

Abstract
Here we report that negatively charged polysulfates can bind to the spike protein of SARS‐CoV‐2 via electrostatic interactions. Using a plaque reduction assay, we compare inhibition of SARS‐CoV‐2 by heparin, pentosan sulfate, linear polyglycerol sulfate (LPGS) and hyperbranched polyglycerol sulfate (HPGS). Highly sulfated LPGS is the optimal inhibitor, with an IC50 of 67 μg mL−1 (approx. 1.6 μm). This synthetic polysulfate exhibits more than 60‐fold higher virus inhibitory activity than heparin (IC50: 4084 μg mL−1), along with much lower anticoagulant activity. Furthermore, in molecular dynamics simulations, we verified that LPGS can bind more strongly to the spike protein than heparin, and that LPGS can interact even more with the spike protein of the new N501Y and E484K variants. Our study demonstrates that the entry of SARS‐CoV‐2 into host cells can be blocked via electrostatic interactions, therefore LPGS can serve as a blueprint for the design of novel viral inhibitors of SARS‐CoV‐2.
Keywords: electrostatic interactions, inhibition, polysulfates, SARS-CoV-2, virus binding
Negatively charged polysulfates can bind to the positively charged patches of RBD of SARS‐CoV‐2 and block viral interaction with host cells for infection inhibition. The synthetic linear polyglycerol sulfate (LPGS) exhibits higher virus inhibitory activity than heparin, but lower anticoagulant activity. Therefore, it is considered to be a promising substitute for heparin in clinical applications.
Introduction
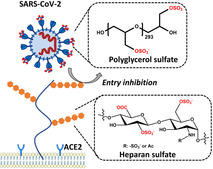
The severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) poses an ongoing major health problem worldwide. [1] Understanding virus attachment and entry into cells is critical for the development of inhibitors. In a number of viruses, electrostatic interactions are essential for the virion's adherence to the cell surface.[ 2 , 3 ] The importance of this process in viral infection has recently been discussed by Cagno et al., [2] who gathered experimental evidence in a large number of viruses (see Table 1 of ref. [2]). Figure 1 schematically displays the first steps of virus entry into cells. Virions first attach to the syndecans and glypicans, which are the most important heparan sulfate proteoglycans (HSPGs) located on the cell surface. [3] Each HSPG consists of a protein and a highly charged glycosaminoglycan (GAG) chain. The negatively charged heparan sulfate (HS) moieties of the HSPG interact with basic patches of the viral capsid proteins. As depicted in Figure 1, viruses exploit this nonspecific electrostatic interaction to increase their concentration at the cell surface and to be transferred to a more specific receptor, that is, the angiotensin‐converting enzyme 2 (ACE2).
Table 1.
Overview of the tested polysulfates.
|
Sample |
Hydrodynamic size [nm][b] |
ζ‐potential [mV] |
IC50 [μg mL−1][c] |
IC50 [μm] |
IC50 [mm ‐SO4 −] |
|---|---|---|---|---|---|
|
LPG20S0.94 [a] |
6.7±3.3 |
−26.1±0.7 |
66.9±32.0 |
1.6±0.8 |
0.4±0.2 |
|
LPG20S0.47 |
6.5±3.8 |
−18.3±0.6 |
679.7±175.7 |
22.7±5.9 |
4.0±1.0 |
|
LPG6S0.81 |
n.d.[d] |
−29.1±0.7 |
>10 000 |
– |
– |
|
HPG10S0.91 |
5.1±2.3 |
−24.9±3.5 |
1909.0±342.3 |
95.4±17.2 |
11.3±2.0 |
|
HPG500S0.85 |
14.3±7.7 |
−21.2±2.3 |
658.5±492.5 |
0.7±0.5 |
3.9±2.9 |
|
HPG2600S0.82 |
34.8±12.2 |
−17.0±1.8 |
>10 000 |
– |
– |
|
Heparin |
10.9±5.3 |
−31.2±1.9 |
4084.0±396.3 |
272.3±26.4 |
2.2±0.6 |
|
Pentosan sulfate |
n.d.[d] |
−29.8±2.3 |
1310.0±292.8 |
– |
2.1±0.5 |
[a] Degree of sulfation determined via elemental analysis. [b] Via DLS. [c] Plaque reduction assay. [d] Not detectable.
Figure 1.
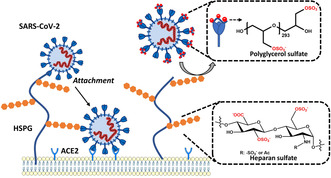
Left: binding of SARS‐CoV‐2 to surface‐exposed heparan sulfate facilitates virus entry; right: competitive binding to soluble synthetic polyglycerol sulfates shields the viral surface and therefore finally reduces infectivity.
Recent studies have furnished evidence that electrostatic interactions are important for the infection of cells by SARS‐CoV‐2. [4] In particular, Kim et al. performed a series of systematic surface plasmon resonance (SPR) studies on the binding of the SARS‐CoV‐2 spike protein to heparin. [5] This research revealed binding constants as low as 40 pm that could be attributed to electrostatic interactions. Moreover, Kwon et al. found that addition of soluble HS inhibits SARS‐CoV‐2 cell infectivity, highlighting the importance of HS for the entry of the virus into host cells. [6] Recent work of Clausen et al. showed that the receptor binding domain (RBD) of SARS‐CoV‐2 exhibits a patch of positive charges on its surface that is considerably larger than in the corresponding RBD of SARS‐CoV. [7] Other recent research demonstrated that the attachment of the spike protein to the HSPG is the first step for virus entry into host cells, as shown in Figure 1. [8] In a second step, the attached virion interacts with ACE2, the actual receptor for the entry of SARS‐CoV‐2.[ 9 , 10 ] The essential role of electrostatic interactions in virus entry provides the principle mode of action for highly charged anionic inhibitors. Heparin was studied intensively in this regard.[ 11 , 12 ] Moreover, synthetic virus inhibitors based on highly charged anionic dendrimers have been investigated intensively.[ 13 , 14 , 15 ]
The work presented here follows our hypothesis that charge–charge interactions are of central importance to inhibit the entry of SARS‐CoV‐2 into cells. As discussed recently,[ 14 , 16 , 17 , 18 ] charge–charge interactions mainly act through counterion release:[ 19 , 20 ] patches of positive charge on the surface of proteins can become multivalent counterions of highly negatively charged polyelectrolytes such as heparin, thus releasing a concomitant number of counterions condensed to the polyelectrolyte into the bulk phase.[ 16 , 17 ] Thus, positively charged patches could be a target for the design of viral‐entry inhibitors. Considering that these positively charged residues are located in close vicinity of the ACE2 binding site of the spike protein, [7] it is envisioned that inhibitors bound to the positively charged patches can interrupt ACE2 binding, leading to virus entry inhibition. [21]
A systematic study of the interactions of the SARS‐CoV‐2 virion with the cell surface requires a detailed investigation of the local interaction of HS with the spike proteins. This problem has become even more urgent considering the new variants of the virus that began to appear in late 2020. It seems that these strains can exhibit a much higher infectivity. [22] The N501Y variant is reported to be more infectious than the wild‐type virus, and a virus carrying this mutation was adapted to infect mice, which cannot be infected by wild‐type SARS‐CoV‐2. [22] The E484K substitution is reported to enable the virus to escape from neutralizing antibodies. [23] Docking studies and MD simulations require only the more easily retrieved data from the spike protein, offering a powerful and accessible tool for assessing these mutations through quantitative computer simulations.
In this study, we systemically assess the potential of different polysulfates as entry inhibitors against SARS‐CoV‐2. Our work combines experimental studies supported by MD simulations: i) Using a plaque reduction assay, we determine the IC50 of various highly sulfated polyelectrolytes.[ 24 , 25 ] We compare two natural polysulfates, namely heparin and pentosan sulfate, with highly sulfated polyglycerols, which present a new class of synthetic inhibitors. By comparing linear polyglycerol sulfate (LPGS) to hyperbranched polyglycerol sulfate (HPGS) at full sulfation, we investigate the influence of molecular weight on inhibitory interaction, as well as the role of architecture in this interaction. ii) MD simulations are used to investigate the binding of mutated spike proteins to LPGS. Here we explore the details of the interaction of HS with the spike protein in order to rationalize our experimental results on inhibition. Moreover, we investigate the consequences of the N501Y and E484K mutations in the spike protein for the virus binding to HS. These investigations aim for a fully quantitative understanding of the inhibition of SARS‐CoV‐2 by polyanions, and further study potential changes of this inhibition that may be caused by novel mutations of the virus's genome.
Results and Discussion
Electrostatic interactions of SARS‐CoV‐2 with host cells. Previous studies have revealed the essential role of electrostatic interactions for SARS‐CoV‐2 infection. In the RBD of SARS‐CoV‐2, five positively charged amino acids are localized next to the ACE2 binding site: R346, R355, K444, R466, and R509. These amino acids form a positively charged patch located at the exterior of the RBD (shown blue in Figure 2 b), which is reported to improve the virus binding affinity to the ACE2 receptor.[ 26 , 27 ] For the new E484K variant, the K484 adds another positive charge to the RBD and is therefore expected to further strengthen viral binding to HS. [23] Recent studies have shown that the positively charged patch contributes to virus binding to cell‐surface HS by facilitating virus docking on the cell surface.[ 3 , 5 ] Based on the finding that cleavage of cell‐surface HS inhibits SARS‐CoV‐2 infection, a two‐step process of SARS‐CoV‐2 was proposed as shown in Figure 1.[ 7 , 8 ] The binding to HS was reported to facilitate the “opening” of the RBD for the binding with ACE2. [7]
Figure 2.
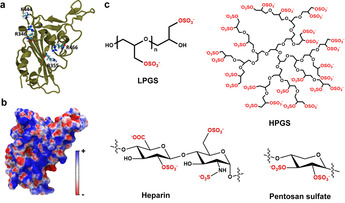
a) Crystal structure of the SARS‐CoV‐2 spike protein RBD (PDB ID: 6M0J) [27] with a few important cationic residues that interact with polyanionic ligands. b) The electrostatic potential map of RBD. c) Schematic illustrations of polyglycerol sulfates in linear and hyperbranched architectures, and of the natural polysulfates, respectively. The negatively charged groups are marked red.
The presence of electrostatic interactions with cell‐surface HS inspired us to test the polysulfates shown in Figure 2 c for SARS‐CoV‐2 inhibition. Heparin is used to prevent blood clotting that is induced by COVID‐19.[ 28 , 29 , 30 ] Evidence has shown that ACE2 binding can be disrupted by heparin, which reveals the potential of heparin as a SARS‐CoV‐2 inhibitor. [21] For virus inhibition, the strong anticoagulant activity of heparin may raise the risk of bleeding for the patients. [31] Heparin‐mimetic polymers with higher virus inhibitory activity and lower anticoagulant activity than heparin are therefore needed for COVID treatment at an earlier stage.
Our group has developed several synthetic polyglycerol sulfates (LPGS and HPGS) as heparin‐mimetic polymers in the past years.[ 32 , 33 ] With similar charge density, the polyglycerol sulfates exhibit lower anticoagulant activity than heparin. [32] Here we studied and compared their activities for the inhibition of SARS‐CoV‐2 with heparin, in an attempt to provide a blueprint for future inhibitor design. The details of synthesis are shown in the Supporting Information. The final product is named as LPG20S0.94 for 94 % sulfated LPG with an approx. 20 kDa precursor.
Inhibition of SARS‐CoV‐2. The inhibition of virus binding was studied by plaque reduction assays with authentic SARS‐CoV‐2 (SARS‐CoV2M; BetaCoV/Germany/BavPat1/2020). [34] In our study, SARS‐CoV‐2 was pre‐treated with the inhibitors and then incubated with Vero E6 cells to assess virus binding. The cells were washed with phosphate‐buffered saline (PBS) to remove unbound virions. Afterwards, the cells were cultured for 48 hours with overlay medium for plaque formation as shown in Figure S3, Supporting Information. Since binding and entry are prerequisite for plaque formation, a plaque reduction assay is an informative way to measure inhibition of virus binding and entry.
Figure 3 a and Table 1 show dose‐dependent virus inhibition activities for the different samples. We first compared virus inhibition between the synthetic polysulfates, the natural polysulfates heparin and pentosan sulfate. As expected, heparin and pentosan sulfate inhibit infection, although the observed inhibitory activity is rather low. The half‐maximal inhibition concentrations (IC50) for heparin and pentosan sulfate are 4084.0±396.3 and 1310±292.8 μg mL−1, respectively. It should also be noted that heparin can completely inhibit blood clotting at levels as low as 5 μg mL−1. LPG20S0.94 shows an IC50 of 66.9±32.0 μg mL−1 (approx. 1.6 μm) and thus a much higher virus inhibitory activity than heparin and pentosan sulfate.
Figure 3.
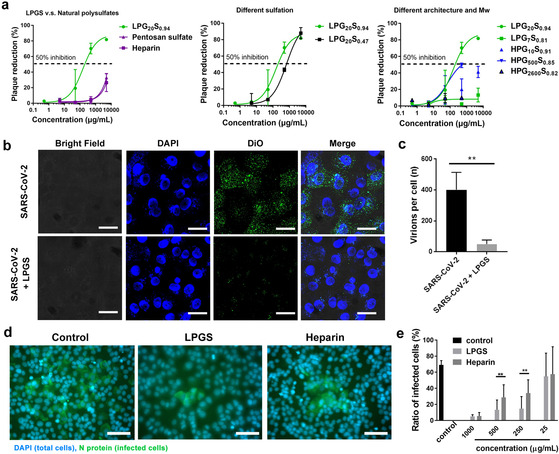
a) Plaque reduction ratios for the samples at different inhibitor doses. Values are expressed as mean ±SD, n=4. Mw shown here refers to the LPG and HPGS precursors. b) CLSM image for the virus binding to Vero E6 cells in presence of LPGS. Scale bar: 10 μm. c) Analysis of virus binding to Vero E6 cells from CLSM images for the number of virions per cells. More detailed images are shown in Figure S4, Supporting Information. d) Immunofluorescent images revealing the infected cells in the post‐infection inhibition assay. The cell nuclei are stained blue, while the infected cells are stained green by antibodies against the nucleocapsid protein (N) of SARS‐CoV‐2. Scale bar: 50 μm. More images are shown in Figure S6, Supporting Information. e) Ratios of infected cells in each group. “LPGS” in (b)–(e) refers to LPG20S0.94. Values are expressed as mean ±SD, n=4. **p<0.01 from Student t‐test.
We also compared the performance of LPG20S with different degrees of sulfation (94 % and 47 %) with respect to virus inhibition. Here, increasing the degree of sulfation increases the inhibitory activity of LPGS to a remarkable extent: LPG20S with 47 % sulfation shows an IC50 of 679.7±175.7 g mL−1, which is 10‐fold higher than the IC50 of almost fully sulfated LPG20S. It is surprising that a two‐fold increase in sulfation caused a ten‐fold improvement in the inhibitory potential. Similar charge‐dependent virus inhibition activity has been reported by Clausen et al. in the study of heparin for SARS‐CoV‐2 inhibition. [7] This strong influence of sulfation on inhibition highlights the importance of the charge density of the inhibitor for virus binding.
To further illustrate the structural factors that affect the virus inhibitory activity, we compared LPGS and HPGS at different molecular weights. We compared the activities of highly sulfated inhibitors (>80 %). For LPGS, only the polysulfates with a starting polymer of 20 kDa inhibit the viruses effectively. LPGS with lower molecular weight shows no virus inhibition, highlighting the importance of inhibitor size for binding the spike protein.
Comparing the activity between LPGS and HPGS with the same molecular weight, it can be concluded that LPGS can inhibit SARS‐CoV‐2 infection more effectively than the hyperbranched polymer. LPG20S0.94 shows the best inhibitory activity among the polysulfates that we tested. Interestingly, LPG20S0.47, which has approx. 137 sulfated groups per molecule, exhibits a two‐fold higher virus inhibitory activity than HPG10S0.91 (approx. 123 sulfated groups per molecule). These two compounds are comparable in size. Zeta potential analysis show that HPG10S0.91 carries even more negative charges than LPG20S0.47. This comparison indicates that molecular architecture is the key parameter for the binding and inhibition of SARS‐CoV‐2, and in our case, LPGS is a more effective compound than HPGS.
For HPGS, we saw maximum inhibition for the 500 kDa molecular weight compound. HPGSs with higher (2600 kDa) and lower (10 kDa) molecular weights show only poor virus inhibition, highlighting the importance of molecular weight for virus binding and inhibition. Even though HPG500S0.85 exhibits a lower IC50 in molar concentration than LPG20S0.94, LPG20S0.94 has a lower IC50 in mass concentration. When normalized to the number of sulfate groups, the IC50 for LPG20S0.94 is 10‐fold lower than HPG500S0.85 (0.4 mm and 3.9 mm for LPG20S0.94 and HPG500S0.85, respectively). Therefore, it is concluded that LPG20S0.94 is better than HPG500S0.85 as a SARS‐CoV‐2 inhibitor.
The structure of HPGS in an aqueous solution can be approximated by a sphere with negative surface charges. [16] LPGS is a linear polymer that can attain multiple conformations and may span larger distances, and can hence conform to larger basic patches on the surface of proteins. Due to its greater backbone flexibility, LPGS can adapt its conformation more easily to the positively charged pockets, resulting in strong binding. HPGS, on the other hand, is a rather rigid spherical structure and cannot adapt its conformation to the binding pocket. Similar results have been obtained in the study of influenza virus and herpes simplex virus inhibitors, where the linear constructs performed much better than hyperbranched constructs at the same molecular weight.[ 32 , 35 ]
A virucidal assay reveals that SARS‐CoV‐2 is not inhibited irreversibly by LPG20S0.94 and heparin (Figure S5, Supporting Information). Upon dilution, the virions are released from the binding, which supports our claim that the LPG20S0.94 inhibits SARS‐CoV‐2 by electrostatic interactions with the spike protein. It is envisioned that a combination with hydrophobic aliphatic chains could result in a virucidal polymer,[ 36 , 37 ] as demonstrated in one of our recent studies. [38]
Competition with host cell for viral binding. After identification of LPG20S0.94 as the most potent inhibitor, we used this compound for further investigations, where we will refer to it simply as LPGS in the following discussions. To demonstrate that polysulfates can compete with cells for viral binding, we acquired fluorescent images of virions binding to Vero E6 cells in the presence of LPGS, as shown in Figure 3 b,c. Without the inhibitor, the viruses bind to cells notably (Figure 3 b). LPGS effectively blocks SARS‐CoV‐2 binding to Vero E6 cells. Automatic image analysis by ImageJ (Figure 3 c) reveals that LPGS causes a >87.5 % inhibition of virus binding. These results confirm the finding of the plaque reduction assays that LPGS can outperform the cell surface for viral binding and can therefore work as a binding decoy to inhibit SARS‐CoV‐2.
Post‐infection study. To demonstrate the therapeutic potential of LPGS, a post‐infection study was carried out. Herein, the cells were first infected by SARS‐CoV‐2 at MOI of 0.01 for 1 h and then cultured in the medium containing LPGS and heparin for 24 h. Finally, the cells were stained by antibodies against the virus as shown in Figure 3 d,e and Figure S6, Supporting Information. At a certain dosage, LPGS and heparin reduce the transmission of SARS‐CoV‐2, because they block the entry of progeny virions into the cells. LPGS shows higher activity than heparin in this test, which is in qualitative agreement with the plaque reduction assays. After all, the fact that LPGS can inhibit viral infection, even when used after the first cycle of infection, supports its potential as therapeutic option of COVID‐19. However, a comprehensive preclinical evaluation is still required for future studies.
Binding with RBD of SARS‐CoV‐2. In order to confirm direct interaction of the inhibitors LPGS and HPGS with the spike protein, we conducted affinity measurements against the RBD using microscale thermophoresis (MST) (Figure 4 a and Table 2). In initial titration experiments against human ACE2, we determined a dissociation constant (K d) of 359 nm. For the synthetic polysulfates LPGS and HPGS, we detected K d values of 5 μm and 141 μm towards the RBD of SARS‐CoV‐2. Heparin showed an affinity of 191 μm. In comparison to the difference in IC50 values for LPGS and heparin, similar K d values for these ligands suggest that the occupation of the HS binding site of the RBD is the inhibitory mechanism. A three times lower affinity of HPGS compared to the IC50 value can be explained by an additional steric contribution of the rather inflexible HPGS sphere.
Figure 4.
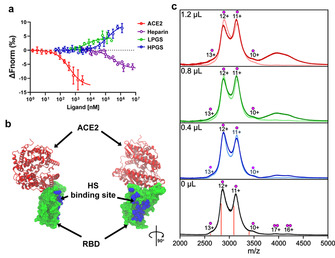
a) Affinity measurements of RBD of wild‐type SARS‐CoV‐2 with LPGS, HPGS, heparin and ACE2 using MST. Each data point represents mean values with N≥4 experiments, and the error bars show the standard deviation. Data points were fitted according to the mass‐action law function to obtain K d values (see Table 1). The differences in the slopes of the dose‐response curves depend on changes of the hydration shell areas and effective charges, but do not affect the determinations of K d‐values from the inflection points of the curves. b) Crystal structure of RBD bound with ACE2 (PDB ID: 6MOJ). ACE2 is shown in secondary structure representation (red), while RBD is shown in surface representation (green). The amino acid residues of RBD (R346, A348, A352, N354, R355, K356, R357, S359, Y396, K444, N450, R466, I468) found in MD simulations to form contacts with the polysulfates are highlighted in VDW representation (blue), denoting the putative HS‐binding site. More detailed images are shown in Figure S7, Supporting Information. c) Mass spectra of 4.0 μL RBD solution mixed with 0, 0.4, 0.8, and 1.2 μL heparin (light traces) or LPGS (dark trace). The charge states are marked with a single dot for the RBD monomer and with a double dot for the RBD dimer, while the calculated m/z for the 10–13+ charge states of the 34 kDa RBD are marked with orange lines.
Table 2.
Summary of dissociation constants (K d) are shown, together with the confidence values (±), indicating with 68 % certainty the range in which K d falls.
|
|
Sample |
K d |
|
|---|---|---|---|
|
|
ACE2 |
359±49 nm |
|
|
|
HPG10S0.91 |
141.9±33.3 μm |
|
|
|
LPG20S0.47 |
5.2±3.6 μm |
|
|
|
Heparin |
191.5±57.7 μm |
It should be noted that LPGS binds to the RBD in close vicinity to the ACE2 binding site. Even though some positively charged amino acids were noticed at the ACE2 binding site, our simulation shows that LPGS binds mostly to the highly positively charged pockets on the side of the RBD, that is, HS binding site (Figure 4 b and Figure S7, Supporting Information).
With LPGS established as the best ligand among the polysulfates tested here, analysis on compound–RBD binding was further conducted using native mass spectrometry, a common technique to study non‐covalent complexes of proteins.[ 39 , 40 ] Figure 4 c shows the results of the mass spectrometry experiments with different amounts of heparin or LPGS added to the RBD solution.
The mass spectrum of the pure RBD exhibits two distinct groups of peaks: the first group in the 2500–3600 m/z range corresponds to the protein monomer in the 10–13+ charge states, while the group in the 3600–4600 m/z range corresponds to the RBD dimer in the 16–17+ charge states. The dimer signals generally exhibit a much lower intensity relative to the region assigned to the RBD monomer (Figure 4 c, bottom spectrum). Furthermore, all peaks are broad and poorly resolved, which suggests heterogeneity that is most probably the result of post‐translational modification. The molecular weight of the pure RBD was calculated to be approx. 34 kDa.
As compared to the pure RBD protein, the addition of unfractionated heparin to the RBD solution did not lead to a substantially different mass spectrum. In a previous native mass spectrometry study, heparin–RBD binding was observed, though not with unfractionated heparin as used here, but rather with a much less heterogeneous, isolated 20mer. [21] However, when we added LPGS to the RBD solution, the overall intensity in the 3600–4600 m/z region increased substantially with increasing LPGS:RBD ratios (Figure 4 c; dark blue, green, and red traces). Given that the addition of LPGS solution lowered the absolute concentration of RBD in the sample, the increased signal intensity in the 3600–4600 m/z region is unlikely the result of increased RBD dimerization. Furthermore, we obtained one large and poorly resolved signal, instead of several at least partially resolved peaks as expected for oligomers. This suggests a high molecular heterogeneity in the species assigned to the same spectral region. The mean molecular weights of LPGS and RBD are approx. 40 kDa and 34 kDa, respectively, and both are highly heterogeneous in weight. The increasing signal intensity in the 3600–4600 m/z region with increasing RBD:LPGS ratios therefore likely arises from the binding of the RBD to LPGS molecules.
MD simulations for investigating interactions of LPGS and heparin with wild‐type RBD. To test the hypothesis that polysulfates inhibit viral infection by electrostatics‐mediated binding to the SARS‐CoV‐2 spike protein, we performed all‐atom MD simulations of the RBD of the spike protein and LPGS/heparin in explicit water (see Figure 5 and Methods in Supporting Information for further details). We found that both LPGS and heparin form complexes with RBD (Figure 5 a,b), the former being completely bound to RBD, whereas a part of the latter is free in solution. The plot of the number of contacts per amino acid residue reveals that both anionic polymers primarily interact with the cationic residues of RBD: namely R355, K356, R357, and R466 (Figure 5 c). Further, we found for LPGS that the total number of contacts with RBD per polymer's molecular weight is 1.6 times the value for heparin (Figure 5 c inset). Normalized to the charge unit, the contact number for LPGS is 3.2 times that of heparin. The absolute value of the total protein–polymer interaction energy is also larger for LPGS as compared to heparin (Figure 5 e). This stronger binding of LPGS to RBD correlates well with its superior virus inhibition efficacy as observed experimentally. It should, however, be noted that because of the well‐known limitations of atomistic simulations, shorter LPGS (undecamer) and heparin (pentamer) were used in simulations compared to the experimental plaque reduction assays (293 and 24 repeating units for LPGS and heparin, respectively). Nevertheless, the simulated systems allow meaningful comparison with the experiments when interpreted in terms of the binding strength per monomer.
Figure 5.
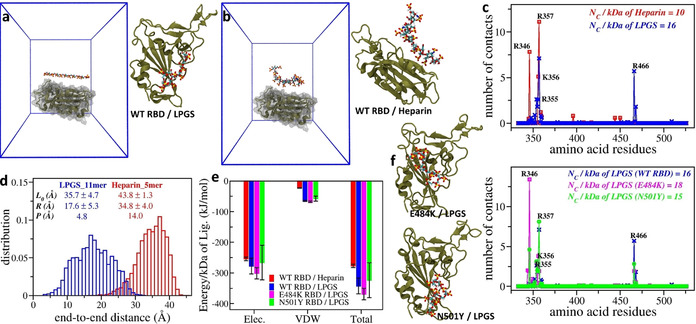
Simulation setup for studying interactions of the RBD of wild‐type SARS‐CoV‐2 with a single a) LPGS undecamer and b) heparin pentamer. The protein is shown in secondary structure representation (tan), whereas polymers are shown in ball and stick representation with each atom type colored differently (hydrogen in white, carbon in cyan, oxygen in red, and sulfur in yellow). Water molecules and ions are omitted for clarity. To the right, snapshots after 500 ns of MD simulations are shown for a) RBD–LPGS and b) RBD–heparin complexes. c) The number of contacts LPGS and heparin forms with each residue of wild‐type RBD averaged over the last 100 ns simulation time. d) End‐to‐end distance distributions for LPGS and heparin free in aqueous solutions, which reveal the different flexibility of the polymers. Relevant parameters of the polymers are given in the inset; see text for details. e) Comparison of interaction energies for the different polymers and RBD variants. The electrostatic (Elec.) and van der Waals (VDW) contributions to the total interaction energy for each protein–polymer complex are given. f) The number of contacts LPGS forms with each residue of the different RBD mutants. To the left, snapshots after 500 ns of MD simulations are provided, representing the complex formation of LPGS with each RBD mutant.
To understand this surprising, stronger binding affinity of LPGS despite its smaller linear charge density relative to heparin, we characterized the flexibility of both polymers in terms of their end‐to‐end distances (R) and persistence lengths (P). As shown in Figure 5 d, the R distribution for LPGS is wider than that for heparin, implying a higher flexibility of LPGS. From the mean values of R and contour length (L 0) of the polymers, we estimated P of LPGS to be 3 times smaller than that of heparin (see Figure 5 d, inset for all the values and see Supporting Information for the calculation details). The higher flexibility of LPGS helps adjust its conformation to the heterogeneous surface topography of RBD, which in turn leads to its enhanced binding. Therefore, in the future design of polymers that bind to SARS‐CoV‐2 spike protein, both the backbone flexibility and the charge density of polymers should be simultaneously optimized for effective binding to spike proteins.
MD simulations of interactions of LPGS with RBD mutants. The successful comparison between experimental and simulated results for wild‐type RBD–LPGS interactions encouraged us to indirectly test the effectiveness of LPGS in inhibiting SARS‐CoV‐2 mutants via simulations, specifically those exhibiting the E484K and N501Y mutations in the RBD. [22] We found that LPGS forms complexes with both RBD mutants (Figure 5 f). As in the case of wild‐type RBD, LPGS interacts mostly with the mutants’ cationic residues, as indicated in the per‐residue contacts plot (Figure 5 f). From the total number of contacts (Figure 5 f, inset) and interaction energies (Figure 5 e), we found that LPGS binds to N501Y RBD as effectively as to wild‐type RBD, but more tightly to E484K RBD, which is consistent with the presence of an extra cationic residue on this mutant surface. The results of our MD simulations thus suggest that LPGS could also work successfully in inhibiting SARS‐CoV‐2 mutants.
Biosafety evaluations. To further exclude the side effects of cellular toxicity for virus inhibition, we tested the inhibitors with three different cell lines, including Vero E6, A549, and human bronchial epithelial (HBE) cells. As shown in Figure S8, Supporting Information, LPGS did not show any cellular toxicity up to a dose of 10 mg mL−1, revealing a half‐maximal cytotoxicity concentration (CC50) value higher than 10 mg mL−1. Selectivity index was calculated by comparing IC50 with CC50. LPGS yielded a selectivity index higher than 150, affirming the potential of LPGS for preclinical testing.
Furthermore, the anticoagulation activity of LPGS was investigated by activated partial thromboplastin time (aPTT), as shown in Figure S9, Supporting Information. With similar charge density, LPGS shows much lower anticoagulation activity than heparin. At a concentration of 5 μg mL−1, heparin leads to complete anticoagulation of plasma, while a concentration of 25 μg mL−1 of LPGS is required to yield a similar effect. This variance is caused by different core structures. Heparin can bind specifically and strongly to antithrombin and inhibit blood coagulation.[ 41 , 42 ] Earlier studies of heparin‐mimicking polymers indicated an important role of saccharide units in anticoagulation activities.[ 43 , 44 ] Glycerol‐based polymers therefore have a weaker anticoagulant effect than heparin.
Conclusion
In this study we investigated the inhibition of SARS‐CoV‐2 by polysulfates of different sources (natural and synthetic), different architectures (linear and hyperbranched), different molecular weights (7 kDa to 2.6 MDa) and different degrees of sulfation (approx. 100 % and 50 %) by authentic SARS‐CoV‐2 plaque reduction assays. Using MD simulations, we demonstrated that the positively charged patch near the RBD of SARS‐CoV‐2 is responsible for the binding of the spike protein to the HSPG located on the cell surface. The optimal inhibitor, LPG20S0.94, showed an IC50 of 67±32 μg mL−1 in a plaque reduction assay. Its inhibitory activity is roughly 61‐fold higher than heparin's.
We also showed that architecture, molecular weight, molecular flexibility, and sulfation can influence SARS‐CoV‐2 binding and inhibition. For future design of SARS‐CoV‐2 inhibitors, these factors should be carefully considered and evaluated for the rational design. With the MD simulations, we were further able to demonstrate that LPGS can bind to the RBD of virus variants, and conclude that LPGS might inhibit infection by variants carrying the E484K and N501Y mutations. Further experiments are needed to elucidate the structural details in RBD variations and their impact on infectivity and inhibitor binding.
The key information from our study is that synthetic polysulfates can inhibit SARS‐CoV‐2 by electrostatic interaction. Our MST and MS analyses, along with MD simulations, show a strong interaction with the spike protein. In our view, the application of polysulfates in the fight against SARS‐CoV‐2 can be diverse. Both the development of a novel pharmaceutical for SARS‐CoV‐2 inhibition and a virus‐clearing surface coating may be possible. For a pharmaceutical application, even though we have shown the in vitro activity of LPGS, the biosafety and effectiveness need a more detailed investigation and evaluation in an animal model. In our previous studies, it has been demonstrated that HPGS is safe for mice, but LPGS has not yet been systemically studied. [45]
There is a growing need for antiviral surfaces. It is envisioned that LPGS coating could help to reduce the transmission of SARS‐CoV‐2 via surfaces. To transfer LPGS onto a surface, layer‐by‐layer assembly via electrostatic interactions can be considered; this process has shown good practicality in the modification of biomedical devices. [46] Another potential method is mussel‐inspired coatings. Our group has developed a mussel‐inspired polyglycerol coating that can be applied to diverse surfaces.[ 47 , 48 ] LPGS can be similarly modified to be a universal coating material.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This work was supported by the Berlin University Alliance (BUA) and by the Focus Area NanoScale (Freie Universität Berlin). The authors thank the Core Facility BioSupraMol for the use of their research facilities. Elisa Quaas, Cathleen Hudziak, and Marleen Selent are thanked for assistance in biological assays, GPC and HPLC measurements, respectively. R.H. acknowledges the support of the Deutsche Forschungsgemeinschaft (DFG) and the Bundesministerium für Bildung und Forschung (BMBF). A.K.S. and R.N. acknowledge support from the Max‐Planck MaxWater initiative. K.P., R.N., M.B., and R.H. acknowledge the support from International Research Training Group (IRTG)‐2262. We also thank Prof. Heberle and Dr. Schlesinger (Freie Universität Berlin) for access to the Monolith NT.115 instrument. We acknowledge the rapid supply of SARS‐CoV‐2 RBD from the lab of Dr. Coskun (TU Dresden), and ACE2 protein from Prof. Bader at MDC Berlin. Open access funding enabled and organized by Projekt DEAL.
C. Nie, P. Pouyan, D. Lauster, J. Trimpert, Y. Kerkhoff, G. P. Szekeres, M. Wallert, S. Block, A. K. Sahoo, J. Dernedde, K. Pagel, B. B. Kaufer, R. R. Netz, M. Ballauff, R. Haag, Angew. Chem. Int. Ed. 2021, 60, 15870.
In memory of Professor Klaus Hafner
A previous version of this manuscript has been deposited on a preprint server (https://doi.org/10.26434/chemrxiv.14074070).
Contributor Information
Dr. Chuanxiong Nie, Email: chuanxnie@zedat.fu-berlin.de.
Dr. Gergo Peter Szekeres, Email: gpszekeres@fhi-berlin.mpg.de.
Dr. Anil Kumar Sahoo, Email: 201992kumarsahoo@gmail.com.
Prof. Dr. Rainer Haag, Email: Haag@zedat.fu-berlin.de.
References
- 1. Zhu N., Zhang D., Wang W., Li X., Yang B., Song J., Zhao X., Huang B., Shi W., Lu R., Niu P., Zhan F., Ma X., Wang D., Xu W., Wu G., Gao G. F., Tan W., N. Engl. J. Med. 2020, 382, 727–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Cagno V., Tseligka E. D., Jones S. T., Tapparel C., Viruses 2019, 11, 596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Koehler M., Delguste M., Sieben C., Gillet L., Alsteens D., Annu. Rev. Virol. 2020, 7, 143–165. [DOI] [PubMed] [Google Scholar]
- 4. Tree J. A., Turnbull J. E., Buttigieg K. R., Elmore M. J., Coombes N., Hogwood J., Mycroft-West C. J., Lima M. A., Skidmore M. A., Karlsson R., Chen Y.-H., Yang Z., Spalluto C. M., Staples K. J., Yates E. A., Gray E., Singh D., Wilkinson T., Page C. P., Carroll M. W., Br. J. Pharmacol. 2021, 178, 626–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kim S. Y., Jin W., Sood A., Montgomery D. W., Grant O. C., Fuster M. M., Fu L., Dordick J. S., Woods R. J., Zhang F., Linhardt R. J., Antiviral Res. 2020, 181, 104873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kwon P. S., Oh H., Kwon S.-J., Jin W., Zhang F., Fraser K., Hong J. J., Linhardt R. J., Dordick J. S., Cell Discovery 2020, 6, 50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Clausen T. M., Sandoval D. R., Spliid C. B., Pihl J., Perrett H. R., Painter C. D., Narayanan A., Majowicz S. A., Kwong E. M., McVicar R. N., Thacker B. E., Glass C. A., Yang Z., Torres J. L., Golden G. J., Bartels P. L., Porell R. N., Garretson A. F., Laubach L., Feldman J., Yin X., Pu Y., Hauser B. M., Caradonna T. M., Kellman B. P., Martino C., Gordts P. L. S. M., Chanda S. K., Schmidt A. G., Godula K., Leibel S. L., Jose J., Corbett K. D., Ward A. B., Carlin A. F., Esko J. D., Cell 2020, 183, 1043–1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chu H., Hu B., Huang X., Chai Y., Zhou D., Wang Y., Shuai H., Yang D., Hou Y., Zhang X., Yuen T. T.-T., Cai J.-P., Zhang A. J., Zhou J., Yuan S., To K. K.-W., Chan I. H.-Y., Sit K.-Y., Foo D. C.-C., Wong I. Y.-H., Ng A. T.-L., Cheung T. T., Law S. Y.-K., Au W.-K., Brindley M. A., Chen Z., Kok K.-H., Chan J. F.-W., Yuen K.-Y., Nat. Commun. 2021, 12, 134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hoffmann M., Kleine-Weber H., Schroeder S., Krüger N., Herrler T., Erichsen S., Schiergens T. S., Herrler G., Wu N.-H., Nitsche A., Müller M. A., Drosten C., Pöhlmann S., Cell 2020, 181, 271–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Shang J., Wan Y., Luo C., Ye G., Geng Q., Auerbach A., Li F., Proc. Natl. Acad. Sci. USA 2020, 117, 11727–11734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Nahmias A. J., Kibrick S., J. Bacteriol. 1964, 87, 1060–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Baba M., Pauwels R., Balzarini J., Arnout J., Desmyter J., De Clercq E., Proc. Natl. Acad. Sci. USA 1988, 85, 6132–6136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Donskyi I., Drüke M., Silberreis K., Lauster D., Ludwig K., Kühne C., Unger W., Böttcher C., Herrmann A., Dernedde J., Adeli M., Haag R., Small 2018, 14, 1800189. [DOI] [PubMed] [Google Scholar]
- 14. Xu X., Ran Q., Dey P., Nikam R., Haag R., Ballauff M., Dzubiella J., Biomacromolecules 2018, 19, 409–416. [DOI] [PubMed] [Google Scholar]
- 15. Ziem B., Rahn J., Donskyi I., Silberreis K., Cuellar L., Dernedde J., Keil G., Mettenleiter T. C., Haag R., Macromol. Biosci. 2017, 17, 1600499. [DOI] [PubMed] [Google Scholar]
- 16. Achazi K., Haag R., Ballauff M., Dernedde J., Kizhakkedathu J. N., Maysinger D., Multhaup G., Angew. Chem. Int. Ed. 2021, 60, 3882–3904; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2021, 133, 3926–3950. [Google Scholar]
- 17. Xu X., Angioletti-Uberti S., Lu Y., Dzubiella J., Ballauff M., Langmuir 2019, 35, 5373–5391. [DOI] [PubMed] [Google Scholar]
- 18. Xu X., Ballauff M., J. Phys. Chem. B 2019, 123, 8222–8231. [DOI] [PubMed] [Google Scholar]
- 19. M. T. Record Jr , Anderson C. F., Lohman T. M., Q. Rev. Biophys. 1978, 11, 103–178. [DOI] [PubMed] [Google Scholar]
- 20. Manning G. S., Q. Rev. Biophys. 1978, 11, 179–246. [DOI] [PubMed] [Google Scholar]
- 21. Yang Y., Du Y., Kaltashov I. A., Anal. Chem. 2020, 92, 10930–10934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gu H., Chen Q., Yang G., He L., Fan H., Deng Y.-Q., Wang Y., Teng Y., Zhao Z., Cui Y., Li Y., Li X.-F., Li J., Zhang N.-N., Yang X., Chen S., Guo Y., Zhao G., Wang X., Luo D.-Y., Wang H., Yang X., Li Y., Han G., He Y., Zhou X., Geng S., Sheng X., Jiang S., Sun S., Qin C.-F., Zhou Y., Science 2020, 369, 1603–1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Andreano E., Piccini G., Licastro D., Casalino L., Johnson N. V., Paciello I., Monego S. D., Pantano E., Manganaro N., Manenti A., Manna R., Casa E., Hyseni I., Benincasa L., Montomoli E., Amaro R. E., McLellan J. S., Rappuoli R., bioRxiv 2020, 10.1101/2020.12.28.424451. [DOI] [Google Scholar]
- 24. Cao Y., Su B., Guo X., Sun W., Deng Y., Bao L., Zhu Q., Zhang X., Zheng Y., Geng C., Chai X., He R., Li X., Lv Q., Zhu H., Deng W., Xu Y., Wang Y., Qiao L., Tan Y., Song L., Wang G., Du X., Gao N., Liu J., Xiao J., Su X.-d., Du Z., Feng Y., Qin C., Qin C., Jin R., Xie X. S., Cell 2020, 182, 73–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wang C., Li W., Drabek D., Okba N. M. A., van Haperen R., Osterhaus A. D. M. E., van Kuppeveld F. J. M., Haagmans B. L., Grosveld F., Bosch B.-J., Nat. Commun. 2020, 11, 2251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Amin M., Sorour M. K., Kasry A., J. Phys. Chem. Lett. 2020, 11, 4897–4900. [DOI] [PubMed] [Google Scholar]
- 27. Lan J., Ge J., Yu J., Shan S., Zhou H., Fan S., Zhang Q., Shi X., Wang Q., Zhang L., Wang X., Nature 2020, 581, 215–220. [DOI] [PubMed] [Google Scholar]
- 28. Thachil J., J. Thromb. Haemostasis 2020, 18, 1020–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Paolisso P., Bergamaschi L., D′Angelo E. C., Donati F., Giannella M., Tedeschi S., Pascale R., Bartoletti M., Tesini G., Biffi M., Cosmi B., Pizzi C., Viale P., Galié N., Front. Pharmacol. 2020, 11, 1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lemos A. C. B., do Espírito Santo D. A., Salvetti M. C., Gilio R. N., Agra L. B., Pazin-Filho A., Miranda C. H., Thromb. Res. 2020, 196, 359–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Oduah E. I., Linhardt R. J., Sharfstein S. T., Pharmaceuticals 2016, 9, 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Pouyan P., Nie C., Bhatia S., Wedepohl S., Achazi K., Osterrieder N., Haag R., Biomacromolecules 2021, 22, 1545–1554. [DOI] [PubMed] [Google Scholar]
- 33. Wallert M., Nie C., Anilkumar P., Abbina S., Bhatia S., Ludwig K., Kizhakkedathu J. N., Haag R., Block S., Small 2020, 16, 2004635. [DOI] [PubMed] [Google Scholar]
- 34. Wölfel R., Corman V. M., Guggemos W., Seilmaier M., Zange S., Müller M. A., Niemeyer D., Jones T. C., Vollmar P., Rothe C., Hoelscher M., Bleicker T., Brünink S., Schneider J., Ehmann R., Zwirglmaier K., Drosten C., Wendtner C., Nature 2020, 581, 465–469. [DOI] [PubMed] [Google Scholar]
- 35. Bhatia S., Lauster D., Bardua M., Ludwig K., Angioletti-Uberti S., Popp N., Hoffmann U., Paulus F., Budt M., Stadtmüller M., Wolff T., Hamann A., Böttcher C., Herrmann A., Haag R., Biomaterials 2017, 138, 22–34. [DOI] [PubMed] [Google Scholar]
- 36. Jones S. T., Cagno V., Janeček M., Ortiz D., Gasilova N., Piret J., Gasbarri M., Constant D. A., Han Y., Vuković L., Král P., Kaiser L., Huang S., Constant S., Kirkegaard K., Boivin G., Stellacci F., Tapparel C., Sci. Adv. 2020, 6, eaax9318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Cagno V., Andreozzi P., D′Alicarnasso M., Jacob Silva P., Mueller M., Galloux M., Le Goffic R., Jones S. T., Vallino M., Hodek J., Weber J., Sen S., Janeček E.-R., Bekdemir A., Sanavio B., Martinelli C., Donalisio M., Rameix Welti M.-A., Eleouet J.-F., Han Y., Kaiser L., Vukovic L., Tapparel C., Král P., Krol S., Lembo D., Stellacci F., Nat. Mater. 2018, 17, 195–203. [DOI] [PubMed] [Google Scholar]
- 38. Donskyi I. S., Nie C., Ludwig K., Trimpert J., Ahmed R., Quaas E., Achazi K., Radnik J., Adeli M., Haag R., Osterrieder K., Small 2021, 17, 2007091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wang G., Chaihu L., Tian M., Shao X., Dai R., de Jong R. N., Ugurlar D., Gros P., Heck A. J. R., Anal. Chem. 2020, 92, 15799–15805. [DOI] [PubMed] [Google Scholar]
- 40. Benesch J. L. P., Ruotolo B. T., Simmons D. A., Robinson C. V., Chem. Rev. 2007, 107, 3544–3567. [DOI] [PubMed] [Google Scholar]
- 41. Rosenberg R. D., N. Engl. J. Med. 1975, 292, 146–151. [DOI] [PubMed] [Google Scholar]
- 42. Jin L., Abrahams J. P., Skinner R., Petitou M., Pike R. N., Carrell R. W., Proc. Natl. Acad. Sci. USA 1997, 94, 14683–14688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Türk H., Haag R., Alban S., Bioconjugate Chem. 2004, 15, 162–167. [DOI] [PubMed] [Google Scholar]
- 44. Ma L., Huang J., Zhu X., Zhu B., Wang L., Zhao W., Qiu L., Song B., Zhao C., Yan F., Int. J. Biol. Macromol. 2019, 122, 784–792. [DOI] [PubMed] [Google Scholar]
- 45. Dernedde J., Rausch A., Weinhart M., Enders S., Tauber R., Licha K., Schirner M., Zügel U., von Bonin A., Haag R., Proc. Natl. Acad. Sci. USA 2010, 107, 19679–19684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Tang Z., Wang Y., Podsiadlo P., Kotov N. A., Adv. Mater. 2006, 18, 3203–3224. [Google Scholar]
- 47. Wei Q., Achazi K., Liebe H., Schulz A., Noeske P.-L. M., Grunwald I., Haag R., Angew. Chem. Int. Ed. 2014, 53, 11650–11655; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 11834–11840. [Google Scholar]
- 48. Wei Q., Haag R., Mater. Horiz. 2015, 2, 567–577. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary