

Abstract
Coronavirus infections are frequent viral infections in several species. As soon as the severe acute respiratory syndrome (SARS) appeared in the early 2000s, most of the research focused on pulmonary disease. However, disorders in immune response and organ dysfunctions have been documented. Elderly individuals with comorbidities exhibit worse outcomes in all the coronavirus that cause SARS. Disease severity in SARS‐CoV‐2 infection is related to severe inflammation and tissue injury, and effective immune response against the virus is still under analysis. ACE2 receptor expression and polymorphism, age, gender and immune genetics are factors that also play an essential role in patients' clinical features and immune responses and have been partially discussed. The present report aims to review the physiopathology of SARS‐CoV‐2 infection and propose new research topics to understand the complex mechanisms of viral infection and viral clearance.
Keywords: complement, coronavirus, cytokine storm, immune response, innate immune response, memory cells, SARS‐CoV‐2 infection
1. INTRODUCTION
Coronaviruses (CoVs) are a collection of enveloped viruses with non‐segmented, positive‐sense single‐stranded RNA genomes with distinctive crown‐like spikes that protrude from the capsid of helical symmetry. 1 They have a remarkably long RNA genome and a particular replication strategy. In this complex family, several members attack different species causing several diseases that can end up in death. In November 2002, in China, a severe acute respiratory syndrome coronavirus (SARS‐CoV) was identified. It promptly spread to other countries. There were around 8000 confirmed cases, and the mortality rate was 9.6%. 2 Then, another member of the family, Middle East respiratory syndrome coronavirus (MERS‐CoV), appeared in Saudi Arabia in 2012 and later emerged in South Korea in 2015. The confirmed cases of MERS‐CoV exceeded 2000, with a mortality rate of ∼35%. 2 In 2019, another member of the family was identified, SARS‐CoV‐2. 3 The number of infected people and the mortality rate still grow continuously. Infected elderly individuals with comorbidities exhibit the worst outcomes. There are now several vaccines against SARS‐CoV‐2 approved for emergency use by the regulatory offices of different countries.
The coronavirus genome is formed by 2 UTR sites, 5′ and 3′, the replicase, the spike (Spike), the envelope E (Envelope), the M (Membrane), the N (Nucleocapsid) and the poly (A) tail. There are additional genes at the end of the genome. The S protein is highly glycosylated, and it is required for infection. 4 Even though the S protein is very similar to SARS (94% nucleotide sequence), a protease‐sensitive site in the SARS‐CoV‐2 is absent in the previous one. 4 The membrane protein and the accessory proteins are non‐essential for replication; however, they have essential viral assembly and pathogenesis roles. Other non‐structural genes, open reading frame (ORF), ORF1ab, ORF3a, ORF6, ORF7a, ORF10 and ORF8, are also transcribed. 5 These proteins' function in the infection, viral replication and host response are still controversial. 6 , 7
The primary transmission is airborne, close and direct human‐to‐human contact, droplets from saliva, sneeze or cough. A less prevalent infection occurs by direct skin contact, faeces or contaminated objects. 8 , 9 The incubation period range is from 2 to 14 days, and the infected person can be asymptomatic during this period. 8 , 9 , 10 SARS‐CoV‐2 has an R0 of 2.2‐2.6, implying that each infected individual has the potential to infect 2.2 other people. 8 , 9 , 10 One key issue is that the virus is detectable by molecular biology early in the nostrils and saliva; however, antigens are reported later at the onset of symptoms. 11 The lag time in detectable antigen can be critical in the pathogenesis and multiorgan infection. Even though most SARS‐CoV‐2‐infected persons are asymptomatic, around 20% of the patients may have severe manifestations, and 5 to 10% require intensive care. 8 , 9 , 10 The severe cases with associated mortality are generally older adults with comorbidities; however, severe forms of infection have been detected in all ages. 12 Moreover, virus variants also impact the rate of infection, disease severity and lethality. 13
The coronavirus infection causes hypoxemia, from mild to severe (SARS), skin rash, fever, anosmia, fatigue, pain chest, muscle, articular and unexpected hyperglycaemia or increase in blood pressure. The virus affects microcirculation; it generates endothelial cell damage, capillary damage and micro thrombosis. 14 The immune response against the virus and the virus's cytopathic effect induces the activation of the innate immune system, protein and cells, which may generate a massive inflammatory reaction (cytokine storm). The hypoxia induces hypoxia transcription‐inducing factor I (HIF‐I), amplifying the inflammatory response by activating myeloid cells and enhancing transcription of proinflammatory cells and oxidative enzymes. 14 Even though it is still controversial, increased BMI has been considered a risk factor for COVID infection. 15 , 16 More studies are required to ascertain the relationship between metabolic syndrome, a subclinical proinflammatory condition and HIF‐I as the triggering factor for massive neutrophil lung recruitment and cytokine storm in SARS‐CoV‐2‐infected patients.
It is unclear how the virus causes neurological effects, and some authors have proposed direct cytopathic effects. 17 Gastrointestinal manifestations, cardiac, kidney and hepatic dysfunctions are observed in human and animals infected by a coronavirus. 1 , 3 Skin manifestations reveal immune complex deposition as it has been recorded in other viral diseases, and it is age‐independent. 18 In a general analysis, Mason 19 described three phases of SARS‐CoV‐2 viral infection. The first phase is the asymptomatic phase since the infection mostly is present in the nose and on the buccal cavity. In the nose, the immune system involves local antibodies' and innate immune cells that may elicit an adaptative immune response. 19 The induction of the adaptative immune response depends on antigen expression, which is low at the early stages of the viral infection. 11 In the oropharyngeal cavity, the innate immune response is prevalent, complement, neutrophils and macrophages, and antibodies, essentially IgA, bactericidal peptides and enzymes that control mostly bacterial infections. 19 There, the virus elicits a minimal innate immune response. In the moderate symptomatic phase, the virus is primarily present in the larger airways' pseudostratified epithelium. 19 There is an excellent innate immune response in these areas with the recruitment of cells and proteins, which may cause damage and obstruction of the airway. However, the damaged epithelial cells can be removed and replenished with basal cells. 7 , 19 There is a more severe disease in the bronchioles where the club cells are usually infected and affect surfactant production and other secretory products. 7 , 19 In severe cases, the alveoli are compromised; the virus targets the epithelial type II cells that express ACE. 15 , 19 The decrease in viable epithelial type II cells is responsible for respiratory insufficiency. The lack of lung surfactant, alveolar flooding and loss of the extracellular matrix generating more viral infection affects the pulmonary parenchyma's standard repair and the inflammation's resolution. 19 The typical active resorption of alveolar fluid and electrolytes is also hampered, resulting in hypokalaemia. 19 Impaired endothelial cells lead to transudation of plasma protein of inflammatory origin and an irregular formation of hyaline membranes. 7 , 19 Residual fibrosis may result after viral infection due to the low resolution of the inflammatory response. 19
The evidence of lung tissue destruction came from an exciting study analysing proteins of organs of autopsies by HPLC/MS. 20 The authors were able to identify high amounts of cathepsin L1, an enzyme involved in intracellular protein catabolism. 20 , 21 The increase in cathepsin L1 is related to extracellular matrix degradation critical in viral infection and release. 19 , 21
Significant alterations in blood electrolytes have been described in hypertensive patients infected with SARS‐CoV‐2, hypokalaemia, hyponatraemia and hypocalcaemia. 22 , 23 Nonetheless, few studies have dealt with the mechanism involving electrolyte imbalance with neural, cardiovascular, gastrointestinal and renal dysfunctions due to either the general renin‐angiotensin system or the virus's cytopathic effect as it occurs in the lung. 22 Electrolyte imbalance induces inflammasome activation and, consequently, exacerbates the inflammatory response; 24 then, multiorgan dysfunction in severe COVID patients may result from multiorgan‐induced inflammatory reaction.
2. ANGIOTENSIN‐CONVERTING ENZYME 2 AND THE SARS‐COV‐2 RECEPTOR
SARS‐CoV‐2 uses the complex angiotensin‐converting enzyme 2 receptor (ACE2), a glycosylated transmembrane protein, to infect and invade the target cell. 25 , 26 Two differentially spliced forms of ACE2 are known and have different substrate specificities. 25 , 26 The maximum expression of ACE2 is observed in the respiratory epithelium, lungs, kidneys, intestines, testis (Sertoli and Leydig cells), uterus, vagina, endothelium and the heart. 25 , 26 , 27 The spike protein protease‐sensitive site is cleaved by a specific transmembrane serine protease 2 (TMPRSS2) and divides the protein in two. The S 1 unit (S1) of the virus binds to the ACE2 as the target receptor. 25 , 26 , 27 Then, it uses the host serine protease TMPRSS2 for S cleavage, allowing the union of viral and cellular membranes and, consequently, viral entry into the cell. 25 , 26 , 27 , 28 The binding of the virus and its internalization leaves the cells without an active ACE2 enzyme. Soluble ACE2 that lacks membrane anchor is found in low levels in the blood. 29 The impact of a small amount of active ACE2 and its role in cardiovascular physiology and SARS‐CoV‐2 physiopathology is still debatable.
Battle and collaborators 29 proposed that soluble ACE2 could act as a competitive receptor of SARS‐CoV and other coronaviruses by avoiding binding the viral particle to the surface‐bound, full‐length ACE2. Therefore, the provision of the recombinant human soluble protein ACE2 could be beneficial as a novel biological therapy to limit the infection's progression caused by coronaviruses that use ACE2 as a receptor. Hoffmann et al 30 presented a study demonstrating that binding the virus to the cell can be inhibited by blocking the cellular serine protease protein TMPRSS2. As aforementioned, the S1 protein/ACE2/ACE receptor complex is responsible for infection and internalization. According to the authors, 30 antibodies may inhibit the binding of S1 to the complex leading to a possible therapeutic target. Johnson et al 31 were able to show that mutation in the furin site in the S protein of the SARS‐CoV‐2 virus is essential in viral pathogenesis. In the animal models, exposure to the mutated virus prevents the infection with the typical virus suggesting a possible role of the mutated protein in therapeutic responses.
Studies involving monoclonal antibodies to decrease viral burden have been published 32 , 33 and several regulatory agencies' have approved its use. However, only a combination of monoclonal antibodies against different S protein domains was shown to be more effective, bamlanivimab and etesevimab. 32 Antibodies and B cells from convalescent patients were used to construct recombinant antibodies; the trials' results are promising. 33 There are, however, some limitations on the use of the therapy as expected. A general question arises from the fact that IVIG has also been shown to be partially protective. 34 In risk populations with initial exposition to the virus, early IVIG therapy could be beneficial to prevent severe disease.
The ACE2 receptor gene is located in the X chromosome, and TMPRSS2 gene is situated in chromosome 21. 35 The ACE2 gene's location may provide an advantage to females (2 copies in duplicate of the same gene); on the other hand, TMPRSS2 is an androgenic stimulated gene. 35 In post‐menopause, as expected, due to the lack of hormones, this protection is less effective.
Zhen and Cao 36 provided evidence of gene polymorphisms of ACE2 in different populations and its importance in SARS‐CoV‐2 infection. In Italy, Asselta et al 37 analysed the Italian population generating an analysis of the critical polymorphism and the role of gender. Most ACE2 genetic variants, although involved in increased hypertension susceptibility, have a similar binding affinity for SARS‐CoV‐2 S protein. Hussain et al 38 were able to demonstrate that ACE2 alleles, rs73635825 (S19P) and rs143936283 (E329G), had a lower affinity to the S1 protein. The relevance of these polymorphisms and their impact on ethical genetic analysis was not demonstrated. 39 It is suggested that admix populations may have an advantage as compared to closed populations. More research is required to ascertain TMPRSS2 allelic variants' role, gene and post‐transcriptional regulation, and the role of furin in SARS‐CoV‐2 infection.
Ziegler et al 40 were able to show that ACE2 is an interferon‐stimulated gene in human cells and tissue; the regulation is not observed in simian or mouse samples. Upon activation of the immune response, more expression of the enzyme on the cell surface may enhance virus infection. Even though these results would suggest limitations of animal models in SARS‐CoV‐2 studies, the models can still be suitable to study immune response against the virus.
3. VIRAL RECOGNITION AND CELL METABOLISM
The immune response to respiratory viral infection has several steps: one humoral component with antibodies and complement, the cellular part that comprises cells that require no antigen presentation (innate response) and the antigen‐specific cells that belong to the adaptive response component. 7 , 41 , 42 , 43 Nevertheless, besides interferons and inflammatory cytokines, the immune response depends upon intracellular transport of pathogen, miRNA from virus and the host, host‐derived machinery and the genetic and metabolic response to the infection. 41
Pattern recognition receptors (PRRs) are cellular membrane and cytoplasmic receptors that recognize viral proteins and nucleic acids. 41 , 42 For example, double‐stranded RNA (dsRNA) or single‐stranded RNA (ssRNA) with a 5′‐triphosphate are sensed by the retinoic acid‐inducible gene I (RIG‐I) cytoplasmic protein. 41 , 42 , 43 These types of RNA are viral replication products not found in the cytoplasm of healthy cells. When RIG‐I recognizes these viral RNAs, cells are activated and start synthesizing interferon and proinflammatory cytokines. 41 , 42 , 43
Other virus detectors are Toll‐like receptors (TLRs), which detect viral dsRNA, ssRNA, CpG sequences and viral glycoproteins. 41 , 42 , 43 Viral genomic RNA and/or replication intermediates (dsRNA) are identified by RIG‐I/MDA and the endosomal RNA receptors, TLR3 and TLR7. 41 , 42 , 43 These events generate the activation of NF‐κB and IRF1, IRF2, IRF5, IRF7, IRF8, IRF9 signalling cascades which induce the transcription and expression of type I interferon (IFN‐α, IFN‐β, IFN‐δ). 43 , 44 , 45 In turn, IFN type I binds to its receptor and triggers the JAK‐STAT pathway, JAK1 and TYK2 kinases phosphorylate STAT1 and STAT2, which then forms a complex with IRF9‐inducing transcription of IFN‐stimulated genes (ISGs) under the control of IFN‐stimulated response element (ISRE)‐containing promoters. 43 , 44
IFN pathway is essential in cell viral response since mutations affecting the cascade are related to the Mendelian predisposition to viral infections. 46 , 47 , 48 Several immune deficiencies have unravelled the importance of several proteins involved in the IFN pathway crucial to antiviral response. 46 , 47 Wang et al 48 described the different proteins involved in the antiviral function of interferon. SARS‐CoV and MERS inhibit IRF3, IRF7 and IRF9 proteins confirmed for SARS‐Cov‐2 infection. 44 , 49 Proteins induced by IFN like the interferon‐induced transmembrane protein 3 (IFITM3) and interferon‐stimulated protein (IFIT1) could interfere with a viral infection of the neighbouring cells. 44 , 48 The blocking effect should be parallel to the impact on cyclin‐dependent kinases (Cdks) on cell proliferation.
Several proteins of SARS‐CoV‐2 have been shown to inhibit IFN signalling. Lei et al 49 screened 23 viral proteins. They found that SARS‐CoV‐2 NSP1, NSP3, NSP12, NSP13, NSP14, ORF3, ORF6 and M protein can inhibit IFN‐β induced Sendai virus promoter activation, suggesting that they may downregulate the production of the cytokine that impedes SARS‐CoV‐2 viral replication. ORF‐3b is a potent interferon antagonist, 50 and ORF6 may also disrupt the IFN type I signal and STAT nuclear import. 51 Protein M (membrane) has also been shown to be a potent inhibitor of interferon type I and III. 52 Interestingly, proteins S and NSP of the virus appear to promote the IFN signal. 49 , 50 , 51
In a genetic study, Zhang et al 53 were able to identify that inborn genetic errors of TLR3 and IRF7, IFN I‐dependent immunity were more susceptible to the virus and may have the worst outcome. New mutations in STAT2 and IFN‐γ have also been reported. 54 These patients could be more susceptible to viral infections and severe disease.
Anti‐IFN antibodies were detected in severe COVID‐19‐infected patients. 55 Autoantibodies would decrease the immune response against the virus and predispose to a higher viral replication and an impaired immune response. Interestingly, the presence of autoantibodies is more frequent in men than in women. 55 The reason for the gender difference is unknown.
In silico and bioinformatics assessments revealed several host binding microRNA (miRNA). 56 , 57 , 58 , 59 , 60 , 61 , 62 , 63 Several host miRNA (15b‐5p, 15a‐5p, 197‐5p, 548c‐5p, 548d‐5p, 409‐3p, 30b‐5p and 505‐3p) may be involved blocking viral replication. 56 , 57 Also, viral miRNA is shared with cells miRNA (8066, 5197, 3611, 3934‐3p, 1307‐3p, 3691‐3p, 1468‐5p), which may modulate cell response facilitating SARS‐CoV‐2 infection. 58 Among the different virus escape mechanisms, inhibition of host miRNA maturation, viral miRNA control of the cellular process and metabolic pathways, and viral miRNA sponges are crucial for viral escape. 56 , 57 , 58 , 59 , 60 , 61 IFN pathway is inhibited or downregulated, and TGF‐β signalling, involved in cell suppression, is enhanced. 49 , 53 , 54 The IFN pathway inhibition affects viral RNA recognition by Toll receptors and RIG. 43 , 44 , 45 , 46 , 47 , 48 , 49 Nonetheless, extracellular vesicles containing non‐coding RNA could be important in controlling viral replication and enhancing an effective immune response. 49
Nersisyan et al, 61 using bioinformatic analysis, identified six hot miRNAs able to bind viral sequences (miR‐21‐3p, miR‐195‐5p, miR‐16‐5p, miR‐3065‐5p, miR‐424‐5p and miR‐421). In the mouse model, miR‐21‐3p is upregulated during SARS‐CoV infection suggesting that it may have an essential role in viral pathogenesis. In the ovalbumin asthma mouse model, miR‐21 was related to M2 macrophage polarization and lung hyperresponsiveness. 62 Thus, miR‐21 is involved in remodelling, which may be crucial for viral infection replication.
An important issue arises concerning the role of miRNA in ACE2 and TMPRSS2 transcripts. There is a controlling system containing let‐7e/miR‐125a/miR‐200 families, histone demethylase JARID1B that regulates ACE2 expression; hsa‐miR‐200c‐3p and hsa‐miR‐141‐3p can bind 3' UTR of ACE2 mRNA and, in consequence, modulate the transcription of the gene. 51 , 61 , 63 Paniri et al 64 proposed that polymorphisms of miR rs3746444 for hsa‐miR‐499a‐3p, rs113808830 for hsa‐miR‐4532, rs3751304 for hsa‐miR‐6763‐3p and hsa‐miR‐26b‐5p were strongly hybridized with ACE2 mRNA and might stimulate its expression.
Recently, Wyler et al, 65 through the analysis of in vitro infection and miRNA, showed that heat‐shock protein (HSP) 90 is crucial in SARS‐CoV‐2 infection. Inhibition of HSP 90 resulted in decreased viral replication and reduced transcription of cytokines. These results generate new research areas and therapeutic options involving chaperone proteins, protein degradation and presentation, which may be necessary to understand viral infection and the immune response against it.
The miRNA reports generate vital information about virus infection and pathogenesis, host immune and metabolic response to infection. This new knowledge can be explored to produce successful therapies for appropriate pharmaceutical intervention.
4. METABOLIC CHANGES IN SARS‐COV‐2 INFECTION
As expected in the hyperinflammatory response, glucose and fatty acid metabolism changes are observed in non‐diabetic patients. The increase in blood glucose may parallel with C‐reactive protein, procalcitonin and lactate suggesting a high metabolic regulation. 66 Most severe cases are observed in the obese and elderly infected patients, presumably due to the subclinical inflammatory condition, which may be prone to generate a deregulated immune response and cytokine storm. Several authors 67 , 68 have suggested that adipokines are responsible for patients' metabolic response against the SARS‐CoV‐2 virus, which, in turn, induces inflammasome NLP3 activation. The inhibition of fatty acid synthase by orlistat and the AMP‐activated protein kinase (AMPK) activator metformin seem to reduce viral replication of the coronavirus along with a decrease in systemic inflammation. 68 These findings and the genetic predisposition of severe SARS‐CoV‐2 infection in Apo E e4e4 homozygotes 69 suggest that host metabolism may be critical in the response against the virus.
Lipidomic changes are also observed upon IFN stimulation in other to protect cells from viral entry. The changes involve modifications at the membrane levels. 70 IFN promotes the increase in cholesterol membrane content. The plasma membrane becomes more rigid, decreasing the possibility of viral infection. 70 Moreover, changes in glycerophospholipids and sphingolipids are also observed upon IFN production. 66 , 67 , 68 Membrane arachidonic acid is augmented, leading to its metabolites' marked production. 70 , 71 , 72 Yan et al 71 reported that arachidonic acid and linoleic acid metabolism are amplified in vitro in HCoV‐229E‐infected epithelial cells, suggesting that the cell response upon viral infection may be combined with the IFN‐induced response. Shen et al 72 reported that arachidonic acid levels in the serum of SARS‐CoV‐2 patients decrease depending on the disease's severity. Phospholipases A2 (secreted, cytosol and membrane) are then the pathway's critical enzymes. 70 , 71 Arachidonic acid and its metabolites could be significant predictors of viral infection and/or replication. An extensive scale lipid plasma analysis reported that two phosphatidylcholine species, 14:0–22:6 and 16:1–22:6, one phosphatidylethanolamine species, 18:1–20:4, plasma triglycerides values, and plasma free fatty acid values, especially arachidonic acid and oleic acid, were positively correlated with SARS‐CoV‐2 infection. 73 Moreover, a more detailed analysis revealed that adiponectin, IL‐26 and ceramides are also involved. 74 Ceramides are essential players of lung inflammation. 75
Assessment of ceramides in several diseases has provided significant evidence of tissue destruction and resolution. 75 In lung inflammation, sphingosine‐1‐phosphate (S1P) has been a substantial marker of resolution of lung inflammation, while ceramide 1 P (C1P) has been the contrary. 76 Prakash et al 77 have proposed that possible treatment with S1 analogue would hamper immune response in SARS, and consequently, C1P can be used to decrease viral replication and enhance T cell response. 77 Furthermore, it has been shown that acid sphingomyelinase activity blockage prevents the uptake of SARS‐CoV‐2 by epithelial cells in vitro. 78 The modification of extracellular matrix and heparan sulphate may also help modulate different enzyme activities that would favour but not necessarily be essential in virus entrance or cytopathogenesis.
Several studies have illustrated ApoE polymorphism's (e4/e4) importance and relevance in virus neurotropism. 69 However, more lipidomic data are required to evaluate the role of Apo E protein and gene polymorphisms and the role of other apolipoproteins, particularly A1 as anti‐inflammatory, in SARS‐CoV‐2 viral infection and resolution.
5. INNATE IMMUNITY
5.1. Complement
Complement is a critical system of the innate immune response. In SARS‐CoV‐2 infection, the activation of complement can be mediated by the lectin pathway and the alternative pathway. The lectin pathway involves the interaction of collectin 11, mannose‐binding protein (MBP) and ficolin, which is associated with the mannose‐binding lectin‐associated serine protease (MASP). MASP activates the coagulation system since it is a serine protease. The alternative pathway involves factor B. 79 Upon tissue destruction with pathogen and danger signals, PAMPs and DAMPs, classical activation of the complement is possible, rendering massive protein‐mediated tissue destruction. 79
Fang et al 80 reported a relationship between C3 levels and patient prognosis. The lower the values, the worse is the outcome. In SARS‐CoV‐2‐infected patients, complement activation is detected on circulating neutrophils, as evidenced by cell surface C3 fragment deposition. 80 , 81 , 82 The activation of C3 requires factor B and Fcγ receptors but not C4. 81 C3 convertase is activated upon priming of neutrophils with immune complex via Fcγ receptors. 81 Moreover, C3 is also involved in NETosis, amplifying neutrophil death and immune response. 82 These two processes were observed studying SARS using the C3 KO mouse. 83
Lin et al 84 and Cugno et al 85 were able to detect increased complement activation during the progression of COVID‐19. This activation is decreased during remission. An association between C3 levels and biomarkers of endothelial damage, soluble von Willebrand factor VIII, tissue injury markers and lower clearance of the virus was observed. On a large scale, multiple analyses performed, 86 complement proteins were found to be overexpressed in COVID‐19 infection, and in severe patients, the levels were the highest. The increase in complement proteins is combined with a decreased apo‐A, and HDL synthesis increased B lymphocyte activation and increased lipoprotein metabolism. 86 The increase in acute‐phase proteins can be partially accounted for the changes; however, their participation in enhancing proinflammatory pathways may generate cytokine storm and, consequently, a dysfunctional immune response.
Intracellular complement 3 has also been involved in protecting airway epithelial cell from pathogen infection and stress. 87 Its content in the lung increases upon inflammation and inflammatory cytokines, and consequently, lung levels of C3 are increased in patients with lung diseases. 87 As expected, SARS‐CoV‐2‐infected patients with lung illness have a higher risk to progress to severe disease and die. 88 Since C3 is involved in airway disease and emphysema, 89 COPD patients with high levels of C3 and infected with SARS‐CoV‐2 may be more susceptible to develop a severe disease that stable COPD patients without exacerbations and treated with anti‐inflammatory therapy. 90
Polycarpou et al 91 have an exciting approach to managing complement and the deleterious effect of the innate immune response in COVID‐19 patients. The goal is to decrease innate cell activation by C3a and C5a; both activated proteins attract and activate immune cells in the inflammatory site. Mastellos et al, 92 treating patients with eculizumab that targets C5 and AMY‐101 that target C3, were able to show a decrease in the inflammatory response in treated patients with a marked reduction in IL‐6 and C‐reactive protein levels an improvement of lung function and then a resolution of the distress syndrome. Interestingly, factor D inhibitor ACH145951 can suppress the complement cascade's activation induced by the virus's S protein; besides, factor H enhanced this inhibition. 93 Besides complement, macrophages' tissue factor upon neutrophil cell death plays a role in distress syndrome. 94 These results suggest that there are still important questions to answer in this field of complement activation and pulmonary distress syndrome.
Holter et al 95 associated the activation of complement to respiratory failure in a hospital screening. The authors were able to show a direct association of sC5b‐9 values with respiratory failure. Interestingly, mannose lectin‐binding protein (MLB) was not altered in patients than controls. 95 The low amount of samples and the lack of MLB deficiencies in the cohort could be responsible for the lack of association. More studies should ascertain la role of collectins, ficolins and MLB in an antibody‐free environment as the first line of defence against SARS‐CoV‐2.
The genetics of complement protein has also been a matter of discussion in the SARS‐CoV‐2 infection. Polymorphisms of the MBP codon 54 variant (A/B) and CCL2 predispose to the severe acute respiratory syndrome in SARS. 96 The antiviral interferon‐induced product myxovirus resistance protein 1 (MxA) may also be involved. 97 Analysis of the chromosome 3p21.31 multigene gene cluster and the ABO blood group revealed that the variant rs11385949 G>GA predisposes to severe infection since it is associated with enhanced complement activation of C5 and terminal activation complex in the non O blood group. 98 These results suggest that both C3 and C5 are involved in the exacerbated immune response.
In conclusion, complement therapeutic may be considered essential in patients with severe disease. Published reports using anti‐C3 and anti‐C5, alone or in combination, in SARS infection have given vital information to design clinical trials. 92 , 98 , 99 , 100 Considering the differences between the convertases C3 and C5, the approach to blocking both pathways or just C5 to prevent terminal activation seems logical. Inhibition of factor D 93 could give an extra advantage of blocking C3b amplification induced by the cell debris generated in the death of neutrophils in the infection and avoid jeopardizing the complement cascade from any opportunistic infection. However, up to date, there are no clinical reports to confirm this hypothesis. More research is needed to generate therapeutic tools to modulate complement protein function.
5.2. Neutrophils, eosinophils, mast cells, macrophages and dendritic cells
Respiratory viruses do not infect neutrophils, yet neutrophils can phagocytose virions, viral particles and apoptotic bodies containing viruses. In the process, neutrophils are recruited by proteins of the complement systems and chemokines to the inflammation site. 7 Activated neutrophils secrete cytokines, antimicrobial peptides and a variety of enzymes. 7 They also produce oxygen radicals and other mediators to kill pathogens. 7
Recruited neutrophils are activated in the lungs and can form and release extracellular traps, neutrophil extracellular traps (NETs), composed of proteases, antimicrobial proteins, and decondensed chromatin, and histones, which, in principle, restrains pathogens and inhibits further dissemination. 101 NETosis, the common apoptotic death of neutrophils, is generally cleared by macrophages, a process that may also hamper local immune response against pathogens. 101 In severe SARS‐CoV‐2‐infected patients, there is an increase in circulating neutrophils in the simple haematological analysis. These neutrophils have been shown as dysfunctional. 102 In essence, the myeloid cells are essential markers of IFN signal with an increase in monocyte HLA‐DR, CD11c and CD16. 102 Remnants of NETs were identified in patients. 101 , 102 , 103 , 104 These remnants contain cell‐free DNA, myeloperoxidase‐DNA complexes, citrullinated histone H3 and calprotectin neutrophil‐derived S100A8/A9. 104 , 105 Consequently, there is an increased risk of hypercoagulability. Lung microbiota controlled by the immune system now, in severe patients, represents a threat for the individual. 104 , 105 , 106 Consequently, the excessive activation of neutrophils may cause damage to the respiratory epithelium amplifying local inflammatory response and decreasing lung function. 103 , 104 , 105 , 106
One of the remarkable yet partially explored findings in leukocyte analysis of SARS‐CoV‐2 patients is the marked decrease in the haemogram's eosinophil cell population. 107 , 108 The reduction in eosinophil number has not been analysed thoroughly. Eosinophils do not seem to be openly involved in human response to respiratory viruses as in other animal species; consequently, they are not considered to be directly involved in SARS pathogenesis. 107 , 108 Since in SARS‐CoV‐2‐infected patients, there is no exacerbation of atopic disease, many researchers discarded its role in the viral infection. 107 However, the number of circulating eosinophils was a significant factor associated with a subprophylactic amount of anti‐factor Xa inhibitor, 109 suggesting that these cells may be involved in tissue destruction or remodelling as a result of the viral infection. Several drugs used to decrease eosinophil migration to the airways in asthma may be useful as palliative treatment in this viral infection.
In post‐mortem studies of patients who have died from SARS‐CoV‐2 infection, an enhanced concentration of perivascular and septal mast cells and a high density of mast cell progenitors recruited in the alveolar were described septa. 110 Mast cell recruitment into the alveolar septa may be a consequence of the initial endothelial and epithelial cell damage of the alveolar septa. However, complement activation may be the key factor involving mast cell migration to the airways and the generation of mast cell extracellular traps generated upon activation. 83 , 84 Also, the degranulation of tryptase and chymase may contribute to more cell death and, consequently, the destruction of lung parenchyma architecture, favouring virus invasion. 83 , 84 In summary, NEToseis, mast extracellular traps and the attraction of other non‐neutrophilic cells along with inflammatory cytokines enhanced activation of complement and coagulation cascades.
Interestingly, the use of antihistamine receptors as adjuvant therapy for SARS‐CoV‐2 patients in the initial schemes seems to have decreased and may have protected patients from the deleterious effect of mast cell activation. 111 Tissue destruction and cell attraction in the inflammatory site may potentiate the cytokine storm observed in SARS‐CoV‐2 patients. Most probably, the use of histamine receptor inhibitors indirectly would protect patients from this event as it has been proposed. 111
Alveolar macrophages are, along with dendritic cells (DC), the lung's sentinels, and their activation is crucial to limit bacterial and viral infections and recruit immune cells to the tissue. 112 Monocytes and macrophages are targets of the SARS‐CoV‐2 virus, 113 and they become nonfunctional after infection; the virus infects but not replicates in these cells. 114 The increase in suppressive myeloid cells with a decrease in normal and inflammatory monocytes and dendritic cells has been reported in COVID patients. 115 The suppressive cells may be crucial in the lack of an effective immune response to the virus.
Inflamed age macrophage is a derived term for alveolar macrophage that distinguish the low responsible macrophage in lung tissue from elderly individuals, which is less active and consequently less prone to eliminate bacterial or viral infections. 113 , 114 These non‐effective cells increase in smokers as compared to non‐smokers suggesting that the impaired response leads to severe lung diseases in these patients. 113 , 114 , 115 Lung microbiota is also vital in SARS‐CoV‐2‐infected patients since dysfunctional myeloid cells may give rise to bacterial co‐infection. 102 , 106
Mature DCs (mDCs) can efficiently activate T cells and maintain specific immune responses, and immature dendritic cells (imDCs) can relocate in different tissues. 116 , 117 Thus, in the absence of mDCs, the adaptive immune response is impaired. 116 , 117 MHC class I and II molecules are immediately regulated with antigens and costimulatory molecules when DC cells are infected with viruses. 118 This activation leads to a prompt and potent stimulation of T lymphocytes activity. 117 , 118 Plasmacytoid dendritic cells (pDC) produce type I IFN and are critical for antiviral response to fight infections. 118 Even though DC cells' role is still debatable in SARS‐CoV‐2 infection by some authors, 119 the viral infection has been shown to modulate DC subclasses and activate pDC in early stages. 120 There is a redistribution of CD1+ cDC cells in severe cases in the infection, along with critical changes in other subpopulations and myeloid suppressor cells. 114 , 115 Thus, it is clear that dysfunctional macrophages and DC affect T activation since proper antigen presentation or activating IFN response is produced. The decrease of co‐receptors as CD40, crucial for DC or macrophage interactions, is not fully expressed. 120 , 121 DC cells' role in the immune response generated by vaccines against SARS‐CoV‐2 will be critical to analyse, especially in those using the attenuated virus.. 114 , 115 , 116 , 117 , 118 , 119
5.3. Inflammasomes and cytokine storm (CS)
Upon cell injury or infection, inflammasomes trigger proinflammatory cytokine production, mainly IL‐1β and IL‐18. 122 PRR receptors, including nucleotide‐binding oligomerization domain‐like receptors (NLR), are critical for inflammasome activation. Excessive activation of inflammasomes may induce cell death (pyroptosis). 122 , 123 , 124 , 125
In several autoinflammatory conditions, the NALP3 inflammasome is uncontrolled. 124 , 125 The balance between beneficial versus detrimental activation of the inflammasome is challenging. 123 , 124 , 125 The inflammasome activity is critical to the host response and microbial pathogens, also vaccine adjuvants, since the production of cytokines by the innate immune system serves to outline the adaptive immune response. 122 , 123 , 124 , 125 The stimulated inflammasomes activate caspase 1, which is involved in the release of bioactive IL‐1β and IL‐18. 122 , 123 , 124 , 125 IL‐1, IL‐1α and IL‐1β then induce robust proinflammatory activities and play an essential role against harmful exogenous or endogenous stimuli. 123 The production of IL‐1 is then critical for the stimulation of the transcription and secretion of other proinflammatory cytokines as IL‐6, TNF‐α, IL‐33, all involved in inflammatory response against pathogens. In genetic hereditary autoinflammatory disorders, there is an uncontrolled production of IL‐1, which is usually treated with biological therapy. 122 , 123 , 124 , 125 There are several therapeutic approaches to modulate inflammasome activation currently available. 124 , 125 , 126
Cytokines and particularly chemokines play an essential role in a robust immune response against coronavirus infections. 83 , 87 However, they are also involved in the immunopathology generated by them. 83 , 87 Even though a fast and coordinated innate immune response is the first line of defence against viral infections, unregulated and disproportional responses may be responsible for disease severity. Several genetic haplotypes have been related to it, although chronic inflammation may predispose a more severe cytokine storm (CS). 124 , 125 Rodrigues et al 126 were able to show the activation of inflammasomes in response to SARS‐CoV‐2 infection and relate the activation to the severity of the disease. As expected, lethality upon virus infection dramatically increases. 127
Several studies have changed our mechanistic understanding of inflammasome genetics, signalling, cell death decisions and cytokine activation and secretion. 128 , 129 , 130 , 131 , 132 , 133 In severe infections, even mild genetic mutations of proteins of the inflammasome affect its activation and regulation. Several mutations have been described that either delay activation or dysregulate its activity. 129 Therefore, activation of the inflammasome cannot be considered destructive, and the therapeutic mechanism of inhibition must be understood to regulate innate immune response properly. 128 , 129 , 130 , 131 , 132 , 133 CS is a common complication in SARS‐CoV‐2, SARS, MERS and other viral respiratory infections. IRF3 and NF‐kB are activated by viral RNA, which induces the transcription of IFN and inflammatory cytokines. 133 Figure 1 illustrates the pathological cytokine storm in SARS‐CoV‐2 infection.
FIGURE 1.
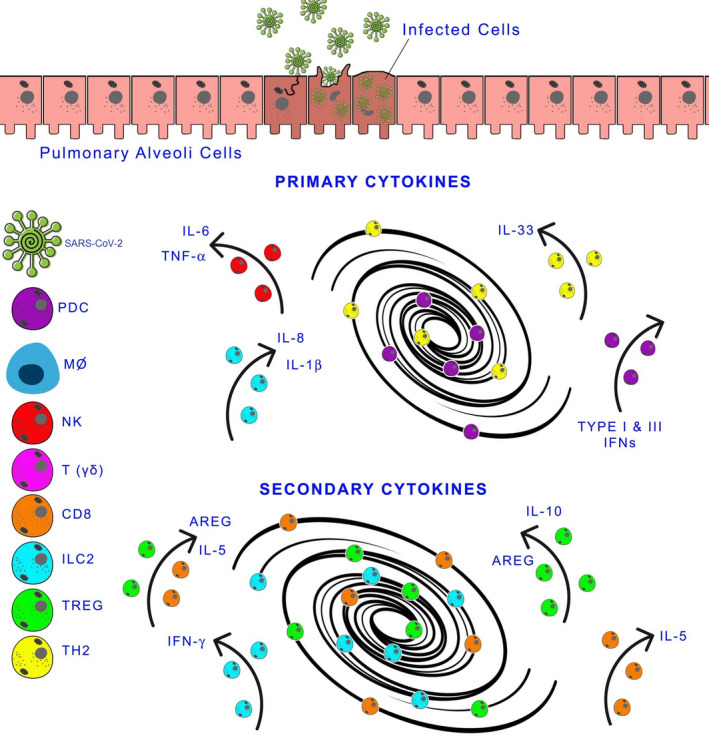
Cytokine Storm and SARS‐CoV‐2 Infection. After infection of the lower respiratory tract's epithelial cells by SARS‐Cov‐2, the innate immune response is initiated by recruiting cells that produce inflammatory cytokines. If the immune response is successful, there is a resolution. On the contrary, an exaggerated inflammatory process is amplified and damages the tissue. In the left‐side panel: SARS‐CoV‐2 and immune cells. In the centre of the figure are the primary and secondary cytokines (players participating in the storm). AREG, amphiregulin; CD8, CD8+T cells; IFN, interferon; IL, interleukin; ILC2, type 2 innate lymphoid cells; MФ, macrophages; NK, natural killer cells; pDC, plasmacytoid dendritic cells; T(γδ), T gamma delta cells; TH2, helper cells; TREG, regulatory T cells
In older adults and patients with comorbidities, including obesity, severe reactions to SARS‐CoV‐2 due to CS activation is observed; inflammaging and subclinical inflammation have been related to severe and worst outcome. 12 , 68 , 131 , 132 , 133 A less developed immune response produces lower levels of inflammation‐driving cytokines. 134 The best model of CS is a severe lung infection and sepsis. Sepsis is characterized by persistent hypotension, hyper‐ or hypothermia, leukocytosis or leukopenia, and thrombocytopenia. Severe lung damage associated with CS resembles sepsis. 126 , 127
Regulatory cytokines present a two‐sided coin in respiratory infection, limiting immunopathology and, at the same time, compromising viral clearance. 135 , 136 The balance between proinflammatory and regulatory cytokines is related to clearance and tolerance of the respiratory infection. Pathogens can modify host regulatory cytokines to promote their persistence, as shown for IL‐10 in Mycobacterium tuberculosis infection and TGF‐β in influenza virus infection. 136
Hyper‐ and hyporesponders to bacterial components are distinguishable in the healthy population. 137 , 138 , 139 The response is partly explained by genetic polymorphisms that affect the structure and function of Toll‐like receptors (TLR). 137 Wurfel et al, 138 in a group of septic patients infected with Gram‐positive bacteria, were able to identify a specific hypermorphic single nucleotide genetic polymorphism of TLR1. This mutation was significantly related to the exacerbated immune response and multiple organ failure and death. TLR2 polymorphism, IL‐4 rs2070874 and the chemokine (C‐C motif) ligand 2 gene (CCL2) also seem to be involved in this phenomenon. 139
In addition to the polymorphisms of PRRs and cytokines, adaptor proteins may be involved in T cells' response upon cytokine storm‐generated in viral infection. 139 The adhesion and degranulation‐promoting adapter protein (ADAP), involved in T cell antigen response and survival, is crucial in controlling influenza in the murine model. 139 Little is known about the importance of more critical proteins of the TLR pathway and inflammasome pathways, modulated by several factors, including ageing. 139 , 140
Cytokine storm appears to affect patients in severe conditions; lymphocytopenia and exhaustion or anergic responses are often reported in critical patients with COVID‐19. 141 Patients who eventually enter the intensive care unit (ICU) have significantly higher plasma levels of IL‐6, and IL‐10, non‐necessarily TNFα and fewer circulating CD4+ and CD8+ T lymphocytes, NK, and B lymphocytes. 141 , 142 , 143 , 144 CD8 and NK cells exhaustion markers indicate progression and prognosis of the viral infection, 142 , 143 , 144 and IL‐6 appears to be the crucial cytokine in the inflammatory process. 144 In animal studies, CS also impaired the development of adequate adaptive immunity against SARS‐CoV infection. 145 Primarily, 14‐month‐old BALB/C mice infected with SARS had a higher inflammatory response (higher biphasic transcription of IL‐6, TNF‐α CCL2, CCL3, CXCL10 and IFN‐γ genes), without resolution of the infection as compared to 8‐week‐old young mice. 145 Molecular analysis showed a wide variety of gene transcription between old and young infected mice, 146 , 147 suggesting that these two different entities cannot be managed with the same therapy.
5.4. NK, NKT, Tγδ cells and Mucosa‐associated invariant T (MAIT) cells
In several viral infections, NK cells, NKT cells and T gamma delta cells may generate a specific memory against the pathogen in a mechanism that differs from adaptive immunity. 148 , 149 , 150 , 151 , 152 , 153 Circulating NK and NKT cells levels were shown to be in very low numbers in both active and severe patients with SARS‐CoV‐2. 148 , 149 , 150 , 151 , 152 , 153 In SARS infection, Vγ9Vδ2 T cells were shown to be protective, and this subpopulation showed a memory cell response to the virus. 149 Higher immunoglobulin G anti‐SARS‐CoV titres were recorded when memory Vγ9Vδ2 T cells were identified. 149 Moreover, stimulated Vγ9Vδ2 T cells, in vitro, secreted interferon‐γ, could activate cytotoxic cells to kill SARS‐CoV–infected cells. 149 , 150 Chen et al 150 encountered a significant inverse correlation between the increase in peripheral nonfunctional immature neutrophils with the circulating numbers of CD8 T and Tγδ cells. Alterations in Tγδ were also recorded by Jouan et al, 151 which demonstrated the importance of CD69 activation marker in patients recovering from the infection. These results suggest that the impaired immune response observed in severe cases is due to the lack of protective antiviral immune response. Any opportunistic pathogen may easily overcome the protective immune response. Following the lack of proper innate immune response, Carissimo et al 152 reported the presence of immature neutrophils, and the ratio of immature neutrophils to VD2 γδ T cells CD8 accurately predicted pneumonia and hypoxia onset.
Maucourant et al 153 analysed different NK populations in infected patients. The authors were able to show that NK cell subpopulations and functions were related to the clinical con the patients. Two critical markers were found: NKG2C, an activation marker, and Ksp37, a secretory protein related to cytotoxicity. In recovered patients, these two markers were important. 153 Nonetheless, a decrease in the expression of the inhibitory receptor CD158b, KIR2DL2/L3 was reported in SARS‐infected patients that recover from the infection. 153 , 154 In animal models, the ligand interaction NKG2D (CD314) protects the animals from neurologic and hepatic damage induced by the coronavirus. 154 Blocking NKG2D made mice more susceptible to tissue damage, suggesting that the receptor may be necessary for immune surveillance. 154 Future studies should assess the importance of killing and KIR receptors and their ligands in this viral disease. More research is required in this field.
The innate immune response is crucial to defining COVID patients' outcome, resolution and viral clearance, severity and chronicity. 18 , 155 , 156 Immune senescence could be a critical issue in generating an effective immune response. 157 An impaired immune response can also be observed in patients infected with other viral infection (influenza, cytomegalovirus) or bacterial infection decreasing immune surveillance and effective antiviral response.
Mucosa‐associated invariant T (MAIT) cells are distinctive innate‐like T cells that facilitate the interaction between innate and adaptive immunity and consequently are important in defence against bacterial and viral infections. 158 MAIT cells express CD8 CD45RO CD161 and can be easily identified by the use of 5‐(2‐oxopropylideneamino)‐6‐D‐ribityl amino uracil (neoantigen of bacterial origin) in tetramers. 158 Tissue activation of MAIT cells predisposes neutrophil attraction which enhances the inflammatory milieu. 158 Recently, it was shown that these cells decrease in peripheral circulation in SARS‐CoV2‐2. 159 However, the circulating cells are activated, and high expression of CD69 and low CXCR3 seems to be related to poor clinical outcome. In convalescence, the number of MAIT cells increases, suggesting a direct relationship with the viral infection resolution. 159 These cells' roles in SARS‐Cov‐2 pathogenesis and other innate lymphoid cells (ILC1, ILC2 and ILC3) are still under investigation since they may be crucial in resolving the viral infection. The use of animal models will improve or vision of these cells in the viral infection.
Multisystemic inflammatory syndrome associated with SARS‐CoV‐2 infection is a new syndrome described in children characterized by fever, rash, conjunctivitis, mucocutaneous involvement and cardiac complications along with gastrointestinal symptoms and coagulopathy. 160 Interestingly, the syndrome affects more Black than Asian descent children suggesting that there may be a genetical relationship related to HLA. In children, the syndrome involves activation of Tγδ and CD4+CCR7+ T cells, high expression of CD64 on neutrophils and monocytes, and a decrease amount of conventional monocytes and antigen‐presenting cells which have low expression of HLA‐DR and CD86. 160 , 161 Some children with a high incidence of respiratory infectious diseases may present temporary hypogammaglobulinaemia or low IgA levels. 162 These patients may be more at risk to develop severe illness from SARS‐CoV‐2 infection. Patients with absent humoral immunity respond well to remdesivir 163 and IL‐6R biological therapy. 160 , 161 Noteworthy, Naito et al 164 proposed an association between IgA deficiency with SARS‐CoV‐2 infection and severity based on the incidence of reported IgA immunodeficiency. It can be concluded that the IgA response induced through MAIT cells can be crucial in an effective and protective response against SARS‐CoV‐2.
6. ANTIGEN PRESENTATION AND ADAPTIVE IMMUNE RESPONSE VIRUS ESCAPE
Antigen presentation in SARS‐CoV‐2 infection has not been well studied as the other members of the coronavirus family SARS and MERS. 165 DC and chemokines such as IP‐10 and MP1 play an essential role in antigen presentation and T cell activation. 165 , 166 Also, antigen presentation by DC is preferential via MHC class I and then class II suggesting that CD8 response may be an earlier event. 116 , 117 Macrophages also present viral antigens that can be obtained by 1) an effective RIG‐TLR dependent response against the virus, 2) the uptake of killed virus‐infected cells or 3) through immune complex binding to Fc receptors as described by other viral diseases 167 and suggested in the data obtained from the analysis of patients´ samples. 114 , 115 The presence of non‐typical monocytes recorded and the decrease in HLA‐DR indicate that one mechanism of viral escape could be through HLA molecules' low expression. 165 , 166 The use of chloroquine as initial treatment in SARS‐CoV‐2 patients may be responsible for decreasing antigen expression and the short‐lasting antibody response in some cases. 168
Both HLA molecules, class I and class II, can bind SARS‐CoV‐2 S peptides with different affinities. 169 , 170 , 171 Sanchez‐Mazas 171 reviewed the link between genetic variability among MHC class I genes (A, B and C) that may affect the predisposition and severity of acute respiratory distress syndrome in coronavirus infection. They found similarities in the previous SARS infection, MERS and SARS‐CoV‐2. In the three reports, 169 , 170 , 171 HLA‐B * 46:01 (a haplotype usually encountered in eastern Asia) had the lowest predicted binding score for S1 peptides, suggesting that people with this allele may be more susceptible to infection, analogous to what occurs in SARS infections. Other alleles encountered HLA‐B*07:03, DRB1*03:01, DRB1 *12:02 are also related to SARS susceptibility. 171 HLA‐B * 15:03 and HLA A 02*01 showed the highest scores for binding highly S peptides and other common human coronaviruses. These results suggest that several HLA haplotypes are associated with different diseases' susceptibilities. 169 , 170 , 171 , 172 , 173 Based on the incidence of SARS‐CoV‐2 infections and death in Africa, Iesa et al 173 encountered common immunodominant regions of Plasmodium falciparum, explaining the lower infection rates. Even though CD8 cells could be responsible for the postulated protective immune response, antibodies that cross‐react against similar epitopes of both pathogens have to be analysed carefully. It is also interesting to examine the effect of hydroxychloroquine treatment on malaria patients exposed to the SARS‐CoV‐2 virus. 168
In a complete and detailed analysis performed with data provided by 98 countries, Leite et al 174 were identified several interesting loci related to mortality. Besides the strong association of polymorphisms of the cytokines IL‐6, IL‐10 and IL‐12B, the authors reported HLA‐B*13:01 as a protective allele. This allele is expressed preferentially in Asiatic populations, and it is linked to dapsone‐induced hypersensitivity reactions. 175 Dapsone was proposed as a potential treatment to decrease cytokine storm induced by SARS‐CoV‐2 infection.
Bruchez et al 176 analysed class II presentation in Ebola infections found that CIIA, an MHC II transactivator, can be crucial for host defence against viruses. CD74 p41 blocks the coronavirus endosomal entry pathway, a process essential for SARS‐CoV‐2 replication. Interestingly, statins suppress CIIA transcription induced by IFN γ 177 and probably affect the mentioned mechanism of protection. More research is required to ascertain the role of MHC and CIIA in viral entry and antigen presentation.
Analysing the changes in circulating cells in SARS‐CoV‐2‐infected patients, García, 156 in a review, outlined the difference between protective immunity and immune dysregulation. In severe patients, the expression of exhaustion markers or inhibitory markers is significant, suggesting that CD8 circulating cells are non‐responsive or anergic. 156 However, other unsolved issues arise from the changes in circulating cells during the infection, the generation of antibodies, cytokine changes and viral clearance. The induction of specific CD8 cells against the virus can be essential to decrease the viral burden. Vibholm et al 178 were able to show that SARS‐CoV‐2 virus persistence was depended on CD8 responses based on a nucleotide‐based screening test. However, the lack of an effective CD8 response is dependent on innate immune cells and MAIT cells. 179 , 180 , 181 Viral shedding, the lack of viral RNA recognition and the impaired generation of IFN I and III production and pathways generated by them are related to viral persistence and chronicity. 179 , 180 , 181 , 182 The adaptive immune response can be long lasting, 181 , 182 suggesting that chronicity may be replaced by an effective innate immune response in the airways for T cells, CD4 and CD8, and B cells. 181 , 182
Chen et al, 183 in BALB/c SARS model, were able to show that the T cell CD4+ population is critical for a viral response as opposed to T cytotoxic CD8+ cells. Antibody response seems to be essential in viral clearance. Likewise, in severely infected patients, IFN‐γ production by CD4+ T cells is significantly lower than controls, 142 , 143 , 156 , 157 and the circulating B lymphocyte number is deficient. The decrease in TH1 and B cells impairs antibody production in these patients. 156 , 157 Wang et al 184 established that post‐treatment, a significant reduction of CD8+ T cells and B cells, and a higher CD4+/CD8+ ratio were markers of unresponsiveness to therapy worse outcome. In a series of experiments ex vivo to understand virus involvement in cell activation, mononuclear cells from patients were stimulated with a mixture of E coli and Candida albicans. 184 The cells secreted more IL‐6 than TNFα and IL‐1β, and the same ratio was observed in the sera. 184 Plasma from patients decreased HLA‐DR expression in stimulated cells, an effect that tocilizumab, anti‐IL‐6R, blocked. 184 It is clear then the dependence of innate immunity and acute‐phase proteins in the infection's adaptive immune response. Once the hyperinflammation is controlled therapeutically, an effective immune response is achieved.
Several bioinformatics studies on COVID‐19 and T and B cell paratopes have been published. 185 , 186 , 187 Two linear epitopes on the SARS‐CoV‐2 spike protein elicit neutralizing antibodies in COVID‐19 patients. 186 Liu et al 187 found potent and neutralizing antibodies against multiple epitopes. Nonetheless, the number of epitopes and the antibody responses of convalescent plasma does not match with the role of CD8 in cell recruitment and cytotoxic response. This paradigm can probably be explained by the longitudinal modulation of T and B cell responses described by Niu et al. 188 The authors showed a diminished TCR repertoire at the beginning of the infection but increased convalescence and recovery. In B cells, clonal expansion occurs in convalescence with a dramatic expansion of IgA positive B cells, and then, other B cells start producing IgG antibodies with neutralizing ability. This process is probably hampered in elderly patients. 189 Nonetheless, important questions also arise in patients with IgA immunodeficiency or mild immunodeficiencies and patients with IgA nephropathy even though they respond to remdesivir therapy. 163
Strong antibody responses against the SARS‐CoV‐2 virus were detected in patients that quickly recover from the infection. 190 In these individuals, the authors were able to detect virus‐specific memory B cells and circulating activated CD4 T cells suggesting that a memory response was responsible for the effective response. In concordance with these studies, in a flow cytometry assessment in our laboratory, 191 we observed that circulating memory T cells do not decrease in infected patients with moderate infection, suggesting a possible response to previous coronavirus infection.
Ferreti et al 192 analysed, doing a genome‐wide screening, the specific antigens recognized by memory CD8 cells against the virus. They identified the epitopes that would bind to the 6 most prevalent MHC class I in the population. Interestingly, of the 29 shared epitopes, only 3 were located in the spike protein. The other epitopes were located in the ORF1ab or in the nucleocapsid protein. 192 The memory response does not seem to be related to other coronaviruses, suggesting that an effective innate response against the virus was crucial in developing CD8 specific T cells. 192
Several studies aiming to analyse the differences between responders and non‐responders to SARS‐CoV‐2 infection have addressed as if there was a protective immune response before infection. 193 , 194 , 195 , 196 It has been shown that there are memory T cell responses against other coronaviruses, which protect from SARS‐CoV‐2 infection. 193 , 194 , 195 , 196 This protection is often encountered in children exposed to other coronaviruses. Eventually, the generation of memory cells and protective antibodies by immunization against other viruses can occur. 196 The protection mechanism may depend on similar epitopes presented by the vaccine or even an induction or immune response stimulation like the BCG vaccine. 197 The vaccine was shown to decrease mortality by activating immune response to activation of innate lymphoid cells (Tγδ, NKT, ILC).
Conti and Younes 198 hypothesized (in an editorial) that the difference in the immune response against coronavirus infection between genders is due to several issues: (a) men are more susceptible to viral infections because men produce a lower amount of antibodies; (b) the amount of circulating T CD4+ cells is smaller in man as compared to women, (c) men produce higher amounts of IL‐6, and consequently, cytokine storm is most probable. In a recent revision, Rahimi et al 199 analyse the different immune responses dependent on gender and the various trials associated with SARS‐CoV‐2. The detailed analysis favours the proposal of several new therapies; however, more research needs to be performed based on virus variations, gender, ethnicity and age. 12 , 13
A relationship between gene clusters and COVID‐19 severity has been postulated since the beginning of the pandemic. Karaderi et al 200 were able to show in an interesting analysis of gene mortality the importance of chromosome 3 cluster and other clusters. In the cluster in chromosome 3.21, six genes, CCR9, CXCR6, FYCO1, LZTFL1, SLC6A20, XCR1, have been associated with severity. The group consists of chemokine receptors, CCR9, CXCR6, XCE1, a protein related to vesicle transport and autophagosome maturation, FYCO1, the leucine zipper transcription factor‐like protein 1 related with ciliary function, LZTFL1, and a sodium and chloride‐dependent transporter, SLC6A20. An additional cluster involved in the viral immune response is the 2'‐5'‐oligoadenylate synthetase 1 cluster, OAS. In this cluster, another six genes are involved: the receptor interferon αβ2, IFNAR2, two chemokine receptors CCR2, CCR3, HLA‐G, a regulator of mRNA, coiled‐coil alpha‐helical rod protein 1, CCHCR1 and NOTCH4. Two independent genes were associated with the finding; the serine protease dipeptidyl peptidase 9 (DPP9) and the kinase TYK2, associated with the IFN α signal. Also, there is an association with ABO genes in various ethnic groups. As stated in the complement section of the review, 98 the chromosome 3 variant rs11385942 is involved in complement activation and makes this cluster a candidate for genetic, biochemical and immunological analysis. Thus, chemokine receptors and IFN receptors are related to the SARS‐CoV‐2 viral infection and therapy response, as has been illustrated through the review. The role of transports involved in electrolyte balance should not be overlooked in patients infected with SARS‐CoV‐2.
In general, we consider that innate immunity, complement, memory T cells, CD4 and CD8 and B cells are crucial to generate an effective immune response against the viral infection independently of ethnicity, gender or ageing. In a proinflammatory environment (inflammaging and metabolic syndrome), the generation of memory cells may be impaired, yet it can be boosted with immune therapy, vaccines or other therapies.
7. CONCLUSIONS
The SARS‐CoV‐2 infection goes through multiple phases. The initial stage involves viral replication, often paired with relatively mild symptoms and innate immune response activation. Subsequently, adaptive immunity is activated, and either the virus is cleared, the infection is solved, or it becomes severe or chronic. The clinical spectrum may vary depending on the viral load and immune status of the patient. Immunocompromised patients or patients with several comorbidities are more susceptible to develop severe disease. Severe pulmonary distress syndrome is often observed in these patients with a long recovery rate. In some patients, lung fibrosis may occur. Young patients with high exposure to the virus may have the same outcome, that is, infected health care personnel.
Clinicians and researchers have learned that severe hyperinflammation (cytokine storm) should be avoided. Genetic polymorphisms and mutations may affect the immune response and viral clearance, essentially IL‐6, IL‐10, IL‐12, chemokine receptors and IFN type I and III response. The role of complement in the disease's immunopathology has provided new insights for treatment and clinical management. The mechanism of the specific generation of memory T CD4 and CD8 cells against the virus seems essential in the production of protective antibodies and virus elimination. The role of NK, NKT, Tγδ and innate cells (MAIT and ILC) on the viral infection is still under scrutiny, although they may play an essential role in the initial infection. Most probably in vaccine studies, their immune vigilance role against the virus could be ascertained in detail. Long‐term memory is the final goal.
Genetic polymorphism and mutations may hamper an effective antiviral immune response, and more research should be done in this area. Even though healthy children are less prone to have severe disease, medical counselling, including up to date vaccine schedule, may help protect this population from SARS infections. Virus infections, common in elders, may also contribute to the impaired response in this population and should be carefully screened. To avoid severity in risk populations, viral and antigen screening should be performed on a large scale. Moreover, continuous screening of recovered patients with comorbidities should be performed to avoid a second infection.
Finally, even though this evolving disease's progress is very fast, the immunologist's role in ascertaining an effective immune response mechanism is still needed.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
AUTHOR CONTRIBUTIONS
JBDS, AG and DM were involved in the review and literature search. The manuscript writing was completed by JBDS, AG and DM and editing by JBDS, AG, DM and MH. MH is responsible for funding.
ACKNOWLEDGEMENTS
The authors wish to thank Andrea García for figure design and Drs. Zury Domínguez, Lérida Padrón, Judith Barroso and Jenny Garmendia for useful discussions.
De Sanctis JB, García AH, Moreno D, Hajduch M. Coronavirus infection: An immunologists' perspective. Scand J Immunol. 2021;93:e13043. 10.1111/sji.13043
Funding information
This review was supported by a grant from European Structural and Investment Operational Funds Program Research entitled: Molecular, cellular, and clinical approach to healthy ageing grant ENOCH; registration number: CZ.02.1.01/0.0/0.0/16_019/0000868 to JBDS, project lieder at the IMTM, Palacky U, MH.
REFERENCES
- 1. Yuen KS, Ye ZW, Fung SY, Chan CP, Jin DY. SARS‐CoV‐2 and COVID‐19: the most critical research questions. Cell Biosci. 2020;10:40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Weber DJ, Rutala WA, Fischer WA, et al. Emerging infectious diseases: focus on infection control issues for novel coronaviruses (Severe Acute Respiratory Syndrome‐CoV and Middle East Respiratory Syndrome‐CoV), hemorrhagic fever viruses (Lassa and Ebola), and highly pathogenic avian influenza viruses, A(H5N1) and A(H7N9). Am J Infect Control. 2016;44(5 Suppl):e91‐e100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ye Z‐W, Yuan S, Yuen K‐S, et al. Zoonotic origins of human coronaviruses. Int J Biol Sci. 2020;16(10):1686‐1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Mousavizadeh L, Ghasemi S. Genotype and phenotype of COVID‐19: Their roles in pathogenesis. J Microbiol Immunol Infect. 2020;S1684‐1182(20):30082–30087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Chen Y, Liu Q, Guo D. Emerging coronaviruses: genome structure, replication, and pathogenesis. J Med Virol. 2020;92(4):418‐423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Fung TS, Liu DX. Human coronavirus: host‐pathogen interaction. Annu. Rev. Microbiol. 2019;73:529‐557. [DOI] [PubMed] [Google Scholar]
- 7. Newton AH, Cardani A, Thomas J. The host immune response in respiratory virus infection: balancing virus clearance and immunopathology. Semin Immunopathol. 2016;38(4):471‐482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Thakur V, Ratho RK, Kumar P, et al. Multi‐organ involvement in COVID‐19: beyond pulmonary manifestations. J Clin Med. 2021;10(3):446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lai C‐C, Liu YH, Wang C‐Y, et al. Asymptomatic carrier state, acute respiratory disease, and pneumonia due to severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2): facts and myths. J Microbiol Immunol Infect. 2020;53(3):404‐412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Jung C‐Y, Park H, Kim DW, et al. Clinical characteristics of asymptomatic patients with COVID‐19: a nationwide Cohort Study in South Korea. Int J Infect Dis. 2020;99:266‐268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Parilli‐Troconis D, Baptista P, Marcano‐Lozada M, et al. COVID‐19 infection and its influence in otorhinolaryngology‐head and neck surgery. Int Arch Otorhinolaryngol. 2020;24(4):e527‐e534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Pepe M, Maroun‐Eid C, Romero R, et al. Clinical presentation, therapeutic approach, and outcome of young patients admitted for COVID‐19, with respect to the elderly counterpart. Clin Exp Med. 2021;8:1‐20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Dumonteil E, Fusco D, Drouin A, Herrera C. Genomic signatures of SARS‐CoV‐2 associated with patient mortality. Viruses. 2021;13(2):227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Østergaard L. SARS CoV‐2 related microvascular damage and symptoms during and after COVID‐19: Consequences of capillary transit‐time changes, tissue hypoxia and inflammation. Physiol Rep. 2021;9(3):e14726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Rechtman E, Curtin P, Navarro E, Nirenberg S, Horton MK. Vital signs assessed in initial clinical encounters predict COVID‐19 mortality in an NYC hospital system. Sci Rep. 2020;10(1):21545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hendren NS, de Lemos JA, Ayers C, et al. Association of body mass index and age with morbidity and mortality in patients hospitalized with COVID‐19: results from the American heart association COVID‐19 cardiovascular disease registry. Circulation. 2021;143(2):135‐144. [DOI] [PubMed] [Google Scholar]
- 17. Pajo AT, Espiritu AI, Apor ADAO, Jamora RDG. Neuropathologic findings of patients with COVID‐19: a systematic review. Neurol Sci. 2021;22:1‐12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Garg S, Garg M, Prabhakar N, Malhotra P, Agarwal R. Unraveling the mystery of Covid‐19 cytokine storm: From skin to organ systems. Dermatol Ther. 2020;33:e13859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mason RJ. Thoughts on the alveolar phase of COVID‐19. Am J Physiol Lung Cell Mol Physiol. 2020;319(1):L115‐L120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Nie X, Qian L, Sun R, et al. Multi‐organ proteomic landscape of COVID‐19 autopsies. Cell. 2021;184(3):775‐791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gomes CP, Fernandes DE, Casimiro F, et al. in COVID‐19: from pharmacological evidences to genetics. Front Cell Infect Microbiol. 2020;10:589505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lim J‐H, Jung H‐Y, Choi J‐Y, et al. Hypertension and electrolyte disorders in patients with COVID‐19. Electrolyte Blood Press. 2020;18(2):23‐30. 10.1007/s11739-021-02632-z. Online ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. De Carvalho H, Richard MC, Chouihed T, et al. Electrolyte imbalance in COVID‐19 patients admitted to the Emergency Department: a case‐control study. Intern Emerg Med. 2021;1‐6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gong T, Yang Y, Jin T, Jiang W, Zhou R. Orchestration of NLRP3 inflammasome activation by ion fluxes. Trends Immunol. 2018;39(5):393‐406. [DOI] [PubMed] [Google Scholar]
- 25. Shang J, Ye G, Shi KE, et al. Structural basis of receptor recognition by SARS‐CoV‐2. Nature. 2020;581(7807):221‐224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Yan R, Zhang Y, Li Y, Xia L, Guo Y, Zhou Q. Structural basis for the recognition of SARS‐CoV‐2 by full‐length human ACE2. Science. 2020;367(6485):1444‐1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Morelli F, Meirelles LEdF, de Souza MVF, et al. COVID‐19 infection in the human reproductive tract of men and Nonpregnant women. Am J Trop Med Hyg. 2021;104:814‐825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Walls AC, Park YJ, Tortorici MA, Wall A, McGuire AT, Veesler D. Structure, function, and antigenicity of the SARS‐CoV‐2 spike glycoprotein. Cell. 2020;181(2):281‐292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Batlle D, Wysocki J, Satchell K. Soluble angiotensin‐converting enzyme 2: a potential approach for coronavirus infection therapy? Clin Sci. 2020;134(5):543‐545. [DOI] [PubMed] [Google Scholar]
- 30. Hoffmann M, Kleine‐Weber H, Schroeder S, et al. SARS‐CoV‐2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. 2020;181:1‐10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Johnson BA, Xie X, Bailey AL, et al. Loss of furin cleavage site attenuates SARS‐CoV‐2 pathogenesis. Nature. 2021;591(7849):293‐299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Gottlieb RL, Nirula A, Chen P, et al. Effect of bamlanivimab as monotherapy or in combination with etesevimab on viral load in patients with mild to moderate COVID‐19: a randomised clinical trial. JAMA. 2021;325(7):632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Rappazzo CG, Tse LV, Kaku CI, et al. Broad and potent activity against SARS‐like viruses by an engineered human monoclonal antibody. Science. 2021;371(6531):823‐829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Jitsuiki K, Katayama I, Iida T, Nagatomo S, Yanagawa Y. Successful treatment of elderly male with COVID‐19 infection with severe acute respiratory distress syndrome using multimodal therapy, including immune modulation therapy. Cureus. 2020;12(12):e12402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Gemmati D, Bramanti B, Serino ML, et al. COVID‐19 and individual genetic susceptibility/receptivity: role of ACE1/ACE2 genes, immunity, inflammation and coagulation. Might the double X‐chromosome in females be protective against SARS‐CoV‐2 compared to the single X‐chromosome in males? Int J Mol Sci. 2020;21(10):E3474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zheng H, Cao JJ. Angiotensin‐converting enzyme gene polymorphism and severe lung injury in patients with coronavirus disease 2019. Am J Pathol. 2020;190(10):2013‐2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Asselta R, Paraboschi EM, Mantovani A, Duga S. ACE2 and TMPRSS2 variants and expression as candidates to sex and country differences in COVID‐19 severity in Italy. Aging. 2020;12(11):10087‐10098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hussain M, Jabeen N, Raza F, et al. Structural variations in human ACE2 may influence its binding with SARS‐CoV‐2 spike protein. J Med Virol. 2020;92(9):1580‐1586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Li Q, Cao Z, Rahman P. Genetic variability of human angiotensin‐converting enzyme 2 (hACE2) among various ethnic populations. Mol Genet Genomic Med. 2020;8(8):e1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ziegler CGK, Allon SJ, Nyquist SK, et al. HCA lung biological network, SARS‐CoV‐2 receptor ACE2 is an interferon‐stimulated gene in human airway epithelial cells and is detected in specific cell subsets across tissues. Cell. 2020;81(5):1016‐1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Xu Q, Tang Y, Huang G. Innate immune responses in RNA viral infection. Front Med. 2020;1‐14. 10.1007/s11684-020-0776-7. Online ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Jones JE, Le Sage V, Lakdawala SS. Viral and host heterogeneity and their effects on the viral life cycle. Nat Rev Microbiol. 2021;19(4):272‐282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Onomoto K, Onoguchi K, Yoneyama M. Regulation of RIG‐I‐like receptor‐mediated signaling: interaction between host and viral factors. Cell Mol Immunol. 2021;18(3):539‐555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Antonczyk A, Krist B, Sajek M, et al. Direct inhibition of IRF‐dependent transcriptional regulatory mechanisms associated with disease. Front Immunol. 2019;10:1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lega S, Naviglio S, Volpi S, Tommasini A. Recent insight into SARS‐CoV2 immunopathology and rationale for potential treatment and preventive strategies in COVID‐19. Vaccines. 2020;8(2):224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Jing H, Su HC. New immunodeficiency syndromes that help us understand the IFN‐mediated antiviral immune response. Curr Opin Pediatr. 2019;31(6):815‐820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Elhabyan A, Elyaacoub S, Sanad E, Abukhadra A, Elhabyan A, Dinu V. The role of host genetics in susceptibility to severe viral infections in humans and insights into host genetics of severe COVID‐19: A systematic review. Virus Res. 2020;289:198163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wang BX, Fish EN. Global virus outbreaks: interferons as 1st responders. Semin Immunol. 2019;43:101300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Lei X, Dong X, Ma R, et al. activation and evasion of type I interferon responses by SARS‐CoV‐2. Nat Commun. 2020;11(1):3810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Konno Y, Kimura I, Uriu K, et al. SARS‐CoV‐2 ORF3b Is a Potent Interferon Antagonist Whose Activity Is Increased by a Naturally Occurring Elongation Variant. Cell Rep. 2020;32(12):108185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Miorin L, Kehrer T, Sanchez‐Aparicio MT, et al. SARS‐CoV‐2 Orf6 hijacks Nup98 to block STAT nuclear import and antagonise interferon signaling. Proc Natl Acad Sci USA. 2020;117(45):28344‐28354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Zheng YI, Zhuang M‐W, Han L, et al. Severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) membrane (M) protein inhibits type I and III interferon production by targeting RIG‐I/MDA‐5 signaling. Signal Transduct Target Ther. 2020;5(1):299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Zhang Q, Bastard P, Liu Z, et al. Inborn errors of type I IFN immunity in patients with life‐threatening COVID‐19. Science. 2020;370:eabd4570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Tangye SG, Al‐Herz W, Bousfiha A, et al. The ever‐increasing array of novel inborn errors of immunity: an interim update by the IUIS committee. J Clin Immunol. 2021;41(3):666‐679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Bastard P, Rosen LB, Zhang Q, et al. Auto‐antibodies against type I IFNs in patients with life‐threatening COVID‐19. Science. 2021;218(4):e20202486. [Google Scholar]
- 56. Fulzele S, Sahay B, Yusufu I, et al. COVID‐19 virulence in aged patients might be impacted by the host cellular MicroRNAs abundance/profile. Aging Dis. 2020;11(3):509‐522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Hosseini Rad Sm A, McLellan AD. Implications of SARS‐CoV2 mutations for genomic RNA structure and host microRNA targeting. Int J Mol Sci. 2020;21(13):4807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Arisan ED, Dart A, Grant GH, et al. The prediction of miRNAs in SARS‐CoV2 genomes: hsa‐miR databases identify 7 Key miRs linked to host responses and virus pathogenicity‐related KEGG pathways significant for comorbidities. Viruses. 2020;12(6):614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Srivastava R, Daulatabad SV, Srivastava M, Janga SC. SARS‐CoV‐2 contributes to altering the post‐transcriptional regulatory networks across human tissues by sponging RNA binding proteins and micro‐RNAs. Int J Mol Sci. 2020;21(19):E7090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Wower IK, Brandebourg TD, Wower J. New insights on the mobility of viral and host non‐coding RNAs reveal extracellular vesicles as intriguing candidate antiviral targets. Pathogens. 2020;9(11):E876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Nersisyan S, Engibaryan N, Gorbonos A, Kirdey K, Makhonin A, Tonevitsky A. Potential role of cellular miRNAs in coronavirus‐host interplay. PeerJ. 2020;8:e9994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Lee HY, Hur J, Kang JY, Rhee CK, Lee SY. MicroRNA‐21 inhibition suppresses alveolar M2 macrophages in an ovalbumin‐induced allergic asthma mice model. Allergy Asthma Immunol Res. 2021;13(2):312‐329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Nersisyan S, Shkurnikov M, Turchinovich A, Knyazev E, Tonevitsky A. Integrative analysis of miRNA and mRNA sequencing data reveals potential regulatory mechanisms of ACE2 and TMPRSS2. PLoS One. 2020;15(7):e0235987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Paniri A, Hosseini MM, Moballegh‐Eslam M, Akhavan‐Niaki H. Comprehensive in silico identification of impacts of ACE2 SNPs on COVID‐19 susceptibility in different populations. Gene Rep. 2021;22:100979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Wyler E, Mösbauer K, Franke V, et al. Transcriptomic profiling of SARS‐CoV‐2 infected human cell lines identifies HSP90 as target for COVID‐19 therapy. iScience. 2021;24(3):102151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Mazori AY, Bass IR, Chan L, et al. Hyperglycemia is associated with increased mortality in critically Ill patients with COVID‐19. Endocr Pract. 2021;27(2):95‐100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. López‐Reyes A, Martinez‐Armenta C, Espinosa‐Velázquez R, et al. NLRP3 inflammasome: the stormy link between obesity and COVID‐19. Front Immunol. 2020;11:570251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Tanner JE, Alfieri C. The fatty acid lipid metabolism nexus in COVID‐19. Viruses. 2021;13(1):90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Kuo C‐L, Pilling LC, Atkins JL, et al. APOE e4 genotype predicts severe COVID‐19 in the UK Biobank community cohort. J Gerontol A Biol Sci Med Sci. 2020;75(11):2231‐2232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Keshavarz M, Solaymani‐Mohammadi F, Namdari H, et al. Metabolic host response and therapeutic approaches to influenza infection. Cell Mol Biol Lett. 2020;25:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Yan B, Chu H, Yang D, et al. Characterisation of the lipidomic profile of human coronavirus‐infected cells: implications for lipid metabolism remodeling upon coronavirus replication. Viruses. 2019;11(1):73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Shen B, Yi X, Sun Y, et al. Proteomic and metabolomic characterisation of COVID‐19 patient sera. Cell. 2020;182(1):59‐72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Barberis E, Timo S, Amede E, et al. Large‐scale plasma analysis revealed new mechanisms and molecules associated with the host response to SARS‐CoV‐2. Int J Mol Sci. 2020;21(22):8623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Caterino M, Gelzo M, Sol S, et al. dysregulation of lipid metabolism and pathological inflammation in patients with COVID‐19. Sci Rep. 2021;11(1):2941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Garić D, De Sanctis JB, Shah J, Dumut DC, Radzioch D. Biochemistry of very‐long‐chain and long‐chain ceramides in cystic fibrosis and other diseases: the importance of side chain. Prog Lipid Res. 2019;74:130‐144. [DOI] [PubMed] [Google Scholar]
- 76. Veltman M, Stolarczyk M, Radzioch D, et al. Correction of lung inflammation in a F508del CFTR murine cystic fibrosis model by the sphingosine‐1‐phosphate lyase inhibitor LX2931. Am J Physiol Lung Cell Mol Physiol. 2016;311(5):L1000‐L1014. [DOI] [PubMed] [Google Scholar]
- 77. Prakash H, Upadhyay D, Bandapalli OR, Jain A, Kleuser B. Host sphingolipids: perspective immune adjuvant for controlling SARS‐CoV‐2 infection for managing COVID‐19 disease. Prostaglandins Other Lipid Mediat. 2021;152:106504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Carpinteiro A, Edwards MJ, Hoffmann M, et al. Pharmacological inhibition of acid sphingomyelinase prevents uptake of SARS‐CoV‐2 by epithelial cells. Cell Rep Med. 2020;1(8):100142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Java A, Apicelli AJ, Liszewski MK, et al. The complement system in COVID‐19: friend and foe? JCI Insight. 2020;5(15):e140711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Fang S, Wang H, Lu L, Jia Y, Xia Z. Decreased complement C3 levels are associated with poor prognosis in patients with COVID‐19: A retrospective cohort study. Int Immunopharmacol. 2020;89(Pt A):107070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Akk A, Springer LE, Yang L, et al. Complement activation on neutrophils initiates endothelial adhesion and extravasation. Mol Immunol. 2019;114:629‐642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Maxwell AJ, Ding J, You Y, et al. Identification of key signaling pathways induced by SARS‐CoV2 that underlie thrombosis and vascular injury in COVID‐19 patients. J Leukoc Biol. 2021;109(1):35‐47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Gralinski LE, Sheahan TP, Morrison TE, et al. Complement activation contributes to severe acute respiratory syndrome coronavirus pathogenesis. MBio. 2018;9(5):e01753‐e1818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Lin P, Chen W, Huang H, et al. Delayed discharge is associated with higher complement C3 levels and a longer nucleic acid‐negative conversion time in patients with COVID‐19. Sci Rep. 2021;11(1):1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Cugno M, Meroni PL, Gualtierotti R, et al. Complement activation and endothelial perturbation parallel COVID‐19 severity and activity. J Autoimmun. 2021;116:102560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Overmyer KA, Shishkova E, Miller IJ, et al. Large‐scale multi‐omic analysis of COVID‐19 severity. Cell Syst. 2021;12(1):23‐40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Kulkarni HS, Elvington ML, Perng YC, et al. Intracellular C3 protects human airway epithelial cells from stress‐associated cell death. Am J Respir Cell Mol Biol. 2019;60(2):144‐157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Kolin DA, Kulm S, Christos PJ, Elemento O. Clinical, regional, and genetic characteristics of Covid‐19 patients from UK Biobank. PLoS One. 2020;15(11):e0241264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. O'Brien ME, Fee L, Browne N, et al. activation of complement component 3 is associated with airways disease and pulmonary emphysema in alpha‐1 antitrypsin deficiency. Thorax. 2020;75(4):321‐330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Rogliani P, Lauro D, di Daniele N, Chetta A, Calzetta L. Reduced risk of COVID‐19 hospitalisation in asthmatic and COPD patients: a benefit of inhaled corticosteroids? Expert Rev Respir Med 2021;15(4):561‐568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Polycarpou A, Howard M, Farrar CA, et al. Rationale for targeting Complement in COVID‐19. EMBO Mol Med. 2020;12(8):e12642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Mastellos DC, Pires da Silva BGP, Fonseca BAL, et al. Complement C3 vs C5 inhibition in severe COVID‐19: early clinical findings reveal differential biological efficacy. Clin Immunol. 2020;220:108598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Yu J, Yuan X, Chen H, Chaturvedi S, Braunstein EM, Brodsky RA. Direct activation of the alternative complement pathway by SARS‐CoV‐2 spike proteins is blocked by factor D inhibition. Blood. 2020;136(18):2080‐2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Skendros P, Mitsios A, Chrysanthopoulou A, et al. Complement and tissue factor‐enriched neutrophil extracellular traps are key drivers in COVID‐19 immunothrombosis. J Clin Invest. 2020;130(11):6151‐6157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Holter JC, Pischke SE, de Boer E, et al. Systemic complement activation is associated with respiratory failure in COVID‐19 hospitalized patients. Proc Natl Acad Sci USA. 2020;117(40):25018‐25025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Tu X, Chong WP, Zhai Y, et al. Functional polymorphisms of the CCL2 and MBL genes cumulatively increase susceptibility to severe acute respiratory syndrome coronavirus infection. J Infect. 2015;71(1):101‐109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Di Maria E, Latini A, Borgiani P, Novelli G. Genetic variants of the human host influencing the coronavirus‐associated phenotypes (SARS, MERS and COVID‐19): rapid systematic review and field synopsis. Hum Genomics. 2020;14(1):30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Valenti L, Griffini S, Lamorte G, et al. Chromosome 3 cluster rs11385942 variant links complement activation with severe COVID‐19. J Autoimmun. 2021;117:102595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Woodruff TM, Shukla AK. The complement C5a–C5aR1 GPCR axis in COVID‐19 therapeutics. Trends Immunol. 2020;41(11):965‐967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Kulasekararaj AG, Lazana I, Large J, et al. Terminal complement inhibition dampens the inflammation during COVID‐19. Br J Haematol. 2020;190(3):e141‐e143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Janiuk K, Jabłońska E, Garley M. Significance of NETs formation in COVID‐19. Cells. 2021;10(1):151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Schulte‐Schrepping J, Reusch N, Paclik D, et al. Severe COVID‐19 Is marked by a dysregulated myeloid cell compartment. Cell. 2020;182(6):1419‐1440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Barnes BJ, Adrover JM, Baxter‐Stoltzfus A, et al. Targeting potential drivers of COVID‐19: neutrophil extracellular traps. J Exp Med. 2020;217(6):e20200652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Zuo Y, Kanthi Y, Knight JS, Kim AHJ. The interplay between neutrophils, complement, and microthrombi in COVID‐19. Best Pract Res Clin Rheumatol. 2021;35(1):101661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Zuo Y, Zuo M, Yalavarthi S, et al. Neutrophil extracellular traps and thrombosis in COVID‐19. J Thromb Thrombolysis. 2021;51(2):446‐453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Bao L, Zhang C, Dong J, Zhao L, Li Y, Sun J. Oral microbiome and SARS‐CoV‐2: beware of lung co‐infection. Front Microbiol. 2020;11:1840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Tan Y, Zhou J, Zhou Q, Hu L, Long Y. Role of eosinophils in the diagnosis and prognostic evaluation of COVID‐19. J Med Virol. 2021;93(2):1105‐1110. [DOI] [PubMed] [Google Scholar]
- 108. Lindsley AW, Schwartz JT, Rothenberg ME. Eosinophil responses during COVID‐19 infections and coronavirus vaccination. J Allergy Clin Immunol. 2020;146(1):1‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Ari S, Can V, Demir ÖF, et al. Elevated eosinophil count is related with lower anti‐factor Xa activity in COVID‐19 patients. J Hematop. 2020;Oct 8 1‐10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Motta Junior JDS, Miggiolaro AFRDS, Nagashima S, et al. Mast cells in alveolar septa of COVID‐19 patients: a pathogenic pathway that may link interstitial edema to immunothrombosis. Front Immunol. 2020;11:574862. 10.1007/s12308-020-00419-3. Online ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Hogan Ii RB, Hogan Iii RB, Cannon T, et al. Dual‐histamine receptor blockade with cetirizine ‐ famotidine reduces pulmonary symptoms in COVID‐19 patients. Pulm Pharmacol Ther. 2020;63:101942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Kopf M, Schneider C, Nobs SP. The development and function of lung‐resident macrophages and dendritic cells. Nat Immunol. 2015;16(1):36‐44. [DOI] [PubMed] [Google Scholar]
- 113. Evren E, Ringqvist E, Willinger T. Origin and ontogeny of lung macrophages: from mice to humans. Immunology. 2020;160(2):126‐138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Boumaza A, Gay L, Mezouar S, et al. Monocytes and macrophages, targets of SARS‐CoV‐2: the clue for Covid‐19 immunoparalysis. J Infect Dis. 2021;jiab044. 10.1093/infdis/jiab044. Online ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Kvedaraite E, Hertwig L, Sinha I, et al. Major alterations in the mononuclear phagocyte landscape associated with COVID‐19 severity. Proc Natl Acad Sci USA. 2021;118(6):e2018587118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Amon L, Lehmann CHK, Heger L, Heidkamp GF, Dudziak D. The ontogenetic path of human dendritic cells. Mol Immunol. 2020;120:122‐129. [DOI] [PubMed] [Google Scholar]
- 117. Reizis B. Plasmacytoid dendritic cells: development, regulation, and function. Immunity. 2019;50(1):37‐50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Freer G, Matteucci D. Influence of dendritic cells on viral pathogenicity. PLoS Pathog. 2009;5(7):e1000384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Campana P, Parisi V, Leosco D, Bencivenga D, Della Ragione F, Borriello A. Dendritic cells and SARS‐CoV‐2 infection: still an unclarified connection. Cells. 2020;9(9):2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Onodi F, Bonnet‐Madin L, Meertens L, et al. SARS‐CoV‐2 induces human plasmacytoid predendritic cell diversification via UNC93B and IRAK4. J Exp Med. 2021;218(4):e20201387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Sánchez‐Cerrillo I, Landete P, Aldave B, et al. COVID‐19 severity associates with pulmonary redistribution of CD1c+ DCs and inflammatory transitional and nonclassical monocytes. J Clin Invest. 2020;130(12):6290‐6300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Guo H, Callaway JB, Ting JP. Inflammasomes: mechanism of action, role in disease, and therapeutics. Nat Med. 2015;21(7):677‐687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Zhao C, Zhao W. NLRP3 Inflammasome‐A Key Player in Antiviral Responses. Front Immunol. 2020;11:211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Krainer J, Siebenhandl S, Weinhäusel A. Systemic autoinflammatory diseases. J Autoimmun. 2020;109:102421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Wang Z, Zhang S, Xiao Y, et al. NLRP3 inflammasome and inflammatory diseases. Oxid Med Cell Longev. 2020;2020:4063562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Rodrigues TS, de Sá KSG, Ishimoto AY, et al. Inflammasomes are activated in response to SARS‐CoV‐2 infection and are associated with COVID‐19 severity in patients. J Exp Med. 2021;218(3):e20201707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Lara PC, Macías‐Verde D, Burgos‐Burgos J. Age‐induced NLRP3 inflammasome over‐activation increases lethality of SARS‐CoV‐2 pneumonia in elderly patients. Aging Dis. 2020;11(4):756‐762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Channappanavar R, Perlman S. Pathogenic human coronavirus infections: causes and consequences of cytokine storm and immunopathology. Semin Immunopathol. 2017;39(5):529‐539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Jacob CO. On the genetics and immunopathogenesis of COVID‐19. Clin Immunol. 2020;220:108591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Ye Q, Wang B, Mao J. The pathogenesis and treatment of the ʽCytokine Storm' in COVID‐19. J Infect. 2020;80(6):607‐613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Meftahi GH, Jangravi Z, Sahraei H, Bahari Z. The possible pathophysiology mechanism of cytokine storm in elderly adults with COVID‐19 infection: the contribution of "inflame‐aging". Inflamm Res. 2020;69(9):825‐839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Schulert GS, Cron RQ. The genetics of macrophage activation syndrome. Genes Immun. 2020;21(3):169‐181. [DOI] [PubMed] [Google Scholar]
- 133. Lee S, Channappanavar R, Kanneganti TD. Coronaviruses: innate immunity, inflammasome activation, inflammatory cell death, and cytokines. Trends Immunol. 2020;41(12):1083‐1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Ovali F. Coronavirus‐2019 disease (COVID‐19) in children. Medeni Med J. 2020;35(3):242‐252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Conti P, Caraffa A, Gallenga CE, et al. Coronavirus‐19 (SARS‐CoV‐2) induces acute severe lung inflammation via IL‐1 causing cytokine storm in COVID‐19: a promising inhibitory strategy. J Biol Regul Homeost Agents. 2020;34(6):1971‐1975. [DOI] [PubMed] [Google Scholar]
- 136. Branchett WJ, Lloyd CM. Regulatory cytokine function in the respiratory tract. Mucosal Immunol. 2019;12:589‐600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Fitzgerald KA, Kagan JC. Toll‐like receptors and the control of immunity. Cell. 2020;180(6):1044‐1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Wurfel MM, Gordon AC, Holden TD, et al. Toll‐like receptor 1 polymorphisms affect innate immune responses and outcomes in sepsis. Am J Respir Crit Care Med. 2008;178(7):710‐720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Patarčić I, Gelemanović A, Kirin M, et al. The role of host genetic factors in respiratory tract infectious diseases: systematic review, meta‐analyses and field synopsis. Sci Rep. 2015;5:16119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Li C, Jiao S, Wang G, et al. The immune adaptor ADAP regulates reciprocal TGF‐β1‐integrin crosstalk to protect from influenza virus infection. PLoS Pathog. 2015;11(4):e1004824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Sebastian‐Valverde M, Pasinetti GM. The NLRP3 inflammasome as a critical actor in the inflammaging process. Cells. 2020;9(6):1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Li M, Guo W, Dong Y, et al. Elevated exhaustion levels of NK and CD8+ T cells as indicators for progression and prognosis of COVID‐19 disease. Front Immunol. 2020;11:580237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Udomsinprasert W, Jittikoon J, Sangroongruangsri S, Chaikledkaew U. Circulating levels of interleukin‐6 and interleukin‐10, but not tumor necrosis factor‐alpha, as potential biomarkers of severity and mortality for COVID‐19: systematic review with meta‐analysis. J Clin Immunol. 2020;41(1):11‐22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Cao X. COVID‐19: immunopathology and its implications for therapy. Nat Rev Immunol. 2020;20(5):269‐270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Roberts A, Paddock C, Vogel L, Butler E, Zaki S, Subbarao K. Aged BALB/c mice as a model for increased severity of severe acute respiratory syndrome in elderly humans. J Virol. 2005;79(9):5833‐5838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Baas T, Roberts A, Teal TH, et al. Genomic analysis reveals age‐dependent innate immune responses to severe acute respiratory syndrome coronavirus. J Virol. 2008;82(19):9465‐9476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Frieman M, Yount B, Agnihothram S, et al. Molecular determinants of severe acute respiratory syndrome coronavirus pathogenesis and virulence in young and aged mouse models of human disease. J Virol. 2012;86(2):884‐897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Masselli E, Vaccarezza M, Carubbi C, et al. NK cells: A double edge sword against SARS‐CoV‐2. Adv Biol Regul. 2020;77:100737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Poccia F, Agrati C, Castilletti C, et al. Anti‐severe acute respiratory syndrome coronavirus immune responses: the role played by V gamma 9V delta 2 T cells. J Infect Dis. 2006;193(9):1244‐1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Chen G, Wu DI, Guo W, et al. Clinical and immunological features of severe and moderate coronavirus disease 2019. J Clin Invest. 2020;130(5):2620‐2629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Jouan Y, Guillon A, Gonzalez L, et al. Phenotypical and functional alteration of unconventional T cells in severe COVID‐19 patients. J Exp Med. 2020;217(12):e20200872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Carissimo G, Xu W, Kwok I, et al. Whole blood immunophenotyping uncovers immature neutrophil‐to‐VD2 T‐cell ratio as an early marker for severe COVID‐19. Nat Commun. 2020;11(1):5243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Maucourant C, Filipovic I, Ponzetta A, et al. Natural killer cell immunotypes related to COVID‐19 disease severity. Sci Immunol. 2020;5(50):eabd6832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. National Research Project for SARS, Beijing Group . The involvement of natural killer cells in the pathogenesis of the severe acute respiratory syndrome. Am J Clin Pathol. 2004;121(4):507‐511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Giamarellos‐Bourboulis EJ, Netea MG, Rovina N, et al. Complex immune dysregulation in COVID‐19 patients with severe respiratory failure. Cell Host Microbe. 2020;27(6):992‐1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. García LF. Immune response, inflammation and the clinical spectrum of COVID‐19. Front. Immunol. 2020;11:1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Hazeldine J, Lord JM. Immunesenescence: a predisposing risk factor for the development of COVID‐19? Front Immunol. 2020;11:573662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Haeryfar SMM. MAIT cells in COVID‐19: heroes, villains, or both? Crit Rev Immunol. 2020;40(2):173‐184. [DOI] [PubMed] [Google Scholar]
- 159. Parrot T, Gorin J‐B, Ponzetta A, et al. MAIT cell activation and dynamics associated with COVID‐19 disease severity. Sci Immunol. 2020;5(51):eabe1670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Carter MJ, Fish M, Jennings A, et al. Peripheral immunophenotypes in children with multisystem inflammatory syndrome associated with SARS‐CoV‐2 infection. Nat Med. 2020;26(11):1701‐1707. [DOI] [PubMed] [Google Scholar]
- 161. Gruber CN, Patel RS, Trachtman R, et al. Mapping systemic inflammation and antibody responses in multisystem inflammatory syndrome in children (MIS‐C). Cell. 2020;183(4):982‐995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Živković J, Lipej M, Banić I, et al. Respiratory and allergic disorders in children with severe and partial immunoglobulin A immunodeficiency. Scand J Immunol. 2019;90(6):e12828. [DOI] [PubMed] [Google Scholar]
- 163. Buckland MS, Galloway JB, Fhogartaigh CN, et al. Treatment of COVID‐19 with remdesivir in the absence of humoral immunity: a case report. Nat Commun. 2020;11:6385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Naito Y, Takagi T, Yamamoto T, Watanabe S. Association between selective IgA deficiency and COVID‐19. J Clin Biochem Nutr. 2020;67(2):122‐125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Kumar S, Nyodu R, Maurya VK, Saxena SK. Host immune response and immunobiology of human SARS‐CoV‐2 infection. Coronavirus Disease 2019 (COVID‐19). 2020;43‐53. [Google Scholar]
- 166. Tomita Y, Ikeda T, Sato R, Sakagami T. Association between HLA gene polymorphisms and mortality of COVID‐19: An in silico analysis. Immun Inflamm Dis. 2020;8(4):684‐694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Marino R, Deibis L, De Sanctis JB, Bianco NE, Toro F. Interaction of immune complexes isolated from hepatitis C virus‐infected individuals with human cell lines. Med Microbiol Immunol. 2005;194(1–2):73‐80. [DOI] [PubMed] [Google Scholar]
- 168. Devarajan A, Vaseghi M. Hydroxychloroquine can potentially interfere with immune function in COVID‐19 patients: Mechanisms and insights. Redox Biol. 2021;38:101810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Baruah V, Bose S. Immunoinformatics‐aided identification of T cell and B cell epitopes in the surface glycoprotein of 2019‐nCoV. J Med Virol. 2020;92(5):495‐500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Li G, Fan Y, Lai Y, et al. Coronavirus infections and immune responses. J Med Virol. 2020;92(4):424‐432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Sanchez‐Mazas A. HLA studies in the context of coronavirus outbreaks. Swiss Med Wkly. 2020;150:w20248. [DOI] [PubMed] [Google Scholar]
- 172. Tavasolian F, Rashidi M, Hatam GR, et al. HLA, Immune Response, and Susceptibility to COVID‐19. Front Immunol. 2021;11:601886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Iesa M, Osman M, Hassan MA, et al. SARS‐CoV‐2 and Plasmodium falciparum common immunodominant regions may explain low COVID‐19 incidence in the malaria‐endemic belt. New Microbes New Infect. 2020;38:100817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Leite MM, Gonzalez‐Galarza FF, Silva BCCD, Middleton D, Santos EJMD. Predictive immunogenetic markers in COVID‐19. Hum Immunol. 2021;S0198‐8859(21):00015‐X. 10.1016/j.humimm.2021.01.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Chen W‐T, Wang C‐W, Lu C‐W, et al. The function of HLA‐B*13:01 involved in the pathomechanism of dapsone‐induced severe cutaneous adverse reactions. J Invest Dermatol. 2018;138(7):1546‐1554. [DOI] [PubMed] [Google Scholar]
- 176. Bruchez A, Sha KY, Johnson J, et al. MHC class II transactivator CIITA induces cell resistance to Ebola virus and SARS‐like coronaviruses. Science. 2020;370(6513):241‐247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Lee SJ, Qin H, Benveniste EN. The IFN‐gamma‐induced transcriptional program of the CIITA gene is inhibited by statins. Eur J Immunol. 2008;38(8):2325‐2336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Vibholm LK, Nielsen SSF, Pahus MH, et al. SARS‐CoV‐2 persistence is associated with antigen‐specific CD8 T‐cell responses. EBioMedicine. 2021;64:103230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Kikkert M. Innate immune evasion by human respiratory RNA viruses. J Innate Immun. 2020;12(1):4‐20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Taefehshokr N, Taefehshokr S, Hemmat N, Heit B. Covid‐19: perspectives on innate immune evasion. Front Immunol. 2020;11:580641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Breton G, Mendoza P, Hägglöf T, et al. Persistent cellular immunity to SARS‐CoV‐2 infection. J Exp Med. 2021;218(4):e20202515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Sokal A, Chappert P, Barba‐Spaeth G, et al. Maturation and persistence of the anti‐SARS‐CoV‐2 memory B cell response. Cell. 2021;184(5):1201‐1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Chen J, Lau YF, Lamirande EW, et al. Cellular immune responses to severe acute respiratory syndrome coronavirus (SARS‐CoV) infection in senescent BALB/c mice: CD4+ T cells are important in control of SARS‐CoV infection. J Virol. 2010;84(3):1289‐1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Wang F, Nie J, Wang H, et al. Characteristics of peripheral lymphocyte subset alteration in COVID‐19 pneumonia. J Infect Dis. 2020;221(11):1762‐1769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185. Kiyotani K, Toyoshima Y, Nemoto K, Nakamura Y. Bioinformatic prediction of potential T cell epitopes for SARS‐Cov‐2. J Hum Genet. 2020;65(7):569‐575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Poh CM, Carissimo G, Wang B, et al. Two linear epitopes on the SARS‐CoV‐2 spike protein that elicit neutralising antibodies in COVID‐19 patients. Nat Commun. 2020;11(1):2806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187. Liu L, Wang P, Nair MS, et al. Potent neutralising antibodies against multiple epitopes on SARS‐CoV‐2 spike. Nature. 2020;584(7821):450‐456. [DOI] [PubMed] [Google Scholar]
- 188. Niu X, Li S, Li P, et al. Longitudinal analysis of T and B cell receptor repertoire transcripts reveal dynamic immune response in COVID‐19 patients. Front Immunol. 2020;11:582010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Covre LP, De Maeyer RPH, Gomes DCO, Akbar AN. The role of senescent T cells in immunopathology. Aging Cell. 2020;19(12):e13272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190. Chen Y, Zuiani A, Fischinger S, et al. Quick COVID‐19 healers sustain Anti‐SARS‐CoV‐2 antibody production. Cell. 2020;183(6):1496‐1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191. Mayora S, Zabaleta‐Lanz M, Martínez W, Toro F, De Sanctis JB, García A. Lymphocyte subpopulations in Venezuelan patients infected with SARS CoV‐2. Gac Méd Caracas. 2020;128(Supl 1):S74‐S78. [Google Scholar]
- 192. Ferretti AP, Kula T, Wang Y, et al. Unbiased screens show CD8+ T cells of COVID‐19 patients recognise shared epitopes in SARS‐CoV‐2 that largely reside outside the spike protein. Immunity. 2020;53(5):1095‐1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193. Robbiani DF, Gaebler C, Muecksch F, et al. Convergent antibody responses to SARS‐CoV‐2 in convalescent individuals. Nature. 2020;584(7821):437‐442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194. Lee CH, Pinho MP, Buckley PR, et al. Potential CD8+ T cell cross‐reactivity against SARS‐CoV‐2 conferred by other coronavirus strains. Front Immunol. 2020;11:579480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195. Schulien I, Kemming J, Oberhardt V, et al. Characterisation of pre‐existing and induced SARS‐CoV‐2‐specific CD8+ T cells. Nat Med. 2021;27(1):78‐85. [DOI] [PubMed] [Google Scholar]
- 196. Reche PA. Potential cross‐reactive immunity to SARS‐CoV‐2 from common human pathogens and vaccines. Front Immunol. 2020;11:586984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197. Marín‐Hernández D, Nixon DF, Hupert N. Anticipated reduction in COVID‐19 mortality due to population‐wide BCG vaccination: evidence from Germany. Hum Vaccin Immunother. 2021;Feb 5 1‐3. 10.1080/21645515.2021.1872344. Online ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198. Conti P, Younes A. Coronavirus COV‐19/SARS‐CoV‐2 affects women less than men: clinical response to viral infection. J Biol Regul Homeost Agents. 2020;34(2):339‐343. [DOI] [PubMed] [Google Scholar]
- 199. Rahimi G, Rahimi B, Panahi M, et al. An overview of Betacoronaviruses‐associated severe respiratory syndromes, focusing on sex‐type‐specific immune responses. Int Immunopharmacol. 2021;92:107365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200. Karaderi T, Bareke H, Kunter I., et al. Host genetics at the intersection of autoimmunity and COVID‐19: a potential key for heterogeneous COVID‐19 severity. Front Immunol. 2020;11:586111. [DOI] [PMC free article] [PubMed] [Google Scholar]