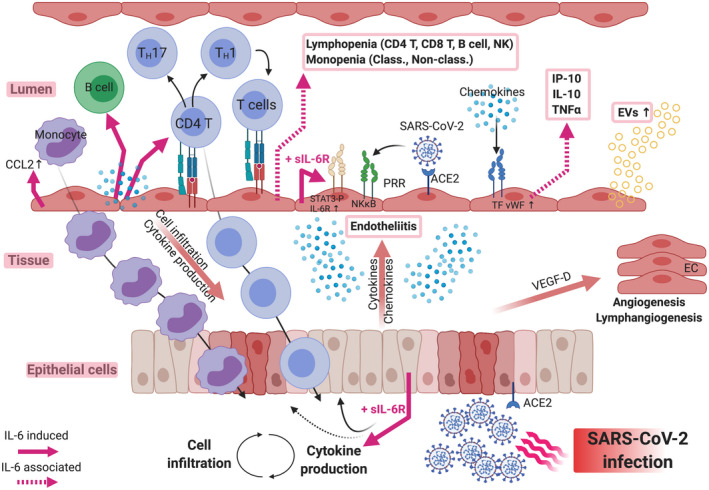
Roles of Interleukin‐6 produced by endothelial cells in inflammation and immune responses in the course of SARS‐CoV‐2 infection. Epithelial cell infection via the ACE2 receptor induces the production of cytokines and chemokines that activate proximal endothelial cells (EC). Activated EC strongly up‐regulate their expression of IL‐6, a pleiotropic alarm signal, which contributes to the development of immune responses (via direct signaling) and also to further inflammation after binding soluble IL‐6R. IL‐6 together with tissue and induced cytokines and chemokines such as CCL2 activate and recruit blood leukocytes. Activated and differentiated cells infiltrate infected tissues and target viruses and infected cells. Failure to eliminate the virus, to down‐regulate cytokine and chemokine expression and to limit tissue damage will result in amplification loops of inflammatory responses with increased endothelial permeability and further recruitment of activated immune cells. IL‐6 and soluble IL‐6R (red arrows) appear as major targets for therapeutic intervention
Endothelial cell activation resulting from signals in the underlying infected and inflamed tissue or from direct infection by the virus induces a pro‐adhesive and chemokine‐producing phenotype that will ultimately recruit circulating blood cells into the tissue. This cell influx, including monocytes and lymphocytes, will also exploit contact‐dependent mechanisms to contribute to innate and adaptative immune responses and viral clearance as well as detrimental inflammatory responses leading to tissue damage.
1.4. Monocyte activation in SARS‐CoV‐2 infected patients and tissue infiltration
During inflammation, large numbers of monocytes are recruited to the tissues and differentiate into inflammatory dendritic cells and macrophages. Recruited cells may also retain their monocyte phenotype after transmigration and are identified as inflammatory monocytes. 51 The proportion of CD16+ monocytes is dramatically increased during inflammation. Their numbers in the blood will determine their availability to interact with endothelial cells in inflamed tissues. Assigning a function to different monocyte populations has been difficult and controversial. Recent data indicate that inflammatory and anti‐inflammatory functions are not exclusive to any of the commonly defined subpopulations. 52
To identify biomarkers of disease progression in COVID‐19 patients, associations between disease severity and blood monocyte phenotypes were analyzed in numerous recent studies. Discrepancies between results were frequent and may be magnified by the lack of clear‐cut markers and boundaries between monocyte subpopulations, by the time at which samples were collected in a new infectious diseases with a highly variable incubation time and disease course, in addition to monocyte heterogeneity in the human population. 52 A study of a cohort of mostly severe COVID‐19 patients found minor modifications in monocyte populations compared to recovered patients and healthy controls. 53 However, others identified dramatic variations in monocyte subpopulation frequencies and phenotypes. CD169, a type I interferon inducible receptor, was overexpressed in monocytes of COVID‐19 patients. 54 CD169+ activated monocytes included classical, intermediate and nonclassical monocytes, and were found exclusively in SARS‐CoV‐2‐infected patients. 55 In mild cases of COVID‐19, the monocyte compartment consisted almost exclusively of CD169+ clusters. Extending these results, SIGLEC‐1, an interferon‐stimulated gene that encodes CD169, was one of the most highly expressed genes in classical monocytes in mild COVID‐19. 56 CD169 expression was noticeable in a fraction of classical monocytes in mild disease and was lower in severe forms. CD169 expression correlated with IFNα plasma levels. Expression of CD169 in monocytes was no longer detected 10( 56 ) and 20 days 55 after the onset of symptoms.
Intermediate CD14+CD16+ monocytes express CCR2 and CX3CR1 and are increased in numerous diseases, including infections, inflammatory syndromes, and auto‐immune diseases. Accordingly, in mildly affected COVID‐19 patients, the proportion of intermediate monocytes was strongly increased 57 , 58 , 59 and they expressed interferon‐stimulated genes. 57 However, no increase of this population was found in severe patients. 57 , 59 In contrast, others described elevated proportions of these cells in moderate and severe patients. 60 , 61 Still, intermediate monocytes were significantly reduced in mild COVID‐19 patients and were found at higher levels in some patients with severe disease compared to healthy controls, 55 an increase also found by others. 62 Intermediate monocyte clusters expressed high CD11c and HLA‐DR. 63
Nonclassical CD14dimCD16+ monocytes express CX3CR1, a receptor for fractalkine (CX3CL1), and include cells monitoring the integrity of the vasculature as well as monocytes capable of extravasation into inflamed tissues. The proportion of nonclassical monocytes was found to be reduced in all COVID‐19 patients 63 , 64 or only in severe COVID‐19 56 , 58 , 59 , 62 and the decrease correlated with high plasma IL‐6 levels. 59 A more in depth analysis identified a reduction only in a specific fraction of nonclassical monocytes in moderate and severe patients. 60 This subset also identified as Slan+ non‐classical monocytes is known as strong activator of immune responses in cooperation with CD4 and CD8 T cells, and with NK cells. In contrast, the remainder of CD14dimCD16+ monocytes were present in severe patients and strongly reduced in moderate patients with higher expression of HLA‐DR, PD‐L1, and PD‐L2. Reduced levels may relate to tissue infiltration or sequestration with a consequent rise in inflammatory responses. In contrast, non‐classical monocytes were found elevated in some severe patients and reduced only in mild COVID‐19. 55
Classical CD14+CD16‐ monocytes strongly express CCR2 which binds MCP‐1 (CCL2), and form the bulk of monocytes in the steady state. The proportion of classical monocytes was higher 65 or lower 55 during mild disease compared to healthy controls and was normal in late recovering severe patients. 65 In all patients, classical monocytes had increased expression of IL1β and a pattern of IFN‐activation. In mild COVID‐19, activated HLA‐DRhigh CD11chigh CD83+ classical monocytes were found in greater numbers, and this phenotype was sustained over 4 weeks together with an interferon‐stimulated gene activation pattern. 63 This contrasted with the profile detected early in severe patients with HLA‐DRdim CD163high classical monocytes (suggestive of anti‐inflammatory potential), that evolved to HLA‐DRdim S100Ahigh in the later phases of disease. This population was highly heterogeneous and seven sub‐clusters were identified. Notably, variable expression of CD62L, CD11c, and Ki67 defined the most abundant clusters. Related HLA class IIdim classical monocytes were found in different cohorts of severe COVID‐19 patients. 56 , 59 , 64 This phenotype was correlated with higher plasma levels of IL‐6. 59 An overall immature monocyte phenotype suggestive of cells produced during emergency myelopoiesis as seen in sepsis was also described. 64 Higher disease severity was associated with stronger CD11b. 56
Cytokine secretion is an essential function of monocytes that can be altered quantitatively, leading to hyper‐ or hypo‐reactivity, and through activation of different gene expression profiles affecting the pro‐ or anti‐inflammatory character of the immune response. Scarce and conflicting data are available in COVID‐19 patients. IL1β and CCL3 were strongly expressed in monocytes. 65 Yet, in severe patients hospitalized in ICU, pro‐inflammatory cytokine genes TNF, IL‐6, IL1β, CCL3, CCL4 or CXCL2 were not expressed by peripheral monocytes. 66 Monocytes also express numerous receptors for cytokines and chemokines that induce specific responses in the cells. Associations between soluble factors present in the environment and the presence of monocyte subpopulations and phenotypes were sought in COVID‐19 patients. At early stages of the infection, the occurrence of CD169+ monocytes correlated with the presence of MCP‐2 and IFNγ. 55 This association persevered during mild disease. At later stages, full recovery of nonclassical and intermediate monocytes was associated with serum CCL3 and CCL4. 55 Type I interferon signature responses in monocytes in milder COVID‐19 was consistently found. 56
Monocyte recruitment in the tissues is dependent on expression of chemokine receptors and their recruitment to the lungs of SARS‐CoV‐2 infected patients has been amply shown. Monocytes were found in higher numbers in bronchoalveolar lavage (BAL) fluids of mild COVID‐19 compared to control and severe groups, and they expressed interferon‐stimulated genes. 56 In severe disease, HLA‐DRlow monocytes expressed NOS and chemotaxis related genes. 56 Non‐classical monocytes with high expression of CD40 were enriched in dense bronchoscopy samples. 59 BAL fluids in severe COVID‐19 patients contained high proportions of activated SPP1+ resident macrophages and of monocyte‐derived FCN1+ macrophages, as well as macrophages of intermediate phenotype. 67 Activated SPP1+ resident macrophages expressed immuno‐modulatory function genes (CCL13/MCP‐4, TGFβ1, protease inhibitor alpha2‐macroglobulin), in contrast to intermediate macrophages which expressed monocyte recruiting chemokines (MCP‐1/CCL2, MIF‐1a/CCL3) and IP10/CXCL10, possibly contributing to an amplification of inflammatory responses. Wauters et al performed an extensive analysis of cells present in BAL fluid from mild and severe COVID‐19 patients by single cell RNAseq, that revealed that moderately inflammatory FCN1+ monocytes, with low MHC class II expression, were increased in severe COVID‐19 patients. In contrast, monocyte‐derived FABP4hi alveolar macrophages were decreased in these patients, suggesting an impediment to differentiation with accumulation of inflammatory cells in the alveolar spaces. 68 Algorithm‐based cell lineage tracing suggested that moderately inflammatory FCN1+ monocytes were close to transmigrated blood monocytes and acted as precursors for monocyte‐derived macrophages. Finally, metatranscriptome sequencing of BAL fluids in 19 patients identified up‐regulation of IL‐1, as well as the chemokines CXCL17, IL‐8, and MCP‐1 in COVID‐19 patients. 69 IL‐8 and MCP‐1 are major chemotactic factors for neutrophils and monocytes, and CXCL17 acts strongly on macrophages. 70 This pattern of responses in COVID‐19 patients was confirmed although not for the presence of IL‐1β, showing in addition a dramatic increase of IL‐6 in BAL fluids. 32 These results show that monocytes and derived cells constitute substantial populations that transmigrate in the inflamed alveolar spaces during SARS‐CoV‐2 infection, amplifying inflammation via cytokine and chemokine production that attracts more inflammatory cells. The monocyte populations involved in lung infiltration are not yet identified.
Although most monocytes respond to tissue inflammation during infection, they may themselves be a target of infection. Monocytes express ACE2 and can be infected by coronaviruses such as SARS‐CoV‐1 and MERS. 71 , 72 However, SARS‐CoV replicated poorly in infected cells. SARS‐CoV‐2 nucleoproteins were detected in lymph node macrophages 73 although it is unclear if this resulted from infection or from phagocytosis of viruses or infected cells. Virus entry in monocytes may also be enhanced by opsonization and Fc receptor internalization. 5 The effects of monocyte infection in virus dissemination remain to be determined.
Together these studies do not yet distinguish a clear pathway of population‐specific monocyte activation and/or loss in SARS‐CoV‐2 infection. This may be due to the heterogeneity of monocyte populations. In addition, the cell fate of differentiated monocytes following their interaction with endothelial cells and exposure to cytokines remains difficult to determine in tissues, muddling the respective contributions of distinct populations in inflammation and tissue repair.
1.5. SARS‐CoV‐2 infection elicits specific and efficient CD4 and CD8 T cell responses
The magnitude of lymphopenia and especially of CD4 and CD8 lymphopenia was initially reported in a small number of SARS‐CoV‐2 infected patients and then confirmed through meta‐analysis gathering 3017 subjects (76.6% classified as “mild/moderate” and 23.4% as “severe/critical”). 74 Severe/critical COVID‐19 patients exhibit 2.1 and 2.2 lower absolute numbers of CD4 and CD8 per µL respectively as compared to patients with moderate presentation of COVID‐19. B cell and NK (CD16+CD56+) lymphopenia was also observed but with a lower magnitude (1.5‐fold reduction). This lymphopenia does not seem to be explained by the recruitment of T cells to inflamed respiratory vascular endothelium or respiratory tract. Indeed, scRNAseq analysis of BAL fluid did not reveal exacerbated infiltration by lymphocytes 67 or in patient’s lung after autopsy. 75 The mechanisms leading to lymphopenia thus remain unidentified and several hypothesis have been proposed (hyperactivation of T cells leading to enhanced expression of pro‐apoptotic molecules, direct impact of IL‐6 and TNF).
The control of many viruses by mounting an effective T cell immune response usually requires 7 to 10 days and this is typically a critical time for COVID‐19 patients regarding development of severe disease or resolution of viral infection. 76 Whereas Spike, M and N proteins of SARS‐CoV‐2 elicit T cells responses, 77 It has been hypothesized that severe COVID‐19 could result from the inability to generate SARS‐CoV‐2 specific T cell responses. For instance, Sattler et al observed that the absence of S‐specific responses was more frequent in deceased patients. 78 Other data do not fully support this idea as the SARS‐CoV‐2 specific CD4 and CD8 T cell responses were robust and of similar magnitude in patients with mild or severe COVID‐19. 79 In support of the hypothesis of a harmful T cell response in patients with severe COVID‐19, the restriction and the quality of T cell responses were broader with higher magnitude in patients with severe disease 80 (see Figure 2).
FIGURE 2.
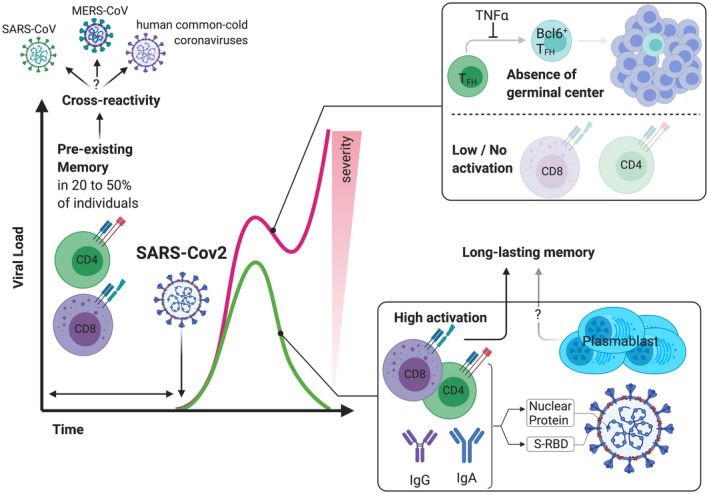
Differential T and B cell immune responses lead to a wide‐range of clinical outcome in patients with COVID‐19. SARS‐CoV‐2 infection results in a lymphopenia and especially of CD4 and CD8 lymphopenia. The control of many virus requires usually 7 to 10 days to mount an effective T cell immune response, and this is typically a critical time for COVID‐19 patients regarding development of severe disease or resolution of viral infection. The ability to generate SARS‐CoV‐2 specific T cell responses and to mount a humoral response (with up to 30% of plasma cells among B cells) has been associated with the ability to clear viral infection whereas patients with severe form of COVID‐19 exhibit low activation of CD4/CD8 T cells and a defect in the differentiation of TFH to Bcl6+ TFH that results in an inability to form germinal center. The CD4 and CD8 specific SARS‐CoV‐2 T cell response is often correlated with the titer of SARS‐CoV‐specific antibodies and protective memory T cells were still detectable even when humoral responses vanished. Finally, pre‐existing SARS‐CoV‐2 specific T cell memory is widely observed in the general population (20 to 50% of patients who had not been infected) suggesting a high cross‐reactivity of T cell responses
The quality of T cell responses may also strongly impact the clinical outcome and COVID‐19 mortality. To test this hypothesis, SARS‐CoV‐2 specific CD4 and CD8 responses were analyzed in patients with mild or severe disease. Longitudinal follow‐up of convalescing patients with mild COVID‐19 demonstrated that SARS‐CoV‐2 specific CD4 T cells were exclusively Th1 and mainly of central memory phenotype whereas SARS‐CoV‐2 specific CD8 T cells were predominantly TEMRA with a less differentiated phenotype (CD27+) than typical TEMRA cells 81 , 82 , 83 Strong production of IFNγ by CD4 and CD8 specific for SARS‐CoV‐2 was observed after stimulation. 80 , 81 SARS‐CoV‐2 specific CD4 T cells with cTfh were also documented. 81 , 82
At the time of SARS‐CoV‐2 clearance, an increase in activated T cells, defined as CD38+HLA‐DR+, especially in the CD8 compartment, was documented. 84 The increase in cells with a highly activated cytotoxic phenotype during the acute phase was later confirmed in a larger study of 206 patients, and they were identified as SARS‐CoV‐2 specific proliferating CD8. 85 High expression of CD38 by T cells is now considered a hallmark of acute COVID‐19 infection. The co‐expression of markers associated with an activated/cycling phenotype is often observed (such as the co‐expression of Ki67 with HLA‐DR and PD‐1). During the recovery phase, the phenotype of SARS‐CoV‐2 specific CD8 and CD4 changed and adopted preferentially a polyfunctional and stem‐like memory phenotype (CCR7+CD127+CD45RA±TCF1+). 85 The high expression of PD1 was found concomitantly with the expression of other inhibitory receptors (LAG3, TIM3, CTLA4, NKG2A, CD39) by CD8 53 , 86 , 87 , 88 , 89 , 90 , 91 and CD4 T cells 87 , 91 , 92 and may reflect their high level of activation or exhaustion. 93 The PDL‐1 protein dampens TCR mediated activation of lymphocytes in an expression‐dependent way and either a very low or a very high expression level has been associated with a loss of PDL‐1 inhibitory function. In the inflammatory setting of COVID‐19, highly elevated endothelial expression of PDL‐1 mediated by pro‐inflammatory cytokines may therefore lack the potential to decrease the pro‐inflammatory T cell response. Using peptide‐loaded HLA class I‐tetramer technology and functional assays, Schulien et al demonstrated a heterologous expansion of pre‐existing and newly induced memory CD8 T cells in patients with mild COVID‐19. 94 Finally, in mild COVID‐19, neither IL‐4 nor IL‐17 was detected in SARS‐CoV‐2 specific CD4+ T cells 81 and IL‐17 levels could serve as a biomarker to discriminate mild from severe COVID‐19 cases. 95
In situ analysis of lung biopsies and in BAL have been performed to question how the observations performed in peripheral blood reflect ongoing and local immune inflammation. Single cell RNA seq analysis of T lymphocytes of the upper respiratory tract of 19 COVID‐19 patients with moderate or critical disease revealed that CTL from patients with severe COVID‐19 had enhanced cytotoxic potential (with high expression of PRF1, GZMA and GZMB, cytotoxic receptors KLRB1, KLRC1 and KLRD1) but lower expression of pro‐inflammatory cytokines IFNγ and TNF. 96 The enhanced cytotoxic function of CTL observed in severe COVID‐19 patients is likely to contribute to damage to epithelial cells (ciliated and secretory cells). Indeed, pro‐apoptotic factors (eg cytochrome C, initiator caspase 8, CASP‐3) are upregulated in ciliated, secretory and FOXN4+ cells, the latter are only found in patients with COVID‐19. 96 The degree to which endothelial activation may directly act upon T lymphocyte recruitment, differentiation or indeed death in SARS‐CoV‐2 remains to be demonstrated although the potential for mediating indirect effects is clear.
1.6. SARS‐CoV‐2 primary responses and generation of protective memory T cells
The ability to mount protective and effective immune responses after primary exposure to SARS‐CoV‐2 is a pre‐requisite for the ongoing development of SARS‐CoV‐2 vaccines. Data from a non‐human primate model of SARS‐CoV‐2 shows that re‐challenge with the same strain of virus one month after primary infection leads to enhanced neutralizing antibody, immune responses, the absence of detectable viral dissemination and clinical symptoms of viral disease. 97 Interestingly, whereas CD4+ and CD8+ T cell responses correlated with the titers of SARS‐CoV‐2 IgG and IgA, 77 , 80 SARS‐CoV‐2 specific CD8+ T cells are more readily detected than antibodies specific for SARS‐CoV‐2‐spike protein 94 as SARS‐CoV‐2 specific CD8+ T cells were detected even in individuals seronegative for anti‐SARS‐CoV‐2 spike antibodies. Whereas COVID‐19 convalescent patients exhibit IgG and IgM responses to SARS‐CoV‐2 proteins (especially S‐RBD and NP), with high titers of neutralizing antibodies, 98 cellular responses against different proteins (Nucleocapside Protein NP, the main protease and receptor binding domain of spike protein S‐RBD) were also detected and the number of NP‐specific T cells correlated with the titer of neutralizing antibodies. 98 The frequent observation of loss of circulating antibodies despite robust memory T cell responses has raised the question of whether protection from severe forms of COVID‐19 can be achieved. Data obtained in the context of MERS and SARS‐CoV‐1 infection indicate that, whereas humoral responses waned, potent memory T cell response persisted for at least 6 years 99 , 100 , 101 and therefore suggest that potent adaptive immunity may protect against severe re‐infection.
The diversity of whole TCR repertoires has been shown to be greater in COVID‐19 patients with mild/moderate disease. 102 Nevertheless, the design of such analysis (ie, comparison of whole TCR repertoire between patients with different clinical outcomes) is unlikely to give meaningful information regarding the ongoing immune response against SARS‐CoV‐2 and the potential link between adaptive immune responses and recovery from viral infection. The dynamic tracking of SARS‐CoV‐2 specific T cell clones may provide more interesting information. Longitudinal monitoring identified CD4 and CD8 clones that contract after recovery as well as a second wave of expansion of CD4 T cell clonotypes. 83
Finally, elevated cross‐reactivity of T cell responses has been speculated upon, especially as the SARS‐CoV‐2 specific T cell response has been identified in 20 to 50% of patients who had not been infected 77 , 82 , 85 , 103 , 104 or in samples collected years before the current pandemic. 77 , 83 SARS‐CoV‐2 specific T cells may derive from memory T cell responses after exposure to common‐cold coronaviruses (HCoV) as nearly all of the human population have detectable IgG antibodies to HCoV in their serum 105 and as anti‐SARS‐CoV‐2 specific T cell responses are found in larger proportions compared with humoral responses defined by anti‐SARS‐CoV‐2 antibodies. The observation of pre‐existing memory raises the question of whether the memory response could provide protection from SARS‐CoV‐2 infection and thereby influence the course of disease. Despite the urgent need to develop a protective vaccine against SARS‐CoV‐2, it should be taken into account that the pre‐existence of memory T cells could be a confounding factor that will need consideration in the evaluation of the tested vaccines.
1.7. B lymphocytes and humoral responses in SARS‐CoV‐2 infection
Screening of 149 COVID‐19‐convalescent patients revealed high heterogeneity of the titers of pseudovirus antibody and one third of the patients had very low titers. 106 Neutralizing activity correlated with the duration and the severity of symptoms. 106 The range of anti‐SARS‐CoV‐2 Ig questions the ability to attain efficient humoral responses in severe forms of infection. Interestingly, the formation of germinal centers (GC), transient microstructures generated after the provision of help by Tfh to antigen‐activated B cells, is lacking in the context of severe/fatal SARS‐CoV‐2 infections, 107 recapitulating observations made during SARS 108 and during malaria infection. 109 The paucity of cTfh as well as the absence of GCs was associated with more‐severe disease. 86 This concords with the observation that an excess of TNFα resulting from enhanced Th1 responses and the blockade of Bcl‐6+ Tfh differentiation prevents the formation of GC. Furthermore, in‐depth characterization of B cell subsets in patients with different presentations of COVID‐19 revealed that naïve B cells, transitional B cells and CXCR5+ follicular B cells were markedly reduced in severely ill COVID‐19 patients. 107 As neutralizing anti‐SARS‐CoV‐2 antibodies are detected in large numbers of COVID‐19 patients, it is possible that the absence of GC reflects an ultimate stage of disease found only in severe forms of COVID‐19. The analysis of a greater number of patients revealed heterogeneity in the B cell response, for some patients, the plasmablast responses represented more than 30% of total cells despite low activated cTfh. 86 This massive plasmablast response was linked with an increase in highly activated CD4 and CD8, activated TEMRA CD8 and an altered cTfh response. 86 The frequency of B cell plasmablasts with high levels of the proliferation marker Ki‐67 and low levels of CXCR5 was significantly higher in patients with severe COVID‐19 53 and directly correlated with the expansion of antibody clones. 53 Finally, several studies have reported neutralizing antibodies in patients after recovery from COVID‐19, demonstrating the ability to mount effective humoral responses. 110 , 111 An original study design using biological samples retrieved from fishermen examined the correlation between protection from re‐infection and the presence of neutralizing antibodies. 112 Indeed, the analysis of the outbreaks on confined shipping vessels was a useful study to investigate protection from SARS‐CoV‐2 infection given the difficulty in implementing social distancing. In the report from Addetia et al, all 120 crew members were virus RT‐PCR negative pre‐departure and 3 individuals had pre‐existing neutralizing anti‐SARS‐CoV‐2 Ab. After a viral infection of than 85% of crew members, none of the three fishermen with pre‐existing antibodies were infected. These data provide evidence of protection against anti‐SARS‐CoV‐2 infection by anti‐SARS‐CoV‐2 neutralizing antibodies in humans 112 (see Figure 2). Little information is available regarding B lymphocyte interactions with the vascular endothelium either in the steady state or in SARS‐CoV‐2 infection. However indirect effects of pro‐inflammatory cytokines may also act on this population in addition to modified responses because of aberrant T lymphocyte activation and/or lymphopenia.
Altogether, the integration of large omics‐data with clinical features confirms the high heterogeneity of the cellular and humoral immune response in COVID‐19 patients 53 , 86 : CD4+ and CD8+ responses (activation and proliferation) were minimal (ie, comparable to control individuals) in about 20% of patients and such an immune profile was associated with less severe pathology. In the remaining 80% of patients, two immune profiles were observed: a rather classical anti‐viral response profile (effector CD8+ T cells, less CD4+ T cells activation and proliferating plasmablasts and memory B cells) or robust CD4+ activation, proliferating activated/exhausted CD8+ and increased Tbet+ PBs. 86
1.8. Future directions
Following infection by SARS‐Cov‐2, endothelial cells have a high and somewhat neglected potential to play a decisive role in the disease process for the following reasons : their number and distribution within different organs, disruption of their ability for immunological homeostasis after activation by cytokines produced in response to infection, their ability for soluble factor production (particularly IL‐6, CCL2 and IL‐8), their pro‐adhesive phenotype in the presence of inflammation that increases their potential to interact with and to regulate cognate responses by B and T lymphocytes as well as their interaction with monocyte sub‐populations; their active role in trans‐endothelial migration of myeloid cells. In the steady‐state, the endothelium can act as a platform for cell‐cell interactions and this particularly merits consideration in the context of SARS‐Cov‐2 infection where altered phenotypes and/or functions of each of the interacting cellular populations are observed. This review aims to document modifications of endothelial and endothelial‐interacting cell populations and to propose how infection could modify their interactions (both cell‐cell and soluble‐factor dependent). Future challenges will include identifying endothelial‐selective therapeutic targets interfering with the mechanisms of aberrant cell‐cell interactions in Covid‐19 disease. The most straightforward approaches to target endothelial interactions may entail a combination of inhibition of endothelial cytokines/chemokines with inhibition of their receptors on leukocytes (eg, IL‐6 and IL‐6R), targeting NF‐kB dependent pathways of endothelial cell activation, and masking of adhesion protein expression (possibly by soluble proteins, eg, sICAM‐1).
Modulation of IL‐6/IL‐6R signaling is an attractive aim for several reasons. In addition to being a key cytokine in endothelial biology, IL‐6 has an amplifying role in inflammation, 113 acts on numerous immune and non‐immune cell types, and contributes to reinforcement loops in the “cytokine storm”. IL‐6 is produced continuously and at high levels during SARS‐CoV‐2 infection, and in the clinical setting therapy can be initiated at any time during the course of the disease, and intervention does not necessarily lead to complete inhibition of the cytokine which may have deleterious effects with respect to secondary infections. 114 Finally, inhibitors of IL‐6/IL‐6R (such as Tocilizumab, Siltuximab, Clazakizumab) are already widely used and/or in advanced trials in other diseases, and are readily available to clinicians. The inhibition of other cytokines leading to endothelial activation is an important objective and relevant monoclonal antibodies are under study (eg, Elupalumab and Anakinra 115 ).
Nitric oxide, which is physiologically produced by endothelial cells, and its derivatives may also provide opportunities for a combined intervention on viral replication, respiratory and immune functions. Inhalation of nitric oxide, administered together with the vasoconstrictor almitrin improved oxygenation 116 and that may result from improved perfusion of ventilated areas of the lungs and gas exchange across the endothelial‐alveolar space in COVID related acute respiratory distress syndrome. Nitric oxide antagonized in vitro replication of closely related SARS‐CoV 117 and of SARS‐CoV‐2 118 in infected cells. In addition, nitric oxide shifts the differentiation of myeloid cells towards an anti‐inflammatory and tissue regenerative M2 spectrum of phenotypes by skewing the energetic metabolism of the cell. 119 Pharmaceutical products are also under investigation with the aim of limiting endothelial‐leukocyte interactions and preventing endothelial damage (eg, Defibrotide). A particular challenge stems from the increasingly reported long term effects of SARS‐CoV‐2 infection and the identification of underlying mechanisms of disease as well as therapeutic targets is crucial in this respect.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
AUTHOR CONTRIBUTIONS
N. Degauque, A. Haziot, S. Brouard, and N. Mooney have written the manuscript.
ACKNOWLEDGMENTS
This work was funded by a grant from the LabEX IGO program supported by the National Research Agency via the “Investment into the Future” program (ANR‐11‐LABX‐0016‐01) (SB, ND), PSPC (Nº DO50121115) (SB, ND), and supported in part by the RTRS Fondation de Coopération Scientifique CENTAURE (SB, ND), by the FP7 VISICORT project, which has received funding from the European Union’s Seventh Framework Programme for research, technological development and demonstration under grant agreement 602470 (SB, ND), and by “European Cooperation in Science & Technology under the COST Action CA17138 (Integrated European Network on Chronic Graft Versus Host Disease: EUROGRAFT) (https://www.gvhd.eu)” (NM). This research has also been funded by “Vaincre la Mucoviscidose” (NM, AH). Figures were created with BioRender.com <http://biorender.com.
Degauque N, Haziot A, Brouard S, Mooney N. Endothelial cell, myeloid, and adaptive immune responses in SARS‐CoV‐2 infection. The FASEB Journal. 2021;35:e21577. 10.1096/fj.202100024R
Alain Haziot, Nicolas Degauque, Nuala Mooney, Sophie Brouard are the authors contributed equally to this work, and are listed by alphabetical order.
Contributor Information
Sophie Brouard, Email: sophie.brouard@univ-nantes.fr.
Nuala Mooney, Email: nuala.mooney@univ-paris-diderot.fr.
REFERENCES
- 1. Andersen KG, Rambaut A, Lipkin WI, Holmes EC, Garry RF. The proximal origin of SARS‐CoV‐2. Nat Med. 2020;26:450‐452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Xia S, Liu M, Wang C, et al. Inhibition of SARS‐CoV‐2 (previously 2019‐nCoV) infection by a highly potent pan‐coronavirus fusion inhibitor targeting its spike protein that harbors a high capacity to mediate membrane fusion. Cell Res. 2020;30:343‐355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Xu X, Chen P, Wang J, et al. Evolution of the novel coronavirus from the ongoing Wuhan outbreak and modeling of its spike protein for risk of human transmission. Sci China Life Sci. 2020;63:457‐460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Walls AC, Park Y‐J, Tortorici MA, Wall A, McGuire AT, Veesler D. Structure, function, and antigenicity of the SARS‐CoV‐2 spike glycoprotein. Cell. 2020;181:281‐292.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wan Y, Shang J, Sun S, et al. Molecular mechanism for antibody‐dependent enhancement of coronavirus entry. J Virol. 2019;94:e02015‐19. /jvi/94/5/JVI.02015‐19.atom. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wrapp D, Wang N, Corbett KS, et al. Cryo‐EM structure of the 2019‐nCoV spike in the prefusion conformation. Science. 2020;367:1260‐1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Yang X‐H, Deng W, Tong Z, et al. Mice transgenic for human angiotensin‐converting enzyme 2 provide a model for SARS coronavirus infection. Comp Med. 2007;57:450‐459. [PubMed] [Google Scholar]
- 8. Zhang H, Rostami MR, Leopold PL, et al. Expression of the SARS‐CoV‐2 ACE2 receptor in the human airway epithelium. Am J Respir Crit Care Med. 2020;202:219‐229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Crackower MA, Sarao R, Oudit GY, et al. Angiotensin‐converting enzyme 2 is an essential regulator of heart function. Nature. 2002;417:822‐828. [DOI] [PubMed] [Google Scholar]
- 10. Danilczyk U, Penninger JM. Angiotensin‐converting enzyme II in the heart and the kidney. Circ Res. 2006;98:463‐471. [DOI] [PubMed] [Google Scholar]
- 11. Nie Y, Wang P, Shi X, et al. Highly infectious SARS‐CoV pseudotyped virus reveals the cell tropism and its correlation with receptor expression. Biochem Biophys Res Commun. 2004;321:994‐1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hamming I, Timens W, Bulthuis M, Lely A, Navis G, van Goor H. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J Pathol. 2004;203:631‐637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ackermann M, Verleden SE, Kuehnel M, et al. Pulmonary vascular endothelialitis, thrombosis, and angiogenesis in covid‐19. N Engl J Med. 2020;383:120‐128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Libby P, Lüscher T. COVID‐19 is, in the end, an endothelial disease. Eur Heart J. 2020;41:3038‐3044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bermejo‐Martin JF, Almansa R, Torres A, González‐Rivera M, Kelvin DJ. COVID‐19 as a cardiovascular disease: the potential role of chronic endothelial dysfunction. Cardiovasc Res. 2020;116:e132‐e133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Varga Z, Flammer AJ, Steiger P, et al. Endothelial cell infection and endotheliitis in COVID‐19. The Lancet. 2020;395:1417‐1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Monteil V, Kwon H, Prado P, et al. Inhibition of SARS‐CoV‐2 infections in engineered human tissues using clinical‐grade soluble human ACE2. Cell. 2020;181:905‐913.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wimmer RA, Leopoldi A, Aichinger M, et al. Human blood vessel organoids as a model of diabetic vasculopathy. Nature. 2019;565:505‐510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Smadja DM, Guerin CL, Chocron R, et al. Angiopoietin‐2 as a marker of endothelial activation is a good predictor factor for intensive care unit admission of COVID‐19 patients. Angiogenesis. 2020;23:611‐620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Pries AR, Secomb TW, Gaehtgens P. The endothelial surface layer. Pflüg Arch ‐ Eur J Physiol. 2000;440:653‐666. [DOI] [PubMed] [Google Scholar]
- 21. Dib H, Chafey P, Clary G, et al. Proteomes of umbilical vein and microvascular endothelial cells reflect distinct biological properties and influence immune recognition. Proteomics. 2012;12:2547‐2555. [DOI] [PubMed] [Google Scholar]
- 22. Muller WA. Transendothelial migration: unifying principles from the endothelial perspective. Immunol Rev. 2016;273:61‐75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Yang L, Froio RM, Sciuto TE, Dvorak AM, Alon R, Luscinskas FW. ICAM‐1 regulates neutrophil adhesion and transcellular migration of TNF‐α‐activated vascular endothelium under flow. Blood. 2005;106:584‐592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. van Buul JD, Allingham MJ, Samson T, et al. RhoG regulates endothelial apical cup assembly downstream from ICAM1 engagement and is involved in leukocyte trans‐endothelial migration. J Cell Biol. 2007;178:1279‐1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Rossaint J, Zarbock A. Tissue‐specific neutrophil recruitment into the lung, liver, and kidney. J Innate Immun. 2013;5:348‐357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hughes CC, Savage CO, Pober JS. Endothelial cells augment T cell interleukin 2 production by a contact‐dependent mechanism involving CD2/LFA‐3 interaction. J Exp Med. 1990;171:1453‐1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Biedermann BC, Pober JS. Human vascular endothelial cells favor clonal expansion of unusual alloreactive CTL. J Immunol Baltim MD 1950. 1999;162:7022‐7030. [PubMed] [Google Scholar]
- 28. Taflin C, Favier B, Baudhuin J, et al. Human endothelial cells generate Th17 and regulatory T cells under inflammatory conditions. Proc Natl Acad Sci. 2011;108:2891‐2896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Jane‐wit D, Manes TD, Yi T, et al. Alloantibody and complement promote T cell‐mediated cardiac allograft vasculopathy through noncanonical nuclear factor‐κB signaling in endothelial cells. Circulation. 2013;128:2504‐2516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lion J, Burbach M, Cross A, et al. Endothelial cell amplification of regulatory T cells is differentially modified by immunosuppressors and intravenous immunoglobulin. Front Immunol. 2017;8:1761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Liu L, Fang C, Fu W, et al. Endothelial cell‐derived interleukin‐18 released during ischemia reperfusion injury selectively expands T peripheral helper cells to promote alloantibody production. Circulation. 2020;141:464‐478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Blanco‐Melo D, Nilsson‐Payant BE, Liu W‐C, et al. Imbalanced host response to SARS‐CoV‐2 drives development of COVID‐19. Cell. 2020;181:1036‐1045.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Gatheral T, Reed DM, Moreno L, et al. A key role for the endothelium in NOD1 mediated vascular inflammation: comparison to TLR4 responses. PLoS One. 2012;7:e42386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Shibamiya A, Hersemeyer K, Schmidt Wöll T, et al. A key role for Toll‐like receptor‐3 in disrupting the hemostasis balance on endothelial cells. Blood. 2009;113:714‐722. [DOI] [PubMed] [Google Scholar]
- 35. Cross AR, Lion J, Poussin K, et al. HLA‐DQ alloantibodies directly activate the endothelium and compromise differentiation of FoxP3high regulatory T lymphocytes. Kidney Int. 2019;96:689‐698. [DOI] [PubMed] [Google Scholar]
- 36. Lomas‐Neira J, Monaghan SF, Huang X, Fallon EA, Chung C‐S, Ayala A. Novel role for PD‐1:PD‐L1 as mediator of pulmonary vascular endothelial cell functions in pathogenesis of indirect ARDS in mice. Front Immunol. 2018;9:3030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Buzhdygan TP, DeOre BJ, Baldwin‐Leclair A, et al. The SARS‐CoV‐2 spike protein alters barrier function in 2D static and 3D microfluidic in‐vitro models of the human blood‐brain barrier. Neurobiol Dis. 2020;146:105131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Mancuso P, Gidaro A, Gregato G, et al. Circulating endothelial progenitors are increased in COVID‐19 patients and correlate with SARS‐CoV‐2 RNA in severe cases. J Thromb Haemost. 2020;18:2744‐2750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Krishnamachary B, Cook C, Spikes L, Chalise P, Dhillon NK The potential role of extracellular vesicles in COVID‐19 associated endothelial injury and pro‐inflammation. medRxiv. 2020;2020.08.27.20182808 [Google Scholar]
- 40. Bikdeli B, Madhavan MV, Jimenez D, et al. COVID‐19 and thrombotic or thromboembolic disease: implications for prevention, antithrombotic therapy, and follow‐up. J Am Coll Cardiol. 2020;75:2950‐2973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hadjadj J, Yatim N, Barnabei L, et al. Impaired type I interferon activity and inflammatory responses in severe COVID‐19 patients. Science. 2020;369:718‐724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Yang Y, Shen C, Li J, et al. Plasma IP‐10 and MCP‐3 levels are highly associated with disease severity and predict the progression of COVID‐19. J Allergy Clin Immunol. 2020;146:119‐127.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Del Valle DM, Kim‐Schulze S, Huang H‐H, et al. An inflammatory cytokine signature predicts COVID‐19 severity and survival. Nat Med. 2020;26:1636‐1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kahn R, Schmidt T, Golestani K, et al. Mismatch between circulating cytokines and spontaneous cytokine production by leukocytes in hyperinflammatory COVID‐19. J Leukoc Biol. 2020;108(1):17–41. 10.1002/JLB.5COVBCR0720-310RR [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lion J, Taflin C, Cross AR, et al. HLA class II antibody activation of endothelial cells promotes Th17 and disrupts regulatory T lymphocyte expansion. Am J Transplant. 2016;16:1408‐1420. [DOI] [PubMed] [Google Scholar]
- 46. Davey MP, Martin TM, Planck SR, Lee J, Zamora D, Rosenbaum JT. Human endothelial cells express NOD2/CARD15 and increase IL‐6 secretion in response to muramyl dipeptide. Microvasc Res. 2006;71:103‐107. [DOI] [PubMed] [Google Scholar]
- 47. Romano M, Sironi M, Toniatti C, et al. Role of IL‐6 and Its soluble receptor in induction of chemokines and leukocyte recruitment. Immunity. 1997;6:315‐325. [DOI] [PubMed] [Google Scholar]
- 48. Zegeye MM, Lindkvist M, Fälker K, et al. Activation of the JAK/STAT3 and PI3K/AKT pathways are crucial for IL‐6 trans‐signaling‐mediated pro‐inflammatory response in human vascular endothelial cells. Cell Commun Signal. 2018;16:55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Xu X, Han M, Li T, et al. Effective treatment of severe COVID‐19 patients with tocilizumab. Proc Natl Acad Sci. 2020;117:10970‐10975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Toniati P, Piva S, Cattalini M, et al. Tocilizumab for the treatment of severe COVID‐19 pneumonia with hyperinflammatory syndrome and acute respiratory failure: a single center study of 100 patients in Brescia, Italy. Autoimmun Rev. 2020;19:102568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Jakubzick CV, Randolph GJ, Henson PM. Monocyte differentiation and antigen‐presenting functions. Nat Rev Immunol. 2017;17:349‐362. [DOI] [PubMed] [Google Scholar]
- 52. Merah‐Mourah F, Cohen SO, Charron D, Mooney N, Haziot A. Identification of novel human monocyte subsets and evidence for phenotypic groups defined by interindividual variations of expression of adhesion molecules. Sci Rep. 2020;10:4397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kuri‐Cervantes L, Pampena MB, Meng W, et al. Comprehensive mapping of immune perturbations associated with severe COVID‐19. Sci Immunol. 2020;5:eabd7114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Bedin A‐S, Makinson A, Picot M‐C, et al. Monocyte CD169 expression as a biomarker in the early diagnosis of COVID‐19. 2020.
- 55. Chevrier S, Zurbuchen Y, Cervia C, et al. A distinct innate immune signature marks progression from mild to severe COVID‐19. 2020. [DOI] [PMC free article] [PubMed]
- 56. Silvin A, Chapuis N, Dunsmore G, et al. Elevated calprotectin and abnormal myeloid cell subsets discriminate severe from mild COVID‐19. Cell. 2020;182:1401‐1418.e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Schulte‐Schrepping J, Reusch N, Paclik D, et al. Deutsche COVID‐19 OMICS Initiative (DeCOI). Suppressive myeloid cells are a hallmark of severe COVID‐19. 2020.
- 58. Gatti A, Radrizzani D, Viganò P, Mazzone A, Brando B. Decrease of non‐classical and intermediate monocyte subsets in severe acute sars‐cov ‐2 infection. Cytometry A. 2020;97:887‐890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Sanchez‐Cerrillo I, Landete P, Aldave B, et al. Differential redistribution of activated monocyte and dendritic cell subsets to the lung associates with severity of COVID‐19. 2020.
- 60. Padgett LE, Dinh HQ, Chee SJ, et al. Interplay of Monocytes and T Lymphocytes in COVID‐19 Severity. 2020.
- 61. Zhou Y, Fu B, Zheng X, et al. Aberrant pathogenic GM‐CSF + T cells and inflammatory CD14 + CD16 + monocytes in severe pulmonary syndrome patients of a new coronavirus. 2020.
- 62. Carsetti R, Zaffina S, Piano Mortari E, et al. Different innate and adaptive immune response to SARS‐CoV‐2 infection of asymptomatic, mild and severe cases. 2020. [DOI] [PMC free article] [PubMed]
- 63. Schulte‐Schrepping J, Reusch N, Paclik D, et al. Severe COVID‐19 Is marked by a dysregulated myeloid cell compartment. Cell. 2020;182:1419‐1440.e23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Reyes M, Filbin MR, Bhattacharyya RP, et al. An immune‐cell signature of bacterial sepsis. Nat Med. 2020;26:333‐340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Wen W, Su W, Tang H, et al. Immune cell profiling of COVID‐19 patients in the recovery stageby single‐cell sequencing. Cell Discov. 2020;6:31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Wilk AJ, Rustagi A, Zhao NQ, et al. A single‐cell atlas of the peripheral immune response in patients with severe COVID‐19. Nat Med. 2020;26:1070‐1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Liao M, Liu Y, Yuan J, et al. Single‐cell landscape of bronchoalveolar immune cells in patients with COVID‐19. Nat Med. 2020;26:842‐844. [DOI] [PubMed] [Google Scholar]
- 68. Wauters E, Van Mol P, Garg AD, et al. Discriminating mild from critical COVID‐19 by innate and adaptive immune single‐cell profiling of bronchoalveolar lavages. 2020. [DOI] [PMC free article] [PubMed]
- 69. Zhou Z, Ren L, Zhang L, et al. Heightened innate immune responses in the respiratory tract of COVID‐19 patients. Cell Host Microbe. 2020;27:883‐890.e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Burkhardt AM, Maravillas‐Montero JL, Carnevale CD, et al. CXCL17 is a major chemotactic factor for lung macrophages. J Immunol. 2014;193:1468‐1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Mesel‐Lemoine M, Millet J, Vidalain P‐O, et al. A human coronavirus responsible for the common cold massively kills dendritic cells but not monocytes. J Virol. 2012;86:7577‐7587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Yilla M, Harcourt BH, Hickman CJ, et al. SARS‐coronavirus replication in human peripheral monocytes/macrophages. Virus Res. 2005;107:93‐101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Feng Z, Diao B, Wang R, et al. The novel severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) directly decimates human spleens and lymph nodes. 2020. 10.1101/2020.03.27.20045427 [DOI]
- 74. Huang W, Berube J, McNamara M, et al. Lymphocyte subset counts in covid ‐19 patients: a meta‐analysis. Cytometry A. 2020;97:772‐776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Wichmann D, Sperhake J‐P, Lütgehetmann M, et al. Autopsy findings and venous thromboembolism in patients with COVID‐19: a prospective cohort study. Ann Intern Med. 2020;173:268‐277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Huang C, Wang Y, Li X, et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. The Lancet. 2020;395:497‐506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Grifoni A, Weiskopf D, Ramirez SI, et al. Targets of T Cell responses to SARS‐CoV‐2 coronavirus in humans with COVID‐19 disease and unexposed individuals. Cell. 2020;181:1489‐1501.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Sattler A, Angermair S, Stockmann H, et al. SARS‐CoV‐2 specific T‐cell responses and correlations with COVID‐19 patient predisposition. J Clin Invest. 2020;130(12):6477–6489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Thieme CJ, Anft M, Paniskaki K, et al. Robust T Cell response toward spike, membrane, and nucleocapsid SARS‐CoV‐2 proteins is not associated with recovery in critical COVID‐19 patients. Cell Rep. Med. 2020;1(6):100092. 10.1101/2020.03.27.20045427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Oxford Immunology Network Covid‐19 Response T cell Consortium , ISARIC4C Investigators , Peng Y, et al. Broad and strong memory CD4+ and CD8+ T cells induced by SARS‐CoV‐2 in UK convalescent individuals following COVID‐19. Nat Immunol. 2020;21:1336‐1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Neidleman J, Luo X, Frouard J, et al. SARS‐CoV‐2‐specific T cells exhibit phenotypic features of helper function, lack of terminal differentiation, and high proliferation potential. Cell Rep. Med. 2020;1:100081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Weiskopf D, Schmitz KS, Raadsen MP, et al. Phenotype and kinetics of SARS‐CoV‐2‐specific T cells in COVID‐19 patients with acute respiratory distress syndrome. Sci Immunol. 2020;5:eabd2071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Minervina AA, Komech EA, Titov A, et al. Longitudinal high‐throughput TCR repertoire profiling reveals the dynamics of T cell memory formation after mild COVID‐19 infection. eLife. 2021;10:e63502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Thevarajan I, Nguyen THO, Koutsakos M, et al. Breadth of concomitant immune responses prior to patient recovery: a case report of non‐severe COVID‐19. Nat Med. 2020;26:453‐455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Sekine T, Perez‐Potti A, Rivera‐Ballesteros O, et al. Robust T cell immunity in convalescent individuals with asymptomatic or mild COVID‐19. Cell. 2020;183:158‐168.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Mathew D, Giles JR, Baxter AE, et al. Deep immune profiling of COVID‐19 patients reveals distinct immunotypes with therapeutic implications. Science. 2020;369:eabc8511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Diao B, Wang C, Tan Y, et al. Reduction and functional exhaustion of T cells in patients with coronavirus disease 2019 (COVID‐19). Front Immunol. 2020;11:827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Laing AG, Lorenc A, Del Barrio IDM, et al. A dynamic COVID‐19 immune signature includes associations with poor prognosis. Nat Med. 2020;26:1623‐1635. 10.1038/s41591-020-1038-6 [DOI] [PubMed] [Google Scholar]
- 89. Zheng M, Gao Y, Wang G, et al. Functional exhaustion of antiviral lymphocytes in COVID‐19 patients. Cell Mol Immunol. 2020;17:533‐535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Zheng H‐Y, Zhang M, Yang C‐X, et al. Elevated exhaustion levels and reduced functional diversity of T cells in peripheral blood may predict severe progression in COVID‐19 patients. Cell Mol Immunol. 2020;17:541‐543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Song J‐W, Zhang C, Fan X, et al. Immunological and inflammatory profiles in mild and severe cases of COVID‐19. Nat Commun. 2020;11:3410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Mazzoni A, Salvati L, Maggi L, et al. Impaired immune cell cytotoxicity in severe COVID‐19 is IL‐6 dependent. J Clin Invest. 2020;130:4694‐4703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Riella LV, Paterson AM, Sharpe AH, Chandraker A. Role of the PD‐1 pathway in the immune response. Am J Transplant. 2012;12:2575‐2587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Schulien I, Kemming J, Oberhardt V, et al. Characterization of pre‐existing and induced SARS‐CoV‐2‐specific CD8+ T cells. Nat Med. 2021;27:78‐85. 10.1038/s41591-020-01143-2 [DOI] [PubMed] [Google Scholar]
- 95. Pacha O, Sallman MA, Evans SE. COVID‐19: a case for inhibiting IL‐17? Nat Rev Immunol. 2020;20:345‐346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Chua RL, Lukassen S, Trump S, et al. COVID‐19 severity correlates with airway epithelium‐immune cell interactions identified by single‐cell analysis. Nat Biotechnol. 2020;38:970‐979. [DOI] [PubMed] [Google Scholar]
- 97. Deng W, Bao L, Liu J, et al. Primary exposure to SARS‐CoV‐2 protects against reinfection in rhesus macaques. Science. 2020;369:818‐823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Ni L, Ye F, Cheng M‐L, et al. Detection of SARS‐CoV‐2‐specific humoral and cellular immunity in COVID‐19 convalescent individuals. Immunity. 2020;52:971‐977.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Tang F, Quan Y, Xin Z‐T, et al. Lack of peripheral memory B Cell responses in recovered patients with severe acute respiratory syndrome: a six‐year follow‐up study. J Immunol. 2011;186:7264‐7268. [DOI] [PubMed] [Google Scholar]
- 100. Shin H‐S, Kim Y, Kim G, et al. Immune responses to Middle East respiratory syndrome coronavirus during the acute and convalescent phases of human infection. Clin Infect Dis. 2019;68:984‐992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Alshukairi AN, Khalid I, Ahmed WA, et al. Antibody response and disease severity in healthcare worker MERS survivors. Emerg Infect Dis. 2016;22(6):1113–1115. 10.3201/eid2206.160010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Huang L, Shi Y, Gong B, et al. Blood single cell immune profiling reveals the interferon‐MAPK pathway mediated adaptive immune response for COVID‐19. 2020. 10.1101/2020.03.15.20033472 [DOI]
- 103. Braun J, Loyal L, Frentsch M, et al. SARS‐CoV‐2‐reactive T cells in healthy donors and patients with COVID‐19. Nature. 2020;587(7833):270‐274. [DOI] [PubMed] [Google Scholar]
- 104. Le Bert N, Tan AT, Kunasegaran K, et al. SARS‐CoV‐2‐specific T cell immunity in cases of COVID‐19 and SARS, and uninfected controls. Nature. 2020;584:457‐462. [DOI] [PubMed] [Google Scholar]
- 105. Gorse GJ, Patel GB, Vitale JN, O’Connor TZ. Prevalence of antibodies to four human coronaviruses is lower in nasal secretions than in serum. Clin Vaccine Immunol. 2010;17:1875‐1880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Robbiani DF, Gaebler C, Muecksch F, et al. Convergent antibody responses to SARS‐CoV‐2 in convalescent individuals. Nature. 2020;584:437‐442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Kaneko N, Kuo H‐H, Boucau J, et al. Loss of Bcl‐6‐expressing T follicular helper cells and germinal centers in COVID‐19. Cell. 2020;183:143‐157.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Gu J, Gong E, Zhang B, et al. Multiple organ infection and the pathogenesis of SARS. J Exp Med. 2005;202:415‐424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Ryg‐Cornejo V, Ioannidis LJ, Ly A, et al. Severe malaria infections impair germinal center responses by inhibiting T follicular helper cell differentiation. Cell Rep. 2016;14:68‐81. [DOI] [PubMed] [Google Scholar]
- 110. Ju B, Zhang Q, Ge J, et al. Human neutralizing antibodies elicited by SARS‐CoV‐2 infection. Nature. 2020;584:115‐119. [DOI] [PubMed] [Google Scholar]
- 111. Wang C, Li W, Drabek D, et al. A human monoclonal antibody blocking SARS‐CoV‐2 infection. Nat Commun. 2020;11:2251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Addetia A, Crawford KH, Dingens A, et al. Neutralizing antibodies correlate with protection from SARS‐CoV‐2 in humans during a fishery vessel outbreak with high attack rate. J Clin Microbiol. 2020;58(11):e02107‐20. 10.1128/JCM.02107-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Hirano T. IL‐6 in inflammation, autoimmunity and cancer. Int Immunol. 2021;33:127‐148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Han Q, Guo M, Zheng Y, et al. Current evidence of interleukin‐6 signaling inhibitors in patients with COVID‐19: a systematic review and meta‐analysis. Front Pharmacol. 2020;11:615972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Swedish Orphan Biovitrum . A phase 2/3, randomized, open‐label, parallel group, 3‐arm, multicenter study investigating the efficacy and safety of intravenous administrations of emapalumab, an anti‐interferon gamma (Anti‐IFNγ) monoclonal antibody, and anakinra, an interleukin‐1(IL‐1) receptor antagonist, versus standard of care, in reducing hyper‐inflammation and respiratory distress in patients with sars‐cov‐2 infection. 2020.
- 116. Bagate F, Tuffet S, Masi P, et al. Rescue therapy with inhaled nitric oxide and almitrine in COVID‐19 patients with severe acute respiratory distress syndrome. Ann Intensive Care. 2020;10:151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Akerström S, Mousavi‐Jazi M, Klingström J, Leijon M, Lundkvist A, Mirazimi A. Nitric oxide inhibits the replication cycle of severe acute respiratory syndrome coronavirus. J Virol. 2005;79:1966‐1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Akaberi D, Krambrich J, Ling J, et al. Mitigation of the replication of SARS‐CoV‐2 by nitric oxide in vitro. Redox Biol. 2020;37:101734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Palmieri EM, McGinity C, Wink DA, McVicar DW. Nitric oxide in macrophage immunometabolism: hiding in plain sight. Metabolites. 2020;10:429. [DOI] [PMC free article] [PubMed] [Google Scholar]