

Abstract
The origin of severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) is the subject of many hypotheses. One of them, proposed by Segreto and Deigin, assumes artificial chimeric construction of SARS‐CoV‐2 from a backbone of RaTG13‐like CoV and receptor binding domain (RBD) of a pangolin MP789‐like CoV, followed by serial cell or animal passage. Here we show that this hypothesis relies on incorrect or weak assumptions, and does not agree with the results of comparative genomics analysis. The genetic divergence between SARS‐CoV‐2 and both its proposed ancestors is too high to have accumulated in a lab, given the timeframe of several years. Furthermore, comparative analysis of S‐protein gene sequences suggests that the RBD of SARS‐CoV‐2 probably represents an ancestral non‐recombinant variant. These and other arguments significantly weaken the hypothesis of a laboratory origin for SARS‐CoV‐2, while the hypothesis of a natural origin is consistent with all available genetic and experimental data.
Keywords: comparative genomics, coronavirus, COVID‐19, evolution, furin cleavage site, SARS‐CoV‐2 laboratory origin, SARS‐CoV‐2
A hypothesis of man‐made SARS‐CoV‐2 origin appeared in the scientific literature. Here we discuss the flaws of this hypothesis and argue that the available comparative genomics data including genetic divergence, distribution of substitution rates, and parsimonious reconstructions of recombination events support a scenario of a natural origin for SARS‐CoV‐2.
Abbreviations
- RBD
receptor binding domain
- CoV
coronavirus
- WIV
Wuhan Institute of Virology
INTRODUCTION
The recent article by Segreto and Deigin advocates the hypothesis of artificial chimeric origin of SARS‐CoV‐2.[ 1 ] According to the authors’ thesis, the virus “could have been synthesized by combining a backbone similar to RaTG13 with the RBD of CoV similar to the one recently isolated from pangolins,” followed by serial cell or animal passage. Here we show that the few supportive arguments presented in that work rely on improbable or incorrect assumptions, while important weaknesses of the hypothesis are completely ignored.
We wish to make explicit that our comment is not about whether SARS‐CoV‐2, regardless of its origin, leaked from a laboratory: this hypothesis cannot be evaluated by analyzing the genetic and phenotypic properties of the virus. Such a leak can only be established by investigating the lab in question. Our comment is about the possible biological origins of SARS‐CoV‐2 in the light of evidence provided by comparative genomics approaches.
POINTS OF DISAGREEMENT WITH THE SEGRETO/DEIGIN HYPOTHESIS
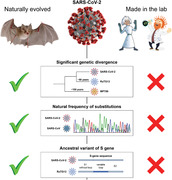
1. The first major problem with Segreto's and Deigin's hypothesis is the significant divergence between the genome sequence of SARS‐CoV‐2 and its proposed ancestor RaTG13. The RaTG13 genome shares only 96.2% similarity with SARS‐CoV‐2.[ 2 ] The estimated divergence timepoint between these two viruses is between 1948 and 1982, indicating that the ancestors of SARS‐CoV‐2 have been circulating unnoticed for decades.[ 3 ] If the RaTG13 genome had been used as the backbone for the creation of SARS‐CoV‐2, the Wuhan Institute of Virology (WIV) group would have needed to passage it in cells or animals for years to accumulate 3.8% sequence divergence.
For example, the mutation rate of SARS‐CoV during passages in cell cultures was found to be 9 × 10–7 substitutions per nucleotide per replication cycle (approximately 12 h).[ 4 ] Serial passage of SARS‐CoV in animals resulted in comparable numbers. Following cultivation in mouse lungs for more than 30 days, the coronavirus accumulated only six nucleotide mutations (the divergence of 0.02%) after 15 passages.[ 5 ] Based on these mutation rate estimates, the accumulation of 3.8% genetic difference via cell or animal passage would require more than 15 years. It is fair to assume that SARS‐CoV‐2 has similar mutation rates. Therefore, given that the RaTG13 virus was discovered in 2013, the accumulation of 3.8% differences in this coronavirus by 2019 seems improbable.
It could be argued that certain laboratory techniques, such as the use of the mutagenic compound ribavirin or inactivation of coronavirus exoribonuclease activity, could be used to increase mutation rates during the passage of SARS‐CoV‐2.[ 4 ] However, to our knowledge, such techniques have not been used previously to enhance coronavirus adaptation. In addition, they seem to produce certain mutational biases,[ 4 , 6 ] which are not presented in SARS‐CoV‐2 when compared to other naturally evolved human‐adapted coronaviruses.[ 7 ] Instead, we observe similar relative frequencies of single nucleotide substitutions distinguishing SARS‐CoV‐2 and SARS‐CoV from their bat relatives RaTG13 and Rs4231, respectively (Figure 1; Pearson‘s chi‐squared p‐value = 0.12, based on data from[ 7 ]), which is consistent with the hypothesis of natural origin of SARS‐CoV‐2.
FIGURE 1.
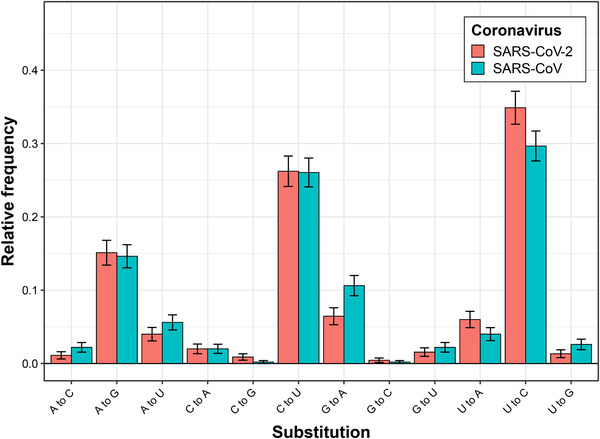
Relative frequencies of different single nucleotide substitutions, which distinguish SARS‐CoV‐2 (red) and SARS‐CoV (blue) from their bat relatives (RaTG13 and Rs4231, respectively).[ 7 ] Differences across substitution frequencies are not significant, as assessed with Pearson's chi‐squared test (p = 0.12)
2. The RBD of pangolin coronavirus MP789 could not have been used for the creation of SARS‐CoV‐2 either. In their paper, Segreto and Deigin state that “the MP789 pangolin strain isolated from Guangdong (GD) pangolins has an almost identical RBD to that of SARS‐CoV‐2″. This claim appears to be true only at the amino acid sequence level. The genetic sequence similarity between MP789 and SARS‐CoV‐2 RBD is only 86.6% (Figure 2), close to that between SARS‐CoV‐2 and RaTG13 RBDs (85.2%), and much lower than the overall genomic similarity between SARS‐CoV‐2 and RaTG13 (96.2%). Cultivation of the murine hepatitis virus (MHV) coronavirus in cell cultures for 5 years (600 passages) resulted in the accumulation of only 63 point mutations across the whole S‐protein gene,[ 8 ] while RBD sequences of MP789 and SARS‐CoV‐2 are separated by 78 nucleotide substitutions. Thus, accumulation of these differences in the lab would also require years of cultivation, a highly unlikely scenario, given that the pangolin CoV was discovered in 2019, the same year in which the COVID‐19 outbreak occurred.[ 9 , 10 ] Moreover, almost all mutations that differ between RBDs of these viruses are synonymous, and thus cannot be explained by site‐directed mutagenesis. Therefore, neither RaTG13, nor MP789 seem to be appropriate candidates for the artificial construction of SARS‐CoV‐2, even if a subsequent passage in cells or animals is considered.
FIGURE 2.

Multiple alignment of S‐protein gene RBD sequences of SARS‐CoV‐2, Pangolin CoV MP789 and RaTG13
3. One of the arguments proposed by Segreto and Deigin in favor of the artificial origin of SARS‐CoV‐2 is the low probability of natural recombination between RaTG13‐ and MP789‐ related strains in pangolins, “considering the low population density of pangolins and the scarce presence of CoVs in their natural populations”. However, many related strains of the pangolin coronavirus have been discovered in bats.[ 9 ] Thus, such recombination events did not need to occur in pangolins for SARS‐CoV‐2 to emerge. Instead, they could happen in bats, followed by a transmission of the resulted virus to a new host.
Segreto and Deigin state that “the most surprising observation was that RaTG13, unlike SARS‐CoV‐2, is unable to bind ACE2 in R. macrotis bats, a close relative of RaTG13's purported host, R. affinis (whose ACE2 receptor has not yet been tested)”. However, in a recent work it was shown that the ACE2 receptor of R. affinis bats can effectively bind and mediate the entry of both RaTG13 and SARS‐CoV‐2 viruses in a pseudovirus assay.[ 11 ] Therefore, the recombination may have occurred in cells of horseshoe bats. Notably, a high frequency of recombination events has been shown between SARS‐like bat coronaviruses presumably involved in the emergence of SARS‐CoV in 2002.[ 12 ]
Finally, the authors failed to mention that a recent analysis of S‐protein gene sequences has demonstrated that it is more likely that the RBD of RaTG13, not SARS‐CoV‐2, is the result of recombination, and that RBDs of SARS‐CoV‐2 and pangolin MP789 represent the original ancestral variant.[ 3 ] This claim is supported by the fact that genetic divergence between MP789 and SARS‐CoV‐2 is similar throughout most of the S‐protein gene, while in the case of recombination at the RBD site one would expect higher similarity in this fragment. This finding also supports bat origin of SARS‐CoV‐2, further weakening the hypothesis of its artificial chimeric construction.
4. Another argument proposed by Segreto and Deigin is based on the presence of FauI restriction site in the SARS‐CoV‐2 12‐nucleotide insertion of the furin cleavage site that is important for the virus's ability to infect human cells. The authors claim that this restriction site may point to the artificial origin of SARS‐CoV‐2, because it “could allow using restriction fragment length polymorphism (RFLP) techniques for cloning or screening for mutations, as the new furin site is prone to deletions in vitro”. However, the presence of a site recognized by some restriction enzyme within a furin cleavage site does not provide evidence for artificial origin, because such sites occur naturally, and the prevalence of different restriction sites through the genome of SARS‐CoV‐2 and other coronaviruses is rather high, as we will demonstrate below.
Using NEBcutter,[ 13 ] we've found that the 500‐nucleotide sequence around the FauI site discussed in the Segreto/Deigin paper includes 287 sites of restriction covering 180 different positions (Figure 3). Therefore, each third nucleotide around this region, on average, may be cut by some restriction enzyme, an observation that makes the probability of finding at least one restriction site within the 12‐nucleotide insertion of the furin cleavage site roughly . Therefore, its presence cannot be considered an argument in favor of the artificial origin hypothesis, especially considering that FauI restriction sites, to our knowledge, have not been mentioned in any works related to cloning or mutation screening of coronaviruses. There are no grounds on which to argue that this restriction site within a furin cleavage site is more suspicious than any other. The reasoning used by the authors is that of the Texas Sharpshooter Fallacy—the emergence of a fortuitous event that is, post hoc, used as evidence of causality or intent.
FIGURE 3.

500‐nucleotide sequence map around the furin cleavage site with some of the restriction sites corresponding to commercially available restriction enzymes. Sites cleaved with blunt, 5′‐extended and 3′‐extended ends are shown in red, blue, and green, respectively
5. When discussing the origin of the furin cleavage site, Segreto and Deigin claim that: “The insertion of the furin cleavage site in SARS‐CoV‐2 is not in frame with the rest of the sequence, when compared with the MP789 and the RaTG13 sequences. Therefore, it is possible to exclude that such an insertion could have originated by polymerase slippage or by releasing and repriming, because insertion mutations generated by these mechanisms have been postulated to maintain the reading frame of the viral sequence”.
Firstly, the length of the 12‐nucleotide insertion discussed by the authors is a multiple of three and, therefore, maintains the reading frame of the S‐protein gene. Secondly, the mechanisms of polymerase slippage or releasing and repriming can produce insertions that are “not in frame with the rest of the sequence” and split a certain codon in two parts. Such examples are well known in influenza viruses.[ 14 ] Ironically, one of the papers demonstrating these cases is referenced in an article of David A. Steinhauer,[ 15 ] which Segreto and Deigin cite as an argument against the possibility of such an event. In addition, the insertion mutations can originate through other mechanisms, besides those described by the authors. Therefore, there is no evidence that the 12‐nucleotide fragment of the SARS‐CoV‐2 furin cleavage site was introduced artificially and did not, instead, emerge in nature.
6. The remaining unfalsifiable scenario of SARS‐CoV‐2′s artificial origin assumes that the WIV used two unknown, unpublished viruses for the chimeric construction of SARS‐CoV‐2. However, this hypothesis is less parsimonious than the scenario of a naturally evolved SARS‐CoV‐2 escaping from the lab, because in the latter case the presence of only one currently unknown virus in the WIV lab is required, while the hypothesis of artificial creation requires the existence of two unknown viruses in the same lab at once (one distinct relative of RaTG13 with SARS‐CoV‐2 backbone and one distinct relative of MP789 with SARS‐CoV‐2 RBD). Taking into account the much higher prevalence of coronaviruses and recombination events in bat populations compared to laboratories,[ 12 , 16 ] and the existence of a more parsimonious hypothesis of SARS‐CoV‐2 origin that doesn't require the recombination at all,[ 3 ] the hypothesis of artificial “recombination” between two unpublished viruses seems unlikely and violates the principle of Occam's razor.
7. At the end of their paper, the authors suggest that “genetic manipulation of SARS‐CoV‐2 may have been carried out in any laboratory in the world with access to the backbone sequence and the necessary equipment and it would not leave any trace. Modern technologies based on synthetic genetics platforms allow the reconstruction of viruses based on their genomic sequence, without the need of a natural isolate”. However, the same argument may be applied to any emerging virus. Why focus only on SARS‐CoV‐2, when “the genetic structure of H1N1/09 does not rule out a laboratory origin” would be another great title?
CONCLUSIONS
In summary, the hypothesis of artificial creation of SARS‐CoV‐2 proposed by Segreto and Deigin is not supported by evidence. Additionally, it does not agree with a number of findings based on genetic analysis of SARS‐CoV‐2 and its relatives. The scenario of chimeric virus combined from RaTG13 and MP789 strains seems incompatible with the high genetic divergence between these coronaviruses and SARS‐CoV‐2. The scenario of SARS‐CoV‐2 synthesis from two still unpublished viruses is not amenable to a test of falsification, as a formal hypothesis should be; furthermore, it does not seem to be likely, given the much higher prevalence of unknown coronaviruses and recombination events in the wild. Moreover, Segreto's and Deigin's hypothesis is significantly weakened by a recent analysis of S‐protein gene divergence suggesting that the most likely explanation for SARS‐CoV‐2 origin doesn't require recombination at all, neither in nature, nor in the lab.[ 3 ]
CONFLICT OF INTEREST
Alexander Panchin and Alexander Tyshkovskiy do not have any conflicts of interest.
ACKNOWLEDGMENTS
Comparative genomics analysis was supported by the Russian Science Foundation grant RSF 20‐14‐00178 to A.T. Sequence analysis of the furin cleavage site was supported by the Russian Foundation for Basic Research grant RFBR 18‐29‐13014 mk to A.Y.P.
Tyshkovskiy, A. , Panchin, AY. (2021). There is no evidence of SARS‐CoV‐2 laboratory origin: Response to Segreto and Deigin (DOI: 10.1002/bies.202000240). BioEssays. 43:e2000325. 10.1002/bies.202000325
DATA AVAILABILITY STATEMENT
All data used is in this study[ 7 ] are publicly available. The SARS‐CoV‐2, Pangolin coronavirus isolate MP789 and Bat coronavirus RaTG13 genomes are available in GenBank and RefSeq with the following identifiers: NC_045512, MT121216 and MN996532 respectively. The relative frequencies of substitutions distinguishing human‐adapted coronaviruses from their bat relatives were calculated based on the data from.
REFERENCES
- 1. Segreto, R. , & Deigin, Y. (2020). The genetic structure of SARS‐CoV‐2 does not rule out a laboratory origin. BioEssays, 2000240. 10.1002/bies.202000240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Zhou, P. , Yang, X.‐L. , Wang, X.‐G. , Hu, B. , Zhang, L. , Zhang, W. , Si, H.‐R. , Zhu, Y. , Li, B. , Huang, C.‐L. , Chen, H.‐D. , Chen, J. , Luo, Y. , Guo, H. , Jiang, R.‐D. , Liu, M.‐Q. , Chen, Y. , Shen, X.‐R. , Wang, X. , …, Shi, Z.‐L. (2020). A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature, 579(7798), 270–273. 10.1038/s41586-020-2012-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Boni, M. F. , Lemey, P. , Jiang, X. , Lam, T. T.‐.Y. , Perry, B. W. , Castoe, T. A. , Rambaut, A. , & Robertson, D. L. (2020). Evolutionary origins of the SARS‐CoV‐2 sarbecovirus lineage responsible for the COVID‐19 pandemic. Nature Microbiology, 5, 1408–1417. 10.1038/s41564-020-0771-4 [DOI] [PubMed] [Google Scholar]
- 4. Eckerle, L. D. , Becker, M. M. , Halpin, R. A. , Li, K. , Venter, E. , Lu, X. , Scherbakova, S. , Graham, R. L. , Baric, R. S. , Stockwell, T. B. , Spiro, D. J. , & Denison, M. R. (2010). Infidelity of SARS‐CoV Nsp14‐exonuclease mutant virus replication is revealed by complete genome sequencing. PLoS Pathogens, 6(5), e1000896. 10.1371/journal.ppat.1000896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Roberts, A. , Deming, D. , Paddock, C. D. , Cheng, A. , Yount, B. , Vogel, L. , Herman, B. D. , Sheahan, T. , Heise, M. , Genrich, G. L. , Zaki, S. R. , Baric, R. , & Subbarao, K. (2007). A mouse‐adapted SARS‐coronavirus causes disease and mortality in BALB/c mice. PLoS Pathogens, 3(1), e5. 10.1371/journal.ppat.0030005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cuevas, J. M. , González‐Candelas, F. , Moya, A. , & Sanjuán, R. (2009). Effect of Ribavirin on the mutation rate and spectrum of Hepatitis C virus in vivo. Journal of Virology, 83(11), 5760–5764. 10.1128/jvi.00201-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Panchin, A. Y. , & Panchin, Y. V. (2020). Excessive G‐U transversions in novel allele variants in SARS‐CoV‐2 genomes. PeerJ, 8(e9648), e9648. 10.7717/peerj.9648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Schickli, J. H. , Thackray, L. B. , Sawicki, S. G. , & Holmes, K. V. (2004). The N‐terminal region of the murine coronavirus spike glycoprotein is associated with the extended host range of viruses from persistently infected murine cells. Journal of Virology, 78(17), 9073–9083. 10.1128/jvi.78.17.9073-9083.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zhang, T. , Wu, Q. , & Zhang, Z. (2020). Probable pangolin origin of SARS‐CoV‐2 associated with the COVID‐19 outbreak. Current Biology, 30, 1346–1351.e2. 10.1016/j.cub.2020.03.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Liu, P. , Chen, W. , & Chen, J.‐P. (2019). Viral metagenomics revealed sendai virus and coronavirus infection of malayan pangolins (Manis javanica). Viruses, 11(11), 979. 10.3390/v11110979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Li, P. , Guo, R. , Liu, Y. , Zhang, Y. , Hu, J. , Ou, X. , Mi, D. , Chen, T. , Mu, Z. , Han, Y. , Chen, Z. , Cui, Z. , Zhang, L. , Wang, X. , Wu, Z. , Wang, J. , Jin, Q. , & Qian, Z. (2021). The Rhinolophus affinis bat ACE2 and multiple animal orthologs are functional receptors for bat coronavirus RaTG13 and SARS‐CoV‐2. Science Bulletin. 10.1016/j.scib.2021.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hu, B. , Zeng, L.‐P. , Yang, X.‐L. , Ge, X.‐Y. , Zhang, W. , Li, B. , Xie, J.‐Z. , Shen, X.‐R. , Zhang, Y.‐Z. , Wang, N. , Luo, D.‐S. , Zheng, X.‐S. , Wang, M.‐N. , Daszak, P. , Wang, L.‐F. , Cui, J. , & Shi, Z.‐L. (2017). Discovery of a rich gene pool of bat SARS‐related coronaviruses provides new insights into the origin of SARS coronavirus. PLoS Pathogens, 13(11), 1–27. 10.1371/journal.ppat.1006698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Vincze, T. (2003). NEBcutter: A program to cleave DNA with restriction enzymes. Nucleic Acids Research, 31(13), 3688–3691. 10.1093/nar/gkg526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Perdue, M. L. , García, M. , Senne, D. , & Fraire, M. (1997). Virulence‐associated sequence duplication at the hemagglutinin cleavage site of avian influenza viruses. Virus Research, 49, 173–186. 10.1016/S0168-1702(97)01468-8 [DOI] [PubMed] [Google Scholar]
- 15. Steinhauer, D. A. (1999). Role of hemagglutinin cleavage for the pathogenicity of Influenza virus. Virology, 258, 1–20. [DOI] [PubMed] [Google Scholar]
- 16. Carroll, D. , Daszak, P. , Wolfe, N. D. , Gao, G. F. , Morel, C. M. , Morzaria, S. , Pablos‐Méndez, A. , Tomori, O. , & Mazet, J. A. K. (2018). The Global Virome Project. Science, 359(6378), 872–874. 10.1126/science.aap7463 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data used is in this study[ 7 ] are publicly available. The SARS‐CoV‐2, Pangolin coronavirus isolate MP789 and Bat coronavirus RaTG13 genomes are available in GenBank and RefSeq with the following identifiers: NC_045512, MT121216 and MN996532 respectively. The relative frequencies of substitutions distinguishing human‐adapted coronaviruses from their bat relatives were calculated based on the data from.