

Abstract
Considering the urgent need for novel therapeutics in ongoing COVID‐19 pandemic, drug repurposing approach might offer rapid solutions comparing to de novo drug design. In this study, we designed an integrative in silico drug repurposing approach for rapid selection of potential candidates against SARS‐CoV‐2 Main Protease (Mpro). To screen FDA‐approved drugs, we implemented structure‐based molecular modelling techniques, physiologically‐based pharmacokinetic (PBPK) modelling of drugs disposition and data mining analysis of drug‐gene‐COVID‐19 association. Through presented approach, we selected the most promising FDA approved drugs for further COVID‐19 drug development campaigns and analysed them in context of available experimental data. To the best of our knowledge, this is unique in silico study which integrates structure‐based molecular modeling of Mpro inhibitors with predictions of their tissue disposition, drug‐gene‐COVID‐19 associations and prediction of pleiotropic effects of selected candidates.
Keywords: COVID-19, drug repurposing, PBPK modeling, drug-gene-disease associations, pleiotropic effects
1. Introduction
Acute respiratory disease with rapidly progressive lung injury was detected in Wuhan, China and reported to the World Health Organization (WHO) in December 2019. In January 2020 a novel β‐coronavirus SARS‐CoV‐2 had been identified. In February 2020, WHO announced a name for the new coronavirus disease – COVID‐19. As of January 15th 2021 there is almost 94 million confirmed cases, more than 2.0 milion confirmed deaths in 196 countries, areas or territories, and this number is rapidly growing. [1] Recent research indicated that COVID‐19 disease affects not only lung tissues, but also other organs such as brain, heart and kidneys,[ 2 , 3 , 4 ] implying that an efficient anti‐COVID‐19 agent should possess a tendency toward these target organs. Several months after the genetic sequence of the novel coronavirus was published, the global race for development of novel vaccine has started. By the end of 2020, two vaccines become available for the prevention of coronavirus disease 2019 (COVID‐19).[ 5 , 6 ]
Since there is an urgent need for novel anti‐COVID‐19 therapy, useful and quick approach would be to identify compounds, which show antiviral activity against SARS‐CoV‐2, by repurposing the existing drugs. Drug repurposing approach implies identification of novel use for approved drugs which are different from their original medical indication.[ 7 , 8 ] Many features of drug repurposing approach (reduced failure rates in early preclinical studies, demonstrated safety profiles of the rediscovered drugs and reduced costs) could accelerate drug development projects during pandemics. Findings observed from drug repurposing studies could also lead to developing innovative hypotheses for de novo design of SARS‐CoV‐2 antiviral drugs.[ 7 , 8 ] Although SARS‐CoV‐2 vaccines have been brought to the market, chemotherapeutic approaches still represent attractive strategy to combat SARS‐CoV‐2. [8] Numerous small molecule drug discovery projects and clinical trials are in progress.[ 9 , 10 ] Clinical studies investigating efficacy and safety of the initially repurposed drugs (remdesivir, hydroxychloroquine, and lopinavir) reported conflicting results which justify further efforts in the field of drug repurposing.[ 11 , 12 , 13 , 14 ]
One of the most attractive protein targets in COVID‐19 repurposing is SARS‐CoV‐2’s main protease (Mpro). Mpro is the key enzyme in viral life cycle involved in the most of the cleavage events on precursor polyproteins (pp1a and pp1ab). This three‐domain (domains I to III) cysteine protease releases functional non‐structural proteins with pivotal role in viral replication and transcription. The substrate binding site of Mpro is located in cleft between domains I and II and consists of four subsites (S1’, S1, S2, and S4). [15] Although Mpro was identified as attractive target for antiviral drug design, recent analyses revealed binding site plasticity and potential of mutations to directly affect plasticity, as major bottlenecks in rational design of Mpro inhibitors. Therefore, structure‐based drug design campaigns aimed to identify novel Mpro inhibitors could greatly benefit from introducing information on binding site plasticity.[ 16 , 17 , 18 ]
Considering the emergency of the situation, many drug repurposing studies on Mpro have been reported so far, including the high throughput screening (HTS) campaign from The National Center for Advancing Translational Sciences (NCATS).[ 19 , 20 , 21 ] Interestingly, some authors reported structure‐based in silico screening protocols with in vitro profiling of Mpro inhibitors resulting in discovery of additional inhibitors previously unseen by HTS campaign.[ 22 , 23 , 24 , 25 ] This adds up to the value of additional in silico evaluation in order to facilitate discovery of potential candidates.
Despite the direct effects on viral proteins, another important aspect of possible repurposable candidates represents evaluation of the effects drug might have on disease mechanism. Regarding the COVID‐19 disease particular emphasis should be paid on amplified immune response and cytokine storm which could lead to severe complications. [26] In this manner, examination of drug‐gene‐disease associations could provide insights into the additional/pleiotropic effects of the candidate drugs and further aid selection of candidates for clinical trials.[ 27 , 28 ] Additionally, when considering potential anti‐COVID‐19 drug candidates, drug affinity to distribute within certain organs/tissues should be considered as well. Namely, COVID‐19 treatment would benefit from favorable drug distribution within target tissues such as the lungs, brain, heart and kidneys to enrich local drug concentration and combat the infection. However, data of drug distribution in various organs/tissues are rarely accessible, and they mostly originate from animal studies. In this context, physiologically‐based pharmacokinetic (PBPK) modeling, coupled with quantitative structure–property relationship (QSPR) predictions, can provide useful information on the expected drug absorption and disposition in humans.[ 29 , 30 ]
The most of the in silico Mpro repurposing studies reported so far, rely solely on structure‐based predictions of drugs binding to the viral protein [20] , neglecting evaluation of additional effects drug could have on mechanism of disease. Herein we present general integrative protocol of drug repurposing of Mpro inhibitors which integrates in silico screening of the FDA‐approved drugs library encompassing structure‐based drug discovery techniques, data mining of drug‐gene‐COVID‐19 associations and QSPR‐PBPK modeling. For the initial screening of the database, we used different structure‐based virtual screening approaches. This was followed by ensemble docking where structural plasticity of studied SARS‐CoV‐2 Mpro was taken into account. Candidates selected as potential SARS‐CoV‐2 Mpro inhibitors were subjected to in silico data mining analysis in order to find drug‐gene‐COVID‐19 associations, construct gene interaction network, single out the most important molecular pathways affected by the investigated drugs and analyze it in the context of potential pleiotropic effects. In order to assess the affinity of each drug to reach the target organs, selected drugs were modeled in terms of their absorption and disposition in humans using PBPK modeling based on QSPR estimated drugs biopharmaceutical properties. Obtained results were used to identify drug candidates with the likelihood of successful COVID‐19 treatment.
2. Materials and Methods
2.1. Database Preparation
The library of 2747 FDA‐approved drugs was downloaded from SelleckChem. [31] The database was pre‐processed by removing duplicates, stripping solvents and salts and selecting the most abundant tautomer species (presented at more than 40 %) at physiological pH=7.4. The molecular mechanics geometry optimization of ligands was performed using Merck molecular force field (MMFF94). [32] All pre‐processing steps were performed using ChemAxon (https://chemaxon.com/) software tools in batch mode. Due to the predefined cutoff value for tautomer selection (40 %) some of the compounds were saved with both of the tautomeric structures. Final number of the compounds used for modeling was 3094.
2.2. Protein Preparation
Several crystal structures of Mpro with co‐crystalized ligands available at the time of accession were obtained from Protein Data Bank (https://www.rcsb.org/). Accession codes of used structures were 5R7Y, 5RYZ, 5R80, 5R81, 5R82, 5R83, and 6LU7. Crystal structures were processed using PlayMolecule's protein prepare wizard. [33] After addition of missing side chains, removal of alternative positions and solvent molecules, and after protonation of residues in respect to pH=7.4, proteins were saved as pdb files for the following virtual screening campaigns.
2.3. Structure‐based Virtual Screening (SBVS)
In order to screen FDA‐approved database, different structure‐based virtual screening approaches were used. Namely, three approaches were applied in initial SBVS: GRID molecular interaction fields (MIFs) fingerprint approach implemented in FLAP v.2.2 software;[ 34 , 35 ] Genetic algorithm based molecular docking in GOLD v.5.8.1; [36] Glide's (Schrödinger Release, 2020–2, limited liability company (LLC)[ 9 , 37 ]) hierarchical docking strategy.
For SBVS in FLAP, 25 conformers were generated for each entry in FDA‐approved database, using RMSD threshold equal to 0.3. MIFs were calculated for each conformer with spatial resolution of 0.75 Å, using 4 GRID MIF probes: H (shape mapping), DRY (evaluating hydrophobic affinities), O and N1 (H‐bond donor and acceptor regions mapping, respectively). For the definition of the Mpro inhibitors binding site, PDB:6LU7 (Mpro in complex with inhibitor N3) was used. The binding pocket was defined from the associated ligand, with pocket point radius of 2 Å around the ligand, and GRID‐MIFs with spatial resolution of 0.75 Å. GRID MIFs of the binding site were calculated using four aforementioned GRID MIF. Obtained maps of GRID MIFs for both, ligands and receptor were converted in quadruplets of pharmacophoric points and a quadruplet matching was used to overlay ligands onto the receptor. Global Sum Score, which represents sums of individual probe scores representing degree of overlap between MIFs, was used as quantitative measure of matching.
For the SBVS in GOLD, the binding pocket was defined within 10.0 Å of the ligand's position in PDB:6LU7. All torsion angles in data set molecules were allowed to rotate freely. The number of genetic algorithm runs was set to 10, and the efficacy was set to 10 %. ChemPLP (Piecewise Linear Potential) scoring function [40] employed in Gold Software was used for ranking of examined compounds.
The third SBVS was performed using Glide from Schrödinger suite. The coordinates of associated ligand from crystal structure (PDB: 6LU7) were taken as a center of the binding pocket. The outer grid box was set to be 20 Å, and inner grid box was set to be 10 Å in all directions. Docking was performed with “Ligand Docking” protocol with following parameters: standard precision (SP), Ligand vdW scale factor: 0.80, cutoff for a good RMSD 2.0 Å. Afterwards, docked compounds were ranked, based on SP scoring function.
2.4. QSPR‐PBPK Modeling
Simulations of drugs absorption and disposition were conducted using GastroPlusTM software (v. 9.7.0009, Simulations Plus Inc., USA). The software operates on the basis of the Advanced Compartment Absorption and Transit (ACAT) model of the human GI tract, coupled with the whole body PBPK model.[ 41 , 42 ] The ACAT model comprises nine consecutive gastrointestinal (GI) compartments i. e., stomach, duodenum, two segments of jejunum, three segments of ileum, caecum and ascending colon, whereat drug transit, dissolution, and absorption are simulated by a series of differential equations. The whole body PBPK model include additional organs/tissues i. e., lung, spleen, liver, adipose, muscle, heart, brain, kidney, skin, reproductive organs, red bone marrow, yellow bone marrow, and rest‐of‐body, and the necessary individual tissue properties (e. g., mass, perfusion rate, fraction unbound in tissue (Fut), drug‐specific tissue‐to‐plasma partition coefficients (Kp)) for the selected human representative are generated by the software. Once dissolved and absorbed, the drug reaches the portal vein and the liver (where it may undergo first pass and/or systemic extraction), and eventually enters into systemic circulation and distributes throughout the body, followed by potential metabolism in individual tissues and concomitant elimination. Each of these processes is described by adequate differential equation, and numerical integration of all the equations enables simulation of the complex interactions among the various processes a drug undergoes in the body.
The selected drug candidates, based on SBVS and ensemble docking results, were used for PBPK modeling, and each model was drug‐specific (based on drug‐specific input data). The preferable administration route (peroral or intravenous) and drug dose were determined based on literature data and used in the simulations (Supp. Table S1). Depending on the administration route, PBPK models considered that the drug was either absorbed from the GI tract and distributed throughout the body, or directly injected into the systemic circulation, followed by tissue distribution and elimination. All drug‐related input model parameters were in silico predicted, based on molecular structure, using the ADMET PredictorTM software (v. 8.5.0, Simulations Plus Inc., USA; https://www.simulations‐plus.com/) [43] (Supp. Table S2). In addition, default software values for particle radius (25 μm), particle density (1.2 g/ml), mean precipitation time (900 s), and volume of liquid taken with orally administered drug (240 ml) were used as inputs. The simulations were performed for single drug dose administered to 70 kg human adult representative in the fasted state. For orally taken drugs, solubility and diffusion coefficient were adjusted to bile salt effect using the software estimate for theoretical solubilization ratio, [44] drug dissolution was modeled using the default Johnson dissolution equation, and software default Opt logD Model SA/V 6.1 was used to scale the differences in drug absorption along the gut. Human intrinsic clearance values were calculated from in silico estimates for the overall intrinsic drug clearance in human liver microsomes (HLM) (Supp. Table S2), and they referred to the liver tissue. In addition, a fraction of drug distributed into kidneys was assumed to undergo passive renal filtration expressed as the product of fraction unbound in plasma (fup) and glomerular filtration rate (GFR). The drugs were assumed to passively perfuse through body tissues, and Kp values were calculated using Lukacova method for perfusion‐limited tissues [45] in conjunction with “S+ v9.5” default software method to calculate Fut values. The simulations were performed for 24 h, and the generated data were used to estimate drug exposure in plasma and target organs.
2.5. In Silico Analysis of Drug‐gene‐disease Associations and Formation of Gene Interaction Network
The Comparative Toxicogenomics Database [46] (CTD; http://CTD.mdibl.org) and Cytoscape [47] software package version 3.8.0 (https://cytoscape.org) were used as the main tools for data mining analysis of drug‐gene‐COVID‐19 association in our research. Genes connected with COVID‐19 disease and associated with the candidate drugs were obtained from the CTD, while the key hub genes were singled out by cytoHubba Cytoscape plug‐in [48] (http://apps.cytoscape.org/apps/cytohubba). Maximal Cilque Centrality (MCC) method, found to be the most accurate of all the methods available in the cytoHubba, was used considering its ability to generate precise predictions of essential proteins. [46] GeneMANIA Cytoscape plug‐in [49] (http://genemania.org/plug‐in/) was used to construct a tight network of genes associated with the drugs selected in this study and to obtain the lists of related genes. ClueGO Cytoscape plug‐in [50] (version 2.5.6), together with CluePedia [51] (version 1.5.6) were used to visualize the molecular pathways associated with the candidate drugs and genes they interact with. For this analysis, two‐sided hypergeometric test+Bonferroni step down correction were used. The list of pathways was extracted from the KEGG, [52] Reactome, [53] and WikiPathways [54] databases in the ClueGO app. The results obtained from the ClueGO were further confirmed by the CTD SetAnalyzer tool (http://ctdbase.org/tools/analyzer.go), which was also used to rank molecular pathways connected with the investigated gene set by the statistical significance (p<0.01).
2.6. Identification of the Most Promising Candidates as Mpro Inhibitors
In order to implement data on different conformational states of Mpro, top scored 10 % in each of SBVS runs was further analyzed using ensemble docking strategy. Among top scored compounds there were 692 unique compounds, while 26 compounds were identified simultaneously using GOLD and Glide SBVS, 17 compounds using Glide and FLAP SBVS, and 7 compounds using FLAP and GOLD SBVS. Only three compounds were identified using all three SBVS approaches in high‐throughput mode. Total number of unique compounds was 692 Ensemble docking was performed using GOLD software v.5.8.1. All Mpro crystal structures were aligned, using PDB:6LU7 as reference structure. The binding pocket was defined within 10 Å of the inhibitor from 6LU7 crystal structure – N3. The number of genetic algorithm runs was set to 30, and the search efficacy was set to 200 %. ChemPLP scoring function was used, for ranking and pose prediction of examined compounds. The final selection of candidates was performed considering obtained score and pose consistency. Additionally, obtained poses with primary interactions outside well established Mpro subsites (S1, S1’, S2 and S4) were discarded. Total of 43 drugs (list of SMILES available in Supporting Information) were selected for further evaluation using pharmacokinetic modelling and network‐based drug‐gene‐disease associations analysis. Additionally, selected candidates were analyzed using Maximum Common Substructure (MCS). MCS clustering was performed usind ChemAxon's heuristic algorithm, implemented in JChem suite. [55]
3. Results
3.1. Integrative Protocol Overview
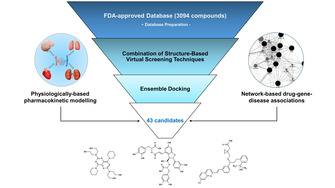
In order to select potential Mpro inhibitors from FDA‐approved database, integrated in silico screening protocol encompassing combination of several techniques was designed. Schematic representation of the protocol is presented on the Figure 1. Firstly, structure‐based molecular modeling techniques were used to select candidates with potential affinity towards Mpro binding site. After ligands’ database preparation, structure‐based virtual screening (SBVS) included screening of FDA‐approved drugs using combination of FLAP's (Fingerprints for Ligands and Proteins) SBVS approach and molecular docking approaches (implemented in Glide and GOLD programs) independently (see Materials and Methods). After selection of top 10 % of ranked compounds in each approach, virtual hits were analyzed using ensemble docking protocol, and final selection of 43 repurposable drugs was made (list of SMILES available in Supporting Information). Network‐based drug‐gene‐disease associations were used to investigate potential for modulating expression of genes associated with COVID‐19 and to gain insight into potential pleiotropic effects of selected candidates. Additionally, selected candidates were further analyzed using in silico QSPR‐PBPK modeling to investigate potential for target organs disposition.
Figure 1.
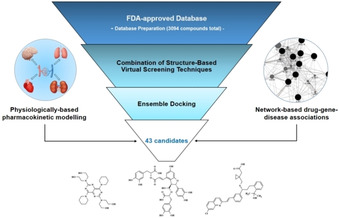
Schematic representation of applied repurposing protocol.
3.2. Structure‐based Screening of FDA‐approved Drugs Against SARS‐CoV‐2 Mpro
According to benchmarking studies, performance of SBVS protocols is highly affected by many factors, including chosen protein conformations, scoring functions and applied SBVS tools. Since there is no universal SBVS tool, general recommendation is to always use several programs and combine their results.[ 56 , 57 , 58 ] Considering aforementioned, we designed our SBVS protocol using combination of SBVS approach from FLAP and two molecular docking tools – GOLD and Glide (see Materials and Methods).
Proteins are dynamical entities and incorporation of proteins’ structural plasticity into SBVS protocols was proven to greatly improve the virtual screening results in the most cases.[ 59 , 60 , 61 ] Structural plasticity of Mpro substrate binding site was recently reported owing to the efforts in the field of structural biology[ 15 , 18 , 62 , 63 , 64 ] and it was implemented in the last step of our screening protocol by introducing ensemble‐docking for analysis of top 10 % of ligands selected by each of used SBVS tools.
Substrate binding site of SARS‐CoV‐2 Mpro is located in cleft between domains I (residues 8–101) and II (residues 102–184) and consists of four subsites: S1’, S1, S2 and S4 (Figure 2A). [15] Our analysis performed on aligned ensemble of available X‐ray structures used in this study (PDB IDs: 5R7Y, 5RYZ, 5R80, 5R81, 5R82, 5R83 and 6LU7) revealed that shape and surface area of the binding site are mainly influenced by following residues: HIS‐41, SER‐46, MET‐49, ASN‐142, MET‐165 and GLN‐189 (Figure 2B). The most of these residues directly shape structure of subsites S2 and S4, which make these subsites largely affected across used ensemble of structures. While the shape of subsite S1 is not affected with prominent changes in structure of surface area, different orientations of sidechain of ASN‐142 bear potential for affecting positioning of hydrogen bond donors or acceptors in putative ligands. Subsite S1’ is the least affected subsite.
Figure 2.
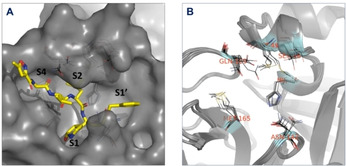
(A) Surface representation (gray) of the substrate binding site of aligned Mpro used in ensemble docking, with designated subsites S1, S1’, S2 and S4. Ligand (gray sticks) and surface are obtained from PDB:6LU7. (B) Aligned Mpro structures used in ensemble docking. The residues involved in detected binding site flexibility are depicted in black lines and labeled with red letters.
Final selection of the 43 ligands through ensemble docking was based on the consensus between ChemPLP [40] score and predicted pose consistency. Conformations of the studied ligands that established interactions with Mpro primarily outside of subsites S1, S1’, S2 and S4 were discarded from discussion. All of the selected candidates for repurposing are presented in the Table 2 and Supp. Figure S1. Accounting for the plasticity of substrate binding site in the virtual screening protocol, we were able to recover several ligands with potential SARS‐CoV‐2 antiviral activity identified previously through cell‐based screening (dipyridamole, lopinavir, velpatasvir, daclatasvir, lapatinib, bazedoxifene, atazanavir) (Table 2).[ 21 , 24 , 65 , 66 , 67 , 68 , 69 ] For some of the selected drugs clinical data for COVID‐19 supports antiviral activity (salvianolic acid B, thymopentin, atorvastatin, montelukast, dipyridamole, lopinavir, ritonavir, telmisartan, daclatasavir, atazanavir, nintedanib, darunavir and ramipril).[ 10 , 70 , 71 , 72 ] Few identified ligands were already confirmed through bioassays on inhibition of Mpro activity (venetoclax, montelukast, dipyridamole, lopinavir, lomitapide, lapatinib, telaprevir, atazanavir, pimozide, darunavir, eltrombopag) (Table 2).[ 21 , 23 , 24 , 25 , 73 ] Under assumption that all of abovementioned drugs achieve their antiviral activity through inhibition of Mpro, our SBVS protocol successfully recovered many of promising repurposable drugs with theoretical hit‐rate of 21/43=48.8 %. This finding validates our SBVS protocol and justifies further experimental validation of selected candidates.
Table 2.
Selected candidates with potential activity on SARS‐CoV‐2 Main Protease, their docking scores, and PBPK predicted disposition data. The most promising candidates for further experimental evaluation are marked in bold letters.
|
Drug |
ChemPLP score |
AUC0→24 h ratio plasma/lungs |
AUC0→24 h ratio plasma/brain |
AUC0→24 h ratio plasma/heart |
AUC0→24 h ratio plasma/kidneys |
Reference (Clinical trial, experimental data) |
|---|---|---|---|---|---|---|
|
Salvianolic acid B† |
121.04 |
4.46 |
16.45 |
5.83 |
7.05 |
[83] |
|
Pralmorelin |
109.38 |
0.19 |
1.34 |
0.32 |
0.15 |
/ |
|
Venetoclax§ |
105.24 |
1.08 |
0.09 |
0.21 |
0.24 |
[21] |
|
Thymopentin† |
104.28 |
0.91 |
0.83 |
0.95 |
1.05 |
[70] |
|
Ombitasvir |
103.24 |
1.06 |
0.09 |
0.21 |
0.24 |
/ |
|
Atorvastatin† |
99.00 |
4.23 |
5.09 |
4.39 |
5.17 |
[10] |
|
Naringin |
98.68 |
3.20 |
5.83 |
3.74 |
4.35 |
/ |
|
Astilbin |
97.36 |
3.43 |
2.24 |
2.83 |
3.25 |
/ |
|
Montelukast§† |
96.67 |
1.13 |
0.10 |
0.23 |
0.26 |
[10, 25] |
|
Dipyridamole§ * † |
95.85 |
1.84 |
0.24 |
0.53 |
0.59 |
[10, 25, 84] |
|
Lopinavir§ * † |
95.68 |
1.63 |
0.17 |
0.39 |
0.44 |
[10, 21, 23, 85] |
|
Fulvestrant |
95.30 |
1.05 |
0.09 |
0.21 |
0.24 |
/ |
|
Revefenacin |
95.11 |
0.35 |
0.24 |
0.32 |
0.22 |
/ |
|
Carfilzomib |
93.55 |
1.71 |
0.19 |
0.43 |
0.49 |
/ |
|
Elbasvir |
93.40 |
1.05 |
0.09 |
0.21 |
0.24 |
/ |
|
Ritonavir† |
92.30 |
1.26 |
0.12 |
0.27 |
0.30 |
[10, 85] |
|
Dabigatran |
92.00 |
1.06 |
0.09 |
0.22 |
0.24 |
/ |
|
Bimatoprost |
89.74 |
2.47 |
0.35 |
0.75 |
0.85 |
/ |
|
Sulfinpyrazone |
89.58 |
4.25 |
6.42 |
4.72 |
5.56 |
/ |
|
Ticagrelor |
89.41 |
1.65 |
0.18 |
0.40 |
0.45 |
/ |
|
Mitoxantrone |
89.14 |
0.10 |
0.54 |
0.16 |
0.08 |
/ |
|
Cilnidipine |
88.95 |
1.06 |
0.09 |
0.22 |
0.24 |
/ |
|
Vilanterol |
87.81 |
0.36 |
0.43 |
0.42 |
0.25 |
/ |
|
Selexipag |
87.68 |
3.21 |
0.74 |
1.42 |
1.62 |
/ |
|
Azilsartan |
87.46 |
3.66 |
1.28 |
2.15 |
2.46 |
/ |
|
Telmisartan† |
87.08 |
2.65 |
0.42 |
0.89 |
1.00 |
[71] |
|
Lifitegrast |
86.09 |
4.44 |
15.36 |
5.81 |
6.94 |
/ |
|
Lomitapide§ |
85.58 |
1.09 |
0.09 |
0.21 |
0.24 |
[21] |
|
Neohesperidin |
85.23 |
2.52 |
3.52 |
2.79 |
3.19 |
/ |
|
Velpatasvir* |
85.03 |
1.06 |
0.09 |
0.22 |
0.24 |
[66] |
|
Daclatasvir*† |
84.72 |
1.26 |
0.12 |
0.27 |
0.30 |
[10, 67] |
|
Lapatinib§ * |
83.89 |
1.10 |
0.10 |
0.22 |
0.25 |
[24] |
|
Bazedoxifene* |
83.55 |
0.84 |
0.09 |
0.21 |
0.22 |
[68] |
|
Telaprevir§ |
81.93 |
2.41 |
0.42 |
0.88 |
1.00 |
[73] |
|
Penfluridol |
81.87 |
0.90 |
0.09 |
0.21 |
0.23 |
/ |
|
Bortezomib |
81.47 |
3.72 |
7.22 |
4.39 |
5.16 |
/ |
|
Repaglinid |
81.40 |
2.38 |
0.36 |
0.77 |
0.85 |
/ |
|
Atazanavir§ * † |
79.43 |
1.58 |
0.17 |
0.38 |
0.43 |
[10, 25, 69] |
|
Nintedanib† |
78.54 |
0.44 |
0.21 |
0.33 |
0.25 |
[10] |
|
Pimozide§ |
77.33 |
1.00 |
0.09 |
0.22 |
0.24 |
[23] |
|
Darunavir§† |
76.71 |
3.31 |
1.57 |
2.32 |
2.66 |
[10, 21] |
|
Eltrombopag§ |
75.04 |
4.39 |
8.62 |
5.19 |
6.17 |
[21] |
|
Ramipril† |
72.04 |
3.38 |
10.22 |
4.46 |
4.25 |
[72] |
§ Reported inhibitory activity in Mpro enzymatic assays; * Reported inhibitory activity in cell‐based assays; † Antiviral activity supported by clinical studies; / There is no experimental or clinical data.
This article is being made freely available through PubMed Central as part of the COVID-19 public health emergency response. It can be used for unrestricted research re-use and analysis in any form or by any means with acknowledgement of the original source, for the duration of the public health emergency.
Analysis of predicted binding modes of selected candidates (Figure 3) revealed three the most common interactions: 1) Van der Waals interaction with MET‐49 in S2 subsite; 2) Van der Waals interaction with MET‐165 at the linkage between S4 and S2 subsites; 3) Hydrogen bonding with GLU‐166 at the linkage between S1 and S4 subsites. According to our results, these three interactions in subsites S1, S2 and S4 were predicted to be of great importance for Mpro inhibitory activity.
Figure 3.
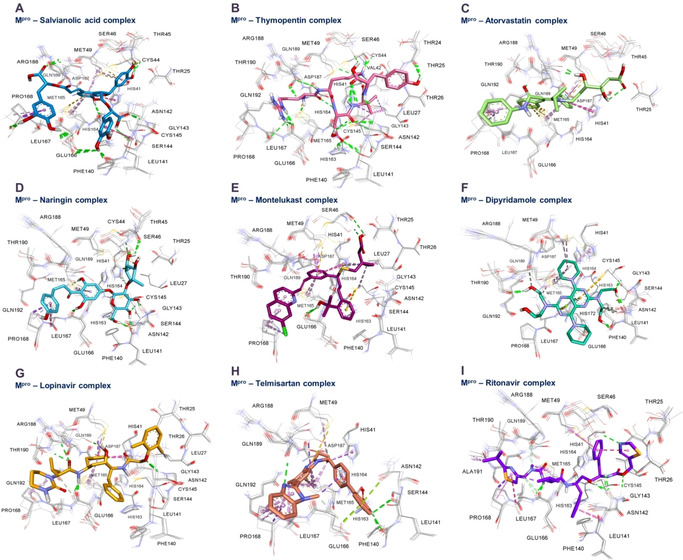
Presentation of predicted intermolecular interactions between SARS‐CoV‐2 main protease (Mpro) and the promising candidates. (A) salvianolic acid (depicted in blue), (B) thymopentin (depicted in magenta), (C) atorvastatin (depicted in light green), (D) naringin (depicted in light blue), (E) montelukast (depicted in dark violet), (F) dipyridamole (depicted in spring green), (G) lopinavir (depicted in bronze), (H) ritonavir (depicted in purple) and (I) telmisartan (depicted in dark brown). Subsites are oriented in the same order as shown in Figure 2 – Residues MET165 and ASN142 border S1 subsite, MET165 and GLN189 border S4 subsite, S2 subsite is located in close proximity of MET49, while S1’ subsite in this representation encompass resides right from ASN142.
In order to further validate our results and provide guidance on rational design of novel candidates, final list of selected candidates was analyzed using maximum common substructure (MCS) hierarchical clustering where fragments shared by the most of the selected hits were identified. Interestingly, the largest clusters – ID1 (counting 12 candidates) and cluster ID2 (counting 3 candidates), which share similar common substructure described the most of candidates with experimental proof of antiviral activity against SARS‐CoV‐2 (Tables 1 and 2). We have further analyzed occurrence of interactions of selected substructures and identified that fragment representatives of clusters ID1 and ID2 interacts mostly with subsites S1 and S4 (Table 1). This finding is in agreement with the latest results from ongoing public fragment‐based drug design (FBDD) campaign at XChem facility of UK's Diamond Light Source.[ 63 , 74 ] Namely, low micromolar ligands (X2646 and X2581) identified through FBDD campaign share similar substructure (arylmethyl carbonylamino group) to representative drugs from ID1 and ID2 cluster. Chemical probes from FBDD campaign as well as virtually clustered compounds interact with Mpro in similar manner (Figure 4A and 4B), primarily affecting S1 and S4 subsites. Additionally, some of the drugs grouped in different clusters, also share abovementioned structural features (e. g. repaglinide from cluster ID4; pimozide from ID3; all representatives of cluster ID5 share similar substructure locked inside flavan moiety). According to our results, moieties capable of interacting through van der Waals interactions with MET‐49 in S2 subsite (S2‐interacting fragment) should have central role in rational drug design. Innovative drug design on COVID‐19 Mpro inhibitors should be guided by introducing diverse arylalkyl moieties onto the S2‐interacting fragment that extend into S1 and S4 subsites (Figure 4C).
Table 1.
Result of Maximum Common Substructure Clustering of 43 virtual hits selected after ensemble docking. Virtual hits, which were not grouped in any of clusters (15 compounds), are omitted from the representation.
|
Substructure |
Number of compounds |
Cluster IDs |
Drugs |
|---|---|---|---|
|
|
|
1 |
Lopinavir, Atazanavir, Ramipril, Montelukast, Lifitegrast, Salvianolic acid B, Pralmorelin, Thymopentin, Naringin, Carfilzomib, Bimatoprost, Penfluridol |
|
|
|
2 |
Ritonavir, Darunavir, Vilanterol |
|
|
|
3 |
Dabigatran, Azilsartan, Telmisartan, Pimozide |
|
|
|
4 |
Repaglinide, Nintendanib, Eltrombopag |
|
|
|
5 |
Astilbin, Neohesperidin |
|
|
|
6 |
Atorvastatin, Bazedoxifene |
|
|
|
7 |
Telaprevir, Bortezomib |






This article is being made freely available through PubMed Central as part of the COVID-19 public health emergency response. It can be used for unrestricted research re-use and analysis in any form or by any means with acknowledgement of the original source, for the duration of the public health emergency.
Figure 4.
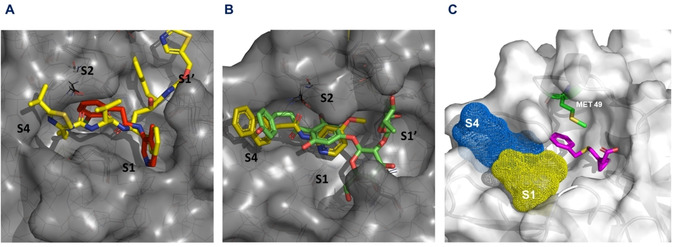
(A) Presentation of low micromolar ligand X2646 (labeled in red) and ritonavir (labeled in yellow, representative of cluster ID2), (B) Presentation of low micromolar ligand X2581 (labeled in yellow) and naringin (labeled in green, representative of cluster ID1), (C) Presentation of S2‐interacting fragment (labeled in magenta) interacting with Met49 from S2 subsite; phenyl group of presented fragment (derived from montelukast) should be substituted with diverse arylalkyl moieties that extend into S1 and S4 subsites.
Taken together, molecular modelling approach presented here could be used not only as a tool for efficient in silico screening of large databases in search for novel Mpro inhibitors, but also as a platform for de novo drug design of COVID‐19 protease inhibitors.
3.3. PBPK Prediction Results
Based on the results of SBVS and ensemble docking, 43 drugs that showed Mpro inhibition potential, were selected for QSPR‐PBPK modeling in order to assess their affinity to distribute within target organs, predominantly lungs, but also brain, heart and the kidneys. The predicted results referring to the ratio of drug exposure in plasma and target organs (expressed as AUC ratio) are presented in Table 2 and Supp. Figure S1, whereas AUC ratio smaller than 1 indicates predominant drug distribution in target organ, AUC close to 1 indicates similar drug distribution in plasma and target organ, and AUC much higher than 1 negligible drug distribution in the organ of interest.
According to the simulation results, pralmorelin, revefenacin, mitoxantrone, vilanterol, nintedanib, are expected to distribute widely into human lungs, with mitoxantrone having the highest potential to reach high drug concentrations in lung tissues. More than a half of selected compounds (60 %) show a tendency to distribute and/or accumulate more widely into other organs/tissues. However, a dozen of selected candidates (e. g. Salvianolic acid B), remain primarily distributed in the circulatory system available to interact with the infected immune cells. [75]
Some of the studied drugs show limited oral absorption[ 76 , 77 , 78 , 79 , 80 , 81 ] due to poor biopharmaceutical or pharmacokinetic properties. This means that alternative dosing routes or higher doses may be considered for future studies, in order to provide appropriate drug concentrations in target tissues. Predicted data regarding the distribution of selected drugs within certain organs/tissues are in agreement with available literature data e. g., published results indicate that dipyridamole is widely distributed into kidneys, heart and lungs [82] , while telmisartan and ritonavir show high partitioning into kidneys and lungs.[ 78 , 80 ] These data support the value of in silico simulation results, and indicate that the predicted data may be useful in decision‐making for suitable candidate compounds.
3.4. Data Mining Analysis of Drug‐gene‐COVID‐19 Association
In order to examine drug‐gene‐COVID‐19 associations of 43 drugs selected after SBVS and ensemble docking, data mining of CTD database was performed. Genes connected with the candidate drugs were extracted from the CTD database. Drugs that contained an insufficient number of gene‐chemical annotations were discarded from the analysis, along with the drugs that were not listed in the CTD. As a result, our data mining analysis has shown that 13 drugs interact with the genes involved in COVID‐19 disease (Table 3).
Table 3.
Candidate drugs for COVID‐19 treatment and genes they interact with (http://ctdbase.org).
|
|
Genes connected with potential COVID‐19 treatment |
||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
Drug |
ACE2 |
AGT |
CCL2 |
CCL3 |
CRP |
CSF3 |
IL1B |
IL2 |
IL2RA |
IL6 |
IL7 |
IL10 |
CXCL8 |
CXCL10 |
TNF |
|
Salvianolic acid B |
|
|
|
|
+ |
|
+ |
|
|
+ |
|
|
|
|
+ |
|
Atorvastatin |
+ |
+ |
+ |
|
+ |
|
+ |
|
+ |
+ |
|
|
+ |
+ |
+ |
|
Naringin |
|
+ |
+ |
+ |
|
|
+ |
+ |
|
+ |
|
+ |
+ |
|
+ |
|
Montelukast |
|
+ |
+ |
|
|
|
+ |
|
|
+ |
|
|
+ |
|
+ |
|
Dipyridamole |
|
|
+ |
|
|
|
+ |
+ |
|
+ |
|
|
|
|
+ |
|
Lopinavir |
|
|
+ |
+ |
|
|
+ |
|
|
+ |
|
|
|
|
+ |
|
Fulvestrant |
|
+ |
+ |
|
|
+ |
+ |
|
|
+ |
|
+ |
+ |
+ |
+ |
|
Ritonavir |
|
|
+ |
+ |
|
|
|
|
|
+ |
+ |
|
+ |
+ |
+ |
|
Telmisartan |
+ |
+ |
+ |
|
+ |
|
+ |
|
|
+ |
|
+ |
+ |
|
+ |
|
Bazedoxifene |
|
|
|
|
|
|
|
|
|
+ |
|
|
|
+ |
|
|
Bortezomib |
|
|
+ |
+ |
|
|
|
+ |
|
+ |
|
+ |
+ |
|
+ |
|
Atazanavir |
|
|
+ |
+ |
|
|
|
|
|
|
|
|
|
|
|
|
Ramipril |
|
+ |
+ |
||||||||||||
This article is being made freely available through PubMed Central as part of the COVID-19 public health emergency response. It can be used for unrestricted research re-use and analysis in any form or by any means with acknowledgement of the original source, for the duration of the public health emergency.
In total, there were 15 genes affected by the drugs selected in this study: ACE2 (encodes the angiotensin‐converting enzyme‐2), AGT (encodes the angiotensinogen), CCL2 (encodes the C−C Motif Chemokine Ligand 2), CCL3 (encodes the C−C Motif Chemokine Ligand 3), CRP (encodes C‐Reactive Protein), CSF3 (encodes Colony Stimulating Factor 3), CXCL10 (encodes the C−X−C Motif Chemokine Ligand 10), IL10 (encodes Interleukin 10), IL1B (encodes Interleukin 1 Beta (IL‐1β)), IL2 (encodes Interleukin 2 (IL‐2)), IL2RA (encodes Interleukin 2 Receptor Alpha Chain), IL6 (encodes Interleukin 6 (IL‐6)), IL7 (encodes Interleukin 7 (IL‐7)), CXCL8 (encodes C−X−C Motif Chemokine Ligand 8), TNF (encodes Tumor Necrosis Factor).
CTD was further manually explored to obtain the exact interactions (mRNA expression, protein expression, gene expression and protein secretion) between the investigated drugs and genes. Binary interactions were identified (one chemical and one gene), while complex interactions (describing more than two molecules and/or genes) were removed, as well as the “no effect” interactions. The exact interaction type could be obtained for 11 out of 13 drugs present in the CTD and 13 genes (Supp. Table S3).
Our gene set (15 genes) was further analyzed by Cytoscape version 3.8.0. Cytohubba plug‐in and MCC method to identify and rank the top 5 genes with the highest inter‐connections, the so‐called hub genes (Figure 5A). Colour of the nodes represents the intensity of the correlation, meaning that yellow represents the lowest and red the highest correlation. It should be noted that IL6 was found to be the gene with the highest correlation among the hub genes and was also among the most common interacting genes for the candidate drugs, since all the investigated drugs affected this gene except ramipril and atazanavir. CCL2 was also both among the hub and the most interacting genes, found to be affected by all the candidate drugs except bazedoxifene and salvianolic acid B. Naringin was the only drug that interacted with all the hub genes, while lopinavir interacted with all the hub genes apart from CXCL8.
Figure 5.
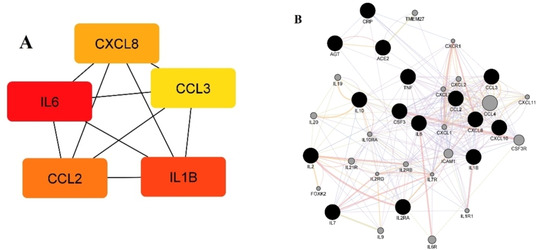
(A) Five hub genes obtained for genes connected with drugs that might be used in COVID‐19 treatment. Colour of the nodes represents the intensity of correlation with yellow being the lowest and red the highest (Cytohubba Cytoscape plug‐in+MCC method), (B) Tight network of genes connected with candidate drugs for COVID‐19 treatment, together with 20 related genes (GeneMANIA Cytoscape plug‐in). Colour legend: co‐expression – purple; co‐localization – blue; physical interaction‐red; pathway – light blue; predicted – orange.
Our further analysis was aimed at constructing a tight network between the 15 genes affected by the investigated drugs, and predict additional 20 related genes that may be important for COVID‐19 therapy (GeneMANIA Cytoscape plug‐in) (Figure 5B). The results revealed that the majority of the genes (70.84 %) were in co‐expression – purple lines, which means that their expression levels are similar under the defined conditions in the gene expression study. 10.07 % of these genes were in co‐localisation (blue lines), which means that they are expressed in the same tissue or their gene products are identified in the same cellular location, while 8.93 % where in physical interactions (red lines), which means that there is an interaction between the protein products of these genes. [49]
In order to further explore the biological importance of our gene set, Pathway enrichment analysis was performed using Cytoscape plug‐in ClueGO+CluePedia version 2.5.6 and 1.5.6. KEGG, Reactome, and WikiPathways databases were selected for the pathway analysis. The results have shown that our set of genes (15 genes associated with the candidate drugs for COVID‐19 treatment+20 related genes obtained by GeneMANIA Cytoscape plug‐in) clustered around 7 (Figure 6A) and hub genes around 4 main pathways (Figure 6B).
Figure 6.
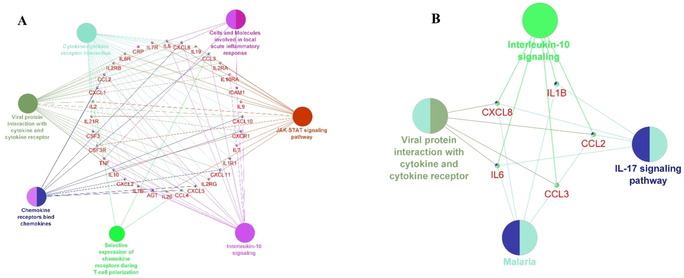
Molecular pathways connected with the candidate drugs. Genes are grouped in the middle and connected to the pathways they regulate (pathways are represented as nodes) (Cytoscape plug‐in ClueGO+CluePedia version 2.5.6 and 1.5.6. The two‐sided hypergeometric test+a Bonferroni step down correction and a κ score=0.3) (A) Molecular pathways connected with the set of 34 genes (15 genes associated with the candidate drugs for COVID‐19 treatment+20 related genes obtained by GeneMANIA Cytoscape plug‐in). Pathways are grouped into 9 clusters, (B) Molecular pathways connected with the 5 hub genes obtained by Cytohubba Cytoscape plug‐in. Pathways are grouped into 3 clusters.
CTD SetAnalyser tool was used to confirm and rank the enriched pathways by the statistical significance (p<0.01). Cytokine‐cytokine receptor interaction, signaling by Interleukins and Cytokine Signaling in Immune system were the top tree molecular pathways connected with our gene set. Other molecular pathways listed among the top 10 in the CTD included Interleukin‐10 (IL‐10) signaling, Immune System, Jak‐STAT signaling pathway, Interleukin‐17 (IL‐17) signaling pathway, Hematopoietic cell lineage, T‐helper cell 17 (Th17) cell differentiation and TNF signaling pathway.
Cytokine‐connected pathways were found significant for the genes affected by the candidate drugs. Among them, IL‐10 signalling could be viewed as particularly important, especially for the hub genes. IL‐10 is an anti‐inflammatory cytokine, produced by T helper 2 lymphocytes (Th2), and found to be increased in severe COVID‐19 cases, along with IL‐6, encoded by the top hub gene in our study, IL6. [86] The Janus kinase (JAK)‐signal transducer and activator of transcription (STAT) pathway (JAK‐STAT signalling pathway) was also found among the top 10 pathways connected with our set of genes, which is significant because this pathway plays critical roles in orchestrating of immune system, especially cytokine receptors. [87]
As seen in the Supp. Table S3, some of the candidate drugs were found to inhibit the expression or secretion of several inflammatory‐connected molecules. For example, Naringin inhibited the expression of IL6 protein and IL1B activity. Salvianoic acid B inhibited protein expression of CRP, one of the main biomarkers of inflammation, while Atorvastatin inhibited not only protein expression of CRP, but also CXCL10, CXCL8 and IL6. Thus, our data mining of drug‐gene‐disease associations has shown that, apart from potentially acting as SARS‐CoV‐2 Mpro inhibitors, some of the selected drugs might have other potential pleiotropic effects. Taking into account that they were found to affect the cytokine signalling pathways and systemic inflammation, and inhibit inflammatory protein secretion and expression, they might also mitigate the severity of the clinical picture in pneumonia known to occur in the severe cases of COVID‐19. Furthermore, Montesarchio et al. (2020) suggested clinical benefit regarding early intervention with IL‐6‐modulatory therapies in COVID‐19 patients, and proposed CRP as a potential biomarker of response to treatment. [88] On the other hand, some of the drugs, like Lopinavir, induced CCL2 mRNA expression and protein secretion, CCL3 protein expression, IL1B mRNA expression, IL6 protein expression and secretion. Similarly, Atazanavir increased CCL2 and CCL3 protein expression, while Ritonavir increased IL6 mRNA expression, protein expression and secretion, CCL2 protein expression and secretion, as well as CCL3 protein expression. [89] Conclusively, several identified drugs (lopinavir, ritonavir, atazanavir) may increase secretion of pro‐inflammatory cytokines, which might aggravate cytokine storm. Another molecular pathway connected with the candidate drugs that was singled out and should be mentioned is IL‐17 signaling pathway. The alterations in Th17/IL‐17 axis are widely recognized as potential targets for therapeutic interventions in COVID‐19. [90]
3.5. Analysis of the Most Promising Candidates
Final analysis of the most promising candidates was performed according to consensus between predicted Mpro affinities, predicted disposition in tissues relevant for COVID‐19, analysis of drug‐gene‐disease associations and analysis of potential pleiotropic effects. Even though drugs singled out through SBVS and ensemble docking (Table 2 and Supp. Figure S1) could be considered as potential Mpro inhibitors, not all of them could be considered as equally involved in other aspects of disease mechanism. Considering the wider context of COVID‐19 disease, after analysis of drug‐gene‐disease associations, following candidates (Table 3) are selected as the most promising for further consideration: salvianolic acid B, atorvastatin, naringin, montelukast, dipyridamole, lopinavir, fulvestrant, ritonavir, telmisartan, bazedoxifene, bortezomib, atazanavir and ramipril. All of the selected candidates were highly scored after ensemble docking which reflects their potential for inhibiting of Mpro. Predicted poses of all of the promising candidates were in agreement with experimental observations, as discussed above. Selected candidate drugs were found to affect cytokine signaling pathways important in systemic inflammation which could indicate their potential pleiotropic effects (Figures 5 and 6). However, additional analysis of currently available data on binary drug‐gene interactions (Supp. Table S3) revealed that lopinavir, atazanavir and ritonavir could even aggravate cytokine storm associated with COVID‐19 which should be considered as caveats in their future chemotherapeutic evaluation. Among the selected drugs with identified pleiotropic effects, atorvastatin, naturally occurring polyphenols (salvianolic acid B and naringin), bortezomib and ramipril show lower tendency to distribute in tissues of interest (Table 2). However, alternative dosing routes or higher doses should be considered for future studies. Overall, the validity of integrated repurposing approach is supported by referred experimental data and clinical trials (Table 2) and lays the foundations for future in silico antiviral drug repurposing studies.
4. Conclusion
In this work, we present in silico repurposing approach for identification of potential inhibitors of SARS‐CoV‐2 Main Protease (Mpro) that integrates diverse structure‐based molecular modeling techniques with QSPR‐PBPK modelling and drug‐gene‐COVID‐19 analysis.
Through stepwise SBVS approach, 43 potential candidates were selected for further analysis. Biological, clinical and structural data on COVID‐19 and Mpro reported so far were in agreement with obtained results, which validate used structure‐based approaches as a practical tool for future screening of larger databases. Selected candidates were further analyzed in terms of their chemical nature and different patterns in Mpro‐ligand intermolecular interactions, which gave us detailed understandings in chemical space of interest for future anti‐COVID‐19 drug discovery protocols. In addition to structure‐based modelling, QSPR‐PBPK approach elicited the drugs with favorable disposition properties, but also indicated the key factors that may limit their systemic and tissue exposure. Complementary, data mining of drug‐gene‐COVID‐19 association singled out candidates which modulate pro‐inflammatory genes expression related to COVID‐19 disease. Pathway enrichment analysis suggested potential pleiotropic effects of selected candidates that could modify severity of COVID‐19 disease. Overall, we demonstrated that repurposing studies should not be based only on structure‐based modeling; future chemotherapeutic repurposing studies should be evaluated in terms of drug‐gene‐disease considerations. Last but not least, adequate tissue distribution is of great importance to be examined for the virus infected tissues.
Conflict of interest
None declared.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
Authors acknowledge project of Ministry of Science and Technological Development of the Republic of Serbia, Contract No. 451‐03‐9/2021‐14/200161. Numerical simulations were run on the PARADOX‐IV supercomputing facility at the Scientific Computing Laboratory, National Center of Excellence for the Study of Complex Systems, Institute of Physics Belgrade, supported in part by the Ministry of Education, Science, and Technological Development of the Republic of Serbia.
N. Djokovic, D. Ruzic, T. Djikic, S. Cvijic, J. Ignjatovic, S. Ibric, K. Baralic, A. Buha Djordjevic, M. Curcic, D. Djukic-Cosic, K. Nikolic, Mol. Inf. 2021, 40, 2000187.
Contributor Information
Dusan Ruzic, Email: dusan.ruzic@pharmacy.bg.ac.rs, Phone: /fax: +381‐11‐3951‐259.
Katarina Nikolic, Email: katarina.nikolic@pharmacy.bg.ac.rs.
References
- 1.“Coronavirus Update (Live): 94,421,017 Cases and 2,020,254 Deaths from COVID-19 Virus Pandemic - Worldometer,” can be found under https://www.worldometers.info/coronavirus/, 2021.
- 2. Wu Y., Xu X., Chen Z., Duan J., Hashimoto K., Yang L., Liu C., Yang C., Brain Behav. Immun. 2020, DOI 10.1016/j.bbi.2020.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Zheng Y.-Y., Ma Y.-T., Zhang J.-Y., Xie X., Nat. Rev. Cardiol. 2020, 17, 259–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. South A. M., Diz D. I., Chappell M. C., Am. J. Physiol. Heart Circ. Physiol. 2020, 318, H1084–H1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.O. of the Commissioner, “Pfizer-BioNTech COVID-19 Vaccine,” can be found under https://www.fda.gov/emergency-preparedness-and-response/coronavirus-disease-2019-covid-19/pfizer-biontech-covid-19-vaccine, 2021.
- 6.O. of the Commissioner, “Moderna COVID-19 Vaccine,” can be found under https://www.fda.gov/emergency-preparedness-and-response/coronavirus-disease-2019-covid-19/moderna-covid-19-vaccine, 2021.
- 7. Pushpakom S., Iorio F., Eyers P. A., Escott K. J., Hopper S., Wells A., Doig A., Guilliams T., Latimer J., McNamee C., Norris A., Sanseau P., Cavalla D., Pirmohamed M., Nat. Rev. Drug Discovery 2019, 18, 41–58. [DOI] [PubMed] [Google Scholar]
- 8. Bellera C. L., Llanos M., Gantner M. E., Rodriguez S., Gavernet L., Comini M., Talevi A., Expert Opin. Drug Discovery 2020, 0, 1–8. [DOI] [PubMed] [Google Scholar]
- 9. Erlanson D. A., Nat. Commun. 2020, 11, 5048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sultana J., Crisafulli S., Gabbay F., Lynn E., Shakir S., Trifirò G., Front. Pharmacol. 2020, 11, DOI 10.3389/fphar.2020.588654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Piscoya A., Ng-Sueng L. F., del Riego A. P., Cerna-Viacava R., Pasupuleti V., Roman Y. M., Thota P., White C. M., Hernandez A. V., PLoS One 2020, 15, e0243705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.WHO Solidarity Trial Consortium, H. Pan, R. Peto, A.-M. Henao-Restrepo, M.-P. Preziosi, V. Sathiyamoorthy, Q. Abdool Karim, M. M. Alejandria, C. Hernández García, M.-P. Kieny, R. Malekzadeh, S. Murthy, K. S. Reddy, M. Roses Periago, P. Abi Hanna, F. Ader, A. M. Al-Bader, A. Alhasawi, E. Allum, A. Alotaibi, C. A. Alvarez-Moreno, S. Appadoo, A. Asiri, P. Aukrust, A. Barratt-Due, S. Bellani, M. Branca, H. B. C. Cappel-Porter, N. Cerrato, T. S. Chow, N. Como, J. Eustace, P. J. García, S. Godbole, E. Gotuzzo, L. Griskevicius, R. Hamra, M. Hassan, M. Hassany, D. Hutton, I. Irmansyah, L. Jancoriene, J. Kirwan, S. Kumar, P. Lennon, G. Lopardo, P. Lydon, N. Magrini, T. Maguire, S. Manevska, O. Manuel, S. McGinty, M. T. Medina, M. L. Mesa Rubio, M. C. Miranda-Montoya, J. Nel, E. P. Nunes, M. Perola, A. Portolés, M. R. Rasmin, A. Raza, H. Rees, P. P. S. Reges, C. A. Rogers, K. Salami, M. I. Salvadori, N. Sinani, J. A. C. Sterne, M. Stevanovikj, E. Tacconelli, K. A. O. Tikkinen, S. Trelle, H. Zaid, J.-A. Røttingen, S. Swaminathan, N. Engl. J. Med. 2020, DOI 10.1056/NEJMoa2023184.
- 13. Rubin D., Chan-Tack K., Farley J., Sherwat A., N. Engl. J. Med. 2020, 383, 2598–2600. [DOI] [PubMed] [Google Scholar]
- 14. Elsawah H. K., Elsokary M. A., Abdallah M. S., ElShafie A. H., Rev. Med. Virol. n.d., n/a, e2187. [DOI] [PubMed] [Google Scholar]
- 15. Jin Z., Du X., Xu Y., Deng Y., Liu M., Zhao Y., Zhang B., Li X., Zhang L., Peng C., Duan Y., Yu J., Wang L., Yang K., Liu F., Jiang R., Yang X., You T., Liu X., Yang X., Bai F., Liu H., Liu X., Guddat L. W., Xu W., Xiao G., Qin C., Shi Z., Jiang H., Rao Z., Yang H., Nature 2020, 1–5. [Google Scholar]
- 16. Pillaiyar T., Manickam M., Namasivayam V., Hayashi Y., Jung S.-H., J. Med. Chem. 2016, 59, 6595–6628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bzówka M., Mitusińska K., Raczyńska A., Samol A., Tuszyński J. A., Góra A., Int. J. Mol. Sci. 2020, 21, DOI 10.3390/ijms21093099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.L. L. Palese, 2020, DOI 10.26434/chemrxiv.12209744.v1.
- 19. Brimacombe K. R., Zhao T., Eastman R. T., Hu X., Wang K., Backus M., Baljinnyam B., Chen C. Z., Chen L., Eicher T., Ferrer M., Fu Y., Gorshkov K., Guo H., Hanson Q. M., Itkin Z., Kales S. C., Klumpp-Thomas C., Lee E. M., Michael S., Mierzwa T., Patt A., Pradhan M., Renn A., Shinn P., Shrimp J. H., Viraktamath A., Wilson K. M., Xu M., Zakharov A. V., Zhu W., Zheng W., Simeonov A., Mathé E. A., Lo D. C., Hall M. D., Shen M., bioRxiv 2020, 2020.06. 04. 135046. [Google Scholar]
- 20. Dotolo S., Marabotti A., Facchiano A., Tagliaferri R., Briefings Bioinf. 2020, DOI 10.1093/bib/bbaa288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.“OpenData Portal-Data Browser,” can be found under https://opendata.ncats.nih.gov/covid19/databrowser, 2021.
- 22. Ghahremanpour M. M., Tirado-Rives J., Deshmukh M., Ippolito J. A., Zhang C.-H., Cabeza de Vaca I., Liosi M.-E., Anderson K. S., Jorgensen W. L., ACS Med. Chem. Lett. 2020, 11, 2526–2533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Vatansever E. C., Yang K., Kratch K. C., Drelich A., Cho C.-C., Mellot D. M., Xu S., Tseng C.-T. K., Liu W. R., bioRxiv 2020, 2020.05. 23. 112235. [Google Scholar]
- 24. Drayman N., Jones K. A., Azizi S.-A., Froggatt H. M., Tan K., Maltseva N. I., Chen S., Nicolaescu V., Dvorkin S., Furlong K., Kathayat R. S., Firpo M. R., Mastrodomenico V., Bruce E. A., Schmidt M. M., Jedrzejczak R., Muñoz-Alía M. Á., Schuster B., Nair V., Botten J. W., Brooke C. B., Baker S. C., Mounce B. C., Heaton N. S., Dickinson B. C., Jaochimiak A., Randall G., Tay S., bioRxiv 2020, 2020.08. 31. 274639. [Google Scholar]
- 25. Li Z., Li X., Huang Y.-Y., Wu Y., Liu R., Zhou L., Lin Y., Wu D., Zhang L., Liu H., Xu X., Yu K., Zhang Y., Cui J., Zhan C.-G., Wang X., Luo H.-B., Proc. Nat. Acad. Sci. 2020, 117, 27381–27387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Krishna S., Augustin Y., Wang J., Xu C., Staines H. M., Platteeuw H., Kamarulzaman A., Sall A., Kremsner P., Trends Parasitol. 2021, 37, 8–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wu Z., Li W., Liu G., Tang Y., Front. Pharmacol. 2018, 9, DOI 10.3389/fphar.2018.01134.29434546 [Google Scholar]
- 28. Cheng F., Li W., Wu Z., Wang X., Zhang C., Li J., Liu G., Tang Y., J. Chem. Inf. Model. 2013, 53, 753–762. [DOI] [PubMed] [Google Scholar]
- 29. Na H., Hm J., Mol. Pharm. 2013, 10, 1207–1215. [DOI] [PubMed] [Google Scholar]
- 30. Miller N. A., Reddy M. B., Heikkinen A. T., Lukacova V., Parrott N., Clin. Pharmacokinet. 2019, 58, 727–746. [DOI] [PubMed] [Google Scholar]
- 31.“Compound Libraries for High Throughput/Content Screening | 96-Well,” can be found under https://www.selleckchem.com/screening/fda-approved-drug-library.html, 2020.
- 32. Halgren T. A., J. Comput. Chem. 1996, 17, 490–519. [Google Scholar]
- 33. Martínez-Rosell G., Giorgino T., De Fabritiis G., J. Chem. Inf. Model. 2017, 57, 1511–1516. [DOI] [PubMed] [Google Scholar]
- 34. Baroni M., Cruciani G., Sciabola S., Perruccio F., Mason J. S., J. Chem. Inf. Model. 2007, 47, 279–294. [DOI] [PubMed] [Google Scholar]
- 35. Cross S., Baroni M., Goracci L., Cruciani G., J. Chem. Inf. Model. 2012, 52, 2587–2598. [DOI] [PubMed] [Google Scholar]
- 36. Jones G., Willett P., Glen R. C., Leach A. R., Taylor R., J. Mol. Biol. 1997, 267, 727–748. [DOI] [PubMed] [Google Scholar]
- 37. Friesner R. A., Murphy R. B., Repasky M. P., Frye L. L., Greenwood J. R., Halgren T. A., Sanschagrin P. C., Mainz D. T., J. Med. Chem. 2006, 49, 6177–6196. [DOI] [PubMed] [Google Scholar]
- 38. Friesner R. A., Banks J. L., Murphy R. B., Halgren T. A., Klicic J. J., Mainz D. T., Repasky M. P., Knoll E. H., Shelley M., Perry J. K., Shaw D. E., Francis P., Shenkin P. S., J. Med. Chem. 2004, 47, 1739–1749. [DOI] [PubMed] [Google Scholar]
- 39. Halgren T. A., Murphy R. B., Friesner R. A., Beard H. S., Frye L. L., Pollard W. T., Banks J. L., J. Med. Chem. 2004, 47, 1750–1759. [DOI] [PubMed] [Google Scholar]
- 40. Korb O., Stützle T., Exner T. E., J. Chem. Inf. Model. 2009, 49, 84–96. [DOI] [PubMed] [Google Scholar]
- 41. Lin L., Wong H., Pharmaceutics 2017, 9, 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Agoram B., Woltosz W. S., Bolger M. B., Adv. Drug Delivery Rev. 2001, 50, S41–S67. [DOI] [PubMed] [Google Scholar]
- 43.“ADMET Property Prediction | QSPR | Physicochemical | ADME,” can be found under https://www.simulations-plus.com/software/admetpredictor/, 2020.
- 44. Mithani S. D., Bakatselou V., TenHoor C. N., Dressman J. B., Pharm. Res. 1996, 13, 163–167. [DOI] [PubMed] [Google Scholar]
- 45.V. Lukacova, N. Parrott, T. Lave, G. Fraczkiewicz, M. Bolger, W. Woltosz, in Poster Sess. Present. 2008 AAPS Natl. Annu. Meet. Expo., 2008.
- 46. Davis A. P., Grondin C. J., Johnson R. J., Sciaky D., McMorran R., Wiegers J., Wiegers T. C., Mattingly C. J., Nucleic Acids Res. 2019, 47, D948–D954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Shannon P., Markiel A., Ozier O., Baliga N. S., Wang J. T., Ramage D., Amin N., Schwikowski B., Ideker T., Genome Res. 2003, 13, 2498–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Chin C.-H., Chen S.-H., Wu H.-H., Ho C.-W., Ko M.-T., Lin C.-Y., BMC Syst. Biol. 2014, 8, S11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Warde-Farley D., Donaldson S. L., Comes O., Zuberi K., Badrawi R., Chao P., Franz M., Grouios C., Kazi F., Lopes C. T., Maitland A., Mostafavi S., Montojo J., Shao Q., Wright G., Bader G. D., Morris Q., Nucleic Acids Res. 2010, 38, W214–W220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Bindea G., Mlecnik B., Hackl H., Charoentong P., Tosolini M., Kirilovsky A., Fridman W.-H., Pagès F., Trajanoski Z., Galon J., Bioinformatics 2009, 25, 1091–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Bindea G., Galon J., Mlecnik B., Bioinformatics 2013, 29, 661–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kanehisa M., Sato Y., Kawashima M., Furumichi M., Tanabe M., Nucleic Acids Res. 2016, 44, D457–D462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Jassal B., Matthews L., Viteri G., Gong C., Lorente P., Fabregat A., Sidiropoulos K., Cook J., Gillespie M., Haw R., Loney F., May B., Milacic M., Rothfels K., Sevilla C., Shamovsky V., Shorser S., Varusai T., Weiser J., Wu G., Stein L., Hermjakob H., D'Eustachio P., Nucleic Acids Res. 2020, 48, D498–D503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Slenter D. N., Kutmon M., Hanspers K., Riutta A., Windsor J., Nunes N., Mélius J., Cirillo E., Coort S. L., Digles D., Ehrhart F., Giesbertz P., Kalafati M., Martens M., Miller R., Nishida K., Rieswijk L., Waagmeester A., Eijssen L. M. T., Evelo C. T., Pico A. R., Willighagen E. L., Nucleic Acids Res. 2018, 46, D661–D667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Englert P., Kovács P., J. Chem. Inf. Model. 2015, 55, 941–955. [DOI] [PubMed] [Google Scholar]
- 56. Yuriev E., Future Med. Chem. 2013, 6, 5–7. [DOI] [PubMed] [Google Scholar]
- 57. Huang S.-Y., Zou X., Proteins Struct. Funct. Bioinf. 2007, 66, 399–421. [DOI] [PubMed] [Google Scholar]
- 58. Rueda M., Bottegoni G., Abagyan R., J. Chem. Inf. Model. 2010, 50, 186–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Craig I. R., Pfleger C., Gohlke H., Essex J. W., Spiegel K., J. Chem. Inf. Model. 2011, 51, 2666–2679. [DOI] [PubMed] [Google Scholar]
- 60. Park S.-J., Kufareva I., Abagyan R., J. Comput.-Aided Mol. Des. 2010, 24, 459–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Korb O., Olsson T. S. G., Bowden S. J., Hall R. J., Verdonk M. L., Liebeschuetz J. W., Cole J. C., J. Chem. Inf. Model. 2012, 52, 1262–1274. [DOI] [PubMed] [Google Scholar]
- 62.W. Dai, B. Zhang, H. Su, J. Li, Y. Zhao, X. Xie, Z. Jin, F. Liu, C. Li, Y. Li, F. Bai, H. Wang, X. Cheng, X. Cen, S. Hu, X. Yang, J. Wang, X. Liu, G. Xiao, H. Jiang, Z. Rao, L.-K. Zhang, Y. Xu, H. Yang, H. Liu, Science 2020, DOI 10.1126/science.abb4489.
- 63.“Main protease structure and XChem fragment screen - - Diamond Light Source,” can be found under https://www.diamond.ac.uk/covid-19/for-scientists/Main-protease-structure-and-XChem.html, 2020.
- 64.T. S. Komatsu, Y. M. Koyama, N. Okimoto, G. Morimoto, Y. Ohno, M. Taiji, 2020, 2, DOI 10.17632/vpps4vhryg.2.
- 65. Liu X., Li Z., Liu S., Sun J., Chen Z., Jiang M., Zhang Q., Wei Y., Wang X., Huang Y.-Y., Shi Y., Xu Y., Xian H., Bai F., Ou C., Xiong B., Lew A. M., Cui J., Fang R., Huang H., Zhao J., Hong X., Zhang Y., Zhou F., Luo H.-B., Acta Pharm. Sin. B 2020, DOI 10.1016/j.apsb.2020.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Nguyenla X., Wehri E., Dis E. V., Biering S. B., Yamashiro L. H., Stroumza J., Dugast-Darzacq C., Graham T., Stanley S., Schaletzky J., bioRxiv 2020, 2020.09.18.302398. [Google Scholar]
- 67. Sacramento C. Q., Fintelman-Rodrigues N., Temerozo J. R., da S. G. Dias S., Ferreira A. C., Mattos M., Pão C. R. R., de Freitas C. S., Soares V. C., Bozza F. A., Bou-Habib D. C., Bozza P. T., Souza T. M. L., bioRxiv 2020, 2020.06. 15. 153411. [Google Scholar]
- 68. Jeon S., Ko M., Lee J., Choi I., Byun S. Y., Park S., Shum D., Kim S., Antimicrob. Agents Chemother. 2020, DOI 10.1128/AAC.00819–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Fintelman-Rodrigues N., Sacramento C. Q., Lima C. R., da Silva F. S., Ferreira A. C., Mattos M., de Freitas C. S., Soares V. C., da S. G. Dias S., Temerozo J. R., Miranda M. D., Matos A. R., Bozza F. A., Carels N., Alves C. R., Siqueira M. M., Bozza P. T., Souza T. M. L., Antimicrob. Agents Chemother. 2020, 64, DOI 10.1128/AAC.00825–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Liu X., Liu Y., Wang L., Hu L., Liu D., Li J., J. Med. Virol. 2020, DOI 10.1002/jmv.26492. [Google Scholar]
- 71.Laboratorio Elea Phoenix S. A., Telmisartan for Treatment of COVID-19 Patients: An Open Label Randomized Trial, Clinicaltrials.Gov, 2020.
- 72.R. Loomba, A Randomized, Double-Blind, Placebo-Controlled Trial to Evaluate the Efficacy of Ramipril to Prevent ICU Admission, Need for Mechanical Ventilation or Death in Persons With COVID-19, Clinicaltrials.Gov, 2020.
- 73. Oerlemans R., Jonathan Ruiz-Moreno A., Cong Y., Kumar N. D., Velasco-Velazquez M. A., Neochoritis C. G., Smith J., Reggiori F., Groves M. R., Dömling A., RSC Med. Chem. 2021, DOI 10.1039/D0MD00367 K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.“XChem @ Diamond,” can be found under https://fragalysis.diamond.ac.uk/viewer/react/landing/, 2020.
- 75. Terpos E., Ntanasis-Stathopoulos I., Elalamy I., Kastritis E., Sergentanis T. N., Politou M., Psaltopoulou T., Gerotziafas G., Dimopoulos M. A., Am. J. Hematol. 2020, 95, 834–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Nassar T., Rohald A., Naraykin N., Barasch D., Amsalem O., Prabhu P., Kotler M., Benita S., J. Drug Targeting 2019, 27, 590–600. [DOI] [PubMed] [Google Scholar]
- 77. Guo F., Zhong H., He J., Xie B., Liu F., Xu H., Liu M., Xu C., Arch. Pharmacal Res. 2011, 34, 1113. [DOI] [PubMed] [Google Scholar]
- 78. Wienen W., Entzeroth M., van Meel J. C. A., Stangier J., Busch U., Ebner T., Schmid J., Lehmann H., Matzek K., Kempthorne-Rawson J., Gladigau V., Hauel N. H., Cardiovasc. Drug Rev. 2000, 18, 127–154. [Google Scholar]
- 79. Gregov D., Jenkins A., Duncan E., Siebert D., Rodgers S., Duncan B., Bochner F., Lloyd J., Br. J. Clin. Pharmacol. 1987, 24, 425–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Denissen J. F., Grabowski B. A., Johnson M. K., Buko A. M., Kempf D. J., Thomas S. B., Surber B. W., Drug Metab. Dispos. Biol. Fate Chem. 1997, 25, 489–501. [PubMed] [Google Scholar]
- 81. Shoenfeld Y., Autoimmun. Rev. 2020, 19, 102538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Nielsen-Kudsk F., Pedersen A. K., Acta Pharmacol. Toxicol. 1979, 44, 391–399. [DOI] [PubMed] [Google Scholar]
- 83. Zhang J., Xie B., Hashimoto K., Brain Behav. Immun. 2020, 87, 59–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Liu X., Li Z., Liu S., Sun J., Chen Z., Jiang M., Zhang Q., Wei Y., Wang X., Huang Y.-Y., Shi Y., Xu Y., Xian H., Bai F., Ou C., Xiong B., Lew A. M., Cui J., Fang R., Huang H., Zhao J., Hong X., Zhang Y., Zhou F., Luo H.-B., Acta Pharm. Sin. B 2020, DOI 10.1016/j.apsb.2020.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Cao B., Wang Y., Wen D., Liu W., Wang J., Fan G., Ruan L., Song B., Cai Y., Wei M., Li X., Xia J., Chen N., Xiang J., Yu T., Bai T., Xie X., Zhang L., Li C., Yuan Y., Chen H., Li H., Huang H., Tu S., Gong F., Liu Y., Wei Y., Dong C., Zhou F., Gu X., Xu J., Liu Z., Zhang Y., Li H., Shang L., Wang K., Li K., Zhou X., Dong X., Qu Z., Lu S., Hu X., Ruan S., Luo S., Wu J., Peng L., Cheng F., Pan L., Zou J., Jia C., Wang J., Liu X., Wang S., Wu X., Ge Q., He J., Zhan H., Qiu F., Guo L., Huang C., Jaki T., Hayden F. G., Horby P. W., Zhang D., Wang C., N. Engl. J. Med. 2020, 382, 1787–1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Gong J., Dong H., Xia S. Q., Huang Y. Z., Wang D., Zhao Y., Liu W., Tu S., Zhang M., Wang Q., Lu F., medRxiv 2020, 2020.02. 25. 20025643. [Google Scholar]
- 87. Seif F., Khoshmirsafa M., Aazami H., Mohsenzadegan M., Sedighi G., Bahar M., Cell Commun. Signaling 2017, 15, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Montesarchio V., Parrella R., Iommelli C., Bianco A., Manzillo E., Fraganza F., Palumbo C., Rea G., Murino P., De Rosa R., Atripaldi L., D'Abbraccio M., Curvietto M., Mallardo D., Celentano E., Grimaldi A. M., Palla M., Trojaniello C., Vitale M. G., Million-Weaver S. L., Ascierto P. A., J. Immunother. Cancer 2020, 8, DOI 10.1136/jitc-2020–001089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Baralić K., Jorgovanović D., Živančević K., Antonijević Miljaković E., Antonijević B., Buha Djordjevic A., Ćurčić M., Đukić-Ćosić D., Toxicol. Appl. Pharmacol. 2020, 406, 115237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Orlov M., Wander P. L., Morrell E. D., Mikacenic C., Wurfel M. M., J. Immunol. 2020, 205, 892–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary