

Abstract
The COVID‐19 pandemic, induced by severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2), has caused great impact on the global economy and people's daily life. In the clinic, most patients with COVID‐19 show none or mild symptoms, while approximately 20% of them develop severe pneumonia, multiple organ failure, or septic shock due to infection‐induced cytokine release syndrome (the so‐called “cytokine storm”). Neutralizing antibodies targeting inflammatory cytokines may potentially curb immunopathology caused by COVID‐19; however, the complexity of cytokine interactions and the multiplicity of cytokine targets make attenuating the cytokine storm challenging. Nonspecific in vivo biodistribution and dose‐limiting side effects further limit the broad application of those free antibodies. Recent advances in biomaterials and nanotechnology have offered many promising opportunities for infectious and inflammatory diseases. Here, potential mechanisms of COVID‐19 cytokine storm are first discussed, and relevant therapeutic strategies and ongoing clinical trials are then reviewed. Furthermore, recent research involving emerging biomaterials for improving antibody‐based and broad‐spectrum cytokine neutralization is summarized. It is anticipated that this work will provide insights on the development of novel therapeutics toward efficacious management of COVID‐19 cytokine storm and other inflammatory diseases.
Keywords: biomaterials, COVID‐19, cytokine release syndrome, cytokine storm, nanoparticles, SARS‐CoV‐2
Enabled by recent advances in materials science and nanotechnology, emerging biomaterials hold great potential to provide better solutions for COVID‐19 cytokine storm and other inflammatory diseases. By reviewing the state‐of‐the‐art cytokine neutralization systems and highlighting the promising technology development, this work intends to spark further research and development activity in this critical research area.
1. Introduction
The COVID‐19 pandemic, induced by a novel severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2; also known as 2019‐nCoV),[ 1 ] has caused more than 83 million infections and 1.8 million deaths globally as of January 1, 2021.[ 2 ] In the past 200 years, multiple epidemics induced by emerging viruses, such as SARS‐CoV, Zika virus, Ebola virus, and recently SARS‐CoV‐2, have posed unprecedented threats to global public health.[ 3 ] Although several vaccine candidates have been approved by the U.S. Food and Drug Administration (FDA) for emergency prevention of SARS‐CoV‐2 infection, so far there is still absence of effective therapeutic options for patients with COVID‐19.[ 4 ] Thus, it is of paramount importance to develop therapeutic approaches for COVID‐19 and other potential pandemics.
Similar to SARS‐CoV infection, SARS‐CoV‐2 relies on spike proteins and angiotensin‐converting enzyme 2 (ACE2) receptors for cell infection.[ 5 , 6 , 7 ] Once the virus enters human body, macrophages and monocytes secrete abundant proinflammatory cytokines, promoting pathogen elimination and tissue recovery. However, an exaggerated release of cytokines, known as a “cytokine storm” (or “cytokine release syndrome”), may worsen inflammatory status and result in immune‐system‐initiated organ damage.[ 8 ] Clinically, the majority of patients with COVID‐19 appear asymptomatic or mildly symptomatic, while ≈20% of COVID‐19 cases develop severe pneumonia, multiple organ failure, and septic shock due to the cytokine storm.[ 9 ] Therefore, in addition to therapies that aim to block viral entry, developing treatment modalities that aim to attenuate aberrant immune responses to viral infection has become one of the major challenges for successful management of COVID‐19.[ 10 ]
Despite progress, effective therapeutic strategies aiming to attenuate cytokine storm in severe COVID‐19 patients are not yet clinically available.[ 8 ] This is partly due to insufficient understanding of the pathological process of the cytokine storm.[ 10 , 11 ] Although various neutralizing antibodies targeting different cytokines are being actively tested, certain limitations in these antibodies need to be seriously considered.[ 12 ] One limiting factor is the dose‐dependent side effects related to the nonspecific in vivo distribution.[ 13 ] More importantly, due to the complexity of cytokine interactions and the multiplicity of cytokine targets, interventions targeting single cytokines may not be sufficient to ease the overwhelming inflammatory response.[ 14 ]
In light of recent advances in materials science and nanotechnology,[ 15 , 16 , 17 , 18 , 19 ] herein, we discuss the evolution of emerging biomaterials for inflammatory diseases, with a specific focus on COVID‐19 cytokine storm. First, potential mechanisms of the COVID‐19 cytokine storm are discussed to design therapeutic strategies, followed by an overview of ongoing and prospective clinical trials that aim to ameliorate aberrant inflammatory responses in patients with severe COVID‐19. Finally, the implications of emerging biomaterials for improving antibody‐based and broad‐spectrum cytokine neutralization is highlighted (Figure 1 ). In short, understanding the structure–activity relationships of emerging biomaterials would result in the development of new therapeutics that effectively address the COVID‐19 cytokine storm and other inflammatory diseases.
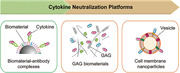
Figure 1.

Therapeutic platforms that capture and neutralize cytokines. A) Single cytokine neutralization platforms, such as neutralizing antibodies and biomaterial–antibody complexes for specific neutralization of one cytokine. B) Multiple cytokine neutralization platforms such as glycosaminoglycan (GAG) biomaterials and cell membrane nanoparticles for simultaneous neutralization of multiple cytokines.
2. Potential Mechanisms of COVID‐19 Cytokine Storm
2.1. The COVID‐19 Cytokine Storm
Inflammatory cytokines, a large group of proteins or peptides secreted by immune cells, play vital roles in inflammatory processes by promoting pathogen recognition, immune cell recruitment, threat elimination, and systemic homeostasis.[ 20 ] Inflammatory cytokines classified into chemokines, interleukins, and growth factors, among which the tumor necrosis factor and interleukin families have been well investigated for their roles in multiple inflammatory responses.[ 21 , 22 ] For example, tumor necrosis factor‐α (TNF‐α) and interleukin‐1β (IL‐1β) increase vascular permeability subsequently easing leukocyte infiltration, while interleukin‐6 (IL‐6) can elicit complement expression, which are essential for innate immune response.[ 21 , 23 ] Yet investigating individual cytokines and their corresponding receptor functions in certain inflammatory conditions remains challenging. Previous studies have illustrated that some chemokine receptors are able to bind multiple ligands, demonstrating considerable redundancy within the chemokine network.[ 24 ]
The term “cytokine storm,” first proposed to characterize the uncontrollable inflammatory states in graft versus host disease (GvHD) in 1993, has now been broadly used to define a situation in which inflammatory cytokines are overly secreted in response to certain diseases.[ 25 ] Cytokine release is a tightly controlled immune process in response to pathogen exposure, with only a few pathogens, such as SARS‐CoV‐2, which can routinely initiate a cytokine storm and consequently lead to organ damage and even death.[ 26 ] A variety of cell types and factors are involved in the initiation and progression of cytokine storm. At the early phase of SARS‐CoV‐2 infection, rapid viral replication triggers a delayed secretion of antiviral interferons (IFNs), meanwhile promoting the release of proinflammatory cytokines, including IL‐6, IL‐1β, and TNF‐α.[ 27 ] Stimulated by IFN signaling, lung macrophages secrete certain chemokines, which recruit other inflammatory immune cells, such as neutrophils, monocytes, and dendritic cells, to the infection sites.[ 28 ] The activated immune cells secret more cytokines, further worsening injury to the lung.[ 26 ]
COVID‐19 provides a compelling example of cytokine storm.[ 8 ] In response to SARS‐CoV‐2 infection, various cytokines, such as IL‐6, TNF‐α, IL‐8, IL‐1, IL‐21, and monocyte chemoattractant protein‐1 (MCP‐1), are upregulated in macrophages and/or monocytes to promote pathogen elimination and tissue repair.[ 9 ] Prolonged high level of cytokines, characterized as the cytokine storm, may exacerbate systemic immune disorder.[ 29 ] Patients with COVID‐19 may develop severe complications due to cytokine storm,[ 30 ] thus immunosuppression has been proposed as an essential means to manage severe COVID‐19 cases.[ 8 ]
2.2. Macrophages in COVID‐19 Cytokine Storm
Macrophages are heterogeneous immune cells that link innate and adaptive immune responses, and are able to initiate rapid protection against pathogens.[ 31 , 32 , 33 , 34 ] In response to infection, monocyte‐derived macrophages are recruited from blood circulation to the site of infection in order to orchestrate prompt immune response.[ 34 , 35 ] Equipped with pattern recognition receptors (PRRs), macrophages can identify pathogen‐associated molecular patterns (PAMPs) and conduct phagocytosis and phagolysosome‐mediated digestion of pathogens, therefore facilitate proinflammatory response.[ 36 , 37 ]
Phagocytosis triggers type I interferon response and release of certain cytokines, including IL‐6, TNF‐α, IL‐1, and IL‐12 (Figure 2A).[ 38 ] These cytokines have local effects that promote vascular permeability and lymphocyte recruitment to the site of infection, and systemic effects that initiate secretion of acute‐phase proteins.[ 10 ] Moderate cytokine release is beneficial for the body, however, potentially harmful if uncontrolled.[ 8 ] For example, an excessive level of IL‐6 is associated with respiratory failure and other adverse outcomes in COVID‐19 patients.[ 9 ]
Figure 2.
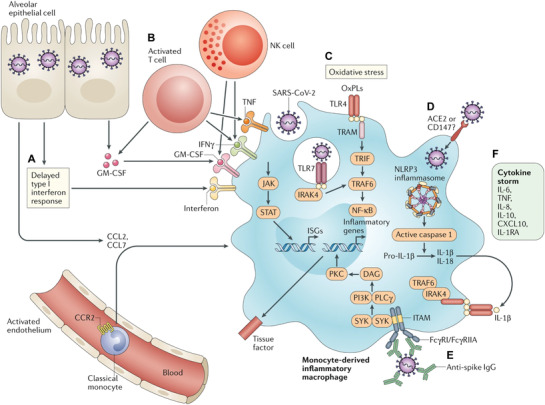
Possible pathways contributing to the COVID‐19 cytokine storm. A) A delayed release of type I IFN promotes the sensing of microbial threats and inhibits the secretion of monocyte chemoattractants by alveolar epithelial cells, resulting in continuous recruitment of circulating monocytes into lungs and differentiation of monocytes into proinflammatory macrophages. B) Activated T cells and NK cells further accelerate sustained recruitment and activation of macrophages via the secretion of GM‐CSF, TNF, and IFN‐γ. C) OxPLs accumulate in infected lungs and activate macrophages. D) Type I IFN may induce the expression of viral entry receptors, enabling SARS‐CoV‐2 to obtain the access to enter into the macrophage cytoplasm and activate the NLRP3 inflammasome, resulting in sustained production of IL‐1β and IL‐18. E). The engagement of FcγRs by antispike protein IgG complexes contributes to sustained activation of proinflammatory macrophages. F) Activated macrophages contribute to the COVID‐19 cytokine storm by secreting elevated levels of cytokines. Reproduced with permission.[ 39 ] Copyright 2020, Springer Nature.
IL‐6, secreted mainly by monocytes and macrophages, activates neutrophils and T cells via the signal transducer and activator of transcription 3 (STAT3) and Janus kinase (JAK) signaling pathways.[ 39 , 40 ] Activated T cells and natural killer (NK) cells further facilitate macrophage activation through secretion of specific cytokines including granulocyte–macrophage colony‐stimulating factor (GM‐CSF), TNF‐α, and IFN‐γ (Figure 2B).[ 11 ] Moreover, oxidized phospholipids (OxPLs) produced in response to oxidative stress in the lungs of SARS‐CoV‐2 patients, may also facilitate local macrophage activation[ 41 ] and stimulate endothelial cells to recruit more monocytes and boost inflammatory response.[ 42 , 43 ] In an acute lung injury (ALI) model, OxPLs trigger recruitment and activation of macrophages (Figure 2C).[ 41 ] An excessive activation of monocytes and endothelial cells by OxPLs may cause thrombotic complications, a life‐threatening event for patients with metabolic and cardiovascular comorbidities.[ 44 ]
Immunostaining of post‐mortem tissues of COVID‐19 patients has revealed that CD169+ macrophages in the spleen and lymph nodes contain viral nucleoproteins and ACE2 receptors, through which viruses enter cells.[ 45 ] Recent studies suggest that the expression of ACE2 on macrophages can be upregulated by proinflammatory factors, such as IFNs (Figure 2D).[ 6 , 39 ] In addition, other receptors, such as CD147 may be also involved in virus entry.[ 46 ] Data from virus‐induced ALI models demonstrated that sustained activation of macrophages and infiltrating monocytes could be driven by the nucleotide‐binding domain, the leucine‐rich‐containing family, pyrin domain‐containing‐3 (NLRP3) inflammasomes, and antispike protein‐IgG complexes engulfed through macrophage FC‐receptors (Figure 2E).[ 47 , 48 ] These activated macrophages lead to the COVID‐19 cytokine storm by secreting excessive inflammatory cytokines (Figure 2F).[ 11 , 35 ]
An immune response to SARS‐CoV‐2 infection includes three crucial phases.[ 39 ] In the first phase, early innate immunity activation is triggered by a powerful IFN response, which is crucial for control of viral replication. In the second phase, a delayed IFN response may result in uncontrolled tissue damage. Ultimately, it may progress to the third phase, a devastating hyperinflammation, characterized by macrophage hyperactivation and agglutination, potentially followed by abnormal tissue repair and fibrosis. Specific mechanisms of the cytokine storm following inflammatory responses in severe COVID‐19 patients need further investigation. It is of great importance to elucidate signaling pathways involved in different phases of COVID‐19 immune response and the interactions between these pathways.[ 10 ] In order to maximize therapeutic efficacy, future studies should aim to figure out not only the corresponding therapeutic strategies but also the optimal timing for those strategies.
3. Potential Therapeutic Targets and Challenges
Although potential mechanisms of the COVID‐19 cytokine storm remain partially understood, clinical trials targeting inflammatory cytokines are in progress (Table 1 ).[ 39 , 49 ] Clinical trials blocking IL‐6 have been initiated all over the world and certain clinical outcomes have been reported in a subgroup of COVID‐19 patients. A study reported that 15 of 20 patients in China benefited significantly from IL‐6 blockade, based on which the U.S. FDA approved the IL‐6 blockade as an emergency treatment for COVID‐19 patients.[ 10 ] Recently, a study from Italy also reported that IL‐6 blockade was effective in 7 out of 21 patients.[ 11 ] Several research groups have also launched clinical trials simultaneously blocking both IL‐1β and IL‐6 in COVID‐19 patients.[ 23 ] In addition, blockade of myeloid‐cell‐derived cytokines, such as GM‐CSF, is promising, and relevant trials have been initiated.[ 50 ] Therapeutic strategies targeting key factors in common inflammatory signaling pathways could significantly suppress excessive inflammatory response. Indeed, several clinical trials exploring the effectiveness of JAK inhibitors in severe COVID‐19 patients are ongoing.[ 50 ]
Table 1.
Possible therapeutic targets and ongoing clinical trials for the cytokine storm in patients with COVID‐19
| Pathway or molecular target a) | Potential roles | Type | Name | Clinical trials reference number b) |
|---|---|---|---|---|
| IL‐6 | Proinflammatory | Anti‐IL‐6 receptor | Tocilizumab |
NCT04320615; NCT04322773; NCT04331795; NCT04331808; NCT04330638; NCT04335071; NCT04333914; NCT04346355 |
| Sarilumab | NCT04315298; NCT04321993; NCT04322773; NCT04341870 | |||
| Anti‐ IL‐6 | Siltuximab | NCT04330638 | ||
| Clazakizumab | NCT04348500; NCT04343989 | |||
| GM‐CSF | Proinflammatory; drives tissue recovery in lungs | Anti‐GM‐CSF | Lenzilumab | NCT04351152 |
| Otilimab | NCT04376684 | |||
| Gimsilumab | NCT04351243 | |||
| TJM2 | NCT04341116 | |||
| Anti‐GM‐CSF receptor | Mavrilimumab | NCT04399980; NCT04397497 | ||
| Recombinant GM‐CSF | Sargramostim | NCT04326920; NCT04400929 | ||
| JAK–STAT | Mediates cytokine regulation | JAK1/JAK2 inhibitors | Baricitinib | NCT04321993; NCT04320277; NCT04340232 |
| Ruxolitinib | NCT04331665; NCT04334044; NCT04348071; NCT04354714 | |||
| JAK1/JAK3 inhibitor | Tofacitinib | NCT04332042 | ||
| IL‐1β | Proinflammatory | IL‐1 receptor antagonist | Anakinra | NCT04324021; NCT04330638; NCT04339712; NCT04341584 |
| CCR5 | Recruits monocytes and T cells | Anti‐CCR5 | Leronlimab | NCT04343651; NCT04347239 |
| IFN‐γ | Proinflammatory | Anti‐IFNγ | Emapalumab | NCT04324021 |
| NLRP3 | Proinflammatory | Anti‐ IL‐1β | Canakinumab | NCT04330638 |
| Complement component C5 | Drives cell death | Anti‐C5 | Eculizumab | NCT04288713 |
IL‐6: interleukin‐6; GM‐CSF: granulocyte–macrophage colony‐stimulating factor; JAK: Janus kinase; STAT: signal transducer and activator of transcription; IL‐1β: interleukin‐1β; CCR5: chemokine receptor‐5; IFN‐γ: interferon‐γ; NLRP3: NACHT, LRR, and PYD domain‐containing protein 3
Data obtained from ClinicalTrials.gov or company public announcements.
This article is being made freely available through PubMed Central as part of the COVID-19 public health emergency response. It can be used for unrestricted research re-use and analysis in any form or by any means with acknowledgement of the original source, for the duration of the public health emergency.
Alternatively, modulation of the release of upstream cytokines represents a promising strategy for managing COVID‐19 cytokine storm.[ 51 ] Recent studies suggest that hyper‐activated macrophages are mostly derived from circulating monocytes rather than tissue‐resident cells,[ 52 , 53 ] thus targeting infiltration of macrophages to the lungs and other affected organs provides a viable treatment option for COVID‐19. Blocking C–C chemokine receptor type 5 (CCR5), which can modulate T cell and monocyte migration, has been actively tested in COVID‐19 patients with mild‐to‐moderate symptoms.[ 39 ] In addition, trials testing the effectiveness of blocking IFN‐γ in COVID‐19 patients suffering from hyperinflammation and respiratory distress have also been launched.[ 54 ]
In general, immunosuppression has been suggested as a promising strategy to attenuate cytokine storm in severe COVID‐19 patients. Utilizing inhibitors of cytokines (e.g., IL‐6, IL‐1β, IL‐17A, IFN‐γ, and TNF‐α) and modulators of innate and adaptive immunity (e.g., CD47, C5, and GM‐CSF) are being actively tested in COVID‐19 patients.[ 4 , 10 , 27 , 39 , 50 ] However, a few limitations remain in these free cytokine‐neutralizing antibodies. One limitation is the dose‐dependent side effects related to the nonspecific biodistribution.[ 11 , 13 ] More importantly, due to the complexity of cytokine interactions and the multiplicity of cytokine targets, immunomodulation of individual cytokines may not be sufficient to suppress systemic inflammatory response.[ 12 ]
Recent advances in biomaterials and nanotechnology have offered many exciting opportunities for treating infectious and inflammatory diseases.[ 15 , 16 ] For instance, neutralizing antibodies conjugated to various biocompatible polymers, scaffolds, and nanoparticles, accumulate better at the sites of infection thereby enhancing cytokine neutralization.[ 12 ] Glycosaminoglycan (GAG)‐based materials and biomimetic cell‐membrane‐based nanoparticles have been developed to mimic intracellular matrix and serve as host cell decoys for broad‐spectrum cytokine neutralization.[ 55 , 56 , 57 ] More importantly, two liposome‐based mRNA vaccines have recently been approved for COVID‐19,[ 58 , 59 , 60 , 61 , 62 ] further highlighting the importance of nanotechnology for the management of infectious diseases.[ 63 ] The following sections will review recent inflammatory disease research involving emerging biomaterials, with a special emphasis on the platforms developed for improving antibody‐based and broad‐spectrum cytokine neutralization.
4. Biomaterial‐Antibody Complexes for Enhanced Cytokine Neutralization
To overcome dose‐limiting side effects of free neutralizing antibodies, biomaterials have been coupled with antibodies to improve their in vivo biodistribution, pharmacokinetics, and possibility to reach the sites where free antibodies cannot reach.[ 64 ] These merits have led to conjugation of drugs to multiple biomaterial platforms, including nanoparticles, polymers, hydrogels, and extracellular vesicles for a series of biomedical applications.[ 12 ]
4.1. Antibody–Polymer Conjugates
Conjugation of antibodies to polymers would be an ideal strategy for the treatment of localized inflammation, thanks to improved molecular weight and reduced diffusion rate of therapeutic antibodies.[ 65 ] High‐molecular‐weight hyaluronic acid (HA) was conjugated with anti‐TNF‐α and anti‐IL‐1β antibodies; the resulting antibody–polymer conjugates were employed for the treatment of burn injury (Figure 3A).[ 66 ] In a rodent model of severe burn injury, HA markedly improved the retention time of free antibodies in the superficial area. Furthermore, the conjugates were efficacious in suppressing acute inflammation and inhibiting secondary necrosis in the burn injury model. There were much fewer infiltrated immune cells in the areas treated with the antibody–polymer conjugates, further suggesting effective remission of the inflammatory status. Notably, the backbones of antibody–polymer conjugates have significant impact on the cytokine–antibody binding affinity. For example, after conjugation to HA or carboxymethylcellulose (CMC), anti‐IL‐1β antibodies preserved similar association kinetics to that of free antibodies. However, the captured cytokines dissociated from CMC conjugates three times faster than from the HA conjugates.[ 67 ] Such dissociation difference occurs due to the conformational alteration of antigen‐binding sites by polysaccharides. Apart from the polymer backbone, the dimension of cytokine targets has profound effect on the binding affinity of antibodies. Conjugation of HA or CMC or both to anti‐TNF‐α antibodies results in decreased TNF‐α adsorption and desorption.[ 65 ] Compared with IL‐1β, TNF‐α has a much larger molecular weight, limiting its binding kinetics.
Figure 3.
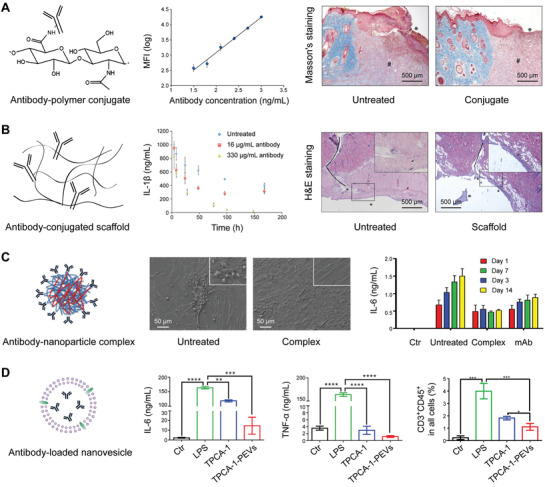
Biomaterial–antibody complexes for enhanced cytokine neutralization and inflammation attenuation. A) Antibody–hyaluronic acid (HA) conjugate maintains superior binding to antibodies and suppresses burn progression in mice. Two panels to left: Adapted with permission.[ 67 ] Copyright 2010, American Chemical Society. Two panels to the right: Reproduced with permission.[ 69 ] Copyright 2012, The Wound Healing Society, published by Wiley. B) Antibody‐conjugated scaffold downregulates IL‐1β level and relieves skin injury in rats. Reproduced with permission.[ 72 ] Copyright 2010, Elsevier. C) Antibody‐conjugated nanoparticles protect cells from morphology shrinkage and inhibit evaluated IL‐6 secretion. Reproduced with permission.[ 73 ] Copyright 2018, American Chemical Society. D) Antibody‐loaded platelet extracellular vesicles (TPCA‐1‐PEVs) reduce the secretion of proinflammatory cytokines and inhibit the proportion of T cells in the lung. Reproduced with permission.[ 13 ] Copyright 2020, Elsevier.
4.2. Antibody‐Conjugated Scaffolds
Another strategy to regulate local inflammation while minimizing the side effects of systemically administered antibodies is conjugation of antibodies to the hydrogels. One classical application of these antibody–hydrogel conjugates is for burn injury, in which local inflammatory cytokines elicit damage progression via complicated cascade reactions (Figure 3B).[ 68 ] In a rat burn injury model, hydrogel‐conjugated anti‐TNF‐α antibodies effectively inhibited the progression of tissue necrosis.[ 69 ] In the same model, the free antibodies hindered macrophage infiltration at the periphery rather than at the surface of wound; in contrast, conjugated antibodies were able to inhibit macrophage infiltration both at the wound surface and periphery.[ 70 ] As demonstrated by local antibody levels, hydrogels significantly improved the residence time of antibodies.[ 71 ] In brief, these results indicate significant benefits of antibody–hydrogel conjugates for the treatment of burn wounds. To investigate the effects of hydrogel crosslinking on inflammation management, HA hydrogels were conjugated with anti‐TNF‐α and anti‐IL‐1β antibodies, and the resulting conjugates were used for local treatment of burn injury.[ 72 ] Intriguingly, the hydrogel conjugates could effectively adsorb and neutralize specific cytokines in vitro; however, they could not suppress the systemic inflammation in vivo. This phenomenon was attributed to the high crosslinking density of HA hydrogels, which hindered cytokine diffusion. Thus, when developing such antibody–hydrogel conjugates, the crosslinking density needs to be considered carefully to preserve gel‐like functions while guaranteeing cytokine mobility for potent cytokine neutralization. Meanwhile, maximizing the interaction time between cytokines and antibodies presents a promising strategy in modulating the overall inflammatory status in acute tissue injury.
4.3. Antibody–Nanoparticle Complexes
Antibodies could also be conjugated to nanoparticles to improve their properties, including enhanced circulation, specific targeting, and prolonged retention after systemic administration. For example, anti‐IL‐6 antibodies were conjugated to chitosan and HA nanoparticles to neutralize cytokines in a model of arthritic joints (Figure 3C).[ 73 ] In this study, nanoparticle surface amine groups reacted with the carboxylic acid groups of anti‐IL‐6 antibodies. Compared to anti‐IL‐6 antibodies, the antibody–nanoparticle conjugates showed better inhibition of macrophage activation for a longer time. The improvement could be attributed to antibody immobilization on the surface of nanoparticles, which decreased the antibody degradation. In another study based on a rat model of acute temporal lobe epilepsy, anti‐IL‐1β antibodies were conjugated to magnetic nanoparticles (MNs) to enhance cytokine neutralization and therapeutic outcomes.[ 74 ] The resulting conjugates not only promoted the neuroprotective effects by blocking IL‐1β but also tied the MNs to the neurons and astrocytes, resulting in better magnetic resonance imaging contrast than unconjugated nanoparticles.
4.4. Drug‐Loaded Extracellular Vesicles
In addition, immunosuppressive drugs can also be loaded into nanovesicles derived from cells for prolonged blood circulation and specific targeting. Inspired by the inflammation targeting property of platelets, their extracellular vesicles (PEVs) were engineered for targeted delivery of antipneumonia drugs (Figure 3D).[ 13 ] In an ALI mouse model, activated platelet‐derived PEVs could selectively accumulate in pneumonia‐affected tissue. PEVs loaded with antipneumonia drug [5‐(p‐fluorophenyl)‐2‐ureido]thiophene‐3‐carboxamide (TPCA‐1), evidently improved therapeutic efficacy by reducing local cytokine levels and preventing the infiltration of immune cells. Moreover, PEVs could serve as a universal platform for inflammation targeting in multiple disease models, such as skin wounds, rheumatoid arthritis, and atherosclerosis.[ 13 ]
5. Biomaterials for Broad‐Spectrum Cytokine Neutralization
In contrast to simplex neutralization agents, multiplex neutralization platforms can neutralize different cytokines concurrently, addressing the issue of multiple cytokine release in different diseases.[ 12 ] In this context, two emerging strategies attract the most attention, including GAG‐based biomaterials and cell‐membrane nanoparticles.[ 55 , 56 , 57 ] The former imitates the intracellular matrix for dynamic cytokine neutralization, while the latter utilizes cell membranes as decoys.
5.1. Hydrogel Scaffolds
Extracellular matrix GAGs, including heparin and heparan sulfate, have been proven to bind multiple inflammatory cytokines primarily through electrostatic interactions between electronegative sulfate residues on the GAGs and electropositive amino acid groups on the cytokines.[ 75 ] By adjusting the composition, concentration, and sulfation degree of GAGs, the extracellular matrix can effectively regulate the transport and bioactivity of cytokines. Inspired by such dynamic binding, GAGs have been applied to construction of biomaterials for cytokine scavenging.[ 76 ] Given their unique capabilities, GAGs have become promising biomaterials for regulating complicated binding events and scavenging various cytokines.[ 77 ]
GAG hydrogels have been further developed to capture multiple cytokines and modulate the inflammatory status in chronic wounds. In one study, hydrogels constructed from multiple desulfated heparin derivatives and star‐shaped poly(ethylene glycol) could effectively attenuate the chemoattractant activities of IL‐8 and MCP‐1 in an excisional wound model (Figure 4A).[ 55 ] This detention effect significantly reduced the mobility of inflammatory cells in the wound sites. Mechanistic studies showed that no heparin‐affine cytokines, such as IL‐6, IL‐1β, and TNF‐α, were trapped by the GAG hydrogels, whereas chemotactic factors such as macrophage inflammatory protein 1α (MIP‐1α) and MIP‐1β interacted and bonded to these hydrogels. Furthermore, decreased mobility of inflammatory cells accompanied the decline of systemic expressions of certain cytokines, such as IL‐1β, TNF‐α, and MCP‐1. Following the decrease of matrix sulfation, the capacity of hydrogels to trap IL‐8 and MCP‐1 was reduced.[ 78 ] In general, the hydrogel promoted wound healing by inhibiting specific inflammatory signaling and promoting tissue vascularization and epithelialization.
Figure 4.
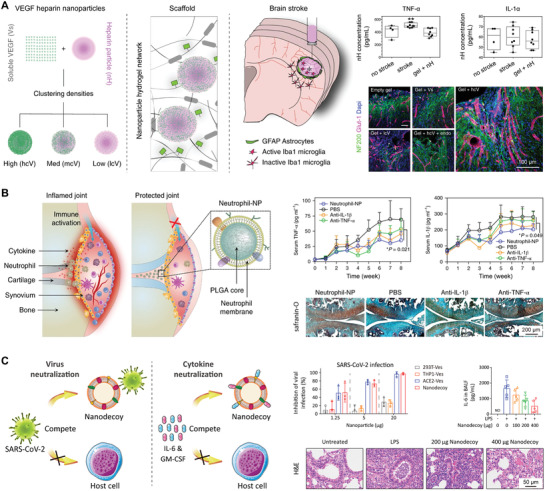
Biomaterials adsorbing broad‐spectrum cytokines for suppression of the cytokine storm. A) Hydrogel scaffold, which contains vascular endothelial growth factor (VEGF) and heparin nanoparticles, adsorbs inflammatory cytokines and repairs poststroke brain damage. Left three panels: Reproduced with permission.[ 82 ] Copyright 2018, The Authors, published by Springer Nature. Four panels to the right: Reproduced with permission.[ 81 ] Copyright 2018, The Authors, published by Springer Nature. B) Neutrophil‐membrane‐coated nanoparticles, which display abundant cytokine receptors on the surface, can neutralize inflammatory cytokines, alleviate synovial inflammation, and provide effective protection against joint damage. Reproduced with permission.[ 93 ] Copyright 2019, The Authors, published by Springer Nature. C) Hybrid genetically edited cell membrane nanodecoys, displaying abundant ACE2 receptors and cytokine receptors, effectively inhibit the SARS‐CoV‐2 infection, neutralize inflammatory cytokines, and suppress lung injury. Reproduced under the terms of the CC‐BY Creative Commons Attribution 4.0 International license (https://creativecommons.org/licenses/by/4.0).[ 57 ] Copyright 2020, The Authors, published by National Academy of Sciences.
Multiple nanoparticles employing GAGs as blockades have been proposed for neutralization of broad‐spectrum cytokines. For example, self‐assembled nanoparticles, made of d‐erythro‐sphingosine‐conjugated heparins, compared with free heparin, exerted much stronger suppression of the release of inflammatory cytokines, including IL‐6, TNF‐α, and IL‐1β, from stimulated macrophages.[ 79 ] These early results encouraged rapid development of nanoparticle conjugates with multiple GAG derivatives, such as HA, chondroitin sulfate (CS), and low‐molecular‐weight heparin. Furthermore, investigations of the relationship between anti‐inflammatory activity and conjugate structure suggested a critical role of the sulfation levels in regulating activity of such antibody–nanoparticle conjugates.[ 80 ] In another study, heparin‐embedded HA hydrogel was used to salvage brain tissue after the onset of stroke.[ 81 , 82 ] The loading of vascular endothelial growth factor (VEGF) in the HA hydrogel reduced levels of TNF‐α in damaged areas of the brain; meanwhile heparin nanoparticles captured cytokines, reduced scar formation, and eventually facilitated tissue repair after the stroke. Moreover, chitosan oligosaccharide was conjugated with inversely charged heparin to formulate uniform polymeric nanoparticles,[ 83 ] and these nanoparticles captured and neutralized cytokines including VEGF and stromal‐cell‐derived factor 1α (SDF‐1α). In another study, polylysine particles were prepared by first coating a polylysine shell onto negatively charged poly(lactic acid)s, followed by depositing heparins on positively charged polylysine.[ 84 ] The resulting particles were further modified with CCR5 fragments, which were responsible for inhibiting leukocyte adhesion mediated by CCR5 ligand. The combination of CCR5 fragments and heparin inhibited adhesion between monocytes and endothelial cells, a process crucial for immune cell infiltration to the inflamed tissue.
5.2. Cell‐Membrane‐Based Nanoparticles
Inspired by abundant biological functions of cellular vesicles, the strategy of wrapping natural cellular vesicle shells onto synthetic nanoparticle cores endowed these nanoparticles with cell‐like functions and extended the applications of nanoparticles.[ 32 , 85 , 86 , 87 , 88 , 89 ] Among these emerging applications, mimicking natural cells to interact with inflammatory cytokines for cytokine neutralization has attracted the most attention.[ 56 , 57 , 90 ] By displaying certain antigens inherited from the source cells, the cell‐membrane‐coated nanoparticles trapped and neutralized cytokines without identifying specific targets.[ 88 , 91 ] More importantly, these cell‐like nanoparticles efficiently trapped cytokines by precisely displaying the diversity and complexity of cytokine receptors.[ 92 ] Given above advantages, these cell‐like particles have emerged as a promising platform for multiplex cytokine neutralization.
Macrophage‐membrane‐capped polymer nanoparticles (known as “MM‐NPs”) were fabricated for sepsis treatment.[ 56 ] MM‐NPs served as decoys to bind cytokines, thus endowing them with the ability to suppress downstream cytokine storm that largely claims sepsis‐caused lethality. Moreover, MM‐NPs displayed complete antigen maps of the source macrophages, preserving the unique macrophage ability to adsorb endotoxins through surface CD14 receptor. These two features of MM‐NPs enable rapid and effective response to uncontrolled inflammation, offering a promising therapeutic intervention for sepsis management. In vitro investigations have demonstrated that MM‐NPs adsorb cytokines along with endotoxins, which otherwise would elicit the sepsis cascade reactions. In a bacteremia mouse model, treatment with MM‐NPs effectively decreased cytokine levels, inhibited bacterial infection, and improved survival of infected mice. The top‐down strategy applied for the preparation of MM‐NPs effectively inherits endotoxin‐binding antigens from the source cell membrane that are otherwise difficult to identify and purify. Furthermore, the surface‐to‐volume ratio of nanoparticles conspicuously increased after the cell‐membrane coating, which was crucial for effective endotoxin neutralization.
In another study, neutrophil‐membrane‐camouflaged particles (known as “Neu‐NPs”) were also developed as a promising therapy for rheumatoid arthritis (RA) (Figure 4B).[ 93 ] Inflammatory injury in RA is regulated by infiltration of inflammatory cells into the joint synovial area.[ 94 ] Among them, neutrophils play a critical role in activating and maintaining the inflammatory status during RA development. By replicating a neutrophil antigenic exterior, Neu‐NPs mimicked the biological functions of neutrophils. Neu‐NPs effectively neutralized multiple types of cytokines, including TNF‐α and IL‐1β, which stimulate and recruit more neutrophils leading to exacerbation of RA. By broad‐spectrum neutralization of cytokines, Neu‐NPs significantly attenuated synovial inflammation and chondrocyte apoptosis. Moreover, Neu‐NPs mimicked the interaction between chondrocytes and neutrophils, thus promoting permeation of Neu‐NPs into the mesochondrium for chondrocyte targeting. In two mouse models of arthritis, Neu‐NPs showed therapeutic effect by suppressing overall inflammatory response and reducing joint damage. The promising results of employing Neu‐NPs for RA treatment indicate that coating nanoparticles with cell membranes could guide the way toward broad‐spectrum cytokine neutralization for inflammatory disorders.
To inhibit SARS‐CoV‐2 infection and suppress downstream COVID‐19 cytokine storm, cellular‐vesicle‐based nanodecoys (known as “COVID‐NDs”) were also developed by merging cellular vesicles derived from monocytes and genetically engineered cells that express ACE2 receptors (Figure 4C).[ 57 ] The COVID‐NDs, which have an antigen map equivalent to that of the source cells, could compete with host cells for viral adsorption and thus protect host cells against the SARS‐CoV‐2 infection. Moreover, depending on surface cytokine receptors, the COVID‐NDs effectively adsorbed and neutralized multiple cytokines, such as IL‐6 and GM‐CSF. In a mouse model of acute pneumonia, the COVID‐NDs significantly suppressed the inflammatory disorder and lung injury. Overall, by taking advantage of viral attachment to host cells, the COVID‐NDs represent a universal neutralization strategy across different viral genus, species, and strains.[ 92 ] The potent two‐step neutralization strategy guarantees effective disruption of viral infection and subsequent inflammatory response, offering a promising theranostic approach for better management of inflammatory and infectious diseases.
6. Summary and Outlook
Due to limited understanding of SARS‐CoV‐2 infection mechanisms and pathological processes, effective attenuation of COVID‐19 cytokine storm is challenging. Although several neutralizing antibodies targeting different cytokines are currently in clinical trials, some limitations in using these antibodies for therapy remain.[ 11 ] Recent advances in materials science and nanotechnology offer many promising opportunities for infectious and inflammatory diseases, including COVID‐19.[ 15 , 18 ] Herein, we first discussed potential mechanisms behind the COVID‐19 cytokine storm and then summarized potential therapeutic strategies and relevant ongoing clinical trials. Finally, the implication of utilizing emerging biomaterials to promote antibody‐based and broad‐spectrum cytokine neutralization was reviewed. In particular, we highlighted three strategies to functionalize synthetic biomaterials for simple, safe, and effective neutralization of inflammatory cytokines: 1) conjugation of antibodies to biomaterials, 2) integration of GAG blockades into biomaterials, and 3) adoption of cell membranes as cytokine decoys. Significant progress in these fields provides many promising opportunities to develop cutting‐edge technologies for inflammatory and infectious diseases, especially to address COVID‐19 cytokine storm.
Despite this progress, drug discovery for inflammatory disorders is still a significant challenge due to limited understanding of specific inflammatory responses in different pathologies.[ 39 , 95 ] For example, the complexity of cytokine network makes neutralization of one or even multiple cytokines insufficient to suppress overall inflammatory response.[ 22 ] Meanwhile, owing to the complexity of feedback pathways, regulation of one single inflammatory signaling pathway might trigger downstream compensatory immune responses.[ 26 ] The complicated inflammatory response network requires balancing the risk‐to‐benefit relationship of these anti‐inflammatory drugs.[ 14 ] In this context, new approaches have emerged to promote biomaterial‐based cytokine detention and neutralization.[ 12 ] For instance, various nanoparticles have been functionalized with high affinity to specific molecular targets, providing alternative options to the biological ligands, such as antibodies and proteins.[ 32 , 33 ] These engineered nanoparticles have been employed to detain and neutralize bacterial toxins, animal venoms, and inflammatory cytokines.[ 56 , 57 , 90 ] Other high‐affinity molecules, such as aptamers, have also been conjugated onto various biomaterials as potential alternatives to free antibodies for cytokine neutralization.[ 96 , 97 ] Collectively, integrating biomaterials for cytokine detention and neutralization represents a universal and promising strategy to address COVID‐19 cytokine storm and other inflammatory disorders.
Other advances in materials science also promise better intervention of inflammatory and infectious diseases.[ 15 ] Considering that neutralizing antibodies have low target efficiency and cause dose‐dependent side effects,[ 12 ] accurate real‐time monitoring of nanoparticles would provide better understanding of in vivo dynamic processes of neutralizing antibodies. Continued development of advanced but also more accessible imaging techniques, such as fluorescence imaging, magnetic resonance imaging (MRI), positron emission tomography (PET), computed tomography (CT), and different combinations of imaging platforms would be of great help.[ 98 ] Moreover, drug development heavily relies on studies in animal models, which cannot entirely recapitulate human pathologies.[ 15 ] Recent microphysiological systems, such as organoid platforms, can provide macroscale models of human tissues featuring the complexity and spatial heterogeneity of human organs.[ 99 ] For example, bat and human intestinal organoids were utilized to confirm the notion that human intestinal tract is a possible transmission route of SARS‐CoV‐2.[ 100 ] To conclude, advances in materials science provide promising opportunities for combating inflammatory and infectious diseases.
Conflict of Interest
The authors declare no conflict of interest.
Acknowledgements
Q.‐F.M. and R.T. contributed equally to this work. The authors would like to thank Dr. Parinaz Fathi at National Institutes of Health (NIH) for her kind help on language editing. This work was supported by the Shenzhen Bay Laboratory Startup Fund (No. 21310071), the National University of Singapore Startup Fund (Nos. R‐180‐000‐017‐133, R‐180‐000‐017‐731, and R‐180‐000‐017‐733), the Singapore Ministry of Health's National Medical Research Council (NMRC/OFYIRG/0081/2018), and the National University of Singapore NanoNASH Program (NUHSRO/2020/002/NanoNash/LOA).
Biographies
Qian‐Fang Meng received her Bachelor's degree in electrical engineering from Wuhan University, China in 2013. She is currently a visiting Ph.D. student in Dr. Lang Rao's Lab at Shenzhen Bay Laboratory (SZBL), China. Her research focuses on the development of smart biomaterials and microdevices for diagnostics and therapeutics of various diseases.

Xiaoyuan Chen received his Ph.D. in chemistry from the University of Idaho (1999). After two postdocs at Syracuse University and Washington University in St. Louis, he started his assistant professorship in 2002 and then moved to Stanford in 2004. He was promoted to associate professor in 2008. He then moved to National Institutes of Health (NIH) in 2009 and became a senior investigator and chief. He is currently Nasrat Muzayyin Professor in the Yong Loo Lin School of Medicine and Faculty of Engineering, National University of Singapore (NUS). His research interests include various forms of theranostics for different types of diseases.

Lang Rao received his Bachelor's and Ph.D. degrees both from Wuhan University, China. After graduation, he started his postdoc carrier at the University of Texas Southwestern Medical Center (UTSW) under the supervision of Dr. Jinming Gao. In the spring of 2019, he moved to National Institutes of Health (NIH) as a research fellow with Dr. Xiaoyuan (Shawn) Chen. In late 2020, he initiated his independent research carrier at Shenzhen Bay Laboratory (SZBL), China. His research aims to create cutting‐edge nanotechnology and exploit them to study and solve complex biological problems that are associated with human diseases.

Meng Q.‐F., Tian R., Long H., Wu X., Lai J., Zharkova O., Wang J.‐W., Chen X., Rao L., Capturing Cytokines with Advanced Materials: A Potential Strategy to Tackle COVID‐19 Cytokine Storm. Adv. Mater. 2021, 33, 2100012. 10.1002/adma.202100012
Contributor Information
Jiong‐Wei Wang, Email: surwang@nus.edu.sg.
Xiaoyuan Chen, Email: chen.shawn@nus.edu.sg.
Lang Rao, Email: lrao@szbl.ac.cn.
References
- 1. Jiang S. B., Shi Z., Shu Y., Song J., Gao G. F., Tan W., Guo D., Lancet 2020, 395, 949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Wang C., Horby P. W., Hayden F. G., Gao G. F., Lancet 2020, 395, 470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Zhu N., Zhang D., Wang W., Li X., Yang B., Song J., Zhao X., Huang B., Shi W., Lu R., Niu P., Zhan F., Ma X., Wang D., Xu W., Wu G., Gao G. F., Tan W., N. Engl. J. Med. 2020, 382, 727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sanders J. M., Monogue M. L., Jodlowski T. Z., Cutrell J. B., JAMA, J. Am. Med. Assoc. 2020, 323, 33. [DOI] [PubMed] [Google Scholar]
- 5. Zhou P., Yang X.‐L., Wang X.‐G., Hu B., Zhang L., Zhang W., Si H.‐R., Zhu Y., Li B., Huang C.‐L., Chen H.‐D., Chen J., Luo Y., Guo H., Jiang R.‐D., Liu M.‐Q., Chen Y., Shen X.‐R., Wang X., Zheng X.‐S., Zhao K., Chen Q.‐J., Deng F., Liu L.‐L., Yan B., Zhan F.‐X., Wang Y.‐Y., Xiao G.‐F., Shi Z.‐L., Nature 2020, 579, 270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hoffmann M., Kleine‐Weber H., Schroeder S., Krüger N., Herrler T., Erichsen S., Schiergens T. S., Herrler G., Wu N.‐H., Nitsche A., Müller M. A., Drosten C., Pöhlmann S., Cell 2020, 181, 271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Xia S., Zhu Y., Liu M., Lan Q., Xu W., Wu Y., Ying T., Liu S., Shi Z., Jiang S., Lu L., Cell. Mol. Immunol. 2020, 17, 765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Moore B. J. B., June C. H., Science 2020, 368, 473. [DOI] [PubMed] [Google Scholar]
- 9. Huang C., Wang Y., Li X., Ren L., Zhao J., Hu Y., Zhang L., Fan G., Xu J., Gu X., Cheng Z., Yu T., Xia J., Wei Y., Wu W., Xie X., Yin W., Li H., Liu M., Xiao Y., Gao H., Guo L., Xie J., Wang G., Jiang R., Gao Z., Jin Q., Wang J., Cao B., Lancet 2020, 395, 497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cao X., Nat. Rev. Immunol. 2020, 20, 269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Liu J., Wan M., Lyon C. J., Hu T. Y., Theranostics 2020, 10, 9591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zhang Q., Gong H., Gao W., Zhang L., CCS Chem. 2020, 2, 376. [Google Scholar]
- 13. Ma Q., Fan Q., Xu J., Bai J., Han X., Dong Z., Zhou X., Liu Z., Gu Z., Wang C., Matter 2020, 3, 287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Dinarello C. A., Cell 2010, 140, 935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Tang Z., Kong N., Zhang X., Liu Y., Hu P., Mou S., Liljeström P., Shi J., Tan W., Kim J. S., Cao Y., Langer R., Leong K. W., Farokhzad O. C., Tao W., Nat. Rev. Mater. 2020, 5, 847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Qin Z., Peng R., Baravik I. K., Liu X., Matter 2020, 3, 628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hu T. Y., Frieman M., Wolfram J., Nat. Nanotechnol. 2020, 15, 247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chen H., Zhang W., Zhu G., Xie J., Chen X., Nat. Rev. Mater. 2017, 2, 17024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Fathi P., Rao L., Chen X., View 2021, 2, 20200187. [Google Scholar]
- 20. Speyer C. L., Ward P. A., J. Invest. Surg. 2011, 24, 18. [DOI] [PubMed] [Google Scholar]
- 21. Medzhitov R., Nature 2007, 449, 819. [DOI] [PubMed] [Google Scholar]
- 22. D'Elia R. V., Harrison K., Oyston P. C., Lukaszewski R. A., Clark G. C., Clin. Vaccine Immunol. 2013, 20, 319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Choy E. H., De Benedetti F., Takeuchi T., Hashizume M., John M. R., Kishimoto T., Nat. Rev. Rheumatol. 2020, 16, 335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zlotnik A., Yoshie O., Nomiyama H., Genome Biol. 2006, 7, 243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ferrara J. L., Abhyankar S., Gilliland D. G., Transplant. Proc. 1993, 25, 1216. [PubMed] [Google Scholar]
- 26. Vaninov N., Nat. Rev. Immunol. 2020, 20, 277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Mehta P., McAuley D. F., Brown M., Sanchez E., Tattersall R. S., Manson J. J., Lancet 2020, 395, 1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Rao L., Bu L.‐L., Meng Q.‐F., Cai B., Deng W.‐W., Li A., Li K., Guo S.‐S., Zhang W.‐F., Liu W., Sun Z.‐J., Zhao X.‐Z., Adv. Funct. Mater. 2017, 27, 1604774. [Google Scholar]
- 29. Chen H., Guo J., Wang C., Luo F., Yu X., Zhang W., Li J., Zhao D., Xu D., Gong Q., Liao J., Yang H., Hou W., Zhang Y., Lancet 2020, 395, 809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Xu Z., Shi L., Wang Y., Zhang J., Huang L., Zhang C., Liu S., Zhao P., Liu H., Zhu L., Tai Y., Bai C., Gao T., Song J., Xia P., Dong J., Zhao J., Wang F.‐S., Lancet Resp. Med. 2020, 8, 420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Shapouri‐Moghaddam A., Mohammadian S., Vazini H., Taghadosi M., Esmaeili S. A., Mardani F., Seifi B., Mohammadi A., Afshari J. T., Sahebkar A., J. Cell Physiol. 2018, 233, 6425. [DOI] [PubMed] [Google Scholar]
- 32. Rao L., Wu L., Liu Z., Tian R., Yu G., Zhou Z., Yang K., Xiong H.‐G., Zhang A., Yu G.‐T., Sun W., Xu H., Guo J., Li A., Chen H., Sun Z.‐J., Fu Y.‐X., Chen X., Nat. Commun. 2020, 11, 4909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Rao L., Zhao S.‐K., Wen C., Tian R., Lin L., Cai B., Sun Y., Kang F., Yang Z., He L., Mu J., Meng Q.‐F., Yao G., Xie N., Chen X., Adv. Mater. 2020, 32, 2004853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Xia Y., Rao L., Yao H., Wang Z., Ning P., Chen X., Adv. Mater. 2020, 32, 2002054. [DOI] [PubMed] [Google Scholar]
- 35. N A. G., Quintana J. A., García‐Silva S., Mazariegos M., González de la Aleja A., Nicolás‐Ávila J. A., Walter W., Adrover J. M., Crainiciuc G., Kuchroo V. K., Rothlin C. V., Peinado H., Castrillo A., Ricote M., Hidalgo A., J. Exp. Med. 2017, 214, 1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Koo S. J., Garg N. J., Redox Biol. 2019, 24, 101198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zhang X., Mosser D. M., J. Pathol. 2008, 214, 161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Channappanavar R., Fehr A. R., Vijay R., Mack M., Zhao J., Meyerholz D. K., Perlman S., Cell Host Microbe 2016, 19, 181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Merad M., Martin J. C., Nat. Rev. Immunol. 2020, 20, 355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kang S., Tanaka T., Narazaki M., Kishimoto T., Immunity 2019, 50, 1007. [DOI] [PubMed] [Google Scholar]
- 41. Imai Y., Kuba K., Neely G. G., Yaghubian‐Malhami R., Perkmann T., van Loo G., Ermolaeva M., Veldhuizen R., Leung Y. H., Wang H., Liu H., Sun Y., Pasparakis M., Kopf M., Mech C., Bavari S., Peiris J. S., Slutsky A. S., Akira S., Hultqvist M., Holmdahl R., Nicholls J., Jiang C., Binder C. J., Penninger J. M., Cell 2008, 133, 235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Berliner J. A., Watson A. D., N. Engl. J. Med. 2005, 353, 9.16000351 [Google Scholar]
- 43. von Brühl M. L., Stark K., Steinhart A., Chandraratne S., Konrad I., Lorenz M., Khandoga A., Tirniceriu A., Coletti R., Köllnberger M., Byrne R. A., Laitinen I., Walch A., Brill A., Pfeiler S., Manukyan D., Braun S., Lange P., Riegger J., Ware J., Eckart A., Haidari S., Rudelius M., Schulz C., Echtler K., Brinkmann V., Schwaiger M., Preissner K. T., Wagner D. D., Mackman N., Engelmann B., Massberg S., J. Exp. Med. 2012, 209, 819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zhou F., Yu T., Du R., Fan G., Liu Y., Liu Z., Xiang J., Wang Y., Song B., Gu X., Guan L., Wei Y., Li H., Wu X., Xu J., Tu S., Zhang Y., Chen H., Cao B., Lancet 2020, 395, 1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Zhou Y., Fu B., Zheng X., Wang D., Zhao C., qi Y., Sun R., Tian Z., Xu X., Wei H., Natl. Sci. Rev. 2020, 7, 998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wang K., Chen W., Zhou Y.‐S., Lian J.‐Q., Zhang Z., Du P., Gong L., Zhang Y., Cui H.‐Y., Geng J.‐J., Wang B., Sun X.‐X., Wang C.‐F., Yang X., Lin P., Deng Y.‐Q., Wei D., Yang X.‐M., Zhu Y.‐M., Zhang K., Zheng Z.‐H., Miao J.‐L., Guo T., Shi Y., Zhang J., Fu L., Wang Q.‐Y., Bian H., Zhu P., Chen Z.‐N., Signal Transduction Targeted Ther. 2020, 5, 283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Liu L., Wei Q., Lin Q., Fang J., Wang H., Kwok H., Tang H., Nishiura K., Peng J., Tan Z., Wu T., Cheung K. W., Chan K. H., Alvarez X., Qin C., Lackner A., Perlman S., Yuen K. Y., Chen Z., JCI Insight 2019, 4, e123158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Vijay R., Hua X., Meyerholz D. K., Miki Y., Yamamoto K., Gelb M., Murakami M., Perlman S., J. Exp. Med. 2015, 212, 1851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Li G., Clercq E. D., Nat. Rev. Drug Discovery 2020, 19, 149. [DOI] [PubMed] [Google Scholar]
- 50. Lang F. M., Lee K. M. C., Teijaro J. R., Becher B., Hamilton J. A., Nat. Rev. Immunol. 2020, 20, 507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Serbina N. V., Pamer E. G., Nat. Immunol. 2006, 7, 311. [DOI] [PubMed] [Google Scholar]
- 52. Blanco‐Melo D., Nilsson‐Payant B. E., Liu W.‐C., Uhl S., Hoagland D., Møller R., Jordan T. X., Oishi K., Panis M., Sachs D., Wang T. T., Schwartz R. E., Lim J. K., Albrecht R. A., tenOever B. R., Cell 2020, 181, 1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Yu G.‐T., Rao L., Wu H., Yang L.‐L., Bu L.‐L., Deng W.‐W., Wu L., Nan X., Zhang W.‐F., Zhao X.‐Z., Liu W., Sun Z.‐J., Adv. Funct. Mater. 2018, 28, 1801389. [Google Scholar]
- 54. Broggi A., Granucci F., Zanoni I., J. Exp. Med. 2020, 217, e20190295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Lohmann N., Schirmer L., Atallah P., Wandel E., Ferrer R. A., Werner C., Simon J. C., Franz S., Freudenberg U., Sci. Transl. Med. 2017, 9, eaai9044. [DOI] [PubMed] [Google Scholar]
- 56. Thamphiwatana S., Angsantikul P., Escajadillo T., Zhang Q., Olson J., Luk B. T., Zhang S., Fang R. H., Gao W., Nizet V., Zhang L., Proc. Natl. Acad. Sci. USA 2017, 114, 11488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Rao L., Xia S., Xu W., Tian R., Yu G., Gu C., Pan P., Meng Q.‐F., Cai X., Qu D., Lu L., Xie Y., Jiang S., Chen X., Proc. Natl. Acad. Sci. USA 2020, 117, 27141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Tang Z., Zhang X., Shu Y., Guo M., Zhang H., Tao W., Nano Today 2021, 36, 101019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Kong N., Tao W., Ling X., Wang J., Xiao Y., Shi S., Ji X., Shajii A., Gan S. T., Kim N. Y., Duda D. G., Xie T., Farokhzad O. C., Shi J., Sci. Transl. Med. 2019, 11, eaaw1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Zhou J., Krishnan N., Jiang Y., Fang R. H., Zhang L., Nano Today 2021, 36, 101031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Tao W., Yurdagul A., Kong N., Li W., Wang X., Doran A. C., Feng C., Wang J., Islam M. A., Farokhzad O. C., Tabas I., Shi J., Sci. Transl. Med. 2020, 12, eaay1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Islam M. A., Xu Y., Tao W., Ubellacker J. M., Lim M., Aum D., Lee G. Y., Zhou K., Zope H., Yu M., Cao W., Oswald J. T., Dinarvand M., Mahmoudi M., Langer R., Kantoff P. W., Farokhzad O. C., Zetter B. R., Shi J., Nat. Biomed. Eng. 2018, 2, 850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Fan W. P., Yung B., Huang P., Chen X. Y., Chem. Rev. 2017, 117, 13566. [DOI] [PubMed] [Google Scholar]
- 64. Langer R., Nature 1998, 392, 5. [PubMed] [Google Scholar]
- 65. Washburn N. R., Prata J. E., Friedrich E. E., Ramadan M. H., Elder A. N., Sun L. T., Biomatter 2013, 3, 25597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Friedrich E. E., Sun L. T., Natesan S., Zamora D. O., Christy R. J., Washburn N. R., J. Biomed. Mater. Res., Part A 2014, 102, 1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Sun L. T., Buchholz K. S., Lotze M. T., Washburn N. R., Mol. Pharmaceutics 2010, 7, 1769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Shupp J. W., Nasabzadeh T. J., Rosenthal D. S., Jordan M. H., Fidler P., Jeng J. C., J. Burn Care Res. 2010, 31, 849. [DOI] [PubMed] [Google Scholar]
- 69. Sun L. T., Friedrich E., Heuslein J. L., Pferdehirt R. E., Dangelo N. M., Natesan S., Christy R. J., Washburn N. R., Wound Repair Regener. 2012, 20, 563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Friedrich E. E., Azofiefa A., Fisch E., Washburn N. R., J. Burn Care Res. 2015, 36, e90. [DOI] [PubMed] [Google Scholar]
- 71. Friedrich E. E., Washburn N. R., Biomaterials 2017, 114, 10. [DOI] [PubMed] [Google Scholar]
- 72. Sun L. T., Bencherif S. A., Gilbert T. W., Lotze M. T., Washburn N. R., Acta Biomater. 2010, 6, 4708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Lima A. C., Cunha C., Carvalho A., Ferreira H., Neves N. M., ACS Appl. Mater. Interfaces 2018, 10, 13839. [DOI] [PubMed] [Google Scholar]
- 74. Fu T., Kong Q., Sheng H., Gao L., Neural Plast. 2016, 2016, 2412958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Campo G. M., Avenoso A., Campo S., D'Ascola A., Traina P., Samà D., Calatroni A., Innate Immun. 2008, 14, 233. [DOI] [PubMed] [Google Scholar]
- 76. Coombe D. R., Immunol. Cell. Biol. 2008, 86, 598. [DOI] [PubMed] [Google Scholar]
- 77. Fan Q., Ma Q., Bai J., Xu J., Fei Z., Dong Z., Maruyama A., Leong K. W., Liu Z., Wang C., Sci. Adv. 2020, 6, eabb4639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Freudenberg U., Atallah P., Limasale Y. D. P., Werner C., Faraday Discuss. 2019, 219, 244. [DOI] [PubMed] [Google Scholar]
- 79. Babazada H., Yamashita F., Yanamoto S., Hashida M., J. Controlled Release 2014, 194, 332. [DOI] [PubMed] [Google Scholar]
- 80. Yanamoto S., Babazada H., Sakai S., Higuchi Y., Yamashita F., Hashida M., Biol. Pharm. Bull. 2017, 40, 540. [DOI] [PubMed] [Google Scholar]
- 81. Nih L. R., Gojgini S., Carmichael S. T., Segura T., Nat. Mater. 2018, 17, 642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Tuladhar A., Shoichet M. S., Nat. Mater. 2018, 17, 573. [DOI] [PubMed] [Google Scholar]
- 83. Wang B., Tan L., Deng D., Lu T., Zhou C., Li Z., Tang Z., Wu Z., Tang H., Int. J. Nanomed. 2015, 10, 3417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Guryanov I., Cipriani S., Fiorucci S., Zashikhina N., Marchianò S., Scarpelli P., Korzhikov‐Vlakh V., Popova E., Korzhikova‐Vlakh E., Biondi B., Formaggio F., Tennikova T., Nanomedicine 2017, 13, 2575. [DOI] [PubMed] [Google Scholar]
- 85. Hu C.‐M. J., Zhang L., Aryal S., Cheung C., Fang R. H., Zhang L., Proc. Natl. Acad. Sci. USA 2011, 108, 10980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Fang R. H., Kroll A. V., Gao W., Zhang L., Adv. Mater. 2018, 30, 1706759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Rao L., Bu L.‐L., Cai B., Xu J.‐H., Li A., Zhang W.‐F., Sun Z.‐J., Guo S.‐S., Liu W., Wang T.‐H., Zhao X.‐Z., Adv. Mater. 2016, 28, 3460. [DOI] [PubMed] [Google Scholar]
- 88. Rao L., Wang W., Meng Q.‐F., Tian M., Cai B., Wang Y., Li A., Zan M., Xiao F., Bu L.‐L., Li G., Li A., Liu Y., Guo S.‐S., Zhao X.‐Z., Wang T.‐H., Liu W., Wu J., Nano Lett. 2019, 19, 2215. [DOI] [PubMed] [Google Scholar]
- 89. Hu C.‐M. J., Fang R. H., Wang K.‐C., Luk B. T., Thamphiwatana S., Dehaini D., Nguyen P., Angsantikul P., Wen C. H., Kroll A. V., Carpenter C., Ramesh M., Qu V., Patel S. H., Zhu J., Shi W., Hofman F. M., Chen T. C., Gao W., Zhang K., Chien S., Zhang L., Nature 2015, 526, 118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Hu C.‐M. J., Fang R. H., Copp J., Luk B. T., Zhang L., Nat. Nanotechnol. 2013, 8, 336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Han X., Shen S., Fan Q., Chen G., Archibong E., Dotti G., Liu Z., Gu Z., Wang C., Sci. Adv. 2019, 5, eaaw6870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Rao L., Tian R., Chen X., ACS Nano 2020, 14, 2569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Zhang Q., Dehaini D., Zhang Y., Zhou J., Chen X., Zhang L., Fang R. H., Gao W., Zhang L., Nat. Nanotechnol. 2018, 13, 1182. [DOI] [PubMed] [Google Scholar]
- 94. Tak P. P., Smeets T. J., Daha M. R., Kluin P. M., Meijers K. A., Brand R., Meinders A. E., Breedveld F. C., Arthritis Rheum. 1997, 40, 217. [DOI] [PubMed] [Google Scholar]
- 95. Rao L., Cai B., Bu L.‐L., Liao Q.‐Q., Guo S.‐S., Zhao X.‐Z., Dong W.‐F., Liu W., ACS Nano 2017, 11, 3496. [DOI] [PubMed] [Google Scholar]
- 96. Hao Z., Wang Z., Li Y., Zhu Y., Wang X., De Moraes C. G., Pan Y., Zhao X., Lin Q., Nanoscale 2018, 10, 21681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Liang C., Guo B., Wu H., Shao N., Li D., Liu J., Dang L., Wang C., Li H., Li S., Lau W. K., Cao Y., Yang Z., Lu C., He X., Au D. W., Pan X., Zhang B. T., Lu C., Zhang H., Yue K., Qian A., Shang P., Xu J., Xiao L., Bian Z., Tan W., Liang Z., He F., Zhang L., Lu A., Zhang G., Nat. Med. 2015, 21, 288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Weissleder R., Pittet M. J., Nature 2008, 452, 580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Yin X., Mead B. E., Safaee H., Langer R., Karp J. M., Levy O., Cell Stem Cell 2016, 18, 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Monteil V., Kwon H., Prado P., Hagelkrüys A., Wimmer R. A., Stahl M., Leopoldi A., Garreta E., Hurtado del Pozo C., Prosper F., Romero J. P., Wirnsberger G., Zhang H., Slutsky A. S., Conder R., Montserrat N., Mirazimi A., Penninger J. M., Cell 2020, 181, 905. [DOI] [PMC free article] [PubMed] [Google Scholar]