

Abstract
Carmi syndrome is a rare and severe disease defined by pyloric atresia and junctional epidermolysis bullosa. There are no clear recommendations when to consider a curative therapy, including surgical repair of pyloric atresia and when to transition to palliative care.
We report the case of a female preterm infant suffering from Carmi syndrome. After definitive diagnosis and appropriate ethical counselling, we decided for surgical repair of the pyloric atresia. Nonetheless, there was no clinical improvement and our patient died after 35 days. Reviewing the literature, we found immunofluorescence microscopy to be most decisive examination to determine the prognosis of this severe disease.
Keywords: JEB-PA, Pyloric atresia, Junctional epidermolysis bullosa, Prognostic factors
Highlights
-
•
Carmi syndrome (JEP-PA) is a rare and severe disease defined by pyloric atresia and junctional epidermolysis bullosa.
-
•
The overall mortality is reported to be 75%.
-
•
Decisive for prognosis is the quantity of integrin evaluated by immunofluorescence microscopy.
-
•
Decision about curative therapy or a palliative approach should be made in an interdisciplinary clinical committee
1. Introduction and importance
Carmi syndrome (JEB-PA) is a rare, autosomal recessive disease. It is defined by pyloric atresia (PA) and junctional epidermolysis bullosa (JEB). About 100 cases have been published so far [7,8,11]. The mortality is reported to be at least 75% [11]. There are no clear recommendations when to consider a curative therapy, including surgical repair of PA, and when to consider a palliative approach.
We present the case of a preterm infant in whom JEB-PA was diagnosed shortly after birth. Pitfalls occurring in practical management and decision making are discussed. This case reports follows the criteria published in the SCARE guidelines [1].
2. Case presentation
2.1. Admission and family history
A female preterm infant of 32 1/7 weeks of gestation and a birth weight of 1400 g was referred to our neonatal intensive care on the first day of life. The girl had been delivered by urgent cesarean section due to placental abruption and was breathing spontaneously. She showed aplasia cutis congenita (ACC) with extensive major skin lesions at all four extremities, the forehead and the periumbilical region (see Fig. 1). Fingers and toes of the left hand and foot, as well as the right ear were dysplastic (see Fig. 2). Antibacterial therapy was initiated. A babygram revealed the typical single bubble sign (see Fig. 3). Additionally, a perimembranous ventricular septal defect, a persistent ductus arteriosus, and unilateral left multicystic kidney with megaureter were also present.
Fig. 1.
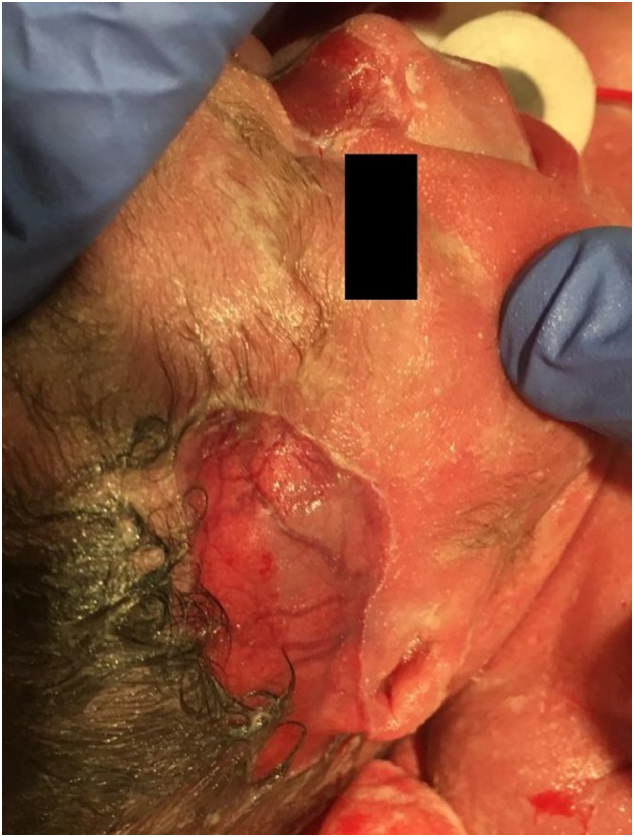
Aplasia cutis congenita on temple and nose and mutilated right ear. The photograph was taken shortly after birth.
Fig. 2.
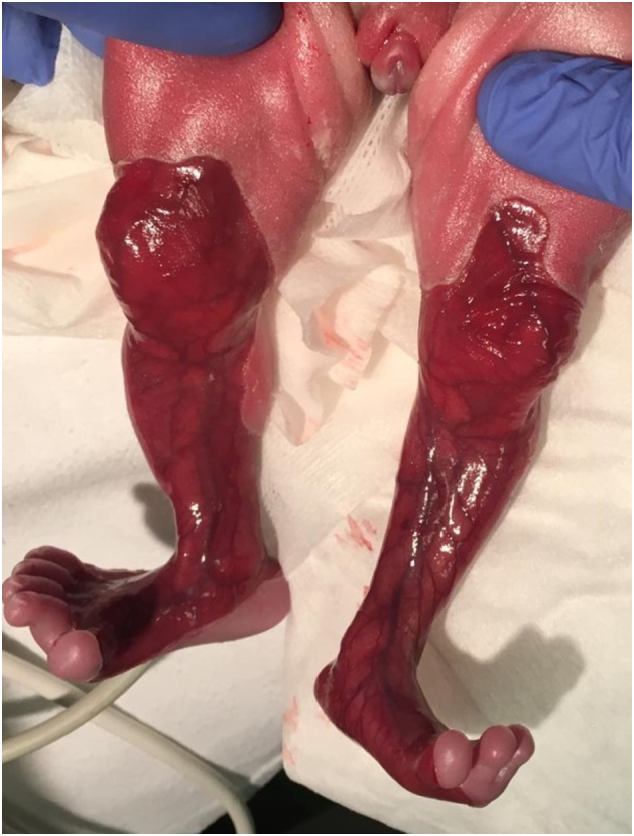
Aplasia cutis congenita on knees, lower legs and feet.
Fig. 3.
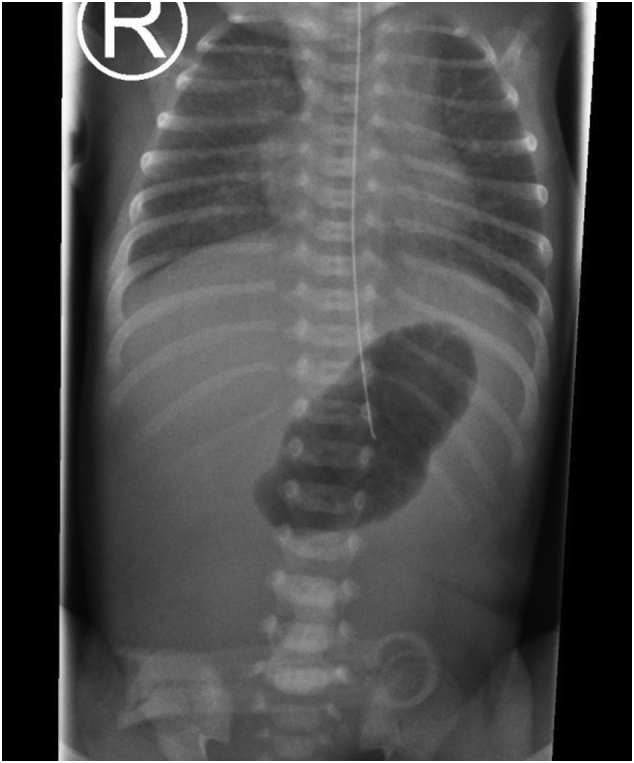
Single bubble sign on the thoracoabdominal radiograph after admission to the neonatal intensive care unit.
The parents were consanguineous and originated from Syria. The mother had had 11 pregnancies, including 5 miscarriages. She had given birth to three healthy boys. Two girls died briefly after birth with similar ACC. The narrative description of the course of one of these girls resembled anencephaly. Medical records from Syria were not available.
2.2. Practical management
After transfer to our center, we established an umbilical venous line for total parenteral nutrition and continued antibacterial therapy. Skin biopsies and blood for genetic testing were obtained on the second day of life.
Practical management was based of the recommendations of the Epidermolysis Bullosa Center Freiburg [4]. Non-adhering silicone-coated dressings were applied for the areas affected by ACC. Open skin lesions on hands and joints were covered with copolymer dressings [13]. Polyhexanide was used for local antisepsis. Upcoming blisters were pricked with sterile needles after local antisepsis. Change of wound dressings was performed under analgesia and sedation when dressings were saturated or became dislodged. On small and dry lesions, dexpanthenol ointment was applied. Our patient was bedded on soft tissue linen in an incubator to prevent cutaneous shear forces and received only minimal handling. Diapers were not closed to prevent shear stress as well. Oxygen saturation and pulse rate were monitored via pulse oxymetry fixed by a loose bandage. Despite fixation of the nasogastric tube with only minimal amount of adhesive tape, blistering and multiple skin lesions on nose, forehead and cheeks became increasingly prevalent.
Every four hours, the neonatal pain, agitation and sedation score (N-PASS) was assessed to evaluate the need for analgesia and sedation. We prescribed non-opioid and opioid analgetics and chloral hydrate for sedation.
2.3. Diagnosis, ethical decision making and operative treatment
Immunofluorescence microscopy (IFM) of the skin showed the total absence of α6β4 integrin, thus diagnosing JEB. After receiving the result on the 7th day of life, we discussed gastrojejunostomy and implantation of a central venous catheter in the context of a clinical ethics committee meeting. According to the literature, no association between surgical intervention and mortality is seen in these patients [7,11,12]. The literature also showed a broad spectrum of JEB without clear prognostic factors for JEB-PA [7,14]. We finally decided upon implantation of a secure tunneled, longterm central venous line and establishment of gastrointestinal continuity for enteral nutrition.
Antecolic jejunogastrostomy was performed by a highly experienced senior consultant pediatric surgeon on the 8th day of life. The intraoperative findings revealed PA with a gap between stomach and duodenum, classified as PA type 3 [12]. Interestingly, most common types in association with JEB are PA type 1 and 2 [11]. After operative therapy, extubation was possible and there were no wound healing anomalies. Nonetheless, insufficient bowel movements complicated enteral nutrition. Due to a coagulase-negative staphylococcus blood stream infection, the antibacterial regime was adapted. Fixation of the nasogastric tube repeatedly provoked painful skin injuries. Our patient showed intermittent breathing difficulties and drops in oxygen saturation. We hypothesized that mucosal wound slough obstructed the airways. Along with upcoming new blisters after local shear stress, there was no significant tendency in improvement of the ACC affected areas.
Genetic testing detected a pathogen homozygous nonsense mutation of the ITGA6 gene. This gene encodes for the α6 integrin subunit. The lack of α6β4 integrin affects the integrity of the hemidesmosome anchoring complex and the thereby the dermal-epidermal adhesion [14].
Since there was no improvement of our patient's condition, we initiated a second meeting of the clinical ethics committee. Together with parent approval, we decided to transition to palliative care. Our patient died on day of life 35 from hypoxemia.
3. Discussion
Most children suffering from JEB-PA are born with congenital absence of skin, which is described as ACC and in association with JEB classified as ACC type 6 [5]. JEB is characterized by blistering after minimal shear stress and is, in most of the cases, a lifelong problem [6]. The short-term prognosis primarily depends on the severity and extension of the ACC-affected areas, along with presence of associated diseases. Involvement of the urothelium and intestinal mucosa, as well as kidney and urinary tract anomalies, are described [11]. Most children die of sepsis, and about half of the children die in the neonatal period [11].
There were no prenatal conspicuities in our patient. Polyhydramnion, enlargement of the gastric bubble or the snowflake sign, describing hyperechoic particles in the amnion fluid, were not present [10]. Elevated levels of alpha-fetoprotein and acetylcholinesterase may hint at JEB-PA, but were not evaluated in our case [3,9].
To our knowledge, guidelines concerning the treatment and estimation of prognosis in cases of JEB-PA do not exist. Mostly, palliative care is provided upfront [11]. Okoye et al. recommend not to preclude patients with JEB-PA from a surgical treatment [12]. Hayashi et al. reported a long-term survival in 4 of 5 patients [7]. We decided to perform gastrojejunostomy to prevent our patient to become subject of a self-fulfilling prophecy by refraining from a curative therapy due to presumed poor prognosis.
However, nonsense mutations of the ITGB4 or of the ITGA6 gene, as found in our patient, are most frequently associated with lethal outcome. On the other hand, cases with only transient blistering are described as well for this mutation [14]. The association with PA is no criterion for prognostication as well. Decisive for the over-all prognosis is the quantity of integrin in the skin evaluated by IFM [11,14]. In particular, the total absence of α6β4 integrin hints at a very poor prognosis and a high probability of a fatal outcome in the first year of life [11,14]. Unfortunately, this was true for our patient but not recognized early on in the course. From a retrospective view, the absence of integrin at IFM could have been reason enough to refrain from surgical repair and to propose a palliative approach to our patient's parents. The small number of published cases and the impression that a curative therapy was withheld from some patients only due to the diagnosis JEB-PA, made it very difficult to support the decision with reliable facts.
4. Conclusion
JEB-PA is a rare disease with a very high mortality. Wound management in JEB is well described in international and national guidelines [2,4]. Concerning the prognosis there is only little and partially inconsistent literature. In awareness of the dilemma to withhold a curative therapy from a patient or to cause unnecessary harm by prolonged futile therapy, we emphasize the value of IFM as a decisive criterion for the burden and prognosis of the JEB. In our center, an interdisciplinary clinical ethic committee is available to discuss such cases. In our opinion, cases as JEB-PA should be discussed with all involved disciplines, i.e. pediatric surgeons, neonatologists and, if available, specialized dermatologists.
Provenance and peer review
Not commissioned, externally peer-reviewed.
Sources of funding
All authors declare that no fundings were received concerning the published case or the decision to publish it.
Ethical approval
N.a.
Consent
Written informed consent was obtained from parents of our patient for publication of this case report and accompanying images.
Guarantor
Schreiner D
Registration of research studies
Does not apply.
CRediT authorship contribution statement
Schreiner D: case presentation, literature research, writing manuscript.
Uebler A: review of manuscript.
Ginghina A: case presentation, review of manuscript.
Muensterer O: review of manuscript.
Has C: literature research, review of manuscript.
Mildenberger E: literature research, review of manuscript.
Declaration of competing interest
All authors state that there was no conflict of interest in writing or submitting the work for publication.
References
- 1.Agha R A, Franchi T, Sohrabi C et al (2020) The SCARE 2020 guideline. Int. J. Surg. 84: 226–230. [DOI] [PubMed]
- 2.DEBRA International International Consensus Best Practice Guidelines Skin and Wound Care in Epidermolysis Bullosa. https://www.debra-international.org/skin-and-wound-care-in-eb-cpg
- 3.Dolan C.R., Smith L.T., Sybert V.P. Prenatal detection of epidermolysis bullosa letalis with pyloric atresia in a fetus by abnormal ultrasound and elevated alpha-fetoprotein. Am. J. Med. Genet. 1993;47(3):395–400. doi: 10.1002/ajmg.1320470320. [DOI] [PubMed] [Google Scholar]
- 4.Epidermolysis bullosa-Zentrum Freiburg Handling von Neugeborenen und Säuglingen mit Verdacht auf Epidermolysis bullosa (EB). https://www.uniklinik-freiburg.de/hautklinik/kompetenzzentrum-fuer-fragile-haut-und-epidermolysis-bullosa/epidermolysis-bullosa-zentrum/handling-von-neugeborenen-und-saeuglingen.html.
- 5.Frieden I J (1986) Aplasia cutis congenita. J. Am. Acad. Dermatol. 14(4): 646–660. [DOI] [PubMed]
- 6.Has C, Bauer J W, Bodemer C et al (2020) Consensus reclassification of inherited epidermolysis bullosa and other disorders with skin fragility. Br. J. Dermatol. [DOI] [PubMed]
- 7.Hayashi A H, Galliani C A, Gillis D A (1991) Congenital pyloric atresia and junctional epidermolysis bullosa. J. Pediatr. Surg. 26(11): 1341–1345. [DOI] [PubMed]
- 8.Hicks T D, Singh H, Mikhael M et al (2018) Carmi syndrome in a preterm neonate. Case Rep. Pediatr. 2018: 4548194. [DOI] [PMC free article] [PubMed]
- 9.Maurice P., Eyrolle-Guignot D., Dhombres F. The key role of ultrasound examination in the prenatal diagnosis of epidermolysis bullosa with pyloric atresia. Prenat. Diagn. 2013;33(9):908–909. doi: 10.1002/pd.4137. [DOI] [PubMed] [Google Scholar]
- 10.Meizner I., Carmi R. The snowflake sign. A sonographic marker for prenatal detection of fetal skin denudation. J. Ultrasound Med. 1990;9(10):607–609. doi: 10.7863/jum.1990.9.10.607. [DOI] [PubMed] [Google Scholar]
- 11.Mylonas K S, Hayes M, Ko L N et al (2019) Clinical outcomes and molecular profile of patients with Carmi syndrome. J. Pediatr. Surg. 54(7): 1351–1358. [DOI] [PubMed]
- 12.Okoye B O, Parikh D H, Buick R G et al (2000) Pyloric atresia. J. Pediatr. Surg. 35(8): 1242–1245. [DOI] [PubMed]
- 13.Sari E, Eryilmaz T, Tetik G et al (2014) Suprathel((R)) -assisted surgical treatment of the hand in a dystrophic epidermolysis bullosa patient. Int. Wound J. 11(5): 472–475. [DOI] [PMC free article] [PubMed]
- 14.Schumann H., Kiritsi D., Pigors M. Phenotypic spectrum of epidermolysis bullosa associated with α6β4 integrin mutations. Br. J. Dermatol. 2013;169(1):115–124. doi: 10.1111/bjd.12317. [DOI] [PubMed] [Google Scholar]